 Ranjani Dhakshinamoorthy
Ranjani Dhakshinamoorthy Moritz Bitzhenner
Moritz Bitzhenner Pierre Cosson
Pierre Cosson Thierry Soldati
Thierry Soldati Matthias Leippe
Matthias Leippe
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Infect. Microbiol. , 15 March 2018
Sec. Molecular Bacterial Pathogenesis
Volume 8 - 2018 | https://doi.org/10.3389/fcimb.2018.00073
This article is part of the Research Topic Amoebae as Host Models to Study the Interaction with Pathogens View all 14 articles
Due to their archaic life style and microbivor behavior, amoebae may represent a source of antimicrobial peptides and proteins. The amoebic protozoon Dictyostelium discoideum has been a model organism in cell biology for decades and has recently also been used for research on host-pathogen interactions and the evolution of innate immunity. In the genome of D. discoideum, genes can be identified that potentially allow the synthesis of a variety of antimicrobial proteins. However, at the protein level only very few antimicrobial proteins have been characterized that may interact directly with bacteria and help in fighting infection of D. discoideum with potential pathogens. Here, we focus on a large group of gene products that structurally belong to the saposin-like protein (SAPLIP) family and which members we named provisionally Apls (amoebapore-like peptides) according to their similarity to a comprehensively studied antimicrobial and cytotoxic pore-forming protein of the protozoan parasite Entamoeba histolytica. We focused on AplD because it is the only Apl gene that is reported to be primarily transcribed further during the multicellular stages such as the mobile slug stage. Upon knock-out (KO) of the gene, aplD− slugs became highly vulnerable to virulent Klebsiella pneumoniae. AplD− slugs harbored bacterial clumps in their interior and were unable to slough off the pathogen in their slime sheath. Re-expression of AplD in aplD− slugs rescued the susceptibility toward K. pneumoniae. The purified recombinant protein rAplD formed pores in liposomes and was also capable of permeabilizing the membrane of live Bacillus megaterium. We propose that the multifarious Apl family of D. discoideum comprises antimicrobial effector polypeptides that are instrumental to interact with bacteria and their phospholipid membranes. The variety of its members would allow a complementary and synergistic action against a variety of microbes, which the amoeba encounters in its environment.
Amoebozoa are interesting models to study the early evolution of innate immunity (Leippe, 1999). The social amoeba Dictyostelium discoideum, a genetically tractable model for the study of cell biology, has recently become a powerful model organism in infection biology. In particular, it has been employed as a surrogate host for human pathogens such as Legionella (Farbrother et al., 2006), Mycobacteria (Hagedorn and Soldati, 2007; Hagedorn et al., 2009), and Pseudomonas (Cosson et al., 2002; Alibaud et al., 2008). In its amoebic stage, D. discoideum phagocytoses microbes and thereby resembles phagocytes of the innate immune system (Cosson and Soldati, 2008). Upon starvation, the amoebae aggregate and undergo a programmed differentiation and morphogenesis. One of the specific stages is the so-called slug, formed by the aggregation of about 100,000 amoebae, which will eventually differentiate into a fruiting body. Among slug cells, Sentinel cells represent a simple and efficient immune system. These phagocytes patrol the slug body to capture toxic compounds and invading bacteria. The Sentinel cells are continuously shed behind in the slime sheath of migrating slugs (Chen et al., 2007). These immune-like phagocytes have recently been reported to possess the capacity to produce extracellular DNA traps around the pathogen/foreign body (Zhang et al., 2016) in a way similar to phagocytes of vertebrates and invertebrates (Brinkmann et al., 2004; Robb et al., 2014). These features make of D. discoideum an even more attractive model to trace back the conserved functions of the innate immune system across evolution from protozoans to metazoans (Chen et al., 2007; Hagedorn et al., 2009; Zhang and Soldati, 2016).
Despite these promising results, the knowledge about the arsenal that D. discoideum employs to kill phagocytosed microbes and to combat potential pathogens is still scarce. The only example of an antimicrobial effector characterized at the protein level is the AlyA lysozyme, which had been isolated from amoebic extracts and found to be able to degrade bacterial cell walls (Müller et al., 2005).
According to the information derived from the genome project, D. discoideum possesses at least 15 genes potentially coding for lysozymes belonging to several different classes of these hydrolytic enzymes (Eichinger and Noegel, 2005; Müller et al., 2005). Beside the family of Alys, putative Entamoeba-type lysozymes, phage-type lysozymes, and C-type lysozymes (LyCs) can be identified in databases (Müller et al., 2005).
Another multifarious gene family of D. discoideum, which products may target bacterial membranes and thereby kill bacteria directly, is the one coding for saposin-like proteins (SAPLIPs). Structurally, SAPLIPs are characterized by four or five compactly packed alpha-helices, and are typically stabilized by three disulfide bonds built by a conserved array of six cysteine residues (Liepinsh et al., 1997). Functionally, SAPLIPs fulfill various biological functions, but the members of this family have in common that they interact with lipids and membranes (Munford et al., 1995; Bruhn, 2005; Kolter et al., 2005). SAPLIPS with antimicrobial activity can be found in phylogenetically diverse organisms ranging from protozoans to mammals (Leippe et al., 1991; Andersson et al., 1995; Peña et al., 1997).
In pathogenic amoebae, saposin-like proteins are well-known as pore-forming proteins that permeabilize the membranes of bacteria and human host cells (Leippe, 2014). The most comprehensively studied member of amoebic SAPLIPs is amoebapore A from E. histolytica, the tertiary structure revealed the characteristic SAPLIP fold (Hecht et al., 2004). We therefore provisionally termed the putative SAPLIPs of which D. discoideum Apls for amoebapore-like peptides.
Dictyostelium discoideum possesses 17 Apl genes that potentially can give rise to 33 SAPLIP peptides given that larger precursor proteins containing more than one SAPLIP domain (also termed saposin B domain) might be processed to release several mature SAPLIPs, as exemplified for the name-giving saposins (O'Brien and Kishimoto, 1991) and for naegleriapores of the free-living amoeba Naegleria fowleri (Herbst et al., 2004). Such an enormous variety of SAPLIPs in one species is only known so far from C. elegans, a nematode that also feeds on microbes (Roeder et al., 2010).
In D. discoideum, one may speculate that these proteins act complementarily and synergistically and constitute an important part of the antimicrobial armamentarium during its unicellular and multicellular stages. Nonetheless, functions other than killing of bacteria by membrane permeabilization have been reported for amoebic SAPLIPs (Michalek and Leippe, 2015).
In the present study, we have chosen AplD among the plethora of potential D. discoideum SAPLIPs for a more detailed functional study because it became apparent that: (i) the primary translation product comprises a single SAPLIP domain preceded by a putative signal peptide as known for amoebapores; (ii) the gene has been reported to be differentially expressed upon bacterial challenge; and (iii) most importantly with respect to immunity, aplD is reported to be primarily transcribed during the multicellular stages (Dicty express: https://dictyexpress.research.bcm.edu/bcm/#/all?genes=DDB_G0293010).
At the protein level, we characterized the ability of AplD to permeabilize the membranes of live bacteria and liposomes by monitoring quantitatively the activity of a recombinantly expressed protein (rAplD). In vivo, we phenotypically analyzed the effect of ablation of aplD on amoebic growth on various bacterial lawns, on bacterial killing in the amoebic stage, and on the slug's capacity to fight a bacterial infection.
Various bacterial strains were used in the study (Supplementary Table 1). The bacterial cultures were grown at 220 rpm at 37 °C for 12 to 14 h in Luria-Bertani medium.
Ax2 obtained from the Dicty stock center was used for generating the apl and lyC KO mutants. Ax2 amoebae were grown in maltose-HL5 medium and blasticidin (8 μg/ml)-containing maltose-HL5 medium was used to select the KOs. The D. discoideum cells transfected with prestalk reporter (ecmAO-RFP), prespore reporter (pSA-RFP) plasmids, and aplD−/[act6]:aplD.FLAG cells [rescue strain; mentioned as aplD−(+)] were cultured in maltose-HL5 medium complemented with G418 (10 μg/ml) and blasticidin (8 μg/ml).
Short gene fragments from the 5′ and 3′ regions of apl and lyC genes were amplified by PCR and ligated appropriately at the 5′ and 3′ flanking ends of the blasticidin resistance (Bsr) gene present in the pLPBLP gene disruption vector as described in Faix et al. (2004). Before transfection, the KO vectors were subjected to DNA sequencing (Eurofins MWG operon, Germany) to verify vector orientations and mutations.
pLPBLP, ecmAO-RFP, pSA-RFP, and pDneo2a-3xFLAG plasmids were received from the Dicty stock center.
Gene disruptions were achieved by homologous recombination between the respective apls and lyCs with their corresponding KO vectors. Ax2 cells (2 × 107) were sedimented at 560 × g at RT for 5 min. The pellet was resuspended in 1 ml ice-cold electroporation (EP) buffer (10 mM K2HPO4, 10 mM KH2PO4, 50 mM sucrose, pH 6.2.), washed at 10,000 × g at RT for 2 min, and briefly incubated on ice. The respective linearized KO vector was resolved in 100 μl ice-cold EP++ buffer (EP buffer containing 1 mM MgSO4, 1 mM NaHCO3, 1 μM CaCl2, and 1 mM ATP, pH 6.2) and subsequently mixed with Ax2. Immediately, this cell mixture was transferred to 2-mm gap electroporation cuvette (BTX electroporation cuvettes, Harvard apparatus) and electroporation was carried out at 300 V, 2 ms time constant with five square pulses, including 5-s intervals inbetween the pulses, using a BTX 830 electroporator (Harvard apparatus). After 10 min incubation on ice, the cell suspension was transferred to Petri dishes that contain 10 ml maltose-HL5 medium and incubated at 22°C. Blasticidin (8 μg/ml) selection was introduced 24 h later. Blasticidin-resistant clones had appeared after a week and were aspirated from the Petri dishes and seeded in 24-well plates (both from Sarstedt, Germany) for PCR analyses.
Cell lysates were prepared from the blasticidin-resistant clones following the method described in Charette and Cosson (2004). As described in Faix et al. (2004) three different PCR analyses were performed to verify the apl and lyC KO vectors insertion in 5′–3′ orientation at the apl and lyC loci. PCR screening results for aplD KO clones are shown in the Supplementary Figure 1. KO vectors (vector control, VC) and Ax2 genomic DNA (wild-type, WT) were used as respective controls for PCR analyses. The KO clones were confirmed for gene ablation at the respective gene locus by performing Southern hybridization (Supplementary Figure 2), using the DIG-High prime DNA labeling and detection starter kit II (Roche), following manufacturer's instructions. A probe was generated for the Bsr-resistance gene sequence, which spans for 249 base pairs and dioxygenin (DIG) was used as the labeling agent.
The cDNA sequence of aplD was amplified by PCR and cloned under the Actin 6 promoter (act6) of pDneo2a-3xFLAG vector (Dubin and Nellen, 2010). The 3′ end of aplD cDNA was fused to the FLAG gene. Finally, the pDneo2a-3xFLAG[act6/aplD] plasmid was transfected into aplD− cells. AplD-FLAG fusion protein expression in the rescue strain [aplD−(+)] was confirmed by Western blot analysis (Supplementary Figure 3). After separation by SDS-PAGE, proteins were transferred onto a polyvynilidene fluoride membrane and the blot was incubated with a mouse anti-FLAG M2 monoclonal antibody (Sigma Life Science) and subsequently developed using a goat anti-mouse IgG (H+L) conjugated to alkaline phosphatase (Jackson ImmunoResearch).
To analyse transcription in axenic cultures, RNA was isolated from axenically grown D. discoideum amoebae (5 × 106 cells) and the first strand cDNA was synthesized using the RNA template (cDNA synthesis kit, Invitrogen). Quantitative RT-PCR (qRT-PCR) was performed using the cDNA template as described by the manufacturer (qRT-PCR mix, TAKARA and Light cycler, Roche). For analysis of xenic cultures, Ax2 cells were mixed with bacteria (ratio 1:10) and the amoebae-bacteria cell suspensions were incubated at 140 rpm and 22°C for 8 h. Subsequently, the xenic Ax2 cells were washed three times at 425 × g at 4°C for 2 min to remove excess bacteria. cDNA was synthesized and qRT-PCR analyses were performed as described above. AplD transcription in axenic Ax2 amoebae was considered as reference to determine the aplD expression in xenic Ax2. Each symbol denotes individual experiment. The house keeping genes tested were Ig7 (rnlA) and Glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The bacterial strains tested include KpLM21, B. subtilis, and PT531 (see Supplementary Table 1). For analysis during development, axenic Ax2 cells were washed with Sörensen's buffer (SB) at 510 × g and 22°C for 7 min. The cell pellets were resuspended in 100 μl SB and deposited on SB agar (1%) plates. Subsequently, the plates were incubated in a dark, moist chamber at 22°C and RNA was isolated from the starving amoebae (0 h), streaming cells (8 h), mounds (12 h), slugs (16 h), and fruiting bodies (24 h) and qRT-PCR was performed in duplicates. AplD expression in the starving amoebae (0 h) was considered as reference to quantify the aplD expression at other stages of development (Sillo et al., 2008). Three independent development experiments were performed and the bars represent standard deviation.
Bacterial growth assays were performed on routinely used SM agar medium, which was devoid of glucose. SM agar plates were prepared as described in Froquet et al. (2009). Varying number (104, 103, 102, and 101) of D. discoideum amoebae were deposited on the bacterial lawn layered on SM agar plates and incubated in the dark at 22°C. Five days later, the KOs were scored for their plaque forming abilities by having Ax2 as comparative controls. Three independent experiments were performed with triplicates (see Supplementary Table 1 for details on the bacterial strains tested).
Dictyostelium discoideum amoebae were mixed with bacteria (100:1) and incubated at 140 rpm, 22°C. The total number of viable bacteria was measured at the indicated time points by following the method detailed in Benghezal et al. (2006). The rate of killing of K. pneumoniae bacteria by amoebae was quantified by counting the number of colony forming units (CFU) at each time point. Three independent experiments were performed.
Dictyostelium discoideum cells were washed with SB and deposited on SB agar (1%) plates at a cell density 5 × 105 cells/cm2. Subsequently, plates were incubated in a dark, moist chamber at 22°C. The morphogenetic stages were imaged under the stereomicroscope (Olympus ULWCD 0.30) at time points mentioned (Supplementary Figure 4). Dictyostelium discoideum development on SB agar was monitored in three independent experiments.
Slug pattern analyses were performed as described in Parkinson et al. (2009). For prestalk reporter (ecmAO-RFP) examination, D. discoideum amoebae carrying the ecmAO-RFP plasmid were mixed with D. discoideum amoebae at 20:80 ratio and seeded on a SB agar (1%) plate at a cell density 5 × 105 cells/cm2. For prespore reporter (pSA-RFP) examination, D. discoideum amoebae (20%) were mixed with D. discoideum amoebae marked with pSA-RFP (80%) and deposited on SB agar plates. The plates were incubated in a dark, moist chamber at 22°C until the migrating slugs were formed and were imaged under the stereomicroscope (Olympus ULWCD 0.30) (Supplementary Figure 4). Two independent experiments were performed in duplicates and at least ten slugs were imaged per plate.
Dictyostelium discoideum cells were harvested from Petri dishes, 2.5 × 107 cells were sedimented, and the pellets were resuspended in 50 μl SB. This high density cell suspension was deposited on SB agar (1%) plates and incubated in a dark, moist chamber with an unidirectional light source until migrating slugs were formed (~18 h). Slug infection experiments were performed following Chen et al. (2007) with some modifications. The slugs were injured with a sterile needle (23G × 11/4, B|BRAUN Injekt F) and a dense bacterial suspension, which was prepared from an overnight culture, was layered on the injured slugs. K. pneumoniae expressing GFP reporter (KpGFP) and E. coli expressing DsRed reporter were used for infecting the injured slugs (see Supplementary Table 1). The slugs infected with Ec DsRed were imaged under the stereo microscope (Olympus ULWCD 0.30) 8 h post infection and those slugs infected with KpGFP were imaged 20 h and 24 h post infection. KpGFP was grown with ampicillin (1 mg/ml) for 12 h at 37°C. Ec DsRed was grown with isopropyl β-D-1-thiogalactopyranoside (IPTG, 100 mM) and kanamycin (150 μg/ml). Three independent experiments were performed and at least ten slugs were imaged per experiment for each strain [Ax2, aplD−, and aplD−(+)].
Dictyostelium discoideum cells were allowed to form slugs on SB agar (1%) plates containing ethidium bromide (EtBr, 3 μg/ml) as described in Chen et al. (2007). After 3 h, Sentinel cells present in the migrating slugs were visualized under the stereomicroscope (Olympus ULWCD 0.30). Three independent experiments were performed and at least ten slugs were imaged per experiment for each strain (Ax2 and aplD−).
The nucleotide sequence encoding putatively mature AplD (DDB0216216) was codon optimized for bacterial expression and synthesized by GeneArt (Regensburg, Germany). The cDNA was ligated into the pET-32a (+) (Novagen) expression vector containing an ampicillin resistance gene using KpnI and XhoI cleavage sites. The resulting plasmid encodes a fusion protein, which comprised an N-terminal thioredoxin followed by a hexahistidine (His6) tag and contained thrombin and tobacco etch virus (TEV) protease cleavage sites preceding the primary structure of AplD. TEV cleavage yielded a product with the amino acid sequence as follows: GEIDNNQCQICELLVKDIIEGLTANQSVEVIEHGLNLICDH
IPLHVRVCKQFVDSNFQKIVQFIENHDDPQEICEKCGVC. The protein was recombinantly expressed in E. coli C 43 at 37°C after induction with 0.5 mM IPTG for 6 h and extracted by sonication on ice (Sonoplus HD 2200 sonicator, MS-73 titanium microtip, Bandelin electronic GmbH, Germany). The fusion protein in the soluble fraction was purified by immobilized-metal affinity chromatography (IMAC) using a TALON resin (Clontech, Saint-Germain-en-Laye, France) and subsequent anion-exchange chromatography (1-ml Resource Q column, GE Healthcare) using an Äkta purifier system (model P-900; GE Healthcare) and 20 mM Tris-HCl, pH 7.0 with a continous linear gradient of 0-1 M NaCl for elution. The N-terminal tag was removed by cleavage with TEV protease (ProTEV Plus protease, Promega) at 30°C overnight. The cleaved product was subjected again to IMAC and anion-exchange chromatography (Mini Q column 4.6/50, GE Healthcare, Solingen, Germany) to remove the fusion partner and the TEV from recombinant AplD. After final purification, the apparently homogeneous protein fraction was lyophilized, redissolved, and dialyzed against 10 mM sodium phosphate, pH 6.8.
Purity was proven by tricine-SDS polyacrylamid gel (SDS PAGE) using 13% separation gels (Schägger and von Jagow, 1987) and SeeBluePlus2 (Invitrogen) as standard protein marker for molecular masses. Protein concentration was determined with the BCA assay (Pierce, Thermo Scientific, Bonn, Germany). The molecular mass of recombinant AplD was determined by mass spectrometry in linear mode using a 4700 Proteomic Analyzer MALDI-TOF/TOF mass spectrometer (Life Technologies, Darmstadt, Germany).
Pore-forming activity using the liposome depolarisation assay (Leippe et al., 1991) and permeabilization of cytoplasmic membranes of live bacteria using the fluorescent dye SYTOX Green (Herbst et al., 2002) were measured as described previously. Briefly, in the liposome-depolarization assay a valinomycin-induced diffusion potential across the membrane of liposomes prepared from asolectin, a crude mixture of soy bean phospholipids, resulted in quenching of the enclosed fluorescent dye. Application of a pore-forming protein disrupts the membrane potential and the fluorescence intensity increases by the release of the dye. An increase of 5% within 1 min after addition of the pore-forming agent at 25°C is defined as one activity unit. In the membrane-permeabilization assay the fluorescent dye SYTOX Green (Invitrogen) is added to Bacillus megaterium, E. coli, Klebsiella aerogenes, K. pneumonia KpLM21, or K. pneumoniae K− as viable target giving a signal while intercalating in DNA. Accordingly, only when the bacterial plasma membrane become compromised, e.g., by an antimicrobial protein, fluorescence appears. The values were expressed as the mean of three independent experiments. Kinetics of the membrane permeabilization of B. megaterium were monitored in that fluorescence was measured for different doses at various time points and presented as a representative example from four replicates with similar results. The synthetic peptides alamethicin and cecropin P1 served as positive controls in the activity assays (Sigma, Taufkirchen, Germany).
In the genome of D. discoideum, we detected 17 genes that may code for Apl proteins containing a single or several SAPLIP domains (Figure 1). The sequences of apl A-R and the corresponding putative proteins can be retrieved from the D. discoideum database (www.dictybase.org) using apl* as a query. Their primary translation products each contain a predicted signal peptide that may allow trafficking of the precursor proteins to lysosomal compartments. Proteolytic processing of larger precursors can result in 33 mature peptides with one SAPLIP domain. An N-terminal glycosylation site can be predicted for nearly all of them.
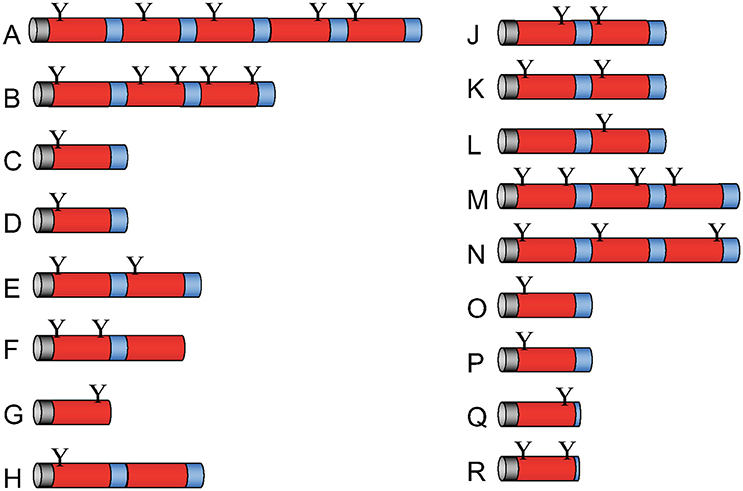
Figure 1. Molecular architecture of Apl precursor proteins. The D. discoideum genome contains 17 genes that potentially code for saposin-like proteins (SAPLIPs) with some similarity to amoebapores provisionally termed Apls. The primary translation products of apls named from A to R (omitting I) contain between one and five SAPLIP domains (also termed saposin B domain). After processing, these preproproteins potentially give rise to 33 different mature polypeptides. Signal peptides (gray), SAPLIP domains (red), putative linker regions and end regions (blue), and potential N-glycosylation sites (Y) are depicted.
The permissiveness of various bacteria for amoebic growth is routinely assessed by testing the ability of D. discoideum mutants to generate growth plaques on bacteria lawns. This strategy has led to the identification of several bacterial virulence genes (Cosson et al., 2002; Pukatzki et al., 2002; Benghezal et al., 2007; Alibaud et al., 2008). Here, we adapted the method to test the growth abilities of D. discoideum Ax2 strain in which genes potentially coding for antimicrobial proteins (apls and lyCs) have been disrupted. To quantitate the degree of bacterial virulence, varying number of D. discoideum cells were spotted on bacterial lawns and the growth plaques were monitored after 5 days. It became apparent that aplD− was defective for growth on KpLM21, a clinical isolate and two apl mutants, aplD− and aplP−, were non-permissive for growth on K−, a capsule defective Kp strain (Figure 2A). In the case of Ax2, even 10 amoebae were sufficient to create growth plaques on K− and KpLM21, whereas at least 10,000 amoebae of aplD− and at least thousand amoebae of aplP− were required to generate visible plaques on K−. Likewise, KpLM21 allowed plaque formation only when 10,000 aplD− amoebae were deposited (Figure 2A). We also found that D. discoideum with a KO of the gene encoding the C-type lysozyme 2, lyC2−, was defective for growth on K− (Supplementary Figure 5). The growth abilities of Apls and lyCs mutants on bacteria such as B. subtilis, E. coli Br, non-pathogenic K. pneumoniae, and P. aeruginosa are summarized in a scheme (Figure 2B).
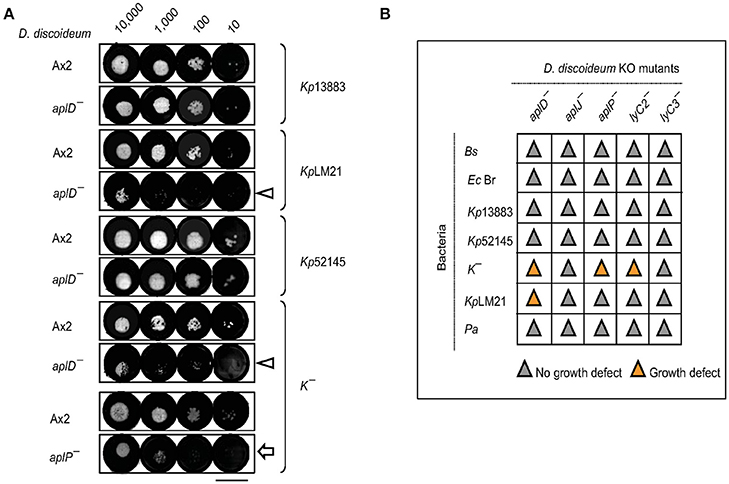
Figure 2. Growth defects of AplD− and aplP− cells on virulent K. pneumoniae. (A) D. discoideum growth abilities on various K. pneumoniae were tested by depositing indicated number of amoebae on K. pneumoniae lawns that were prepared on SM agar. D. discoideum amoebae were fed with Kp13883, KpLM21, Kp52145, and K−. Arrowheads indicate aplD− cells growth defects on KpLM21 and K−. Arrow represents aplP− growth impairment on K−. Scale bar, 1.5 cm. (B) Growth abilities of aplD−, aplJ−, aplP−, lyC2− (see Supplementary Figure 5), and lyC3− were tested on B. subtilis (Bs), E. coli B/r (Ec Br), K. pneumoniae strains (Kp13883, Kp52145, K−, and KpLM21), and P. aeruginosa (Pa). Gray and yellow colors demonstrate the absence and presence of D. discoideum growth plaques on bacteria (see Supplementary Table 1 for bacterial strain details).
As described in Benghezal et al. (2006), we performed a specific assay to investigate whether aplD− amoebae are defective in phagocytosis and/or killing of KpLM21. These assays measure the total number of live bacteria remaining at indicated time points. We found that aplD− amoebae were able to phagocytose and kill Klebsiella aerogenes and KpLM21 as efficiently as the wild-type Ax2 (Figure 3). This indicates that AplD is not essential for intracellular killing of K. pneumoniae bacteria in free-living Dictyostelium amoebae.
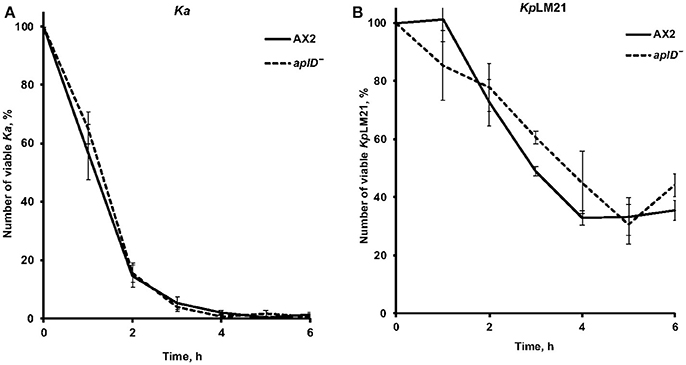
Figure 3. AplD− cells kill efficiently Klebsiella pneumoniae. (A) The ability of D. discoideum to ingest and kill non-virulent K. aerogenes Ka was tested by mixing amoebae and bacteria and measuring the total number of viable bacteria at the indicated time points. (B) The same assay was performed using virulent K. pneumoniae KpLM21. Ax2 was used as comparative control. Each curve is the average of three independent experiments, bars represent standard error of the mean.
To examine whether the expression of aplD is regulated by exposure to various bacteria, we measured the level of the aplD mRNA by qRT-PCR. Contact with KpLM21 increased aplD at least three-fold, but contact with Bacillus subtilis did not induce a significant change. In contrast, exposure to Pseudomonas aeruginosa PT531 downregulated aplD on average about 6-fold (Figure 4A). Analyses of aplD regulation during D. discoideum development on non-nutrient KK2 agar showed that aplD was weakly expressed in axenic conditions, but was strongly upregulated during late development stages, starting from the mound stage [12 h] and slug stage [16 h], and peaking at fruiting body stage [24 h] (Figure 4B). This profile is comparable to the aplD transcriptional regulation observed in Ax4 grown on bacteria and subjected to development on SB agar (Dicty express, Supplementary Figure 6). The transcriptional regulation of antimicrobial genes during the growth phase of D. discoideum might be essential to adapt to various food sources, but also combat various pathogens. It has been reported that amoebae stop feeding during development. Therefore, the main purpose for the aplD upregulation might be an extracellular role in cellular defenses inside the multicellular structures or inside phagocytic Sentinel cells. Interestingly, upregulation of aplD during development, in particular at late stages, is several folds higher than during exposure to KpLM21.
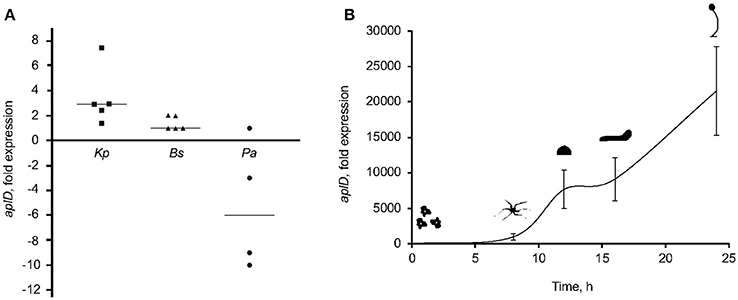
Figure 4. AplD upregulation upon K. pneumoniae feeding and during late development. (A) AplD transcription profile in xenic Ax2 cultures were examined by mixing amoebae with bacteria at an 1:10 ratio and measuring aplD transcript levels by qRT-PCR. AplD fold expression in xenic Ax2 cultures were determined based on aplD transcription in axenic Ax2 cultures. Bacterial strains tested were K. pneumoniae (KpLM21), B. subtilis (Bs), and P. aeruginosa (Pa). Each symbol denotes the data from an individual experiment and the bar represents the median thereof. (B) AplD regulation during Ax2 development was tested by subjecting Ax2 for development on KK2 agar plates and quantifying aplD transcription at different morphogenetic stages by qRT-PCR. AplD expression at 0 h was considered as reference to determine aplD fold expression at subsequent development stages. n = 3, bars represent standard deviation.
To test the idea that AplD is primarily instrumental in multicellular stages of D. discoideum, we infected slugs derived from aplD− amoebae with a K. pneumoniae (Kp) strain that expressed GFP. Unlike wild type slugs, at 20 h post Kp infection, aplD- slugs showed bacterial deposits both on their surface and in their body. At 24 h post Kp infection, aplD− slugs showed Kp clumps mainly inside. Additionally, aplD− slugs were not successful in sloughing off Kp in their slime sheath (Figure 5). By contrast, aplD− slugs were able to shed E. coli in their slime sheath as early as 8 h post infection (Figure 5). As the major defense mechanism reported so far in D. discoideum slugs is based on the phagocytic nature of Sentinel cells, we examined whether aplD− slugs possess a functional population of these cells. As evidenced by the uptake of the fluid-phase tracer EtBr, aplD- slugs were capable of generating Sentinel cells that internalized the toxic dye and were sloughed off during migration (Figure 6) and aplD− did not depict any prominent developmental defect (Supplementary Figure 4).
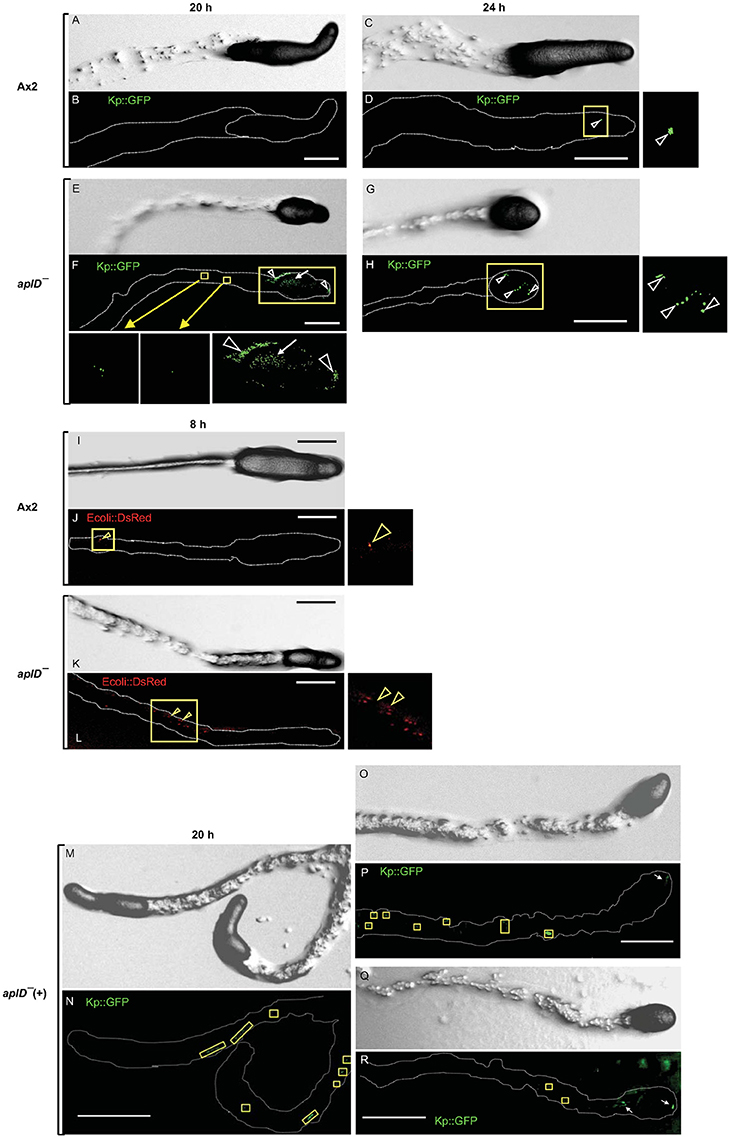
Figure 5. Susceptibility of slugs to infection with Klebsiella and E. coli. Migrating D. discoideum slugs were injured with a fine needle and infected with GFP-expressing K. pneumoniae (KpGFP). Slugs were imaged at 20 and 24 h post KpGFP infection. Differential Interference Contrast (DIC) images of Ax2 (A,C) and aplD− slugs (E,G) at 20 and 24 h post infection are represented. KpGFP infections in Ax2 (B,D) and aplD− slugs (F,H) at 20 and 24 h post infection are shown under green fluorescence filter. KpGFP infection in Ax2 slug imaged 24 h post KpGFP infection (D) is also magnified. KpGFP infection on slug surface (arrowheads) and slug interior (arrow) and in the slime sheath of aplD− slug at 20 h post infection are magnified in the insets below. KpGFP clumps inside aplD− slug 24 h post infection (H) is magnified in the adjacent panel. AplD− slugs sloughed off E. coli in their slime sheaths. DIC images of Ax2 and aplD− slugs (I,K) 8 h post infection with E. coli DsRed are represented. E. coli DsRed clumps (arrowheads) in Ax2 (J) and aplD− (L) slime sheaths (boxes) are shown under red fluorescence filter and are also magnified in the respective insets. AplD-(+) slugs sloughed off KpGFP in their slime sheaths. AplD−(+) slugs were infected with KpGFP as described earlier and were imaged 20 h post KpGFP infection. DIC images of aplD−(+) slugs are shown in (M,O,Q). K. pneumoniae infections in aplD−(+) slugs are shown under green fluorescence filter in (N,P,R). AplD−(+) slime sheaths showing KpGFP deposits (boxes) and clumps in the slug interior (arrows) at 20 h post infection are depicted. Scale bars, 100 μm.
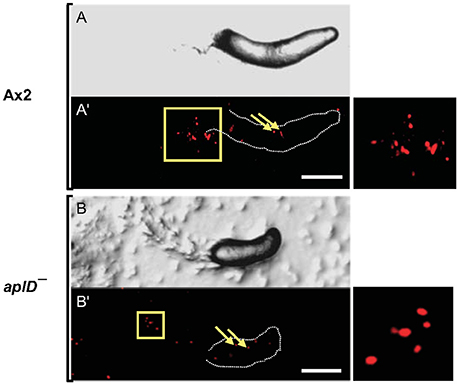
Figure 6. Sentinel cells were unaffected in aplD− slugs. Ax2 and aplD− slugs were allowed to migrate on EtBr-containing (3 μg/ml) agar for 3 h. DIC images of Ax2 and aplD− slugs (A,B) imaged under a stereomicroscope are represented. Sentinel cells inside Ax2 and aplD− slugs (A',B') are shown under red fluorescence filter (arrows). Sentinel cells detected in slime sheaths are magnified (boxes, right). Scale bars, 100 μm.
As described above, aplD− amoebae were restricted for growth on K. pneumoniae, K−, and KpLM21, and aplD- slugs were vulnerable to Kp infections. We tested whether restoration of aplD expression in aplD− would rescue the phenotypic defects. Constitutive expression of aplD in aplD− amoebae, termed aplD−(+), under the control of the act6 promoter of an extra-chromosomal vector rescued the formation of plaques on K− but showed only partial rescue on KpLM21 lawns (Figure 7). Bacterial clearing was observed on KpLM21 lawns when 104 and 103 amoebae were deposited, but not with 102 and 101 amoebae. This observation might indicate that spatio-temporal regulation of aplD expression is crucial for full functionality. AplD−(+) slugs infected with Kp were able to clear off Kp by sloughing them off in their slime sheath. It appeared that the interior of aplD−(+) slugs was virtually free of Kp deposits and clumps (Figure 5).
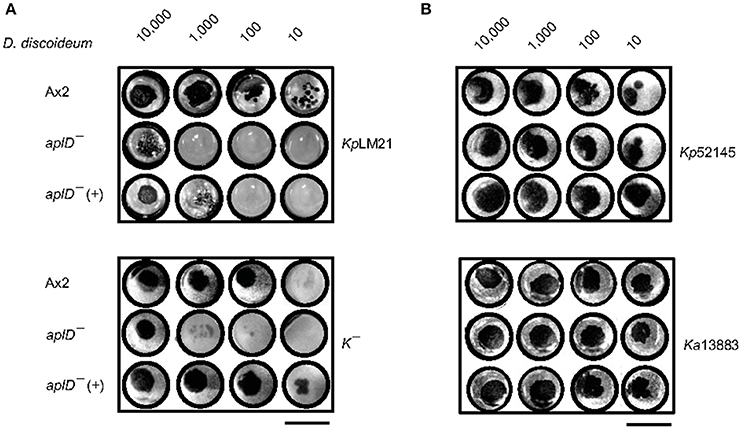
Figure 7. AplD−(+) strain growth on K. pneumoniae. Growth abilities of aplD−(+) cells were tested on K. pneumoniae KpLM21 and K− (A) and on K. pneumoniae Kp52145 and Ka 13883 (B). Growth abilities of Ax2 and aplD− cells are also represented for comparison. Scale bar, 1.5 cm.
AplD was heterologously synthesized in E. coli as a fusion protein and subsequently the thioredoxin-His6 tag was removed by proteolytic cleavage. The final product was purified to apparent homogeneity by repeated steps of IMAC and anion-exchange chromatography (Figure 8A). Analysis by MALDI-TOF mass spectrometry confirmed the molecular identity of rAplD. It revealed an experimental average mass of 9,070.8 Da, which is in good agreement with the calculated molecular mass (9,070.3 Da) provided that six cysteine residues are involved in disulfide bonds. The recombinant protein was tested for its pore-forming and bacterial-membrane permeabilizing activities in vitro. AplD formed pores in liposomes composed of asolectin. At mildly acidic pH (5.2), AplD depolarized liposomal membranes with similar activity as the prototype of a pore-forming peptide, alamethicin (Figure 8B). AplD acted in a pH-dependent manner. The protein displayed the highest pore-forming activity at pH 4.4 and gave decreasing values with increasing pH (Figure 8C). In the Sytox-Green assay, performed exemplarily with B. megaterium, it became apparent that AplD is capable of permeabilizing the cytoplasmic membranes of live bacteria. Membrane-permeabilizing activity increased with time. After 2 min, virtually no activity was detectable, but already after 5 min bacteria with compromised membranes appeared, particularly at higher protein concentrations. After 1 h, approximately 50% of the bacteria were permeabilized by AplD at 2.5 μM (Figure 8D). We could not detect bacterial membrane permeabilization with up to 5 μM AplD in the same assay when we used Gram-negative bacteria, K. pneumoniae LM21, K. pneumoniae K−, K. aerogenes, or E. coli, as target cells (data not shown) indicating that the outer membrane of these species constitutes an additional barrier for AplD for reaching the bacterial cytoplasmic membrane.
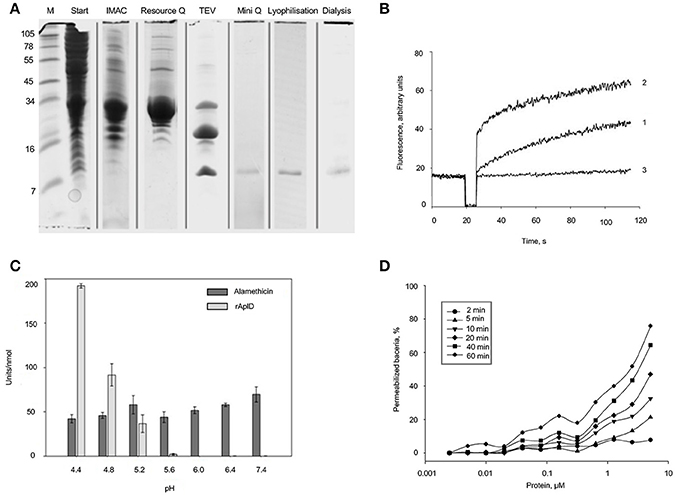
Figure 8. Purification and functional in-vitro analysis of recombinant AplD. (A) Purification steps of AplD after recombinant synthesis evidenced by SDS PAGE and Coomassie staining of the gel: Extracts of E. coli C43 transformed with aplD in pET-32a (+) in TBS, pH 7.0 (start), enrichment with IMAC, further purification with anion-exchange chromatography (Resource Q), proteolytic cleavage with TEV, and final purification via a Mini Q column. Purity of AplD after lyophylisation and dialysis is also shown at the right. All samples were reduced and alkylated before separation. At the very left, marker proteins (M) were separated and the molecular masses of these are depicted in kDa. (B) Time course of pore formation induced by AplD. The dissipation of a valinomycin-induced diffusion potential in vesicles of asolectin after addition of AplD (80 pmol) (trace 1), control peptide alamethicin (100 pmol) (trace 2), and the peptide solvent (trace 3) were recorded at pH 5.2. Pore-forming activity is reflected by the increase of fluorescence as a function of time. (C) pH dependence of pore-forming activity of AplD. Activity of AplD in comparison with that of alamethicin was measured in six independent experiments in Tris-maleate buffer (50 mM Tris-maleate/50 mM sodium sulfate/0.5 mM EDTA/0.02 % sodium azide) adjusted to various pH. Mean and standard deviation are shown. (D) Kinetics of membrane permeabilization by AplD. Membrane permeabilization of viable B. megaterium was measured as an increase in fluorescence of the DNA intercalating dye SYTOX Green at pH 5.2 induced by AplD and monitored after different time points at various concentrations.
Dictyostelium discoideum is a non-pathogenic, unicellular host used for bacterial and fungal risk assessment studies (Cosson et al., 2002; Alibaud et al., 2008; Koller et al., 2016) and the outcomes from such studies remain surprisingly similar to the observations made in animal hosts (Benghezal et al., 2006; Hagedorn et al., 2009). Although D. discoideum serves as a versatile system to explore bacterial virulence genes and their mechanisms of action, not much is known about the effector molecules of D. discoideum and their mode of action to defend against pathogens. In D. discoideum, host-pathogen interaction studies can be attempted both at the single-cell phagocyte stage and during the multicellular stages of its life cycle. Interestingly, the single-cell and multicellular phases are clearly separated and involve differential regulation of gene expression. During the growth phase, D. discoideum internalizes microbes by phagocytosis and the resulting phagosome fuses with lysosome, a compartment that houses hydrolytic enzymes and antimicrobial peptides, leading to efficient killing and degradation of the microbes to fulfill the nutritional need of the growing amoeba.
However, several intracellular pathogens have evolved mechanisms to block phago-lysosome fusion and to exploit the phagosomal compartment as their replication hub (Hagedorn et al., 2009; Shevchuk et al., 2014). During the multicellular stages of development, the antibacterial defense mechanisms are even more sophisticated. For example, in the slug formed mainly of non-phagocytic prespore and prestalk cells, about 1% of the cells, called the Sentinel cells retain their phagocytic ability. The foreign bodies and poisonous compounds that enter the slug are swallowed by these cells and finally get sloughed off the migrating slug and remain in the slime sheath, where they are entangled within extracellular DNA traps (Zhang and Soldati, 2016; Zhang et al., 2016). Mechanistically, this helps to restrict microbial infection and/or the accumulation of toxins. The present study reveals for the first time the specific involvement of the AplD gene product in antimicrobial defense during multicellular stages of development. In addition, aplD expression is differentially regulated by exposure to different bacteria, e.g., induced in the presence of K. pneumoniae and repressed in the presence of P. aeruginosa.
These in vivo evidences strongly suggest that Apl peptides are involved in antimicrobial defense, but a direct membrane-lysing activity had not been reported so far. Here, we show that purified, recombinant AplD displayed pore-forming activity in the minimalistic system of liposomes and also readily permeabilized the membranes of the live Gram-positive bacterium B. megaterium as monitored by the Sytox green assay. However, an activity against Gram-negative bacteria was not detected at the concentrations employed. One might speculate that AplD can also permeabilize Klebsiella, provided that the outer membrane does not hamper the access to the primary target, the bacterial plasma cell membrane. The antimicrobial efficacy of the relatively negatively charged AplD might be substantially enhanced by the synergistic action of other antimicrobial proteins, e.g., Apls with a more positive net charge, or when other Dictyostelium factors that are not bactericidal per se but are capable of perturbing the outer membrane structure act in concert with AplD.
There are several examples of SAPLIPs including the saposins themselves that have been found to be glycosylated in their natural form. The vast majority of the potential peptides released from Apl precursors bear a potential N-glycosylation site. This also holds true for AplD. In amoebae, we have previously compared the pore-forming activity toward liposomes of glycosylated and deglycosylated naegleriapores and could not detect a substantial negative effect upon glycan removal (Herbst et al., 2002). However, we cannot exclude that a natural AplD, glycosylated specifically by D. discoideum, might have a different activity spectrum against natural targets than the recombinant unglycosylated version. A single glycosylation motif may determine a particular oligomeric structure, e.g., dimerization, to generate a more active protein, it may mediate an interaction with a partner molecule that helps to overcome the outer membrane of Gram-negative bacteria such as Klebsiella, or it may simply facilitate binding to bacterial targets, e.g., by shielding the negative charge of particular regions of the protein.
Apart from Apls, there are two other proteins that harbor a SAPLIP domain, i.e., acyloxyacyl hydrolase (AOAH) and countin (Ctn). The enzyme AOAH deacylates lipopolysaccharides in vertebrates, but has not been characterized in amoebae so far (Munford et al., 2009). Countin is a well-studied major component of a protein complex called counting factor, known to play a crucial role in aggregate size determination during development (Brock and Gomer, 1999). When recombinant countin was monitored for pore-forming activity in a liposome-depolarization assay, it exhibited only marginal activity (Gao et al., 2002). In our study, we found that aplD− amoebae showed mild defects at early development, such as stream breaks and developmental delay, but finally were able to form slugs and fruiting bodies, although they were smaller than from wild-type Ax2 cells.
We found that aplD− amoebae are specifically impaired for growth on one virulent strain of K. pneumoniae. Although aplD is poorly transcribed in the amoebic stage under axenic growth conditions, it appears as if its gene product is nevertheless instrumental to allow growth in the presence of some K. pneumoniae strains. The plethora of Klebsiella bacteria present over a longer period in the growth assay, different from the short exposure to K. pneumoniae in a killing assay, may change gene expression of several genes including aplD. More impressively, we found that aplD− slugs are immune-compromised specifically for K. pneumoniae. During that multicellular stage, we observed a dramatic increase of aplD transcript abundance. Notably, complementation of aplD in aplD− rescued the capacity of slugs to effectively capture and slough off K. pneumoniae in their slime sheath in a way similar to wild-type Ax2.
With respect to the evolution of innate immunity, the antimicrobial capacities of D. discoideum are still a relatively unexplored area of research. On the one hand, the amoeba is an interesting model because it resembles in its unicellular stage mammalian macrophages in several aspects (Bozzaro et al., 2008; Cosson and Soldati, 2008; Dunn et al., 2018). On the other hand, the developmental cycle of the social amoeba represents an intriguing example in biology of appearance of multicellularity before the advent of true metazoans. In particular, the slug, which is composed of about 100,000 prespore and prestalk cells, hosts about 1% of Sentinel cells. These phagocytic cells reminiscent of patrolling neutrophils and tissue macrophages of animals, represent an extraordinarily appealing model system for comparative immunologists, as they defend the slug against infection using bactericidal phagocytosis and DNA extracellular traps (Chen et al., 2007; Zhang et al., 2016). Here, we reveal the involvement of an amoebic SAPLIP in the struggle of the slug stage against virulent bacteria, as an early example of a molecular defense implemented at the transition from unicellular to multicellular organisms.
Conceived the study: ML. Designed the experiments: RD, PC, TS, and ML. Performed the experiments: RD and MB. Analyzed the data: RD, MB, PC, TS, and ML. Wrote the paper: RD, PC, TS, and ML.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This study was supported by the German Research Foundation (DFG); CRC 1182 Function and Origin of Metaorganisms (project A1). TS is a member of iGE3 (http://www.ige3.unige.ch); is supported by an RTD grant from SystemsX.ch, and by multiple grants from the Swiss National Science Foundation. The authors thank the Dicty stock center for providing the plasmids/strains used in this study. RD is supported by DST, INDIA. RD thanks Baskar Ramamurthy for providing laboratory space to conduct the rescue experiments. The authors thank Heidrun Lieβegang for technical assistance in the activity assays with the recombinant protein, Judith Bossen for performing Southern hybridization, Christoph Gelhaus for mass spectrometry measurements and Rosa Herbst for creating Figure 1.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2018.00073/full#supplementary-material
Alibaud, L., Köhler, T., Coudray, A., Prigent-Combaret, C., Bergeret, E., Perrin, J., et al. (2008). Pseudomonas aeruginosa virulence genes identified in a Dictyostelium host model. Cell. Microbiol. 10, 729–740. doi: 10.1111/j.1462-5822.2007.01080.x
Andersson, M., Gunne, H., Agerberth, B., Boman, A., Bergman, T., Sillard, R., et al. (1995). NK-lysin, a novel effector peptide of cytotoxic T and NK cells. Structure and cDNA cloning of porcine form, induction by interleukin 2, antibacterial and antitumour activity. EMBO J. 14, 1615–1625.
Balestrino, D., Haagensen, J. A. J., Rich, C., and Forestier, C. (2005). Characterization of type 2 quorum sensing in Klebsiella pneumoniae and relationship with biofilm formation. J. Bacteriol. 187, 2870–2880. doi: 10.1128/JB.187.8.2870-2880.2005
Benghezal, M., Adam, E., Lucas, A., Burn, C., Orchard, M. G., Deuschel, C., et al. (2007). Inhibitors of bacterial virulence identified in a surrogate host model. Cell. Microbiol. 9, 1336–1342. doi: 10.1111/j.1462-5822.2006.00877.x
Benghezal, M., Fauvarque, M. O., Tournebize, R., Froquet, R., Marchetti, A., Bergeret, E., et al. (2006). Specific host genes required for the killing of Klebsiella bacteria by phagocytes. Cell. Microbiol. 8, 139–148. doi: 10.1111/j.1462-5822.2005.00607.x
Bozzaro, S., Bucci, C., and Steinert, M. (2008). Phagocytosis and host-pathogen interactions in Dictyostelium with a look at macrophages. Int. Rev. Cell Mol. Biol. 271, 253–300. doi: 10.1016/S1937-6448(08)01206-9
Brinkmann, V., Reichard, U., Goosmann, C., Fauler, B., Uhlemann, Y., Weiss, D. S., et al. (2004). Neutrophil extracellular traps kill bacteria. Science 303, 1532–1535. doi: 10.1126/science.1092385
Brock, D. A., and Gomer, R. H. (1999). A cell-counting factor regulating structure size in Dictyostelium. Genes Dev. 13, 1960–1969.
Bruhn, H. (2005). A short guided tour through functional and structural features of saposin-like proteins. Biochem. J. 389, 249–257. doi: 10.1042/BJ20050051
Charette, S. J., and Cosson, P. (2004). Preparation of genomic DNA from Dictyostelium discoideum for PCR analysis. BioTechniques 36, 574–575.
Chen, G., Zhuchenko, O., and Kuspa, A. (2007). Immune-like phagocyte activity in the social amoeba. Science 317, 678–681. doi: 10.1126/science.1143991
Cosson, P., and Soldati, T. (2008). Eat, kill or die: when amoeba meets bacteria. Curr. Opin. Microbiol. 11, 271–276. doi: 10.1016/j.mib.2008.05.005
Cosson, P., Zulianello, L., Join-lambert, O., Faurisson, F., Gebbie, L., Benghezal, M., et al. (2002). Pseudomonas aeruginosa virulence analyzed in a Dictyostelium discoideum host system. J. Bacteriol. 184, 3027–3033. doi: 10.1128/JB.184.11.3027-3033.2002
Dubin, M., and Nellen, W. (2010). A versatile set of tagged expression vectors to monitor protein localisation and function in Dictyostelium. Gene 465, 1–8. doi: 10.1016/j.gene.2010.06.010
Dunn, J. D., Bosmani, C., Barisch, C., Raykov, L., Lefrançois, L. H., Cardenal-Muñoz, E., et al. (2018). Eat prey, live: Dictyostelium discoideum as a model for cell-autonomous defenses. Front. Immunol. 8:1906. doi: 10.3389/fimmu.2017.01906
Eichinger, L., and Noegel, A. A. (2005). Comparative genomics of Dictyostelium discoideum and Entamoeba histolytica. Curr. Opin. Microbiol. 8, 606–611. doi: 10.1016/j.mib.2005.08.009
Faix, J., Kreppel, L., Shaulsky, G., Schleicher, M., and Kimmel, A. R. (2004). A rapid and efficient method to generate multiple gene disruptions in Dictyostelium discoideum using a single selectable marker and Cre-loxP system. Nucleic Acids Res. 32:e143. doi: 10.1093/nar/gnh136
Farbrother, P., Wagner, C., Na, J., Tunggal, B., Morio, T., Urushihara, H., et al. (2006). Dictyostelium transcriptional host cell response upon infection with Legionella. Cell. Microbiol. 8, 438–456. doi: 10.1111/j.1462-5822.2005.00633.x
Favre-Bonté, S., Licht, T. R., Forestier, C., and Krogfelt, K. A. (1999). Klebsiella pneumoniae capsule expression is necessary for colonization of large intestines of streptomycin-treated mice. Infect. Immun. 67, 6152–6156.
Froquet, R., Lelong, E., Marchetti, A., and Cosson, P. (2009). Dictyostelium discoideum a model host to measure bacterial virulence. Nat. Protoc. 4, 25–30. doi: 10.1038/nprot.2008.212
Gao, T., Ehrenman, K., Tang, L., Leippe, M., Brock, D. A., and Gomer, R. H. (2002). Cells respond to and bind countin, a component of a multisubunit cell number counting factor. J. Biol. Chem. 277, 32596–32605. doi: 10.1074/jbc.M203075200
Hagedorn, M., and Soldati, T. (2007). Flotillin and RacH modulate the intracellular immunity of Dictyostelium to Mycobacterium marinum infection. Cell Microbiol. 9, 2716–2733. doi: 10.1111/j.1462-5822.2007.00993.x
Hagedorn, M., Rohde, K. H., Russell, D. G., and Soldati, T. (2009). Infection by tubercular mycobacteria is spread by nonlytic ejection from their amoeba hosts. Science 323, 1729–1733. doi: 10.1126/science.1169381
Hecht, O., Van Nuland, N. A., Schleinkofer, K., Dingley, A. J., Bruhn, H., Leippe, M., et al. (2004). Solution structure of the pore-forming protein of Entamoeba histolytica. J. Biol. Chem. 279, 17834–17841. doi: 10.1074/jbc.M312978200
Herbst, R., Marciano-Cabral, F., and Leippe, M. (2004). Antimicrobial and pore-forming peptides of free-living and potentially highly pathogenic Naegleria fowleri are released from the same precursor molecule. J. Biol. Chem. 279, 25955–25958. doi: 10.1074/jbc.M401965200
Herbst, R., Ott, C., Jacobs, T., Marti, T., Marciano-Cabral, F., and Leippe, M. (2002). Pore-forming polypeptides of the pathogenic protozoon Naegleria fowleri. J. Biol. Chem. 277, 22353–22360. doi: 10.1074/jbc.M201475200
Koller, B., Schramm, C., Siebert, S., Triebel, J., Deland, E., Pfefferkorn, A. M., et al. (2016). Dictyostelium discoideum as a novel host system to study the interaction between phagocytes and yeasts. Front. Microbiol. 7:1665. doi: 10.3389/fmicb.2016.01665
Kolter, T., Winau, F., Schaible, U. E., Leippe, M., and Sandhoff, K. (2005). Lipid-binding proteins in membrane digestion, antigen presentation, and antimicrobial defense. J. Biol. Chem. 280, 41125–41128. doi: 10.1074/jbc.R500015200
Lee, A. K., and Falkow, S. (1998). Constitutive and inducible green fluorescent protein expression in Bartonella henselae. Infect. Immun. 66, 3964–3967.
Leippe, M. (1999). Antimicrobial and cytolytic polypeptides of amoeboid protozoa-effector molecules of primitive phagocytes. Dev. Comp. Immunol. 23, 267–279. doi: 10.1016/S0145-305X(99)00010-5
Leippe, M. (2014). Pore-forming toxins from pathogenic amoebae. Appl. Microbiol. Biotechnol. 98, 4347–4353. doi: 10.1007/s00253-014-5673-z
Leippe, M., Ebel, S., Schoenberger, O. L., Horstmann, R. D., and Müller-Eberhard, H. J. (1991). Pore-forming peptide of pathogenic Entamoeba histolytica. Proc. Natl. Acad. Sci. U.S.A. 88, 7659–7663. doi: 10.1073/pnas.88.17.7659
Liepinsh, E., Andersson, M., Ruysschaert, J. M., and Otting, G. (1997). Saposin fold revealed by the NMR structure of NK-lysin. Nat. Struct. Biol. 4, 793–795. doi: 10.1038/nsb1097-793
Michalek, M., and Leippe, M. (2015). Mechanistic insights into the lipid interaction of an ancient saposin-like protein. Biochemistry 54, 1778–1786. doi: 10.1021/acs.biochem.5b00094
Müller, I., Subert, N., Otto, H., Herbst, R., Rühling, H., Maniak, M., et al. (2005). A Dictyostelium mutant with reduced lysozyme levels compensates by increased phagocytic activity. J. Biol. Chem. 280, 10435–10443. doi: 10.1074/jbc.M411445200
Munford, R., Lu, M., and Varley, A. (2009). Chapter 2: Kill the bacteria…and also their messengers? Adv. Immunol. 103, 29–48. doi: 10.1016/S0065-2776(09)03002-8
Munford, R. S., Sheppard, P. O., and O'Hara, P. J. (1995). Saposin-like proteins (SAPLIP) carry out diverse functions on a common backbone structure. J. Lipid Res. 36, 1653–1663.
Nassif, X., Fournier, J. M., Arondel, J., and Sansonetti, P. J. (1989). Mucoid phenotype of Klebsiella pneumoniae is a plasmid-encoded virulence factor. Infect. Immun. 57, 546–552.
O'Brien, J. S., and Kishimoto, Y. (1991). Saposin proteins: structure, function, and role in human lysosomal storage disorders. FASEB J. 5, 301–308. doi: 10.1096/fasebj.5.3.2001789
Parkinson, K., Bolourani, P., Traynor, D., Aldren, N. L., Kay, R. R., Weeks, G., et al. (2009). Regulation of Rap1 activity is required for differential adhesion, cell-type patterning and morphogenesis in Dictyostelium. J. Cell Sci. 122, 335–344. doi: 10.1242/jcs.036822
Peña, S. V., Hanson, D. A., Carr, B. A., Goralski, T. J., and Krensky, A. M. (1997). Processing, subcellular localization, and function 519 (granulysin), a human late T cell activation molecule with homology to small, lytic, granule proteins. J. Immunol. 158, 2680–2688.
Pukatzki, S., Kessin, R. H., and Mekalanos, J. J. (2002). The human pathogen Pseudomonas aeruginosa utilizes conserved virulence pathways to infect the social amoeba Dictyostelium discoideum. Proc. Natl. Acad. Sci. U.S.A. 99, 3159–3164. doi: 10.1073/pnas.052704399
Robb, C. T., Dyrynda, E. A., Gray, R. D., Rossi, A. G., and Smith, V. J. (2014). Invertebrate extracellular phagocyte traps show that chromatin is an ancient defence weapon. Nat. Commun. 5:4627. doi: 10.1038/ncomms5627
Roeder, T., Stanisak, M., Gelhaus, C., Bruchhaus, I., Grötzinger, J., and Leippe, M. (2010). Caenopores are antimicrobial peptides in the nematode Caenorhabditis elegans instrumental in nutrition and immunity. Dev. Comp. Immunol. 34, 203–209. doi: 10.1016/j.dci.2009.09.010
Schägger, H., and von Jagow, G. (1987). Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem. 166, 368–379 doi: 10.1016/0003-2697(87)90587-2
Shevchuk, O., Pägelow, D., Rasch, J., Döhrmann, S., Günther, G., Hoppe, J., et al. (2014). Polyketide synthase (PKS) reduces fusion of Legionella pneumophila-containing vacuoles with lysosomes and contributes to bacterial competitiveness during infection. Int. J. Med. Microbiol. 304, 1169–1181. doi: 10.1016/j.ijmm.2014.08.010
Sillo, A., Bloomfield, G., Balest, A., Balbo, A., Pergolizzi, B., Peracino, B., et al. (2008). Genome-wide transcriptional changes induced by phagocytosis or growth on bacteria in Dictyostelium. BMC Genomics 9:291. doi: 10.1186/1471-2164-9-291
Zhang, X., and Soldati, T. (2016). Of amoebae and men: extracellular DNA traps as an ancient cell-intrinsic defense mechanism. Front. Immunol. 7:269. doi: 10.3389/fimmu.2016.00269
Keywords: amoebapore, antimicrobial peptides, Dictyostelium discoideum, host-pathogen interactions, saposin-like proteins, slugs, Sentinel cells, innate immunity
Citation: Dhakshinamoorthy R, Bitzhenner M, Cosson P, Soldati T and Leippe M (2018) The Saposin-Like Protein AplD Displays Pore-Forming Activity and Participates in Defense Against Bacterial Infection During a Multicellular Stage of Dictyostelium discoideum. Front. Cell. Infect. Microbiol. 8:73. doi: 10.3389/fcimb.2018.00073
Received: 22 November 2017; Accepted: 27 February 2018;
Published: 15 March 2018.
Edited by:
Patricia Ann Champion, University of Notre Dame, United StatesReviewed by:
Falk Hillmann, Leibniz-Institut für Naturstoff-Forschung und Infektionsbiologie, Hans Knöll Institut, GermanyCopyright © 2018 Dhakshinamoorthy, Bitzhenner, Cosson, Soldati and Leippe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matthias Leippe, bWxlaXBwZUB6b29sb2dpZS51bmkta2llbC5kZQ==
†Present Address: Ranjani Dhakshinamoorthy, Department of Biotechnology, Bhupat and Jyoti Mehta School of Biosciences, Indian Institute of Technology Madras, Chennai, India
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.