 Sharvan Sehrawat
Sharvan Sehrawat Dhaneshwar Kumar
Dhaneshwar Kumar Barry T. Rouse
Barry T. Rouse
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Infect. Microbiol., 25 May 2018
Sec. Virus and Host
Volume 8 - 2018 | https://doi.org/10.3389/fcimb.2018.00177
Most vertebrates are infected with one or more herpesviruses and remain so for the rest of their lives. The relationship of immunocompetent healthy host with herpesviruses may sometime be considered as harmonious. However, clinically severe diseases can occur when host immunity is compromised due to aging, during some stress response, co-infections or during neoplastic disease conditions. Discord can also occur during iatrogenic immunosuppression used for controlling graft rejection, in some primary genetic immunodeficiencies as well as when the virus infects a non-native host. In this review, we discuss such issues and their influence on host-herpesvirus interaction.
The members of herpesviridae family are categorized into alpha (α), beta (β), and gamma (γ) herpesviruses based on their host range, genetic organization and replication strategies (Whitley, 1996). Herpes simplex virus (HSV) 1, 2 and varicella zoster virus (VZV) are α-herpesviruses, cytomegalovirus (CMV), human herpesvirus (HHV)−6 and 7 are β-herpesviruses while Epstein Barr Virus (EBV) and human herpesvirus 8 (HHV8) are γ-herpesviruses infecting humans. All humans become infected with one or more herpesviruses during their life span (Boshoff and Weiss, 2001; Virgin et al., 2009). Characteristically, herpesviruses persist in the host for an extended duration following a primary infection, but severe disease and mortality in healthy immunocompetent individuals caused by α- and γ-herpesviruses are rare. However, CMV infection involving critical organs of nervous system, hematological and vascular system, gastrointestinal system may be accompanied by severe disease outcome in apparently healthy individuals (Rafailidis et al., 2008). The influence of any unaccounted for critical conditions remain a possibility in such cases. The general perception is that herpesviruses are innocuous pathogens, a status that can probably be attributed to their long association with mankind (Parrish et al., 2008). In some instances, the persisting herpesvirus infections might even provide some benefits to the host against other infections and clinical conditions such as malignancies (Barton et al., 2007; White et al., 2012; Furman et al., 2015; Litjens et al., 2018). Humans not infected with any of the herpesviruses however, represent a rare subset; therefore in comparison to infected individuals their ability to handle other infections is not well-established. The outcome of herpesvirus infection is severe in genetically immunodeficient, very young or aged individuals as well as when the virus gains entry to certain anatomical locations such as central nervous system or other immunoprivileged sites. In addition, when herpesviruses infect either a non-native susceptible host or those organisms that harbor other concurrent infections, severe disease may occur (Figure 1). Evidence that co-infection of HSV and human immunodeficiency virus (HIV) can result in more severe disease outcome is well established (Freeman et al., 2006; Des Jarlais et al., 2014; Looker et al., 2017). In this review, we discuss situations and underlying cellular and molecular mechanisms where the disease pattern caused by herpesviruses is changed. We focus our analysis on α-herpesviruses while briefly alluding to the other members of the herpesviridae family throughout the text.
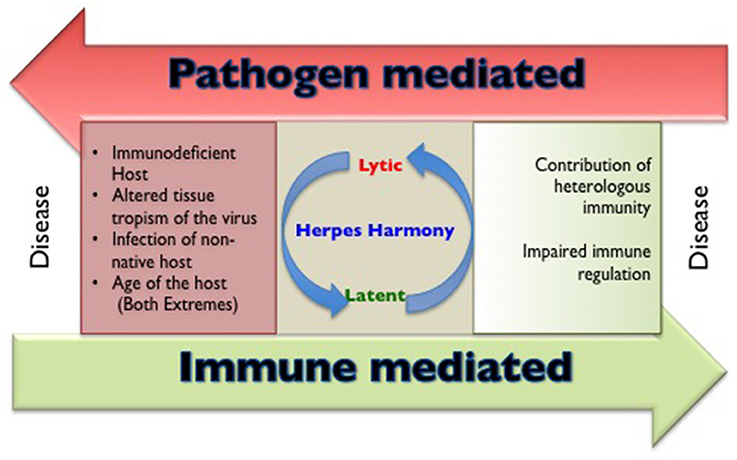
Figure 1. A cartoon to show the circumstances that are responsible for making and breaking herpesviruses and host harmony.
Herpesviruses are considered as highly successful pathogens. These viruses might have originated from those viruses that infected a common ancestor of mammals, birds and reptiles (McGeoch et al., 1995; Virgin et al., 2009; Virgin, 2014). The evolutionary processes led to the selection of variants with altered infectivity and tissue tropism probably to ensure survival and propagation. The sequence analyses of HSV1 and HSV2 revealed that HSV1 might have infected the ancestors of humans much earlier than HSV2 and therefore has evolved better to persist in human host while HSV2 was introduced in humans at a later time point through an intermediate host. Therefore HSV2 may not had enough time to evolve with the host (Parrish et al., 2008; Wertheim et al., 2014; Underdown et al., 2017). This temporally distinct but longer association of HSV1 with human host could possibly explain why HSV1 is less pathogenic than HSV2 (Sedarati and Stevens, 1987; Smith T. J. et al., 2002). In general, herpesviruses have better adapted for their human host as compared to many other categories of viruses and therefore can persist in the host.
Numerous properties of herpesviruses contribute to their success and these have been discussed in detail by others (Whitley, 1996; Ploegh, 1998; Kapadia et al., 2002; Orange et al., 2002). The most important characteristic is their ability to adopt two different modes of life cycle; the latency and the lytic cycles. Herpesviruses after a primary productive infection resort to latency, a transcriptional and translational suppressed state. However, the latent stage is frequently interrupted by clinically asymptomatic reactivation episodes (Stevens, 1989; Wald et al., 2002; Kelly et al., 2006; Mark et al., 2008; Nicoll et al., 2012; Roizman and Whitley, 2013; Uppal et al., 2014; Virgin, 2014). Neurotropic viruses such as HSV 1, 2 and VZV preferentially establish latency in neuronal cells but one particular region of the virus genome remains transcriptionally active and produces latency-associated transcripts (LATs) (Roizman and Whitley, 2013). LATs are well characterized for HSV 1 and 2 but not so well for VZV (Leib et al., 1989; Strelow and Leib, 1995; Depledge et al., 2017). Functional protein products are rarely detected during latency (Simmons et al., 1992; Roizman and Whitley, 2013). Moreover most neurons lack MHC molecules and hence are unlikely to stimulate the immune system even when limited viral proteins are made under some circumstances (Maehlen et al., 1989). The immune cells or other non-neuronal cells in the vicinity of infected neurons could still acquire such antigens and help trigger or maintain immune reactivity. The overall kinetics, the magnitude and the contribution of these immune induction processes are not well understood. The primary infection of neurons by α-herpesviruses, results in the lysis of a significant proportion of neurons that may cause neuritis or post herpetic neuralgia in some infected individuals (Tontodonati et al., 2012). Fewer neurons are damaged during intermittent reactivation or abortive replication events in immunocompetent host and in such situations clinical symptoms usually do not occur (Antinone and Smith, 2010; Sawtell and Thompson, 2016).
Unlike α-herpesviruses, β- and γ-herpesviruses such as CMV and EBV, establish latency in non-neuronal cells that include macrophages and B cells (Klein et al., 1989; Slobedman and Mocarski, 1999; Kelly et al., 2006; Sinclair and Sissons, 2006; Khairallah et al., 2017). Murine cytomegalovirus (MCMV) infection in mice represents one of the well-studied β-HV model system to understand the host pathogen interaction. This infection effectively induces both innate and adaptive immunity. One of the proteins (m157) encoded by MCMV serves as a ligand for receptor (Ly49H) expressed by NK cells. This interaction induces the activation and differentiation of such cells in a manner CD4+ and CD8+ T cells are signaled. Some of the responding NK cells can even form a memory pool (Sun et al., 2009). The conventional αβ- as well as a less-well characterized γδ-T cell response help control the balance of lytic and latent virus (Steffens et al., 1998; Couzi et al., 2015; Khairallah et al., 2017). The entire repertoire of γδ-T cells is not exhaustively studied but nonetheless include antigenic moieties from diverse sources such as proteins, lipids, lapidated peptides, small molecules and antigen recognition by these cells may be dependent on class I MHC molecules or other non-classical MHC like molecules such as CD1c (Chien et al., 2014). Such cells were also shown to provide protection to MCMV infected mice independently of αβ-T cells (Khairallah et al., 2015). Many viral proteins are expressed in EBV infected B cells during the non-productive cycle. Many excellent reviews and research articles can be referred to for further insights (Kelly et al., 2006; Speck and Ganem, 2010; Nicoll et al., 2012; Roizman and Whitley, 2013; Khairallah et al., 2017). We highlight some of the generally accepted key points during herpesvirus latency.
The microenvironment, cell autonomous factors as well as viral elements may all contribute to the latency establishment, maintenance and reactivation of herpesviruses. Among viral factors, LATs predominantly regulate herpes simplex virus latency. LATs can actively regulate the expression of viral lytic genes such as immediate early protein encoding genes (ICP0, ICP4) and thymidine kinase of HSV to limit reactivation (Kramer and Coen, 1995; Chen et al., 1997; Roizman and Whitley, 2013). LATs can also interfere with cellular metabolism and inhibit the caspases dependent apoptosis of infected cells (Perng et al., 2000; Henderson et al., 2002; Roizman and Whitley, 2013). Some have suggested that epigenetic modifications in HSV genome differ during latency and lytic cycles (Bloom et al., 2010). In general, the association of HSV genome with nucleosomes and modification of histones by methylation and acetylation at specific lysine residues were shown to determine the lytic and latent viral replication cycles (Knipe and Cliffe, 2008; Bloom et al., 2010). The sampled tissues invariably contain both latently infected and some neurons undergoing recent reactivation of viral genome. Therefore, the results thus obtained are difficult to interpret but the technologies that offer analyses on identifiable single cells might provide better insights.
Nerve growth factors (NGF) produced by multiple cell types could also be involved in latency (Wilcox et al., 1990). NGF signaling up-regulates Bcl2 to promote cellular proliferation and survival (Finkbeiner, 2000; Biswas and Greene, 2002). Nerve termini express NGFs and physical damage could change NGF levels and possibly precipitate viral reactivation (Wilcox et al., 1990; Wilson and Mohr, 2012). This could possibly explain why some people suffer from frequent HSV reactivation upon mechanical or infection induced injuries to the skin which is an extensively innervated organ (Hsieh et al., 1997). The herpesvirus encoded miRNAs or a modulation of host miRNAs by herpesvirus infection can influence different aspects of latency (Rezaee et al., 2006; Umbach et al., 2008; Du et al., 2015; Grey, 2015). Herpesvirus encoded miRNAs help restrict replication and favor latency (Umbach et al., 2008). Examples include miR-UL112-1 of HCMV and miR-K12-9, which target the immediate early transactivator, IE72 of HCMV and RTA of KSHV, respectively, to interfere with viral replication (Grey et al., 2007; Bellare and Ganem, 2009). This interference with viral replication ensures that the latency is maintenained. Several cellular miRNAs that are either expressed at a basal level or induced upon infection may also influence viral gene expression (Lecellier et al., 2005). An intriguing example is miR138, which is mainly expressed by neurons and targets ICP0 of HSV to block the replication cycle and thereby facilitating latency (Pan et al., 2014). There are also examples of host miRNA such as miR23a which promotes the lytic life cycle by interfering with the activity of interferon regulatory factors (IRFs) to compromise the antiviral state in infected cell (Ru et al., 2014). Some host miRNAs could interfere with cellular metabolism. Example is miR101, whose ectopic expression in latently infected cells blocked HSV1 replication while its depletion promoted viral replication. This miRNA directly interacted with one of the genes whose product is required for ATP production (Zheng et al., 2011). Therefore, the expression kinetics of different miRNAs in infected cells may help decide between a lytic cycle and latency.
Upon viral infection both effector and regulatory cells are recruited in the response. The magnitude and nature of such cells may influence the switch between the lytic cycle and latency. How these cells are maintained in infected ganglionic tissues is still not clearly understood (Lund et al., 2008; Veiga-Parga et al., 2013). Thus, during latency, the first signal (peptide-MHC) required to activate T cells may not be available to maintain the pool of viral reactive T cells. Some studies suggest that even during latency, a limited number of neurons permit viral replication to provide antigenic stimulation for T cells (Wald et al., 2002; Wald and Corey, 2007). The role of T cells in regulating herpesvirus latency is frequently studied in mice models as samples obtained from human cadavers might display pronounced viral reactivity (Ouwendijk et al., 2012). However, one of the key HSV immune modulators, ICP47, fails to block the antigen presentation machinery by interacting with mouse transporter of antigen processing and presentation (TAP) molecules unlike its human counterpart (Tomazin et al., 1998; Verweij et al., 2011). This suggests for a limited direct applicability of mice studies in humans. Extensive studies supporting a role for CD8+ and perhaps CD4+ T cells have mainly come from the Hendricks laboratory (Liu et al., 2000, 2001; Knickelbein et al., 2008). Basically, those studies implicate the role for granzyme B and interferon gamma (IFN-γ) produced by virus specific CD8+ T cells with the major viral glycoprotein, gB and some early replication proteins providing the peptides for T cell recognition. Both these immune mediators are responsible for inhibiting viral replication (Cantin et al., 1995; Liu et al., 2000, 2001; Knickelbein et al., 2008; Treat et al., 2017). Granzyme B cleaves viral protein ICP4 to cause abortive replication events (Knickelbein et al., 2008). In fact, an abundance of virus-specific activated CD8+ T cells that produce effector molecules were found in latently infected ganglionic tissues and a large majority of these produced effector molecules such as granzyme B and IFN-γ (Wilson and Mohr, 2012). Whether local microenvironment provides sufficient signals to maintain viral reactive T cells or secondary lymphoid organs are continuously feeding infected ganglionic tissues remain poorly understood. Regulatory T cells are also found in latently infected ganglionic tissues and these cells probably help control the hyper reactivity of immune cells that may kill irreparable neurons (Suvas et al., 2006). However, some studies have suggested that the microenvironment within the infected ganglionic tissues might help induce the expression of inhibitory ligands to dampen the activity of resident CD8+ T cells. An impaired activity of CD8+ T cells may help explain the frequent reactivation episodes. Some of the inhibitory receptor and their ligands that have been investigated in infected ganglions include PD1-PDL1, TIM-3-Galectin-9 (Frank et al., 2010; Reddy et al., 2011). IFN-γ presumably produced by resident T cells could help induce the ligands (Garcia-Diaz et al., 2017).
Stronger evidence for CD8+ T cells involvement in maintaining latency came from studies focusing on EBV latency (Dunne et al., 2002; Angelini et al., 2013). With EBV latency, some viral proteins are made which can provide the peptides for T cell recognition. Evidence indicates that the IFN-γ producing CD8+ T cells are critically involved for controlling γ-herpesviruses such as MHV68 (Steed et al., 2006). Infection of mice with MHV68 has provided useful insights into the contribution of viral reactive CD8+ T cells in the pathogenesis of γ-herpesviruses (Husain et al., 1999; Gredmark-Russ et al., 2008; Nash and Dutia, 2008; Freeman et al., 2010). Additional information on the role of CD8+ T cells in γ-herpesviruses' latency in vivo may come from the newly developed TCR transnuclear mouse model for MHV68 (Sehrawat et al., 2012).
Herpesviruses evade immune destruction using a number of strategies. These include infection of tissues with limited accessibility to immune mediators particularly for α-herpesviruses, establishment of latency that allows minimal immune recognition and numerous active immunomodulatory procedures intrinsic to herpesviruses. Many excellent reviews have discussed immune evasive or immune managemental properties of different herpesviruses (Ploegh, 1998; Tortorella et al., 2000; Orange et al., 2002; Hewitt, 2003; Rezaee et al., 2006). Table 1 summarizes many such properties. We briefly discuss how herpesviruses can manage the immune system to ensure their persistence.
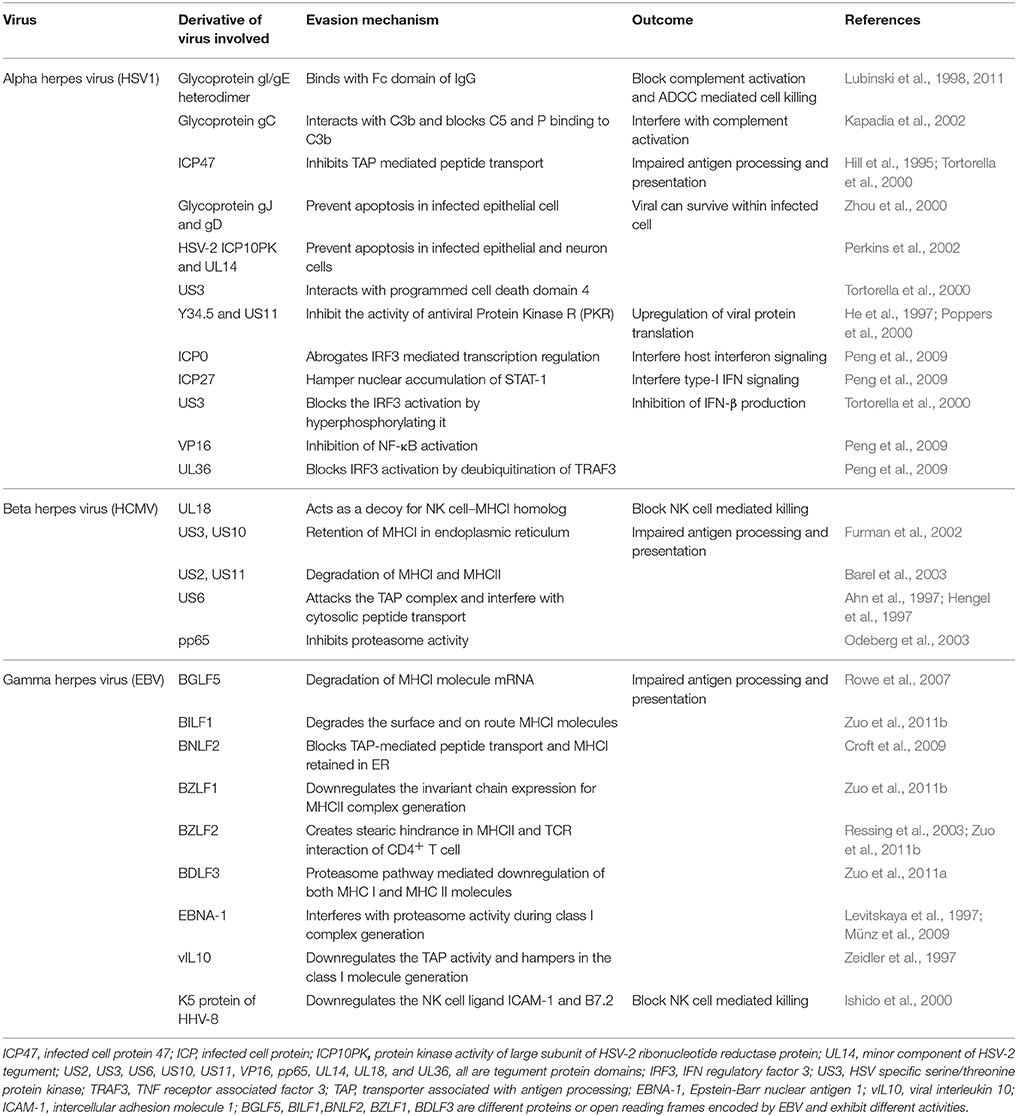
Table 1. Immune management strategies used by herpesviruses.
Herpesviruses can even establish a productive infection in the immune host. This fact reduces enthusiasm for vaccination strategies and most vaccine candidates have had only limited success. All herpesviruses blunt immunity either by interfering with immune induction or by producing anti-inflammatory molecules. For example, ICP47 of HSV and US6 of HCMV interact with the transporter of antigen presentation and processing (TAP) molecule thereby blocking efficient transportation of viral derived cytosolic peptides for loading on class I MHC (Hill et al., 1995; Ahn et al., 1997; Hengel et al., 1997). The HCMV protein pp65 can induce retention of class II MHC molecules in lysosomes for its destruction thereby minimizing its availability for surface display and limiting the T cell response (Ploegh, 1998; Odeberg et al., 2003). MCMV and HCMV can destabilize surface class I MHC by interacting with surface displayed β2 microglobulin leading to its down regulation (Jones and Sun, 1997; Halenius et al., 2015). A low level of MHC expression by infected cells could activate NK cells, but some herpesviruses have devised strategies to counteract NK cell mediated lysis of infected cells (Jonjić et al., 2008). These include modulation of host cell protein expression, encoding host homologs as well as viral proteins that could dampen NK cell responses (Grauwet et al., 2014; Campbell et al., 2015). The EBNA1 protein of EBV is not efficiently recognized by CD8+ T cells owing to its glycine-alanine repeat sequences which hinder the generation of immunogenic epitopes (Levitskaya et al., 1997; Münz and Moormann, 2008). Similarly, latency associated nuclear antigen (LANA) of KSHV and LANA-homologs of other γ-herpesviruses also interfere with antigen presentation (Coscoy, 2007). KSHV exhibit numerous immunomodulatory activities that range from interfering with components of the complement system, type I IFN signaling, in addition to impairing T and B cell responses as well as blocking apoptosis (Moore and Chang, 2003; Rezaee et al., 2006). Some herpesviruses encode for anti-inflammatory molecules (Spencer et al., 2002; Coscoy, 2007). For example, homologs of host receptors and anti-inflammatory molecules such as IL-10 and IL-35 are encoded by CMV and EBV, respectively (Birkenbach et al., 1993; Spencer et al., 2002; Rezaee et al., 2006; Rouse and Sehrawat, 2010).
Herpesviruses in general are excellent managers of the immune response and can successfully survive in the immune host. Since herpesviruses infect most individuals, an idea worth considering is how herpesviruses influence the outcome of other concurrent infections, cancers and grafts. Recent studies have shown that herpesviruses may, in fact, help fine-tune host immunity. This occurs by modulating one or more types of immune responses to make the host either more resistant or susceptible to other disease situations (Barton et al., 2007; White et al., 2010; Furman et al., 2015; Litjens et al., 2018) For example, some studies measured the influence of persisting β-herpesvirus (MCMV) or a γ-herpesvirus (MHV68) infection on the subsequent infection by Listeria monocytogenes and Yersinia pestis. Previously infected mice controlled these infections better than those not infected with MHV68 (Barton et al., 2007). The enhanced IFN-γ production, presumably by virus reactive T cells, activated macrophages, which then controlled bacterial growth. Enhanced NK cell responses as a result of MHV68 infection might also have participated in the control of these secondary infections (White et al., 2010). Prior infection with herpesviruses in humans as well as mice was also shown to promote immune response to a subsequent infection or the vaccination (Furman et al., 2015). CMV positive adults mounted a stronger anti-influenza virus CD8+ T cell response as compared to CMV negative individuals (Furman et al., 2015). Genital HSV2 infection promotes colonization of group B streptococcus locally in the genital tract (Cherpes et al., 2005). However, the molecular and cellular mechanisms involved have not been investigated but an anti-inflammatory milieu induced by the virus might have facilitated subsequent bacterial colonization. Another well established example of herpesvirus infection promoting secondary bacterial infection is VZV and invasive Group A streptococcus infections (Wilson et al., 1995; Laupland et al., 2000; Oyake et al., 2000). Severe lesions such as cellulitis, necrotizing fasciitis and sometimes endocarditis occur in people infected with VZV and Group A streptococcus infection (Zachariadou et al., 2014). A generalized immunosuppression caused by VZV infection seems to explain the outcome (Laskey et al., 2000). Cellular and molecular mechanisms for enhanced disease due to double infection are yet to be fully investigated.
Herpesviruses may also exhibit altered dynamics with their host during transplantation. Many humans receive transplantation to ameliorate malignant diseases or to restore organ functions. Iatrogenic immunosuppression is usually induced in patients undergoing solid organ transplants or hematopoietic stem cell transplants (HSCT). Such procedures often reactivate any persistent herpesvirus. Depending on the herpesvirus involved, the outcome of transplantation procedures might vary. For example, HSCT aimed at improving the prognosis of acute myeloid leukemia (AML), induced reactivation of CMV which then expanded a subset of donor derived NKG2C expressing NK cells as well as γδ-T cells of a particular phenotype (Vδ2−ve γδ-T cells) (Litjens et al., 2018). These cells helped control the relapse of leukemic episodes thereby benefiting the host. CMV in transplant patients can also cause harmful effects such as genital tract disease, hepatitis, encephalitis or retinitis. In some patients undergoing HSCT to improve the outcome of Hodgkin lymphoma, HHV-6 gets reactivated (Drobyski et al., 1994). The reactivated virus eventually resulted in meningioencephalitis and death. Many a time transplant tissues could also serve as the source of transmitting herpesviruses in the recipients (Openshaw et al., 1995; Remeijer et al., 2001). Therefore, in patients undergoing transplantation, anti-herpesviral drugs are usually infused (McIntosh et al., 2016).
The large majority of people harbor herpesviruses as part of their virome (Virgin et al., 2009) and these infections usually do not cause major harm in healthy adult host. The scenario can change, however, under several circumstances such as when the host has defects in innate or adaptive immunity. Such problems normally become evident in neonates, children and elderly; transplant patients as well as cancer patients or patients having one or more concurrent infections. Such issues are discussed subsequently.
A compromised or dysfunctional immune system invariably results in severe disease outcome due to herpesvirus infections. Individuals with defects of innate immunity usually fail to control most herpesviruses (Fitzgerald et al., 1985; Krug et al., 2004). The activity of NK cells and more importantly signaling through type I interferons (IFNs) are critical in providing anti-herpesvirus defense (Fitzgerald et al., 1985; Smith H. R. C. et al., 2002; Krug et al., 2004; Takeuchi and Akira, 2009). Most studies have come from animal models, but evidence from human studies also suggest for the role of innate immune mediators in defense against herpesvirus infections (Dupuis et al., 2003; Jost and Altfeld, 2013). Genetic deficiency or mutations in TLR3 and UNC93B can result in herpes simplex virus encephalitis and mortality due to impaired type I IFN response (Casrouge et al., 2006; Zhang et al., 2007; Iwasaki, 2012; Rosato et al., 2015). One of the typical clinical presentations of HSV 1 infection is an occurrence of herpes simplex labialis (HLS). However, only 20-30% of infected individuals exhibit HLS, therefore attempts were made to identify genetic susceptibility loci (Kriesel et al., 2011). Specifically, two single nucleotide polymorphism (SNP) within the chromosome 21orf 91 (C21orf91) exhibited a strong association with disease development. This reading frame, also called as cold sore susceptibility gene (CSSG1), encoded for a cytosolic expressing protein whose function still remains unknown. The transcripts for this gene were also recorded in other cells such as immune cells and ganglionic cells (Kriesel et al., 2011). Therefore, there is possibility that the protein encoded might have critical function in deciding the outcome of herpesvirus-mediated encephalitis which also exhibit some associations with genetic immunodeficiencies. Additional genetic mutations responsible for defective innate immunity against herpesviruses include mutations of the NF-kB essential modulator gene as well as mutation in GATA2 (Zandi et al., 1997; Dropulic and Cohen, 2011). An extended list of genes responsible for severe herpesvirus infections is provided in Table 2. Many of these deficiencies result in uncontrolled and aberrant immune responses in infected individuals leading to immunopathologies (Parvaneh et al., 2013). Some of these conditions can also cause neoplastic transformations, especially in γ-herpesvirus infected individuals (Parvaneh et al., 2013; Ruffner et al., 2017). Defects in some anti-inflammatory response such as the inability of host to mount an appropriate anti-inflammatory response can also result in severe immunopathologies caused by some herpesviruses. Extensively studied anti-inflammatory mechanisms include the cytokine IL-10 and regulatory T cells expressing transcription factor Foxp3 (Lund et al., 2008; Sarangi et al., 2008; Rouse and Sehrawat, 2010; Veiga-Parga et al., 2013; Sehrawat and Rouse, 2017).
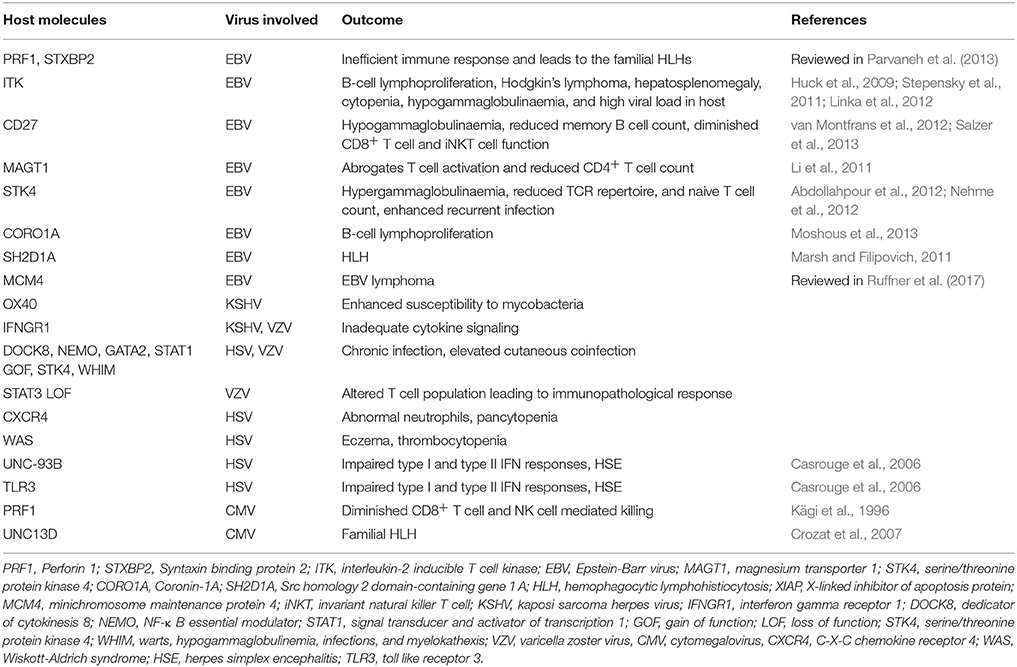
Table 2. Primary immunodeficiencies and the outcome of herpesviral infections.
All primary immunodeficiencies invariably result in severe disease resulting from primary herpesvirus infections before the onset of a harmonious relationship with virus.
The emergence of human immunodeficiency virus - acquired immunodeficiency syndrome (HIV-AIDS) 40 years ago seems to have altered the relationship of several herpesviruses with their host. Indeed, co-infection involving HIV and herpesviruses are frequent. Kaposi sarcomas on the skin caused by HHV-8 or KSHV readily became evident in HIV infected individuals in the pre-treatment era (Sepkowitz, 2001). Another common problem in the pre-treatment era was retinitis caused by HCMV infection, a lesion almost never seen in treated AIDS patients (Salzberger et al., 2005). Enhanced expression of proinflammatory molecules such as IL-1β, TNF-α, CCR5, vCXCL1, vCXCL2 in concurrently infected individuals facilitated the recruitment of more inflammatory cells that precipitated HCMV retinitis (Safdar et al., 2002; Heiden et al., 2007; Lichtner et al., 2015). Similarly individuals receiving profound iatrogenic immunosuppressive therapies to achieve acceptance of graft tissues also tend to develop severe HCMV infections (Dowling et al., 1976). Zostriform infection (commonly known as shingles) caused by the reactivation of VZV infection is usually a problem of aged individuals but untreated younger AIDS patients also exhibit severe manifestation (Buchbinder et al., 1992; Leppard and Naburi, 1998). There are instances where co-infection of herpesvirus and HIV can aggravate the outcome of HIV infections (Kucera et al., 1990; Regezi et al., 1996; Tobian and Quinn, 2009). Multiple explanations have been proposed. For example, genital ulcers caused by herpesviruses (particularly HSV 2) may disrupt the integrity of the mucosa which can facilitate HIV infection of infiltrating T cells and macrophages (Kucera et al., 1990; Tobian and Quinn, 2009). Although HIV can infect resting immune cells, activated CD4+ T cells are more prone to infection (Okoye and Picker, 2013). Episodic reactivation of HSV expands activated CD4+ T cell population which can be easily infected by HIV (Okoye and Picker, 2013). With the loss of T cells, host's susceptibility to HSV and other opportunistic infections increases (Bartlett et al., 2012; Okoye and Picker, 2013).
Another idea proposed for enhanced susceptibility of HIV by prior herpesvirus infections included an accelerated phagocytosis and internalization of HIV (Takeda et al., 1988; McKeating et al., 1990). Thus, HSV 1 or CMV infections were shown to enhance Fc receptor expression on some cells (Westmoreland and Watkins, 1974; McKeating et al., 1990). Elevated FcR expression can efficiently bind to the Fc portion of an antibody molecule some of which can also form complex with HIV. The immune complexes thus formed were internalized more efficiently. However, this could only be a factor when the pre-existing anti-HIV antibodies unable to neutralize the virus are present in the host. In addition this phenomenon may be more relevant to amplify the infection. Some suggest that the direct interaction of components of herpesviruses with HIV can enhance HIV replication in infected cells (Ostrove et al., 1987; Gimble et al., 1988; Albrecht et al., 1989; Margolis et al., 1992). For instance, ICP0 and ICP4 of HSV can interact with the long terminal repeats (LTRs) of HIV to enhance its replication (Gimble et al., 1988; Margolis et al., 1992). Another theory proposes that concurrent herpesviruses and HIV infections result in the selection of new HIV variants with greater infectivity toward new cell types (Grivel et al., 2001). For example, HIV variants (X4) that predominantly infect CD4+ T cells emerge more frequently in patients co-infected with HHV-6 and HHV-7 than in those infected with HIV alone (Grivel et al., 2001). Although detailed molecular mechanisms to explain such observations are lacking but some studies suggested the involvement of tissue resident inflammatory cells that included CD4+ T cells and dendritic cells expressing receptor for HIV such as DC-SIGN or CD123 in previously HSV 2 infected individuals (Zhu et al., 2009). Intriguingly such cells persisted at tissue sites even after virus is controlled effectively by anti-viral treatment (Zhu et al., 2009). The prolonged persistence of immune cells in healed genital ulcer could therefore also help facilitate HIV infection. An association between EBV induced Burkitt's lymphoma (BL) and HIV is reasonably well established (Beral, 1991). BL is a rare cancer of children and is endemic in some African countries. HIV induces hyper proliferation of B cells which then can be infected by EBV (Grogg et al., 2007). This cellular mechanism could enhance lymphoma development.
We can conclude that individuals having co-infection with one or more herpesviruses and HIV exhibit more severe disease by either agent. Accordingly, the clinical management of such cases would require treating both infections simultaneously.
The severe consequence of co-infection involving the γ-herpesvirus (EBV) and malaria is well established (Epstein et al., 1964). The interaction could promote the development of Burkitt's lymphoma (BL). Antigens derived from Plasmodium falciparum (Pf), the causative agent of a severe form of malaria, induce polyclonal B cell activation (Chêne et al., 2007). B cells thus activated are more permissive to EBV infection and undergo hyper proliferation. Pf infection is also known to down regulate the expression of activation induced cytidine deaminase, an enzyme responsible for c-myc translocation in latently EBV infected B cells (Torgbor et al., 2014). This step is considered critical for the development of cancer. A possible role of upregulated TLR9 by EBV latently infected B cells and its subsequent activation by Pf derived agonists is also proposed as a mechanism for hyperproliferation of B cells (Crompton et al., 2009; Iskra et al., 2010). Co-infection of EBV and Pf, especially in children, creates an anti-inflammatory environment with enhanced IL-10 production. This could blunt the activity of CD8+ T cells which would fail to clear infected B cells (Peyron et al., 1994; Medina et al., 2011). In addition, EBV EBNA1 protein is not a potent stimulator of protective CD8+ T cells and this effect is further enhanced by co-infection with Pf (Levitskaya et al., 1997). Some studies were performed in mice to understand the cellular and molecular mechanism of enhanced severity of coinfection involving γ-herpesvirus and Plasmodium. Mice coinfected with MHV68 and Plasmodium displayed severe disease (Matar et al., 2015). An acute but not latent infection of mice with γ-herpesvirus failed to control the subsequent non-lethal P. yoelii infection (Haque et al., 2004; Matar et al., 2015). A compromised anti-plasmodial humoral immune response mediated by the M2 protein of MHV68 was suggested to explain the phenomenon (Matar et al., 2015). Other factors such as the altered responses of immune cells other than those of the adaptive arm were not investigated and could be also involved.
An accumulating body of evidence shows that the outcome of a herpesvirus infection can be influenced by the host's past experience with other infectious agents, as well as the microflora, which colonize the gut, skin and other sites (Robinson and Pfeiffer, 2014; Shannon et al., 2017). Cross-reactive B and or T cell responses to unrelated pathogens can be responsible for the altered outcome of a subsequent infection. This is known as heterologous immunity. A series of elegant studies in mice with multiple infecting viruses was done by Welsh and Selin group (Welsh and Selin, 2002; Clute et al., 2005; Welsh and Fujinami, 2007; Welsh et al., 2010). Their studies provided some rules to explain why a second infection has variable outcome in terms of disease expression. Accordingly, one of the influenza virus epitope (M158−66) and EBV derived epitopes (BMLF1280−288 and BRLF1109−117) were shown to be cross-reactive. The severity of mononucleosis correlated with specific expansion of BMLF1280−288 specific CD8+ T cells (Clute et al., 2005). The presence of a particular MHC haplotype (HLA-A2+) was suggested to account for the skewed response and lymphocytosis. The possibility of clinical mononucleosis (kissing disease) occurring after primary infection with EBV is more common in young adults who have been previously infected with influenza virus and generated a particular type of antibody response (McClain et al., 2003). Additional evidence where herpesvirus infections could influence responses to heterologous infections as well as to other diseases such as allergies, metabolic diseases and perhaps some cancers are awaited.
During the course of evolution, most herpesviruses have adapted to a single or a limited number of host species. Nevertheless, when for a variety of reasons they happen to infect non-native species, aberrant and severe disease can result. A common example is HSV in mice, a popular model to study HSV pathogenesis. In mice, HSV does not behave as it does in its natural human host. Many strains are highly virulent in mice and cause lethal encephalitis particularly when higher doses are used for infection. Encephalitis is a very rare outcome in healthy humans and not influenced by viral strain type. When some herpesviruses infect a non-native species, encephalitis can result. For example humans accidentally infected with herpes B virus, an α-herpesvirus of monkeys, can suffer from lethal encephalitis (Wozniakowski and Samorek-Salamonowicz, 2015). Similarly pseudorabies virus of pigs causes encephalitis in cows (Crandell et al., 1982). Another example include a β-herpesvirus, elephant endotheliotropic herpesvirus (EEHV), which normally infects African elephants but is usually innocuous in the adult animals (Richman et al., 1999; Fickel et al., 2001). When the virus happens to infect young animals or Asian elephants, the disease is invariably severe (Richman et al., 2000; Fickel et al., 2001). Why encephalitis occurs so commonly in non-native species as compared to the native species is not known. The virus may be arrested in ganglionic neurons in latent form and fail to spread anterograde to the CNS. This retention may not happen in a non-native host for reasons still not clear.
Multiple events contribute to the onset, progression and severity of autoimmune diseases. Many have advocated that herpesviruses can be associated with several autoimmune diseases, perhaps acting as triggering agents for their onset (Münz et al., 2009; Getts et al., 2013). However, few if any investigators subscribe to the hypothesis that some human autoimmunities are directly caused by herpesviruses or any other virus infection. Circumstantial evidence suggest that several herpesviruses, particularly EBV and HHV-6 could initiate the onset of multiple sclerosis, but such ideas have never been confirmed independently (Wucherpfennig and Strominger, 1995; Poole et al., 2006; Lünemann et al., 2007, 2008; Münz et al., 2009). It was advocated that the HSV induced ocular lesion, stromal keratitis, represented an autoimmune lesion during its chronic phase (Zhao et al., 1998). Evidence to support this hypothesis was presented using a mouse model in which molecular mimicry between a protein of HSV (UL-6) and an auto-antigen expressed in the cornea was suggested to cause the disease (Zhao et al., 1998). Such ideas were never independently confirmed and some data strongly argued against this hypothesis (Deshpande et al., 2001). At this stage of investigation, it is probably safe to assume that while there is no compelling evidence that herpesviruses can directly cause one or more autoimmune diseases, the viruses might serve as cofactors in the pathogenesis of autoimmune diseases.
For most of us with a normally functioning immune system, exposure to and living with many herpesviruses has no major consequences. We usually develop a harmonious relationship with multiple herpesviruses that persistently infect us. Problems arise mainly with immune immaturity or when it declines with age or is dysregulated. The latter occurs most commonly as a consequence of cancer, infections with some pathogens, or immunosuppressive therapy to control transplants and tissue damaging lesions. A particular problem that changed the face of herpesvirus infections was the emergence of HIV, especially in the era before effective antiviral therapy. Fortunately, we now have better mechanistic understanding of the circumstances, which disrupt herpesvirus-host-harmony and may well be poised to exploit such information for better management of such situations. It has also become evident that herpesviruses form part of our virome and this can impact on susceptibility to other infections and disease producing agencies. Whether or not the composition of our virome can help explain the variability of the outcome of many herpetic diseases such as development of zosteriform infection, post-herpetic neuralgia as well as the severity of eye disease remains investigated.
A contentious issue in the herpesvirus field is the development of vaccines against member viruses. Indeed, studies in mice with almost any form of HSV vaccine protect them from disease, yet no vaccine against HSV in humans induces effective immunity, at least when subjected to double bind independent evaluation (Koelle and Corey, 2003). There are many enthusiastic advocates for universal vaccines against herpesviruses but our own view is that the prophylactic vaccines against some herpesviruses could be useful only under special circumstances. These include transplant patients treated with immunosuppressant drugs and defects in one or more aspects of immunity. What might really be useful against some herpesviruses is a therapeutic vaccine that could rewrite the language of immune responsiveness. This is because herpesviruses are so ubiquitous and their unmatched prevalence in general population. Making an inflammatory tissue damaging response into one that is far more benign particularly against pathogenic epitopes represents an appealing idea. However, this would require the identification of those antigens, which are predominantly pathogenic rather than protective. Progress in this area has been less than impressive, but we expect useful discovery in this field.
SS has collected data, surveyed literature, written, and edited the MS. DK has surveyed literature, collected information and written the MS. BR help logically present ideas, written, and edited the MS.
The work was supported by Department of Science and Technology, Govt. of India (Project no. EMR/2015/000367) to SS and NIH (grant no. EY 005093) to BR.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abdollahpour, H., Appaswamy, G., Kotlarz, D., Diestelhorst, J., Beier, R., Schaffer, A. A., et al. (2012). The phenotype of human STK4 deficiency. Blood 119, 3450–3457. doi: 10.1182/blood-2011-09-378158
Ahn, K., Gruhler, A., Galocha, B., Jones, T. R., Wiertz, E. J. H., Ploegh, H. L., et al. (1997). The ER-luminal domain of the HCMV glycoprotein US6 inhibits peptide translocation by TAP. Immunity 6, 613–621. doi: 10.1016/S1074-7613(00)80349-0
Albrecht, M. A., DeLuca, N. A., Byrn, R. A., Schaffer, P. A., and Hammer, S. M. (1989). The herpes simplex virus immediate-early protein, ICP4, is required to potentiate replication of human immunodeficiency virus in CD4+ lymphocytes. J. Virol. 63, 1861–1868.
Angelini, D. F., Serafini, B., Piras, E., Severa, M., Coccia, E. M., Rosicarelli, B., et al. (2013). Increased CD8+ T cell response to Epstein-Barr virus lytic antigens in the active phase of multiple sclerosis. PLoS Pathog. 9:e1003220. doi: 10.1371/journal.ppat.1003220
Antinone, S. E., and Smith, G. A. (2010). Retrograde axon transport of herpes simplex virus and pseudorabies virus: a live-cell comparative analysis. J. Virol. 84, 1504–1512. doi: 10.1128/JVI.02029-09
Barel, M. T., Ressing, M., Pizzato, N., van Leeuwen, D., Le Bouteiller, P., Lenfant, F., et al. (2003). Human cytomegalovirus-encoded US2 differentially affects surface expression of MHC class I locus products and targets membrane-bound, but not soluble HLA-G1 for degradation. J. Immunol. 171, 6757–6765. doi: 10.4049/jimmunol.171.12.6757
Bartlett, J., Shao, J., Schechter, M., Balestre, E., and Myer, L. (2012). CD4 cell count and the risk of AIDS or death in HIV-infected adults on combination antiretroviral therapy with a suppressed viral load: a longitudinal cohort study from COHERE. PLoS Med. 9:e1001194. doi: 10.1371/journal.pmed.1001194
Barton, E. S., White, D. W., Cathelyn, J. S., Brett-McClellan, K. A., Engle, M., Diamond, M. S., et al. (2007). Herpesvirus latency confers symbiotic protection from bacterial infection. Nature 447, 326–329. doi: 10.1038/nature05762
Bellare, P., and Ganem, D. (2009). Regulation of KSHV lytic switch protein expression by a virus-encoded microRNA: an evolutionary adaptation that fine-tunes lytic reactivation. Cell Host Microbe 6, 570–575. doi: 10.1016/j.chom.2009.11.008
Beral, P. (1991). AIDS-associated non-Hodgkin lymphoma. Lancet 337, 805–809. doi: 10.1016/0140-6736(91)92513-2
Birkenbach, M., Josefsen, K., Yalamanchili, R., Lenoir, G., and Kieff, E. (1993). Epstein-Barr virus-induced genes: first lymphocyte-specific G protein-coupled peptide receptors. J. Virol. 67, 2209–2220.
Biswas, S. C., and Greene, L. A. (2002). Nerve growth factor (NGF) down-regulates the Bcl-2 homology 3 (BH3) domain-only protein bim and suppresses its proapoptotic activity by phosphorylation. J. Biol. Chem. 277, 49511–49516. doi: 10.1074/jbc.M208086200
Bloom, D. C., Giordani, N. V., and Kwiatkowski, D. L. (2010). Epigenetic regulation of latent HSV-1 gene expression. Biochim. Biophys. Acta 1799, 246–256. doi: 10.1016/j.bbagrm.2009.12.001
Boshoff, C., and Weiss, R. A. (2001). Epidemiology and pathogenesis of Kaposi's sarcoma-associated herpesvirus. Philos. Trans. R. Soc. Lond. B Biol. Sci. 356, 517–534. doi: 10.1098/rstb.2000.0778
Buchbinder, S. P., Katz, M. H., Hessol, N. A., Liu, J. Y., O'Malley, P. M., Underwood, R., et al. (1992). Herpes zoster and human immunodeficiency virus infection. J. Infect. Dis. 166, 1153–1156.
Campbell, T. M., McSharry, B. P., Steain, M., Slobedman, B., and Abendroth, A. (2015). Varicella-zoster virus and herpes simplex virus 1 differentially modulate NKG2D ligand expression during productive infection. J. Virol. 89, 7932–7943. doi: 10.1128/JVI.00292-15
Cantin, E. M., Hinton, D. R., Chen, J., and Openshaw, H. (1995). Gamma interferon expression during acute and latent nervous system infection by herpes simplex virus type 1. J. Virol. 69, 4898–4905.
Casrouge, A., Zhang, S.-Y., Eidenschenk, C., Jouanguy, E., Puel, A., Yang, K., et al. (2006). Herpes simplex virus encephalitis in human UNC-93B deficiency. Science 314, 308–312. doi: 10.1126/science.1128346
Chêne, A., Donati, D., Guerreiro-Cacais, A. O., Levitsky, V., Chen, Q., Falk, K. I., et al. (2007). A molecular link between malaria and Epstein–Barr virus reactivation. PLoS Pathog. 3:e80. doi: 10.1371/journal.ppat.0030080
Chen, S. H., Kramer, M. F., Schaffer, P. A., and Coen, D. M. (1997). A viral function represses accumulation of transcripts from productive-cycle genes in mouse ganglia latently infected with herpes simplex virus. J. Virol. 71, 5878–5884.
Cherpes, T. L., Melan, M. A., Kant, J. A., Cosentino, L. A., Meyn, L. A., and Hillier, S. L. (2005). Genital tract shedding of herpes simplex virus type 2 in women: effects of hormonal contraception, bacterial vaginosis, and vaginal group B Streptococcus colonization. Clin. Infect. Dis. 40, 1422–1428. doi: 10.1086/429622
Chien, Y., Meyer, C., and Bonneville, M. (2014). γδ T cells: first line of defense and beyond. Annu. Rev. Immunol. 32, 121–155. doi: 10.1146/annurev-immunol-032713-120216
Clute, S. C., Watkin, L. B., Cornberg, M., Naumov, Y. N., Sullivan, J. L., Luzuriaga, K., et al. (2005). Cross-reactive influenza virus-specific CD8+ T cells contribute to lymphoproliferation in Epstein-Barr virus-associated infectious mononucleosis. J. Clin. Invest. 115, 3602–3612. doi: 10.1172/JCI25078
Coscoy, L. (2007). Immune evasion by Kaposi's sarcoma-associated herpesvirus. Nat. Rev. Immunol. 7, 391–401. doi: 10.1038/nri2076
Couzi, L., Pitard, V., Moreau, J.-F., Merville, P., and Déchanet-Merville, J. (2015). Direct and indirect effects of cytomegalovirus-induced T cells after kidney transplantation. Front. Immunol. 6, 3. doi: 10.3389/fimmu.2015.00003
Crandell, R. A., Mesfin, G. M., and Mock, R. E. (1982). Horizontal transmission of pseudorabies virus in cattle. Am. J. Vet. Res. 43, 326–328.
Croft, N. P., Shannon-Lowe, C., Bell, A. I., Horst, D., Kremmer, E., Ressing, M. E., et al. (2009). Stage-specific inhibition of MHC class I Presentation by the Epstein-Barr virus BNLF2a protein during virus lytic cycle. PLoS Pathog. 5:e1000490. doi: 10.1371/journal.ppat.1000490
Crompton, P. D., Mircetic, M., Weiss, G., Baughman, A., Huang, C.-Y., Topham, D. J., et al. (2009). The TLR9 ligand CpG promotes the acquisition of Plasmodium falciparum-specific memory B cells in malaria-naive individuals. J. Immunol. 182, 3318–3326. doi: 10.4049/jimmunol.0803596
Crozat, K., Hoebe, K., Ugolini, S., Hong, N. A., Janssen, E., Rutschmann, S., et al. (2007). Jinx, an MCMV susceptibility phenotype caused by disruption of Unc13d: a mouse model of type 3 familial hemophagocytic lymphohistiocytosis. J. Exp. Med. 204, 853–863. doi: 10.1084/jem.20062447
Depledge, D. P., Ouwendijk, W. J. D., Sadaoka, T., Braspenning, S. E., Mori, Y., Cohrs, R., et al. (2017). Newly identified varicella-zoster virus latency transcript inhibits viral replication. bioRxiv 174797. doi: 10.1101/174797
Deshpande, S., Zheng, M., Lee, S., Banerjee, K., Gangappa, S., Kumaraguru, U., et al. (2001). Bystander activation involving T lymphocytes in herpetic stromal keratitis. J. Immunol. 167, 2902–2910. doi: 10.4049/jimmunol.167.5.2902
Des Jarlais, D. C., Arasteh, K., McKnight, C., Perlman, D. C., Feelemyer, J., Hagan, H., et al. (2014). HSV-2 co-infection as a driver of HIV transmission among heterosexual non-injecting drug users in New York city. PLoS ONE 9:e87993. doi: 10.1371/journal.pone.0087993
Dowling, J. N., Saslow, A. R., Armstrong, J. A., and Ho, M. (1976). Cytomegalovirus infection in patients receiving immunosuppressive therapy for rheumatologic disorders. J. Infect. Dis. 133, 399–408. doi: 10.1093/infdis/133.4.399
Drobyski, W. R., Knox, K. K., Majewski, D., and Carrigan, D. R. (1994). Fatal encephalitis due to variant B human herpesvirus-6 infection in a bone marrow-transplant recipient. N. Engl. J. Med. 330, 1356–1360. doi: 10.1056/NEJM199405123301905
Dropulic, L. K., and Cohen, J. I. (2011). Severe viral infections and primary immunodeficiencies. Clin. Infect. Dis. 53, 897–909. doi: 10.1093/cid/cir610
Du, T., Han, Z., Zhou, G., Roizman, B., and Roizman, B. (2015). Patterns of accumulation of miRNAs encoded by herpes simplex virus during productive infection, latency, and on reactivation. Proc. Natl. Acad. Sci. U.S.A. 112, E49–E55. doi: 10.1073/pnas.1422657112
Dunne, P. J., Faint, J. M., Gudgeon, N. H., Fletcher, J. M., Plunkett, F. J., Vieira, M., et al. (2002). Epstein-Barr virus–specific CD8(+) T cells that re-express CD45RA are apoptosis-resistant memory cells that retain replicative potential. Blood 100, 933–940. doi: 10.1182/blood-2002-01-0160
Dupuis, S., Jouanguy, E., Al-Hajjar, S., Fieschi, C., Al-Mohsen, I. Z., Al-Jumaah, S., et al. (2003). Impaired response to interferon-α/β and lethal viral disease in human STAT1 deficiency. Nat. Genet. 33, 388–391. doi: 10.1038/ng1097
Epstein, M., Achong, B., and Barr, Y. (1964). Virus particles in cultured lymphoblasts from Burkitt's lymphoma. Lancet 283, 702–703.
Fickel, J., Richman, L. K., Montali, R., Schaftenaar, W., Göritz, F., Hildebrandt, T. B., et al. (2001). A variant of the endotheliotropic herpesvirus in Asian elephants (Elephas maximus) in European zoos. Vet. Microbiol. 82, 103–109. doi: 10.1016/S0378-1135(01)00363-7
Finkbeiner, S. (2000). CREB couples neurotrophin signals to survival messages. Neuron 25, 11–14. doi: 10.1016/S0896-6273(00)80866-1
Fitzgerald, P. A., Mendelsohn, M., and Lopez, C. (1985). Human natural killer cells limit replication of herpes simplex virus type 1 in vitro. J. Immunol. 134, 2666–2672.
Frank, G. M., Lepisto, A. J., Freeman, M. L., Sheridan, B. S., Cherpes, T. L., and Hendricks, R. L. (2010). Early CD4+ T cell help prevents partial CD8+ T cell exhaustion and promotes maintenance of herpes simplex virus 1 latency. J. Immunol. 184, 277–286. doi: 10.4049/jimmunol.0902373
Freeman, E. E., Weiss, H. A., Glynn, J. R., Cross, P. L., Whitworth, J. A., and Hayes, R. J. (2006). Herpes simplex virus 2 infection increases HIV acquisition in men and women: systematic review and meta-analysis of longitudinal studies. AIDS 20, 73–83. doi: 10.1097/01.aids.0000198081.09337.a7
Freeman, M. L., Lanzer, K. G., Cookenham, T., Peters, B., Sidney, J., Wu, T.-T., et al. (2010). Two kinetic patterns of epitope-specific CD8 T-cell responses following murine gammaherpesvirus 68 infection. J. Virol. 84, 2881–2892. doi: 10.1128/JVI.02229-09
Furman, D., Jojic, V., Sharma, S., Shen-Orr, S. S., Angel, C. J., Onengut-Gumuscu, S., et al. (2015). Cytomegalovirus infection enhances the immune response to influenza. Sci. Transl. Med. 7:281ra43. doi: 10.1126/scitranslmed.aaa2293
Furman, M. H., Dey, N., Tortorella, D., and Ploegh, H. L. (2002). The human cytomegalovirus US10 gene product delays trafficking of major histocompatibility complex class I molecules. J. Virol. 76, 11753–11756. doi: 10.1128/JVI.76.22.11753-11756.2002
Garcia-Diaz, A., Shin, D. S., Moreno, B. H., Saco, J., Escuin-Ordinas, H., Rodriguez, G. A., et al. (2017). Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. 19, 1189–1201. doi: 10.1016/j.celrep.2017.04.031
Getts, D. R., Chastain, E. M. L., Terry, R. L., and Miller, S. D. (2013). Virus infection, antiviral immunity, and autoimmunity. Immunol. Rev. 255, 197–209. doi: 10.1111/imr.12091
Gimble, J. M., Duh, E., Ostrove, J. M., Gendelman, H. E., Max, E. E., and Rabson, A. B. (1988). Activation of the human immunodeficiency virus long terminal repeat by herpes simplex virus type 1 is associated with induction of a nuclear factor that binds to the NF-kappa B/core enhancer sequence. J. Virol. 62, 4104–4112.
Grauwet, K., Cantoni, C., Parodi, M., De Maria, A., Devriendt, B., Pende, D., et al. (2014). Modulation of CD112 by the alphaherpesvirus gD protein suppresses DNAM-1-dependent NK cell-mediated lysis of infected cells. Proc. Natl. Acad. Sci. U.S.A. 111, 16118–16123. doi: 10.1073/pnas.1409485111
Gredmark-Russ, S., Cheung, E. J., Isaacson, M. K., Ploegh, H. L., and Grotenbreg, G. M. (2008). The CD8 T-cell response against murine gammaherpesvirus 68 is directed toward a broad repertoire of epitopes from both early and late antigens. J. Virol. 82, 12205–12212. doi: 10.1128/JVI.01463-08
Grey, F. (2015). Role of microRNAs in herpesvirus latency and persistence. J. Gen. Virol. 96, 739–751. doi: 10.1099/vir.0.070862-0
Grey, F., Meyers, H., White, E. A., Spector, D. H., and Nelson, J. (2007). A human cytomegalovirus-encoded microRNA Regulates expression of multiple viral genes involved in replication. PLoS Pathog. 3:e163. doi: 10.1371/journal.ppat.0030163
Grivel, J.-C., Ito, Y., Fagà, G., Santoro, F., Shaheen, F., Malnati, M. S., et al. (2001). Suppression of CCR5-but not CXCR4-tropic HIV-1 in lymphoid tissue by human herpesvirus 6. Nat. Med. 7, 1232–1235. doi: 10.1038/nm1101-1232
Grogg, K. L., Miller, R. F., and Dogan, A. (2007). HIV infection and lymphoma. J. Clin. Pathol. 60, 1365–1372. doi: 10.1136/jcp.2007.051953
Halenius, A., Gerke, C., and Hengel, H. (2015). Classical and non-classical MHC I molecule manipulation by human cytomegalovirus: so many targets—but how many arrows in the quiver? Cell. Mol. Immunol. 12, 139–153. doi: 10.1038/cmi.2014.105
Haque, A., Rachinel, N., Quddus, M. R., Haque, S., Kasper, L. H., and Usherwood, E. (2004). Co-infection of malaria and gamma-herpesvirus: exacerbated lung inflammation or cross-protection depends on the stage of viral infection. Clin. Exp. Immunol. 138, 396–404. doi: 10.1111/j.1365-2249.2004.02652.x
He, B., Gross, M., and Roizman, B. (1997). The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc. Natl. Acad. Sci. U.S.A. 94, 843–848. doi: 10.1073/pnas.94.3.843
Heiden, D., Ford, N., Wilson, D., Rodriguez, W. R., Margolis, T., Janssens, B., et al. (2007). Cytomegalovirus retinitis: the neglected disease of the AIDS pandemic. PLoS Med. 4:e334. doi: 10.1371/journal.pmed.0040334
Henderson, G., Peng, W., Jin, L., Perng, G.-C., Nesburn, A., Wechsler, S., et al. (2002). Regulation of caspase 8- and caspase 9-induced apoptosis by the herpes simplex virus type 1 latency-associated transcript. J. Neurovirol. 8, 103–111. doi: 10.1080/13550280290101085
Hengel, H., Koopmann, J. O., Flohr, T., Muranyi, W., Goulmy, E., Hämmerling, G. J., et al. (1997). A viral ER-resident glycoprotein inactivates the MHC-encoded peptide transporter. Immunity 6, 623–632. doi: 10.1016/S1074-7613(00)80350-7
Hewitt, E. W. (2003). The MHC class I antigen presentation pathway: strategies for viral immune evasion. Immunology 110, 163–169. doi: 10.1046/j.1365-2567.2003.01738.x
Hill, A., Jugovic, P., York, lan, Russ, G., Bennink, J., Yewdell, J., et al. (1995). Herpes simplex virus turns off the TAP to evade host immunity. Nature 375, 411–415. doi: 10.1038/375411a0
Hsieh, S.-T., Lin, W.-M., Chiang, H.-Y., Huang, I.-T., Ko, M.-H., Chang, Y.-C., et al. (1997). Skin innervation and its effects on the epidermis. J. Biomed. Sci. 4, 264–268. doi: 10.1007/BF02253428
Huck, K., Feyen, O., Niehues, T., Rüschendorf, F., Hübner, N., Laws, H.-J., et al. (2009). Girls homozygous for an IL-2-inducible T cell kinase mutation that leads to protein deficiency develop fatal EBV-associated lymphoproliferation. J. Clin. Invest. 119, 1350–1358. doi: 10.1172/JCI37901
Husain, S. M., Usherwood, E. J., Dyson, H., Coleclough, C., Coppola, M. A., Woodland, D. L., et al. (1999). Murine gammaherpesvirus M2 gene is latency-associated and its protein a target for CD8(+) T lymphocytes. Proc. Natl. Acad. Sci. USA. 96, 7508–7513. doi: 10.1073/pnas.96.13.7508
Ishido, S., Choi, J. K., Lee, B. S., Wang, C., DeMaria, M., Johnson, R. P., et al. (2000). Inhibition of natural killer cell-mediated cytotoxicity by Kaposi's sarcoma-associated herpesvirus K5 protein. Immunity 13, 365–374. doi: 10.1016/S1074-7613(00)00036-4
Iskra, S., Kalla, M., Delecluse, H.-J., Hammerschmidt, W., and Moosmann, A. (2010). Toll-like receptor agonists synergistically increase proliferation and activation of B cells by epstein-barr virus. J. Virol. 84, 3612–3623. doi: 10.1128/JVI.01400-09
Iwasaki, A. (2012). A virological view of innate immune recognition. Annu. Rev. Microbiol. 66, 177–196. doi: 10.1146/annurev-micro-092611-150203
Jones, T. R., and Sun, L. (1997). Human cytomegalovirus US2 destabilizes major histocompatibility complex class I heavy chains. J. Virol. 71, 2970–2979.
Jonjić, S., Babić, M., Polić, B., and Krmpotić, A. (2008). Immune evasion of natural killer cells by viruses. Curr. Opin. Immunol. 20, 30–38. doi: 10.1016/j.coi.2007.11.002
Jost, S., and Altfeld, M. (2013). Control of human viral infections by natural killer cells. Annu. Rev. Immunol. 31, 163–194. doi: 10.1146/annurev-immunol-032712-100001
Kägi, D., Ledermann, B., Bürki, K., Zinkernagel, R. M., and Hengartner, H. (1996). Molecular mechanisms of lymphocyte-mediated cytotoxicity and their role in immunological protection and pathogenesis in vivo. Annu. Rev. Immunol. 14, 207–232. doi: 10.1146/annurev.immunol.14.1.207
Kapadia, S. B., Levine, B., Speck, S. H., and Virgin, H. W. (2002). Critical role of complement and viral evasion of complement in acute, persistent, and latent gamma-Herpesvirus Infection. Immunity 17, 143–155. doi: 10.1016/S1074-7613(02)00369-2
Kelly, G. L., Milner, A. E., Baldwin, G. S., Bell, A. I., and Rickinson, A. B. (2006). Three restricted forms of Epstein-Barr virus latency counteracting apoptosis in c-myc-expressing Burkitt lymphoma cells. Proc. Natl. Acad. Sci. U.S.A. 103, 14935–14940. doi: 10.1073/pnas.0509988103
Khairallah, C., Déchanet-Merville, J., and Capone, M. (2017). γδ T cell-mediated immunity to cytomegalovirus infection. Front. Immunol. 8:105. doi: 10.3389/fimmu.2017.00105
Khairallah, C., Netzer, S., Villacreces, A., Juzan, M., Rousseau, B., Dulanto, S., et al. (2015). γδ T cells confer protection against murine cytomegalovirus (MCMV). PLoS Pathog. 11:e1004702. doi: 10.1371/journal.ppat.1004702
Klein, G., Rotbard, J., Gotch, F. M., Bahadur, G., Wraith, D., McMichael, A. J., et al. (1989). Viral latency and transformation: the strategy of Epstein-Barr virus. Cell 58, 5–8. doi: 10.1016/0092-8674(89)90394-2
Knickelbein, J. E., Khanna, K. M., Yee, M. B., Baty, C. J., Kinchington, P. R., and Hendricks, R. L. (2008). Noncytotoxic lytic granule-mediated CD8+ T cell inhibition of HSV-1 reactivation from neuronal latency. Science 322, 268–271. doi: 10.1126/science.1164164
Knipe, D. M., and Cliffe, A. (2008). Chromatin control of herpes simplex virus lytic and latent infection. Nat. Rev. Microbiol. 6, 211–221. doi: 10.1038/nrmicro1794
Koelle, D. M., and Corey, L. (2003). Recent progress in herpes simplex virus immunobiology and vaccine research. Clin. Microbiol. Rev. 16, 96–113. doi: 10.1128/CMR.16.1.96-113.2003
Kramer, M. F., and Coen, D. M. (1995). Quantification of transcripts from the ICP4 and thymidine kinase genes in mouse ganglia latently infected with herpes simplex virus. J. Virol. 69, 1389–1399.
Kriesel, J. D., Jones, B. B., Matsunami, N., Patel, M. K., St Pierre, C. A., Kurt-Jones, E. A., et al. (2011). C21orf91 genotypes correlate with herpes simplex labialis (cold sore) frequency: description of a cold sore susceptibility gene. J. Infect. Dis. 204, 1654–1662. doi: 10.1093/infdis/jir633
Krug, A., Luker, G. D., Barchet, W., Leib, D. A., Akira, S., and Colonna, M. (2004). Herpes simplex virus type 1 activates murine natural interferon-producing cells through toll-like receptor 9. Blood 103, 1433–1437. doi: 10.1182/blood-2003-08-2674
Kucera, L. S., Leake, E., Iyer, N., Raben, D., and Myrvik, Q. N. (1990). Human Immunodeficiency Virus Type 1 (HIV-1) and Herpes Simplex Virus Type 2 (HSV-2) can coinfect and simultaneously replicate in the same human CD4+ cell: effect of coinfection on infectious HSV-2 and HIV-1 replication. AIDS Res. Hum. Retroviruses 6, 641–647. doi: 10.1089/aid.1990.6.641
Laskey, A. L., Johnson, T. R., Dagartzikas, M. I., and Tobias, J. D. (2000). Endocarditis attributable to group A beta-hemolytic streptococcus after uncomplicated varicella in a vaccinated child. Pediatrics 106:E40. doi: 10.1542/peds.106.3.e40
Laupland, K. B., Davies, H. D., Low, D. E., Schwartz, B., Green, K., and McGeer, A. (2000). Invasive group A streptococcal disease in children and association with varicella-zoster virus infection. Ontario Group A Streptococcal Study Group. Pediatrics 105:E60. doi: 10.1542/peds.105.5.e60
Lecellier, C.-H., Dunoyer, P., Arar, K., Lehmann-Che, J., Eyquem, S., Himber, C., et al. (2005). A cellular microRNA mediates antiviral defense in human cells. Science 308, 557–560. doi: 10.1126/science.1108784
Leib, D. A., Bogard, C. L., Kosz-Vnenchak, M., Hicks, K. A., Coen, D. M., Knipe, D. M., et al. (1989). A deletion mutant of the latency-associated transcript of herpes simplex virus type 1 reactivates from the latent state with reduced frequency. J. Virol. 63, 2893–2900.
Leppard, B., and Naburi, A. E. (1998). Herpes zoster: an early manifestation of HIV infection. Afr. Health 21, 5–6.
Levitskaya, J., Sharipo, A., Leonchiks, A., Ciechanover, A., and Masucci, M. G. (1997). Inhibition of ubiquitin/proteasome-dependent protein degradation by the Gly-Ala repeat domain of the Epstein-Barr virus nuclear antigen 1. Proc. Natl. Acad. Sci. U.S.A. 94, 12616–12621. doi: 10.1073/pnas.94.23.12616
Li, F.-Y., Chaigne-Delalande, B., Kanellopoulou, C., Davis, J. C., Matthews, H. F., Douek, D. C., et al. (2011). Second messenger role for Mg2+ revealed by human T-cell immunodeficiency. Nature 475, 471–476. doi: 10.1038/nature10246
Lichtner, M., Cicconi, P., Vita, S., Cozzi-Lepri, A., Galli, M., Lo Caputo, S., et al. (2015). Cytomegalovirus coinfection is associated with an increased risk of severe non–AIDS-defining events in a large cohort of HIV-infected patients. J. Infect. Dis. 211, 178–186. doi: 10.1093/infdis/jiu417
Linka, R. M., Risse, S. L., Bienemann, K., Werner, M., Linka, Y., Krux, F., et al. (2012). Loss-of-function mutations within the IL-2 inducible kinase ITK in patients with EBV-associated lymphoproliferative diseases. Leukemia 26, 963–971. doi: 10.1038/leu.2011.371
Litjens, N. H. R., van der Wagen, L., Kuball, J., and Kwekkeboom, J. (2018). Potential Beneficial effects of cytomegalovirus infection after transplantation. Front. Immunol. 9:389. doi: 10.3389/fimmu.2018.00389
Liu, T., Khanna, K. M., Carriere, B. N., and Hendricks, R. L. (2001). Gamma interferon can prevent herpes simplex virus type 1 reactivation from latency in sensory neurons. J. Virol. 75, 11178–11184. doi: 10.1128/JVI.75.22.11178-11184.2001
Liu, T., Khanna, K. M., Chen, X., Fink, D. J., and Hendricks, R. L. (2000). CD8(+) T cells can block herpes simplex virus type 1 (HSV-1) reactivation from latency in sensory neurons. J. Exp. Med. 191, 1459–1466. doi: 10.1084/jem.191.9.1459
Looker, K. J., Elmes, J. A. R., Gottlieb, S. L., Schiffer, J. T., Vickerman, P., Turner, K. M. E., et al. (2017). Effect of HSV-2 infection on subsequent HIV acquisition: an updated systematic review and meta-analysis. Lancet Infect. Dis. 17, 1303–1316. doi: 10.1016/S1473-3099(17)30405-X
Lubinski, J. M., Lazear, H. M., Awasthi, S., Wang, F., and Friedman, H. M. (2011). The herpes simplex virus 1 IgG fc receptor blocks antibody-mediated complement activation and antibody-dependent cellular cytotoxicity in vivo. J. Virol. 85, 3239–3249. doi: 10.1128/JVI.02509-10
Lubinski, J. M., Wang, L., Soulika, A. M., Burger, R., Wetsel, R. A., Colten, H., et al. (1998). Herpes simplex virus type 1 glycoprotein gC mediates immune evasion in vivo. J. Virol. 72, 8257–8263.
Lund, J. M., Hsing, L., Pham, T. T., and Rudensky, A. Y. (2008). Coordination of early protective immunity to viral infection by regulatory T cells. Science 320, 1220–1224. doi: 10.1126/science.1155209
Lünemann, J. D., Jelcić, I., Roberts, S., Lutterotti, A., Tackenberg, B., Martin, R., et al. (2008). EBNA1-specific T cells from patients with multiple sclerosis cross react with myelin antigens and co-produce IFN-gamma and IL-2. J. Exp. Med. 205, 1763–1773. doi: 10.1084/jem.20072397
Lünemann, J. D., Kamradt, T., Martin, R., and Münz, C. (2007). Epstein-barr virus: environmental trigger of multiple sclerosis? J. Virol. 81, 6777–6784. doi: 10.1128/JVI.00153-07
Maehlen, J., Olsson, T., Zachau, A., Klareskog, L., and Kristensson, K. (1989). Local enhancement of major histocompatibility complex (MHC) class I and II expression and cell infiltration in experimental allergic encephalomyelitis around axotomized motor neurons. J. Neuroimmunol. 23, 125–132. doi: 10.1016/0165-5728(89)90031-3
Margolis, D. M., Rabson, A. B., Straus, S. E., and Ostrove, J. M. (1992). Transactivation of the HIV-1 LTR by HSV-1 immediate-early genes. Virology 186, 788–791. doi: 10.1016/0042-6822(92)90048-T
Mark, K. E., Wald, A., Magaret, A. S., Selke, S., Olin, L., Huang, M., et al. (2008). Rapidly cleared episodes of herpes simplex virus reactivation in immunocompetent adults. J. Infect. Dis. 198, 1141–1149. doi: 10.1086/591913
Marsh, R. A., and Filipovich, A. H. (2011). Familial hemophagocytic lymphohistiocytosis and X-linked lymphoproliferative disease. Ann. N. Y. Acad. Sci. 1238, 106–121. doi: 10.1111/j.1749-6632.2011.06265.x
Matar, C. G., Anthony, N. R., O'Flaherty, B. M., Jacobs, N. T., Priyamvada, L., Engwerda, C. R., et al. (2015). Gammaherpesvirus co-infection with malaria suppresses anti-parasitic humoral immunity. PLoS Pathog. 11:e1004858. doi: 10.1371/journal.ppat.1004858
McClain, M. T., Rapp, E. C., Harley, J. B., and James, J. A. (2003). Infectious mononucleosis patients temporarily recognize a unique, cross-reactive epitope of Epstein-Barr virus nuclear antigen-1. J. Med. Virol. 70, 253–257. doi: 10.1002/jmv.10385
McGeoch, D. J., Cook, S., Dolan, A., Jamieson, F. E., and Telford, E. A. R. (1995). Molecular phylogeny and evolutionary timescale for the family of mammalian herpesviruses. J. Mol. Biol. 247, 443–458. doi: 10.1006/jmbi.1995.0152
McIntosh, M., Hauschild, B., and Miller, V. (2016). Human cytomegalovirus and transplantation: drug development and regulatory issues. J. virus Erad. 2, 143–148.
McKeating, J. A., Griffiths, P. D., and Weiss, R. A. (1990). HIV susceptibility conferred to human fibroblasts by cytomegalovirus-induced Fc receptor. Nature 343, 659–661. doi: 10.1038/343659a0
Medina, T. S., Costa, S. P. T., Oliveira, M. D., Ventura, A. M., Souza, J. M., Gomes, T. F., et al. (2011). Increased interleukin-10 and interferon-γ levels in Plasmodium vivax malaria suggest a reciprocal regulation which is not altered by IL-10 gene promoter polymorphism. Malar. J. 10:264. doi: 10.1186/1475-2875-10-264
Moore, P. S., and Chang, Y. (2003). Kaposi's sarcoma–associated herpesvirus immunoevasion and tumorigenesis: two sides of the same coin? Annu. Rev. Microbiol. 57, 609–639. doi: 10.1146/annurev.micro.57.030502.090824
Moshous, D., Martin, E., Carpentier, W., Lim, A., Callebaut, I., Canioni, D., et al. (2013). Whole-exome sequencing identifies Coronin-1A deficiency in 3 siblings with immunodeficiency and EBV-associated B-cell lymphoproliferation. J. Allergy Clin. Immunol. 131, 1594.e9–1603.e9. doi: 10.1016/j.jaci.2013.01.042
Münz, C., Lünemann, J. D., Getts, M. T., and Miller, S. D. (2009). Antiviral immune responses: triggers of or triggered by autoimmunity? Nat. Rev. Immunol. 9, 246–258. doi: 10.1038/nri2527
Münz, C., and Moormann, A. (2008). Immune escape by Epstein-Barr virus associated malignancies. Semin. Cancer Biol. 18, 381–387. doi: 10.1016/j.semcancer.2008.10.002
Nash, A. A., and Dutia, B. M. (2008). “Murine Gammaherpesvirus 68,” in Encyclopedia of Virology, eds B. W. J. Mahy and M. H. V. van Regenmortel (Elsevier), 372–378.
Nehme, N. T., Schmid, J. P., Debeurme, F., Andre-Schmutz, I., Lim, A., Nitschke, P., et al. (2012). MST1 mutations in autosomal recessive primary immunodeficiency characterized by defective naive T-cell survival. Blood 119, 3458–3468. doi: 10.1182/blood-2011-09-378364
Nicoll, M. P., Proença, J. T., and Efstathiou, S. (2012). The molecular basis of herpes simplex virus latency. FEMS Microbiol. Rev. 36, 684–705. doi: 10.1111/j.1574-6976.2011.00320.x
Odeberg, J., Plachter, B., Brandén, L., and Söderberg-Nauclér, C. (2003). Human cytomegalovirus protein pp65 mediates accumulation of HLA-DR in lysosomes and destruction of the HLA-DR-chain. Blood 101, 4870–4877. doi: 10.1182/blood-2002-05-1504
Okoye, A. A., and Picker, L. J. (2013). CD4(+) T-cell depletion in HIV infection: mechanisms of immunological failure. Immunol. Rev. 254, 54–64. doi: 10.1111/imr.12066
Openshaw, H., McNeill, J. I., Lin, X. H., Niland, J., and Cantin, E. M. (1995). Herpes simplex virus DNA in normal corneas: persistence without viral shedding from ganglia. J. Med. Virol. 46, 4675–4680. doi: 10.1002/jmv.1890460116
Orange, J. S., Fassett, M. S., Koopman, L. A., Boyson, J. E., and Strominger, J. L. (2002). Viral evasion of natural killer cells. Nat. Immunol. 3, 1006–1012. doi: 10.1038/ni1102-1006
Ostrove, J. M., Leonard, J., Weck, K. E., Rabson, A. B., and Gendelman, H. E. (1987). Activation of the human immunodeficiency virus by herpes simplex virus type 1. J. Virol. 61, 3726–3732.
Ouwendijk, W. J. D., Choe, A., Nagel, M. A., Gilden, D., Osterhaus, A. D. M. E., Cohrs, R. J., et al. (2012). Restricted varicella-zoster virus transcription in human trigeminal ganglia obtained soon after death. J. Virol. 86, 10203–10206. doi: 10.1128/JVI.01331-12
Oyake, S., Ohi, T., and Koga, M. (2000). A case of varicella complicated by cellulitis and scarlet fever due to Streptococcus pyogenes. J. Dermatol. 27, 750–752. doi: 10.1111/j.1346-8138.2000.tb02272.x
Pan, D., Flores, O., Umbach, J. L., Pesola, J. M., Bentley, P., Rosato, P. C., et al. (2014). A neuron-specific host microrna targets herpes simplex virus-1 ICP0 expression and promotes latency. Cell Host Microbe 15, 446–456. doi: 10.1016/j.chom.2014.03.004
Parrish, C. R., Holmes, E. C., Morens, D. M., Park, E.-C., Burke, D. S., Calisher, C. H., et al. (2008). Cross-species virus transmission and the emergence of new epidemic diseases. Microbiol. Mol. Biol. Rev. 72, 457–470. doi: 10.1128/MMBR.00004-08
Parvaneh, N., Filipovich, A. H., and Borkhardt, A. (2013). Primary immunodeficiencies predisposed to Epstein-Barr virus-driven haematological diseases. Br. J. Haematol. 162, 573–586. doi: 10.1111/bjh.12422
Peng, T., Zhu, J., Klock, A., Phasouk, K., Huang, M.-L., Koelle, D. M., et al. (2009). Evasion of the mucosal innate immune system by herpes simplex virus type 2. J. Virol. 83, 12559–12568. doi: 10.1128/JVI.00939-09
Perkins, D., Pereira, E. F. R., Gober, M., Yarowsky, P. J., and Aurelian, L. (2002). The herpes simplex virus type 2 R1 protein kinase (ICP10 PK) blocks apoptosis in hippocampal neurons, involving activation of the MEK/MAPK survival pathway. J. Virol. 76, 1435–1449. doi: 10.1128/JVI.76.3.1435-1449.2002
Perng, G. C., Jones, C., Ciacci-Zanella, J., Stone, M., Henderson, G., Yukht, A., et al. (2000). Virus-induced neuronal apoptosis blocked by the herpes simplex virus latency-associated transcript. Science 287, 1500–1503. doi: 10.1126/science.287.5457.1500
Peyron, F., Burdin, N., Ringwald, P., Vuillez, J. P., Rousset, F., and Banchereau, J. (1994). High levels of circulating IL-10 in human malaria. Clin. Exp. Immunol. 95, 300–303. doi: 10.1111/j.1365-2249.1994.tb06527.x
Ploegh, H. L. (1998). Viral strategies of immune evasion. Science 280, 248–253. doi: 10.1126/science.280.5361.248
Poole, B. D., Scofield, R. H., Harley, J. B., and James, J. A. (2006). Epstein-Barr virus and molecular mimicry in systemic lupus erythematosus. Autoimmunity 39, 63–70. doi: 10.1080/08916930500484849
Poppers, J., Mulvey, M., Khoo, D., and Mohr, I. (2000). Inhibition of PKR activation by the proline-rich RNA binding domain of the herpes simplex virus type 1 Us11 protein. J. Virol. 74, 11215–21. doi: 10.1128/JVI.74.23.11215-11221.2000
Rafailidis, P. I., Mourtzoukou, E. G., Varbobitis, I. C., and Falagas, M. E. (2008). Severe cytomegalovirus infection in apparently immunocompetent patients: a systematic review. Virol. J. 5, 47. doi: 10.1186/1743-422X-5-47
Reddy, P. B. J., Sehrawat, S., Suryawanshi, A., Rajasagi, N. K., Mulik, S., Hirashima, M., et al. (2011). Influence of galectin-9/Tim-3 interaction on herpes simplex virus-1 latency. J. Immunol. 187, 5745–5755. doi: 10.4049/jimmunol.1102105
Regezi, J. A., Eversole, L. R., Barker, B. F., Rick, G. M., and Silverman, S. (1996). Herpes simplex and cytomegalovirus coinfected oral ulcers in HIV-positive patients. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 81, 55–62. doi: 10.1016/S1079-2104(96)80149-1
Remeijer, L., Maertzdorf, J., Doornenbal, P., Verjans, G. M., and Osterhaus, A. D. (2001). Herpes simplex virus 1 transmission through corneal transplantation. Lancet 357:442. doi: 10.1016/S0140-6736(00)04011-3
Ressing, M. E., van Leeuwen, D., Verreck, F. A. W., Gomez, R., Heemskerk, B., Toebes, M., et al. (2003). Interference with T cell receptor-HLA-DR interactions by Epstein-Barr virus gp42 results in reduced T helper cell recognition. Proc. Natl. Acad. Sci. U.S.A. 100, 11583–11588. doi: 10.1073/pnas.2034960100
Rezaee, S. A. R., Cunningham, C., Davison, A. J., and Blackbourn, D. J. (2006). Kaposi's sarcoma-associated herpesvirus immune modulation: an overview. J. Gen. Virol. 87, 1781–1804. doi: 10.1099/vir.0.81919-0
Richman, L. K., Montali, R. J., Cambre, R. C., Schmitt, D., Hardy, D., Hildbrandt, T., et al. (2000). Clinical and pathological findings of a newly recognized disease of elephants caused by endotheliotropic herpesviruses. J. Wildl. Dis. 36, 1–12. doi: 10.7589/0090-3558-36.1.1
Richman, L. K., Montali, R. J., Garber, R. L., Kennedy, M. A., Lehnhardt, J., Hildebrandt, T., et al. (1999). Novel endotheliotropic herpesviruses fatal for Asian and African elephants. Science 283, 1171–1176. doi: 10.1126/science.283.5405.1171
Robinson, C. M., and Pfeiffer, J. K. (2014). Viruses and the Microbiota. Annu. Rev. Virol. 1, 55–69. doi: 10.1146/annurev-virology-031413-085550
Roizman, B., and Whitley, R. J. (2013). An inquiry into the molecular basis of HSV latency and reactivation. Annu. Rev. Microbiol. 67, 355–374. doi: 10.1146/annurev-micro-092412-155654
Rosato, P. C., Leib, D. A., Saun, M., van, Ying, G.-S., Oh, J., and Fraser, N. (2015). neuronal interferon signaling is required for protection against herpes simplex virus replication and pathogenesis. PLoS Pathog. 11:e1005028. doi: 10.1371/journal.ppat.1005028
Rouse, B. T., and Sehrawat, S. (2010). Immunity and immunopathology to viruses: what decides the outcome? Nat. Rev. Immunol. 10, 514–526. doi: 10.1038/nri2802
Rowe, M., Glaunsinger, B., van Leeuwen, D., Zuo, J., Sweetman, D., Ganem, D., et al. (2007). Host shutoff during productive Epstein-Barr virus infection is mediated by BGLF5 and may contribute to immune evasion. Proc. Natl. Acad. Sci. U.S.A. 104, 3366–3371. doi: 10.1073/pnas.0611128104
Ru, J., Sun, H., Fan, H., Wang, C., Li, Y., Liu, M., et al. (2014). MiR-23a facilitates the replication of HSV-1 through the suppression of interferon regulatory factor 1. PLoS ONE 9:e114021. doi: 10.1371/journal.pone.0114021
Ruffner, M. A., Sullivan, K. E., and Henrickson, S. E. (2017). Recurrent and sustained viral infections in primary immunodeficiencies. Front. Immunol. 8:665. doi: 10.3389/fimmu.2017.00665
Safdar, A., Rubocki, R. J., Horvath, J. A., Narayan, K. K., and Waldron, R. L. (2002). Fatal immune restoration disease in human immunodeficiency virus type 1–infected patients with progressive multifocal leukoencephalopathy: impact of antiretroviral therapy–associated immune reconstitution. Clin. Infect. Dis. 35, 1250–1257. doi: 10.1086/344056
Salzberger, B., Hartmann, P., Hanses, F., Uyanik, B., Cornely, O. A., Wöhrmann, A., et al. (2005). Incidence and prognosis of CMV disease in HIV-infected patients before and after introduction of combination antiretroviral therapy. Infection 33, 345–349. doi: 10.1007/s15010-005-5050-z
Salzer, E., Daschkey, S., Choo, S., Gombert, M., Santos-Valente, E., Ginzel, S., et al. (2013). Combined immunodeficiency with life-threatening EBV-associated lymphoproliferative disorder in patients lacking functional CD27. Haematologica 98, 473–478. doi: 10.3324/haematol.2012.068791
Sarangi, P. P., Sehrawat, S., Suvas, S., and Rouse, B. T. (2008). IL-10 and natural regulatory T cells: two independent anti-inflammatory mechanisms in herpes simplex virus-induced ocular immunopathology. J. Immunol. 180, 6297–6306. doi: 10.4049/jimmunol.180.9.6297
Sawtell, N. M., and Thompson, R. L. (2016). De novo herpes simplex virus VP16 expression gates a dynamic programmatic transition and sets the latent/lytic balance during acute infection in trigeminal ganglia. PLoS Pathog. 12:e1005877. doi: 10.1371/journal.ppat.1005877
Sedarati, F., and Stevens, J. G. (1987). Biological basis for virulence of three strains of herpes simplex virus type 1. J. Gen. Virol. 68, 2389–2395. doi: 10.1099/0022-1317-68-9-2389
Sehrawat, S., Kirak, O., Koenig, P.-A., Isaacson, M. K., Marques, S., Bozkurt, G., et al. (2012). CD8+ T cells from mice transnuclear for a TCR that recognizes a single H-2Kb-restricted MHV68 epitope derived from gB-ORF8 help control infection. Cell Rep. 1, 461–471. doi: 10.1016/j.celrep.2012.03.009
Sehrawat, S., and Rouse, B. T. (2017). Interplay of regulatory T cell and Th17 cells during infectious diseases in humans and animals. Front. Immunol. 8:341. doi: 10.3389/fimmu.2017.00341
Sepkowitz, K. A. (2001). AIDS — the first 20 years. N. Engl. J. Med. 344, 1764–1772. doi: 10.1056/NEJM200106073442306
Shannon, B., Gajer, P., Yi, T. J., Ma, B., Humphrys, M. S., Thomas-Pavanel, J., et al. (2017). Distinct effects of the cervicovaginal microbiota and herpes simplex type 2 infection on female genital tract immunology. J. Infect. Dis. 215, 1366–1375. doi: 10.1093/infdis/jix088
Simmons, A., Slobedman, B., Speck, P., Arthur, J., and Efstathiou, S. (1992). Two patterns of persistence of herpes simplex virus DNA sequences in the nervous systems of latently infected mice. J. Gen. Virol. 73, 1287–1291. doi: 10.1099/0022-1317-73-5-1287
Sinclair, J., and Sissons, P. (2006). Latency and reactivation of human cytomegalovirus. J. Gen. Virol. 87, 1763–1779. doi: 10.1099/vir.0.81891-0
Slobedman, B., and Mocarski, E. S. (1999). Quantitative analysis of latent human cytomegalovirus. J. Virol. 73, 4806–4812.
Smith, H. R. C., Heusel, J. W., Mehta, I. K., Kim, S., Dorner, B. G., Naidenko, O. V, et al. (2002). Recognition of a virus-encoded ligand by a natural killer cell activation receptor. Proc. Natl. Acad. Sci. U.S.A. 99, 8826–8831. doi: 10.1073/pnas.092258599
Smith, T. J., Morrison, L. A., and Leib, D. A. (2002). Pathogenesis of herpes simplex virus type 2 virion host shutoff (VHS) mutants. J. Virol. 76, 2054–2061. doi: 10.1128/jvi.76.5.2054-2061.2002
Speck, S. H., and Ganem, D. (2010). Viral latency and its regulation: lessons from the γ-herpesviruses. Cell Host Microbe 8, 100–115. doi: 10.1016/j.chom.2010.06.014
Spencer, J. V., Lockridge, K. M., Barry, P. A., Lin, G., Tsang, M., Penfold, M. E. T., et al. (2002). Potent immunosuppressive activities of cytomegalovirus-encoded interleukin-10. J. Virol. 76, 1285–1292. doi: 10.1128/JVI.76.3.1285-1292.2002
Steed, A. L., Barton, E. S., Tibbetts, S. A., Popkin, D. L., Lutzke, M. L., Rochford, R., et al. (2006). Gamma interferon blocks gammaherpesvirus reactivation from latency. J. Virol. 80, 192–200. doi: 10.1128/JVI.80.1.192-200.2006
Steffens, H. P., Kurz, S., Holtappels, R., and Reddehase, M. J. (1998). Preemptive CD8 T-cell immunotherapy of acute cytomegalovirus infection prevents lethal disease, limits the burden of latent viral genomes, and reduces the risk of virus recurrence. J. Virol. 72, 1797–1804.
Stepensky, P., Weintraub, M., Yanir, A., Revel-Vilk, S., Krux, F., Huck, K., et al. (2011). IL-2-inducible T-cell kinase deficiency: clinical presentation and therapeutic approach. Haematologica 96, 472–476. doi: 10.3324/haematol.2010.033910
Stevens, J. G. (1989). Human herpesviruses: a consideration of the latent state. Microbiol. Rev. 53, 318–332.
Strelow, L. I., and Leib, D. A. (1995). Role of the virion host shutoff (vhs) of herpes simplex virus type 1 in latency and pathogenesis. J. Virol. 69, 6779–6786.
Sun, J. C., Beilke, J. N., and Lanier, L. L. (2009). Adaptive immune features of natural killer cells. Nature 457, 557–561. doi: 10.1038/nature07665
Suvas, S., Azkur, A. K., and Rouse, B. T. (2006). Qa-1b and CD94-NKG2a interaction regulate cytolytic activity of herpes simplex virus-specific memory CD8+ T cells in the latently infected trigeminal ganglia. J. Immunol. 176, 1703–1711. doi: 10.4049/jimmunol.176.3.1703
Takeda, A., Tuazon, C. U., and Ennis, F. A. (1988). Antibody-enhanced infection by HIV-1 via Fc receptor-mediated entry. Science 242, 580–583. doi: 10.1126/science.2972065
Takeuchi, O., and Akira, S. (2009). Innate immunity to virus infection. Immunol. Rev. 227, 75–86. doi: 10.1111/j.1600-065X.2008.00737.x
Tobian, A. A., and Quinn, T. C. (2009). Herpes simplex virus type 2 and syphilis infections with HIV: an evolving synergy in transmission and prevention. Curr. Opin. HIV AIDS 4, 294–299. doi: 10.1097/COH.0b013e32832c1881
Tomazin, R., van Schoot, N. E., Goldsmith, K., Jugovic, P., Semp,é, P., Früh, K., et al. (1998). Herpes simplex virus type 2 ICP47 inhibits human TAP but not mouse TAP. J. Virol. 72, 2560–2563.
Tontodonati, M., Ursini, T., Polilli, E., Vadini, F., Di Masi, F., Volpone, D., et al. (2012). Post-herpetic neuralgia. Int. J. Gen. Med. 5, 861–871. doi: 10.2147/IJGM.S10371
Torgbor, C., Awuah, P., Deitsch, K., Kalantari, P., Duca, K. A., and Thorley-Lawson, D. A. (2014). A multifactorial role for P. falciparum malaria in endemic Burkitt's lymphoma pathogenesis. PLoS Pathog. 10, e1004170. doi: 10.1371/journal.ppat.1004170
Tortorella, D., Gewurz, B. E., Furman, M. H., Schust, D. J., and Ploegh, H. L. (2000). Viral subversion of the immune system. Annu. Rev. Immunol. 18, 861–926. doi: 10.1146/annurev.immunol.18.1.861
Treat, B. R., Bidula, S. M., Ramachandran, S., St Leger, A. J., Hendricks, R. L., and Kinchington, P. R. (2017). Influence of an immunodominant herpes simplex virus type 1 CD8+ T cell epitope on the target hierarchy and function of subdominant CD8+ T cells. PLoS Pathog. 13:e1006732. doi: 10.1371/journal.ppat.1006732
Umbach, J. L., Kramer, M. F., Jurak, I., Karnowski, H. W., Coen, D. M., and Cullen, B. R. (2008). MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 454, 780–783. doi: 10.1038/nature07103
Underdown, S. J., Kumar, K., and Houldcroft, C. (2017). Network analysis of the hominin origin of Herpes Simplex virus 2 from fossil data. Virus Evol. 3:vex026. doi: 10.1093/ve/vex026
Uppal, T., Banerjee, S., Sun, Z., Verma, S. C., and Robertson, E. S. (2014). KSHV LANA–the master regulator of KSHV latency. Viruses 6, 4961–4998. doi: 10.3390/v6124961
van Montfrans, J. M., Hoepelman, A. I. M., Otto, S., van Gijn, M., van de Corput, L., de Weger, R. A., et al. (2012). CD27 deficiency is associated with combined immunodeficiency and persistent symptomatic EBV viremia. J. Allergy Clin. Immunol. 129, 787.e6–793.e6. doi: 10.1016/j.jaci.2011.11.013
Veiga-Parga, T., Sehrawat, S., and Rouse, B. T. (2013). Role of regulatory T cells during virus infection. Immunol. Rev. 255, 182–196. doi: 10.1111/imr.12085
Verweij, M. C., Ressing, M. E., Knetsch, W., Quinten, E., Halenius, A., van Bel, N., et al. (2011). Inhibition of mouse TAP by immune evasion molecules encoded by non-murine herpesviruses. Mol. Immunol. 48, 835–845. doi: 10.1016/j.molimm.2010.12.008
Virgin, H. W. (2014). The virome in mammalian physiology and disease. Cell 157, 142–150. doi: 10.1016/j.cell.2014.02.032
Virgin, H. W., Wherry, E. J., and Ahmed, R. (2009). Redefining chronic viral infection. Cell 138, 30–50. doi: 10.1016/j.cell.2009.06.036
Wald, A., and Corey, L. (2007). “Persistence in the population: epidemiology, transmission,” in Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis, eds A. Arvin, G. Campadelli-Fiume, E. Mocarski, P. S. Moore, B. Roizman, R. Whitley, and K. Yamanishi (Cambridge: Cambridge University Press).
Wald, A., Zeh, J., Selke, S., Warren, T., Ashley, R., and Corey, L. (2002). Genital shedding of herpes simplex virus among men. J. Infect. Dis. 186, S34–S39. doi: 10.1086/342969
Welsh, R. M., Che, J. W., Brehm, M. A., and Selin, L. K. (2010). Heterologous immunity between viruses. Immunol. Rev. 235, 244–266. doi: 10.1111/j.0105-2896.2010.00897.x
Welsh, R. M., and Fujinami, R. S. (2007). Pathogenic epitopes, heterologous immunity and vaccine design. Nat. Rev. Microbiol. 5, 555–563. doi: 10.1038/nrmicro1709
Welsh, R. M., and Selin, L. K. (2002). No one is naive: the significance of heterologous T-cell immunity. Nat. Rev. Immunol. 2, 417–426. doi: 10.1038/nri820
Wertheim, J. O., Smith, M. D., Smith, D. M., Scheffler, K., and Kosakovsky Pond, S. L. (2014). Evolutionary origins of human herpes simplex viruses 1 and 2. Mol. Biol. Evol. 31, 2356–2364. doi: 10.1093/molbev/msu185
Westmoreland, D., and Watkins, J. F. (1974). The IgG receptor induced by herpes simplex virus: studies using radioiodinated IgG. J. Gen. Virol. 24, 167–178. doi: 10.1099/0022-1317-24-1-167
White, D. W., Keppel, C. R., Schneider, S. E., Reese, T. A., Coder, J., Payton, J. E., et al. (2010). Latent herpesvirus infection arms NK cells. Blood 115, 4377–4383. doi: 10.1182/blood-2009-09-245464
White, D. W., Suzanne Beard, R., and Barton, E. S. (2012). Immune modulation during latent herpesvirus infection. Immunol. Rev. 245, 189–208. doi: 10.1111/j.1600-065X.2011.01074.x
Wilcox, C. L., Smith, R. L., Freed, C. R., and Johnson, E. M. (1990). Nerve growth factor-dependence of herpes simplex virus latency in peripheral sympathetic and sensory neurons in vitro. J. Neurosci. 10, 1268–1275. doi: 10.1523/JNEUROSCI.10-04-01268.1990
Wilson, A. C., and Mohr, I. (2012). A cultured affair: HSV latency and reactivation in neurons. Trends Microbiol. 20, 604–611. doi: 10.1016/j.tim.2012.08.005
Wilson, G. J., Talkington, D. F., Gruber, W., Edwards, K., and Dermody, T. S. (1995). Group A streptococcal necrotizing fasciitis following varicella in children: case reports and review. Clin. Infect. Dis. 20, 1333–8. doi: 10.1093/clinids/20.5.1333
Wozniakowski, G., and Samorek-Salamonowicz, E. (2015). Animal herpesviruses and their zoonotic potential for cross-species infection. Ann. Agric. Environ. Med. 22, 191–194. doi: 10.5604/12321966.1152063
Wucherpfennig, K. W., and Strominger, J. L. (1995). Molecular mimicry in T cell-mediated autoimmunity: viral peptides activate human T cell clones specific for myelin basic protein. Cell 80, 695–705. doi: 10.1016/0092-8674(95)90348-8
Zachariadou, L., Stathi, A., Tassios, P. T., Pangalis, A., Legakis, N. J., Papaparaskevas, J., et al. (2014). Differences in the epidemiology between paediatric and adult invasive Streptococcus pyogenes infections. Epidemiol. Infect. 142, 512–519. doi: 10.1017/S0950268813001386
Zandi, E., Rothwarf, D. M., Delhase, M., Hayakawa, M., and Karin, M. (1997). The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell 91, 243–252. doi: 10.1016/S0092-8674(00)80406-7
Zeidler, R., Eissner, G., Meissner, P., Uebel, S., Tamp,é, R., Lazis, S., et al. (1997). Downregulation of TAP1 in B Lymphocytes by Cellular and Epstein-Barr Virus–Encoded Interleukin-10. Blood 90, 2390–2397.
Zhang, S.-Y., Jouanguy, E., Ugolini, S., Smahi, A., Elain, G., Romero, P., et al. (2007). TLR3 deficiency in patients with herpes simplex encephalitis. Sci. 317, 1522–1527. doi: 10.1126/science.1139522
Zhao, Z.-S., Granucci, F., Yeh, L., Schaffer, P. A., and Cantor, H. (1998). Molecular mimicry by herpes simplex virus-type 1: autoimmune disease after viral infection. Science 279. doi: 10.1126/science.279.5355.1344
Zheng, S., Li, Y., Zhang, Y., Li, X., and Tang, H. (2011). MiR-101 regulates HSV-1 replication by targeting ATP5B. Antiviral Res. 89, 219–226. doi: 10.1016/j.antiviral.2011.01.008
Zhou, G., Galvan, V., Campadelli-Fiume, G., and Roizman, B. (2000). Glycoprotein D or J delivered in trans blocks apoptosis in SK-N-SH cells induced by a herpes simplex virus 1 mutant lacking intact genes expressing both glycoproteins. J. Virol. 74, 11782–11791. doi: 10.1128/JVI.74.24.11782-11791.2000
Zhu, J., Hladik, F., Woodward, A., Klock, A., Peng, T., Johnston, C., et al. (2009). Persistence of HIV-1 receptor–positive cells after HSV-2 reactivation is a potential mechanism for increased HIV-1 acquisition. Nat. Med. 15, 886–892. doi: 10.1038/nm.2006
Zuo, J., Quinn, L. L., Tamblyn, J., Thomas, W. A., Feederle, R., Delecluse, H.-J., et al. (2011a). The Epstein-barr virus-encoded BILF1 protein modulates immune recognition of endogenously processed antigen by targeting major histocompatibility complex class I molecules trafficking on both the exocytic and endocytic pathways. J. Virol. 85, 1604–1614. doi: 10.1128/JVI.01608-10
Keywords: herpesviruses, cofactors, host–pathogen interactions, disease outcome, virome, coinfections
Citation: Sehrawat S, Kumar D and Rouse BT (2018) Herpesviruses: Harmonious Pathogens but Relevant Cofactors in Other Diseases? Front. Cell. Infect. Microbiol. 8:177. doi: 10.3389/fcimb.2018.00177
Received: 14 March 2018; Accepted: 08 May 2018;
Published: 25 May 2018.
Edited by:
Rachel L. Roper, The Brody School of Medicine at East Carolina University, United StatesReviewed by:
Bin Su, Beijing Youan Hospital, Capital Medical University, ChinaCopyright © 2018 Sehrawat, Kumar and Rouse. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sharvan Sehrawat, c2hhcnZhbkBpaXNlcm1vaGFsaS5hYy5pbg==
Barry T. Rouse, YnRyQHV0ay5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.