
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Infect. Microbiol. , 10 April 2018
Sec. Molecular Bacterial Pathogenesis
Volume 8 - 2018 | https://doi.org/10.3389/fcimb.2018.00114
This article is part of the Research Topic Cellular and Molecular Mechanisms of Mycobacterium tuberculosis Virulence View all 9 articles
 H. M. Adnan Hameed1,2
H. M. Adnan Hameed1,2 Md Mahmudul Islam1,2
Md Mahmudul Islam1,2 Chiranjibi Chhotaray1,2
Chiranjibi Chhotaray1,2 Changwei Wang1Yang Liu1,3Yaoju Tan4Xinjie Li4Shouyong Tan4
Changwei Wang1Yang Liu1,3Yaoju Tan4Xinjie Li4Shouyong Tan4 Vincent Delorme5Wing W. Yew6Jianxiong Liu4*
Vincent Delorme5Wing W. Yew6Jianxiong Liu4* Tianyu Zhang1,2*
Tianyu Zhang1,2*Tuberculosis (TB) is a formidable infectious disease that remains a major cause of death worldwide today. Escalating application of genomic techniques has expedited the identification of increasing number of mutations associated with drug resistance in Mycobacterium tuberculosis. Unfortunately the prevalence of bacillary resistance becomes alarming in many parts of the world, with the daunting scenarios of multidrug-resistant tuberculosis (MDR-TB), extensively drug-resistant tuberculosis (XDR-TB) and total drug-resistant tuberculosis (TDR-TB), due to number of resistance pathways, alongside some apparently obscure ones. Recent advances in the understanding of the molecular/ genetic basis of drug targets and drug resistance mechanisms have been steadily made. Intriguing findings through whole genome sequencing and other molecular approaches facilitate the further understanding of biology and pathology of M. tuberculosis for the development of new therapeutics to meet the immense challenge of global health.
Tuberculosis (TB) is an airborne infectious disease caused by Mycobacterium tuberculosis (Arnold, 2007; Lillebaek et al., 2016). During the eighteenth and nineteenth centuries, this disease widely plagued Europe and North America and was known as “Captain among these men of Death.” In the 1800's, a person with TB was advised to “take slumber and ingest nutritious diet,” due to lack of knowledge about this disease and the absence of medication (Keshavjee and Farmer, 2012). Later, the discoveries of tuberculin in 1890, Bacille-Calmette-Guérin (BCG) vaccine in 1908, streptomycin (STR) in 1944 and isoniazid (INH) in 1952 (Daniel, 2006) were all perceived as scientific revolutions that could finally lead to the eradication of this deadly disease (Fogel, 2015). The emergence of drug-resistance as well as infection and transmission of the human-immunodeficiency virus (HIV) further demonstrated the need of better weapons to eliminate TB. Soon after, researchers realized that more advanced investigations would be required for development of rapid diagnostic tools and effective chemotherapy to reduce drug resistant-TB morbidity, mortality and risk of transmission.
According to the World Health Organization (WHO), 10.4 million TB cases were reported in 2016, including 1 million people co-infected with HIV, 1.9 million were attributable to malnutrition, 0.8 million to diabetes and 0.8 million to smoking. The same year, TB caused 1.3 million deaths among HIV-negative patients and additionally 374,000 deaths among HIV-positive patients (WHO, 2017). M. tuberculosis, unlike other pathogens, requires an extended anti-TB therapy treatment of at least 6 months in order to achieve complete sterilization and prevent relapse. Chemotherapy of TB is required to tackle growing and semi-dormant/dormant bacilli, as well as to restrain the emergence of drug resistance. Unfortunately, development of drug resistance in the form of monoresistant-TB or MDR-/XDR-/TDR-TB critically obstructs the efficacy of currently available drug regimens. Improper use of antibiotics, lack of treatment adherence, mutational modifications, and limited access to drugs or diagnostic tools all are risk factors associated with the emergence of resistant forms of TB (Lange et al., 2014; Georghiou et al., 2017; Manson et al., 2017). Besides, acquired drug-resistant TB and transmitted drug-resistant TB pose a serious threat to global TB control and population health. To meet the evolving challenge of TB today, newly developed anti-TB drugs and vaccines are urgently needed.
Furthermore, several studies strongly emphasize that pathoadaptive mutations and the mutations in drug-related genes (e.g., genes responsible for drug activation/conversion) are mainly responsible for numerous pathways in the evolution of pathogenic organisms and emergence of drug-resistant strains, respectively (Day et al., 2001). In this review we mainly focused on molecular targets containing mutations which are found to be involved in development of drug resistance in M. tuberculosis.
WHO identified that TB is among the top 10 leading causes of death around the globe and caused more deaths than HIV in 2015 and 2016. Monoresistance to STR in M. tuberculosis was first reported in 1947-48 (Crofton and Mitchison, 1948). To overcome drug resistance development, initially, combined therapy including INH and para-aminosalicylic acid (PAS) along with STR, and subsequently addition of pyrazinamide (PZA) and rifampicin (RIF) had been practiced. However due to poor physician prescription, and/or poor patient adherence, and/or poor drug quality/supply, and factors not commonly encountered or well known, drug resistance can be acquired and escalated to involve both RIF and INH–MDR-TB (Frieden et al., 1993). In 2006, Centres for Disease Control and Prevention (CDC) and WHO jointly reported XDR-TB (MDR-TB with additional bacillary resistance to any fluoroquinolone (FQ) and at least one of the three second-line injectable drugs [i.e., amikacin (AMK), kanamycin (KAN), and capreomycin (CAP); Gandhi et al., 2006]. In the recent past, XDR strains of M. tuberculosis when tested to be resistant against rifabutin (RFB), clofazimine (CLO), dapsone, clarithromycin (CLR), and thiacetazone (THZ) alongside conventional first-line drugs (FLD) and second-line drugs (SLD), were proposed as “XXDR-TB” in Italy (Migliori et al., 2007). The term “TDR-TB” was proposed by Iranian researchers to describe the “XXDR-TB” (Velayati et al., 2009). Later, TDR-TB was also reported in India (Udwadia et al., 2012) and South Africa (Klopper et al., 2013).
In addition, two new drugs bedaquiline (BDQ) and delamanid (DMD) were also found to be ineffective against TDR-TB (Maeurer et al., 2014). Moreover, molecular analysis identified the mutations in the genomic sequence are the core cause of drug-resistant TB. MDR and XDR-TB are extremely serious disease worldwide as these can progress to TDR-TB with seemingly higher mortality than cancer by 2050 (Roca et al., 2015; Furin et al., 2016). WHO report of 2016 publicized the global extent of TB/HIV, MDR-, XDR-, and TDR-TB (Table 1). The collected data suggests the burden of TB in India is significantly higher than previously estimated, and slightly rising trends for the North Korea and the Philippines were also noticed. Moreover, India, China and the Russian Federation accounted altogether for 45% of the total RIF-resistant/MDR-TB burden. By the end of 2015, a total of 7579 XDR-TB cases were reported from 74 countries, which were more than twice the sum of 2014 report (WHO, 2016). The CDC stated in 2013 that XDR-TB patients have been identified in most of the regions of the world, including the United States. Interestingly, Australia was comparatively protected from DR-TB because of its strict public health policies as nearly 90% of TB cases occurred in the immigrant communities (Toms et al., 2015).
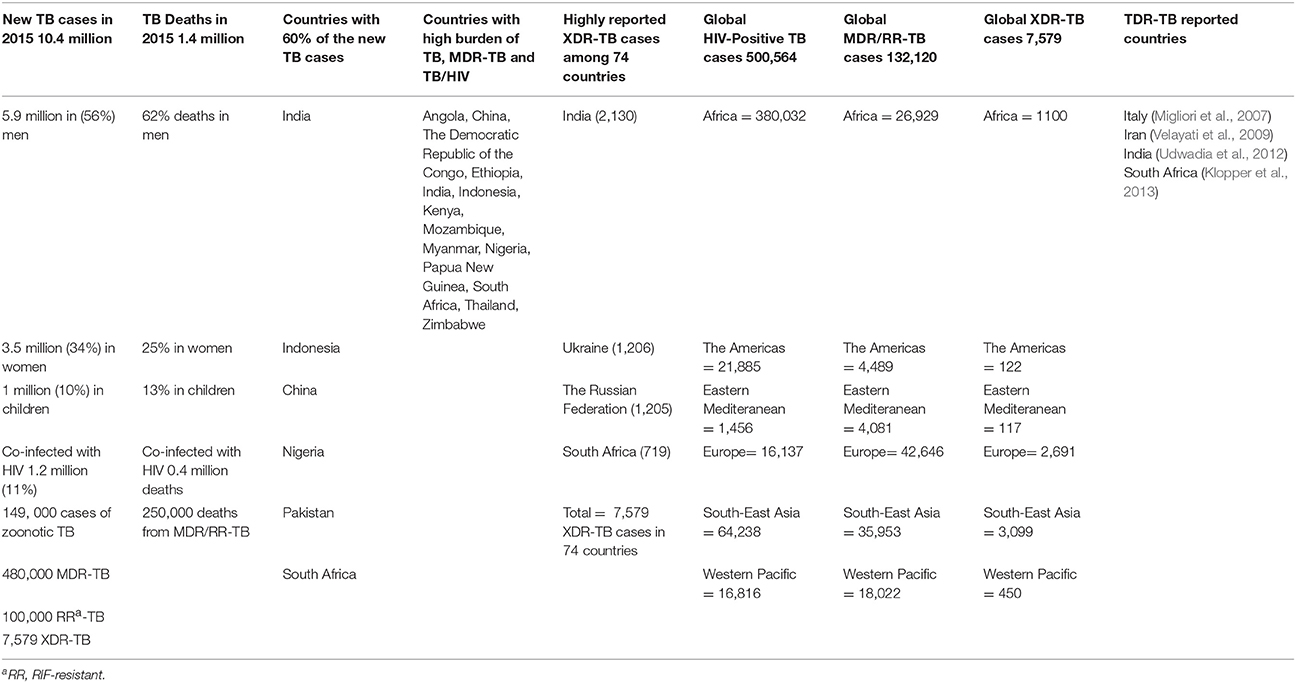
Table 1. Global Extent of MDR-, XDR-, and TDR-TB (WHO, 2016).
M. tuberculosis is a slow growing mycobacterium, which results in prolonged duration of drug susceptibility test (DST) on solid media from 4 to 6 weeks whereas in liquid media from 1 to 2 weeks. The lack of rapid diagnostic tools can be proposed as a risk factor for the prevalence of MDR-, XDR-, and TDR-TB, because the use of inefficient drugs during the initial treatment phase may promote the appearance of drug-resistant profiles. In this context, the WHO recommend the rapid and more advanced in vitro growth monitoring system, MGIT 960, as well as nucleic acid amplification method, Xpert MTB/RIF for early diagnosis of PZA-resistant and RIF-resistant TB, respectively (Lawn et al., 2013). Other devices using line probe assay (LPA) technology are now available, like Genotype MTBDRplus for rapid analysis of MDR-TB (Jacobson et al., 2013) and its advanced version Genotype MTBDRsl (Barnard et al., 2012) to diagnose XDR-TB.
New approaches have also been reported, like FASTPlaque-response bacteriophage assay (Minion and Pai, 2010), microscopic observation drug susceptibility assay (MODS) (Peter et al., 2016), use of dyes to monitor bacterial growth (Coban et al., 2016), sequencing and hybridization, reverse hybridization, direct sequencing (Deggim-Messmer et al., 2016), TB Biochip platform (Xue et al., 2016) and other molecular approaches. Each method was proposed to provide faster as well as more reliable results for early detection of drug-resistance. An important consideration is the cost and the infrastructure requirements of each technique, as this is the key factor that will ultimately limit their clinical use in many regions around the world.
Drug-resistant TB is mainly associated with chromosomal mutation mechanisms, in particular single nucleotide polymorphisms (SNP). The resistance causing factors that affect the mutation rate can be divided mainly into two groups; (i) cellular mechanisms, for example, inefficiency of mismatch repair, microsatellites, inadequate translations and error-prone DNA polymerases and (ii) external stress factors, including absence of rapid diagnostic facilities, improper anti-TB drugs prescribing practices, host environment and exposure to smoking or pollution (McGrath et al., 2014). Additionally, clinical outcomes of antibiotic combinations may also be influenced by inter-individual heterogeneities in drug pharmacokinetic and pharmacodynamics (Srivastava et al., 2011). Poor adherence of TB patients and costs required to achieve complete cure are probably the two main factors responsible for emergence of drug-resistance.
Indeed, most of the MDR-/XDR-TB patients had been treated previously either with ineffective anti-TB drugs or for suboptimal duration, resulting in an incomplete sterilization of the lungs and re-growth of “persisters” that became more resistant to the drugs which had been formerly practiced and turned them into XDR-/TDR-TB, respectively (Dalton et al., 2012). According to another study, development of drug resistance is mainly because of spontaneous mutations in drug targets which ensure the survival of TB bacilli at very low pH through induction of acid resistance, drugs adaptations, inactivity or degradation of drugs through enzymes encoded by genes of M. tuberculosis, molecular simulation of drug targets, and epigenetic drug tolerance (Jenkins et al., 2009; Smith et al., 2013).
Some previous anti-TB drugs are also found to be ineffective in preventing mycolic acid synthesis in DR-TB bacilli due to mutations in drug targets (Telenti et al., 1993; Ramaswamy and Musser, 1998; Gillespie, 2002; Watanabe et al., 2002). Therefore, other new targets should also be considered to control the pathways of drug resistance by new and more effective antituberculars. However, two new drugs, DMD and pretomanid may have better action on cell wall synthesis (Stover et al., 2000; Gler et al., 2012). Besides this, the role of efflux-pumps in drug-resistance in TB should not be underestimated, as reported for INH (Machado et al., 2012) and indicated by recent reports involving the membrane transporter MmpL5 in resistance to BDQ (Hartkoorn et al., 2014). Finally, concomitant bacterial infections and lack of new pharmacophore could be additional causes for the rapid emergence of MDR/XDR and TDR-TB (Srivastava et al., 2011; Vadwai et al., 2011; Machado et al., 2012; Dharmadhikari et al., 2013; Grossman et al., 2014; Lange et al., 2014). It is also worth noticing that the term “resistance” should be used carefully because misconception or misapplication of this may devise an idea that resistance is a binary phenotype, whereas it can be evaluated at multiple levels, i.e., low, moderate and high-level drug resistance (Böttger, 2011).
Several factors are associated with a worsening of TB infection. These include HIV (Das and Horton, 2010), diabetes mellitus (Jeon and Murray, 2008), cancer (Vento and Lanzafame, 2011), solid organ transplantation (Skrahina et al., 2012), renal disease (Wu et al., 2013) tumor necrosis factor alpha (TNF-α) antagonist treatment (Kisacik et al., 2016), alcohol abuse (Stoffels et al., 2013), tobacco use (Glickman and Schluger, 2016), air pollution, malignancies and an aging population (Negin et al., 2015). The development of TB disease is estimated to be 26- to 31-fold higher in people living with HIV than those without HIV infection in 2015 (WHO, 2016). In 2015, more than 28% of TB deaths were HIV positive, which describes a strong link between the two infections (HIV and M. tuberculosis).
A major feature of HIV infection is the chronic T-cell activation and progressive loss of CD4+ T-lymphocytes, which predispose the host to active TB. Depletion of CD4+ T-cells in HIV subjects with latent TB infection disrupts the steadiness and structure of TB granulomas in the lung, thereby promoting the progression of infection to disease by 20-fold (Geldmacher et al., 2012). Recently, the emerging facts indicated that clinical findings including lower lung field lesions, cavities, and acid-fast bacilli (AFB) smear positivity were at higher frequencies among TB patients having comorbidity with diabetes mellitus and suffered increased risk of treatment failure, relapse, and death (Carreira et al., 2012; Workneh et al., 2016). The WHO report signifies both TB and HIV infections were influenced by the increased burden of diabetes mellitus in Sub-Saharan African countries (WHO, 2016).
Whole-genome sequencing markedly increase the detection capability to find out the mutations in molecular targets which play significant role in development of resistance against anti-TB drugs and lead toward MDR/XDR/TDR-TB (Figure 1). Concurrently, it also facilitates in rapid and precise identification of virulence factors of pathogen and can be used to explore the pathways of disease transmission (Gilchrist et al., 2015). Though, antimicrobial resistance and bacterial virulence have been considered as different aspects but there is a composite relationship between antibiotic resistance and virulence as they share few common characteristics. A detailed study by Beceiro et al. (2013) has been published to explain how bacterial virulence and fitness can be affected by drug resistance and the relationship between resistance and virulence can be influenced by different genetic mechanisms (e.g., co-selection and compensatory mutations). Several observational studies have verified that emergence of drug resistance and increased virulence often arise almost simultaneously; but, their genetic association has been relatively ignored (Schroeder et al., 2017). However, uncovering the complexities of genetic modifications and drug resistance mechanisms may recognize the new drug targets which ultimately provides the more opportunities for discovery and development of new anti-TB drugs (Vincent et al., 2012). This review mainly considers the molecular targets and mutations involved in altering the metabolic activities to develop drug resistance which directs toward severe resistant strains of M. tuberculosis.
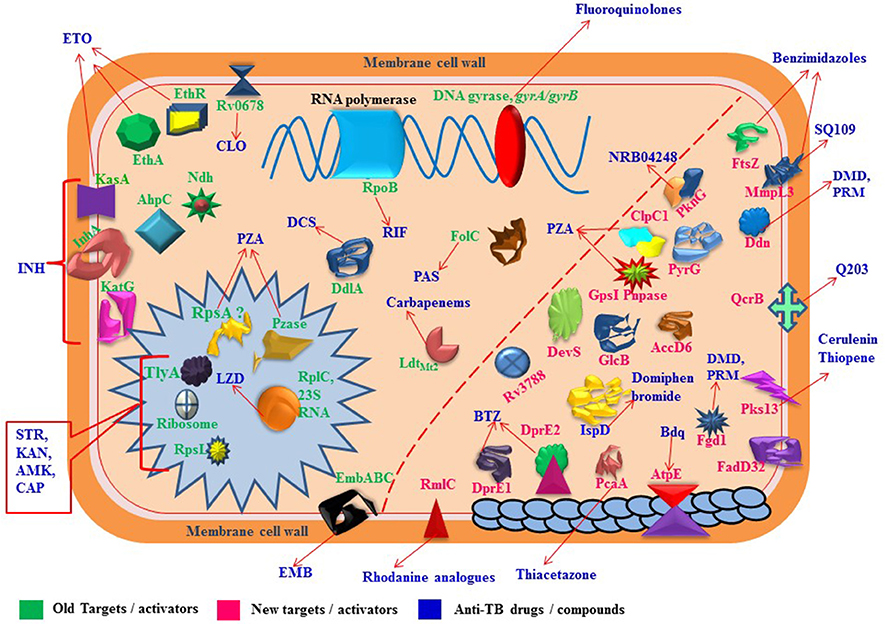
Figure 1. Proteins/RNAs/genes related to anti-TB drugs. 2'-O-methyltransferase TlyA (tlyA), Ribosomal RNA 16S (rrs), 30S ribosomal protein S12 RpsL (rpsL), 50S ribosomal protein L3 RplC (rplC), arabinosyltransferases EmbABC (embABC), DNA topoisomerase II /DNA gyrases GyrA, GyrB (gyrA, gyrB), RNA polymerase β-subunit RpoB (rpoB), catalase-peroxidase KatG (katG), alkyl hydroperoxidase C AhpC (ahpC), NADH dehydrogenase Ndh (ndh), enoyl-acyl-carrier protein-reductase InhA (inhA), β-ketoacyl ACP synthase KasA (kasA), Monooxygenase EthA (ethA), Transcriptional regulatory repressor protein EthR (ethR), Pyrazinamidase/nicotinamidase PZase PncA (pncA), 30S ribosomal protein S1 RpsA (rpsA), GpsI (polynucleotide phosphorylase, gpsI) and Pnpase (Rv2783c), ClpC1 (ATP-dependent protease ATP-binding subunit, clpC1), Conserved protein (Rv0678, Rv0678), folate synthase FolC (folC), nonclassical transpeptidase (LdtMt2), D-alanyl-D-alanine ligase DdlA (ddlA), transmembrane transport protein MmpL3 (mmpL3), Cell division protein FtsZ (ftsZ), dTDP-4-dehydrorhamnose 3,5-epimerase RmlC (rmlC), Acetyl/propionyl-CoA carboxylase (β-subunit) AccD6 (accD6), Deazaflavin-dependent nitroreductase Ddn (ddn), Mycolic acid synthase PcaA (cyclopropane synthase) (pcaA), CTP synthetase for Pyrimidine biosynthesis PyrG (pyrG), Serine/threonine-protein kinase PknG (pknG), Two component sensor histidine kinase DevS (devS), Malate synthase GlcB (glcB), ATP synthase C chain AtpE (atpE), MEP cytidylyltransferase IspD (ispD), Polyketide synthase Pks13 (pks13), Fatty-acid-AMP ligase FadD32 (fatty-acid-AMP synthetase) (fadD32), Hypothetical protein (Rv3788), ubiquinol-cytochrome C reductase QcrB (qcrB).
M. tuberculosis H37Rv has a genome comprising 4,411,532 bp with high GC contents (65.9 %) containing around 4,000 protein-coding genes as well as 13 pseudogenes, 45 tRNA genes, 3 rRNA genes, 30 ncRNA genes, and 2 miscRNA genes, indicating a high protein coding percentage (91.2%) as gene density is measured 0.91 genes per Kb, so the average length is 1,002 bases per gene (Data collected from, http://genolist.pasteur.fr/TubercuList). Mutations are very important in determining the transmissibility of specific genotypes (de Vos et al., 2013). Molecular and epidemiological data assists to evaluate the transmission rate of resistance genotypes (Dye et al., 2002; Cohen et al., 2003) which actually report that fitness rate may be affected by epistasis, the phenotypic effect of a mutation depends on the presence or absence of other mutations in the same genome (Borrell and Gagneux, 2011). In fact, drug resistance based on genetic mutations is more challenging which leads toward unknown resistance mechanisms (Zhang et al., 2013). A summary of different molecular targets and the related drugs is presented in Table 2. Each target is briefly described below.
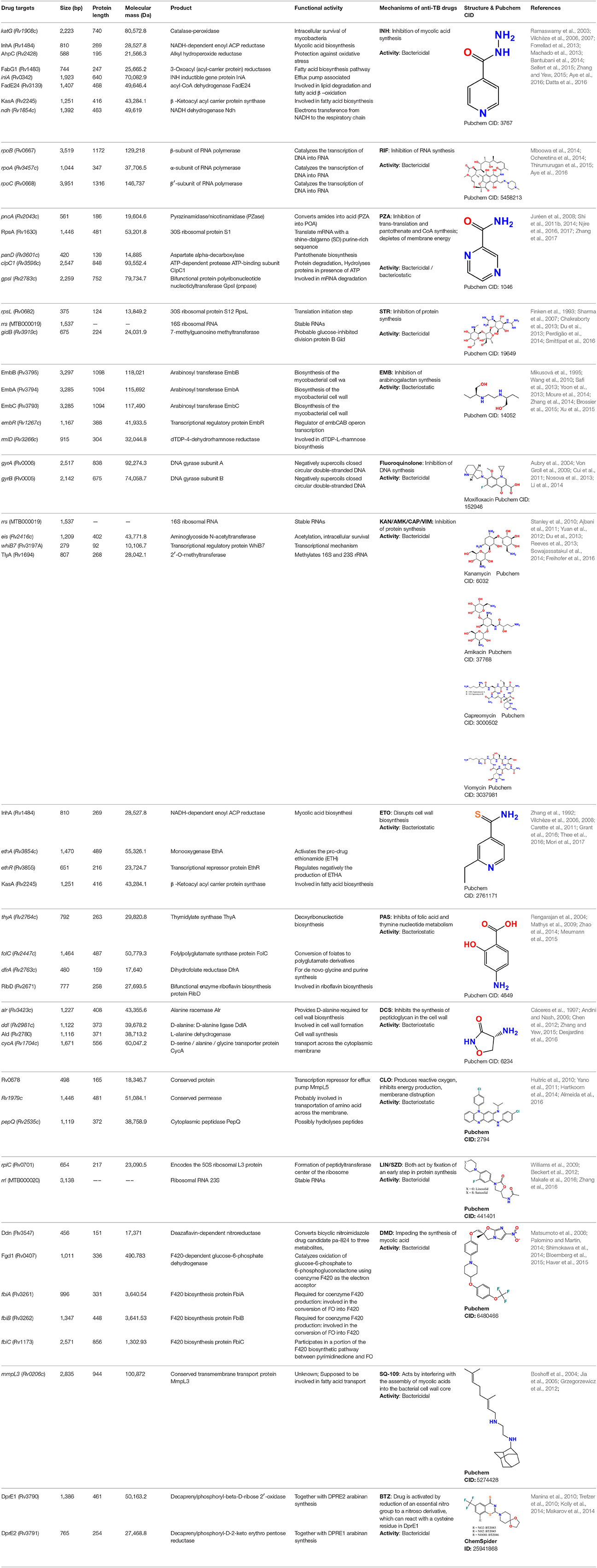
Table 2. Drug targets (genes/proteins), their products and functional activities and mode of action of anti-TB drugs.
RpoB is the target of RIF, a derivative of rifamycin. RpoB catalyzes the transcription of DNA into mRNA by using the four ribonucleoside triphosphates as substrates. In M. tuberculosis RIF binds with β-subunit of the RNA polymerase, encoded by rpoB, and inhibits the elongation of messenger RNA, thus interfering with transcription (Carlos and Martin, 2013; Piccaro et al., 2014). RpoB has a gene length of 3519 bp and conformational changes caused by mutations in rpoB can lead to RIF-resistance (Telenti et al., 1993). Ninety-six percent of RIF-resistance occurs within the “hot-spot region” (of 81 bp), also known as RIF-resistance determining region (RRDR), covering codons 507–533 of rpoB gene (Ramaswamy et al., 2003). Several studies have reported mutations in codons 516, 526 and 531 are most commonly found in RIF-resistant isolates (Ocheretina et al., 2014; Thirumurugan et al., 2015).
In particular, mutation in the rpoB gene at 531 codon (serine to leucine) is highly significant as it confers cross-resistance to rifabutin (Mboowa et al., 2014; Thirumurugan et al., 2015; Aye et al., 2016). whereas mutations at codons 516, 518, 526, and 529 are associated with low-level resistance to RIF and conserved susceptibility to other rifamycins, e.g., rifabutin or rifalazil (Cavusoglu et al., 2004; Tan et al., 2012). However, compensatory mutations were also identified in rpoA and rpoC encoding respectively for α and β' subunits of RNA polymerase (Comas et al., 2012). These compensatory mutations could be important for reinstating the fitness and emergence of MDR strains, and their transmissibility in vivo (Brandis and Hughes, 2013). Further studies are required to better understand their specific roles.
Mutations in katG and inhA are the main cause of resistance to INH in M. tuberculosis. KatG has a molecular mass of 80,572.8 Da and gene size of 2,223 bp while InhA has 28,527.8 Da molecular mass and 810 bp gene size, respectively. katG encodes multifunctional enzymes that exhibit both catalase-peroxidase and peroxynitritase activities, the former being important for activating the prodrug, and the latter in association with pathways involving reactive nitrogen and oxygen intermediates (Zhang et al., 1992). On the other hand, AhpC, SodC, KatG, and TpX are recognized as essential for virulence of the M. tuberculosis (Forrellad et al., 2013). NADH-dependent enoyl-acyl carrier protein (ACP)-reductase, encoded by inhA, is involved in the biosynthesis of mycolic acids and participates in the second reductive step in fatty acid biosynthesis (Rozwarski et al., 1998; Vilchèze et al., 2006). Mutational changes in these two genes, katG and inhA, are mainly associated with the resistance mechanism of INH (Ramaswamy et al., 2003).
Conversely, mutations in Rv0340-0343, fadE24, efpA, and kasA were identified in both INH-resistant and INH-susceptible strains, so their association with INH-resistance requires further clarification (Vilchèze et al., 2007). Among the katG mutations, S315T is considered as the most pervasive mutation that accounts for 40~94% resistance in MDR strains and results in reducing the ability of KatG to convert INH into isonicotinic acid, a precursor for the formation of INH-NAD adduct (Bantubani et al., 2014; Seifert et al., 2015; Aye et al., 2016). In addition, another newly reported katG mutation L101R identified in clinical isolates, changing an hydrophobic leucine to hydrophilic arginine, possibly involved in alteration of the conformation of binding protein near the active site and inhibited its bio-activation (Datta et al., 2016).
The second main cause of resistance to INH is due to mutations in the promoter region of inhA, resulting in the overexpression of inhA. The most frequently observed mutation in inhA regulatory region at position −15C/T is more generally associated with low level resistance to INH (MIC < 1 μg/ml) (Banerjee et al., 1994; Fenner et al., 2012; Aye et al., 2016). Moreover, double mutations at −8T/C, −15/T (Zhang and Yew, 2015), and −17C/T (Müller et al., 2011) in the inhA promoter region were also found to be associated with INH-resistance. Some recent studies have described that a mutation in the regulatory region of inhA, together with a mutation in inhA coding region, results in high-level resistance against INH as well as cross-resistance against the structurally related ethionamide (Machado et al., 2013).
EmbB and EmbC have molecular masses of 118,021 Da, and 117,490 Da, respectively, and gene sizes of 3,297 bp, 3,285 bp, respectively. Mutations in embB and embC cause resistance against ethambutol [EMB; dextro-2,2′-(ethylenediimino)-DI-1-butanol], through restricting the action of drug to cease the biosynthesis of mycobacterial cell wall. In M. tuberculosis, the genes embCAB, are organized as an operon that encodes arabinosyl transferases, involved in the synthesis of arabinogalactan. (Mikusová et al., 1995). EMB plays intrusive role in biosynthesis of arabinogalactan in the cell wall to convert multiplying bacilli into a bacteriostatic phase (Wang et al., 2010). Mutations in the embCAB operon were described to cause alterations in the drug-protein binding site (Xu et al., 2015). Numerous studies concluded that a mutation at codon306 in embB was the cause of EMB-resistance (Shi et al., 2011a; Yoon et al., 2013; Moure et al., 2014).
It was also reported that mutations in genes of the decaprenylphosphoryl-beta-D-arabinose (DPA) biosynthesis and utilization pathway genes, Rv3806c and Rv3792, combined with mutations in embB and embC genes increased the MICs ranges of EMB, depending on mutation type and number (Shi et al., 2011a; Safi et al., 2013). A strong association was also noticed between the Met306Val and Met306Leu at emb306 and EMB-resistance (Zhang et al., 2014), which suggests the usefulness of the embB306 mutation to serve as potential molecular markers for EMB resistance. However, in 30% EMB-resistant M. tuberculosis strains there was no mutation in embB, alluding to the need to intensively search for other mechanisms of EMB resistance (Shen et al., 2007; Perdigão et al., 2009). Interestingly, in another study, 70% mutations were found in 306, 406 or 497 codons, 13% mutations outside the three regions between codons 296 and 426 and 15% mutations in the embC-embA intergenic region among the total 98% of mutations in the embCAB locus (Brossier et al., 2015). Recently, mutations in embB along with mutations in ubiA, encoding for a decaprenylphosphoryl-5-phosphoribose (DPPR) synthase associated with arabinogalactan synthesis pathway, were associated with high level of resistance to EMB (Tye et al., 2015).
Pyrazinamide (PZA) is an important front-line anti-TB drug. In M. tuberculosis, resistance to PZA is mainly associated with pncA, rpsA, and panD. However, some other proteins such as ppsA encoding polyketide synthase involved in phthiocerol dimycocerosate (PDIM) synthesis, cell division protein FtsH, TetR family transcriptional regulator (3R)-hydroxyacyl-ACP dehydratase subunit HadC, phosphate ABC transporter permease protein PstC2 and transmembrane transport protein MmpL4 found to be responsible for PZA resistance (Zhang et al., 2017). Pyrazinamidase (Pzase), encoded by the 561-bp pncA, converts the pro-drug PZA into pyrazinoic acid (POA, its active form). PZA resistance in M. tuberculosis is most commonly associated with mutations in the whole open reading frame or 82–262 bp regulatory region of pncA gene (Juréen et al., 2008).
POA interrupts the bacterial membrane activities. POA and its n-propyl ester may have the ability to constrain the fatty acid synthase type I in replicating M. tuberculosis bacilli, ineffective efflux pump also may result in accumulation of protonated POA during acidic conditions resulting in bacterial cell damage (Scorpio and Zhang, 1996; Zimhony et al., 2007; Shi et al., 2011b, 2014; Njire et al., 2016). The 481-amino acid-long 30S ribosomal protein S1 is encoded by rpsA. It plays a significant role in mRNA translation (involving a short shine-dalgarno (SD) purine-rich sequence), and in trans-translation, a unique process that utilizes transfer-messenger RNA (tmRNA) molecule to rescue stalled ribosomes. POA was also associated with disruption of the trans-translation process through binding to the 30S ribosomal protein S1 (RpsA) (Shi et al., 2011b). In the work of Shi et al. (2011b), deletion of alanine, resulting from a GCC deletion at 438 bp (C-terminus) of rpsA, was found in PZA-resistant strains lacking mutations in pncA gene. Such mutation was thought to induce resistance against POA and, accordingly, increased level of resistance were observed after overexpression of rpsA (Shi et al., 2011b).
Later, many studies presented disparate results about the mutations in rpsA in both PZA-resistant and PZA-susceptible isolates (Bhuju et al., 2013; Maslov et al., 2015). However, only synonymous mutations were reported in rpsA (A636C and G960A) for both PZA-resistant and PZA-susceptible clinical strains (Alexander et al., 2012). In another study, mutations in rpsA gene were found in 3/52 PZA-resistant and 1/108 PZA-susceptible clinical isolates (Tan et al., 2014). This absence of clinical isolates presenting rpsA mutations may indicate a strong fitness cost associated with such mutations, although more investigations are required to fully understand the link between RpsA and the activity of PZA. The third gene panD, of 420 bp in size, is putatively involved in pantothenate biosynthesis through converting L-aspartate into beta-alanine (Shi et al., 2014). Some studies found mutations in panD gene of PZA-resistant isolates lacking mutations in pncA and rpsA (Pandey et al., 2016).
Among the three genes identified so far as potentially linked with resistance to PZA, i.e., pncA, rpsA, and panD, the most frequent mutations (>80%) were found to occur in the pncA gene (Xia et al., 2015; Xu et al., 2016). Recently, a new target of PZA clpC1 (Rv3596c) was identified, encodes an ATP-dependent ATPase which is involved in protein degradation by forming a complex with protease ClpP1 and ClpP2 (Zhang et al., 2017). ClpC1 is an 848-amino acids containing protein and the mutation (G296T) change amino acid G99D cause resistance to PZA. Interestingly, mutations in clpC1 has also been reported as a resistance factor against three new cyclic peptide antibiotics including cyclomarin A (Schmitt et al., 2011), lassomycin at the N-terminal repeat region at Q17R, R21S, and P79T (Gavrish et al., 2014) and ecumicin at L92S or F or L96P sites (Gao et al., 2015). The three new drug candidates appear to bind at the different sites of the ClpC1. The gene Rv2783c encoding a bifunctional enzyme was indicated as a new target very recently (Njire et al., 2017). Rv2783 was proved to be able not only to catalyze metabolism of RNA and single-stranded DNA, but also to metabolize ppGpp, an important signal transducer involved in the stringent response in bacteria.
The rpsL gene is very small, only 375-bp, while rrs gene is much larger, with 1,537 bp but both are associated with resistance to STR. The rpsL encodes 30S ribosomal protein S12 RpsL involved in the initiation step of RNA translation, while rrs encodes 16S rRNA. Mutations in rpsL and rrs are known to cause resistance against STR (Finken et al., 1993), the first anti-TB drug discovered. STR is an aminoglycoside and targets the actively growing bacteria by hindering the translation of protein synthesis (Chakraborty et al., 2013). More precisely, STR interacts with formyl-methionyl-tRNA to bind the 30S subunit of the ribosome at the ribosomal protein S12 encoded by rpsL gene and also at 16S rRNA encoded by rrs gene, preventing normal functioning of the ribosome (Sharma et al., 2007).
The most commonly reported mutation in rpsL is the replacement of lysine into arginine at positions 43 and 88. Regarding rrs, mutations around nucleotides 530–915 make M. tuberculosis strains highly resistant to STR (Jagielski et al., 2014; Zhao et al., 2014). Additionally, a 675 bp gene, gidB, encoding for a conserved 7-methylguanosine methyltransferase specific for the 16S rRNA, was found to confer intermediate-level of resistance toward STR by conferring the A80P mutation to the gene product GidB (Perdigão et al., 2014). Generally, mutations in the rpsL gene covers around 80% of STR-resistance (Jnawali et al., 2013). More precisely, the mutations at codons 43 and 88 in rpsL gene were identified in more than 60% of STR-resistance cases, while 17% contained rrs mutations and 14% were observed with mutations in gidB, in a panel of 161 clinical isolates (Smittipat et al., 2016).
Some STR-resistant M. tuberculosis strains also conferred cross-resistance with kanamycin (KAN) and amikacin (AMK). The mutation at the codon A1401G in the rrs gene in particular was associated with high-level resistance to KAN and AMK, along with cross-resistance to capreomycin (CAP). Whereas mutations C1402T or G1484T were also associated with CAP resistance in addition cross-resistance with KAN or viomycin (VIM) (Du et al., 2013; Sowajassatakul et al., 2014). Similarly, mutations at position 1,400, 1,401, and 1,483 bp in the rrs gene were linked with high-level resistance to both KAN and AMK in KAN-resistant strains (Ajbani et al., 2011; Yuan et al., 2012). Moreover, mutations in the promoter region of the whiB7 gene, with a role in transcriptional mechanisms, were shown to be associated with resistance to an array of antibiotics, and increased expression of the efflux enhanced by atpE and the eis promoter region was also observed (Reeves et al., 2013).
Further, eis and tlyA (1,209 bp and 807 bp in size, respectively) were also found to have putative roles in resistance. eis encodes an aminoglycoside acetyltransferase and mutation at position −10 and −35 of the eis promoter has been associated with the low-level resistance to KAN (Ajbani et al., 2011; Yuan et al., 2012; Du et al., 2013; Reeves et al., 2013; Sowajassatakul et al., 2014). tlyA encodes rRNA methyltransferase, specific for 2′-O-methylation of ribose in rRNA. Expression of tlyA gene (commonly A1408G) affects both drug susceptibility and fitness cost of drug resistance through methylation mechanisms (Freihofer et al., 2016). However, another study did not find any mutation in the tlyA gene (Du et al., 2013). Besides, overexpression of Rv0148 protein play a vital role in three-fold increase in MIC of AMK and two-fold of KAN (Sharma et al., 2015). While overexpression of ferritin (Rv3841/bfrB) showed an alternate behavior, there was two-fold increase in MIC of AMK and three-fold of KAN (Sharma et al., 2016). Regarding to above discussed genes and their role in development of resistance, it is important to note that KAN and AMK are aminoglycosides and CAP and VIM are cyclic peptide antibiotics, they have similar structures and bind on the same location in the ribosome, at the interface of the small and large subunits (Stanley et al., 2010). The similarity of the mechanisms of actions and their resistance development simplifies understanding to some extent.
gyrA and gyrB are the two main genes with mutations linked to resistance against fluoroquinolones (FQ's). gyrA is 2517-bp long while gyrB gene is 2,028-bp. Mutations in the quinolone resistance-determining region (QRDR) of gyrA (74–113 codons) and gyrB (codon 500–540) in M. tuberculosis are largely responsible for generating the resistance to FQ's (Pantel et al., 2012; Nosova et al., 2013). Two subunits α and β each of gyrA and gyrB, that encode topoisomerase II (DNA gyrase) catalyzing the supercoiling of DNA, furnishes the most important target of FQ action (Aubry et al., 2004). The most commonly found mutations at position Ala-74, Gly-88 Ala-90, Ser-91, and Asp-94 of gyrA generally result in high-level resistance against FQ (Li et al., 2014).
A natural polymorphism at position 95 (Ser or Thr) in gyrA is not related to FQ's-resistance since it is also found in FQ-sensitive strains (Nosova et al., 2013). Usually, mutations in gyrB (10–15%), being less commonly found among clinical isolates, induce low-level resistance to FQ's (Cui et al., 2011). However, the synchronized mutations in both gyrA and gyrB, such as Ala543Val (gyrB)-Asp94Asn/Asp94Gly (gyrA) and Asn538lle (gyrB)-Asp94Ala (gyrA) persuaded very high resistance against FQ's (Long et al., 2012). An interested study identified that presence of mutations T80A and A90G in gyrA led to hypersusceptibility to several quinolones (Aubry et al., 2006). Similarly, most of the mutations in the gyrA Asn538Asp and Asp500His in gyrB are shown to be associated with cross-resistance among the FQs, whereas in gyrB the mutation Arg485His does not confer any resistance (Nosova et al., 2013). One intriguing study found a M. tuberculosis strain with Asn533Thr mutation in gyrB showing susceptibility to ofloxacin (OFX) but resistance to moxifloxacin (MOX) and gatifloxacin (Von Groll et al., 2009).
Proteomic comparative study enlightened the 11 proteins (Rv1080c, Rv1827, Rv2623, Rv1636, Rv0952, Rv1932, Rv0009, Rv0054, Rv2889c, Rv3418c, and Rv3914) that were overexpressed in the presence of OFX and MOX. Among them, Rv1636, Rv2623, and Rv1827 were reported with unidentified functions. Molecular docking and InterProScan investigation explored the interaction of conserved domain of hypothetical proteins with these both drugs which possibly directs the inhibition of functions of these proteins that might be overexpressed to repress this effect (Lata et al., 2015b).
In another study, the same author reported 14 proteins with more intensities in OFX resistant isolates as compare to susceptible. Interaction of conserved domains and motifs of hypothetical proteins (Rv3551, Rv2744c, Rv0148, and Rv2140c) and OFX was revealed through molecular docking, these findings indicated the role of these proteins in resistance mechanisms as well as prospective drug targets (Lata et al., 2015a). Drug efflux mechanisms or changes in the membrane permeability to agents might constitute alternative resistance mechanisms regarding FQ resistance in M. tuberculosis (Escribano et al., 2007). A study corroborating such ideas has found that 30% of FQ's-R strains do not harbor any mutation in the QRDRs of both gyrA and gyrB genes (Alvarez et al., 2014). In a recent study by Pucci et al., M. tuberculosis GyrB inhibitors (SPR-720, SPR-750) have proved to be attractive for developing novel drugs against TB without cross-resistance to known quinolones and showing very good activity in vivo.
ethA and ethR are associated with resistance to ethionamide (ETH) (2-ethylpyridine-4-carbothioamide) and prothionamide (2-propylpyridine-4-carbothioamide) which are important drugs for the treatment of MDR-TB and TB meningitis in both adults and children (Thee et al., 2016). The 1,470-bp long ethA encodes the monooxygenase EthA which bio-activates the pro-drug ETH into its active form (Grant et al., 2016). EthA is negatively regulated by the transcriptional repressor EthR (Carette et al., 2011). ETH is derived from nicotinic acid having a similar structure to INH. The action of ETH is to interrupt mycolic acid synthesis by establishing an adduct with NAD that inhibits the enoyl-ACP reductase InhA (Mori et al., 2017). Mutations in ethA/ethR, as well as mutations in inhA or its promoter cause the resistance to both INH and ETH (Carette et al., 2011). Accordingly, −15C to −15T mutation in the promoter region of inhA and S94A (serine to alanine) and I194T (isoleucine to threonine) mutations in the gene product InhA were found to confer high-level resistance against INH and ETH in MDR-TB (Machado et al., 2013). Furthermore, mshA, a gene encoding a glycosyltransferase involved in mycothiol biosynthesis, has also been considered as a possible resistance target for ETH (Vilchèze et al., 2008).
The 792-bp long thyA, encoding thymidylate synthase A, has an important role in deoxyribonucleotide biosynthesis, by providing the sole de novo source of dTMP through conversion of deoxyuridine monophosphate (dUMP) to deoxythymidine monophosphate (dTMP). Interacting with folate metabolism is essential, especially involving tetrahydrofolate as a “methyl” donor. In an interesting study, mutations in thyA gene were identified by transposon mutagenesis were associated with resistance to PAS (Rengarajan et al., 2004). Some studies have affirmed Thr202Ala in thyA as the most common mutation associated with PAS-resistance, though a few susceptible isolates containing the same mutation have also been observed (Meumann et al., 2015). According to recent studies, the 1464-bp long folC gene (encoding FolC-dihydrofolate synthase/ folylpolyglutamate synthase), with various missense mutations were found to confer resistance to PAS in laboratory isolates of M. tuberculosis. The main function of FolC, the bifunctional enzyme, is to convert folates into polyglutamate derivatives, in simplistic terms. Bacteria require folate for the biosynthesis of glycine, methionine, formyl-met-tRNA, thymidylates, purines and pantothenate. In 85 MDR-TB clinical isolates, mutations in folC were identified in five PAS-resistant isolates (Zhao et al., 2014). In another study, only 37% of PAS-resistant strains had mutations in thyA, suggesting that other mechanisms of resistance to PAS would need to be explored (Mathys et al., 2009).
rplC and rrl genes have been associated with different levels of phenotypic resistance to Linezolid (LZD) and Sutezolid (PNU-100480, SZD) in M. tuberculosis (Makafe et al., 2016; Zhang et al., 2016). rplC gene has 654 bp in length and encodes the 50S ribosomal L3 protein, and may contribute in the synthesis of the ribosomal peptidyltransferase. Whereas, rrl is 3138 bp long and encodes 23S ribosomal RNA. LZD and SZD belonging to the oxazolidinone class, act by fixation of an early step in protein synthesis, through binding of the assembly initiator protein directly near the 3′-end of 23S rRNA, where nucleation of the assembly of the 50S subunit is performed (Williams et al., 2009). However, recently, the mutation T460C in rplC of LZD-resistant strain (Cys154Arg) has been repeatedly shown to be of considerable importance (Williams et al., 2009; Beckert et al., 2012; Makafe et al., 2016; Zhang et al., 2016). Moreover, in vitro selected LZD-resistant mutants with G2576T and G2061T mutations in rrl gene had MICs of 16–32 μg/ml, while susceptible strains without mutations showed MICs of 4–8 μg/ml (Hillemann et al., 2008; Zhang et al., 2016). The mutations in these two genes are associated with only 29.4% of LZD-resistance in M. tuberculosis (Islam et al., 2017). So, further resistance mechanisms need to be explored.
The 498-bp Rv0678 encodes a conserved protein with a molecular mass of 18,346.7 (Da). Some recent studies recommended that mutations in the transcriptional regulator of Rv0678 up-regulated MmpL5, a multi-substrate efflux pump, causing resistance not only to CLO (a riminophenazine compound) but also to BDQ, a diarylquinoline (Andries et al., 2014). Some studies suggested the outer membrane of M. tuberculosis as the possible target of CLO (Yano et al., 2011). The main factor for resistance to CLO other than mutations in the transcriptional regulator of Rv0678 is not yet fully characterized (Hartkoorn et al., 2014). The atpE gene is known to encode the subunit C of the ATP synthase, AtpE (lipid-binding protein) (dicyclohexylcarbodiimide-binding protein), a complex structure that generates the ATP needed by the mycobacterial cell.
Mutations in the 246-bp long atpE gene (Rv1305) are thought to be related to the resistance to BDQ with the most frequently detected mutations including A63P and I66M (Andries et al., 2005; Koul et al., 2007). BDQ has a favored specificity toward the mycobacterial ATP synthase, as compared to mitochondrial ATP synthase in exerting its therapeutic action (Haagsma et al., 2009), but the mutations in AtpE (A63P and I66M) disturbs such inhibition against bacterial ATP synthase C. However, out of 53 strains of M. tuberculosis only 15 displayed mutations while 38 strains lacked mutations in atpE or even in the F0, F1 operons (Huitric et al., 2010), which strongly implicates that some other resistance mechanisms against BDQ might be present. Recently, mutations in pepQ (Rv2535c, a putative Xaa-Pro aminopeptidase) were proposed to be able to confer cross-resistance between BDQ and CLO. Whereas mutations in Rv0678 and pepQ were noticed at a similar rate in a wild-type population treated with BDQ or CLO in mice. Mutations in Rv0678 and pepQ regulate the significant mechanisms of clinical resistance to BDQ and CFZ (Almeida et al., 2016).
alrA encodes D-alanine racemase AlrA, required for the conversion of L-alanine to D-alanine (Palomino and Martin, 2014). D-alanine ligase inhibits the production of peptidoglycan conductive to cell wall formation in bacteria. D-cycloserine (DCS) is a time-honored oral bacteriostatic anti-TB drug used in treatment of TB, especially MDR-TB and XDR-TB (Zhang and Yew, 2015). The absolute target of DCS in M. tuberculosis is still not fully identified. However, according to some prior studies, overexpression of AlrA triggered resistance to DCS in recombinant mutants of Mycobacterium smegmatis (Cáceres et al., 1997). Interestingly, a very recent study reported that loss-of-function mutations in ald (Rv2780), encoding L-alanine dehydrogenase, were also associated with resistance to DCS (Desjardins et al., 2016).
Another gene cycA, of 1,671 bp in size, encodes a D-alanine transporter. The point mutation in cycA has been considered a possible cause for resistance in M. bovis against DCS (Chen et al., 2012). Low cell wall permeability and the expression of emr37, a gene that collates methylase at a specific site in the 23S rRNA, hindering the binding of the antibiotic has been ascribed as the key factor of resistance to macrolides leading four to eight-fold rise in MIC values (Andini and Nash, 2006).
ddn has a gene size of 456 bp and encodes deazaflavin-dependent nitroreductase, while fgd1 with a size of 1,011 bp encodes F420-dependent glucose-6-phosphate dehydrogenase Fgd1. fbiA/B/C is a complex/operon of three genes with 996, 1,347, and 2,571 bp gene sizes, respectively. These three genes are mainly required for coenzyme F420 production for the biosynthesis of protein FbiA, FbiB, and FbiC sequentially. ddn, fgd1, and fbiA/B/C gene products were proposed as the targets of DMD and pretomanid (PA-824, PRM). DMD is a derivative of nitro-dihydro-imidazooxazole and acts by impeding the synthesis of mycolic acid in a distinct fashion compared to INH, as it only inhibits methoxy- and keto-mycolic acid synthesis while INH also inhibits α-mycolic acid (Matsumoto et al., 2006; Palomino and Martin, 2014). Mutations in ddn, fgd1, and fbiA/B/C have been proposed to be a mechanism of resistance to DMD and PRM in M. tuberculosis (Shimokawa et al., 2014).
A very recent study reported mutations in the fbiA and fgd1 genes associated with M. tuberculosis resistance to DMD in a patient with formidable drug-resistant TB (Bloemberg et al., 2015). PRM is a nitroimidazole derivative with activity against both latent TB and active TB as tested under aerobic and anaerobic conditions (Stover et al., 2000). PRM is a prodrug that needs a metabolic activation principally by a deazaflavin (cofactor F420)-dependent nitroreductase (Ddn) (Manjunatha et al., 2006). It also acts by inhibiting mycolic acid synthesis. A very recent assay of drug metabolism genes found potential resistance to PRM, and detected mutations frequency higher than those previously reported (Stover et al., 2000; Haver et al., 2015). However, further studies are essential to better delineate the mechanisms underlying bacillary resistance.
Mutations in mmpL3 were attributed to resistance in M. tuberculosis against SQ-109 (1,2-ethylenediamine), a synthetic analog of EMB (Tahlan et al., 2012). The 2,835-bp MmpL3 is a transmembrane transport protein presumably involved in fatty acid transport. SQ109 acts by interfering with the assembly of mycolic acids into the bacterial cell wall, resulting in accumulation of trehalose monomycolate, a precursor of the trehalose dimycolate. Similarly other cell wall inhibitors such as INH and EMB, SQ-109 induces the transcription of iniBAC operon required for efflux pump functioning (Boshoff et al., 2004). Its synergistic effects with BDQ in vitro and interactions with SZD were also observed. Additionally, mutations in the mmpL3 gene of spontaneously generated SQ-109-resistant mutants has suggested mmpL3 as the target of SQ-109 by virtue of the transporter mechanism discussed above (Grzegorzewicz et al., 2012). In M. tuberculosis strains resistant to INH, EMB and SQ109, there is an up-regulation of ahpC, signifying a possible role of this gene in the development of resistance against these drug(s) (Jia et al., 2005).
dprE1 (Rv3790) and dprE2 (Rv3791) encode decaprenylphosphoryl-beta-D-ribose 2′-oxidase and decaprenylphosphoryl-D-2-keto-beta-erythro-pentose reductase, respectively. These genes encode proteins that catalyze the two-step epimerization of decaprenylphosphoryl ribose (DPR) to decaprenylphosphoryl arabinose (DPA) in the arabinan synthesis pathway, which is essential for building the bacterial cell wall (Kolly et al., 2014). dprE1 and dprE2 were the possible targets of 1,3-benzothiazin-4-one or benzothiazinone (BTZ) (Makarov et al., 2014). BTZ is activated in the bacteria by reduction of an essential nitro group to a nitroso derivative, which can react with a key cysteine residue in DprE1 and form a covalent adduct (Trefzer et al., 2010). In spontaneous BTZ-resistant mutants, amino-acid Cys387 of DprE1 was replaced by Ser or Gly. On the other hand, in M. avium, which is naturally resistant to BTZ, amino-acid Cys387 was replaced by an Ala. An alternative BTZ-resistance mechanism has recently been described in M. smegmatis. Overexpression of nitroreductase NfnB indeed inactivated the drug by reducing its critical nitro-group into an amino-group. This was facilitated by a common amino acid stretch between NfnB and DprE1 (Manina et al., 2010). M. tuberculosis, however, seems to lack nitroreductases which enables it to inactivate this drug. Nevertheless, this finding could be important for development of new BTZ analogs with improved activity.
There are some new findings about therapeutic drug targets of new anti-TB drugs. Very recently, VCC234718, a molecule found to be very active against M. tuberculosis with growth inhibition activity. In a VCC234718-resistant mutant, a Y487C resistance-conferring substitution was identified in the inosine monophosphate dehydrogenase, GuaB2, which was subsequently recognized to be the prime molecular target of VCC234718 (Singh et al., 2017). Similarly, another study reported a potential promoter mutation in upstream of guaB2 in a non-cytotoxic indazole sulfonamide resistant mutant (Park et al., 2017).
Conversely, 11 eukaryotic-like serine/threonine protein kinases (STPKs) were observed in M. tuberculosis, which are considered as key components in cell growth, signal transduction and pathogenesis. An interesting study discovered the binding proteins in M. tuberculosis for all of the STPKs, and constructed the first STPK protein interaction (KPI) map that includes 492 binding proteins and 1,027 interactions. Functional investigations confirmed that PknG regulates cell wall integrity through peptidoglycan (PG) biosynthesis, e.g., MurC (Wu et al., 2017). A better understanding of biochemical pathways in M. tuberculosis is essential for the development of new and efficient chemotherapeutics.
Two important enzymes, isocitrate lyase (ICL) and malate synthase (MS) play vital role in regulation of glyoxylate shunt, a pathway required by M. tuberculosis to metabolize fatty acids (FAs). ICL may facilitates in survival of M. tuberculosis during the acute or chronic phases of infection through physiologic activities apart from fatty acid metabolism. MS mediates growth and survival on fatty acids through its potential ability to simultaneously detoxify a metabolic byproduct arising from the initial assimilation of acetyl coenzyme A (acetyl-CoA), glyoxylate, while assimilating a second molecule of acetyl-CoA. Depletion of MS during acute or chronic phase infections kills TB bacilli. These studies recommend MS as a potential drug target in M. tuberculosis (Puckett et al., 2017).
The cytoplasmic phases in peptidoglycan biosynthetic pathway are catalyzed by the Mur (A-F) enzymes. The imperative role of Mur enzymes in cell integrity and the minimum availability of their complements in eukaryotes highlighted them as promising anti-TB drug targets (Eniyan et al., 2016). Besides these, several other potential targets have also been proposed, such as, MbtA—involved in the iron metabolism of M. tuberculosis; cytochrome b subunit (QcrB) and type II NADH dehydrogenase—involved in energy generation; fatty acid synthases (FASs) and polyketide synthases (PKSs) involved in cell wall biosynthesis (Fernandes et al., 2015). Phenyl-diketo acid (PDKA) predicted to be the inhibitor for malate synthase (glcB) that characterize fatty acid metabolism (Krieger et al., 2012). Cyclospropane synthase (pcaA) involved in mycolic acid synthesis, presumed to be the significant target of sinefungin, thiacetazone, s-adenosyl-N-decylaminoethyl (Vaubourgeix et al., 2009). Furthermore, polyketide synthase (pks13) and acyl-AMP ligase (fadD32) are also responsible in mycolic acid synthesis, however, they are considered as the imperative targets of Cerulenin; Thiopene and 4,6-diaryl-5,7-dimethyl coumarin, respectively (Gavalda et al., 2009; Stanley et al., 2013; Wilson et al., 2013). Propanamide [5-methyl-N-(4-nitrophenyl) thiophene-2- carboxamide and 3-phenyl-N-(4-piperidin-1- ylphenyl) carbamothioyl] was approved as an efficient inhibitor against CTP synthase (pyrG) pyrimidine which is responsible to regulate metabolic processes (Mori et al., 2015).
Similarly, in another study Rhodanine analogs provided promising results by inhibiting mAGP complex synthesis directing through dTDP-keto-deoxyglucose epimerase (rmlC) (Ren et al., 2015). 4-diphosphocytidyl-2-Cmethylerythritol synthetase (ispD) involved in isopentenyl diphosphate biosynthesis assumed to be inhibited by Domiphen bromide (Gao et al., 2012). Consequently, shortening the TB therapy, simplifying DS-TB as well as DR-TB regimens and increasing the cure rates are the prime goals of newly developed or repurposed compounds (Brigden et al., 2014).
In conclusion, DR-TB (MDR, XDR, and TDR) is an intensifying health crisis around the globe, mainly distressing economically active young adults and responsible for high mortality rate regardless of HIV status (Dheda et al., 2014). The genetic modifications underlying the profound drug resistance of M. tuberculosis immensely require insight investigations to unravel the complex pathways which hamper not only the clinical application of available drugs but also the development of new anti-TB drugs. The frequency of drug resistance continues toward upset level when TB regimens are practiced without fully exploration of resistance mechanisms. Likewise, the absence of effective drugs is the prime reason for rapid emergence of DR-TB. Hopefully the new knowledge along with vigilant understanding of alterations in genomics of drug targets and drug resistance mechanisms in M. tuberculosis would be tremendously helpful in development of new and highly effective anti-TB drugs.
HH, MI, CC, CW, and TZ: Conceived and designed the initial draft to discuss the issues; HH, MM, and TZ: Wrote the manuscript; HH, YL, YT, XL, ST, and TZ: Assisted in evaluation of the article; WY, VD, HH, JL, and TZ: Critically assessed and guided up to final version. All the authors contributed to highlight related concerns and approved the final version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by the National Natural Science Foundation of China (81572037), the Chinese Academy of Sciences (154144KYSB20150045, KFZD-SW-207), the National Mega-project of China for Innovative Drugs (2018ZX09721001-003-003), the National Mega-project of China for Main Infectious Diseases (2017ZX10302301-003-002), the Key Project Grant (SKLRD2016ZJ003) and the Open Project Grant (2014SKLRD-O06) from the State Key Lab of Respiratory Disease, Guangzhou Institute of Respiratory Disease, First Affiliated Hospital of Guangzhou Medical University, the UCAS Ph.D. Fellowship Program (to HH) and CAS-TWAS President's Ph.D. Fellowship Program (to MI and CC) for International Students, and partially supported by Guangzhou Municipal Industry and Research Collaborative Innovation Program (201508020248, 201604020019) and Guangzhou Municipal Clinical Medical Center Program (155700012). TZ received Science and Technology Innovation Leader of Guangdong Province (2016TX03R095). VD was supported by the National Research Foundation in Korea (NRF), funded by the Ministry of Science, ICT and future Planning (MSIP, grant numbers NRF-2017M3A9G6068246 and 2015R1C1A1A01053355), as well as the French ministry of Foreign Affairs. We acknowledge the support of Valery N. Danilenko, Vavilov Institute of General Genetics, Russian Academy of Sciences.
TB, Tuberculosis; M. tuberculosis, Mycobacterium tuberculosis; BCG, Bacille-Calmette-Guérin; M. smegmatis, Mycobacterium smegmatis; M. bovis, Mycobacterium bovis; M. avium, Mycobacterium avium; FLD's, First line drugs; SLD's, Second line drugs; STR, Streptomycin; INH, Isoniazid; PZA, Pyrazinamide; RIF, Rifampicin; FQ, Fluoroquinolone; AMK, Amikacin; KAN, Kanamycin; CAP, Capreomycin; RFB, Rifabutin; CLO, Clofazimine; CLR, Clarithromycin; THZ, Thiacetazone; BDQ, Bedaquiline; DMD, Delamanid; EMB, Ethambutol; POA, Pyrazinoic acid; VIM, Viomycin; LZD, Linezolid; SZD, Sutezolid; DCS, D-cycloserine; PRM, Pretomanid; BTZ, Benzothiazinone; ETH, Ethionamide; PAS, Para-aminosalicylic acid; MDR-TB, Multidrug resistance tuberculosis; XDR-TB, Extensively drug resistance tuberculosis; TDR-TB, Totally drug resistance tuberculosis; HIV, Human-immunodeficiency virus; WHO, World health organization; MIC, Minimum inhibitory concentration; TEM, Transmission electron microscopy; AFM, Atomic force microscopy; MGIT96, Mycobacteria Growth Indicator Tube; LPA, Line probe assay; MODS, Microscopic observation drug susceptibility assay; AFB, Acid-fast bacilli; SNP, Single nucleotide polymorphisms; ETL, Electron-transparent layer; EOL, Outer electron-opaque layer; TNF-α, Tumour necrosis factor alpha; RRDR, RIF-resistance determining region; ACP, Enoyl-acyl carrier protein; DPPR, Decaprenylphosphoryl-5-phosphoribose; PDIM, Phthiocerol dimycocerosate; QRDR, Quinolone resistance determining region; dUMP, deoxyuridine monophosphate; dTMP, deoxythymidine monophosphate.
Ajbani, K., Rodrigues, C., Shenai, S., and Mehta, A. (2011). Mutation detection and accurate diagnosis of extensively drug-resistant tuberculosis: report from a tertiary care center in India. J. Clin. Microbiol. 49, 1588–1590. doi: 10.1128/JCM.00113-11
Alexander, D. C., Ma, J. H., Guthrie, J. L., Blair, J., Chedore, P., and Jamieson, F. B. (2012). Gene sequencing for routine verification of pyrazinamide resistance in Mycobacterium tuberculosis: a role for pncA but not rpsA. J. Clin. Microbiol. 50, 3726–3728. doi: 10.1128/JCM.00620-12
Almeida, D., Ioerger, T., Tyagi, S., Li, S. -Y., Mdluli, K., Andries, K., et al. (2016). Mutations in pepQ confer low-level resistance to Bedaquiline and Clofazimine in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 60, 4590–4599. doi: 10.1128/AAC.00753-16
Alvarez, N., Zapata, E., Mejía, G., Realpe, T., Araque, P., Peláez, C., et al. (2014). The structural modeling of the interaction between levofloxacin and the Mycobacterium tuberculosis gyrase catalytic site sheds light on the mechanisms of fluoroquinolones resistant tuberculosis in Colombian clinical isolates. Biomed. Res. Int. 2014:367268. doi: 10.1155/2014/367268
Andini, N., and Nash, K. A. (2006). Intrinsic macrolide resistance of the Mycobacterium tuberculosis complex is inducible. Antimicrob. Agents Chemother. 50, 2560–2562. doi: 10.1128/AAC.00264-06
Andries, K., Verhasselt, P., Guillemont, J., Göhlmann, H. W., Neefs, J. -M., Winkler, H., et al. (2005). A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 307, 223–227. doi: 10.1126/science.1106753
Andries, K., Villellas, C., Coeck, N., Thys, K., Gevers, T., Vranckx, L., et al. (2014). Acquired resistance of Mycobacterium tuberculosis to bedaquiline. PLoS ONE 9:e102135. doi: 10.1371/journal.pone.0102135
Arnold, C. (2007). Molecular evolution of Mycobacterium tuberculosis. Clin. Microbiol. Infect. 13, 120–128. doi: 10.1111/j.1469-0691.2006.01637.x
Aubry, A., Pan, X. -S., Fisher, L. M., Jarlier, V., and Cambau, E. (2004). Mycobacterium tuberculosis DNA gyrase: interaction with quinolones and correlation with antimycobacterial drug activity. Antimicrob. Agents Chemother. 48, 1281–1288. doi: 10.1128/AAC.48.4.1281-1288.2004
Aubry, A., Veziris, N., Cambau, E., Truffot-Pernot, C., Jarlier, V., and Fisher, L. M. (2006). Novel gyrase mutations in quinolone-resistant and-hypersusceptible clinical isolates of Mycobacterium tuberculosis: functional analysis of mutant enzymes. Antimicrob. Agents Chemother. 50, 104–112. doi: 10.1128/AAC.50.1.104-112.2006
Aye, K. S., Nakajima, C., Yamaguchi, T., Win, M. M., Shwe, M. M., Win, A. A., et al. (2016). Genotypic characterization of multi-drug-resistant Mycobacterium tuberculosis isolates in Myanmar. J. Infect. Chemother. 22, 174–179. doi: 10.1016/j.jiac.2015.12.009
Banerjee, A., Dubnau, E., Quemard, A., Balasubramanian, V., Um, K. S., Wilson, T., et al. (1994). inhA, a gene encoding a target for isoniazid and ethionamide in Mycobacterium tuberculosis. Science 263, 227–229. doi: 10.1126/science.8284673
Bantubani, N., Kabera, G., Connolly, C., Rustomjee, R., Reddy, T., Cohen, T., et al. (2014). High rates of potentially infectious tuberculosis and multidrug-resistant tuberculosis (MDR-TB) among hospital inpatients in KwaZulu Natal, South Africa indicate risk of nosocomial transmission. PLoS ONE 9:e90868. doi: 10.1371/journal.pone.0090868
Barnard, M., Warren, R., Van Pittius, N. G., Van Helden, P., Bosman, M., Streicher, E., et al. (2012). Genotype MTBDR sl line probe assay shortens time to diagnosis of extensively drug-resistant tuberculosis in a high-throughput diagnostic laboratory. Am. J. Respir. Crit. Care Med. 186, 1298–1305. doi: 10.1164/rccm.201205-0960OC
Beceiro, A., Tomás, M., and Bou, G. (2013). Antimicrobial resistance and virulence: a successful or deleterious association in the bacterial World. Clin. Microbiol. Rev. 26, 185–230. doi: 10.1128/CMR.00059-12
Beckert, P., Hillemann, D., Kohl, T. A., Kalinowski, J., Richter, E., Niemann, S., et al. (2012). rplC T460C identified as a dominant mutation in linezolid-resistant Mycobacterium tuberculosis strains. Antimicrob. Agents Chemother. 56, 2743–2745. doi: 10.1128/AAC.06227-11
Bhuju, S., Fonseca, L., de, S., Marsico, A. G., De Oliveira Vieira, G. B., Sobral, L. F., Stehr, M., et al. (2013). Mycobacterium tuberculosis isolates from Rio de Janeiro reveal unusually low correlation between pyrazinamide resistance and mutations in the pncA gene. Infect. Genet. Evol. 19, 1–6. doi: 10.1016/j.meegid.2013.06.008
Bloemberg, G. V., Keller, P. M., Stucki, D., Trauner, A., Borrell, S., Latshang, T., et al. (2015). Acquired resistance to bedaquiline and delamanid in therapy for tuberculosis. N. Engl. J. Med. 373, 1986–1988. doi: 10.1056/NEJMc1505196
Borrell, S., and Gagneux, S. (2011). Strain diversity, epistasis and the evolution of drug resistance in Mycobacterium tuberculosis. Clin. Microbiol. Infect. 17, 815–820. doi: 10.1111/j.1469-0691.2011.03556.x
Boshoff, H. I., Myers, T. G., Copp, B. R., McNeil, M. R., Wilson, M. A., and Barry, C. E. (2004). The transcriptional responses of Mycobacterium tuberculosis to inhibitors of metabolism novel insights into drug mechanisms of action. J. Biol. Chem. 279, 40174–40184. doi: 10.1074/jbc.M406796200
Böttger, E. C. (2011). The ins and outs of Mycobacterium tuberculosis drug susceptibility testing. Clin. Microbiol. Infect. 17, 1128–1134. doi: 10.1111/j.1469-0691.2011.03551.x
Brandis, G., and Hughes, D. (2013). Genetic characterization of compensatory evolution in strains carrying rpoB Ser531Leu, the rifampicin resistance mutation most frequently found in clinical isolates. J. Antimicrob. Chemother. 68, 2493–2497. doi: 10.1093/jac/dkt224
Brigden, G., Nyang'wa, B. -T., Du Cros, P., Varaine, F., Hughes, J., Rich, M., et al. (2014). Principles for designing future regimens for multidrug-resistant tuberculosis. Bull. World Health. Organ. 92, 68–74. doi: 10.2471/BLT.13.122028
Brossier, F., Sougakoff, W., Bernard, C., Petrou, M., Adeyema, K., Pham, A., et al. (2015). Molecular analysis of the embCAB locus and embR gene involved in ethambutol resistance in clinical isolates of Mycobacterium tuberculosis in France. Antimicrob. Agents Chemother. 59, 4800–4808. doi: 10.1128/AAC.00150-15
Cáceres, N. E., Harris, N. B., Wellehan, J. F., Feng, Z., Kapur, V., and Barletta, R. G. (1997). Overexpression of the D-alanine racemase gene confers resistance to D-cycloserine in Mycobacterium smegmatis. J. Bacteriol. 179, 5046–5055. doi: 10.1128/jb.179.16.5046-5055.1997
Carette, X., Blondiaux, N., Willery, E., Hoos, S., Lecat-Guillet, N., Lens, Z., et al. (2011). Structural activation of the transcriptional repressor EthR from Mycobacterium tuberculosis by single amino acid change mimicking natural and synthetic ligands. Nucleic Acids Res. 40, 3018–3030. doi: 10.1093/nar/gkr1113
Carlos, J. C., and Martin, A. (2013). Tuberculosis clinical trial update and the current anti-tuberculosis drug portfolio. Curr. Med. Chem. 20, 3785–3796. doi: 10.2174/09298673113209990166
Carreira, S., Costeira, J., Gomes, C., André, J. M., and Diogo, N. (2012). Impact of diabetes on the presenting features of tuberculosis in hospitalized patients. Rev. Port. Pneumol. 18, 239–243. doi: 10.1016/j.rppneu.2012.04.001
Cavusoglu, C., Karaca-Derici, Y., and Bilgic, A. (2004). In-vitro activity of rifabutin against rifampicin-resistant Mycobacterium tuberculosis isolates with known rpoB mutations. Clin. Microbiol. Infect. 10, 662–665. doi: 10.1111/j.1469-0691.2004.00917.x
Chakraborty, S., Gruber, T., Barry, C. E., Boshoff, H. I., and Rhee, K. Y. (2013). Para-aminosalicylic acid acts as an alternative substrate of folate metabolism in Mycobacterium tuberculosis. Science 339, 88–91. doi: 10.1126/science.1228980
Chen, J. M., Uplekar, S., Gordon, S. V., and Cole, S. T. (2012). A point mutation in cycA partially contributes to the D-cycloserine resistance trait of Mycobacterium bovis BCG vaccine strains. PLoS ONE 7:e43467. doi: 10.1371/journal.pone.0043467
Coban, A. Y., Akbal, A. U., Bicmen, C., Albay, A., Sig, A. K., Uzun, M., et al. (2016). Multicenter evaluation of crystal violet decolorization assay (CVDA) for rapid detection of isoniazid and rifampicin resistance in Mycobacterium tuberculosis. Sci. Rep. 6:39050. doi: 10.1038/srep39050
Cohen, T., Sommers, B., and Murray, M. (2003). The effect of drug resistance on the fitness of Mycobacterium tuberculosis. Lancet Infect. Dis. 3, 13–21. doi: 10.1016/S1473-3099(03)00483-3
Comas, I., Borrell, S., Roetzer, A., Rose, G., Malla, B., Kato-Maeda, M., et al. (2012). Whole-genome sequencing of rifampicin-resistant Mycobacterium tuberculosis strains identifies compensatory mutations in RNA polymerase genes. Nat. Genet. 44, 106–110. doi: 10.1038/ng.1038
Crofton, J., and Mitchison, D. A. (1948). Streptomycin resistance in pulmonary tuberculosis. Br. Med. J. 2, 1009–1015. doi: 10.1136/bmj.2.4588.1009
Cui, Z., Wang, J., Lu, J., Huang, X., and Hu, Z. (2011). Association of mutation patterns in gyrA/B genes and ofloxacin resistance levels in Mycobacterium tuberculosis isolates from East China in 2009. BMC Infect. Dis. 11:78. doi: 10.1186/1471-2334-11-78
Dalton, T., Cegielski, P., Akksilp, S., Asencios, L., Campos Caoili, J., Cho, S. N., et al. (2012). Prevalence of and risk factors for resistance to second-line drugs in people with multidrug-resistant tuberculosis in eight countries: a prospective cohort study. Lancet 380, 1406–1417. doi: 10.1016/S0140-6736(12)60734-X
Daniel, T. M. (2006). The history of tuberculosis. Resp. Med. 100, 1862–1870. doi: 10.1016/j.rmed.2006.08.006
Das, P., and Horton, R. (2010). Tuberculosis—time to accelerate progress. Lancet 375, 1755–1757. doi: 10.1016/S0140-6736(10)60600-9
Datta, G., Nieto, L. M., Davidson, R. M., Mehaffy, C., Pederson, C., Dobos, K. M., et al. (2016). Longitudinal whole genome analysis of pre and post drug treatment Mycobacterium tuberculosis isolates reveals progressive steps to drug resistance. Tuberculosis 98, 50–55. doi: 10.1016/j.tube.2016.02.004
Day, W. A., Fernández, R. E., and Maurelli, A. T. (2001). Pathoadaptive mutations that enhance virulence: genetic organization of the cadA regions of Shigella spp. Infect. Immun. 69, 7471–7480. doi: 10.1128/IAI.69.12.7471-7480.2001
Deggim-Messmer, V., Bloemberg, G. V., Ritter, C., Voit, A., Hömke, R., Keller, P. M., et al. (2016). Diagnostic molecular mycobacteriology in regions with low tuberculosis endemicity: combining real-time PCR assays for detection of multiple mycobacterial pathogens with line probe assays for identification of resistance mutations. EBioMedicine 9, 228–237. doi: 10.1016/j.ebiom.2016.06.016
Desjardins, C. A., Cohen, K. A., Munsamy, V., Abeel, T., Maharaj, K., Walker, B. J., et al. (2016). Genomic and functional analyses of Mycobacterium tuberculosis strains implicate ald in D-cycloserine resistance. Nat. Genet. 48, 544–551. doi: 10.1038/ng.3548
de Vos, M., Müller, B., Borrell, S., Black, P. A., Van Helden, P. D., Warren, R. M., et al. (2013). Putative compensatory mutations in the rpoC gene of rifampin-resistant Mycobacterium tuberculosis are associated with ongoing transmission. Antimicrob. Agents Chemother. 57, 827–832. doi: 10.1128/AAC.01541-12
Dharmadhikari, A. S., Kabadi, M., Gerety, B., Hickey, A. J., Fourie, P. B., and Nardell, E. (2013). Phase, I., single-dose, dose-escalating study of inhaled dry powder Capreomycin: a new approach to therapy of drug-resistant Tuberculosis. Antimicrob. Agents Chemother. 57, 2613–2619. doi: 10.1128/AAC.02346-12
Dheda, K., Gumbo, T., Gandhi, N. R., Murray, M., Theron, G., Udwadia, Z., et al. (2014). Global control of tuberculosis: from extensively drug-resistant to untreatable tuberculosis. Lancet Respir. Med. 2, 321–338. doi: 10.1016/S2213-2600(14)70031-1
Du, Q., Dai, G., Long, Q., Yu, X., Dong, L., Huang, H., et al. (2013). Mycobacterium tuberculosis rrs A1401G mutation correlates with high-level resistance to kanamycin, amikacin, and capreomycin in clinical isolates from mainland China. Diagn. Microbiol. Infect. Dis. 77, 138–142. doi: 10.1016/j.diagmicrobio.2013.06.031
Dye, C., Williams, B. G., Espinal, M. A., and Raviglione, M. C. (2002). Erasing the World's slow stain: strategies to beat multidrug-resistant Tuberculosis. Science 295, 2042–2046. doi: 10.1126/science.1063814
Eniyan, K., Kumar, A., Rayasam, G. V., Perdih, A., and Bajpai, U. (2016). Development of a one-pot assay for screening and identification of Mur pathway inhibitors in Mycobacterium tuberculosis. Sci. Rep. 6:35134. doi: 10.1038/srep35134
Escribano, I., Rodríguez, J., Llorca, B., García-Pachon, E., Ruiz, M., and Royo, G. (2007). Importance of the efflux pump systems in the resistance of Mycobacterium tuberculosis to fluoroquinolones and linezolid. Chemotherapy 53, 397–401. doi: 10.1159/000109769
Fenner, L., Egger, M., Bodmer, T., Altpeter, E., Zwahlen, M., Jaton, K., et al. (2012). Effect of mutation and genetic background on drug resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 56, 3047–3053. doi: 10.1128/AAC.06460-11
Fernandes, G., Chin, C., and Santos, J. (2015). Screening and identification of new potential targets against Mycobacterium tuberculosis. Biochem. Pharmacol. 4:e178. doi: 10.4172/2167-0501.1000e178
Finken, M., Kirschner, P., Meier, A., Wrede, A., and Böttger, E. C. (1993). Molecular basis of streptomycin resistance in Mycobacterium tuberculosis: alterations of the ribosomal protein S12 gene and point mutations within a functional 16S ribosomal RNA pseudoknot. Mol. Microbiol. 9, 1239–1246. doi: 10.1111/j.1365-2958.1993.tb01253.x
Fogel, N. (2015). Tuberculosis: a disease without boundaries. Tuberculosis 95, 527–531. doi: 10.1016/j.tube.2015.05.017
Forrellad, M. A., Klepp, L. I., Gioffré, A., Sabio y Garcia, J., Morbidoni, H. R., Santangelo, M. D. L. P., et al. (2013). Virulence factors of the Mycobacterium tuberculosis complex. Virulence 4, 3–66. doi: 10.4161/viru.22329
Freihofer, P., Akbergenov, R., Teo, Y., Juskeviciene, R., Andersson, D. I., and Böttger, E. C. (2016). Nonmutational compensation of the fitness cost of antibiotic resistance in mycobacteria by overexpression of tlyA rRNA methylase. RNA 22, 1836–1843. doi: 10.1261/rna.057257.116
Frieden, T. R., Sterling, T., Pablos-Mendez, A., Kilburn, J. O., Cauthen, G. M., and Dooley, S. W. (1993). The emergence of drug-resistant tuberculosis in New York City. N. Engl. J. Med. 328, 521–526. doi: 10.1056/NEJM199302253280801
Furin, J., Brigden, G., Lessem, E., Rich, M., Vaughan, L., and Lynch, S. (2016). Global progress and challenges in implementing new medications for treating multidrug-resistant Tuberculosis. Emerg. Infect. Dis. 22:e151430. doi: 10.3201/eid2203.151430
Gandhi, N. R., Moll, A., Sturm, A. W., Pawinski, R., Govender, T., Lalloo, U., et al. (2006). Extensively drug-resistant tuberculosis as a cause of death in patients co-infected with tuberculosis and HIV in a rural area of South Africa. Lancet 368, 1575–1580. doi: 10.1016/S0140-6736(06)69573-1
Gao, P., Yang, Y., Xiao, C., Liu, Y., Gan, M., Guan, Y., et al. (2012). Identification and validation of a novel lead compound targeting 4-diphosphocytidyl-2-C-methylerythritol synthetase (IspD) of mycobacteria. Eur. J. Pharmacol. 694, 45–52. doi: 10.1016/j.ejphar.2012.08.012
Gao, W., Kim, J. Y., Anderson, J. R., Akopian, T., Hong, S., Jin, Y. Y., et al. (2015). The cyclic peptide ecumicin targeting ClpC1 is active against Mycobacterium tuberculosis in vivo. Antimicrob. Agents Chemother. 59, 880–889. doi: 10.1128/AAC.04054-14
Gavalda, S., Léger, M., Van Der Rest, B., Stella, A., Bardou, F., Montrozier, H., et al. (2009). The Pks13/FadD32 crosstalk for the biosynthesis of mycolic acids in Mycobacterium tuberculosis. J. Biol. Chem. 284, 19255–19264. doi: 10.1074/jbc.M109.006940
Gavrish, E., Sit, C. S., Cao, S., Kandror, O., Spoering, A., Peoples, A., et al. (2014). Lassomycin, a ribosomally synthesized cyclic peptide, kills Mycobacterium tuberculosis by targeting the ATP-dependent protease ClpC1P1P2. Chem. Biol. 21, 509–518. doi: 10.1016/j.chembiol.2014.01.014
Geldmacher, C., Zumla, A., and Hoelscher, M. (2012). Interaction between HIV and Mycobacterium tuberculosis: HIV-1-induced CD4 T-cell depletion and the development of active tuberculosis. Curr. Opin. HIV AIDS 7, 268–274. doi: 10.1097/COH.0b013e3283524e32
Georghiou, S. B., Seifert, M., Catanzaro, D. G., Garfein, R. S., and Rodwell, T. C. (2017). Increased tuberculosis patient mortality associated with Mycobacterium tuberculosis mutations conferring resistance to second-line antituberculous drugs. J. Clin. Microbiol. 55, 1928–1937. doi: 10.1128/JCM.00152-17
Gilchrist, C. A., Turner, S. D., Riley, M. F., Petri, W. A., and Hewlett, E. L. (2015). Whole-genome sequencing in outbreak analysis. Clin. Microbiol. Rev. 28, 541–563. doi: 10.1128/CMR.00075-13
Gillespie, S. H. (2002). Evolution of drug resistance in Mycobacterium tuberculosis: clinical and molecular perspective. Antimicrob. Agents Chemother. 46, 267–274. doi: 10.1128/AAC.46.2.267-274.2002
Gler, M. T., Skripconoka, V., Sanchez-Garavito, E., Xiao, H., Cabrera-Rivero, J. L., Vargas-Vasquez, D. E., et al. (2012). Delamanid for multidrug-resistant pulmonary tuberculosis. N. Engl. J. Med. 366, 2151–2160. doi: 10.1056/NEJMoa1112433
Glickman, M. S., and Schluger, N. (2016). Adding insult to injury: exacerbating TB risk with smoking. Cell Host Microbe 19, 432–433. doi: 10.1016/j.chom.2016.04.001
Grant, S. S., Wellington, S., Kawate, T., Desjardins, C. A., Silvis, M. R., Wivagg, C., et al. (2016). Baeyer-Villiger Monooxygenases EthA and MymA are required for activation of replicating and non-replicating Mycobacterium tuberculosis inhibitors. Cell Chem. Biol. 23, 666–677. doi: 10.1016/j.chembiol.2016.05.011
Grossman, R. F., Hsueh, P.-R., Gillespie, S. H., and Blasi, F. (2014). Community-acquired pneumonia and tuberculosis: differential diagnosis and the use of fluoroquinolones. Int. J. Infect. Dis. 18, 14–21. doi: 10.1016/j.ijid.2013.09.013
Grzegorzewicz, A. E., Korduláková, J., Jones, V., Born, S. E., Belardinelli, J. M., Vaquié, A., et al. (2012). A common mechanism of inhibition of the Mycobacterium tuberculosis mycolic acid biosynthetic pathway by isoxyl and thiacetazone. J. Biol. Chem. 287, 38434–38441. doi: 10.1074/jbc.M112.400994
Haagsma, A. C., Abdillahi-Ibrahim, R., Wagner, M. J., Krab, K., Vergauwen, K., Guillemont, J., et al. (2009). Selectivity of TMC207 towards mycobacterial ATP synthase compared with that towards the eukaryotic homologue. Antimicrob. Agents Chemother. 53, 1290–1292. doi: 10.1128/AAC.01393-08
Hartkoorn, R. C., Uplekar, S., and Cole, S. T. (2014). Cross-resistance between clofazimine and bedaquiline through upregulation of MmpL5 in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 58, 2979–2981. doi: 10.1128/AAC.00037-14
Haver, H. L., Chua, A., Ghode, P., Lakshminarayana, S. B., Singhal, A., Mathema, B., et al. (2015). Mutations in genes for the F420 biosynthetic pathway and a nitroreductase enzyme are the primary resistance determinants in spontaneous in vitro-selected PA-824-resistant mutants of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 59, 5316–5323. doi: 10.1128/AAC.00308-15
Hillemann, D., Rüsch-Gerdes, S., and Richter, E. (2008). In vitro-selected linezolid-resistant Mycobacterium tuberculosis mutants. Antimicrob. Agents Chemother. 52, 800–801. doi: 10.1128/AAC.01189-07
Huitric, E., Verhasselt, P., Koul, A., Andries, K., Hoffner, S., and Andersson, D. I. (2010). Rates and mechanisms of resistance development in Mycobacterium tuberculosis to a novel diarylquinoline ATP synthase inhibitor. Antimicrob. Agents Chemother. 54, 1022–1028. doi: 10.1128/AAC.01611-09
Islam, M. M., Hameed, H. M., Mugweru, J., Chhotaray, C., Wang, C., Tan, Y., et al. (2017). Drug resistance mechanisms and novel drug targets for tuberculosis therapy. J. Genet. Genomics 44, 21–37. doi: 10.1016/j.jgg.2016.10.002
Jacobson, K. R., Theron, D., Kendall, E. A., Franke, M. F., Barnard, M., Van Helden, P. D., et al. (2013). Implementation of genotype MTBDRplus reduces time to multidrug-resistant tuberculosis therapy initiation in South Africa. Clin. Infect. Dis. 56, 503–508. doi: 10.1093/cid/cis920
Jagielski, T., Ignatowska, H., Bakuła, Z., Dziewit, Ł., Napiórkowska, A., Augustynowicz-Kopeć, E., et al. (2014). Screening for streptomycin resistance-conferring mutations in Mycobacterium tuberculosis clinical isolates from Poland. PLoS ONE 9:e100078. doi: 10.1371/journal.pone.0100078
Jenkins, C., Bacon, J., Allnutt, J., Hatch, K. A., Bose, A., O'sullivan, D. M., et al. (2009). Enhanced heterogeneity of rpoB in Mycobacterium tuberculosis found at low pH. J. Antimicrob. Chemother. 63, 1118–1120. doi: 10.1093/jac/dkp125
Jeon, C. Y., and Murray, M. B. (2008). Diabetes mellitus increases the risk of active Tuberculosis: a systematic review of 13 observational studies. PLoS Med. 5:e152. doi: 10.1371/journal.pmed.0050152
Jia, L., Coward, L., Gorman, G. S., Noker, P. E., and Tomaszewski, J. E. (2005). Pharmacoproteomic effects of isoniazid, ethambutol, and N-geranyl-N′-(2-adamantyl) ethane-1, 2-diamine (SQ109) on Mycobacterium tuberculosis H37Rv. J. Pharm. Exp. Ther. 315, 905–911. doi: 10.1124/jpet.105.087817
Jnawali, H. N., Hwang, S. C., Park, Y. K., Kim, H., Lee, Y. S., Chung, G. T., et al. (2013). Characterization of mutations in multi-and extensive drug resistance among strains of Mycobacterium tuberculosis clinical isolates in Republic of Korea. Diagn. Microbiol. Infect. Dis. 76, 187–196. doi: 10.1016/j.diagmicrobio.2013.02.035
Juréen, P., Werngren, J., Toro, J.-C., and Hoffner, S. (2008). Pyrazinamide resistance and pncA gene mutations in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 52, 1852–1854. doi: 10.1128/AAC.00110-08
Keshavjee, S., and Farmer, P. E. (2012). Tuberculosis, drug resistance, and the history of modern medicine. N. Engl. J. Med. 367, 931–936. doi: 10.1056/NEJMra1205429
Kisacik, B., Pamuk, O. N., Onat, A. M., Erer, S. B., Hatemi, G., Ozguler, Y., et al. (2016). Characteristics predicting tuberculosis risk under tumor necrosis factor-α inhibitors: report from a large multicenter cohort with high background prevalence. J. Rheumatol. 43, 524–529. doi: 10.3899/jrheum.150177
Klopper, M., Warren, R. M., Hayes, C., Gey Van Pittius, N. C., Streicher, E. M., Müller, B., et al. (2013). Emergence and spread of extensively and totally drug-resistant tuberculosis, South Africa. Emerg. Infect. Dis. 19, 449–455. doi: 10.3201/eid1903.120246
Kolly, G. S., Boldrin, F., Sala, C., Dhar, N., Hartkoorn, R. C., Ventura, M., et al. (2014). Assessing the essentiality of the decaprenyl-phospho-d-arabinofuranose pathway in Mycobacterium tuberculosis using conditional mutants. Mol. Microbiol. 92, 194–211. doi: 10.1111/mmi.12546
Koul, A., Dendouga, N., Vergauwen, K., Molenberghs, B., Vranckx, L., Willebrords, R., et al. (2007). Diarylquinolines target subunit c of mycobacterial ATP synthase. Nat. Chem. Biol. 3, 323–324. doi: 10.1038/nchembio884
Krieger, I. V., Freundlich, J. S., Gawandi, V. B., Roberts, J. P., Gawandi, V. B., Sun, Q., et al. (2012). Structure-guided discovery of phenyl-diketo acids as potent inhibitors of M. tuberculosis malate synthase. Chem. Biol. 19, 1556–1567. doi: 10.1016/j.chembiol.2012.09.018
Lange, C., Abubakar, I., Alffenaar, J. W., Bothamley, G., Caminero, J. A., Carvalho, A. C., et al. (2014). Management of patients with multidrug-resistant/extensively drug-resistant tuberculosis in Europe: a TBNET consensus statement. Eur. Respir. J. 44, 23–63. doi: 10.1183/09031936.00188313
Lata, M., Sharma, D., Deo, N., Tiwari, P. K., Bisht, D., and Venkatesan, K. (2015a). Proteomic analysis of ofloxacin-mono resistant Mycobacterium tuberculosis isolates. J. Proteomics 127, 114–121. doi: 10.1016/j.jprot.2015.07.031
Lata, M., Sharma, D., Kumar, B., Deo, N., Tiwari, P. K., Bisht, D., et al. (2015b). Proteome analysis of ofloxacin and moxifloxacin induced Mycobacterium tuberculosis isolates by proteomic approach. Protein. Pept. Lett. 22, 362–371. doi: 10.2174/0929866522666150209113708
Lawn, S. D., Mwaba, P., Bates, M., Piatek, A., Alexander, H., Marais, B. J., et al. (2013). Advances in tuberculosis iagnostics: the Xpert MTB/RIF assay and future prospects for a point-of-care test. Lancet Infect. Dis. 13, 349–361. doi: 10.1016/S1473-3099(13)70008-2
Li, J., Gao, X., Luo, T., Wu, J., Sun, G., Liu, Q., et al. (2014). Association of gyrA/B mutations and resistance levels to fluoroquinolones in clinical isolates of Mycobacterium tuberculosis. Emerg. Microbes Infect. 3:e19. doi: 10.1038/emi.2014.21
Lillebaek, T., Norman, A., Rasmussen, E. M., Marvig, R. L., Folkvardsen, Å. B., Andersen, A. B., et al. (2016). Substantial molecular evolution and mutation rates in prolonged latent Mycobacterium tuberculosis infection in humans. Int. J. Med. Microbiol. 306, 580–585. doi: 10.1016/j.ijmm.2016.05.017
Long, Q., Li, W., Du, Q., Fu, Y., Liang, Q., Huang, H., et al. (2012). gyrA/B fluoroquinolone resistance allele profiles amongst Mycobacterium tuberculosis isolates from mainland China. Int. J. Antimicrob. Agents 39, 486–489. doi: 10.1016/j.ijantimicag.2012.02.015
Machado, D., Couto, I., Perdigão, J., Rodrigues, L., Portugal, I., Baptista, P., et al. (2012). Contribution of efflux to the emergence of isoniazid and multidrug resistance in Mycobacterium tuberculosis. PLoS ONE 7:e34538. doi: 10.1371/journal.pone.0034538
Machado, D., Perdigão, J., Ramos, J., Couto, I., Portugal, I., Ritter, C., et al. (2013). High-level resistance to isoniazid and ethionamide in multidrug-resistant Mycobacterium tuberculosis of the Lisboa family is associated with inhA double mutations. J. Antimicrob. Chemother. 68, 1728–1732. doi: 10.1093/jac/dkt090
Maeurer, M., Schito, M., and Zumla, A. (2014). Totally-drug-resistant tuberculosis: hype versus hope. Lancet Respir. Med. 2, 256–257. doi: 10.1016/S2213-2600(14)70020-7
Makafe, G. G., Cao, Y., Tan, Y., Julius, M., Liu, Z., Wang, C., et al. (2016). Oxazolidinone resistance in Mycobacterium tuberculosis: what is the role of Cys154Arg mutation in the ribosomal protein L3? Antimicrob. Agents Chemother. 60, 3202–3206. doi: 10.1128/AAC.00152-16
Makarov, V., Lechartier, B., Zhang, M., Neres, J., Van Der Sar, A. M., Raadsen, S. A., et al. (2014). Towards a new combination therapy for tuberculosis with next generation benzothiazinones. EMBO Mol. Med. 6, 372–383. doi: 10.1002/emmm.201303575
Manina, G., Bellinzoni, M., Pasca, M. R., Neres, J., Milano, A., De Jesus Lopes Ribeiro, A. L., et al. (2010). Biological and structural characterization of the Mycobacterium smegmatis nitroreductase NfnB, and its role in enzothiazinone resistance. Mol. Microbiol. 77, 1172–1185. doi: 10.1111/j.1365-2958.2010.07277.x
Manjunatha, U. H., Boshoff, H., Dowd, C. S., Zhang, L., Albert, T. J., Norton, J. E., et al. (2006). Identification of a nitroimidazo-oxazine-specific protein involved in PA-824 resistance in Mycobacterium tuberculosis. Proc. Nat. Acad. Sci. U.S.A. 103, 431–436. doi: 10.1073/pnas.0508392103
Manson, A. L., Cohen, K. A., Abeel, T., Desjardins, C. A., Armstrong, D. T., Barry Iii, C. E., et al. (2017). Genomic analysis of globally diverse Mycobacterium tuberculosis strains provides insights into emergence and spread of multidrug resistance. Nat. Genet. 49, 395–402. doi: 10.1038/ng.3767
Maslov, D. A., Zaichikova, M. V., Chernousova, L. N., Shur, K. V., Bekker, O. B., Smirnova, T. G., et al. (2015). Resistance to pyrazinamide in Russian Mycobacterium tuberculosis isolates: pncA sequencing versus Bactec MGIT 960. Tuberculosis 95, 608–612. doi: 10.1016/j.tube.2015.05.013
Mathys, V., Wintjens, R., Lefevre, P., Bertout, J., Singhal, A., Kiass, M., et al. (2009). Molecular genetics of para-aminosalicylic acid resistance in clinical isolates and spontaneous mutants of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 53, 2100–2109. doi: 10.1128/AAC.01197-08
Matsumoto, M., Hashizume, H., Tomishige, T., Kawasaki, M., Tsubouchi, H., Sasaki, H., et al. (2006). OPC-67683, a nitro-dihydro-imidazooxazole derivative with promising action against tuberculosis in vitro and in mice. PLoS. Med. 3:e466. doi: 10.1371/journal.pmed.0030466
Mboowa, G., Namaganda, C., and Ssengooba, W. (2014). Rifampicin resistance mutations in the 81 bp RRDR of rpoB gene in Mycobacterium tuberculosis clinical isolates using Xpert® MTB/RIF in Kampala, Uganda: a retrospective study. BMC Infect. Dis. 14:481. doi: 10.1186/1471-2334-14-481
McGrath, M., Gey van Pittius, N. C., Van Helden, P. D., Warren, R. M., and Warner, D. F. (2014). Mutation rate and the emergence of drug resistance in Mycobacterium tuberculosis. J. Antimicrob. Chemother. 69, 292–302. doi: 10.1093/jac/dkt364
Meumann, E. M., Globan, M., Fyfe, J. A., Leslie, D., Porter, J. L., Seemann, T., et al. (2015). Genome sequence comparisons of serial multi-drug-resistant Mycobacterium tuberculosis isolates over 21 years of infection in a single patient. Microb. Genom. 1:e000037. doi: 10.1099/mgen.0.000037
Migliori, G. B., Ortmann, J., Girardi, E., Besozzi, G., Lange, C., Cirillo, D. M., et al. (2007). Extensively drug-resistant tuberculosis, Italy and Germany. Emerg. Infect. Dis. 13, 780–782. doi: 10.3201/eid1305.070200
Mikusová, K., Slayden, R. A., Besra, G. S., and Brennan, P. J. (1995). Biogenesis of the mycobacterial cell wall and the site of action of ethambutol. Antimicrob. Agents Chemother. 39, 2484–2489. doi: 10.1128/AAC.39.11.2484
Minion, J., and Pai, M. (2010). Bacteriophage assays for rifampicin resistance detection in Mycobacterium tuberculosis: updated meta-analysis. Int. J. Tuberc. Lung. Dis. 14, 941–951.
Mori, G., Chiarelli, L. R., Esposito, M., Makarov, V., Bellinzoni, M., Hartkoorn, R. C., et al. (2015). Thiophenecarboxamide derivatives activated by EthA kill Mycobacterium tuberculosis by inhibiting the CTP synthetase PyrG. Chem. Biol. 22, 917–927. doi: 10.1016/j.chembiol.2015.05.016
Mori, G., Chiarelli, L. R., Riccardi, G., and Pasca, M. R. (2017). New prodrugs against tuberculosis. Drug Discov. Today 22, 519–525. doi: 10.1016/j.drudis.2016.09.006
Moure, R., Español, M., Tudó, G., Vicente, E., Coll, P., Gonzalez-Martin, J., et al. (2014). Characterization of the embB gene in Mycobacterium tuberculosis isolates from Barcelona and rapid detection of main mutations related to ethambutol resistance using a low-density DNA array. J. Antimicrob. Chemother. 69, 947–954. doi: 10.1093/jac/dkt448
Müller, B., Streicher, E., Hoek, K., Tait, M., Trollip, A., Bosman, M., et al. (2011). inhA promoter mutations: a gateway to extensively drug-resistant tuberculosis in South Africa? Int. J. Tuberc. Lung Dis. 15, 344–351.
Negin, J., Abimbola, S., and Marais, B. J. (2015). Tuberculosis among older adults – time to take notice. Int. J. Infect. Dis. 32, 135–137. doi: 10.1016/j.ijid.2014.11.018
Njire, M., Tan, Y., Mugweru, J., Wang, C., Guo, J., Yew, W., et al. (2016). Pyrazinamide resistance in Mycobacterium tuberculosis: review and update. Adv. Med. Sci. 61, 63–71. doi: 10.1016/j.advms.2015.09.007
Njire, M., Wang, N., Wang, B., Tan, Y., Cai, X., Liu, Y., et al. (2017). Pyrazinoic acid inhibits a bifunctional enzyme in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 61:e00070-17. doi: 10.1128/AAC.00070-17
Nosova, E. Y., Bukatina, A. A., Isaeva, Y. D., Makarova, M. V., Galkina, K. Y., and Moroz, A. M. (2013). Analysis of mutations in the gyrA and gyrB genes and their association with the resistance of Mycobacterium tuberculosis to levofloxacin, moxifloxacin and gatifloxacin. J. Med. Microbiol. 62, 108–113. doi: 10.1099/jmm.0.046821-0
Ocheretina, O., Escuyer, V. E., Mabou, M.-M., Royal-Mardi, G., Collins, S., Vilbrun, S. C., et al. (2014). Correlation between genotypic and phenotypic testing for resistance to rifampin in Mycobacterium tuberculosis clinical isolates in Haiti: investigation of cases with discrepant susceptibility results. PLoS ONE 9:e90569. doi: 10.1371/journal.pone.0090569
Palomino, J. C., and Martin, A. (2014). Drug resistance mechanisms in Mycobacterium tuberculosis. Antibiotics 3, 317–340. doi: 10.3390/antibiotics3030317
Pandey, B., Grover, S., Tyagi, C., Goyal, S., Jamal, S., Singh, A., et al. (2016). Molecular principles behind pyrazinamide resistance due to mutations in panD gene in Mycobacterium tuberculosis. Gene 581, 31–42. doi: 10.1016/j.gene.2016.01.024
Pantel, A., Petrella, S., Veziris, N., Brossier, F., Bastian, S., Jarlier, V., et al. (2012). Extending the definition of the GyrB quinolone resistance-determining region in Mycobacterium tuberculosis DNA gyrase for assessing fluoroquinolone resistance in M. tuberculosis. Antimicrob. Agents Chemother. 56, 1990–1996. doi: 10.1128/AAC.06272-11
Park, Y., Pacitto, A., Bayliss, T., Cleghorn, L. A., Wang, Z., Hartman, T., et al. (2017). Essential but not vulnerable: indazole Sulfonamides targeting inosine monophosphate dehydrogenase as potential leads against Mycobacterium tuberculosis. ACS. Infect. Dis. 3, 18–33. doi: 10.1021/acsinfecdis.6b00103
Perdigão, J., Macedo, R., Machado, D., Silva, C., Jordão, L., Couto, I., et al. (2014). GidB mutation as a phylogenetic marker for Q1 cluster Mycobacterium tuberculosis isolates and intermediate-level streptomycin resistance determinant in Lisbon, Portugal. Clin. Microbiol. Infect. 20, O278–O284. doi: 10.1111/1469-0691.12392
Perdigão, J., Macedo, R., Ribeiro, A., Brum, L., and Portugal, I. (2009). Genetic characterisation of the ethambutol resistance-determining region in Mycobacterium tuberculosis: prevalence and significance of embB306 mutations. Int. J. Antimicrob. Agents 33, 334–338. doi: 10.1016/j.ijantimicag.2008.09.021
Peter, S., Cernomaz, A.-T., Rusu-Cordunean, F., Macovei, I.-I., Pavel, I., Zonda, R., et al. (2016). Rapid detection of Mycobacterium tuberculosis from MODS culture by PCR. Eur. Respir. Soc. 48:PA1908. doi: 10.1183/13993003.congress-2016.PA1908
Piccaro, G., Pietraforte, D., Giannoni, F., Mustazzolu, A., and Fattorini, L. (2014). Rifampin induces hydroxyl radical formation in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 58, 7527–7533. doi: 10.1128/AAC.03169-14
Puckett, S., Trujillo, C., Wang, Z., Eoh, H., Ioerger, T. R., Krieger, I., et al. (2017). Glyoxylate detoxification is an essential function of malate synthase required for carbon assimilation in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 114, E2225–E2232. doi: 10.1073/pnas.1617655114
Ramaswamy, S., and Musser, J. M. (1998). Molecular genetic basis of antimicrobial agent resistance in Mycobacterium tuberculosis. Tuber. Lung. Dis. 79, 3–29. doi: 10.1054/tuld.1998.0002
Ramaswamy, S. V., Reich, R., Dou, S.-J., Jasperse, L., Pan, X., Wanger, A., et al. (2003). Single nucleotide polymorphisms in genes associated with isoniazid resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 47, 1241–1250. doi: 10.1128/AAC.47.4.1241-1250.2003
Reeves, A. Z., Campbell, P. J., Sultana, R., Malik, S., Murray, M., Plikaytis, B. B., et al. (2013). Aminoglycoside cross-resistance in Mycobacterium tuberculosis due to mutations in the 5′ untranslated region of whiB7. Antimicrob. Agents Chemother. 57, 1857–1865. doi: 10.1128/AAC.02191-12
Ren, J.-X., Qian, H.-L., Huang, Y.-X., Zhu, N.-Y., Si, S.-Y., and Xie, Y. (2015). Virtual screening for the identification of novel inhibitors of Mycobacterium tuberculosis cell wall synthesis: inhibitors targeting RmlB and RmlC. Comput. Biol. Med. 58, 110–117. doi: 10.1016/j.compbiomed.2014.12.020
Rengarajan, J., Sassetti, C. M., Naroditskaya, V., Sloutsky, A., Bloom, B. R., and Rubin, E. J. (2004). The folate pathway is a target for resistance to the drug para-aminosalicylic acid (PAS) in mycobacteria. Mol. Microbiol. 53, 275–282. doi: 10.1111/j.1365-2958.2004.04120.x
Roca, I., Akova, M., Baquero, F., Carlet, J., Cavaleri, M., Coenen, S., et al. (2015). The global threat of antimicrobial resistance: science for intervention. New Microbes. New Infect. 6, 22–29. doi: 10.1016/j.nmni.2015.02.007
Rozwarski, D. A., Grant, G. A., Barton, D. H., Jacobs, W. R., and Sacchettini, J. C. (1998). Modification of the NADH of the isoniazid target (InhA) from Mycobacterium tuberculosis. Science 279, 98–102. doi: 10.1126/science.279.5347.98
Safi, H., Lingaraju, S., Amin, A., Kim, S., Jones, M., Holmes, M., et al. (2013). Evolution of high-level ethambutol-resistant tuberculosis through interacting mutations in decaprenylphosphoryl-[beta]-D-arabinose biosynthetic and utilization pathway genes. Nat. Genet. 45, 1190–1197. doi: 10.1038/ng.2743
Schmitt, E. K., Riwanto, M., Sambandamurthy, V., Roggo, S., Miault, C., Zwingelstein, C., et al. (2011). The natural product cyclomarin kills Mycobacterium tuberculosis by targeting the ClpC1 subunit of the caseinolytic protease. Angew. Chem. Int. Ed. Engl. 50, 5889–5891. doi: 10.1002/anie.201101740
Schroeder, M., Brooks, B. D., and Brooks, A. E. (2017). The complex relationship between virulence and antibiotic resistance. Genes 8:39. doi: 10.3390/genes8010039
Scorpio, A., and Zhang, Y. (1996). Mutations in pncA, a gene encoding pyrazinamidase/nicotinamidase, cause resistance to the antituberculous drug pyrazinamide in tubercle bacillus. Nat. Med. 2, 662–667. doi: 10.1038/nm0696-662
Seifert, M., Catanzaro, D., Catanzaro, A., and Rodwell, T. C. (2015). Genetic mutations associated with isoniazid resistance in Mycobacterium tuberculosis: a systematic review. PLoS ONE 10:e0119628. doi: 10.1371/journal.pone.0119628
Sharma, D., Cukras, A. R., Rogers, E. J., Southworth, D. R., and Green, R. (2007). Mutational analysis of S12 protein and implications for the accuracy of decoding by the ribosome. J. Mol. Biol. 374, 1065–1076. doi: 10.1016/j.jmb.2007.10.003
Sharma, D., Lata, M., Faheem, M., Khan, A. U., Joshi, B., Venkatesan, K., et al. (2016). M. tuberculosis ferritin (Rv3841): potential involvement in amikacin (AK) andamp; kanamycin (KM) resistance. Biochem. Biophys. Res. Commun. 478, 908–912. doi: 10.1016/j.bbrc.2016.08.049
Sharma, D., Lata, M., Faheem, M., Ullah Khan, A., Joshi, B., Venkatesan, K., et al. (2015). Cloning, expression and correlation of Rv0148 to amikacin and kanamycin resistance. Curr. Proteomics 12, 96–100. doi: 10.2174/157016461202150903113053
Shen, X., Shen, G.-M., Wu, J., Gui, X.-H., Li, X., Mei, J., et al. (2007). Association between embB codon 306 mutations and drug resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 51, 2618–2620. doi: 10.1128/AAC.01516-06
Shi, D., Li, L., Zhao, Y., Jia, Q., Li, H., Coulter, C., et al. (2011a). Characteristics of embB mutations in multidrug-resistant Mycobacterium tuberculosis isolates in Henan, China. J. Antimicrob. Chemother. 66, 2240–2247. doi: 10.1093/jac/dkr284
Shi, W., Chen, J., Feng, J., Cui, P., Zhang, S., Weng, X., et al. (2014). Aspartate decarboxylase (PanD) as a new target of pyrazinamide in Mycobacterium tuberculosis. Emerg. Microbes Infect. 3:e58. doi: 10.1038/emi.2014.61
Shi, W., Zhang, X., Jiang, X., Yuan, H., Lee, J. S., Barry, C. E., et al. (2011b). Pyrazinamide inhibits trans-translation in Mycobacterium tuberculosis. Science 333, 1630–1632. doi: 10.1126/science.1208813
Shimokawa, Y., Sasahara, K., Yoda, N., Mizuno, K., and Umehara, K. (2014). Delamanid does not inhibit or induce cytochrome p450 enzymes in vitro. Biol. Pharm. Bull. 37, 1727–1735. doi: 10.1248/bpb.b14-00311
Singh, V., Donini, S., Pacitto, A., Sala, C., Hartkoorn, R. C., Dhar, N., et al. (2017). The inosine monophosphate dehydrogenase, GuaB2, is a vulnerable new bactericidal drug target for Tuberculosis. ACS Infect. Dis. 3, 5–17. doi: 10.1021/acsinfecdis.6b00102
Skrahina, A., Hurevich, H., Zalutskaya, A., Sahalchyk, E., Astrauko, A., van Gemert, W., et al. (2012). Alarming levels of drug-resistant tuberculosis in Belarus: results of a survey in Minsk. Eur. Respir. J. 39, 1425–1431. doi: 10.1183/09031936.00145411
Smith, T., Wolff, K. A., and Nguyen, L. (2013). Molecular biology of drug resistance in Mycobacterium tuberculosis. Curr. Top. Microbiol. Immunol. 374, 53–80. doi: 10.1007/82_2012_279
Smittipat, N., Juthayothin, T., Billamas, P., Jaitrong, S., Rukseree, K., Dokladda, K., et al. (2016). Mutations in rrs, rpsL and gidB in streptomycin-resistant Mycobacterium tuberculosis isolates from Thailand. J. Global. Antimicrob. Resist. 4, 5–10. doi: 10.1016/j.jgar.2015.11.009
Sowajassatakul, A., Prammananan, T., Chaiprasert, A., and Phunpruch, S. (2014). Molecular characterization of amikacin, kanamycin and capreomycin resistance in M/XDR-TB strains isolated in Thailand. BMC Microbiol. 14:165. doi: 10.1186/1471-2180-14-165
Srivastava, S., Pasipanodya, J. G., Meek, C., Leff, R., and Gumbo, T. (2011). Multidrug-resistant Tuberculosis not due to noncompliance but to between-patient pharmacokinetic variability. J. Infect. Dis. 204, 1951–1959. doi: 10.1093/infdis/jir658
Stanley, R. E., Blaha, G., Grodzicki, R. L., Strickler, M. D., and Steitz, T. A. (2010). The structures of the anti-tuberculosis antibiotics viomycin and capreomycin bound to the 70S ribosome. Nat. Struct. Mol. Biol. 17, 289–293. doi: 10.1038/nsmb.1755
Stanley, S. A., Kawate, T., Iwase, N., Shimizu, M., Clatworthy, A. E., Kazyanskaya, E., et al. (2013). Diarylcoumarins inhibit mycolic acid biosynthesis and kill Mycobacterium tuberculosis by targeting FadD32. Proc. Natl. Acad. Sci. U.S.A. 110, 11565–11570. doi: 10.1073/pnas.1302114110
Stoffels, K., Allix-Béguec, C., Groenen, G., Wanlin, M., Berkvens, D., Mathys, V., et al. (2013). From multidrug- to extensively drug-resistant Tuberculosis: upward trends as seen from a 15-Year Nationwide study. PLoS ONE 8:e63128. doi: 10.1371/journal.pone.0063128
Stover, C. K., Warrener, P., Vandevanter, D. R., Sherman, D. R., Arain, T. M., Langhorne, M. H., et al. (2000). A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature 405, 962–966. doi: 10.1038/35016103
Tahlan, K., Wilson, R., Kastrinsky, D. B., Arora, K., Nair, V., Fischer, E., et al. (2012). SQ109 targets MmpL3, a membrane transporter of trehalose monomycolate involved in mycolic acid donation to the cell wall core of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 56, 1797–1809. doi: 10.1128/AAC.05708-11
Tan, Y., Hu, Z., Zhang, T., Cai, X., Kuang, H., Liu, Y., et al. (2014). Role of pncA and rpsA gene sequencing in detection of pyrazinamide resistance in Mycobacterium tuberculosis isolates from southern China. J. Clin. Microbiol. 52, 291–297. doi: 10.1128/JCM.01903-13
Tan, Y., Hu, Z., Zhao, Y., Cai, X., Luo, C., Zou, C., et al. (2012). The beginning of the rpoB gene in addition to the rifampin resistance determination region might be needed for identifying rifampin/rifabutin cross-resistance in multidrug-resistant Mycobacterium tuberculosis isolates from Southern China. J. Clin. Microbiol. 50, 81–85. doi: 10.1128/JCM.05092-11
Telenti, A., Imboden, P., Marchesi, F., Matter, L., Schopfer, K., Bodmer, T., et al. (1993). Detection of rifampicin-resistance mutations in Mycobacterium tuberculosis. Lancet 341, 647–651. doi: 10.1016/0140-6736(93)90417-F
Thee, S., Garcia-Prats, A., Donald, P., Hesseling, A., and Schaaf, H. (2016). A review of the use of ethionamide and prothionamide in childhood tuberculosis. Tuberculosis 97, 126–136. doi: 10.1016/j.tube.2015.09.007
Thirumurugan, R., Kathirvel, M., Vallayyachari, K., Surendar, K., Samrot, A. V., and Muthaiah, M. (2015). Molecular analysis of rpoB gene mutations in rifampicin resistant Mycobacterium tuberculosis isolates by multiple allele specific polymerase chain reaction in Puducherry, South India. J. Infect. Public. Health. 8, 619–625. doi: 10.1016/j.jiph.2015.05.003
Toms, C., Stapledon, R., Waring, J., Douglas, P., National Tuberculosis Advisory Committee FTCDNA The Australian Mycobacterium Reference Laboratory Network (2015). Tuberculosis notifications in Australia, 2012 and 2013. Commun. Dis. Intell. Q. Rep. 39, E217–E235.
Trefzer, C., Rengifo-Gonzalez, M., Hinner, M. J., Schneider, P., Makarov, V., Cole, S. T., et al. (2010). Benzothiazinones: prodrugs that covalently modify the decaprenylphosphoryl-β-D-ribose 2′-epimerase DprE1 of Mycobacterium tuberculosis. J. Am. Chem. Soc. 132, 13663–13665. doi: 10.1021/ja106357w
Tye, G. J., Lew, M. H., Choong, Y. S., Lim, T. S., Sarmiento, M. E., Acosta, A., et al. (2015). Vaccines for TB: lessons from the past translating into future potentials. J. Immunol. Res. 2015:916780. doi: 10.1155/2015/916780
Udwadia, Z. F., Amale, R. A., Ajbani, K. K., and Rodrigues, C. (2012). Totally drug-resistant Tuberculosis in India. Clin. Infect. Dis. 54, 579–581. doi: 10.1093/cid/cir889
Vadwai, V., Daver, G., Udwadia, Z., Sadani, M., Shetty, A., and Rodrigues, C. (2011). Clonal population of Mycobacterium tuberculosis strains reside within multiple lung cavities. PLoS ONE 6:e24770. doi: 10.1371/journal.pone.0024770
Vaubourgeix, J., Bardou, F., Boissier, F., Julien, S., Constant, P., Ploux, O., et al. (2009). S-Adenosyl-N-decyl-aminoethyl, a potent bisubstrate inhibitor of Mycobacterium tuberculosis mycolic acid methyltransferases. J. Biol. Chem. 284, 19321–19330. doi: 10.1074/jbc.M809599200
Velayati, A. A., Masjedi, M. R., Farnia, P., Tabarsi, P., Ghanavi, J., Ziazarifi, A. H., et al. (2009). Emergence of new forms of totally drug-resistant tuberculosis bacilli: super extensively drug-resistant tuberculosis or totally drug-resistant strains in iran. Chest 136, 420–425. doi: 10.1378/chest.08-2427
Vento, S., and Lanzafame, M. (2011). Tuberculosis and cancer: a complex and dangerous liaison. Lancet Oncol. 12, 520–522. doi: 10.1016/S1470-2045(11)70105-X
Vilchèze, C., Av-Gay, Y., Attarian, R., Liu, Z., Hazbón, M. H., Colangeli, R., et al. (2008). Mycothiol biosynthesis is essential for ethionamide susceptibility in Mycobacterium tuberculosis. Mol. Microbiol. 69, 1316–1329. doi: 10.1111/j.1365-2958.2008.06365.x
Vilchèze, C., Jacobs, J., and William, R. (2007). The mechanism of isoniazid killing: clarity through the scope of genetics. Annu. Rev. Microbiol. 61, 35–50. doi: 10.1146/annurev.micro.61.111606.122346
Vilchèze, C., Wang, F., Arai, M., Hazbón, M. H., Colangeli, R., Kremer, L., et al. (2006). Transfer of a point mutation in Mycobacterium tuberculosis inhA resolves the target of isoniazid. Nat. Med. 12, 1027–1029. doi: 10.1038/nm1466
Vincent, V., Rigouts, L., Nduwamahoro, E., Holmes, B., Cunningham, J., Guillerm, M., et al. (2012). The TDR Tuberculosis Strain Bank: a resource for basic science, tool development and diagnostic services. Int. J. Tuberc. Lung. Dis. 16, 24–31. doi: 10.5588/ijtld.11.0223
Von Groll, A., Martin, A., Jureen, P., Hoffner, S., Vandamme, P., Portaels, F., et al. (2009). Fluoroquinolone resistance in Mycobacterium tuberculosis and mutations in gyrA and gyrB. Antimicrob. Agents Chemother. 53, 4498–4500. doi: 10.1128/AAC.00287-09
Wang, F., Jain, P., Gulten, G., Liu, Z., Feng, Y., Ganesula, K., et al. (2010). Mycobacterium tuberculosis dihydrofolate reductase is not a target relevant to the antitubercular activity of isoniazid. Antimicrob. Agents Chemother. 54, 3776–3782. doi: 10.1128/AAC.00453-10
Watanabe, M., Aoyagi, Y., Mitome, H., Fujita, T., Naoki, H., Ridell, M., et al. (2002). Location of functional groups in mycobacterial meromycolate chains; the recognition of new structural principles in mycolic acids. Microbiology 148, 1881–1902. doi: 10.1099/00221287-148-6-1881
Williams, K., Stover, C., Zhu, T., Tasneen, R., Tyagi, S., Grosset, J., et al. (2009). Promising antituberculosis activity of the oxazolidinone PNU-100480 relative to that of linezolid in a murine model. Antimicrob. Agents Chemother. 53, 1314–1319. doi: 10.1128/AAC.01182-08
Wilson, R., Kumar, P., Parashar, V., Vilchèze, C., Veyron-Churlet, R., Freundlich, J. S., et al. (2013). Antituberculosis thiophenes define a requirement for Pks13 in mycolic acid biosynthesis. Nat. Chem. Biol. 9, 499–506. doi: 10.1038/nchembio.1277
Workneh, M. H., Bjune, G. A., and Yimer, S. A. (2016). Diabetes mellitus is associated with increased mortality during tuberculosis treatment: a prospective cohort study among tuberculosis patients in South-Eastern Amahra Region, Ethiopia. Infect. Dis. Poverty 5:22. doi: 10.1186/s40249-016-0115-z
Wu, F.-L., Liu, Y., Jiang, H.-W., Luan, Y.-Z., Zhang, H.-N., He, X., et al. (2017). The Ser/Thr protein kinase protein-protein interaction map of M. tuberculosis. Mol. Cell. Proteomics 16, 1491–1506. doi: 10.1074/mcp.M116.065771
Wu, V.-C., Wang, C.-Y., Shiao, C.-C., Chang, C.-H., Huang, H.-Y., Huang, T.-M., et al. (2013). Increased risk of active tuberculosis following acute kidney injury: a nationwide, population-based study. PLoS ONE 8:e69556. doi: 10.1371/journal.pone.0069556
Xia, Q., Zhao, L.-L., Li, F., Fan, Y.-M., Chen, Y.-Y., Wu, B.-B., et al. (2015). Phenotypic and genotypic characterization of pyrazinamide resistance among multidrug-resistant Mycobacterium tuberculosis isolates in Zhejiang, China. Antimicrob. Agents Chemother. 59, 1690–1695. doi: 10.1128/AAC.04541-14
Xu, P., Wu, J., Yang, C., Luo, T., Shen, X., Zhang, Y., et al. (2016). Prevalence and transmission of pyrazinamide resistant Mycobacterium tuberculosis in China. Tuberculosis. 98, 56–61. doi: 10.1016/j.tube.2016.02.008
Xue, W., Peng, J., Yu, X., Zhang, S., Zhou, B., Jiang, D., et al. (2016). A color-reaction-based biochip detection assay for RIF and INH resistance of clinical mycobacterial specimens. J. Microbiol. Biotechnol. 26, 180–189. doi: 10.4014/jmb.1501.01015
Xu, Y., Jia, H., Huang, H., Sun, Z., and Zhang, Z. (2015). Mutations found in embCAB, embR, and ubiA genes of ethambutol-sensitive and -resistant Mycobacterium tuberculosis clinical isolates from China. BioMed. Res. Int. 2015:951706. doi: 10.1155/2015/951706
Yano, T., Kassovska-Bratinova, S., Teh, J. S., Winkler, J., Sullivan, K., Isaacs, A., et al. (2011). Reduction of clofazimine by mycobacterial Type 2 NADH: quinone oxidoreductase a pathway for the generation of bactericidal levels of reactive oxygen species. J. Biol. Chem. 286, 10276–10287. doi: 10.1074/jbc.M110.200501
Yoon, J.-H., Nam, J.-S., Kim, K.-J., and Ro, Y.-T. (2013). Simple and rapid discrimination of embB codon 306 mutations in Mycobacterium tuberculosis clinical isolates by a real-time PCR assay using an LNA-TaqMan probe. J. Microbiol. Methods 92, 301–306. doi: 10.1016/j.mimet.2012.12.014
Yuan, X., Zhang, T., Kawakami, K., Zhu, J., Li, H., Lei, J., et al. (2012). Molecular characterization of multidrug and extensively drug-resistant Mycobacterium tuberculosis strains in Jiangxi, China. J. Clin. Microbiol. 50, 2404–2413. doi: 10.1128/JCM.06860-11
Zhang, H., Li, D., Zhao, L., Fleming, J., Lin, N., Wang, T., et al. (2013). Genome sequencing of 161 Mycobacterium tuberculosis isolates from China identifies genes and intergenic regions associated with drug resistance. Nat. Genet. 45, 1255–1260. doi: 10.1038/ng.2735
Zhang, S., Chen, J., Cui, P., Shi, W., Shi, X., Niu, H., et al. (2016). Mycobacterium tuberculosis mutations associated with reduced susceptibility to linezolid. Antimicrob. Agents Chemother. 60, 2542–2544. doi: 10.1128/AAC.02941-15
Zhang, S., Chen, J., Shi, W., Cui, P., Zhang, J., Cho, S., et al. (2017). Mutation in clpC1 encoding an ATP-dependent ATPase involved in protein degradation is associated with pyrazinamide resistance in Mycobacterium tuberculosis. Emerg. Microbes Infect. 6:e8. doi: 10.1038/emi.2017.1
Zhang, Y., Heym, B., Allen, B., Young, D., and Cole, S. (1992). The catalase-peroxidase gene and isoniazid resistance of Mycobacterium tuberculosis. Nature 358, 591–593. doi: 10.1038/358591a0
Zhang, Y., and Yew, W. (2015). Mechanisms of drug resistance in Mycobacterium tuberculosis: update 2015. Int. J. Tuberc. Lung. Dis. 19, 1276–1289. doi: 10.5588/ijtld.15.0389
Zhang, Z., Wang, Y., Pang, Y., and Kam, K. M. (2014). Ethambutol resistance as determined by broth dilution method correlates better than sequencing results with embB mutations in multidrug-resistant Mycobacterium tuberculosis isolates. J. Clin. Microbiol. 52, 638–641. doi: 10.1128/JCM.02713-13
Zhao, F., Wang, X.-D., Erber, L. N., Luo, M., Guo, A.-Z., Yang, S.-S., et al. (2014). Binding pocket alterations in dihydrofolate synthase confer resistance to para-aminosalicylic acid in clinical isolates of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 58, 1479–1487. doi: 10.1128/AAC.01775-13
Keywords: Mycobacterium tuberculosis, drug resistance, molecular, comorbidities, therapeutic, drug targets
Citation: Hameed HMA, Islam MM, Chhotaray C, Wang C, Liu Y, Tan Y, Li X, Tan S, Delorme V, Yew WW, Liu J and Zhang T (2018) Molecular Targets Related Drug Resistance Mechanisms in MDR-, XDR-, and TDR-Mycobacterium tuberculosis Strains. Front. Cell. Infect. Microbiol. 8:114. doi: 10.3389/fcimb.2018.00114
Received: 20 August 2017; Accepted: 23 March 2018;
Published: 10 April 2018.
Edited by:
Jianjun Sun, University of Texas at El Paso, United StatesReviewed by:
Jianping Xie, Southwest University, ChinaCopyright © 2018 Hameed, Islam, Chhotaray, Wang, Liu, Tan, Li, Tan, Delorme, Yew, Liu and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianxiong Liu, bGp4ZXI2NEBxcS5jb20=
Tianyu Zhang, emhhbmdfdGlhbnl1QGdpYmguYWMuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.