 Joseph D. Lykins1†‡
Joseph D. Lykins1†‡ Ekaterina V. Filippova2†‡
Ekaterina V. Filippova2†‡ Andrei S. Halavaty2†George Minasov2†Ying Zhou3†Ievgeniia Dubrovska2Kristin J. Flores2Ludmilla A. Shuvalova2Jiapeng Ruan2‡
Andrei S. Halavaty2†George Minasov2†Ying Zhou3†Ievgeniia Dubrovska2Kristin J. Flores2Ludmilla A. Shuvalova2Jiapeng Ruan2‡ Kamal El Bissati3Sarah Dovgin4‡
Kamal El Bissati3Sarah Dovgin4‡ Craig W. Roberts5Stuart Woods5
Craig W. Roberts5Stuart Woods5 Jon D. Moulton6Hong Moulton7
Jon D. Moulton6Hong Moulton7 Martin J. McPhillie8‡
Martin J. McPhillie8‡ Stephen P. Muench9Colin W. G. Fishwick10Elisabetta Sabini2Dhanasekaran Shanmugam11
Stephen P. Muench9Colin W. G. Fishwick10Elisabetta Sabini2Dhanasekaran Shanmugam11 David S. Roos12Rima McLeod3,13*Wayne F. Anderson2*
David S. Roos12Rima McLeod3,13*Wayne F. Anderson2* Huân M. Ngô2,14*
Huân M. Ngô2,14*- 1Pritzker School of Medicine, University of Chicago, Chicago, IL, United States
- 2Center for Structural Genomics of Infectious Diseases and the Department of Biochemistry and Molecular Genetics, Feinberg School of Medicine, Northwestern University, Chicago, IL, United States
- 3Department of Ophthalmology and Visual Sciences, University of Chicago, Chicago, IL, United States
- 4Illinois Math and Science Academy, Aurora, IL, United States
- 5Strathclyde Institute of Pharmacy and Biomedical Sciences, University of Strathclyde, Glasgow, United Kingdom
- 6Gene Tools, LLC, Philomath, OR, United States
- 7Department of Biomedical Sciences, College of Veterinary Medicine, Oregon State University, Corvallis, OR, United States
- 8Department of Molecular Biology and Biotechnology, University of Sheffield, Sheffield, United Kingdom
- 9School of Biomedical Sciences, Faculty of Biological Sciences, and Astbury Centre for Structural Molecular Biology, University of Leeds, Leeds, United Kingdom
- 10School of Chemistry and Astbury Centre for Structural Molecular Biology, University of Leeds, Leeds, United Kingdom
- 11Biochemical Sciences Division, CSIR National Chemical Laboratory, Pune, India
- 12Department of Biology, University of Pennsylvania, Philadelphia, PA, United States
- 13Department of Pediatrics (Infectious Diseases), Institute of Genomics, Genetics, and Systems Biology, Global Health Center, Toxoplasmosis Center, CHeSS, The College, University of Chicago, Chicago, IL, United States
- 14BrainMicro LLC, New Haven, CT, United States
Toxoplasma gondii, an Apicomplexan parasite, causes significant morbidity and mortality, including severe disease in immunocompromised hosts and devastating congenital disease, with no effective treatment for the bradyzoite stage. To address this, we used the Tropical Disease Research database, crystallography, molecular modeling, and antisense to identify and characterize a range of potential therapeutic targets for toxoplasmosis. Phosphoglycerate mutase II (PGMII), nucleoside diphosphate kinase (NDK), ribulose phosphate 3-epimerase (RPE), ribose-5-phosphate isomerase (RPI), and ornithine aminotransferase (OAT) were structurally characterized. Crystallography revealed insights into the overall structure, protein oligomeric states and molecular details of active sites important for ligand recognition. Literature and molecular modeling suggested potential inhibitors and druggability. The targets were further studied with vivoPMO to interrupt enzyme synthesis, identifying the targets as potentially important to parasitic replication and, therefore, of therapeutic interest. Targeted vivoPMO resulted in statistically significant perturbation of parasite replication without concomitant host cell toxicity, consistent with a previous CRISPR/Cas9 screen showing PGM, RPE, and RPI contribute to parasite fitness. PGM, RPE, and RPI have the greatest promise for affecting replication in tachyzoites. These targets are shared between other medically important parasites and may have wider therapeutic potential.
Introduction
Toxoplasma gondii is one of the most significant parasites that impacts human health, with estimates that as many as one third to one half of the human population are infected (Montoya and Liesenfeld, 2004; Furtado et al., 2011; Torgerson and Mastroiacovo, 2013; Flegr et al., 2014; McLeod et al., 2014; Lykins et al., 2016). A relative of the parasite that causes malaria, T. gondii is an intracellular parasite that has two major life stages in humans, tachyzoites and bradyzoites. Tachyzoites cause acute infection, while bradyzoites are the encysted, dormant life stage responsible for reactivation disease. While treatment is available for the acute infection, there is currently no effective medication for the bradyzoite stage (McLeod et al., 2014). Additionally, parasites can be passed to a fetus in utero when a pregnant woman is acutely infected during gestation. This can cause chorioretinitis and neurological complications in the fetus (McLeod et al., 2012). Moreover, there is increasing understanding of the potential long-term sequelae of chronic infection with T. gondii on risk of neurodegenerative disease and malignancy (Ngô et al., 2017). Treatment for active infection exists but is limited by toxicity and hypersensitivity. Thus, new therapeutic targets and medicines are needed, with several potential solutions in development (Zhou et al., 2014; McPhillie et al., 2016; Sidik et al., 2016).
At the Center for Structural Genomics of Infectious Diseases (CSGID), the first Toxoplasma Structural Genomics Pipeline was established. Subsequently, CSGID began selecting parasite proteins for structural characterization using established approaches capable of successful identification of potential drug targets, coupled with the Tropical Diseases Research (TDR) Database (Anderson, 2009; Crowther et al., 2010; Magariños et al., 2012). Herein, 5 soluble enzymes were selected for further study. This process was made possible due to the integration of large amounts of genomic, biochemical, and pharmacological data by the TDR Database, which provides evidence collectively generated by the scientific community concerning potential molecular targets and inhibitory compounds that have properties consistent with Lipinski's rules for orally available drugs (Lipinski, 2004). The targets studied herein were crystallized and their structures characterized, as structural studies have potential to inform molecular targeting and medicinal chemistry can facilitate development of novel anti-parasitic compounds.
We further hypothesized that using phosphorodiamidate morpholino oligomers linked to a cellular delivery moiety, such as either an octaguanidinium dendrimer [Vivo-Morpholinos (vivoPMOs)], or arginine-rich peptide, we would decrease expression of these enzymes, identified as potential drug targets by the Toxoplasma Structural Genomic Pipeline, in YFP-expressing T. gondii tachyzoites, and that down-regulation of these enzymes would result in decreased replication as quantified by fluorescent intensity. The approach of using morpholinos to target specific parasitic enzymes has been successful in previous studies (Lai et al., 2012; McPhillie et al., 2016). VivoPMOs are typically used to decrease gene expression by one of two different mechanisms, namely mechanical disruption of interactions between RNA and snRNP, thereby preventing splicing of introns, resulting in nonsense-mediated decay of the transcript and/or defective protein upon translation, and through direct prevention of translation by blocking interactions between mature mRNA and the ribosome. In preventing effective protein expression, we could determine whether a particular enzyme contributed to parasite replication, suggesting its potential as a therapeutic target.
Molecular transporters can deliver PMOs and small inhibitory molecules of therapeutic value. Transductive peptides or octaguanidinium dendrimer of a Vivo-Morpholino (Gene Tools, Philomath, Oregon) deliver PMOs or other molecules across cell membranes. Octaarginine can carry small molecules into the retina (McLeod et al., 2013). Similar arginine-rich cell-penetrating peptides can access other places where medication transport is problematic; for example, rabies virus glycoprotein-tagged small molecules are capable of passing through the blood-brain barrier and octaarginine-conjugated small molecules, for example, cross into encysted bradyzoites (Samuel et al., 2003; Liu et al., 2009).
The enzymes selected from the TDR database as small and tractable for expression and crystallization included: phosphoglycerate mutase II (hereafter referred to as PGM), nucleotide diphosphate kinase (NDK), ribulose phosphate 3-epimerase (RPE), ribose-5-phosphate isomerase (RPI), and ornithine aminotransferase (OAT). Information about candidate inhibitors of these apicomplexan enzymes is summarized in Table 1.
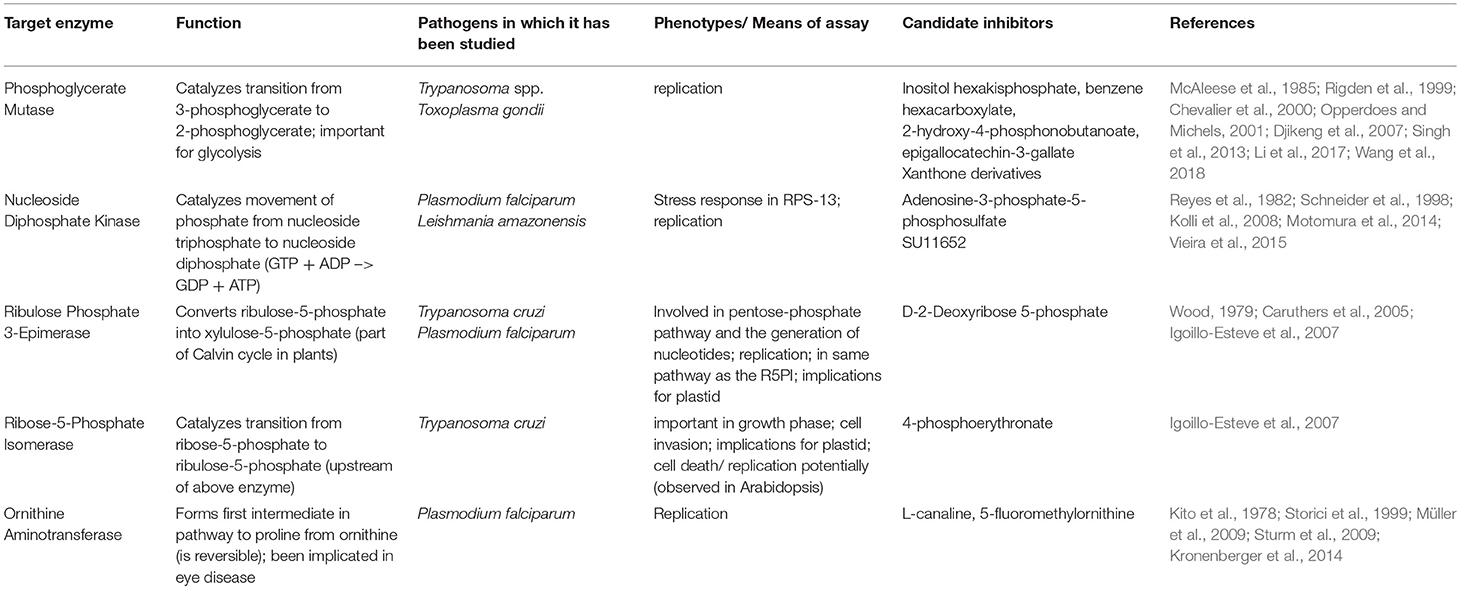
Table 1. Target enzyme characterization and candidate inhibitors.
Methods
Cloning, Expression, and Purification
Five genes from T. gondii ME49 (GI: 237843677, 237844373, 237835673, 237834547, and 237832613) corresponding to a putative phosphoglycerate mutase II (TgPGM; residues 1–265 of the original CSGID entry; this entry was modified in 2012; see original NCBI Reference Sequence: XP_002371136.1), a putative nucleoside diphosphate kinase (TgNDK) (residues 1–155), ribulose phosphate 3-epimerase (TgRPE) (residues 1–230), a putative ribose 5-phosphate isomerase (TgRPI) (residues 1–259), and a putative ornithine aminotransferase (TgOAT) (residues 17–441), respectively, were cloned into the ligation-independent-cloning (Aslanidis and de Jong, 1990) isopropyl β-D-1-thiogalactopyranoside (IPTG)-inducible pMCSG28 vector for recombinant bacterial expression in BL21(DE3)/pMagic Escherichia coli (E. coli). All clones were obtained from J. Craig Venter Institute, a CSGID partner, and submitted into the CSGID protein production pipeline (Figure 1) as IDP92076 (TgPGM), IDP92074 (TgNDK), IDP92047 (TgRPE), IDP92040 (TgRPI), and IDP92102 (TgOAT). The pMCSG28 vector possesses a purification tag including 6 × His affinity sequence and a Tobacco Etch Virus (TEV) protease cleavage site. The E. coli cells were induced with 1 mM IPTG at 25°C after the optical density of cells in culture flasks reached 0.6 at 600 nm under 37°C and constant aeration at 200 rpm. Terrific Broth (TB) (PGM, NDK, and RPE) and the Se-Met MCSG-M9 (Medicilon Inc.) (RPI) medium was used. Overnight induction was completed by collecting cells at 6,000 rpm, 4°C for 10 min. Cells' paste was resuspended in chilled Lysis Buffer [43 mM Na2HPO4, 3.25 mM citric acid, 250 mM NaCl, 100 mM ammonium sulfate, 5% glycerol, 5 mM imidazole, 1.5 mM magnesium acetate, 1 mM CaCl2, 0.08% n-Dodecyl β-D-maltoside (DDM), 5 mM β-mercaptoethanol (BME)] pH 7.8 followed by sonication on ice. Crude sonication mixture was centrifuged at 19,000 rpm, 4°C for 40 min to obtain soluble fraction containing target protein, which was applied onto a 5-ml Ni-NTA column (GE Healthcare, Piscataway, NJ) for purification. The column was washed with buffer containing 10 mM Tris-HCl pH 8.3, 500 mM NaCl, 25 mM imidazole and 5 mM BME to remove non-specifically bound E. coli proteins, followed by elution of target protein with 500 mM imidazole in the 10 mM Tris-HCl buffer pH 8.3 containing 500 mM NaCl and 5 mM BME (buffer A). A HiLoad™ 26/60 Superdex™ 200 column (GE Healthcare, Piscataway, NJ) was used to further purify target protein in the buffer A. Purity of all proteins was analyzed by SDS-PAGE. Pooled fractions were concentrated and stored at −80°C for further use or used in screening crystallization conditions.
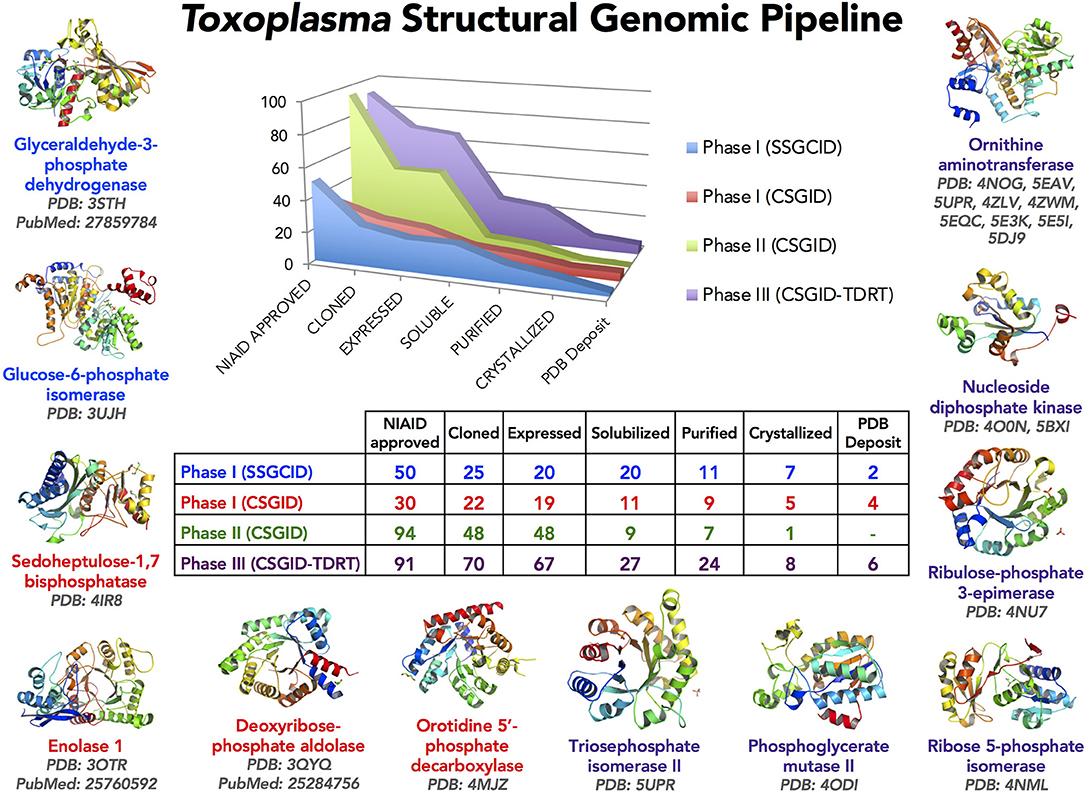
Figure 1. The Toxoplasma Structural Genomics Pipeline identified promising targets with good predicted druggability (Phase III) and outstanding interest to the Toxoplasma research community (Phase I, II). Toxoplasma proteins were selected for Phase I from published work up to 2009 and communicated with the research laboratories. Phase II included proteins from published and unpublished studies suggested by international researchers at the end of 2011. Phase III candidates were selected from the Tropical Diseases Research Database (TDRT). Proteins judged very unlikely to crystallize based on analysis by XtalPred were eliminated (Slabinski et al., 2007). Several structures solved have already been described (Ruan et al., 2015; Tonkin et al., 2015; Dubey et al., 2017). This crystallography pipeline remains available for production and solution of structures of proteins for scientists in the Toxoplasma research community.
Crystallization, X-Ray Data Collection, and Structure Determination
Each target protein was crystallized by the vapor-diffusion sitting-drop method mixing 1 μL of the protein in buffer A and 1 μL of a crystallization screen solution at 22°C. Single crystals were soaked in a crystallization condition for cryoprotection and flash frozen in liquid nitrogen for monochromatic X-ray data collection. Data were collected at 100 K from a single frozen crystal at the LS-CAT beamline 21-ID-F (λ = 0.97872 Å) at the Argonne National Laboratory (ANL), Advanced Photon Source (APS). Diffraction images were collected in oscillation mode and processed with HKL-3000 (Minor et al., 2006). Crystal structures of TgPGM, TgNDK, TgRPE, and TgOAT were determined by molecular replacement using Phaser (McCoy et al., 2007) from CCP4 suite (Winn et al., 2011). Initial molecular replacement solutions were rebuilt with ARP/wARP (Morris et al., 2003). Crystal structure of TgRPI was determined by single-wavelength anomalous dispersion (SAD) method in HKL-3000 (Minor et al., 2006). Non-Crystallographic Symmetry (NCS) restrains and Translation-Libration-Screw (TLS) groups refinement in REFMAC v.5.7 (Murshudov et al., 2011) were used to improve the quality of the structures. Coot (Emsley and Cowtan, 2004; Emsley et al., 2010) was used to manually check structures after each cycle of refinement in REFMAC and correct for side chain rotamers and fitting. The final models were validated with the Protein Data Bank (PDB) validation server (ADIT validation server; https://validate-rcsb-2.wwpdb.org/) and MolProbity (http://molprobity.biochem.duke.edu/) (Davis et al., 2007; Chen et al., 2010). Figures presenting crystal structures were prepared in the graphical program PyMol (The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC). Crystallization conditions, data-collection, structure determination and refinement statistics are summarized in Table 2. Crystal structures are described in Figures 2–6.
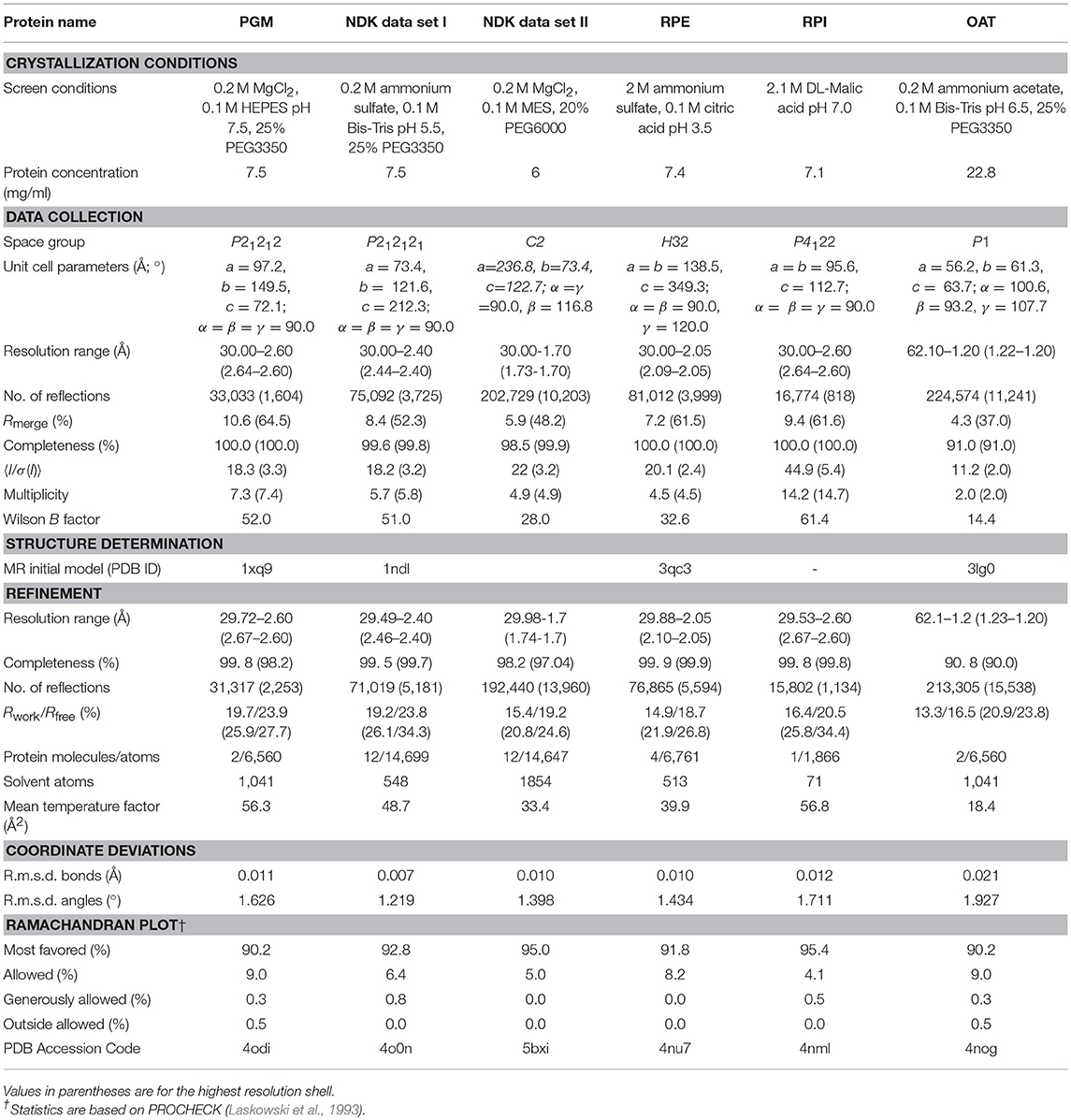
Table 2. Crystallization conditions, data-collection, structure determination and refinement statistics of T. gondii proteins.
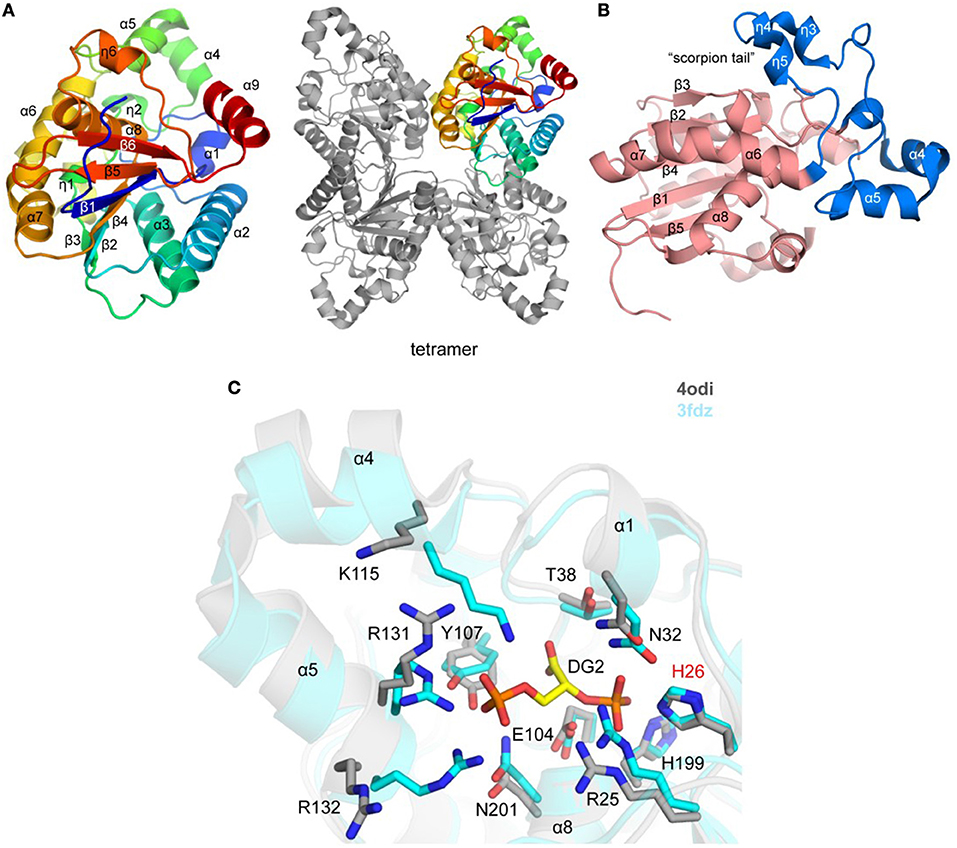
Figure 2. Crystal structure of TgPGM (A). Ribbon representation of TgPGM monomer (left side) colored blue (N-terminus) to red (C-terminus) and tetramer of TgPGM (right side) (B). Domain structure of TgPGM [red—an α/β/α-sandwich domain (residues 12–107 and 169–257) and blue—a domain without any defined folding motif (residues 108–168)] (C). Pairwise structural alignment of TgPGM (gray) and BpPGM (cyan) showing active site with residues of TgPGM shown in sticks and labeled in one-letter code. Equivalent residues of BpPGM are displayed. BpPGM binds (2R)-2,3-diphosphoglyceric acid, DG2, in the active site.
Data Accessibility
Coordinates and structure factors of the determined crystal structures were deposited to Protein Data Bank (www.rcsb.org) (Berman et al., 2000). Diffraction images for the deposited crystal structures can be found at the CSGID website (http://www.csgid.org/csgid/pages/home).
Crystal Structure Data Analysis
We used CATH/Gene3D v4.1 (http://www.cathdb.info/) (Sillitoe et al., 2015; Lam et al., 2016) to assess domain composition of studied proteins. Oligomeric state of the proteins and buried surface area (BSA) of multimers were estimated from crystal structure coordinates using PISA (http://www.ebi.ac.uk/pdbe/pisa/pistart.html) (Krissinel and Henrick, 2005, 2007; Krissinel, 2009). The number of intermolecular bonds that may contribute to protein quaternary structure was estimated from the structure coordinates using PDBsum (http://www.ebi.ac.uk/thornton-srv/databases/cgi-bin/pdbsum/GetPage.pl?pdbcode=index.html) (de Beer et al., 2014). Assessment of metal binding sites was done with help of the CheckMyMetal server (http://csgid.org/csgid/metal_sites) (Zheng et al., 2017). Protein structure comparison service PDBeFold at European Bioinformatics Institute (http://www.ebi.ac.uk/msd-srv/ssm) was used to identify structural homologs (Krissinel and Henrick, 2004). Pair-wise structural alignments were performed with DaliLite v.3 (https://www.ebi.ac.uk/Tools/structure/dalilite/) (Hasegawa and Holm, 2009).
Molecular Modeling
Schrödinger 2015-3, maestro v10.3 was used for binding site identification (SiteMap) on the five enzymes, and therefore, predict “druggability” by small molecules (Schrödinger Release 2015-3, 2018). A SiteScore threshold of 0.80 (with > 0.80 suggesting druggability) is used in this regard (Halgren, 2007).
Cell Culture
Human foreskin fibroblasts (HFF) were maintained in Iscove's Modified Dulbecco's Medium (IMDM) supplemented with 10% fetal bovine serum (FBS) and PENSTRA (penicillin and streptomycin). Cells were maintained at 37°C and 5% CO2.
VivoPMO Design
The vivoPMOs were designed using genomic DNA sequences obtained from ToxoDB (accession numbers for genomic DNA sequences: PGM-TGME49_297060, NDK-TGME49_295350, RPE-TGME49_047670, RPI- TGME49_039310, OAT-TGME49_069110) with exon/intron junctions identified. One of these junctions was identified in each target gene and a vivoPMO was designed to be complementary to nucleotides on both sides of said junction. A diagrammatic representation of vivoPMO structure and RNA binding is in Figure 7A. The sequences of these morpholinos can be found in Figure 7B.
VivoPMO Efficacy Assay
HFFs were grown in black, flat-bottomed 96-well microplates. HFFs were infected with 3,200 Type I RH parasites expressing yellow fluorescent protein (YFP). This allowed quantification of parasites in vitro post-treatment with vivoPMO. The parasites were incubated with the cells for 1 h, to allow sufficient time for invasion of HFFs, and were then treated with vivoPMO. Control triplicates with only fibroblasts and with pyrimethamine and sulfadiazine (the current standard of treatment for T. gondii infection) were also conducted. A concentration gradient of YFP parasites was also established, allowing quantification of knockdown. Several replicates of this efficacy assay were completed applying different concentrations of vivoPMO (2.5, 5, 10, and 20 μM). The cells and parasites were then incubated at 37°C for 72 h. This timing was previously established in other work. Fluorescence was measured using a Bio-Tek SynergyTM H4 Hybrid Multi-Mode Microplate Reader. This methodology was consistent with previous work using morpholinos in T. gondii (Lai et al., 2012).
VivoPMO Toxicity Assay
HFFs were grown in 96-well microplates, as in the efficacy assay. A gradient of dimethyl sulfoxide (DMSO) was used to quantify the amount of cell death caused by the vivoPMO in vitro. Different concentrations of vivoPMO (3.5, 5, 10, and 20 μM) were used to identify the level at which toxicity occurred. Following 72 h of incubation at 37°C, each well was treated with 10 μL WST-1, which reacts in metabolically active, viable cells through a complex set of chemical reactions dependent upon glycolytic NADPH production to form formazan dyes, which can be detected via a colorimeter. This method is also consistent with previous work (Lai et al., 2012).
Data Analysis of Knockdown With vivoPMO
Knockdown was analyzed statistically using student T-test comparing parasite fluorescence between enzyme-specific vivoPMO and off-target vivoPMO. Additionally, the enzyme-specific vivoPMOs were compared to the levels of fluorescence at the standardized parasite load (2000 YFP-expressing parasites per well). Student T-test was also used to analyze toxicity data, comparing levels of optical density at 420 nm for untreated fibroblasts to treated cells. Statistical analysis was performed using STATA. Results were considered significant with p < 0.05.
Antibody Production
Antibodies were raised in mice at the University of Strathclyde (CR, SW). Briefly, mice were given two injections of formulated protein with NISV (non-ionic surfactant vesicle). The vesicles were made by melting mono-palmitoyl glycerol, cholesterol and dicetyl-phosphate (All from Sigma, UK) in a molar ratio of 5:4:1. 10 days after the final injection, serum was collected and tested by Western Blot using recombinant protein and Toxoplasma lysate.
Immunofluorescence Assay (IFA)
HFF cells were grown to confluence on sterilized coverslips in 24-well plates. Cells were fixed in 3% paraformaldehyde ~20–24 h after infection with Type I RH-strain tachyzoites and permeabilized in 0.25% Triton X-100. Serum from immunized mice, coupled with another primary antibody, RPS13, was applied at 1:500 dilution in PBS 1x/3% BSA/Triton-X-100 and detected using either Texas Red-conjugated goat anti-mouse antibody, or Alexa-488–conjugated goat anti-rabbit antibody. DAPI was used to stain for DNA. Coverslips were mounted with Antifade (Molecular Probes, Eugene, OR), and images were analyzed by high-resolution fluorescence using deconvolution protocols. Microscopy was performed with an inverted microscope (IX81; Olympus).
Comparison to Predicted Essentiality Via Literature CRISPR/Cas9 Screen
As an approach to identify whether target enzymes might be essential to parasite viability and fitness, a survey was done of phenotypic scores previously published in a genome-wide CRISPR/Cas9 screen (Sidik et al., 2016). Gene IDs were used and the average of the three available scores was taken. Negative scores were considered likely to be significant contributors to parasite fitness. Data of the genome-wide CRISPR/Cas9 screen are subsequently annotated in ToxoDB (http://toxodb.org/toxo/), including these gene IDs: TgPGM: TGME49_297060, TgNDK: TGME49_295350, TgRPE: TGME49_247670, TgRPI: TGME49_239310, TgOAT: TGME49_269110.
Results
Structural Genomic Pipeline for Toxoplasma
The NIAID Structural Genomics Centers selected 265 proteins for the Toxoplasma Structural Genomic Pipeline in three phases (Figure 1). Phase I and II were a collaborative effort between CSGID and the Toxoplasma research community. Proteins were selected from published and unpublished work up to 2011. Selection was based on the mechanisms and pathways that are important for parasite infection and survival in human and animal hosts. Protein sequences were analyzed by XtalPred and final selection was also based on their crystallization feasibility (Slabinski et al., 2007).
Phase III utilized the TDR Database (Anderson, 2009; Crowther et al., 2010; Magariños et al., 2012). TDR integrated and weighed candidate drug targets based on extensive genetic, biochemical, pharmacologic, compound desirability and computationally-predicted druggability characteristics. Herein, 5 soluble enzymes were selected for further study. The genes encoding these enzymes were cloned, proteins expressed and purified, crystallized, and crystal structures were determined (Figures 2–6). The crystal structures have been deposited in the PDB database in accordance with CSGID and NIH policies. PDB codes can be found in Figure 1 and Table 2.
Phosphoglycerate Mutase II (PGM)
The crystal structure of a putative TgPGM1 was determined and refined to 2.6 Å resolution (PDB 4odi) (Table 2 and Figure 2A). The P21212 asymmetric unit comprises four PGM chains [~0.4 Å root-mean-square-deviation (r.m.s.d.) over 240 Cα atoms] assembled in two dimers with BSA of ~1,900 Å2, while four chains bury a surface area of ~ 6,800 Å2 (Figure 2A). Eleven salt bridges, 15 hydrogen-bonded and 236 van der Waals interactions hold the tetramer in place. A sodium ion-binding site (e.g., NA 301/A) was identified during structure refinement. Tyr155, Val158, and Asn160 coordinate NA 301/A. Residues 12–257 (chain A), 15–259 (chain B), 15–257 (chain C), and 17–248 (chain D) are present in the deposited structure.
TgPGM has an α/β/α-sandwich domain that comprises residues 12–107 and 169–257 (Figure 2A). The domain has a six-stranded central β-sheet flanked by helices α1, α2, α3, α6, α7, α8, and α9, and 310-helices η1 and η6. Residues 108–168 (helices α4 and α5, and four 310 helices; η2–η5) flank the α/β/α domain without any defined folding motif (Figure 2B). Lys115 and Arg132 of this motif comprise a portion of the active site and together with Arg25, His26, Asn32, Thr38, Glu104, Tyr107, His199, and Asn201of the α/β/α domain shape the entrance to the active site (Figure 2C). An interesting feature of the structure is a “scorpion tail” segment (residues 136–168 belonging to 310-helices η3, η4, and η5) that lies on top of the α/β/α domain (Figure 2B). Position of this segment within the crystal lattice suggests that it affects oligomerization and crystal packing. The tip of the tail possesses the sodium ion-binding site.
The structure of the Plasmodium falciparum PGM (Pf PGM; PDB 3kkk; DOI: 10.2210/pdb3KKK/pdb) is the closest homolog of TgPGM with estimated 75% sequence identity and ~0.6 Å r.m.s.d. over 229 Cα atoms. The structure of the homologous bacterial enzyme from Burkholderia pseudomallei (PDB 3fdz; 67% sequence identity; r.m.s.d. ~1.0 Å over 230 Cα atoms) determined in the complex with (2R)-2,3-diphosphoglyceric reveals that TgPGM possesses identical active site residues with an essential phospho-acceptor residue His26 (Davies et al., 2011). Preliminary validation of this active site in Schrödinger SiteMap gave a favorable score of 0.98. Structural alignment of BpPGM and TgPGM demonstrates significant main- and side-chain conformational changes at the protein active site that would most likely take place in TgPGM during the interconversion of its substrate 3-phosphoglycerate to 2-phosphoglycerate (Figure 2C). Conformational differences were also observed for residues located on the tip of the scorpion-like tail.
Nucleoside Diphosphate Kinase (NDK)
Crystal structures of a putative TgNDK were determined and refined to 2.4 Å (PDB 4o0n) and 1.7 Å (PDB 5bxi) resolution (Figure 3A and Table 2). Monomers in both TgNDK structures are superimposed with ~0.2–0.7 Å r.m.s.d. over 153 Cα atoms, while monomers in 4o0n align with ~0.2–0.7 Å r.m.s.d (153 Cα atoms) and monomers in 5bxi align with ~0.2–0.6 Å r.m.s.d (153 Cα atoms). In both structures, TgNDK forms a hexamer with an average BSA of ~18,900 Å2 (Figure 3A). Comparison of TgNDK with known crystal structures of homologs proteins suggests that the TgNDK hexamer is likely a biologically functional assembly (Min et al., 2002; Vieira et al., 2015).
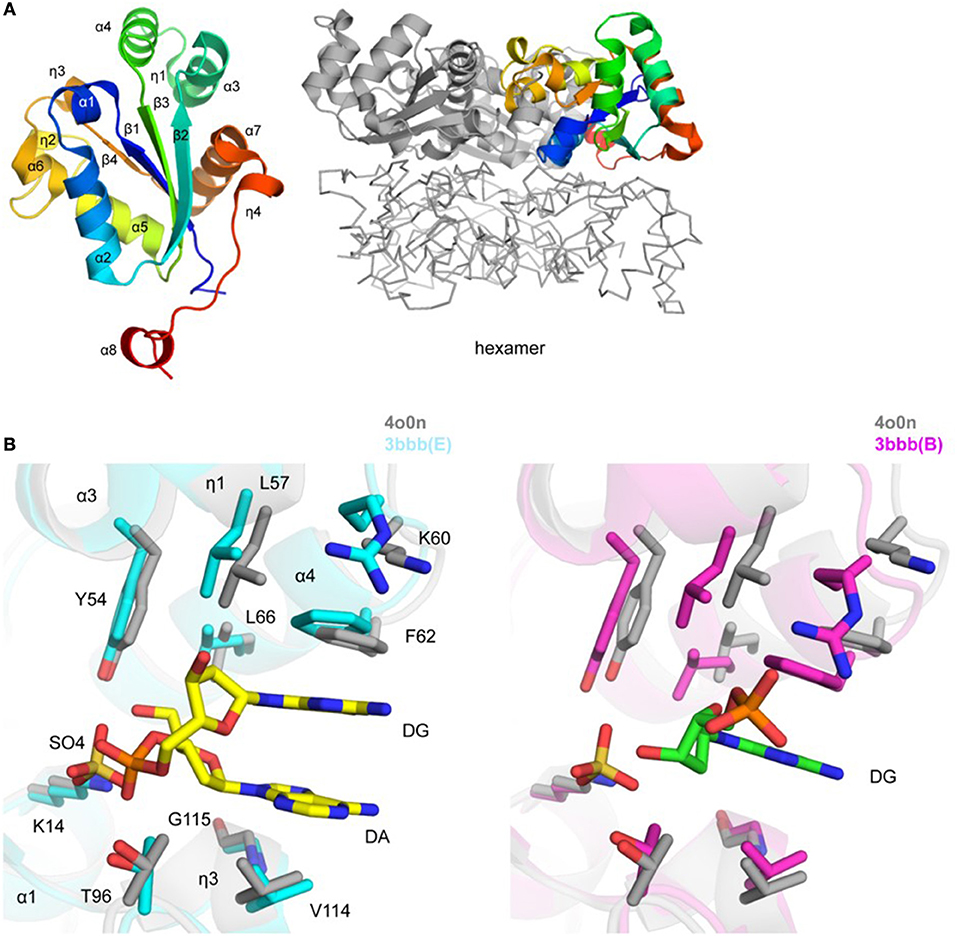
Figure 3. Crystal structure of TgNDK (A). Ribbon representation of TgNDK monomer (left side) colored blue (N-terminus) to red (C-terminus) and trimeric and hexameric assemblies of TgNDK (middle and right side) (B). Pairwise structural alignment of TgPGM (gray; SO4 is shown in sticks) and human NM23-H2 transcription factor [cyan (chain E with bound 2′-deoxyguanosine-5′-monophosphate (DG) and 2′-deoxyadenosine-5′-monophosphate (DA)] and magenta [chain B with bound 2′-deoxyguanosine-5′-monophosphate (DG)] showing active site with residues of TgNDK shown in sticks and labeled in one-letter code. Equivalent residues of human NM23-H2 transcription factor are displayed.
Residues 156–160 (GENLY) of the C-terminal tag in chains D and G only of the 5bxi structure were modeled, while they are absent in all chains of the 4o0n structure. These residues protrude to the solvent and make contacts with symmetry-related hexamer. The side chain of Tyr160 is spatially positioned in a nucleotide base-binding pocket of the active site of a symmetry-related molecule (not shown). In addition, residues 56–64 (DLKGKPFFP; chain C of 5bxi) that belong to the C-terminus of helix α3, 310 helix η1, η1–α4 loop, and the N-terminus of helix α4 are disordered. This peptide stretch may constitute a portion of a nucleotide base-binding pocket of the active site. Superposition of all twelve chains in 5bxi and 4o0n structures revealed that this segment has similar secondary structure conformations. Presumably, lack of favorable crystal contacts resulted in its disorder in chain C of the 5bxi structure.
TgNDK adopts an α/β/α sandwich fold with four β strands and eight surrounding α helices (Figure 3A). The putative active site of TgNDK was identified based on a pairwise structural alignment with the crystal structure of human NM23-H2 transcription factor in complex with the dinucleotide d(AG) (PDB 3bbb; Dexheimer et al., 2009). The active site comprises residues Lys14, Tyr54, Leu57, Lys60, Phe62, Leu66, Thr96, Val114, and Gly115 (Figure 3B), where analysis by Schrödinger SiteMap revealed a score of 0.99 (using PDB 4o0n), indicating a “druggable” pocket. Crystals of TgNDK (PDB 4o0n) grew in the presence of ammonium sulfate and, thus, several sulfate ions were identified and modeled during refinement. Superposition of TgNDK with the structure of human NM23-H2 transcription factor (~67% homology; ~0.6 Å r.m.s.d. over 148 Cα atoms) revealed that a sulfate ion (e.g., SO4 201/A) binds close to a phosphate-binding pocket of the active site (Figure 3B). A bicarbonate ion (e.g., BCT 201/A) was modeled at a similar location in the 5bxi structure. Similar main- and side-chain rearrangements of residues of the active site as seen in NM23-H2 are expected in TgNDK upon binding its substrate.
Ribulose Phosphate 3-Epimerase (RPE)
Crystal structure of TgRPE was determined and refined to 2.05 Å resolution (PDB 4nu7; Table 2 and Figure 4A). TgRPE adopts an aldolase-type TIM-barrel α/β fold resembling homologous RPEs. Four chains of TgRPE (~0.3–0.5 Å r.m.s.d. over 225 Cα atoms) in the H32 asymmetric unit form two dimers (A–C and B–D; Figure 4A) each burying a surface area of ~3,500 Å2. The dimer is similar to one observed for known RPEs (Williamson and Wood, 1966; Jelakovic et al., 2003; Caruthers et al., 2005; Liang et al., 2011). Helices α1 and α2, and the β2–α2 and η1– α1 loops of TgRPE comprise the dimer interface with 4 salt bridges, 8 hydrogen-bonded and 130 van der Waals contacts (e.g., between chains A and C). PISA prediction and crystal packing analysis revealed a hexameric assembly of TgRPE (BSA is ~13,300 Å2; not shown) that has been reported previously for homologous RPEs (Kopp et al., 1999; Wise et al., 2004; Akana et al., 2006).
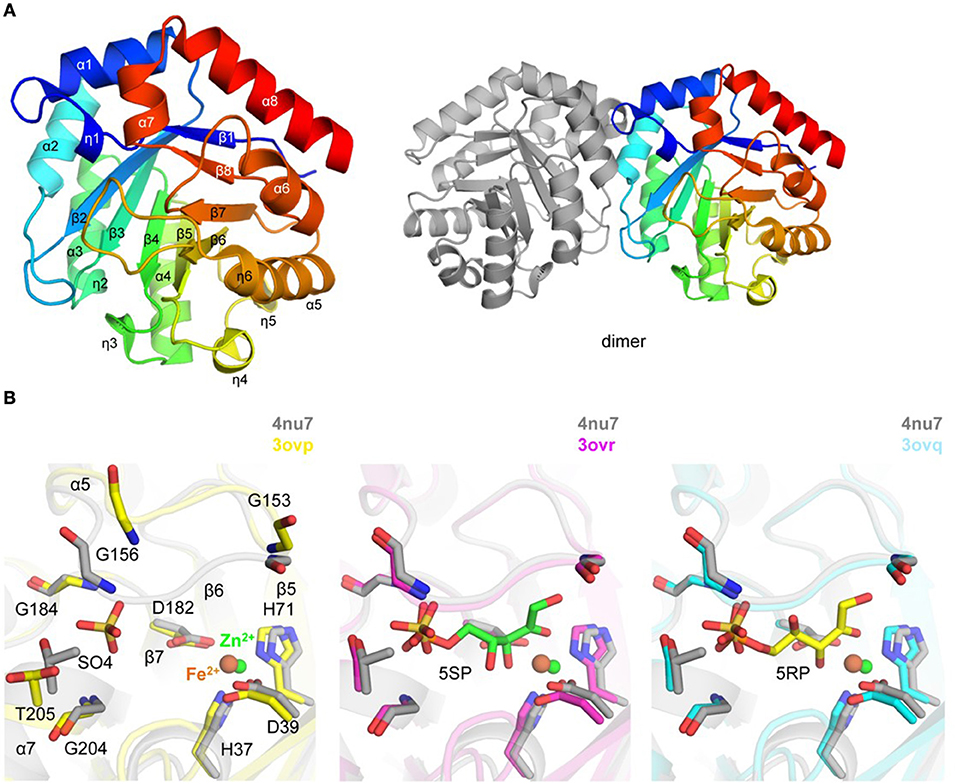
Figure 4. Crystal structure of TgRPE (A). Ribbon representation of TgRPE monomer (left side) colored blue (N-terminus) to red (C-terminus) and TgRPE dimer (right side) (B). Pairwise structural alignment of TgRPE (gray; SO4 is shown in sticks and Zn2+ as green sphere) and human RPE with bound Fe2+ (yellow ribbon), 5-O-phosphono-D-xylulose (5SR; magenta ribbon), and ribulose-5-phosphate (5RP; cyan ribbon) showing active site with residues of TgRPE are shown in sticks and labeled in one-letter code. Equivalent residues of human RPE are displayed.
A Zn2+ (e.g., ZN 303/A) ion was modeled per each chain of TgRPE, with His37, Asp39, His71, Asp182 and a water molecule coordinating the metal (Figure 4B). Similar coordination has been reported for D-xylulose 5-phosphate co-crystallized with human RPE enzyme (PDB: 3ovr) (Liang et al., 2011) (Figure 4B). PDBeFold identified human RPE as the closest homolog of TgRPE (~0.9 Å r.m.s.d. over 215 Cα atoms; 52% sequence identity). The CheckMyMetal analysis identified that Co2+ and Cu2+ may also bind in a similar position as zinc in TgRPE. It is known that the RPE enzymes can utilize Fe2+, Co2+, and Mn2+ for catalysis (Jelakovic et al., 2003; Wise et al., 2004; Caruthers et al., 2005; Akana et al., 2006; Liang et al., 2011). Residues of the metal binding site are strictly conserved in the RPE enzymes. Structural superposition revealed that Fe2+ in ferrous-, substrate- and product-bound human RPE structures is ~1.0 Å away from the position of Zn2+ in TgRPE. We also compared TgRPE structure with two other RPEs structures (PDBs 1tqx and 1h1z) (Jelakovic et al., 2003; Caruthers et al., 2005) that have zinc and bound in similar locations as TgRPE. Positions of zinc and side chains of the Zn2+-coordinating residues are similar in the three compared structures (not shown). Other RPE structures with bound Zn2+ and other than ligands (e.g., PDBs 2fli, 3qc3, and 5umf) have similar positions of Zn2+ and side chains of the metal-binding residues (Akana et al., 2006; Joint Center for Structural Genomics, 2011; Dranow et al., 2017). Additional experiments are needed to confirm the biological importance of zinc in the TgRPE-based catalysis. This metal-binding region was analyzed for “druggability” and scored 1.04 (Schrödinger SiteMap).
Crystals of TgRPE grew in the presence of 500 mM NaCl and 2 M ammonium sulfate and, thus multiple sulfate and chloride ions were modeled to interpret additional electron density. Structure comparison analysis revealed that a sulfate ion (e.g., SO4 304/A) in TgRPE occupies the binding site of a phosphate moiety of substrate or product observed in the structure of the human RPE enzyme (PDBs 3ovq and 3ovr) (Figure 4B). In all ligand-bound human RPE structures, a loop (e.g., the β6–α5 loop in TgRPE) supports position of a ligand at the active site, i.e., moves from its ligand-free conformation to cap a ligand bound to the enzyme. Thus, similar loop movement is expected for TgRPE upon binding of the substrate, i.e., a ligand. Apparently, binding of sufficed the loop repositioning in TgRPE (Figure 4B).
Ribose 5-Phosphate Isomerase (RPI)
Crystal structure of a putative TgRPI was determined and refined to 2.6 Å resolution with one protein molecule per asymmetric unit (PDB 4nml; Figure 5A and Table 2). TgRPI has a larger α/β/α sandwich catalytic domain (residues 1–127 and residues 225–259) with the Rossmann topology and a smaller α/β sandwich oligomerization domain (residues 128–224) (Figure 5A). Residues 180–186 between strand β8 and a 310-helix (η3), Arg258, and Lys259 are absent in the structure due to their disorder. BME was used in buffers during purification and crystallization and is covalently linked to Cys82 in the structure. One D-malate molecule and two chloride ions were modeled to interpret additional electron density (Figures 5A,C). Applying crystal symmetry operations PISA predicted that TgRPI might exist as unstable dimer (BSA of ~2,430 Å2) or stable dimer (BSA of ~3,340 Å2) in the crystal environment (Figure 5B). Visualization of both dimers revealed that the first assembly is held in place primarily by hydrogen-bonded interactions between main-chain atoms of strand β13 of each monomer's catalytic domain (Figure 5B). The second dimer is stabilized by multiple intramolecular contacts between residues from both TgRPI domains. Homologous RPIs form similar stable dimers and, thus stable TgRPI dimer may be considered biologically relevant.
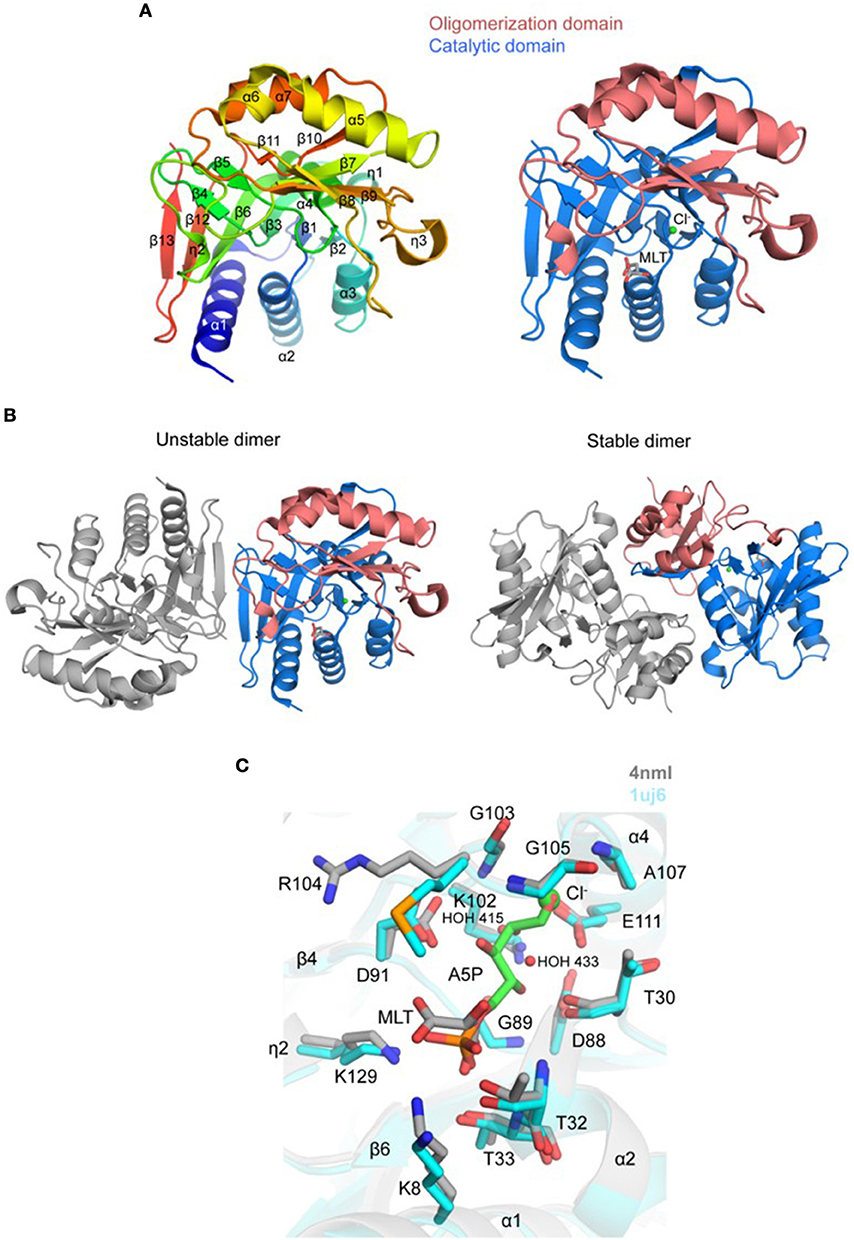
Figure 5. Crystal structure of TgRPI (A). Ribbon representation of TgRPI monomer (left side) colored blue (N-terminus) to red (C-terminus). Catalytic domain and oligomerization domain of TgRPI with bound D-malate (MLT; sticks) and chloride ion (Cl−; green sphere) are shown on the right. (B). Unstable and stable TgRPI dimers according to PISA prediction (C). Pairwise structural alignment of TgRPI (gray; MLT is shown in sticks and Cl− as green sphere) and TtRPI (cyan ribbon) with bound arabinose 5-phosphate (A5P; sticks) showing active site with residues of TgRPI are shown in sticks and labeled in one-letter code. Equivalent residues of TtRPI are displayed. Water molecules are shown as small red spheres.
PDBeFold identified several homologs of TgRPI, and we used the crystal structure of RPI from Thermus thermophilus (TtRPI; PDB 1uj6) (Hamada et al., 2003) determined in complex with arabinose 5-phosphate to identify the active site of TgRPI (Figure 5C). Both proteins share 45% sequence homology and are superimposed with ~1.0 Å r.m.s.d. over 218 Cα atoms. This structural alignment revealed that D-malate and chloride ion (e.g., CL 301/A) in TgRPI are bound in the protein's active site, mimicking parts of arabinose 5-phosphate in 1uj6 (Figure 5C). D-malate makes hydrogen bonds with side chains of Lys8, Thr32, and Thr33, and several van der Waals contacts with residues of the phosphate-binding pocket of the active site (Schrödinger SiteMap score 0.97). CL 301/A is bound in the oxyanion hole of the active site and coordinated by main-chain nitrogen atoms of Gly105 and Ala107, side chain carboxyl group of Glu111, and two water molecules, HOH 415 and HOH 433 (Figure 5C). The second chloride ion (e.g., CL 302/A) in TgRPI binds near disordered residues 179–185 and makes bonds with main-chain nitrogen atoms of Phe172 and Ile190, and the ε-amino group of Lys154 (not shown).
The catalytic domain of homologous RPIs align well, while the oligomerization domain has some distinct structural differences among various species. For example, a peptide stretch between residues Leu160 and Arg169 of the oligomerization domain of TgRPI is longer than the equivalent region in TtRPI. In the TgRPI, structure this segment is helix α6. We have found that a similar helical element is present in E. coli RPI (Zhang et al., 2003) (PDB 1o8b; 45% sequence homology; ~1.4 Å r.m.s.d. over 164 Cα atoms) and P. falciparum RPI (Holmes et al., 2006) (PDB 2f8m; 45% sequence homology; ~1.1 Å r.m.s.d. over 233 Cα atoms). Another example, strands β8 and β9 in TgRPI are separated by a 310-helix (η3) and disordered residues 179–185, while similar region in TtRPI, EcRPI, and Pf RPI is shorter, ordered, and extends toward the N-terminus of helix α4 of the superimposed TgRPI. Thus, η3 and residues 179–185 seem to be unique to TgRPI. Its length, proximity to the phosphate-binding pocket, and potential flexibility suggest that it may participate in the catalysis.
Ornithine Aminotransferase (OAT)
The crystal structure of TgOAT in complex with pyrodoxal-5′-phosphate (PLP) was determined at 1.2 Å resolution (PDB 4nog; Figure 6A and Table 2) and contains two protein chains in the asymmetric unit (~0.3 Å r.m.s.d. over 422 Cα atoms) that form a well-studied functional dimer with BSA of ~5,770 Å2 (Shah et al., 1997; Shen et al., 1998; Markova et al., 2005; Vedadi et al., 2007; Jortzik et al., 2010) (Figure 6B). Each monomer has a mixed α/β structural fold and consists of the cofactor PLP-binding domain (residues 87–336), an N-terminal domain (residues 17–86), and a C-terminal domain (residues 337–441) (Figure 6A). The PLP-binding domain comprises the eight-stranded central β-sheet linked to 10 α-helices and 4 short α-helical segments. The N- and C-terminal domain comprises three α-helices bounded to a β-sheet of three and four β strands, respectively. The difference between TgOAT monomers is observed at the position of N-terminal α-helix and is likely affected by crystal packing. BME used in the protein buffer solutions forms a covalent bond to Cys353 in both TgOAT monomers. One 1,3,5-Tris(4-carboxyphenyl)benzene (BTB), polyethylene glycol (PEG) and acetate (ACT) molecule were modeled to interpret additional electron density in chain B.
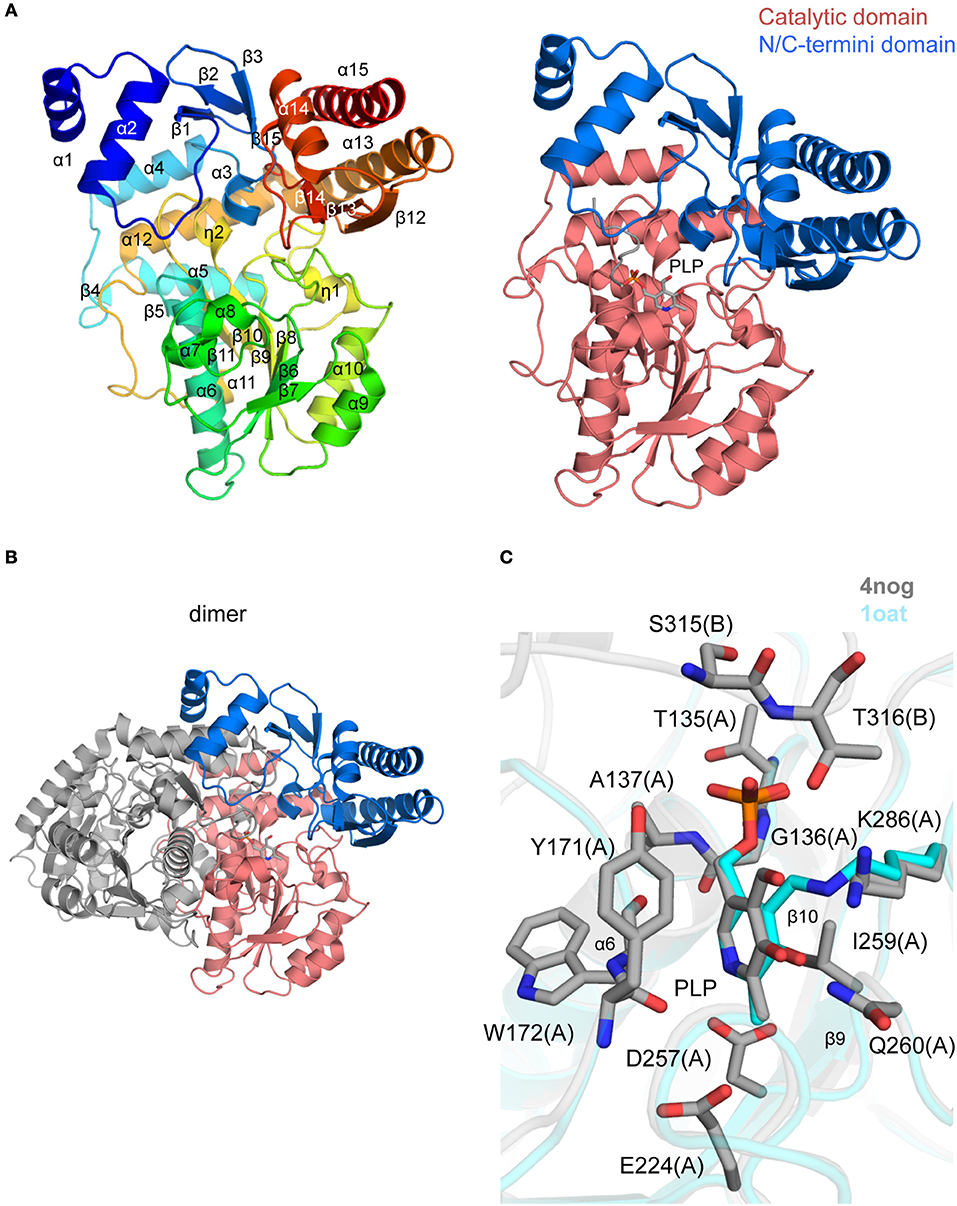
Figure 6. Crystal structure of TgOAT (A). Ribbon representation of TgOAT monomer (left side) colored blue (N-terminus) to red (C-terminus). Catalytic and N/C-termini domains with bound PLP are shown on the right (B). Dimer of TgOAT (C). Pairwise structural alignment of TgOAT (gray; PLP is shown in sticks) and hOAT with bound PLP (cyan ribbon; PDB 1oat). Residues of TgOAT are shown in sticks and labeled in one-letter code. Equivalent lysine residues of hOAT are displayed. Chain identifier in parentheses.
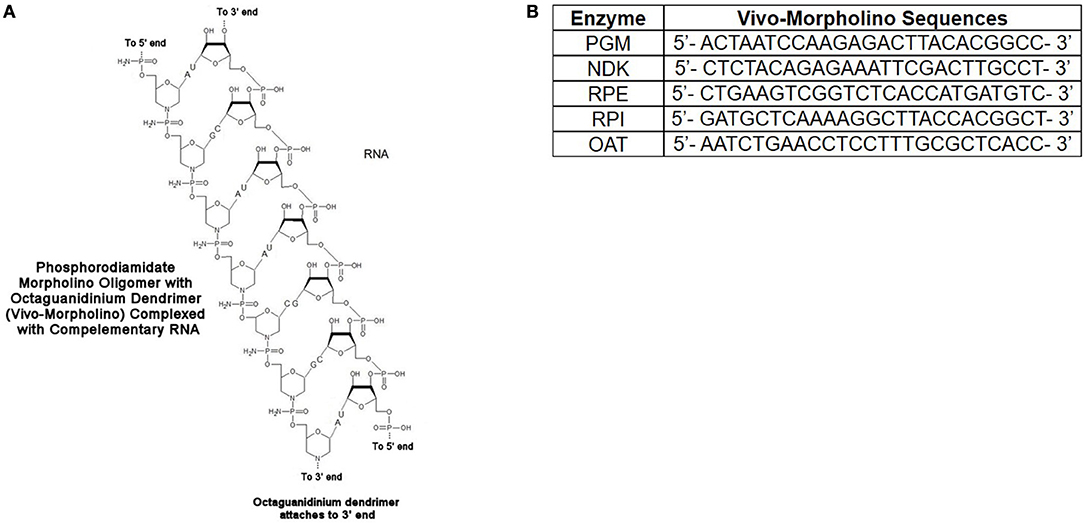
Figure 7. (A) Schematic of Interaction Between vivoPMO and RNA. (B) Sequences for PMO. Sequences for all enzymes were designed to be complementary to regions overlapping splice sites so as to prevent appropriate interaction between mRNA and the spliceosome predicted to lead to the production of a non-functional protein product.
The dimerization interface is primarily formed between residues of the PLP-binding and N-terminal domains of TgOAT. The dimer is held by 3 salt bridges, Cys96–Cys96 disulfide bond, 63 hydrogen bonds and 782 van der Waals interactions. The TgOAT dimer bears structural similarities to homologous enzymes from the aminotransferase class-III protein family (Christen and Metzler, 1985; Mehta et al., 1993) and, thus, may be considered biologically relevant. The list of homologous structures with sequence identity >40% encloses structure of OAT from human (r.m.s.d. ~0.9 Å over 404 Cα atoms) (Shah et al., 1997; Storici et al., 1999), OAT from P. falciparum (r.m.s.d. ~1.2 Å over 384 Cα atoms) (Vedadi et al., 2007; Jortzik et al., 2010) and OAT from P. yoelii (r.m.s.d. ~1.0 Å over 369 Cα atoms) (Vedadi et al., 2007). Among structures with similar secondary structure fold and high r.m.s.d. value (~1.9 Å over 395 Cα atoms) is the structure of GABA-OAT from E. coli with sequence identity 31%. Recent reports indicate that TgOAT shows a dual N-acetyl-ornithine (AcOrn) and γ-aminobutyric acid (GABA) transaminase activity and may function in both arginine and GABA metabolism (Astegno et al., 2017).
The cofactor-binding domain of TgOAT non-covalently binds PLP molecule, i.e., PLP does not form the Schiff base with conserve Lys286 (Figures 6A,C). Similarly to homologous OAT structures obtained in complex with the cofactor (Shah et al., 1997; Shen et al., 1998; Storici et al., 1999; Vedadi et al., 2007; Jortzik et al., 2010), PLP interacts with Thr135, Gly136, Ala137, Tyr171, Trp172, Glu224, Asp257, Ile259, Gln260, Lys286, Ser315, and Thr316 (Figure 6C). Most of these residues are highly conserved among TgOAT homologs and represent a viable, “druggable” pocket (SiteMap score 1.03).
In vitro Studies of Targeted vivoPMO on T. gondii Replication
HFFs were infected with YFP-expressing T. gondii tachyzoites and treated with vivoPMOs targeted against all five enzymes of interest. When treated at 10 μM concentrations, each morpholino resulted in approximately 50% reduction in fluorescence in vitro (between 44% for PGM-targeted vivoPMO to 56% reduction for OAT-targeted vivoPMO). This was compared to the off-target, control morpholino, which resulted in < 10% fluorescence reduction. When compared statistically using student T-test, levels of fluorescence in vitro were statistically decreased (p < 0.05) when treating with targeted vivoPMO relative to untreated fibroblasts infected with 2000 YFP-expressing tachyzoites as well as when compared to infected cells treated with off-target morpholino. A representative experiment showing these findings is in Figure 8A. Data from all replicate experiments of the morpholino efficacy assays, as well as statistical calculations and ratios reflecting efficacy of targeted morpholino to off-target can be found in Supplementary Table 1.
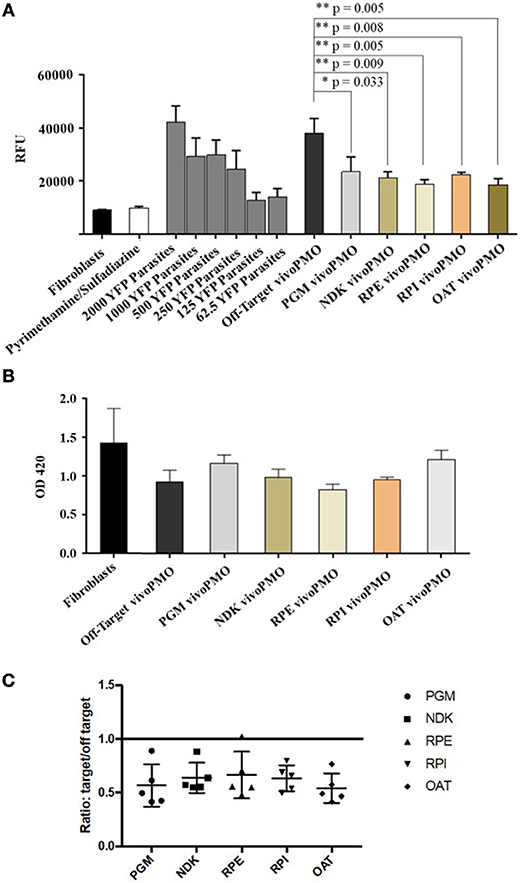
Figure 8. (A) YFP/vivoPMO Efficacy Assay at 10 μM. Targeted vivoPMOs show statistically significant knockdown relative to off-target and to a standardized parasite concentration as determined by student t-test. (B) Wst-1 Cell Viability/ vivoPMO Toxicity Assay at 10 μM. Targeted vivoPMOs do not show statistically significantly (p < 0.05 by student T-test) different levels of optical density relative to off-target vivoPMO or uninfected fibroblasts at 10 μM concentrations. (C) Ratio of On- to Off-Target vivoPMO for Replicate Experiments Using 10 μM of Targeted vivoPMO. The data presented here represent five replicate experiments. Lower individual values suggest a larger effect of the morpholino.
To confirm that these findings of decreased fluorescence in vitro was not secondary to host-cell toxicity from the vivoPMOs, a WST-1 assay was used. HFFs treated with 10 μM concentrations of enzyme-targeted vivoPMOs were statistically indistinguishable (p > 0.05) from untreated cells by Student T-test. Toxicity was demonstrated at higher vivoPMO concentrations of 20 μM. A representative experiment demonstrating the absence of host-cell toxicity with vivoPMO treatment at 10 μM is in Figure 8B.
The ratio of targeted to off-target vivoPMO are displayed in Figure 8C. This is shown for five replicate experiments (Supplementary Table 1). This demonstrates that, in cultures treated with on-target vivoPMO, parasitic replication was reduced.
These findings were compared with the effect on tachyzoites in a previously published full genome screen using CRISPR/Cas9 (Sidik et al., 2016). Three of the target proteins (TgPGM, TgRPE, and TgRPI) that had a significantly reduced replication phenotype in tachyzoites caused by vivoPMO knockdown were found to contribute to parasite fitness when studied using CRISPR/Cas9. The CRISPR/Cas9 data was annotated and graphically presented in ToxoTB (http://toxodb.org/toxo). TgPGM, TgRPE, and TgRPI show negative phenotypic/fitness scores (−4.45, −0.31, −1.92, respectively) suggesting essentiality. TgOAT appeared dispensable (fitness score of +1.2) in the tachyzoite CRISPR/Cas9 assays consistent with TgOAT appeared to be expressed predominantly in the oocyst stage (ToxoDB). TgNDK (+2.45) also appeared to be dispensable in tachyzoites.
Immunofluorescence Assay
Recombinant protein for the five targets was also used to produce antibodies in mice, which were used for immunostaining to determine expression patterns within the parasite. Immunolocalization was not successful with tachyzoites of Toxoplasma for four of the five enzymes. For NDK, enzyme was present in a granular pattern in the cytoplasm of tachyzoites, located around the perimeter of the parasite and the posterior part of the parasite. This can be seen in Figure 9.
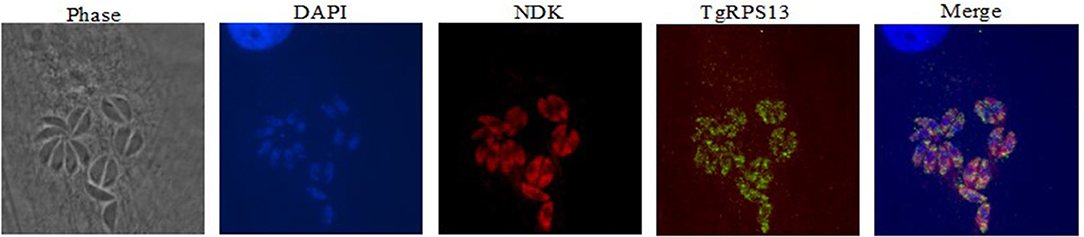
Figure 9. Immunofluorescence Assay. This series of images depict the localization of nucleoside diphosphate kinase, one of the molecular targets herein discussed. Note the concentration of red fluorescence at the periphery of the parasite. This is consistent with the localization of secreted proteins like Gra1, suggesting that NDK could be secreted by the parasite. This has precedents in other pathogens, including M. tuberculosis and Leishmania.
Discussion
The work characterized herein presents a detailed characterization of enzyme structure that can be used for modeling inhibitors of these targets and also presents approaches for studying target phenotype with vivoPMO and CRISPR/Cas9 which in combination develops a system to move forward candidate targets (Figure 10). The targeted vivoPMOs demonstrated statistically significant perturbation of parasitic replication when compared to off-target morpholinos, without concomitant host-cell toxicity, confirmed for some of these with CRISPR/CAS9 screen. Inhibition of target enzymatic function via small molecules or anti-sense might be a novel therapeutic modality, should such small molecules exist. Given the current limitations of anti-parasitic medicines for the treatment of toxoplasmosis, new pharmacotherapy is of significant interest. The multi-step approach, detailed here, of in vitro inhibition through anti-sense techniques accompanied by detailed structural characterization to identify possible exploitable differences between host and parasite enzymes. Confirmation by referring to a recently published CRISPR/Cas9-based analysis, can also help to suggest importance of targets in T. gondii tachyzoites (Sidik et al., 2016). This provides another level of evidence that particular targets may be important for the parasites replication. This approach was performed for each of five enzymes identified as having potential biologic importance and with favorable predicted druggability using the TDR methodology. This favorable druggability was also confirmed with in silico modeling using Schrödinger SiteMap, which indicated each target demonstrated “druggable” pockets. Each enzyme will be considered sequentially hereafter.
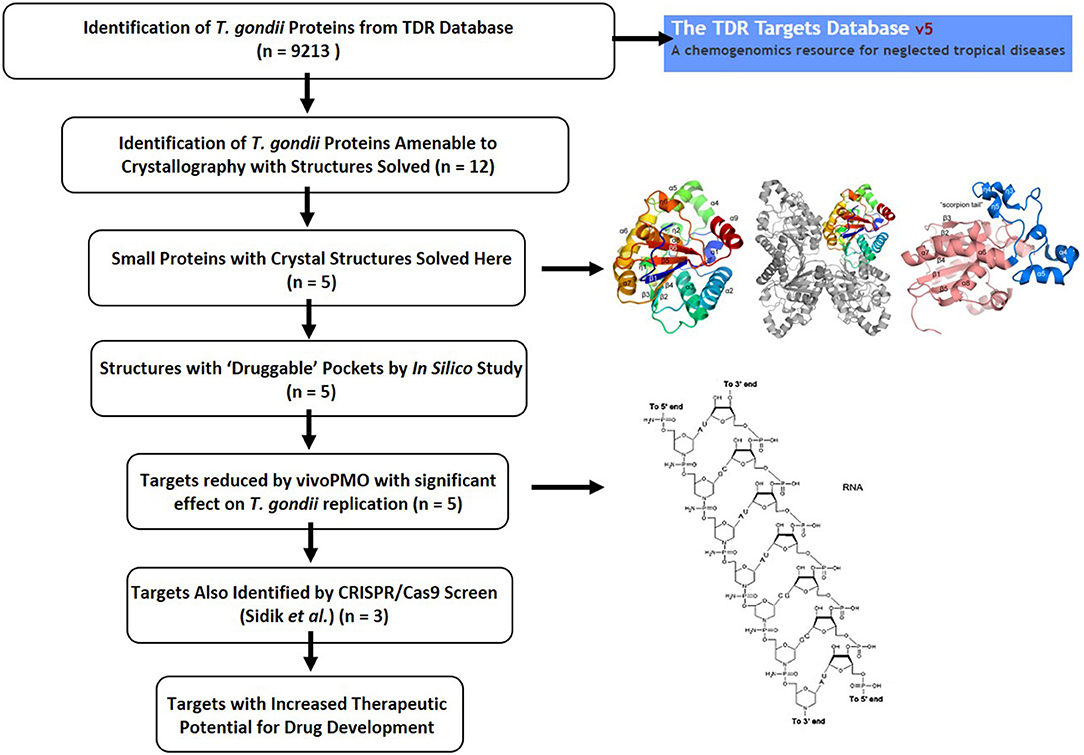
Figure 10. Summary figure showing the T. gondii drug discovery pipeline as described herein. The TDR Database provided important initial insights into potentially important parasite proteins (Agüero et al., 2008; Magariños et al., 2012). Identification of proteins amenable to crystallographic analysis, solution of their structures, abrogation with vivoPMO and confirmation of essentiality by querying the CRISPR/Cas9 database completed the pathway, identifying enriched targets with improved therapeutic potential.
Phosphoglycerate mutase II, an enzyme at the core of the glycolytic pathway, catalyzes the transition from 3-phosphoglycerate to 2-phosphoglycerate, an important preparatory step upstream of enolase and pyruvate kinase. Although little work had been done on this Apicomplexan enzyme as a potential drug target, we inferred that interrupting glycolysis would prevent the production of pyruvate by glycolysis, and thereby reduce input into the Kreb's Cycle. This, in turn, will reduce the number of electron carrying compounds (NADH, FADH2), which would markedly reduce ATP production. Interestingly, glycolysis has been targeted successfully in other parasites, and also has been demonstrated to have importance to host cell egress, as well as maintenance of energy reserves when the parasite is found outside of host cells (Ananvoranich et al., 2006; Fleige et al., 2007; Pomel et al., 2008; Lin et al., 2011; Singh et al., 2013). These observations, coupled with our own, are consistent with the observed effect on parasite replication. It appears that putative inhibitors could make this a robust tachyzoite molecular target with a compound easily available to test. With the present crystal structure, further structure-based molecular design approaches (such as virtual screening) is possible given the favorable SiteMap score (0.98).
Nucleotide diphosphate kinase catalyzes the movement of phosphate from a nucleoside triphosphate to a nucleoside diphosphate (e.g., GTP + ADP –> GDP + ATP). Naturally, disruption of this process could have an impact on the energy economy within the parasite, as ATP would not be available for important cellular tasks related to DNA replication and the production of more parasites. Additionally, this is a stress kinase, and thus its impact on the stressed organism (through a tetracycline-dependent gene expression construct, RPS-13) would be useful to explore. An immunofluorescence assay revealed peripheral and posterior concentration of staining. Interestingly, another intracellular pathogen, Leishmania amazonensis, has been shown to secrete NDK to prevent host-cell autolysis (Kolli et al., 2008). It would be of interest to determine whether this enzyme plays a similar role in Apicomplexans like T. gondii. Of note, NDK inhibition by candidate compounds has been demonstrated to have efficacy against certain species of Leishmania in vitro (Vieira et al., 2015; Mishra et al., 2017). Crystal structure of TgNDK suggests that the protein forms a hexamer with conserved nucleotide binding sites. Pairwise structural alignment revealed that active site of TgNDK may undergo similar conformational changes as its closest homolog, humanNM23-H2 transcription factor (Figure 3B). High structural homology to human transcription factor (NM23-H2) in the residues in the catalytic site indicate that selective inhibitors that do not act on human NM23-H2 will be needed. Possible strategies that will facilitate such selectivity include antisense, CRISPR, aptamer-based approaches where the DNA/RNA sequences are divergent, among others. When the dinucleotide is present in the active site, the residues in both Tg and Hs structures have similar side chain orientations (Figure 3B), suggesting inhibitor selectivity between the species might be difficult to achieve. On the other hand, when the dinucleotide is absent, the active site loop comprising of residues G59-F62 (Tg numbering) is shifted significantly (3–4 Å) between species. Additionally, TgNDK residue K60 is residue R58 in the human ortholog, offering different hydrogen-bond capacities to putative ligands. These observations suggest opportunities for the selective design of TgNDK inhibitors using structure-based molecular modeling techniques. Wang et al. reported selective NM23-H2 (human ortholog) inhibitors based on an isaindigotone scaffold (Wang et al., 2017). These compounds were subjected to molecular docking studies and were predicted to bind to the dinucleotide pocket. More so, they offer an obvious starting point for biological evaluation in in vitro parasite models and the subsequent design of selective TgNDK chemical probes/ligands.
The next two enzymes, due to their sequential placement within the pentose-phosphate pathway, should be considered together. Ribulose phosphate 3-epimerase functions in the conversion of ribulose-5-phosphate into xylulose-5-phosphate, which is a reaction in the Calvin cycle. It is just downstream of the next target enzyme, ribose-5-phosphate isomerase, and is important for the development of a pool of NADPH, as well as in the pentose phosphate pathway that can convert monosaccharides like glucose into nucleotide precursor pentose sugars. This pathway has been of interest in targeting various organisms, including P. falciparum and Trypanosoma cruzi (Barrett, 1997; Bozdech and Ginsburg, 2005; Igoillo-Esteve et al., 2007). The biology of RPE has proven to be of particular interest in T. cruzi (Gonzalez et al., 2017). Indeed, the structure of RPE had already been characterized in P. falciparum (Caruthers et al., 2005). Determined crystal structure of TgRPE revealed that Zn2+ binds in a putative active site. In addition, the presence of in the active site induces the β6–α5 loop to move from its presumed apo conformation in the absence of ligand, as seen in the human RPE homolog, to holo conformation in the ligand bound state (Figure 4B). Ribose-5-phosphate isomerase is the enzyme in the pentose phosphate pathway just upstream of ribulose phosphate 3-epimerase, and it has similar functions, though it catalyzes the transition from ribose-5-phosphate to ribulose-5-phosphate. It has been identified as a potential drug target in P. falciparum, one of the causative agents of malaria, due to its necessity in creating nucleotide precursors for DNA synthesis and for maintaining a large pool of NADPH for rapid replication and the metabolism necessary for the maintenance thereof (Holmes et al., 2006). It has also been of interest and targeted in Mycobacterium tuberculosis where inhibitors have been identified (Roos et al., 2005). Additionally, this pathway has been suggested as a drug target in trypanosomes, with identified compounds and multiple mechanisms of inhibition having been demonstrated, with current patents existing (de V. C. Sinatti et al., 2017). Perhaps the most interesting feature of the determined crystal structure of TgRPI is the disordered region between residues 179–185 that comprises a part of what may appear to be an active site loop. These residues and 310-helix η3 are unique to TgRPI and might be important for catalysis (Figure 5A).
Ornithine aminotransferase, the final enzyme considered herein, is an enzyme involved in the urea cycle, TCA cycle, polyamine synthesis, and other pathways. It catalyzes a reversible reaction allowing interconversion of intermediates from ornithine to amino acids. Other enzymes which act in this pathway have been suggested as potential drug targets, and it is critical for maintaining proper amounts of free amino acids, so it was a good candidate for further study. Herein we found a modest phenotype on replication but raised antibody did not immunostain tachyzoites. Ornithine aminotransferase will be studied further in the future to resolve some of these questions and attempt to develop effective inhibitors, were it to prove to be essential. Structural analysis reveals that TgOAT shares similar structural folds to known OAT enzymes (Figure 6). We have identified that TgOAT shares a conserved cofactor and substrate-binding site with its closest homolog human GABA-AT. To gain insights into function of TgOAT, we attempted to crystallize this enzyme with several different inhibitors and inactivators. As a result, three additional crystal structures of TgOAT in complex with gabaculine (a potent inhibitor of human GABA-AT) and (S)-4-amino-5-fluoropentanoic acid have been determined (PDBs 5DJ9, 5E5I, and 5E3K). Therefore, we have performed a full kinetic and structural analysis. The details of the structure, substrate binding site, kinetic mechanism and function of TgOAT will be described in our subsequent works.
Our approach described herein is a productive way to identify molecular targets and could potentially be useful for identifying small molecule inhibitors. In silico analysis of each enzyme's surface pockets/active sites suggest “druggable” areas for binding of putative small molecules. The recent discovery that CRISPR/Cas9 has future potential for treating HIV (Yin et al., 2017) also raises the possibility that expression of simply validated molecular targets can be eliminated by CRISPR/Cas9. SiRNA is another possible therapeutic modality, and is being studied in several infectious diseases, including Hepatitis C, Ebola, and viral encephalitis, among others (Kumar et al., 2006; Wan et al., 2014; Watanabe et al., 2014; Thi et al., 2015). As our studies suggest, vivoPMO inhibited T. gondii replication; antisense PMO is, therefore, another potential therapeutic modality that might effectively treat parasitic infection. The data presented in Figure 8 and CRISPR/Cas9 screen suggest a phenotype for TgPGM, TgRPE, and TgRPI, but do not yet prove the targets are essential. With confirmation in the future, vivoPMO-based therapy with a less toxic molecular transporter has promise. The safety of PMO is well-documented in several clinical trials in treating genetic, cardiovascular, and infectious diseases including Duchenne muscular dystrophy (DMD), restenosis, and Marburg and Ebola hemorrhagic fevers (Kipshidze et al., 2007; Kinali et al., 2009; Cirak et al., 2011; Heald et al., 2014). It has even shown efficacy against a relative of T. gondii, the causative agent of malaria, P. falciparum (Augagneur et al., 2012). Eteplirsen (ExonDys51) is an FDA-approved PMO drug for treatment of DMD in patients who have confirmed mutation of the DMD gene that is amenable to exon 51 skipping. A clinical trial is underway using a cell-penetrating peptide conjugated eteplirsen (the PPMO technology) to increase intracellular delivery of eteplirsen for greater efficacy, lower dose and less frequent dosing (clincialtrials.gov: NCT03375255). With the advent of novel modalities, including antisense and small molecule inhibition, for the treatment of both active and latent infection, it may be possible to eradicate human T. gondii infection, and the successes achieved by physicians and scientists in combatting smallpox and dracunculiasis might extend to toxoplasmosis as well.
Ethics Statement
This study was carried out in accordance with the recommendations of The Home Office of the UK Government under the Animals [Scientific Procedures] Act 1986. All work was covered by License PPL60/4568, Treatment and Prevention of Toxoplasmosis with approval by the University of Strathclyde ethical review board.
Author Contributions
1JL, HN, and RM: Conceptualization. JL, HN, EF, AH, GM, YZ, ID, KF, LS, JR, KE, SD, CR, SW, MM, SM, CF, ES, RM, and WA: Data curation. JL, EF, AH, GM, YZ, and RM: Formal analysis. CR, RM, and WA: Funding acquisition. JL, HN, EF, AH, GM, YZ, ID, KF, LS, JR, KE, SD, CR, SW, MM, SM, CF, ES, RM, and WA: Investigation. JL, HN, EF, AH, GM, YZ, ID, KF, LS, JR, KE, SD, CR, SW, JM, HM, MM, SM, CF, ES, DS, DR, RM, and WA: Methodology. RM and WA: Project administration. CR, RM, and WA: Resources. RM and WA: Supervision. JL, EF, AH, GM, YZ, RM, and WA: Validation. JL, EF, AH, GM, YZ, and KE: Visualization. JL, EF, AH, GM, and RM: Writing-original draft. JL, HN, EF, AH, GM, YZ, ID, KF, LS, JR, KE, SD, CR, SW, JM, HM, MM, SM, CF, ES, DS, DR, RM, and WA: Writing-review and editing.
Conflict of Interest Statement
RM, KE, HM, and JM are inventors on a U.S. patent (US8575126B2) that is focused on delivery of morpholino oligomers as a treatment for Toxoplasma gondii infection. JM is employed by Gene Tools, LLC, the company that manufactured the vivoPMO for this study. However, he took no part in data collection or analysis. RM has completed an unrelated literature review for Sanofi-Pasteur.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported in part by the NIH NIDDK grant #2T35DK062719-27. The data collection was performed at the LS-CAT Sector 21 at the Advanced Photon Source supported by the Argonne National Laboratory operated by the University of Chicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357. The LS-CAT is supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817). This work has been funded by NIAID, NIH, Department of HHS, under Contracts No. HHSN272200700058C, HHSN272201200026C, and HHSN272201700060C (WA).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2018.00352/full#supplementary-material
Supplementary Table 1. Data from replicate experiments demonstrate the efficacy of target-specific morpholino on parasite replication in vitro. Ratios of target-specific to off-target morpholino for all targets reflect statistically significant perturbation of parasite replication.
References
Agüero, F., Al-Lazikani, B., Aslett, M., Berriman, M., Buckner, F. S., Campbell, R. K., et al. (2008). Genomic-scale prioritization of drug targets: the TDR Targets database. Nat. Rev. Drug Discov. 7, 900–907. doi: 10.1038/nrd2684
Akana, J., Fedorov, A. A., Fedorov, E., Novak, W. R. P., Babbitt, P. C., Almo, S. C., et al. (2006). D-Ribulose 5-phosphate 3-epimerase: functional and structural relationships to members of the ribulose-phosphate binding (beta/alpha)8-barrel superfamily. Biochemistry 45, 2493–2503. doi: 10.1021/bi052474m
Ananvoranich, S., Al Rayes, M., Al Riyahi, A., and Wang, X. (2006). RNA silencing of glycolysis pathway in Toxoplasma gondii. J. Eukaryot. Microbiol. 53(Suppl. 1), S162–S163. doi: 10.1111/j.1550-7408.2006.00216.x
Anderson, W. F. (2009). Structural genomics and drug discovery for infectious diseases. Infect. Disord. 9, 507–517. doi: 10.2174/187152609789105713
Aslanidis, C., and de Jong, P. J. (1990). Ligation-independent cloning of PCR products (LIC-PCR). Nucleic Acids Res. 18, 6069–6074. doi: 10.1093/nar/18.20.6069
Astegno, A., Maresi, E., Bertoldi, M., Verde, V. L., Paiardini, A., and Dominici, P. (2017). Unique substrate specificity of ornithine aminotransferase from Toxoplasma gondii. Biochem. J. 474, 939–955. doi: 10.1042/BCJ20161021
Augagneur, Y., Wesolowski, D., Tae, H. S., Altman, S., and Mamoun, C. B. (2012). Gene selective mRNA cleavage inhibits the development of Plasmodium falciparum. Proc. Natl. Acad. Sci. U.S.A. 109, 6235–6240. doi: 10.1073/pnas.1203516109
Barrett, M. P. (1997). The pentose phosphate pathway and parasitic protozoa. Parasitol. Today 13, 11–16. doi: 10.1016/S0169-4758(96)10075-2
Berman, H. M., Bhat, T. N., Bourne, P. E., Feng, Z., Gilliland, G., Weissig, H., et al. (2000). The protein data bank and the challenge of structural genomics. Nat. Struct. Mol. Biol. 7, 957–959. doi: 10.1038/80734
Bozdech, Z., and Ginsburg, H. (2005). Data mining of the transcriptome of Plasmodium falciparum: the pentose phosphate pathway and ancillary processes. Malar. J. 4:17. doi: 10.1186/1475-2875-4-17
Caruthers, J., Bosch, J., Buckner, F., Van Voorhis, W., Myler, P., Worthey, E., et al. (2005). Structure of a ribulose 5-phosphate 3-epimerase from Plasmodium falciparum. Proteins Struct. Funct. Bioinforma. 62, 338–342. doi: 10.1002/prot.20764
Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., et al. (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21. doi: 10.1107/S0907444909042073
Chevalier, N., Rigden, D. J., Van Roy, J., Opperdoes, F. R., and Michels, P. A. (2000). Trypanosoma brucei contains a 2,3-bisphosphoglycerate independent phosphoglycerate mutase. Eur. J. Biochem. FEBS 267, 1464–1472. doi: 10.1046/j.1432-1327.2000.01145.x
Cirak, S., Arechavala-Gomeza, V., Guglieri, M., Feng, L., Torelli, S., Anthony, K., et al. (2011). Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet 378, 595–605. doi: 10.1016/S0140-6736(11)60756-3
Crowther, G. J., Shanmugam, D., Carmona, S. J., Doyle, M. A., Hertz-Fowler, C., Berriman, M., et al. (2010). Identification of attractive drug targets in neglected-disease pathogens using an in silico approach. PLoS Negl. Trop. Dis. 4:e804. doi: 10.1371/journal.pntd.0000804
Davies, D. R., Staker, B. L., Abendroth, J. A., Edwards, T. E., Hartley, R., Leonard, J., et al. (2011). An ensemble of structures of Burkholderia pseudomallei 2,3-bisphosphoglycerate-dependent phosphoglycerate mutase. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 67, 1044–1050. doi: 10.1107/S1744309111030405
Davis, I. W., Leaver-Fay, A., Chen, V. B., Block, J. N., Kapral, G. J., Wang, X., et al. (2007). MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383. doi: 10.1093/nar/gkm216
de Beer, T. A., Berka, K., Thornton, J. M., and Laskowski, R. A. (2014). PDBsum additions. Nucleic Acids Res. 42, D292–D296. doi: 10.1093/nar/gkt940
de V. C. Sinatti, V., R Baptista, L. P., Alves-Ferreira, M., Dardenne, L., Hermínio Martins da Silva, J., and Guimarães, A. C. (2017). In silico identification of inhibitors of ribose 5-phosphate isomerase from Trypanosoma cruzi using ligand and structure based approaches. J. Mol. Graph. Model. 77, 168–180. doi: 10.1016/j.jmgm.2017.08.007
Dexheimer, T. S., Carey, S. S., Zuohe, S., Gokhale, V. M., Hu, X., Murata, L. B., et al. (2009). NM23-H2 may play an indirect role in transcriptional activation of c-myc gene expression but does not cleave the nuclease hypersensitive element III1. Mol. Cancer Ther. 8, 1363–1377. doi: 10.2210/pdb3bbb/pdb
Djikeng, A., Raverdy, S., Foster, J., Bartholomeu, D., Zhang, Y., El-Sayed, N. M., et al. (2007). Cofactor-independent phosphoglycerate mutase is an essential gene in procyclic form Trypanosoma brucei. Parasitol. Res. 100, 887–892. doi: 10.1007/s00436-006-0332-7
Dranow, D. M., Conrady, D. G., Lorimer, D. D., and Edwards, T. E. (2017). Crystal Structure of a ribulose-phosphate 3-epimerase from Neisseria gonorrhoeae with bound phosphate. BE Publ. doi: 10.2210/pdb5umf/pdb
Dubey, R., Staker, B. L., Foe, I. T., Bogyo, M., Myler, P. J., Ngô, H. M., et al. (2017). Membrane skeletal association and post-translational allosteric regulation of Toxoplasma gondii GAPDH1. Mol. Microbiol. 103, 618–634. doi: 10.1111/mmi.13577
Emsley, P., and Cowtan, K. (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132. doi: 10.1107/S0907444904019158
Emsley, P., Lohkamp, B., Scott, W. G., and Cowtan, K. (2010). Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501. doi: 10.1107/S0907444910007493
Flegr, J., Prandota, J., Sovičková, M., and Israili, Z. H. (2014). Toxoplasmosis – A Global Threat. Correlation of latent toxoplasmosis with specific disease burden in a set of 88 countries. PLoS ONE 9:e90203. doi: 10.1371/journal.pone.0090203
Fleige, T., Fischer, K., Ferguson, D. J. P., Gross, U., and Bohne, W. (2007). Carbohydrate metabolism in the Toxoplasma gondii apicoplast: localization of three glycolytic isoenzymes, the single pyruvate dehydrogenase complex, and a plastid phosphate translocator. Eukaryot. Cell 6, 984–996. doi: 10.1128/EC.00061-07
Furtado, J. M., Smith, J. R., Belfort, R., Gattey, D., and Winthrop, K. L. (2011). Toxoplasmosis: a global threat. J. Glob. Infect. Dis. 3, 281–284. doi: 10.4103/0974-777X.83536
Gonzalez, S. N., Valsecchi, W. M., Maugeri, D., Delfino, J. M., and Cazzulo, J. J. (2017). Structure, kinetic characterization and subcellular localization of the two ribulose 5-phosphate epimerase isoenzymes from Trypanosoma cruzi. PLoS ONE 12:e0172405. doi: 10.1371/journal.pone.0172405
Halgren, T. (2007). New method for fast and accurate binding-site identification and analysis. Chem. Biol. Drug Des. 69, 146–148. doi: 10.1111/j.1747-0285.2007.00483.x
Hamada, K., Ago, H., Sugahara, M., Nodake, Y., Kuramitsu, S., and Miyano, M. (2003). Oxyanion hole-stabilized stereospecific isomerization in ribose-5-phosphate isomerase (Rpi). J. Biol. Chem. 278, 49183–49190. doi: 10.1074/jbc.M309272200
Hasegawa, H., and Holm, L. (2009). Advances and pitfalls of protein structural alignment. Curr. Opin. Struct. Biol. 19, 341–348. doi: 10.1016/j.sbi.2009.04.003
Heald, A. E., Iversen, P. L., Saoud, J. B., Sazani, P., Charleston, J. S., Axtelle, T., et al. (2014). Safety and pharmacokinetic profiles of phosphorodiamidate morpholino oligomers with activity against ebola virus and marburg virus: results of two single ascending dose studies. Antimicrob. Agents Chemother. 58, 6639–47. doi: 10.1128/AAC.03442-14
Holmes, M. A., Buckner, F. S., Van Voorhis, W. C., Verlinde, C. L. M. J., Mehlin, C., Boni, E., et al. (2006). Structure of ribose 5-phosphate isomerase from Plasmodium falciparum. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 62, 427–431. doi: 10.1107/S1744309106010876
Igoillo-Esteve, M., Maugeri, D., Stern, A. L., Beluardi, P., and Cazzulo, J. J. (2007). The pentose phosphate pathway in Trypanosoma cruzi: a potential target for the chemotherapy of Chagas disease. An. Acad. Bras. Ciênc. 79, 649–663. doi: 10.1590/S0001-37652007000400007
Jelakovic, S., Kopriva, S., Süss, K.-H., and Schulz, G. E. (2003). Structure and catalytic mechanism of the cytosolic d-ribulose-5-phosphate 3-epimerase from rice. J. Mol. Biol. 326, 127–135. doi: 10.1016/S0022-2836(02)01374-8
Joint Center for Structural Genomics (2011). Crystal Structure of a D-ribulose-5-phosphate-3-epimerase (NP_954699) from HOMO SAPIENS at 2.20 A Resolution. Available online at: http://www.rcsb.org/pdb/explore.do?structureId=3qc3
Jortzik, E., Fritz-Wolf, K., Sturm, N., Hipp, M., Rahlfs, S., and Becker, K. (2010). Redox regulation of Plasmodium falciparum ornithine δ-aminotransferase. J. Mol. Biol. 402, 445–459. doi: 10.1016/j.jmb.2010.07.039
Kinali, M., Arechavala-Gomeza, V., Feng, L., Cirak, S., Hunt, D., Adkin, C., et al. (2009). Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: a single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 8, 918–928. doi: 10.1016/S1474-4422(09)70211-X
Kipshidze, N., Iversen, P., Overlie, P., Dunlap, T., Titus, B., Lee, D., et al. (2007). First human experience with local delivery of novel antisense AVI-4126 with Infiltrator catheter in de novo native and restenotic coronary arteries: 6-month clinical and angiographic follow-up from AVAIL study. Cardiovasc. Revasc. Med. Mol. Interv. 8, 230–235. doi: 10.1016/j.carrev.2007.04.002
Kito, K., Sanada, Y., and Katunuma, N. (1978). Mode of inhibition of ornithine aminotransferase by L-canaline. J. Biochem. 83, 201–206.
Kolli, B. K., Kostal, J., Zaborina, O., Chakrabarty, A. M., and Chang, K.-P. (2008). Leishmania-released nucleoside diphosphate kinase prevents ATP-mediated cytolysis of macrophages. Mol. Biochem. Parasitol. 158, 163–175. doi: 10.1016/j.molbiopara.2007.12.010
Kopp, J., Kopriva, S., Süss, K. H., and Schulz, G. E. (1999). Structure and mechanism of the amphibolic enzyme D-ribulose-5-phosphate 3-epimerase from potato chloroplasts. J. Mol. Biol. 287, 761–771. doi: 10.1006/jmbi.1999.2643
Krissinel, E. (2009). Crystal contacts as nature's docking solutions. J. Comput. Chem. 31, 133–143. doi: 10.1002/jcc.21303
Krissinel, E., and Henrick, K. (2004). Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 60, 2256–2268. doi: 10.1107/S0907444904026460
Krissinel, E., and Henrick, K. (2005). “Detection of protein assemblies in crystals,” in Computational Life Sciences Lecture Notes in Computer Science, eds M. R. Berthold, R. C. Glen, K. Diederichs, O. Kohlbacher, and I. Fischer (Berlin; Heidelberg: Springer), 163–174.
Krissinel, E., and Henrick, K. (2007). Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797. doi: 10.1016/j.jmb.2007.05.022
Kronenberger, T., Lindner, J., Meissner, K. A., Zimbres, F. M., Coronado, M. A., Sauer, F. M., et al. (2014). Vitamin B6-dependent enzymes in the human malaria parasite Plasmodium falciparum: a druggable target? BioMed Res. Int. 2014:108516. doi: 10.1155/2014/108516
Kumar, P., Lee, S. K., Shankar, P., and Manjunath, N. (2006). A single siRNA suppresses fatal encephalitis induced by two different flaviviruses. PLoS Med. 3:e96. doi: 10.1371/journal.pmed.0030096
Lai, B.-S., Witola, W. H., Bissati, K. E., Zhou, Y., Mui, E., Fomovska, A., et al. (2012). Molecular target validation, antimicrobial delivery, and potential treatment of Toxoplasma gondii infections. Proc. Natl. Acad. Sci. U.S.A. 109, 14182–7. doi: 10.1073/pnas.1208775109
Lam, S. D., Dawson, N. L., Das, S., Sillitoe, I., Ashford, P., Lee, D., et al. (2016). Gene3D: expanding the utility of domain assignments. Nucleic Acids Res. 44, D404–409. doi: 10.1093/nar/gkv1231
Laskowski, R. A., MacArthur, M. W., Moss, D. S., and Thornton, J. M. (1993). PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291.
Li, X., Tang, S., Wang, Q.-Q., Leung, E. L.-H., Jin, H., Huang, Y., et al. (2017). Identification of epigallocatechin-3- gallate as an inhibitor of phosphoglycerate mutase 1. Front. Pharmacol. 8:325. doi: 10.3389/fphar.2017.00325
Liang, W., Ouyang, S., Shaw, N., Joachimiak, A., Zhang, R., and Liu, Z.-J. (2011). Conversion of D-ribulose 5-phosphate to D-xylulose 5-phosphate: new insights from structural and biochemical studies on human RPE. FASEB J. 25, 497–504. doi: 10.1096/fj.10-171207
Lin, S. S., Blume, M., von Ahsen, N., Gross, U., and Bohne, W. (2011). Extracellular Toxoplasma gondii tachyzoites do not require carbon source uptake for ATP maintenance, gliding motility and invasion in the first hour of their extracellular life. Int. J. Parasitol. 41, 835–841. doi: 10.1016/j.ijpara.2011.03.005
Lipinski, C. A. (2004). Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov. Today Technol. 1, 337–341. doi: 10.1016/j.ddtec.2004.11.007
Liu, Y., Huang, R., Han, L., Ke, W., Shao, K., Ye, L., et al. (2009). Brain-targeting gene delivery and cellular internalization mechanisms for modified rabies virus glycoprotein RVG29 nanoparticles. Biomaterials 30, 4195–4202. doi: 10.1016/j.biomaterials.2009.02.051
Lykins, J., Wang, K., Wheeler, K., Clouser, F., Dixon, A., Bissati, K. E., et al. (2016). Understanding toxoplasmosis in the united states through “large data” analyses. Clin. Infect. Dis. 63, 468–475. doi: 10.1093/cid/ciw356
Magariños, M. P., Carmona, S. J., Crowther, G. J., Ralph, S. A., Roos, D. S., Shanmugam, D., et al. (2012). TDR Targets: a chemogenomics resource for neglected diseases. Nucleic Acids Res. 40, D1118–D1127. doi: 10.1093/nar/gkr1053
Markova, M., Peneff, C., Hewlins, M. J. E., Schirmer, T., and John, R. A. (2005). Determinants of substrate specificity in ω-aminotransferases. J. Biol. Chem. 280, 36409–36416. doi: 10.1074/jbc.M506977200
McAleese, S. M., Fothergill-Gilmore, L. A., and Dixon, H. B. (1985). The phosphonomethyl analogue of 3-phosphoglycerate is a potent competitive inhibitor of phosphoglycerate mutases. Biochem. J. 230, 535–542.
McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C., and Read, R. J. (2007). Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674. doi: 10.1107/S0021889807021206
McLeod, R., Boyer, K. M., Lee, D., Mui, E., Wroblewski, K., Karrison, T., et al. (2012). Prematurity and severity are associated with Toxoplasma gondii alleles (NCCCTS, 1981-2009). Clin. Infect. Dis. 54, 1595–1605. doi: 10.1093/cid/cis258
McLeod, R., Lai, B. S., Witola, W., Bissati, K. E., Mui, E., Moulton, H., et al. (2013). Conjugate Constructs, Delivery, and Use for Treatment of Disease. Available online at: http://www.google.com/patents/US8575126 (Accessed May 8, 2017).
McLeod, R., Lykins, J., Noble, A. G., Rabiah, P., Swisher, C. N., Heydemann, P. T., et al. (2014). Management of congenital toxoplasmosis. Curr. Pediatr. Rep. 2, 166–194. doi: 10.1007/s40124-014-0055-7
McPhillie, M., Zhou, Y., El Bissatai, K., Dubey, J., Lorenzi, H., Capper, M., et al. (2016). New paradigms for understanding and step changes in treating active and chronic, persistent apicomplexan infections. Sci. Rep. 6:29179. doi: 10.1038/srep29179
Mehta, P. K., Hale, T. I., and Christen, P. (1993). Aminotransferases: demonstration of homology and division into evolutionary subgroups. Eur. J. Biochem. 214, 549–561. doi: 10.1111/j.1432-1033.1993.tb17953.x
Min, K., Song, H. K., Chang, C., Kim, S. Y., Lee, K.-J., and Suh, S. W. (2002). Crystal structure of human nucleoside diphosphate kinase A, a metastasis suppressor. Proteins Struct. Funct. Bioinform. 46, 340–342. doi: 10.1002/prot.10038
Minor, W., Cymborowski, M., Otwinowski, Z., and Chruszcz, M. (2006). HKL-3000: the integration of data reduction and structure solution – from diffraction images to an initial model in minutes. Acta Crystallogr. D Biol. Crystallogr. 62, 859–866. doi: 10.1107/S0907444906019949
Mishra, A. K., Singh, N., Agnihotri, P., Mishra, S., Singh, S. P., Kolli, B. K., et al. (2017). Discovery of novel inhibitors for Leishmania nucleoside diphosphatase kinase (NDK) based on its structural and functional characterization. J. Comput. Aided Mol. Des. 31, 547–562. doi: 10.1007/s10822-017-0022-9
Montoya, J. G., and Liesenfeld, O. (2004). Toxoplasmosis. Lancet 363, 1965–1976. doi: 10.1016/S0140-6736(04)16412-X
Morris, R. J., Perrakis, A., and Lamzin, V. S. (2003). “ARP/wARP and automatic interpretation of protein electron density maps,” in Macromolecular Crystallography, Part D., ed. B.-M. Enzymology (Academic Press), 229–244. Available online at: http://www.sciencedirect.com/science/article/pii/S0076687903740117 (Accessed May 5, 2017).
Motomura, K., Hirota, R., Okada, M., Ikeda, T., Ishida, T., and Kuroda, A. (2014). A new subfamily of polyphosphate kinase 2 (Class III PPK2) catalyzes both nucleoside monophosphate phosphorylation and nucleoside diphosphate phosphorylation. Appl. Environ. Microbiol. 80, 2602–2608. doi: 10.1128/AEM.03971-13
Müller, I. B., Wu, F., Bergmann, B., Knöckel, J., Walter, R. D., Gehring, H., et al. (2009). Poisoning pyridoxal 5-phosphate-dependent enzymes: a new strategy to target the malaria parasite Plasmodium falciparum. PLoS ONE 4:e4406. doi: 10.1371/journal.pone.0004406
Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., et al. (2011). REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367. doi: 10.1107/S0907444911001314
Ngô, H. M., Zhou, Y., Lorenzi, H., Wang, K., Kim, T.-K., Zhou, Y., et al. (2017). Toxoplasma modulates signature pathways of human epilepsy, neurodegeneration & cancer. Sci. Rep. 7:11496. doi: 10.1038/s41598-017-10675-6
Opperdoes, F. R., and Michels, P. A. (2001). Enzymes of carbohydrate metabolism as potential drug targets. Int. J. Parasitol. 31, 482–490. doi: 10.1016/S0020-7519(01)00155-2
Pomel, S., Luk, F. C. Y., and Beckers, C. J. M. (2008). Host cell egress and invasion induce marked relocations of glycolytic enzymes in Toxoplasma gondii tachyzoites. PLoS Pathog 4:e1000188. doi: 10.1371/journal.ppat.1000188
Reyes, P., Rathod, P. K., Sanchez, D. J., Mrema, J. E., Rieckmann, K. H., and Heidrich, H. G. (1982). Enzymes of purine and pyrimidine metabolism from the human malaria parasite, Plasmodium falciparum. Mol. Biochem. Parasitol. 5, 275–290.
Rigden, D. J., Walter, R. A., Phillips, S. E. V., and Fothergill-Gilmore, L. A. (1999). Polyanionic inhibitors of phosphoglycerate mutase: combined structural and biochemical analysis. J. Mol. Biol. 289, 691–699. doi: 10.1006/jmbi.1999.2848
Roos, A. K., Burgos, E., Ericsson, D. J., Salmon, L., and Mowbray, S. L. (2005). Competitive inhibitors of Mycobacterium tuberculosis ribose-5-phosphate isomerase B reveal new information about the reaction mechanism. J. Biol. Chem. 280, 6416–6422. doi: 10.1074/jbc.M412018200
Ruan, J., Mouveaux, T., Light, S. H., Minasov, G., Anderson, W. F., Tomavo, S., et al. (2015). The structure of bradyzoite-specific enolase from Toxoplasma gondii reveals insights into its dual cytoplasmic and nuclear functions. Acta Crystallogr. D Biol. Crystallogr. 71, 417–426. doi: 10.1107/S1399004714026479
Samuel, B. U., Hearn, B., Mack, D., Wender, P., Rothbard, J., Kirisits, M. J., et al. (2003). Delivery of antimicrobials into parasites. Proc. Natl. Acad. Sci. U.S.A. 100, 14281–14286. doi: 10.1073/pnas.2436169100
Schneider, B., Xu, Y. W., Janin, J., Véron, M., and Deville-Bonne, D. (1998). 3′-Phosphorylated nucleotides are tight binding inhibitors of nucleoside diphosphate kinase activity. J. Biol. Chem. 273, 28773–28778. doi: 10.1074/jbc.273.44.28773
Schrödinger Release 2015-3. (2018). Small-Molecule Drug Discovery Suite 2018-3. New York, NY: Schrödinger, LLC.
Shah, S. A., Shen, B. W., and Brünger, A. T. (1997). Human ornithine aminotransferase complexed with L-canaline and gabaculine: structural basis for substrate recognition. Structure 5, 1067–1075. doi: 10.1016/S0969-2126(97)00258-X
Shen, B. W., Hennig, M., Hohenester, E., Jansonius, J. N., and Schirmer, T. (1998). Crystal structure of human recombinant ornithine aminotransferase. J. Mol. Biol. 277, 81–102. doi: 10.1006/jmbi.1997.1583
Sidik, S. M., Huet, D., Ganesan, S. M., Huynh, M.-H., Wang, T., Nasamu, A. S., et al. (2016). A genome-wide CRISPR screen in toxoplasma identifies essential apicomplexan genes. Cell 166, 1423–1435.e12. doi: 10.1016/j.cell.2016.08.019
Sillitoe, I., Lewis, T. E., Cuff, A., Das, S., Ashford, P., Dawson, N. L., et al. (2015). CATH: comprehensive structural and functional annotations for genome sequences. Nucleic Acids Res. 43, D376–D381. doi: 10.1093/nar/gku947
Singh, P. K., Kushwaha, S., Mohd, S., Pathak, M., and Misra-Bhattacharya, S. (2013). in vitro gene silencing of independent phosphoglycerate mutase (iPGM) in the filarial parasite Brugia malayi. Infect. Dis. Poverty 2:5. doi: 10.1186/2049-9957-2-5
Slabinski, L., Jaroszewski, L., Rychlewski, L., Wilson, I. A., Lesley, S. A., and Godzik, A. (2007). XtalPred: a web server for prediction of protein crystallizability. Bioinform. Oxf. Engl. 23, 3403–3405. doi: 10.1093/bioinformatics/btm477
Storici, P., Capitani, G., Müller, R., Schirmer, T., and Jansonius, J. N. (1999). Crystal structure of human ornithine aminotransferase complexed with the highly specific and potent inhibitor 5-fluoromethylornithine11. J. Mol. Biol. 285, 297–309. doi: 10.1006/jmbi.1998.2289
Sturm, N., Jortzik, E., Mailu, B. M., Koncarevic, S., Deponte, M., Forchhammer, K., et al. (2009). Identification of proteins targeted by the thioredoxin superfamily in Plasmodium falciparum. PLoS Pathog. 5:e1000383. doi: 10.1371/journal.ppat.1000383
Thi, E. P., Mire, C. E., Lee, A. C. H., Geisbert, J. B., Zhou, J. Z., Agans, K. N., et al. (2015). Lipid nanoparticle siRNA treatment of Ebola-virus-Makona-infected nonhuman primates. Nature 521, 362–365. doi: 10.1038/nature14442
Tonkin, M. L., Halavaty, A. S., Ramaswamy, R., Ruan, J., Igarashi, M., Ngô, H. M., et al. (2015). Structural and functional divergence of the aldolase fold in Toxoplasma gondii. J. Mol. Biol. 427, 840–852. doi: 10.1016/j.jmb.2014.09.019
Torgerson, P. R., and Mastroiacovo, P. (2013). The global burden of congenital toxoplasmosis: a systematic review. Bull. World Health Organ. 91, 501–508. doi: 10.2471/BLT.12.111732
Vedadi, M., Lew, J., Artz, J., Amani, M., Zhao, Y., Dong, A., et al. (2007). Genome-scale protein expression and structural biology of Plasmodium falciparum and related Apicomplexan organisms. Mol. Biochem. Parasitol. 151, 100–110. doi: 10.1016/j.molbiopara.2006.10.011
Vieira, P. S., de Giuseppe, P. O., Murakami, M. T., and de Oliveira, A. H. C. (2015). Crystal structure and biophysical characterization of the nucleoside diphosphate kinase from Leishmania braziliensis. BMC Struct. Biol. 15:2. doi: 10.1186/s12900-015-0030-8
Wan, C., Allen, T. M., and Cullis, P. R. (2014). Lipid nanoparticle delivery systems for siRNA-based therapeutics. Drug Deliv. Transl. Res. 4, 74–83. doi: 10.1007/s13346-013-0161-z
Wang, P., Jiang, L., Cao, Y., Zhang, X., Chen, B., Zhang, S., et al. (2018). Xanthone derivatives as phosphoglycerate mutase 1 inhibitors: design, synthesis, and biological evaluation. Bioorg. Med. Chem. 26, 1961–1970. doi: 10.1016/j.bmc.2018.02.044
Wang, Y.-Q., Huang, Z.-L., Chen, S.-B., Wang, C.-X., Shan, C., Yin, Q.-K., et al. (2017). Design, synthesis, and evaluation of new selective NM23-H2 binders as c-MYC transcription inhibitors via disruption of the NM23-H2/G-quadruplex interaction. J. Med. Chem. 60, 6924–6941. doi: 10.1021/acs.jmedchem.7b00421
Watanabe, T., Hatakeyama, H., Matsuda-Yasui, C., Sato, Y., Sudoh, M., Takagi, A., et al. (2014). in vivo therapeutic potential of Dicer-hunting siRNAs targeting infectious hepatitis C virus. Sci. Rep. 4:4750. doi: 10.1038/srep04750
Williamson, W. T., and Wood, W. A. (1966). “[108] d-Ribulose 5-phosphate 3-epimerase,” in Methods in Enzymology Carbohydrate Metabolism, ed W. Wood (East Lansing: Academic Press), 605–608. Available online at: http://www.sciencedirect.com/science/article/pii/0076687966091225
Winn, M. D., Ballard, C. C., Cowtan, K. D., Dodson, E. J., Emsley, P., Evans, P. R., et al. (2011). Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242. doi: 10.1107/S0907444910045749
Wise, E. L., Akana, J., Gerlt, J. A., and Rayment, I. (2004). Structure of d-ribulose 5-phosphate 3-epimerase from synechocystis to 1.6 Å resolution. Acta Crystallogr. D Biol. Crystallogr. 60, 1687–1690. doi: 10.1107/S0907444904015896
Wood, T. (1979). Purification and properties of d-ribulose-5-phosphate 3-epimerase from calf liver. Biochim. Biophys. Acta 570, 352–362. doi: 10.1016/0005-2744(79)90155-4
Yin, C., Zhang, T., Qu, X., Zhang, Y., Putatunda, R., Xiao, X., et al. (2017). in vivo excision of HIV-1 provirus by saCas9 and Multiplex Single-Guide RNAs in animal models. Mol. Ther. 25, 1168–1186. doi: 10.1016/j.ymthe.2017.03.012
Zhang, R., Andersson, C. E., Savchenko, A., Skarina, T., Evdokimova, E., Beasley, S., et al. (2003). Structure of Escherichia coli Ribose-5-phosphate isomerase: a ubiquitous enzyme of the pentose phosphate pathway and the calvin cycle. Struct. Lond. Engl. 11, 31–42. doi: 10.1016/S0969-2126(02)00933-4
Zheng, H., Cooper, D. R., Porebski, P. J., Shabalin, I. G., Handing, K. B., and Minor, W. (2017). CheckMyMetal: a macromolecular metal-binding validation tool. Acta Crystallogr. Sect. Struct. Biol. 73, 223–233. doi: 10.1107/S2059798317001061
Keywords: Toxoplasma gondii, PPMO, phosphoglycerate mutase, nucleoside diphoshate kinase, ribulose-3-phosphate epimerase, ribose-5-phosphate isomerase, ornithine aminotransferase, crystallography
Citation: Lykins JD, Filippova EV, Halavaty AS, Minasov G, Zhou Y, Dubrovska I, Flores KJ, Shuvalova LA, Ruan J, El Bissati K, Dovgin S, Roberts CW, Woods S, Moulton JD, Moulton H, McPhillie MJ, Muench SP, Fishwick CWG, Sabini E, Shanmugam D, Roos DS, McLeod R, Anderson WF and Ngô HM (2018) CSGID Solves Structures and Identifies Phenotypes for Five Enzymes in Toxoplasma gondii. Front. Cell. Infect. Microbiol. 8:352. doi: 10.3389/fcimb.2018.00352
Received: 30 July 2018; Accepted: 14 September 2018;
Published: 05 October 2018.
Edited by:
Nicolas Blanchard, INSERM U1043 Centre de Physiopathologie de Toulouse Purpan, FranceReviewed by:
Qian Han, Hainan University, ChinaAndrés Palencia, INSERM U1209 Institut pour l'Avancée des Biosciences (IAB), France
Copyright © Lykins, Filippova, Halavaty, Minasov, Zhou, Dubrovska, Flores, Shuvalova, Ruan, El Bissati, Dovgin, Roberts, Woods, Moulton, Moulton, McPhillie, Muench, Fishwick, Sabini, Shanmugam, Roos, McLeod, Anderson and Ngô. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rima McLeod, cm1jbGVvZEBic2QudWNoaWNhZ28uZWR1
Wayne F. Anderson, d2YtYW5kZXJzb25Abm9ydGh3ZXN0ZXJuLmVkdQ==
Huân M. Ngô, ZHIuaHVhbi5uZ29AZ21haWwuY29t
† These authors have contributed equally to this work
‡ Present Address: Joseph Lykins, Department of Internal Medicine and Department of Emergency Medicine, Virginia Commonwealth University Health System, Richmond, VA, United States
Ekaterina V. Filippova, Department of Biochemistry and Molecular Biology, Knapp Center for Biomedical Discovery, University of Chicago, IL, United States
Jiapeng Ruan, Department of Internal Medicine, Yale University School of Medicine, New Haven, CT, United States
Sarah Dovgin, Case Western Reserve University, Cleveland, OH, United States
Martin McPhillie, School of Chemistry and Astbury Centre for Structural Molecular Biology, University of Leeds, Leeds, United Kingdom