 Filipe Almeida
Filipe Almeida Maria P. Luís
Maria P. Luís Inês Serrano Pereira
Inês Serrano Pereira Sara V. Pais
Sara V. Pais Luís Jaime Mota
Luís Jaime Mota- 1Research Unit on Applied Molecular Biosciences (UCIBIO) – REQUIMTE, Departamento de Ciências da Vida, Faculdade de Ciências e Tecnologia, Universidade Nova de Lisboa, Costa da Caparica, Portugal
- 2Instituto de Tecnologia Química e Biológica António Xavier, Universidade Nova de Lisboa, Oeiras, Portugal
Chlamydia trachomatis is an obligate intracellular human pathogen causing mainly ocular and genital infections of significant clinical and public health impact. C. trachomatis multiplies intracellularly in a membrane bound vacuole, known as inclusion. Both extracellularly and from within the inclusion, C. trachomatis uses a type III secretion system to deliver several effector proteins into the cytoplasm of host cells. A large proportion of these effectors, the inclusion membrane (Inc) proteins, are exposed to the host cell cytosol but possess a characteristic hydrophobic domain mediating their insertion in the inclusion membrane. By yeast two-hybrid, we found that C. trachomatis Inc CT288 interacts with the human centrosomal protein CCDC146 (coiled-coil domain-containing protein 146). The interaction was also detected by co-immunoprecipitation in mammalian cells either ectopically expressing CCDC146 and CT288 or ectopically expressing CCDC146 and infected by a C. trachomatis strain expressing epitope-tagged and inclusion membrane-localized CT288. In uninfected mammalian cells, ectopically expressed full-length CCDC146 (955 amino acid residues) localized at the centrosome; but in cells infected by wild-type C. trachomatis, its centrosomal localization was less evident and CCDC146 accumulated around the inclusion. Recruitment of CCDC146 to the inclusion periphery did not require intact host Golgi, microtubules or microfilaments, but was dependent on chlamydial protein synthesis. Full-length CCDC146 also accumulated at the periphery of the inclusion in cells infected by a C. trachomatis ct288 mutant; however, a C-terminal fragment of CCDC146 (residues 692–955), which interacts with CT288, showed differences in localization at the periphery of the inclusion in cells infected by wild-type or ct288 mutant C. trachomatis. This suggests a model in which chlamydial proteins other than CT288 recruit CCDC146 to the periphery of the inclusion, where the CT288-CCDC146 interaction might contribute to modulate the function of this host protein.
Introduction
Chlamydiae are a large group of obligate intracellular bacteria, including human and animal pathogens, and symbionts of free-living amoebae. Among Chlamydiae, C. trachomatis is an important human pathogen causing ocular and genital infections. C. trachomatis ocular strains (serovars A–C) are the leading cause of infectious blindness (trachoma; Taylor et al., 2014), urogenital strains (serovars D–K) are the most common cause of bacterially sexually transmitted diseases worldwide, and lymphogranuloma venereum (LGV) strains (serovars L1–L3) cause invasive urogenital or anorectal infection (O'Connell and Ferone, 2016).
As all Chlamydiae, C. trachomatis undergoes a developmental cycle involving the inter-conversion between an infectious form, the elementary body (EB), and a non-infectious form, the reticulate body (RB). Attachment of EBs to host cells leads to bacterial invasion with the formation of a Chlamydia-containing vacuole known as inclusion. Early after uptake, EBs start differentiating into RBs and the inclusion segregates from normal endosomal maturation. Through the activity of cytoplasmic dynein, the nascent inclusion migrates along microtubules toward a centrosomal region. RBs then divide, while membrane and nutrients are routed to the inclusion by interaction with host cell vesicular and non-vesicular transport pathways. During the cycle, the inclusion remains near the host centrosome and nucleus. Eventually, most RBs differentiate back into EBs, which exit the host cell and can infect neighboring cells [reviewed by (Elwell et al., 2016) and references therein].
Throughout the developmental cycle, Chlamydiae manipulate host cells by using a type III secretion (T3S) system to deliver several effector proteins into host cells (Mueller et al., 2014). Chlamydial T3S substrates include inclusion membrane (Inc) proteins (Subtil et al., 2001), a family of Chlamydiae-unique proteins characterized by a bilobed hydrophobic motif thought to mediate their insertion into the inclusion membrane (Rockey et al., 2002). Bioinformatics indicates that C. trachomatis encodes about 60 Inc proteins (Bannantine et al., 2000; Dehoux et al., 2011; Lutter et al., 2012), and 37 of them have been experimentally detected in the inclusion membrane (Weber et al., 2015).
Considering that Inc proteins reside at the interface of the inclusion and the host cell cytosol, it is not surprising that many Inc proteins have been shown to play a role in mediating Chlamydia-host cell interactions. For example, C. trachomatis CT005/IncV tethers the inclusion to host the endoplasmic reticulum (Stanhope et al., 2017); CT101/MrcA and CT228 regulate the process of C. trachomatis release from host cells (Lutter et al., 2013; Nguyen et al., 2018); CT115/IncD interferes with host non-vesicular transport (Derré et al., 2011); CT116/IncE (Mirrashidi et al., 2015) and CT119/IncA (Delevoye et al., 2008) modulate host cell vesicular trafficking; CT223/IPAM and CT813/InaC are involved in the manipulation of host cell microtubules (Dumoux et al., 2015; Wesolowski et al., 2017); CT813/InaC also promotes actin assembly and Golgi redistribution around the inclusion (Kokes et al., 2015); and CT850 binds dynein light chain and thereby mediates transport of the inclusion toward the centrosome (Mital et al., 2015). In addition, CT229/CpoS inhibits host cell death in C. trachomatis-infected cells (Sixt et al., 2017; Weber et al., 2017). This could be related to its potential capacity to modulate host cell vesicular trafficking (Rzomp et al., 2006; Mirrashidi et al., 2015; Sixt et al., 2017), or to its role in the stability of the inclusion membrane (Weber et al., 2017). CT233/IncC and CT383 are also essential for the stability of the inclusion membrane (Weber et al., 2017). Together with the observation that some Inc proteins form homo- and heterooligomers (Gauliard et al., 2015), this is in line with the idea that some Inc proteins could have mainly a structural role (Mital et al., 2013; Moore and Ouellette, 2014). Finally, a large proteomics study identified interactions of 38 Incs with several host proteins (Mirrashidi et al., 2015). However, the function of many Inc proteins remains undefined.
In this work, we identified the host centrosomal protein CCDC146 (coiled-coil domain-containing protein 146) (Firat-Karalar et al., 2014) as an interacting partner of C. trachomatis Inc CT288 and found that in infected cells CCDC146 is recruited to the periphery of the inclusion in a CT288-independent fashion. However, CT288 might modulate the function of CCDC146 at the inclusion.
Materials and Methods
Mammalian Cell Lines
HeLa 229, HEK293T and Vero cells (all from the European Collection of Cell Culture; ECACC) were maintained in high-glucose Dulbecco's modified Eagle Medium (DMEM; Thermo Fisher Scientific) supplemented with heat-inactivated 10% (v/v) fetal bovine serum (FBS; Thermo Fisher Scientific) at 37°C in a humidified atmosphere of 5% (v/v) CO2. Cells were checked for Mycoplasma by conventional PCR either using the Venor® GeM Advance kit (Minerva Biolabs) or as described (Uphoff and Drexler, 2011).
Bacterial Strains and Growth Conditions
Escherichia coli TOP10 (Thermo Fisher Scientific) or NEB® 10β (New England Biolabs) were used for construction and purification of plasmids, and E. coli ER2925 (New England Biolabs) to amplify and purify plasmids for transformation of C. trachomatis. E. coli strains were routinely grown in liquid or solid lysogeny broth (LB) medium with the appropriate antibiotics and supplements. Plasmids were introduced into E. coli by electroporation.
Chlamydia trachomatis LGV serovar L2 strain 434/Bu (L2/434; from ATCC) was propagated in HeLa 229 cells using standard techniques (Scidmore, 2005). Throughout this work we used the nomenclature of the annotated C. trachomatis D/UW3 strain (Stephens et al., 1998). Transformation of C. trachomatis, first reported by the laboratory of Ian Clarke (Wang et al., 2011), was done essentially as described (Agaisse and Derre, 2013). The optimal antibiotic concentrations to select transformants were 1 U/ml of penicillin G, or 250 μg/ml of spectinomycin. Once established, the transformed strains were cultured in the presence of 10 U/ml of penicillin, or 500 μg/ml of spectinomycin, and plaque purified using Vero cells, as described (Nguyen and Valdivia, 2013). Infection of mammalian cells by C. trachomatis and quantification of infection progeny was done as previously described (da Cunha et al., 2017).
DNA Manipulation, Plasmids, and Primers
The plasmids used in this work, their main characteristics and construction details, are described in Table S1. The DNA primers used in their construction are listed in Table S2. Plasmids were constructed and purified using standard molecular biology procedures with proof-reading Phusion DNA polymerase (Thermo Fisher Scientific), restriction enzymes (Thermo Fisher Scientific), T4 DNA Ligase (Thermo Fisher Scientific), DreamTaq DNA polymerase (Thermo Fisher Scientific), DNA clean and concentrator™-5 kit and Zymoclean™ gel DNA recovery kit (Zymo Research), and GeneElute Plasmid Miniprep kit (Sigma Aldrich) or NZYMidiprep kit (NZYtech), according to the instructions of the manufacturers. The backbone plasmids used in this work were: pGADT7 (Clontech) and pGBKT7 (Clontech), used for yeast two-hybrid assays; pEGFP-C1 (Clontech), and pEF6/myc-His C (Thermo Fisher Scientific), used to generate mammalian transfection plasmids; and pSVP247 (da Cunha et al., 2017), a derivative of p2TK2—SW2 (Agaisse and Derre, 2013), used to generate a C. trachomatis expression plasmid bearing CT288 with a double hemagglutinin epitope tag (2HA) at its C-terminus (CT288-2HA). The accuracy of the nucleotide sequence of all the inserts in the constructed plasmids was confirmed by DNA sequencing.
Construction of a C. trachomatis ct288::aadA Mutant Strain
A C. trachomatis ct288::aadA mutant was generated using group II intron-based insertional mutagenesis, as previously described (Johnson and Fisher, 2013; Key and Fisher, 2017). Briefly, intron-insertion sites in the C. trachomatis L2/434 ctl0540 gene (ortolog of ct288 in C. trachomatis strain D/UW3) were identified using the TargeTron algorithm (Sigma). Then, the intron in pDFTT3 aadA (Table S1; Key and Fisher, 2017) was retargeted for ctl0540 using standard molecular biology procedures and the DNA primers listed in Table S2. The ctl0540 mutator plasmid (pML2; Table S1) was then used to transform C. trachomatis L2/434, as described above (in bacterial strains and growth conditions).
Yeast Two-Hybrid (Y2H) Screen
The Matchmaker® Gold Yeast Two-Hybrid System (Clontech) was used to screen of a pre-transformed Mate and Plate™ Library—normalized Universal Human HeLa cDNA library (Clontech), following the instructions of the manufacturer. In summary, Saccharomyces cerevisiae Y2HGold (Clontech) was transformed with plasmid pFA147, a derivative of pGBKT7 (Table S1) and mated with yeast strain Y187 (Clontech) carrying the HeLa cDNA library cloned into pGADT7. The two strains were mated and plated in lower stringency or double dropout (DDO) media (SD/–Leu/–Trp) supplemented with X-α-Galactosidase (X) and Aureobasidin A (A) (all from Clontech) and incubated 3–5 days at 28–30°C. The blue colonies grown on DDO/X/A media were patched into a higher stringency or quadruple dropout (QDO) media (SD/–Ade/–His/–Leu/–Trp) supplemented with X-α-Galactosidase and Aureobasidin A and incubated 3–5 days at 28–30°C. The blue colonies that grew in QDO/X/A were further analyzed for autonomous system activation, and for identification of the target protein, by sequencing the plasmid DNA (primers listed in Table S2). We analyzed 1.43 × 108 clones, patched 60 blue colonies and recovered 41 from high stringency media for further analysis.
Preparation of Yeasts Extracts
Protein extracts of the S. cerevisiae Y2HGold strains expressing Gal4 DNA binding domain fusion proteins were prepared by the trichloroacetic acid (TCA) method, as described in the supporting protocols of the Matchmaker® Gold Yeast Two-Hybrid System (Clontech).
Transient Transfection of Mammalian Cells
HeLa 229 and HEK293T cells were transfected with plasmid DNA by using the jetPEI reagent (Polyplus-Transfection) as detailed in the instructions of the manufacturer but using 250 ng of DNA per well of a 24-well tissue culture plate or 1,250 ng of DNA per well of a 6-well tissue culture plate. When seeding HEK293T cells, the tissue culture plates were previously coated with 0.001% (v/v) poly-L-lysine (Sigma Aldrich) in phosphate buffered saline (PBS). Cells were transfected immediately after infection with C. trachomatis and incubated at 37°C in a humidified atmosphere of 5% (v/v) CO2, prior to collection and immunoblotting, or fixation and immunolabeling.
Preparation of Cell Lysates, Fractionation and Co-immunoprecipitation (co-IP)
For the co-IP, we used GFP-Trap kit (ChromoTek), according to the protocol of the manufacturer with minor adjustments. In experiments involving only co-transfection, 5 × 105 HEK293T cells per well of a 6-well tissue culture plate, in a total of 2 wells per condition, were transfected with combinations of two plasmids, one encoding an EGFP fusion protein and the other encoding a HA-tagged protein. In experiments involving simultaneous infection and transfection, 5 × 105 HEK293T cells per well of a 6-well tissue culture plate, in a total of 12 wells per condition, were infected with C. trachomatis transformed with pSVP255 (pCT288-2HA; Table S1) at a multiplicity of infection of 2, and transfected with plasmids encoding for EGFP, EGFP-CCDC146692−955, or EGFP-CCDC1461−955 (Table S1). In both cases, after 24 h of incubation at 37°C in a humidified atmosphere of 5% (v/v) CO2, the cells were collected by trypsinization, pelleted by centrifugation and washed with PBS. In the case of the simultaneous infection and transfection experiments, an additional crosslinking step was done. For that, the cell pellets were resuspended in 1 ml of paraformaldehyde 1% (w/v) in PBS per 1 × 107 cells and incubated 7 min at room temperature. The cells were immediately centrifuged for 3 min at 1,800 × g at room temperature. Then, the pellets were washed twice with 500 μl 1.25 M glycine, and centrifugations of 3 min, 1,800 × g, room temperature. The remainder of the procedure was identical for co-transfection, and simultaneous infection and transfection experiments. The cell pellets were lysed in 200 μl ice-cold co-IP lysis buffer (20 mM Tris-HCl pH 7.5, 137 mM NaCl, 2 mM EDTA, 1% [v/v] NP40, freshly added protease inhibitors [Amresco] and phenylmethylsulfonyl fluoride [PMSF]) for 30 min on ice, and mixed by pipetting every 10 min. The lysates were centrifuged for 10 min, 17,000 × g, 4°C, and the supernatants were either used for analysis in SDS-PAGE (input of co-IP) or added to 800 μl of ice-cold GFP Trap buffer (10 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.5 mM EDTA). Then, 750 μl of the diluted supernatant were added to previously washed GFP-Trap beads and incubated overnight at 4°C with end-over-end mixing. The beads were washed six times with GFP-Trap buffer with centrifugations of 2 min, 2,000 × g, 4°C. Finally, the pelleted beads (output of co-IP) and the input of co-IP (20 ul) were resuspended in 20 μl of SDS-PAGE buffer 2X (100 mM Tris-HCl, pH 6.8, 4.0% [w/v] SDS, 20% [v/v] glycerol, 0.2 M β-mercaptoethanol, 0.2% [w/v] bromophenol blue). The samples were incubated for 10 min at 100°C and the proteins were analyzed by immunoblotting.
Antibodies and Fluorescent Dyes
For immunoblotting, mouse anti-myc antibody (Calbiochem) was used at 1:1,000, mouse anti-α-tubulin (Sigma-Aldrich) was used at 1:1,000, rat anti-HA 3F10 (Roche) was used at 1:1,000, goat anti-GFP (Sicgen) was used at 1:1,000, goat anti-MOMP (Abcam) was used at 1:1,000, and mouse anti-CT288 (a gift from Guangming Zhong; Li et al., 2008) was used at 1:1,000. Horseradish peroxidase-conjugated secondary antibody anti-mouse (GE Healthcare) was used at 1:10,000, anti-rat (Sigma-Aldrich) was used at 1:10,000, and anti-goat (Jackson ImmunoResearch Laboratories) was used at 1:10,000. For immunofluorescence, mouse anti-γ-tubulin antibody (Sigma-Aldrich) was used at 1:200, mouse anti-CT442 antibody (a gift from Guangming Zhong; Li et al., 2008) was used at 1:200, rat anti-HA 3F10 antibody (Roche) was used at 1:200, goat anti-GFP antibody (Sicgen) was used at 1:200, and goat anti-MOMP antibody (Abcam) was used at 1:200. Secondary antibody donkey anti-goat conjugated to cyanine 5 (Jackson ImmunoResearch Laboratories) was used at 1:200, anti-rat conjugated to rhodamine RedX (Jackson ImmunoResearch Laboratories) was used at 1:200, and goat anti-mouse AF568 (Invitrogen) was used at 1:200. 4′,6-diamidino-2-phenylindole (DAPI; Thermo Fisher Scientific) was used to stain DNA.
Immunoblotting
To prepare total mammalian cell extracts, HeLa 229 cells were trypsinized, collected and centrifuged 5 min, 2,300 × g, 4°C. The pellet was washed twice with ice-cold PBS and resuspended in 50 μl of SDS-PAGE buffer 1X (50 mM Tris-HCl 50 mM, pH 6.8, 2.0% [w/v] SDS, 10% [v/v] glycerol, 0.1 M β-mercaptoethanol, 0.1% [w/v] bromophenol blue) and 1 μl benzonase (Novagen). The samples were incubated 10 min at 100°C. Total yeast cell extracts, total mammalian cell extracts, and fractionated co-IP samples were resolved in 12% SDS-PAGE, followed by immunobloting. Proteins in the gel were transferred onto nitrocellulose membranes (Bio-Rad) and blocked in 4% (w/v) dried skimmed milk diluted in PBS containing 0.1% (v/v) Tween-20. The membranes were probed with primary and horseradish peroxidase-conjugated secondary antibodies and generally detected using SuperSignal™ West Pico Chemiluminescent Substrate (Thermo Fisher Scientific), or SuperSignal™ West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific), in a Chemidoc XRS+ system (Bio-Rad) or by exposure to Amersham Hyperfilm ECL (GE Healthcare).
Immunofluorescence Microscopy
For immunofluorescence microscopy, cells were either fixed with 4% (w/v) paraformaldehyde for 15 min at room temperature or with methanol for 5 min at −20°C. Antibodies were diluted in PBS with 0.1% (v/v) Triton-X100 and 10% (v/v) horse serum. The cells were washed in PBS with 0.1% (v/v) Triton-X100 and incubated for 1 h with primary antibodies. The cells were washed again in PBS with 0.1% (v/v) Triton-X100 and incubated with appropriate secondary antibodies for another 1 h. The cells were washed again first in PBS with 0.1% (v/v) Triton-X100, then PBS, and finally in H2O. In the end, coverslips were mounted onto glass slides using Aqua-poly/Mount mounting medium (Polysciences). Samples were analyzed using widefield fluorescence microscopes (Leica DMRA2 or Zeiss AxioImager D2) or a confocal laser scanning microscope (Zeiss LSM 710 META). Unless otherwise indicated, images were obtained by confocal microscopy and processed using Zeiss LSM Image Browser and Adobe Photoshop software or Fiji (Schindelin et al., 2012).
Drug Treatments
To disrupt the host Golgi complex, microtubules, or microfilaments, C. trachomatis-infected cells were incubated with 1 μg/ml of Brefeldin A (BFA; Sigma; stock solution at 5 mg/ml in dimethyl sulfoxide [DMSO]), 1 μg/ml nocodazole (stock solution at 5 mg/ml in DMSO; Sigma), or 2 μM cytochalasin D (stock solution at 5 mg/ml in DMSO; Sigma), respectively. To inhibit bacterial protein synthesis, C. trachomatis infected cells were incubated with 200 μg/ml of chloramphenicol (Carl Roth; stock solution at 34 mg/ml in ethanol).
Statistical Analyses
Statistical analyses were done using GraphPad Prism, version 5.04 for Windows, GraphPad Software, San Diego California, USA (www.graphpad.com)]. Differences between data sets were considered significant if P < 0.05.
Results
C. trachomatis Inc Protein CT288 Binds the Human Centrosomal Protein CCDC146 in a Y2H Assay
Chlamydia trachomatis Inc protein CTL0540 (herein CT288) from L2/434 strain is composed of 564 amino acid (aa) residues and encompasses bilobed hydrophobic motifs between residues 36–88, and 242–291 (Figure 1A). CT288 is different from most Inc proteins in that it contains more than one bilobed hydrophobic motif (Dehoux et al., 2011). To identify host cell interacting partners of CT288, we performed a Y2H screen using CT288 deleted of its first 35 aa residues and of its bilobed hydrophobic regions (CT288ΔNΔTMD; Figures 1A,B) as bait for proteins expressed from a human HeLa cell line cDNA library. After discarding false positives (i.e., autonomously activating the system or out-of-frame relative to the coding region of the Gal4 activation domain; GAL4AD), this resulted in the identification of 29 interacting protein fragments (Table S3). Among these CT288-interacting protein fragments, 9 (31%) were the C-terminal region (residues 692–955 or 721–955) of the 955 residues-long CCDC146 (Figure 1C and Table S3), which has been identified as a centrosomal protein (Firat-Karalar et al., 2014). Additional assays showed that CT288ΔNΔTMD can bind CCDC146692−955 (Figures 1D,E). We next used Y2H to identify the region of CT288 involved in binding to CCDC146692−955. For this, Y2H plasmids encoding CT28889−241 and CT288292−564 were constructed (Figures 1A,B). Using these plasmids, we observed that CT288292−564, but not CT28889−241, binds to CCDC146692−955 (Figure 1F). We also tested if CT288ΔNΔTMD could bind to full-length CCDC146 (CCDC146FL) and to the N-terminal region of CCDC146 (CCDC1461−691) by Y2H (Figures 1C,D). However, in this assay, we did not detect binding of CT288ΔNΔTMD to either CCDC146FL or CCDC1461−691 (Figure 1G).
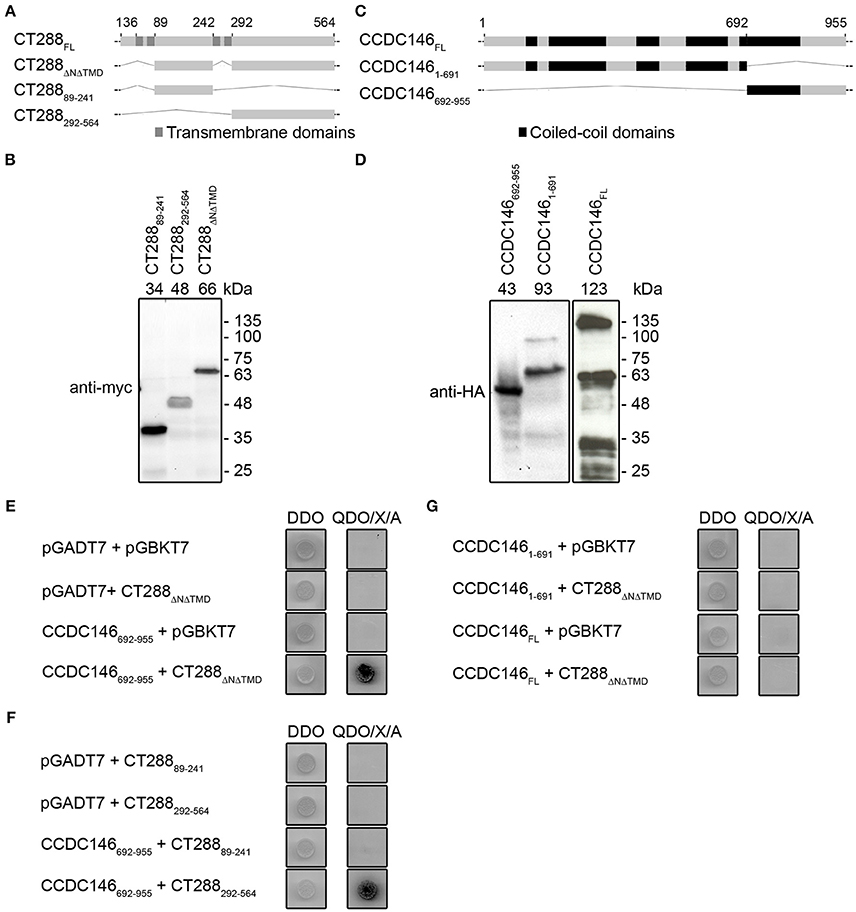
Figure 1. Chlamydia trachomatis Inc CT288 binds to human centrosomal protein CCDC146 by yeast two-hybrid (Y2H). (A,C) Schematic representation of CT288 and CCDC146 and of the fragments of these proteins used in Y2H assays. The predicted transmembrane domains of CT288 and coiled-coil domains of CCDC146 (as indicated in UniProt Q8IYE0; The UniProt, 2017) are highlighted. (B,D) Immunoblots of protein extracts of S. cerevisiae Y2HGold strains producing (B) myc-tagged fusions of CT288 fragments to the Gal4 DNA-binding domain or (D) HA-tagged fusions of CCDC146 to the Gal4 activation domain. The numbers above the blot indicate the predicted molecular mass of the corresponding fusion proteins. (E) Interaction between CT288ΔNΔTMD and CCDC146692−955 by Y2H. (F) Interaction between CT288292−564 and CCDC146692−955 by Y2H, but not between CT28889−241 and CCDC146692−955. (G) CCDC146FL and CCDC1461−691 do not bind CT288ΔNΔTMD by Y2H. In (E–G), DDO, double dropout media; QDO/X/A, quadruple dropout media supplemented with X-α-Galactosidase and Aureobasidin A (see Materials and Methods;) yeast growth as blue colonies (dark in the image) in high stringency QDO/X/A media indicates a protein-protein interaction. The empty pGADT7 and pGBKT7 plasmids were used as controls in the Y2H assays.
As we previously showed that CT288 from C. trachomatis LGV strains contains 18 unique aa residues that are different from its orthologues in ocular and urogenital strains (Almeida et al., 2012), we tested if CT288 from C. trachomatis serovar C (ocular strain TW3) and from serovar E (urogenital strain BOUR) were able to bind CCDC146692−955. For this, plasmids encoding CT288ΔNΔTMD from these strains were constructed (Figure S1A). Using these plasmids, we showed that CT288ΔNΔTMD from C. trachomatis ocular and urogenital strains also binds CCDC146692−955 in a Y2H assay (Figure S1B).
Overall, these experiments showed that C. trachomatis Inc CT288 can bind the human centrosomal protein CCDC146 in a Y2H assay.
CT288 and CCDC146 Interact After Their Ectopic Expression in Human Cells
To further test the interaction between CT288 and CCDC146, we performed co-immunoprecipitation (co-IP) assays after transient expression of the proteins in HEK293T cells. For this, we used plasmids encoding CT288ΔNΔTMD, CCDC146FL, and CCDC146692−955 either fused to the C-terminus of EGFP (EGFP-CT288ΔNΔTMD, EGFP- CCDC146FL, and EGFP-CCDC146692−955) or with a HA epitope tag at their C-termini (CT288ΔNΔTMD-HA, CCDC146FL-HA, and CCDC146692−955-HA). For the co-IP assays, we transfected HEK293T cells with combinations of two plasmids (one encoding an HA-tagged protein and one encoding either a EGFP fusion protein or EGFP alone, as control). The transfected cells were lysed and EGFP or EGFP fusion proteins were immunoprecipitated. This revealed that CT288ΔNΔTMD-HA was pulled-down by the IP of EGFP-CCDC146692−955 or of EGFP-CCDC146FL, but not by the IP of EGFP alone (Figure 2A and Figure S2). Similarly, CCDC146692−955-HA or CCDC146FL-HA were pulled-down by the IP of EGFP-CT288ΔNΔTMD but not by EGFP alone (Figure 2B and Figure S3). Therefore, CT288 and CCDC146 can also interact after their ectopic expression in HEK293T cells. Furthermore, in these conditions, we could detect an interaction between CT288ΔNΔTMD and CCDC146FL.
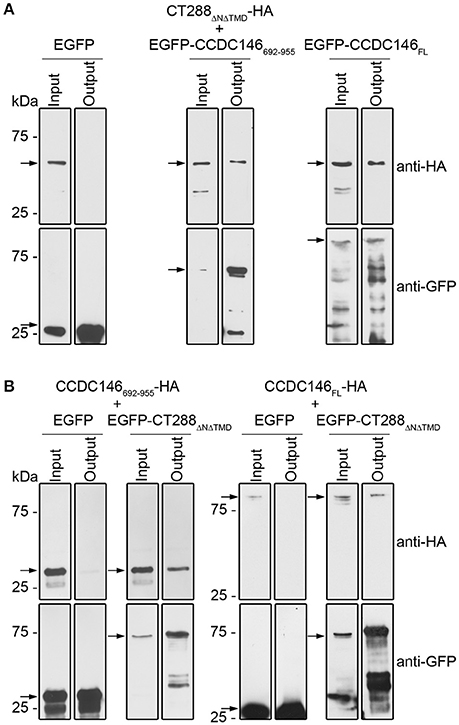
Figure 2. CT288 and CCDC146 interact after ectopic expression in mammalian cells. Plasmids encoding the indicated proteins were used to transfect HEK293T cells for 24 h. The cells were then lysed and EGFP proteins were pulled-down using GFP-Trap (ChromoTek). Proteins in the input lysate and in the pull-down output fractions were analyzed by immunoblotting using anti-HA and anti-GFP antibodies. (A) EGFP-CCDC146692−955 and EGFP-CCDC146FL, but not EGFP alone, co-immunoprecipitate CT288ΔNΔTMD-HA. (B) EGFP-CT288ΔNΔTMD, but not EGFP alone, co-immunoprecipitates CCDC146FL-HA and CCDC146692−955-HA. The arrows indicate the position in the blots of the relevant proteins (CT288ΔNΔTMD-HA, predicted molecular mass of 50 kDa; EGFP, 28 kDa; EGFP-CCDC146692−955, 60 kDa; EGFP-CCDC146FL, 140 kDa; EGFP-CT288ΔNΔTMD, 80 kDa; CCDC146FL-HA, 110 kDa; CCDC146692−955-HA, 30 kDa). The whole blots are shown in Figures S2, S3.
CT288 Produced by C. trachomatis During Infection Interacts With Ectopically Expressed EGFP-CCDC146692−955
We next tested if CT288 and CCDC146 could interact during C. trachomatis infection of HeLa cells. For this, we first constructed a C. trachomatis L2/434 strain carrying a plasmid encoding full-length CT288 with a double HA epitope tag at its C-terminus (pCT288-2HA). In this strain, expression of ct288-2HA is directed by the promoter of the gene encoding C. trachomatis IncD (Figure S4A). Immunoblotting of whole cell extracts of HeLa 229 cells left uninfected or infected by the C. trachomatis L2/434 strain or by L2/434 harboring pCT288-2HA revealed that CT288FL-2HA was expressed and migrated on SDS-PAGE as expected from its predicted molecular mass of 63 kDa (Figure S4B). Furthermore, indirect immunofluorescence microscopy using antibodies against HA and C. trachomatis major outer membrane protein (MOMP), or Inc protein CT442, of HeLa 229 cells infected for 24 h by C. trachomatis L2/434 harboring pCT288-2HA confirmed the localization of CT288FL-2HA at the inclusion membrane (Figure S4C). The intracellular multiplication of C. trachomatis L2/434 harboring pCT288-2HA was slightly, but significantly, affected relative to C. trachomatis L2/434, as judged by quantification of the number of infectious progeny of either strain at different times of infection of HeLa cells (Figure S4D). By comparison to C. trachomatis L2/434, chlamydial intracellular multiplication was also similarly affected in L2/434 strains transformed with plasmids expressing CT288FL-2HA from the tetracycline-inducible tetA promoter or from the ct288 promoter (data not shown).
To test if CT288 and CCDC146 could interact during infection, we infected HEK293T cells with C. trachomatis L2/434 harboring pCT288-2HA. After the inoculation with C. trachomatis, HEK293T cells were also transfected with plasmids encoding EGFP or EGFP-CCDC146692−955. After 24 h of infection, the cells were incubated with 1% (w/v) paraformaldehyde as a crosslinking agent, lysed, and EGFP or EGFP-CCDC146692−955 were immunoprecipitated. Subsequent immunoblotting revealed that CT288FL-2HA was pulled-down by the IP of EGFP-CCDC146692−955 but not by EGFP (Figure 3 and Figure S5). Despite of multiple attempts, we were unable to detect an interaction between CT288FL-2HA and ectopically expressed EGFP-CCDC146FL in Chlamydia-infected cells. Furthermore, as the endogenous levels of CCDC146 could not be detected in either HeLa or HEK293T cells by immunoblotting using a commercially available antibody that detected ectopically expressed EGFP-CCDC146FL or CCDC146FL-HA (Figure S6), we could not test the interaction of endogenous CCDC146 with CT288.
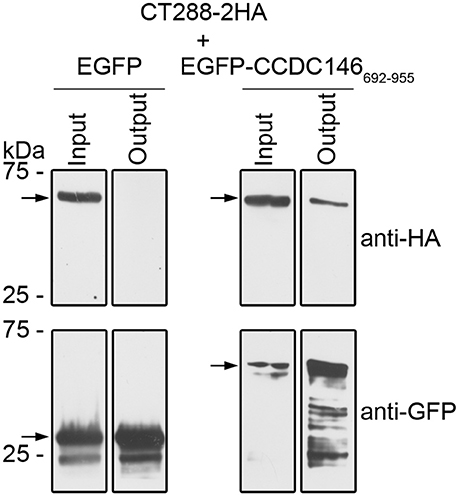
Figure 3. CT288-2HA produced by C. trachomatis during infection interacts with EGFP-CCDC146692−955. Plasmids encoding EGFP or EGFP-CCDC146692−955 were used to transfect HEK293T cells that were also infected for 24 h with C. trachomatis harboring pCT288-2HA. The cells were then lysed and EGFP proteins were pulled-down using GFP-Trap (ChromoTek). Proteins in the input lysate and in the pull-down output fractions were analyzed by immunoblotting using anti-HA and anti-GFP antibodies. The arrows indicate the position in the blots of the relevant proteins (CT288-2HA, predicted molecular mass of 66 kDa; EGFP, 28 kDa; EGFP-CCDC146692−955, 60 kDa). The whole blots are shown in Figure S5.
Overall, these experiments support that C. trachomatis Inc CT288 can interact with the human centrosomal protein CCDC146.
Ectopically Expressed CCDC146 Is Recruited to the Periphery of the Inclusion Membrane
To analyse the subcellular localization of ectopically expressed CCDC146, HeLa 229 cells were transiently transfected with plasmids encoding EGFP-CCDC146FL or CCDC146FL-HA. Analysis of the transfected cells by indirect immunofluorescence microscopy with antibodies against GFP or HA and γ-tubulin (to label the centrosomes) revealed that in ~ 90% of the cells both EGFP-CCDC146FL (Figures 4A,B) and CCDC146FL-HA (Figures S7A,B) could be co-localized with γ-tubulin, although the proteins were also seen dispersed in the cytosol. However, in cells transfected with plasmids encoding EGFP-CCDC146FL or CCDC146FL-HA and infected for 24 h by C. trachomatis L2/434, the co-localization between γ-tubulin and EGFP-CCDC146FL (Figures 4A,B) or CCDC146FL-HA (Figures S7A,B) was less evident and could only be seen in about 40% of the cases. Instead, in virtually all infected cells, ectopically expressed EGFP-CCDC146FL or CCDC146FL-HA showed a ring-like appearance suggestive of their accumulation in the periphery of the inclusion (Figure 4A and Figure S7A). To further analyse this, cells transiently expressing EGFP-CCDC146FL and infected by the L2/434 strain were analyzed by indirect confocal immunofluorescence microscopy using anti-MOMP antibodies or anti-Inc CT442 antibodies, which confirmed that EGFP-CCDC146FL was around the inclusion (Figure 4C) and co-localized with CT442 (Figure 4D). Similar observations were made in cells expressing CCDC146FL-HA and infected by the L2/434 strain (Figures S7C,D). We also examined the localization of ectopically expressed EGFP-CCDC146FL, or EGFP alone as control, in cells infected by C. trachomatis L2/434 harboring pCT288-2HA. As expected from the previous analyses (Figures 4A–C), EGFP-CCDC146FL, but not EGFP alone, localized at the periphery of the inclusion membrane and showed colocalization with bacterially translocated CT288-2HA (Figure 4E).
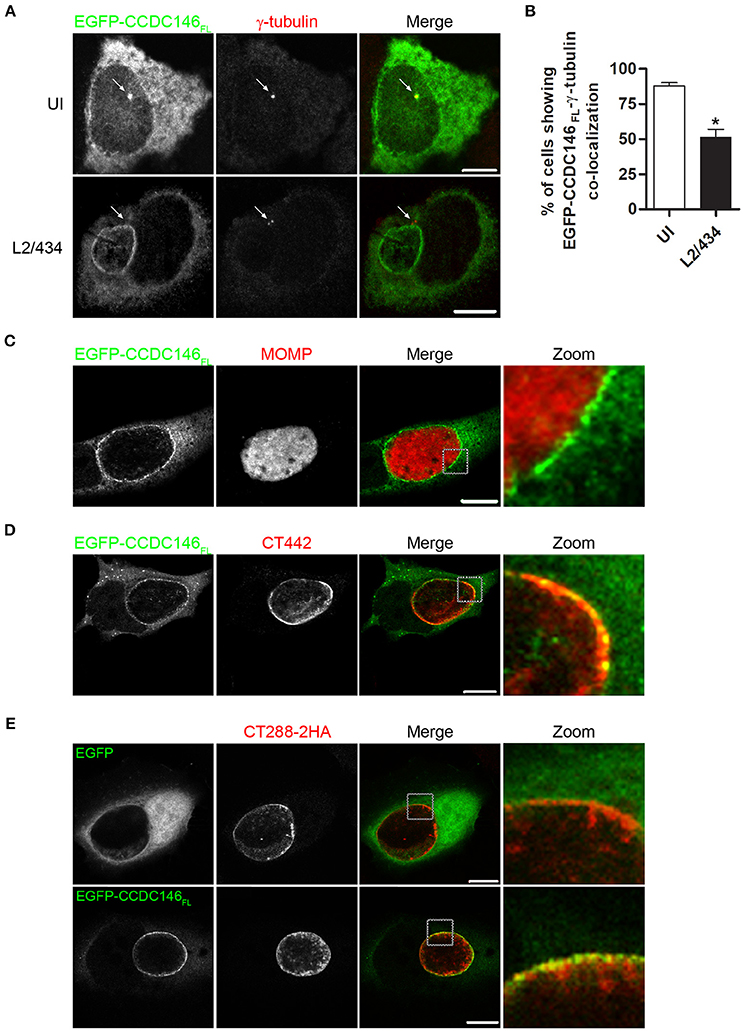
Figure 4. EGFP-CCDC146 is recruited to the periphery of the inclusion membrane in C. trachomatis infected cells. HeLa cells transfected with plasmids encoding either EGFP-CCDC146FL or EGFP were left uninfected (UI) or infected for 24 h with C. trachomatis L2/434 (A–D) or C. trachomatis L2/434 harboring pCT288-2HA (E). (A) The cells were fixed with methanol, immunolabeled with anti-GFP and anti-γ-tubulin antibodies, and appropriate fluorophore-conjugated secondary antibodies, and analyzed by confocal immunofluorescence microscopy. The arrows in each panel highlight the γ-tubulin-labeled centrosome. (B) Percentage of uninfected or C. trachomatis-infected HeLa 229 cells showing co-localization between EGFP-CCDC146FL and γ-tubulin. Data represents three independent experiments (100 cells counted per experiment). P-values were calculated by a two-tailed unpaired Student's t-test relative to UI cells. *P < 0.05. (C–E) The cells were fixed with paraformaldehyde 4% (w/v), immunolabeled with anti-MOMP (C), anti-CT442 (D), or anti-HA (E) antibodies, and appropriate fluorophore-conjugated secondary antibodies, and analyzed by confocal immunofluorescence microscopy. All scale bars, 10 μm.
In summary, these experiments showed that in Chlamydia-infected cells ectopically expressed CCDC146 is recruited to the periphery of the inclusion membrane.
Recruitment of Full-Length CCDC146 to the Periphery of the Inclusion Membrane Is Not Dependent on CT288
To analyse if recruitment of CCDC146 to the periphery of the inclusion membrane is dependent on CT288, we constructed a C. trachomatis L2/434-derived strain where the ct288 gene was inactivated by insertion between nucleotides 114 and 115 of a modified group II intron containing a spectinomycin-resistance gene (aadA) (Figure 5A). The correct insertion of the intron in the ct288::aadA mutant strain was verified by PCR and locus-specific DNA sequencing (Figure S8). This insertion could lead to the production of a truncated peptide of 49 amino acid residues, lacking the transmembrane regions of CT288 and with 11 of the residues encoded by the intron nucleotide sequence. To further characterize the insertional mutant strain, immunoblotting of extracts of HeLa cells infected by L2/434 or by ct288::aadA confirmed that insertion of the intron prevented detectable production of full-length CT288 (Figure 5B). A previous report where a similar penicillin-resistant ct288::bla mutant was constructed and analyzed, revealed a slight intracellular growth defect associated with the ct288 mutant strain (Weber et al., 2017). However, the intracellular growth of C. trachomatis ct288::aadA was not significantly different from the parental L2/434 strain (Figure 5C). In HeLa cells transfected with the plasmid encoding EGFP-CCDC146FL and infected for 24 h either by the L2/434 strain or by the ct288::aadA mutant strain, we observed recruitment of EGFP-CCDC146FL to the periphery of the inclusion membrane in both cases (Figure 5D). EGFP-CCDC146FL was first detected around the inclusion at 16 h p.i., in cells infected by the L2/434 strain (Figure S9A), and the same was observed in cells infected by the ct288::aadA mutant strain (Figure S9B). We also tested if presence of ectopically expressed EGFP-CCDC146FL at the periphery of the inclusion would be affected by disrupting host cell microtubules (with nocodazole), microfilaments (with cytochalasin D), or the Golgi complex (with BFA), and whether this would depend on CT288. However, in Chlamydia-infected cells, regardless of the presence of intact ct288, EGFP-CCDC146FL remained at the periphery of the inclusion after disruption of the cytoskeleton or Golgi (Figure S10A). Likewise, when adding the same drugs at 8 h p.i., and fixing the cells at 16 h p.i., we observed that EGFP-CCDC146FL was recruited to the periphery of the inclusion in cells infected by the parental or mutant strain (Figure S10B). However, when chloramphenicol was added to block protein bacterial synthesis at 8 h p.i., then EGFP-CCDC146FL was not detected around the inclusion at 16 h p.i., in cells infected by either of the strains (Figure S10C).
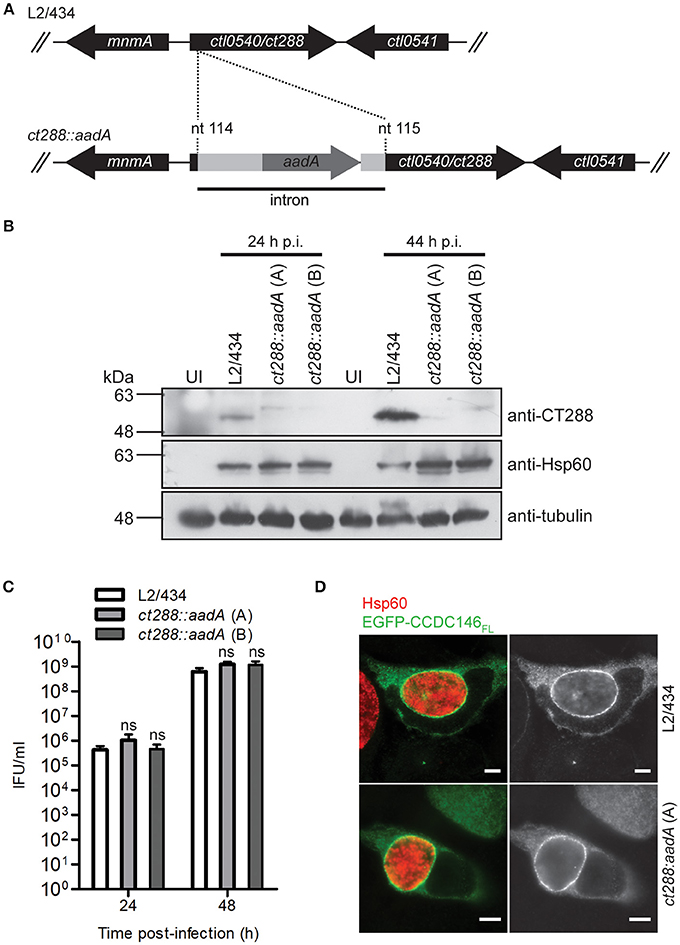
Figure 5. Recruitment of full-length CCDC146 to the periphery of the C. trachomatis inclusion does not depend on CT288. (A) A C. trachomatis ctl0540 (ct288 in C. trachomatis strain D/UW3) mutant was generated in strain L2/434 by the targeted insertion of a modified group II intron carrying the aadA gene, conferring spectinomycin resistance. (B) HeLa cells were infected for 24 or 44 h (p.i., post-infection) with C. trachomatis L2/434 or with two C. trachomatis ct288:aadA insertional mutant plaque-purified clones (A,B). Whole cell lysates were analyzed by immunoblotting with antibodies against CT288, C. trachomatis Hsp60 (bacterial loading control) and α-tubulin (loading control for host cells). (C) HeLa cells were infected with the indicated strains at a multiplicity of infection of 5 and recoverable inclusion forming units (IFUs) were determined at 24 and 48 h p.i., Data are mean and standard error of the mean of 4 independent experiments. For each time-point, P-values were calculated by a two-tailed unpaired Student's t-test relative to the L2/434 strain; ns, not significant. (D) HeLa cells transfected with plasmids encoding EGFP or EGFP-CCDC146FL, and infected for 24 h by C. trachomatis L2/434 or ct288:aadA (clone A) were fixed with methanol, immunolabeled with anti-GFP and anti-Hsp60 antibodies, and appropriate fluorophore-conjugated secondary antibodies, and analyzed by immunofluorescence microscopy. Identical observations were made with clone B (data not shown). Scale bars, 5 μm.
In summary, despite the ability of CT288 to interact with CCDC146 (Figures 1–3), recruitment of ectopically expressed CCDC146 to the periphery of the inclusion in C. trachomatis-infected cells does not depend on the presence of CT288 (Figures 4, 5). Recruitment of ectopically expressed EGFP-CCDC146FL to the periphery of the inclusion does not require intact host Golgi, microtubules, or microfilaments, but depends on chlamydial protein synthesis.
CT288 Modulates the Recruitment of Ectopically Expressed EGFP-CCDC146692−955 to the Periphery of the Inclusion
A fragment of CCDC146 between amino acids residues 692 and 955 was the minimal region of the protein that we have found to bind CT288 (Figures 1–3). We therefore tested if ectopically expressed EGFP-CCDC146692−955 was recruited to the periphery of the inclusion and if this was dependent on CT288. In C. trachomatis-infected cells, EGFP-CCDC146692−955 was more frequently found surrounding the inclusion of the L2/434 strain (28 ± 4%) than of the ct288::aadA mutant strain (11 ± 1%, clone A; 13 ± 2%, clone B) (Figures 6A,B). However, localization of ectopically expressed EGFP-CCDC146692−955 around the inclusion (Figure 6A) was much less evident than with EGFP-CCDC146FL (Figures 4, 5D). No detectable recruitment of EGFP-CCDC146692−955 to the inclusion periphery was also more frequently seen in cells infected by the parental strain (53 ± 7%) than by the ct288 mutant strain (17 ± 4%, clone A; 18 ± 1%, clone B) (Figures 6A,B). This is because in C. trachomatis-infected cells, EGFP-CCDC146692−955 was seen in patches near the inclusion (Figure 6A), which was much more frequent in cells infected by the ct288::aadA mutant (72 ± 4%, clone A; 69 ± 4%, clone B) than by the L2/434 strain (18 ± 6%; Figure 6B). This localization of EGFP-CCDC146692−955 in patches resembled the previously described inclusion microdomains that localize near host cell centrosomes (Mital et al., 2010). In fact, the patches of EGFP-CCDC146692−955 detected in cells infected by C. trachomatis were always very near of, or even co-localizing with, γ-tubulin-labeled centrosomes (Figure 6C).
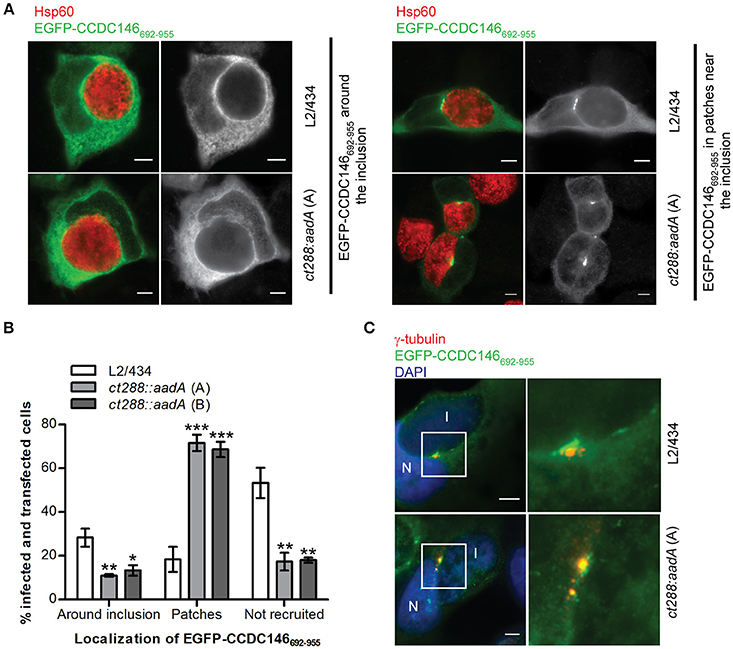
Figure 6. Recruitment of EGFP-CCDC146652−955 to the periphery of the C. trachomatis inclusion is modulated by CT288. HeLa cells transfected with a plasmid encoding EGFP- CCDC146652−955 were infected for 24 h with C. trachomatis L2/434 or the ct288::aadA mutant (clones A and B; see Figure 5). The cells were fixed with methanol, immunolabelled with anti-GFP, anti-γ-tubulin, or anti-Hsp60 antibodies, and appropriate fluorophore-conjugated secondary antibodies, and/or stained with DAPI (as indicated), and analyzed by immunofluorescence microscopy. (A) Representative images of the localization of ectopically expressed EGFP-CCDC146652−955 surrounding the inclusion (left-side panels), or in patches near the inclusion (right-side panels), in Chlamydia-infected cells. (B) Enumeration of cells infected by either C. trachomatis L2/434 or the ct288::aadA mutant showing ectopically expressed EGFP-CCDC146652−955 surrounding the inclusion, in patches near the inclusion, or not recruited to the inclusion periphery. Data represents three independent experiments (at least 50 infected and transfected cells were counted per experiment). P-values were obtained by one-way ANOVA and Dunnett post-hoc analyses relative to cells infected by the L2/434 strain in each class (around inclusion, in patches, or not recruited). *P < 0.05; **P < 0.01; ***P < 0.001. (C) Co-localization between EGFP-CCDC146652−955 in patches near the inclusion and γ-tubulin, in cells infected by C. trachomatis L2/434 (less frequently observed; see B) or the ct288::aadA mutant (more frequently observed; see B). I, inclusion; N, host cell nucleus. Scale bars, 5 μm.
In summary, these results indicate that: (i) EGFP-CCDC146692−955 can be recruited to the inclusion periphery partly, but not only, by CT288; (ii) the EGFP-CCDC146692−955-CT288 interaction is not sufficient to fully recruit EGFP-CCDC146692−955, either because EGFP-CCDC146692−955 is not always recruited or dissociates after recruitment; (iii) Chlamydia infection of HeLa cells promotes the accumulation of EGFP-CCDC146692−955 in patches near the inclusion but this is inhibited by CT288. Therefore, CT288 modulates the recruitment of EGFP-CCDC146692−955 to the periphery of the inclusion, which also suggests it could control the function of CCDC146 at the inclusion membrane during infection.
Discussion
We found that the human centrosomal protein CCDC146 can bind the C. trachomatis Inc protein CT288 and is recruited to the periphery of the inclusion membrane in cells infected by C. trachomatis. This recruitment of CCDC146 is not CT288-dependent because it was observed both in cells infected by C. trachomatis strain L2/434 (producing CT288) and by a C. trachomatis mutant (ct288::aadA) that does not produce CT288. Furthermore, recruitment of CCDC146 did not require intact host Golgi, microtubules or microfilaments, but was dependent on chlamydial protein synthesis. This indicates that CCDC146 is recruited to the periphery of the inclusion membrane either directly or indirectly by other Chlamydia proteins. CCDC146 could bind directly other Inc proteins or interact with host cell proteins recruited by them to the inclusion membrane or to its periphery. 14-3-3β is another host protein recruited to the inclusion membrane that binds more than one Inc (at least, IncG and CT813/InaC) (Scidmore and Hackstadt, 2001; Kokes et al., 2015). Another example indicating the existence of multiple interactions, or redundant mechanisms, between Incs and host proteins is CT005/IncV, which binds and recruits VAPs (vesicle-associated membrane protein-associated proteins) to the inclusion membrane, but an incV mutant displays a moderate defect in VAP recruitment (Stanhope et al., 2017). In our case, we propose a model in which the described interaction between CT288 and CCDC146 would function to modulate the activity of CCDC146 at the inclusion membrane. This hypothesis is supported by the observations indicating that recruitment of ectopically expressed EGFP-CCDC146692−955 to the inclusion periphery is modulated by CT288. However, a role of CT288 in contributing to the recruitment of full-length CCDC146 to the inclusion membrane cannot be ruled out, as we analyzed ectopically expressed EGFP-CDCC146FL and hypothetical CT288-dependent subtle effects might only be detectable by looking at the endogenous CCDC146.
CCDC146 has been shown to localize at the centrosome, in particular at the mother centriole (Firat-Karalar et al., 2014), and we could observe the centrosomal localization by ectopic expression of CCDC146 in HeLa cells and its co-localization with γ-tubulin. Although CCDC146 (NCBI Reference Sequence: NP_065930.2) possesses a predicted SMC (structural maintenance of chromosomes) region, the exact cellular roles of the protein are unknown. Unfortunately, it was not possible to address the physiological and functional relevance of the recruitment of CCDC146 to the periphery of the inclusion, because we could not detect production of the protein in the mammalian cell lines used (HeLa and HEK293T). This did not enable us to test if the CT288-CCDC146 interaction occurs under near physiological conditions, where both proteins are produced at endogenous levels, and prevented us from testing the consequences of depleting CCDC146 on the intracellular multiplication of C. trachomatis. The presumably very low levels of CCDC146 in HeLa and HEK293T cells also explain why the protein was not found in a previous quantitative proteomics analysis of the inclusion (Aeberhard et al., 2015), and why the CT288-CCDC146 interaction was not detected in the proteomics-based Inc-human interactome (Mirrashidi et al., 2015). However, a RNA sequencing (RNA-Seq) analysis of tissue samples from several human individuals revealed the highest levels of CCDC146 RNA in testis and above-average levels of CCDC146 RNA in the endometrium (Fagerberg et al., 2014), which suggests that the protein should be present in tissues related to C. trachomatis genital infections. Furthermore, CT288 is a core Inc protein (Lutter et al., 2012), with orthologues in different Chlamydia species, but not in environmental Chlamydiae, and CCDC146 is conserved among a large number of eukaryotes, including mammals, birds, and amphibians. Therefore, while this remains to be directly analyzed, it is possible that the CT288-CCDC146 interaction and recruitment of CCDC146 to the periphery of the inclusion are relevant in the general context of Chlamydia infections.
We constructed a C. trachomatis ct288::aadA mutant using the same parental strain, methodology and intron insertion site that had been previously used to generate a ct288::bla mutant (Weber et al., 2017). By comparison to the parental strain, we did not detect an intracellular growth defect for the ct288::aadA mutant. In contrast, a slight defect in intracellular growth has been reported for the ct288::bla mutant (Weber et al., 2017). Without a side-by-side comparison, the reason for the discrepancy in the analyses of the intracellular growth of the two ct288 mutants are unclear, but they could be related to the different antibiotic resistance genes in the inserted intron, different media and serum composition, or other slightly different experimental procedures. Weber et al. also reported a defect of the ct288::bla during in vivo infection of mice (Weber et al., 2017), which we did not analyze for the ct288::aadA mutant. Nevertheless, either considering our observations or those from Weber et al, it appears that CT288 is largely dispensable for C. trachomatis intracellular growth in a tissue culture infection system. This could be thought as somewhat surprising knowing that C. trachomatis have undergone a process of genome reduction (Nunes and Gomes, 2014), which suggests that the large majority of its actual genes should have an important role during infection. However, other C. trachomatis mutants defective for Inc proteins or other chlamydial T3S effectors that have been characterized (e.g., ct005/incV, ct179, ct224, ct695/tmeB, ct813/inaC, ct850) also did not reveal an intracellular growth defect in tissue culture infection models (McKuen et al., 2017; Stanhope et al., 2017; Weber et al., 2017). This suggests that the essentiality of a C. trachomatis gene for infection could extend beyond a role in intracellular growth in tissue culture and that a more detailed phenotypic analysis of the ct288 mutant is required to clarify the function(s) of the protein during infection.
We detected the interaction between CT288 and CCDC146 using different methods (Y2H and co-immunoprecipitations), different experimental systems (yeast, transfected cells, infected cells), and different tagged versions (e.g., GFP- or 2HA-tagged) of the full-length or truncated proteins. However, regarding CT288, we only used a full-length version (containing its transmembrane domains) produced by C. trachomatis in infected cells because we expected that in the other experimental systems the presence of these hydrophobic regions would direct the protein to membranes (or lead to protein aggregation) and prevent the detection of protein-protein interactions. For CCDC146, when we ectopically expressed the C-terminal region of the protein (CCDC146692−955), the CT288-CCDC146 interaction was detected by all methods and in all experimental systems. However, this does not necessarily mean that the CT288-CCDC146 interaction is only mediated by this C-terminal region of CCDC146, as we did not perform a detailed mapping of the regions (and predicted coiled-coil domains) of the protein that are involved in the interaction. The interaction between CT288 and CCDC146 using full-length CCDC146 could only be detected when both proteins were ectopically expressed in HEK293T cells. It is possible that the topology of the fusion proteins in the Y2H assay prevented the detection of this interaction. Regarding the co-immunoprecipitation assay from infected cells, the detection of the CT288-CCDC146FL interaction was probably hampered by the low amounts of CT288-2HA that could be solubilized from the protein inserted in the inclusion membrane and the lower level of expression of EGFP-CCDC146FL relative to EGFP-CCDC146692−955. It should also be noted that prolonged ectopic expression of CCDC146FL or CCDC146692−955 was toxic to mammalian cells and for that reason we did not analyze the presence of the protein at the periphery of the inclusion for longer than 24 h of C. trachomatis infection.
In previous studies, plasmid-encoded and FLAG-tagged CT288 (expressed from the tetracycline promoter) delivered by C. trachomatis into the inclusion membrane has been shown to localize in inclusion microdomains (Weber et al., 2015). These microdomains are characterized by the presence of phosphorylated Src family kinases (seen as patches, by immunofluorescence microscopy) nearby host centrosomes (Mital et al., 2010). We consistently observed a homogenous distribution of both Chlamydia-delivered CT288-2HA and ectopically-expressed EGFP-CCDC146FL around the inclusion, which did not suggest their localization at inclusion microdomains. However, we did detect ectopically expressed EGFP-CCDC146692−955 in patches near the inclusion and of host centrosomes labeled with γ-tubulin. This localization of EGFP-CCDC146692−955 was much more frequently seen in cells infected by the ct288::aadA mutant than by the parental L2/434 strain, indicating that CT288 interferes with the localization of the protein at the microdomains. Because recruitment of EGFP-CCDC146FL, and possibly also of EGFP-CCDC146692−955, to the inclusion periphery depends on other chlamydial proteins, this further suggests a complex and dynamic recruitment of CCDC146 to the inclusion that might involve its transient localization at microdomains in a process modulated by CT288.
The CT288-CCDC146 interaction can be integrated in a series of previous observations that establish an intricate relationship between C. trachomatis and its virulence proteins and the host cell centrosome, the main microtubule-organizing center in mammalian cells. It has been known that the C. trachomatis inclusion migrates toward the centrosome in a dynein-dependent, but dynactin-independent, manner and that this inclusion-centrosome association is maintained throughout infection (Grieshaber et al., 2003). Notably, infection of host cells by C. trachomatis leads to different types of centrosomal or mitotic spindle abnormalities (Grieshaber et al., 2006; Johnson et al., 2009; Knowlton et al., 2011), which have been suggested to be related with the known epidemiological link between C. trachomatis infection and cervical cancer (Knowlton et al., 2011). Furthermore, C. trachomatis host cell infection causes alterations in the microtubule network (Al-Zeer et al., 2014; Dumoux et al., 2015; Wesolowski et al., 2017). Some of the molecular players involved in these C. trachomatis inclusion-centrosome/microtubule interactions have been revealed: Inc proteins IncB, CT101, CT222, and CT850 associate with Src family kinases in inclusion microdomains that associate with centrosomes (Mital et al., 2010); the chlamydial protease CPAF is responsible for defects in the regulation of centrosome duplication and for inducing early mitotic exit (Brown et al., 2014); Inc CT850 binds dynein light chain, which likely promotes the positioning of the inclusion at the centrosome (Mital et al., 2015); and Inc CT223/IPAM binds the host centrosomal protein 170 kDa (CEP170) to subvert microtubule functions (Dumoux et al., 2015). In addition, in a large proteomics screen several Inc proteins have been shown to be capable of binding to host cell proteins with functions related to the centrosome (CT005, CT192, CT383) and with the eukaryotic cell division cycle (CT036, CT135, CT249, CT324, CT449, CT556, CT728, CT788). At the present, it is generally unclear how all these centrosome-related phenotypes and C. trachomatis proteins are related with each other to contribute to chlamydial infection. Therefore, it is also unknown how the CT288-CCDC146 interaction relates with all these previous observations. A more detailed future characterization of the C. trachomatis ct288 mutant could provide insights regarding this issue.
In summary, we found a novel interaction between a C. trachomatis Inc protein (CT288) and a host cell protein (CCDC146), which is recruited to the periphery of the inclusion membrane and thus could play an important role during Chlamydia host cell infection. However, the relevance of our findings remains to be validated in more physiological infection models.
Author Contributions
FA performed the experiments, interpreted the data, wrote the article. MPL and ISP performed the experiments, interpreted the data. SVP performed the experiments. LJM conceived and designed the project, interpreted the data, wrote the article.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by Fundação para a Ciência e a Tecnologia (FCT/MCTES) through grant PTDC/IMI-MIC/1300/2014 and by the Applied Molecular Biosciences Unit (UCIBIO), which is financed by national funds from FCT/MCTES (UID/Multi/04378/2013) and co-financed by the European Regional Development Fund (ERDF) under the PT2020 Partnership Agreement (POCI-01-0145-FEDER-007728). FA, ISP, and SVP were supported by PhD fellowships (SFRH/BD/73545/2010, SFRH/BD/129756/2017, and PD/BD/52210/2013, respectively) from FCT/MCTES. Fellowship PD/BD/52210/2013 is within the scope of the PhD program Molecular Biosciences (PD/00133/2012) funded by FCT/MCTES.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2018.00254/full#supplementary-material
Figure S1. CT288 from different C. trachomatis strains (L2/434, C/TW3, and E/Bour) also bind CCDC146 by Y2H. (A) The DNA encoding CT288 from C. trachomatis strains L2/434, C/TW3, and E/Bour was used to construct plasmids enabling production of myc-tagged fusions of CT288ΔNΔTMD to the Gal 4 DNA-binding domain. The numbers above the blot indicate the predicted molecular mass of the corresponding fusion proteins. (B) Interaction between CT288ΔNΔTMD from the indicated C. trachomatis strain and CCDC146692−955 by Y2H. Yeast growth as blue colonies (dark in the image) in high stringency QDO/X/A media indicates a protein-protein interaction. pGADT7 is the empty plasmid (used to generate the construct encoding CCDC146692−955) used as control in the Y2H assays.
Figure S2. Whole blots of the co-immunoprecipitation experiments to test if CT288ΔNΔTMD is pulled-down by EGFP-CCDC146 fusion proteins after ectopic expression in mammalian cells. For details see Figure 2 legend. The arrows indicate the position in the blots of the relevant proteins. I, input fractions; O, output fractions.
Figure S3. Whole blots of the co-immunoprecipitation experiments to test if CCDC146-derived proteins are pulled-down by EGFP-CT288ΔNΔTMD after ectopic expression in mammalian cells. For details see Figure 2 legend. The arrows indicate the position in the blots of the relevant proteins. I, input fractions; O, output fractions.
Figure S4. Characterization of a C. trachomatis strain harboring a plasmid encoding CT288-2HA expressed from the promoter of the inclusion membrane protein gene incD. (A) Schematic representation of the ct288 gene present in the plasmid (pSVP255) introduced C. trachomatis L2/434 strain. PincD, incD promoter; TincD, incD terminator. (B) HeLa cells were either left uninfected (UI) or infected by the indicated C. trachomatis strains for 14, 24, or 40 h. Whole cell lysates were analyzed by immunoblotting with antibodies against HA, C. trachomatis MOMP (bacterial loading control) and α-tubulin (loading control for host cells). (C) Hela cells infected by C. trachomatis expressing CT288-2HA for 24 h were fixed with paraformaldehyde 4% (w/v), immunolabeled with anti-HA and anti-MOMP antibodies, or anti-Inc CT442 antibodies, and adequate fluorescent-conjugated secondary antibodies, and analyzed by immunofluorescence microscopy. Scale bar 10 μm. (D) HeLa cells were infected with the indicated strains at a multiplicity of infection of 5 and recoverable inclusion forming units (IFUs) were determined at 20, 24, 30, and 40 h p.i., Data are mean and standard error of the mean of 3 independent experiments. P-values were calculated by a two-tailed unpaired Student's t-test. *P < 0.05; ns, not significant.
Figure S5. Whole blots of the co-immunoprecipitation experiments to test if C. trachomatis-produced CT288-2HA is pulled-down by EGFP- CCDC146692−955. For details see Figure 3 legend. The arrows indicate the position in the blots of the relevant proteins. I, input fractions; O, output fractions.
Figure S6. Endogenous CCDC146 cannot be detected in whole-cell extracts of HeLa or HEK293T cells. Plasmids encoding the indicated proteins were used to transfect HeLa 229 (A) or HEK293T cells (B) for 24 h. Whole cell extracts were collected, and proteins were analyzed by immunoblotting using anti-CCDC146, anti-HA, anti-GFP, and anti-α-tubulin antibodies.
Figure S7. CCDC146-HA is recruited to the periphery of the inclusion membrane in C. trachomatis infected cells. HeLa cells transfected with a plasmid encoding CCDC146FL-HA were either left uninfected (UI) or infected for 24 h with C. trachomatis L2/434. (A) The cells were fixed with methanol, immunolabeled with anti-HA and anti-γ-tubulin antibodies, and appropriate fluorophore-conjugated secondary antibodies, and analyzed by confocal immunofluorescence microscopy. The arrows in each panel highlight the γ-tubulin-labeled centrosome. (B) Percentage of uninfected or C. trachomatis-infected HeLa 229 cells showing co-localization between CCDC146FL-HA and γ-tubulin. Data is from one experiment (100 cells counted). (C) and (D) The infected cells were fixed with paraformaldehyde 4% (w/v), immunolabeled with anti-HA and anti-MOMP (C), or anti-CT442 (D) antibodies, and analyzed by confocal immunofluorescence microscopy. All scale bars, 10 μm.
Figure S8. Verification of intron insertion in the C. trachomatis ct288:aadA mutant strain. (A) Representation of the ctl0540 (ortolog of ct288 in strain D/UW3) locus in C. trachomatis LGV serovar L2 strain 434/Bu (L2/434). (B) Representation of the ctl0540 locus in the ct288:aadA mutant derivative of C. trachomatis L2/434. In (A) and (B) the arrows and numbers indicate the approximate hybridization position of DNA primers (Table S2) used in PCR reactions, yielding DNA products of the indicated length in base pairs (bp). (C) Agarose gel displaying the result from the PCR with the indicated primers (Table S2) and DNA templates; pML2 is the plasmid containing the intron targeting ct288 (Table S1), used to generate the ct288:aadA strain; bp, base pairs.
Figure S9. Comparison of the localization of ectopically expressed full-length EGFP-CCDC146 in cells infected by C. trachomatis L2/434 or ct288::aadA mutant strains. HeLa cells transfected with a plasmid encoding full-length EGFP-CCDC146 (EGFP-CCDC146FL) were infected for 8, 16, or 24 h by C. trachomatis L2/434 (A) or ct288:aadA (clone A; Figure 5) (B). The cells were fixed with methanol, immunolabeled with anti-GFP and anti-Hsp60 antibodies, and appropriate fluorophore-conjugated secondary antibodies, and analyzed by immunofluorescence microscopy. Scale bars, 5 μm.
Figure S10. Localization of full-length EGFP-CCDC146 at the periphery of the C. trachomatis inclusion does not require intact host Golgi, microtubules or microfilaments, but depends on chlamydial protein synthesis. HeLa cells transfected with a plasmid encoding full-length EGFP-CCDC146 (EGFP-CCDC146FL) were infected for 24 h (A) or 16 h (B,C) by C. trachomatis L2/434 or ct288:aadA (clone A; Figure 5). The cells were fixed with methanol, immunolabeled with anti-GFP and anti-Hsp60 antibodies, and appropriate fluorophore-conjugated secondary antibodies, and analyzed by immunofluorescence microscopy. At 23 h p.i. (A) or 8 h p.i., (B), the cells were incubated in the presence of 1 μg/ml nocodazole (to depolymerize microtubules), 2 μM cytochalasin D (to depolymerize microfilaments), or 1 μg/ml brefeldin A (BFA; to disrupt the Golgi complex). (C) At 8 h p.i., the cells were incubated in the presence of 100 μg/ml chloramphenicol (to inhibit bacterial protein synthesis). The solvents (dimethyl sulfoxide or ethanol) did not affect the localization of EGFP-CCDC146FL at the inclusion periphery, and the disrupting effect of nocodazole, cytochalasin D, and BFA was confirmed by fluorescence microscopy (not shown). Scale bars, 5 μm.
Table S1. Plasmids used in this work.
Table S2. DNA primers used in this work.
Table S3. Candidate binding partners of C. trachomatis Inc protein CT288 identified in a yeast two-hybrid screen using a HeLa cDNA library as bait.
References
Aeberhard, L., Banhart, S., Fischer, M., Jehmlich, N., Rose, L., Koch, S., et al. (2015). The proteome of the isolated Chlamydia trachomatis containing vacuole reveals a complex trafficking platform enriched for retromer components. PLoS Pathog. 11:e1004883. doi: 10.1371/journal.ppat.1004883
Agaisse, H., and Derre, I. (2013). A C. trachomatis cloning vector and the generation of C. trachomatis strains expressing fluorescent proteins under the control of a C. trachomatis promoter. PLoS ONE 8:e57090. doi: 10.1371/journal.pone.0057090
Almeida, F., Borges, V., Ferreira, R., Borrego, M. J., Gomes, J. P., and Mota, L. J. (2012). Polymorphisms in inc proteins and differential expression of inc genes among Chlamydia trachomatis Strains correlate with invasiveness and tropism of lymphogranuloma venereum isolates. J. Bacteriol. 194, 6574–6585. doi: 10.1128/JB.01428-12
Al-Zeer, M. A., Al-Younes, H. M., Kerr, M., Abu-Lubad, M., Gonzalez, E., Brinkmann, V., et al. (2014). Chlamydia trachomatis remodels stable microtubules to coordinate Golgi stack recruitment to the chlamydial inclusion surface. Mol. Microbiol. 94, 1285–1297. doi: 10.1111/mmi.12829
Bannantine, J. P., Griffiths, R. S., Viratyosin, W., Brown, W. J., and Rockey, D. D. (2000). A secondary structure motif predictive of protein localization to the chlamydial inclusion membrane. Cell. Microbiol. 2, 35–47. doi: 10.1046/j.1462-5822.2000.00029.x
Brown, H. M., Knowlton, A. E., Snavely, E., Nguyen, B. D., Richards, T. S., and Grieshaber, S. S. (2014). Multinucleation during C. trachomatis infections is caused by the contribution of two effector pathways. PLoS ONE 9:e100763. doi: 10.1371/journal.pone.0100763
da Cunha, M., Pais, S. V., Bugalhao, J. N., and Mota, L. J. (2017). The Chlamydia trachomatis type III secretion substrates CT142, CT143, and CT144 are secreted into the lumen of the inclusion. PLoS ONE 12:e0178856. doi: 10.1371/journal.pone.0178856
Dehoux, P., Flores, R., Dauga, C., Zhong, G., and Subtil, A. (2011). Multi-genome identification and characterization of chlamydiae-specific type III secretion substrates: the Inc proteins. BMC Genomics 12:109. doi: 10.1186/1471-2164-12-109
Delevoye, C., Nilges, M., Dehoux, P., Paumet, F., Perrinet, S., Dautry-Varsat, A., et al. (2008). SNARE protein mimicry by an intracellular bacterium. PLoS Pathog. 4:e1000022. doi: 10.1371/journal.ppat.1000022
Derré, I., Swiss, R., and Agaisse, H. (2011). The lipid transfer protein CERT interacts with the Chlamydia inclusion protein IncD and participates to ER-Chlamydia inclusion membrane contact sites. PLoS Pathog. 7:e1002092. doi: 10.1371/journal.ppat.1002092
Dumoux, M., Menny, A., Delacour, D., and Hayward, R. D. (2015). A Chlamydia effector recruits CEP170 to reprogram host microtubule organization. J. Cell Sci. 128, 3420–3434. doi: 10.1242/jcs.169318
Elwell, C., Mirrashidi, K., and Engel, J. (2016). Chlamydia cell biology and pathogenesis. Nat. Rev. Microbiol. 14, 385–400. doi: 10.1038/nrmicro.2016.30
Fagerberg, L., Hallstrom, B. M., Oksvold, P., Kampf, C., Djureinovic, D., Odeberg, J., et al. (2014). Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteomics 13, 397–406. doi: 10.1074/mcp.M113.035600
Firat-Karalar, E. N., Sante, J., Elliott, S., and Stearns, T. (2014). Proteomic analysis of mammalian sperm cells identifies new components of the centrosome. J. Cell Sci. 127(Pt 19), 4128–4133. doi: 10.1242/jcs.157008
Gauliard, E., Ouellette, S. P., Rueden, K. J., and Ladant, D. (2015). Characterization of interactions between inclusion membrane proteins from Chlamydia trachomatis. Front. Cell. Infect. Microbiol. 5:13. doi: 10.3389/fcimb.2015.00013
Grieshaber, S. S., Grieshaber, N. A., and Hackstadt, T. (2003). Chlamydia trachomatis uses host cell dynein to traffic to the microtubule-organizing center in a p50 dynamitin-independent process. J. Cell. Sci. 116(Pt 18), 3793–3802. doi: 10.1242/jcs.00695
Grieshaber, S. S., Grieshaber, N. A., Miller, N., and Hackstadt, T. (2006). Chlamydia trachomatis causes centrosomal defects resulting in chromosomal segregation abnormalities. Traffic 7, 940–949. doi: 10.1111/j.1600-0854.2006.00439.x
Johnson, C. M., and Fisher, D. J. (2013). Site-specific, insertional inactivation of incA in Chlamydia trachomatis using a group II intron. PLoS ONE 8:e83989. doi: 10.1371/journal.pone.0083989
Johnson, K. A., Tan, M., and Sutterlin, C. (2009). Centrosome abnormalities during a Chlamydia trachomatis infection are caused by dysregulation of the normal duplication pathway. Cell. Microbiol. 11, 1064–1073. doi: 10.1111/j.1462-5822.2009.01307.x
Key, C. E., and Fisher, D. J. (2017). Use of group II Intron technology for targeted mutagenesis in Chlamydia trachomatis. Methods Mol. Biol. 1498, 163–177. doi: 10.1007/978-1-4939-6472-7_11
Knowlton, A. E., Brown, H. M., Richards, T. S., Andreolas, L. A., Patel, R. K., and Grieshaber, S. S. (2011). Chlamydia trachomatis infection causes mitotic spindle pole defects independently from its effects on centrosome amplification. Traffic 12, 854–866. doi: 10.1111/j.1600-0854.2011.01204.x
Kokes, M., Dunn, J. D., Granek, J. A., Nguyen, B. D., Barker, J. R., Valdivia, R. H., et al. (2015). Integrating chemical mutagenesis and whole-genome sequencing as a platform for forward and reverse genetic analysis of Chlamydia. Cell Host Microbe 17, 716–725. doi: 10.1016/j.chom.2015.03.014
Li, Z., Chen, C., Chen, D., Wu, Y., Zhong, Y., and Zhong, G. (2008). Characterization of fifty putative inclusion membrane proteins encoded in the Chlamydia trachomatis genome. Infect Immun. 76, 2746–2757. doi: 10.1128/IAI.00010-08
Lutter, E. I., Barger, A. C., Nair, V., and Hackstadt, T. (2013). Chlamydia trachomatis inclusion membrane protein CT228 recruits elements of the myosin phosphatase pathway to regulate release mechanisms. Cell Rep. 3, 1921–1931. doi: 10.1016/j.celrep.2013.04.027
Lutter, E. I., Martens, C., and Hackstadt, T. (2012). Evolution and conservation of predicted inclusion membrane proteins in chlamydiae. Comp. Funct. Genomics 2012:362104. doi: 10.1155/2012/362104
McKuen, M. J., Mueller, K. E., Bae, Y. S., and Fields, K. A. (2017). Fluorescence-reported allelic exchange mutagenesis reveals a role for Chlamydia trachomatis TmeA in invasion that is independent of host AHNAK. Infect. Immun. 85:e00640–17. doi: 10.1128/IAI.00640-17
Mirrashidi, K. M., Elwell, C. A., Verschueren, E., Johnson, J. R., Frando, A., Von Dollen, J., et al. (2015). Global mapping of the inc-human interactome reveals that retromer restricts chlamydia infection. Cell Host Microbe 18, 109–121. doi: 10.1016/j.chom.2015.06.004
Mital, J., Lutter, E. I., Barger, A. C., Dooley, C. A., and Hackstadt, T. (2015). Chlamydia trachomatis inclusion membrane protein CT850 interacts with the dynein light chain DYNLT1 (Tctex1). Biochem. Biophys. Res. Commun. 462, 165–170. doi: 10.1016/j.bbrc.2015.04.116
Mital, J., Miller, N. J., Dorward, D. W., Dooley, C. A., and Hackstadt, T. (2013). Role for chlamydial inclusion membrane proteins in inclusion membrane structure and biogenesis. PLoS ONE 8:e63426. doi: 10.1371/journal.pone.0063426
Mital, J., Miller, N. J., Fischer, E. R., and Hackstadt, T. (2010). Specific chlamydial inclusion membrane proteins associate with active Src family kinases in microdomains that interact with the host microtubule network. Cell. Microbiol. 12, 1235–1249. doi: 10.1111/j.1462-5822.2010.01465.x
Moore, E. R., and Ouellette, S. P. (2014). Reconceptualizing the chlamydial inclusion as a pathogen-specified parasitic organelle: an expanded role for Inc proteins. Front. Cell. Infect. Microbiol. 4:157. doi: 10.3389/fcimb.2014.00157
Mueller, K. E., Plano, G. V., and Fields, K. A. (2014). New frontiers in type III secretion biology: the Chlamydia perspective. Infect. Immun. 82, 2–9. doi: 10.1128/IAI.00917-13
Nguyen, B. D., and Valdivia, R. H. (2013). Forward genetic approaches in Chlamydia trachomatis. J. Vis. Exp. e50636. doi: 10.3791/50636
Nguyen, P. H., Lutter, E. I., and Hackstadt, T. (2018). Chlamydia trachomatis inclusion membrane protein MrcA interacts with the inositol 1,4,5-trisphosphate receptor type 3 (ITPR3) to regulate extrusion formation. PLoS Pathog. 14:e1006911. doi: 10.1371/journal.ppat.1006911
Nunes, A., and Gomes, J. P. (2014). Evolution, phylogeny, and molecular epidemiology of Chlamydia. Infect. Genet. Evol. 23, 49–64. doi: 10.1016/j.meegid.2014.01.029
O'Connell, C. M., and Ferone, M. E. (2016). Chlamydia trachomatis genital infections. Microb Cell 3, 390–403. doi: 10.15698/mic2016.09.525
Rockey, D. D., Scidmore, M. A., Bannantine, J. P., and Brown, W. J. (2002). Proteins in the chlamydial inclusion membrane. Microbes Infect. 4, 333–340. doi: 10.1016/S1286-4579(02)01546-0
Rzomp, K. A., Moorhead, A. R., and Scidmore, M. A. (2006). The GTPase Rab4 interacts with Chlamydia trachomatis inclusion membrane protein CT229. Infect. Immun. 74, 5362–5373. doi: 10.1128/IAI.00539-06
Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. doi: 10.1038/nmeth.2019
Scidmore, M. A. (2005). Cultivation and Laboratory Maintenance of Chlamydia trachomatis. Curr. Protoc. Microbiol. 11:Unit 11A.1. doi: 10.1002/9780471729259.mc11a01s00
Scidmore, M. A., and Hackstadt, T. (2001). Mammalian 14-3-3beta associates with the Chlamydia trachomatis inclusion membrane via its interaction with IncG. Mol. Microbiol. 39, 1638–1650. doi: 10.1046/j.1365-2958.2001.02355.x
Sixt, B. S., Bastidas, R. J., Finethy, R., Baxter, R. M., Carpenter, V. K., Kroemer, G., et al. (2017). The Chlamydia trachomatis inclusion membrane protein CpoS counteracts STING-mediated cellular surveillance and suicide programs. Cell Host Microbe 21, 113–121. doi: 10.1016/j.chom.2016.12.002
Stanhope, R., Flora, E., Bayne, C., and Derre, I. (2017). IncV, a FFAT motif-containing Chlamydia protein, tethers the endoplasmic reticulum to the pathogen-containing vacuole. Proc. Natl. Acad. Sci. U.S.A. 114, 12039–12044. doi: 10.1073/pnas.1709060114
Stephens, R. S., Kalman, S., Lammel, C., Fan, J., Marathe, R., Aravind, L., et al. (1998). Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science 282, 754–759.
Subtil, A., Parsot, C., and Dautry-Varsat, A. (2001). Secretion of predicted Inc proteins of Chlamydia pneumoniae by a heterologous type III machinery. Mol. Microbiol. 39, 792–800. doi: 10.1046/j.1365-2958.2001.02272.x
Taylor, H. R., Burton, M. J., Haddad, D., West, S., and Wright, H. (2014). Trachoma. 384, 2142–2152. Lancet. doi: 10.1016/S0140-6736(13)62182-0
The UniProt, C. (2017). UniProt: the universal protein knowledgebase. Nucleic Acids Res. 45, D158–D169. doi: 10.1093/nar/gkw1099
Uphoff, C. C., and Drexler, H. G. (2011). Detecting mycoplasma contamination in cell cultures by polymerase chain reaction. Methods Mol. Biol. 731, 93–103. doi: 10.1007/978-1-61779-080-5_8
Wang, Y., Kahane, S., Cutcliffe, L. T., Skilton, R. J., Lambden, P. R., and Clarke, I. N. (2011). Development of a transformation system for Chlamydia trachomatis: restoration of glycogen biosynthesis by acquisition of a plasmid shuttle vector. PLoS Pathog. 7:e1002258. doi: 10.1371/journal.ppat.1002258
Weber, M. M., Bauler, L. D., Lam, J., and Hackstadt, T. (2015). Expression and localization of predicted inclusion membrane proteins in Chlamydia trachomatis. Infect. Immun. 83, 4710–4718. doi: 10.1128/IAI.01075-15
Weber, M. M., Lam, J. L., Dooley, C. A., Noriea, N. F., Hansen, B. T., Hoyt, F. H., et al. (2017). Absence of specific Chlamydia trachomatis inclusion membrane proteins triggers premature inclusion membrane lysis and host cell death. Cell Rep. 19, 1406–1417. doi: 10.1016/j.celrep.2017.04.058
Keywords: host-pathogen interactions, Chlamydia trachomatis, type III secretion, Inc proteins, centrosome
Citation: Almeida F, Luís MP, Pereira IS, Pais SV and Mota LJ (2018) The Human Centrosomal Protein CCDC146 Binds Chlamydia trachomatis Inclusion Membrane Protein CT288 and Is Recruited to the Periphery of the Chlamydia-Containing Vacuole. Front. Cell. Infect. Microbiol. 8:254. doi: 10.3389/fcimb.2018.00254
Received: 21 March 2018; Accepted: 04 July 2018;
Published: 26 July 2018.
Edited by:
Kenneth Fields, University of Kentucky, United StatesReviewed by:
Scott Grieshaber, University of Idaho, United StatesTravis Jewett, University of Central Florida, United States
Copyright © 2018 Almeida, Luís, Pereira, Pais and Mota. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luís Jaime Mota, bGptb3RhQGZjdC51bmwucHQ=