
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell. Infect. Microbiol. , 30 November 2012
Sec. Molecular Bacterial Pathogenesis
Volume 2 - 2012 | https://doi.org/10.3389/fcimb.2012.00149
This article is part of the Research Topic The pathogenic Yersiniae – advances in the understanding of physiology and virulence View all 17 articles
 Naomi H. Philip1,2
Naomi H. Philip1,2 Igor E. Brodsky1,2,3*
Igor E. Brodsky1,2,3*Cell death plays a central role in host-pathogen interactions, as it can eliminate the pathogen's replicative niche and provide pro-inflammatory signals necessary for an effective immune response; conversely, cell death can allow pathogens to eliminate immune cells and evade anti-microbial effector mechanisms. In response to developmental signals or cell-intrinsic stresses, the executioner caspases-3 and -7 mediate apoptotic cell death, which is generally viewed as immunologically silent or immunosuppressive. A proinflammatory form of cell death that requires caspase-1, termed pyroptosis, is activated in response to microbial products within the host cytosol or disruption of cellular membranes by microbial pathogens. Infection by the bacterial pathogen Yersinia has features of both apoptosis and pyroptosis. Cell death and caspase-1 processing in Yersinia-infected cells occur in response to inhibition of NF-κB and MAPK signaling by the Yersinia virulence factor YopJ. However, the molecular basis of YopJ-induced cell death, and the role of different death pathways in anti-Yersinia immune responses remain enigmatic. Here, we discuss the role that cell death may play in inducing specific pro-inflammatory signals that shape innate and adaptive immune responses against Yersinia infection.
Cell death plays a key role in maintaining tissue homeostasis by eliminating stressed, damaged, and infected cells. Cell death is an evolutionarily conserved immune response to microbial infection, as it prevents pathogen replication and can provide pro-inflammatory signals necessary for an effective immune response. Distinct cell death pathways that result in distinct downstream outcomes are induced under different circumstances (Kono and Rock, 2008; Green et al., 2009; Zitvogel et al., 2010). Apoptosis is traditionally viewed as an immunomodulatory form of cell death characterized by cell shrinkage, while necrosis and pyroptosis are pro-inflammatory forms of death associated with rapid loss of membrane integrity and release of intracellular contents. How these distinct cell death pathways contribute to antibacterial responses remains an important unanswered question. Recent studies indicate that caspase-1-dependent pyroptosis can promote antibacterial responses against intracellular pathogens independent of production of the caspase-1 dependent cytokines IL-1β and IL-18 (Miao et al., 2010). Similarly, RIP3-dependent necrosis, a recently described form of programmed necrosis (Cho et al., 2009; He et al., 2009; Zhang et al., 2009; Oberst et al., 2011), promotes control of Vaccinia and CMV viral infections (Upton et al., 2010). Both pyroptosis and programmed necrosis can occur in the context of pathological conditions that are not directly associated with microbial infections (Martinon et al., 2006; Welz et al., 2011; Inoue et al., 2012). However, how different cell death pathways contribute to cytokine production, and orchestrate activation of innate and adaptive cells during bacterial infections remains a major unresolved question for understanding of anti-Yersinia immunity.
The three pathogenic Yersinia spp., Y. pestis, Y. pseudotuberculosis, and Y. enterocolitica, share a virulence plasmid encoding a conserved Type Three Secretion System (T3SS) and virulence factors, known as Yersinia outer proteins (Yops) (Viboud and Bliska, 2005). T3SS-mediated injection of Yops into infected cells enables Yersinia to modulate host signaling pathways and suppress innate and adaptive immunity (Cornelis, 2006). YopJ of Y. pestis and Y. pseudotuberculosis, termed YopP in Y. enterocolitica, blocks NF-κB and MAPK signaling, thereby inhibiting cytokine production and triggering death of Yersinia infected cells (Mills et al., 1997; Monack et al., 1997; Ruckdeschel et al., 1998). Among the sequenced strains of pathogenic Yersiniae, YopJ and YopP share 95–98% identity across the full length of the protein sequence, but key polymorphisms have been identified that impact both enzymatic activity and translocation of the protein, which affect the outcome of Yersinia infection (Ruckdeschel et al., 2001b; Zauberman et al., 2006; Brodsky and Medzhitov, 2008; Zheng et al., 2011).
Interestingly, Yersinia-infected cells exhibit features of apoptosis, pyroptosis, or necrosis, depending on the state of the cells and the cell type involved (Monack et al., 1997; Ruckdeschel et al., 1997, 1998, 2001a; Bergsbaken and Cookson, 2007; Zheng et al., 2012). However, whether YopJ-induced cell death promotes host defense or bacterial virulence during in vivo infection remains unclear. A number of studies have revealed key players in cell death pathways during Yersinia infection, providing some insight into mechanisms of Yersinia-induced cell death, but key questions about the nature and role of Yersinia death in vivo remain. Yersinia is thought to primarily replicate as an extracellular pathogen that evades phagocytosis by neutrophils and monocytic cells in lymphoid tissues. Cell death has therefore been viewed as a strategy for Yersinia to eliminate host phagocytes (Monack et al., 1998). However, several studies suggest that host cell death during Yersinia infection may promote anti-Yersinia immunity, although the precise mechanisms are not entirely clear (Brodsky and Medzhitov, 2008; Bergman et al., 2009; Zauberman et al., 2009). An alternative possibility is that the Yersiniae are capable of intracellular replication, suggesting the existence of an intracellular stage during the Yersinia lifecycle in vivo (Grabenstein et al., 2004).
Early studies observed that macrophages and dendritic cells infected by Yersinia exhibit characteristics of apoptotic cells, specifically membrane blebbing, nuclear condensation, DNA fragmentation, and formation of large cytoplasmic vacuoles (Monack et al., 1997; Ruckdeschel et al., 1997). Apoptosis has been viewed as immunologically silent, but growing evidence suggests that during infection, apoptosis may promote inflammatory responses (Green et al., 2009; Torchinsky et al., 2009). Furthermore, apoptotic cells can be phagocytosed, and their associated microbial antigens used to prime CD8+ T cell responses (Heath and Carbone, 2001). Therefore, while cell death during Yersinia infection is thought to be apoptotic, it may not be immunologically silent. Below, we discuss the nature of Yersinia-induced cell death and its contribution to bacterial virulence or host defense.
The cysteine protease YopJ, called YopP in Y. enterocolitica, induces cell death during Yersinia infection (Mills et al., 1997; Monack et al., 1997, 1998). YopJ is a potent inhibitor of MAPK and NF-κB signaling, and blocks proinflammatory cytokine production by infected cells (Ruckdeschel et al., 1998, 2001a; Palmer et al., 1999; Orth et al., 2000) (Figure 1). YopJ has been reported to function as an ubiquitin-like protein protease (Orth et al., 2000), and as a deubiquitinase (Zhou et al., 2005; Sweet et al., 2007). YopJ is also reported to be an acyl transferase that acetylates serine residues in the activation loop of MKK family proteins and prevents their activation (Mittal et al., 2006; Mukherjee et al., 2006). A recent study has also indicated that the sensitivity of NF-κB signaling pathways to YopJ-mediated inhibition occurs at the level of TAK1 and is evolutionarily conserved from Drosophila to mammalian cells (Paquette et al., 2012).
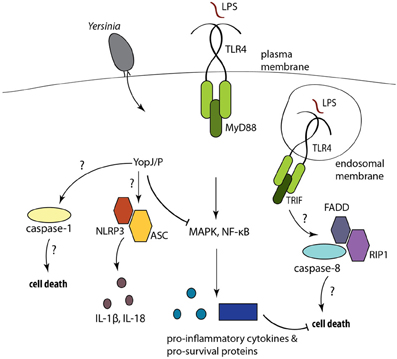
Figure 1. Yops are injected into phagocytes during Yersinia infection. YopJ/P inhibits NF-κB and MAPK signaling, preventing the expression of pro-inflammatory cytokines and pro-survival molecules, resulting in cell death. YopJ also activates caspase-1 processing and IL-1β and IL-18 release. The mechanisms of cell death and inflammasome activation are not well-understood.
Macrophages stimulated with LPS in the presence of inhibitors of protein synthesis or components of NF-κB signaling also undergo cell death (Ruckdeschel et al., 2004; Zhang et al., 2005). Consistent with this, Tlr4−/− macrophages are resistant to YopJ-dependent apoptosis, as are cells deficient in the TLR3/4 adaptor TRIF, but not MyD88 (Haase et al., 2003; Zhang and Bliska, 2003; Ruckdeschel et al., 2004). Moreover, infection of dendritic cells with Yersinia leads to the formation of a FADD/caspase-8/RIP1 complex and caspase-8 activation (Figure 1) (Grobner et al., 2007). Cytochrome-c release and caspase-9 cleavage were observed downstream of Yersinia-induced cleavage of the pro-apoptotic Bid protein (Denecker et al., 2001). Additionally, treatment with broad-spectrum caspase inhibitors reduced the number of TUNEL+ cells during Yersinia infection (Monack et al., 1998; Denecker et al., 2001). Collectively, these data indicate that Yersinia activates an extrinsic pathway of apoptosis. Interestingly, Yersinia infection of dendritic cells exposed to a pan-caspase inhibitor still induced a cell death that is presumably caspase-independent and exhibited morphological features of necrosis (Grobner et al., 2006). Recent studies have described a caspase-independent pathway of programmed necrosis linked to TRIF that involves signaling through the RIP1 and RIP3 kinases (Vandenabeele et al., 2010; Feoktistova et al., 2011; Tenev et al., 2011), but how this pathway functions during Yersinia infection and the link between programmed necrosis and other forms of Yersinia-induced death is not known.
Pathogens express pore-forming toxins and virulence proteins that disrupt membrane integrity and modulate signaling networks (Vance et al., 2009). Consequently, mammalian hosts have evolved mechanisms, including the activation of a multiprotein complex called the inflammasome, that detect these virulence activities (Schroder and Tschopp, 2010). Inflammasomes are activated by Nod-like Receptors (NLRs) that contain a leucine-rich-repeat (LRR) sensor domain, a nucleotide-binding oligomerization domain (NOD) and a pyrin- or CARD-containing signaling domain (Davis et al., 2011). Inflammasome activation is thought to require two signals, the first one involving sensing of conserved PAMPs that induce expression of certain inflammasome components and cytokines, and a second signal involving disruption of cellular membranes or signaling pathways (Mariathasan and Monack, 2007). Inflammasomes form a platform for the autoprocessing and activation of the cysteine protease caspase-1, resulting in caspase-1-dependent secretion of IL-1α, IL-1β, and IL-18, and caspase-1-dependent cell death termed pyroptosis (Mariathasan et al., 2004; Martinon et al., 2006; Sutterwala et al., 2006; Bergsbaken et al., 2009; Hornung et al., 2009). Yersinia expresses a conserved T3SS that activates the NLRP3 and NLRC4 inflammasomes, triggering pyroptosis (Brodsky et al., 2010). However, the virulence factor YopK prevents this inflammasome activation, and promotes bacterial replication and dissemination in vivo (Brodsky et al., 2010). In the presence of YopJ, YopK-sufficient bacteria still induce cell death. Thus, a key question is how these two seemingly contradictory outcomes are controlled during Yersinia infection. Yersinia expressing YopK but lacking YopJ, do not induce T3SS-dependent inflammasome activation or cell death. Thus, YopK likely limits inflammasome activation under conditions where YopJ expression or translocation is reduced, as may happen during infection of systemic sites (discussed further below). Nevertheless, macrophages primed by inflammatory stimuli still undergo pyroptosis in response to YopJ-deficient Yersinia (Bergsbaken and Cookson, 2007), which is greatly enhanced in the additional absence of YopK (Brodsky et al., 2010). As LPS priming upregulates NLRP3 inflammasome components (Bauernfeind et al., 2009), the threshold for NLRP3 inflammasome activation could be lowered, even in the presence of YopK.
Interestingly, YopJ-dependent apoptosis is also associated with caspase-1 activation (Lilo et al., 2008; Brodsky et al., 2010; Zheng et al., 2011), and the extent of YopJ-mediated NF-κB inhibition correlates with the degree of caspase-1 activation (Zheng et al., 2011), consistent with the finding that deletion of IKKβ in macrophages induces spontaneous inflammasome activation (Greten et al., 2007). Although NLRP3 and the adaptor ASC are required for YopJ-dependent secretion of IL-1β and IL-18 (Zheng et al., 2011), the mechanism by which YopJ activates caspase-1 is unclear, as caspase-1 processing and YopJ-dependent cell death still occur in cells lacking ASC, NLRC4, or NLRP3 (Brodsky et al., 2010). Distinct inflammasome complexes with different functions have been identified, and could potentially account for these observations (Figure 1). Caspase-1 could be recruited to an NLRP3/ASC complex that regulates IL-1β and IL-18 production, and to a separate complex that activates cell death. A complex containing catalytically active caspase-1, but not ASC triggers cell death but not cytokine secretion, while a distinct ASC-containing focus mediates caspase-1 processing and cytokine secretion, during Salmonella infection (Broz et al., 2010). NLRP12 was also recently found to induce inflammasome activation in response to Y. pestis infection, and both NLRP3 and NLRP12 contributed to host defense against Yersinia infection, presumably via induction of caspase-1-dependent IL-1β and IL-18 (Vladimer et al., 2012). YopJ may activate this NLRP12 inflammasome, although this remains to be demonstrated. Finally, a non-canonical inflammasome pathway involving caspase-11, TRIF, and type I IFN signaling has been described that responds to Gram-negative intracellular bacteria independently of T3SS activity (Kayagaki et al., 2011; Sander et al., 2011; Broz et al., 2012; Rathinam et al., 2012). Whether this pathway contributes to anti-Yersinia host defense remains unknown.
A number of studies indicate that YopJ/P promotes Yersinia virulence. Oral infection with Y. pseudotuberculosis and Y. enterocolitica demonstrate that YopJ/P contributes to systemic disease and barrier dysfunction (Monack et al., 1998; Jung et al., 2012; Meinzer et al., 2012). YopJ is dispensable for colonization of the Peyer's patches (PPs) and mesenteric lymph nodes (mLNs), especially at higher infectious doses; however, YopJ-deficient Yersinia had significantly reduced levels of spleen colonization (Monack et al., 1998). Furthermore, spleens and mLNs from mice infected with YopJ-sufficient bacteria had a higher percentage of Mac1+ TUNEL+ and total TUNEL+ cells compared to YopJ-deficient bacteria, consistent with the role of YopJ in apoptosis in vivo. Furthermore, in competitive index experiments, YopJ-deficient Yersinia showed colonization defects in PPs, mLNs, and spleen. YopJ-deficient Yersinia were not defective for splenic replication following intraperitoneal infection, indicating that YopJ primarily regulates dissemination from mucosal tissues, rather replication at systemic sites (Monack et al., 1998) (Figure 2). Consistently, YopJ-deficient Y. pestis are still able to cause systemic infection in a rat model of bubonic plague, despite a defect in induction of apoptosis and cytokine inhibition (Lemaitre et al., 2006). These initial findings thus implied that apoptosis may be utilized by Yersinia to eliminate immune cells and dampen anti-Yersinia immunity during infection of peripheral or mucosal tissues.
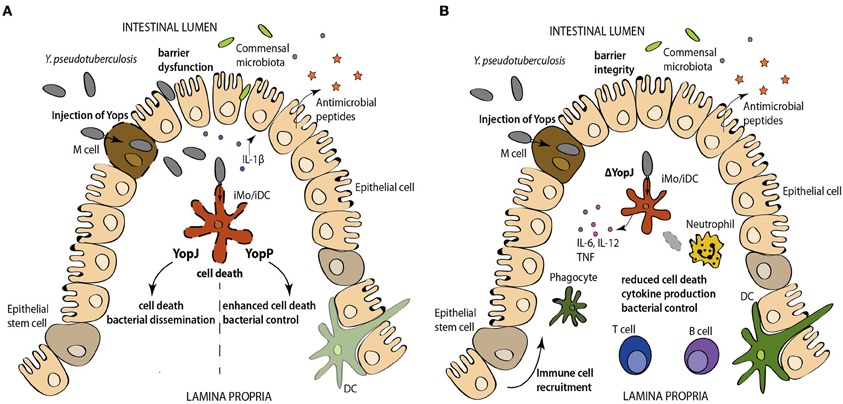
Figure 2. (A) Injection of YopJ/P activates caspase-1 and the release of the caspase-1-dependent cytokines. However, the hypercytotoxic YopP-expressing Y. pseudotuberculosis induces enhanced levels of cell death and promotes bacterial control, relative to infection with YopJ-expressing Y. pseudotuberculosis. (B) Conversely infection with YopJ-deficient Y. pseudotuberculosis results in robust cytokine production, intact barrier function and control of bacterial spread. Abbreviations: DC, dendritic, iMo/iDC, intestinal macrophage/intestinal DC.
Paradoxically, ectopic expression of a hypercytotoxic YopP from Y. enterocolitica in Y. pseudotuberculosis results in its attenuation in oral mouse infection (Brodsky and Medzhitov, 2008) (Figure 2). While both Y. pseudotuberculosis and Y. enterocolitica cause cell death in cultured macrophages and infected tissues, infection with YopJ- vs. YopP-expressing Y. pseudotuberculosis showed a significant increase in TUNEL+ CD11b+, CD11c+ and B220+ cells in mLNs in mice infected with the YopP-expressing strain (Brodsky and Medzhitov, 2008). Similarly, Y. pestis strains expressing YopP had higher cytotoxic potency than strains expressing YopJ, both in vitro and in tissues of infected mice; furthermore, expression of YopP in Y. pestis also resulted in lower virulence following subcutaneous, but not intranasal or intravenous routes of infection (Zauberman et al., 2009). Interestingly, subcutaneous administration of Y. pestis expressing YopP protected against infection with virulent Y. pestis, regardless of the route of challenge. These observations suggest that YopJ contributes to dissemination of Yersinia from barrier surfaces, but may be less important once bacteria have spread to systemic sites. Whether YopJ or additional immunosuppressive virulence mechanisms play a role in dampening the early inflammatory response to Yersinia infection in pneumonic plague (Lathem et al., 2007) also remains to be determined.
Consistent with observations that YopJ promotes systemic dissemination following oral infection, YopJ contributes to gut barrier disruption (Jung et al., 2012; Meinzer et al., 2012). Specifically, YopJ can induce TLR2-dependent IL-1β secretion in PPs, which was associated with increased barrier permeability, suggesting that TLR2 signaling mediates YopJ-dependent gut disruption (Jung et al., 2012). However, the actual bacterial loads in these mice were not measured. Conversely, TLR2-deficient mice have been reported to be more susceptible to oral infection by Y. pseudotuberculosis, due to a loss of TLR2-dependent Reg3β expression in the gut epithelium (Dessein et al., 2009). Thus, the precise role of TLR2 in Yersinia infection remains to be further dissected. Notably, IL-1α is associated with pathological intestinal inflammation and increased dissemination of Y. enterocolitica (Dube et al., 2001), but the role of YopP or TLR2 in this context has not been examined.
In contrast to cell death induced by the activity of a bacterial virulence factor, CD8+ cytotoxic T cells also induce death of Yersinia-infected cells, and are important for control of Yersinia infection, as demonstrated by the more severe disease in infected β2m−/−, anti-CD8α-treated, or perforin-deficient mice (Bergman et al., 2009). CD8+ T cell-mediated killing of bacteria-associated cells targeted them for phagocytosis by uninfected macrophages, and could bypass the anti-phagocytic activity of Yersinia Yops (Bergman et al., 2009). Notably, CD8+ T cells were not responsible for resistance to YopP-expressing Y. pestis, (Zauberman et al., 2009). The more cytotoxic YopP may bypass the requirement for CD8+ T cell-mediated killing due to elevated cytotoxicity induced by the bacteria themselves. These studies collectively suggest that regulation of cytotoxicity during Yersinia infection impacts virulence, and that a balance between the cytokine-blocking and death-inducing functions of YopJ is required for optimal virulence. Specifically, absence of YopJ results in failure of Yersinia to suppress cytokine production or induce cell death and causes a defect in dissemination. However, Y. pseudotuberculosis expressing YopP, which enables stronger inhibition of cytokine production and elevated levels of cell death, are also significantly attenuated in vivo (Figure 2). Thus, while the relative contributions of bacteria-induced and T cell-induced cell death during Yersinia infection in vivo are not yet defined, activation of cell death in vivo either in response to YopJ activity, or as a consequence of T-cell-mediated cytotoxicity likely promotes immune responses against Yersinia.
Apoptotic cell death is generally viewed as non-inflammatory or immunosuppressive, and YopJ-dependent cell death has been characterized as apoptosis (Mills et al., 1997; Monack et al., 1997, 1998; Ruckdeschel et al., 1997, 1998). However, apoptotic cell death can also promote immune responses. For example, anthracyclin treatment of tumor cells causes apoptosis that leads to exposure of calreticulin on the surface, which acts as a signal to induce phagocytosis and promote anti-tumor T cell responses in vivo (Kepp et al., 2009). Similarly, the dendritic cell C-type lectin receptor, DNGR-1, can promote cross-presentation of apoptotic cell antigens by CD8α+ DCs to CD8+ T cells in both non-infectious and infectious settings (Sancho et al., 2009; Ahrens et al., 2012). Dendritic cells that phagocytose bacterially-infected apoptotic cells produce both the immunoregulatory cytokine TGFβ, and the inflammatory cytokine IL-6, which together promote the differentiation of naïve CD4+ T cells into TH17 cells (Torchinsky et al., 2009). These cells play a critical role in anti-bacterial immunity and pathological inflammatory responses at mucosal barrier surfaces (Ye et al., 2001; O'Connor et al., 2009; Sonnenberg et al., 2010). Investigating whether these mechanisms promote immune responses against Yersinia infection may provide new insights into both anti-Yersinia immunity and potential bacterial evasion strategies (Lin et al., 2011; Smiley, 2008).
Understanding the control of programmed cell death remains essential for understanding anti-bacterial immunity. Appropriately distinguishing pathogens and non-pathogens is critical both for host defense and the maintenance of tissue homeostasis. The ability to detect conserved bacterial virulence activities, such as the disruption of actin cytoskeleton or inhibition of core signaling pathways, provides a mechanism for rapidly sensing the presence of pathogens. Induction of cell death in response to microbial virulence activities is an evolutionarily conserved response from plants to higher eukaryotes that limits the spread of infection. In higher organisms, distinct forms of programmed cell death, including apoptosis, pyroptosis, and necrosis, can have differential effects on downstream responses. Various cell types may have differing thresholds for activation of these distinct death pathways due to differential expression of pro-survival, pro-apoptotic, and immune sensor proteins. Thus, understanding how cells die and how cell death influences both the local microenvironment and ensuing systemic response may provide key insight into new approaches to modulate both antimicrobial immune responses and immunopathologies that result from dysregulation of these death pathways.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Sunny Shin for scientific discussion and editorial suggestions. Work in the Brodsky lab is supported by grants from the University Research Foundation, McCabe Fund, NIH/NIDDK Center for Molecular Studies in Digestive and Liver Diseases (P30-DK050306), and The Mari Lowe Center for Comparative Oncology.
Ahrens, S., Zelenay, S., Sancho, D., Hanc, P., Kjaer, S., Feest, C., et al. (2012). F-actin is an evolutionarily conserved damage-associated molecular pattern recognized by DNGR-1, a receptor for dead cells. Immunity 36, 635–645.
Bauernfeind, F. G., Horvath, G., Stutz, A., Alnemri, E. S., MacDonald, K., Speert, D., et al. (2009). Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 183, 787–791.
Bergman, M. A., Loomis, W. P., Mecsas, J., Starnbach, M. N., and Isberg, R. R. (2009). CD8(+) T cells restrict Yersinia pseudotuberculosis infection: bypass of anti-phagocytosis by targeting antigen-presenting cells. PLoS Pathog. 5:e1000573. doi: 10.1371/journal.ppat.1000573
Bergsbaken, T., and Cookson, B. T. (2007). Macrophage activation redirects Yersinia-infected host cell death from apoptosis to caspase-1-dependent pyroptosis. PLoS Pathog. 3:e161. doi: 10.1371/journal.ppat.0030161
Bergsbaken, T., Fink, S. L., and Cookson, B. T. (2009). Pyroptosis: host cell death and inflammation. Nat. Rev. Microbiol. 7, 99–109.
Brodsky, I. E., and Medzhitov, R. (2008). Reduced secretion of YopJ by Yersinia limits in vivo cell death but enhances bacterial virulence. PLoS Pathog. 4:e1000067. doi: 10.1371/journal.ppat.1000067
Brodsky, I. E., Palm, N. W., Sadanand, S., Ryndak, M. B., Sutterwala, F. S., Flavell, R. A., et al. (2010). A Yersinia effector protein promotes virulence by preventing inflammasome recognition of the type III secretion system. Cell Host Microbe 7, 376–387.
Broz, P., Ruby, T., Belhocine, K., Bouley, D. M., Kayagaki, N., Dixit, V. M., et al. (2012). Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature 490, 288–291.
Broz, P., von Moltke, J., Jones, J. W., Vance, R. E., and Monack, D. M. (2010). Differential requirement for Caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe 8, 471–483.
Cho, Y. S., Challa, S., Moquin, D., Genga, R., Ray, T. D., Guildford, M., et al. (2009). Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137, 1112–1123.
Davis, B. K., Wen, H., and Ting, J. P. (2011). The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu. Rev. Immunol. 29, 707–735.
Denecker, G., Declercq, W., Geuijen, C. A., Boland, A., Benabdillah, R., van Gurp, M., et al. (2001). Yersinia enterocolitica YopP-induced apoptosis of macrophages involves the apoptotic signaling cascade upstream of bid. J. Biol. Chem. 276, 19706–19714.
Dessein, R., Gironella, M., Vignal, C., Peyrin-Biroulet, L., Sokol, H., Secher, T., et al. (2009). Toll-like receptor 2 is critical for induction of Reg3 beta expression and intestinal clearance of Yersinia pseudotuberculosis. Gut 58, 771–776.
Dube, P. H., Revell, P. A., Chaplin, D. D., Lorenz, R. G., and Miller, V. L. (2001). A role for IL-1 alpha in inducing pathologic inflammation during bacterial infection. Proc. Natl. Acad. Sci. U.S.A. 98, 10880–10885.
Feoktistova, M., Geserick, P., Kellert, B., Dimitrova, D. P., Langlais, C., Hupe, M., et al. (2011). cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol. Cell 43, 449–463.
Grabenstein, J. P., Marceau, M., Pujol, C., Simonet, M., and Bliska, J. B. (2004). The response regulator PhoP of Yersinia pseudotuberculosis is important for replication in macrophages and for virulence. Infect. Immun. 72, 4973–4984.
Green, D. R., Ferguson, T., Zitvogel, L., and Kroemer, G. (2009). Immunogenic and tolerogenic cell death. Nat. Rev. Immunol. 9, 353–363.
Greten, F. R., Arkan, M. C., Bollrath, J., Hsu, L. C., Goode, J., Miething, C., et al. (2007). NF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell 130, 918–931.
Grobner, S., Adkins, I., Schulz, S., Richter, K., Borgmann, S., Wesselborg, S., et al. (2007). Catalytically active Yersinia outer protein P induces cleavage of RIP and caspase-8 at the level of the DISC independently of death receptors in dendritic cells. Apoptosis 12, 1813–1825.
Grobner, S., Autenrieth, S. E., Soldanova, I., Gunst, D. S., Schaller, M., Bohn, E., et al. (2006). Yersinia YopP-induced apoptotic cell death in murine dendritic cells is partially independent from action of caspases and exhibits necrosis-like features. Apoptosis 11, 1959–1968.
Haase, R., Kirschning, C. J., Sing, A., Schrottner, P., Fukase, K., Kusumoto, S., et al. (2003). A dominant role of Toll-like receptor 4 in the signaling of apoptosis in bacteria-faced macrophages. J. Immunol. 171, 4294–4303.
He, S., Wang, L., Miao, L., Wang, T., Du, F., Zhao, L., et al. (2009). Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 137, 1100–1111.
Heath, W. R., and Carbone, F. R. (2001). Cross-presentation, dendritic cells, tolerance and immunity. Annu. Rev. Immunol. 19, 47–64.
Hornung, V., Ablasser, A., Charrel-Dennis, M., Bauernfeind, F., Horvath, G., Caffrey, D. R., et al. (2009). AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458, 514–518.
Inoue, M., Williams, K. L., Gunn, M. D., and Shinohara, M. L. (2012). NLRP3 inflammasome induces chemotactic immune cell migration to the CNS in experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. U.S.A. 109, 10480–10485.
Jung, C., Meinzer, U., Montcuquet, N., Thachil, E., Chateau, D., Thiebaut, R., et al. (2012). Yersinia pseudotuberculosis disrupts intestinal barrier integrity through hematopoietic TLR-2 signaling. J. Clin. Invest. 122, 2239–2251.
Kayagaki, N., Warming, S., Lamkanfi, M., Van de Walle, L., Louie, S., Dong, J., et al. (2011). Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121.
Kepp, O., Tesniere, A., Zitvogel, L., and Kroemer, G. (2009). The immunogenicity of tumor cell death. Curr. Opin. Oncol. 21, 71–76.
Kono, H., and Rock, K. L. (2008). How dying cells alert the immune system to danger. Nat. Rev. Immunol. 8, 279–289.
Lathem, W. W., Price, P. A., Miller, V. L., and Goldman, W. E. (2007). A plasminogen-activating protease specifically controls the development of primary pneumonic plague. Science 315, 509–513.
Lemaitre, N., Sebbane, F., Long, D., and Hinnebusch, B. J. (2006). Yersinia pestis YopJ suppresses tumor necrosis factor alpha induction and contributes to apoptosis of immune cells in the lymph node but is not required for virulence in a rat model of bubonic plague. Infect. Immun. 74, 5126–5131.
Lilo, S., Zheng, Y., and Bliska, J. B. (2008). Caspase-1 activation in macrophages infected with Yersinia pestis KIM requires the type III secretion system effector YopJ. Infect. Immun. 76, 3911–3923.
Lin, J. S., Kummer, L. W., Szaba, F. M., and Smiley, S. T. (2011). IL-17 contributes to cell-mediated defense against pulmonary Yersinia pestis infection. J. Immunol. 186, 1675–1684.
Mariathasan, S., and Monack, D. M. (2007). Inflammasome adaptors and sensors: intracellular regulators of infection and inflammation. Nat. Rev. Immunol. 7, 31–40.
Mariathasan, S., Newton, K., Monack, D. M., Vucic, D., French, D. M., Lee, W. P., et al. (2004). Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 430, 213–218.
Martinon, F., Petrilli, V., Mayor, A., Tardivel, A., and Tschopp, J. (2006). Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440, 237–241.
Meinzer, U., Barreau, F., Esmiol-Welterlin, S., Jung, C., Villard, C., Leger, T., et al. (2012). Yersinia pseudotuberculosis effector YopJ subverts the Nod2/RICK/TAK1 pathway and activates caspase-1 to induce intestinal barrier dysfunction. Cell Host Microbe 11, 337–351.
Miao, E. A., Leaf, I. A., Treuting, P. M., Mao, D. P., Dors, M., Sarkar, A., et al. (2010). Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat. Immunol. 11, 1136–1142.
Mills, S. D., Boland, A., Sory, M. P., van der Smissen, P., Kerbourch, C., Finlay, B. B., et al. (1997). Yersinia enterocolitica induces apoptosis in macrophages by a process requiring functional type III secretion and translocation mechanisms and involving YopP, presumably acting as an effector protein. Proc. Natl. Acad. Sci. U.S.A. 94, 12638–12643.
Mittal, R., Peak-Chew, S. Y., and McMahon, H. T. (2006). Acetylation of MEK2 and I kappa B kinase (IKK) activation loop residues by YopJ inhibits signaling. Proc. Natl. Acad. Sci. U.S.A. 103, 18574–18579.
Monack, D. M., Mecsas, J., Bouley, D., and Falkow, S. (1998). Yersinia-induced apoptosis in vivo aids in the establishment of a systemic infection of mice. J. Exp. Med. 188, 2127–2137.
Monack, D. M., Mecsas, J., Ghori, N., and Falkow, S. (1997). Yersinia signals macrophages to undergo apoptosis and YopJ is necessary for this cell death. Proc. Natl. Acad. Sci. U.S.A. 94, 10385–10390.
Mukherjee, S., Keitany, G., Li, Y., Wang, Y., Ball, H. L., Goldsmith, E. J., et al. (2006). Yersinia YopJ acetylates and inhibits kinase activation by blocking phosphorylation. Science 312, 1211–1214.
Oberst, A., Dillon, C. P., Weinlich, R., McCormick, L. L., Fitzgerald, P., Pop, C., et al. (2011). Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 471, 363–367.
O'Connor, W. Jr., Kamanaka, M., Booth, C. J., Town, T., Nakae, S., Iwakura, Y., et al. (2009). A protective function for interleukin 17A in T cell-mediated intestinal inflammation. Nat. Immunol. 10, 603–609.
Orth, K., Xu, Z., Mudgett, M. B., Bao, Z. Q., Palmer, L. E., Bliska, J. B., et al. (2000). Disruption of signaling by Yersinia effector YopJ, a ubiquitin-like protein protease. Science 290, 1594–1597.
Palmer, L. E., Pancetti, A. R., Greenberg, S., and Bliska, J. B. (1999). YopJ of Yersinia spp. is sufficient to cause downregulation of multiple mitogen-activated protein kinases in eukaryotic cells. Infect. Immun. 67, 708–716.
Paquette, N., Conlon, J., Sweet, C., Rus, F., Wilson, L., Pereira, A., et al. (2012). Serine/threonine acetylation of TGFbeta-activated kinase (TAK1) by Yersinia pestis YopJ inhibits innate immune signaling. Proc. Natl. Acad. Sci. U.S.A. 109, 12710–12715.
Rathinam, V. A., Vanaja, S. K., Waggoner, L., Sokolovska, A., Becker, C., Stuart, L. M., et al. (2012). TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell 150, 606–619.
Ruckdeschel, K., Harb, S., Roggenkamp, A., Hornef, M., Zumbihl, R., Kohler, S., et al. (1998). Yersinia enterocolitica impairs activation of transcription factor NF-kappaB: involvement in the induction of programmed cell death and in the suppression of the macrophage tumor necrosis factor alpha production. J. Exp. Med. 187, 1069–1079.
Ruckdeschel, K., Mannel, O., Richter, K., Jacobi, C. A., Trulzsch, K., Rouot, B., et al. (2001a). Yersinia outer protein P of Yersinia enterocolitica simultaneously blocks the nuclear factor-kappa B pathway and exploits lipopolysaccharide signaling to trigger apoptosis in macrophages. J. Immunol. 166, 1823–1831.
Ruckdeschel, K., Richter, K., Mannel, O., and Heesemann, J. (2001b). Arginine-143 of Yersinia enterocolitica YopP crucially determines isotype-related NF-kappaB suppression and apoptosis induction in macrophages. Infect. Immun. 69, 7652–7662.
Ruckdeschel, K., Pfaffinger, G., Haase, R., Sing, A., Weighardt, H., Hacker, G., et al. (2004). Signaling of apoptosis through TLRs critically involves toll/IL-1 receptor domain-containing adapter inducing IFN-beta, but not MyD88, in bacteria-infected murine macrophages. J. Immunol. 173, 3320–3328.
Ruckdeschel, K., Roggenkamp, A., Lafont, V., Mangeat, P., Heesemann, J., and Rouot, B. (1997). Interaction of Yersinia enterocolitica with macrophages leads to macrophage cell death through apoptosis. Infect. Immun. 65, 4813–4821.
Sancho, D., Joffre, O. P., Keller, A. M., Rogers, N. C., Martinez, D., Hernanz-Falcon, P., et al. (2009). Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature 458, 899–903.
Sander, L. E., Davis, M. J., Boekschoten, M. V., Amsen, D., Dascher, C. C., Ryffel, B., et al. (2011). Detection of prokaryotic mRNA signifies microbial viability and promotes immunity. Nature 474, 385–389.
Sonnenberg, G. F., Nair, M. G., Kirn, T. J., Zaph, C., Fouser, L. A., and Artis, D. (2010). Pathological versus protective functions of IL-22 in airway inflammation are regulated by IL-17A. J. Exp. Med. 207, 1293–1305.
Sutterwala, F. S., Ogura, Y., Szczepanik, M., Lara-Tejero, M., Lichtenberger, G. S., Grant, E. P., et al. (2006). Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity 24, 317–327.
Sweet, C. R., Conlon, J., Golenbock, D. T., Goguen, J., and Silverman, N. (2007). YopJ targets TRAF proteins to inhibit TLR-mediated NF-kappaB, MAPK and IRF3 signal transduction. Cell. Microbiol. 9, 2700–2715.
Tenev, T., Bianchi, K., Darding, M., Broemer, M., Langlais, C., Wallberg, F., et al. (2011). The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol. Cell 43, 432–448.
Torchinsky, M. B., Garaude, J., Martin, A. P., and Blander, J. M. (2009). Innate immune recognition of infected apoptotic cells directs T(H)17 cell differentiation. Nature 458, 78–82.
Upton, J. W., Kaiser, W. J., and Mocarski, E. S. (2010). Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe 7, 302–313.
Vance, R. E., Isberg, R. R., and Portnoy, D. A. (2009). Patterns of pathogenesis: discrimination of pathogenic and nonpathogenic microbes by the innate immune system. Cell Host Microbe 6, 10–21.
Vandenabeele, P., Declercq, W., Van Herreweghe, F., and Vanden Berghe, T. (2010). The role of the kinases RIP1 and RIP3 in TNF-induced necrosis. Sci. Signal. 3, re4.
Viboud, G. I., and Bliska, J. B. (2005). Yersinia outer proteins: role in modulation of host cell signaling responses and pathogenesis. Annu. Rev. Microbiol. 59, 69–89.
Vladimer, G. I., Weng, D., Paquette, S. W., Vanaja, S. K., Rathinam, V. A., Aune, M. H., et al. (2012). The NLRP12 inflammasome recognizes Yersinia pestis. Immunity 37, 96–107.
Welz, P. S., Wullaert, A., Vlantis, K., Kondylis, V., Fernandez-Majada, V., Ermolaeva, M., et al. (2011). FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature 477, 330–334.
Ye, P., Rodriguez, F. H., Kanaly, S., Stocking, K. L., Schurr, J., Schwarzenberger, P., et al. (2001). Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J. Exp. Med. 194, 519–527.
Zauberman, A., Cohen, S., Mamroud, E., Flashner, Y., Tidhar, A., Ber, R., et al. (2006). Interaction of Yersinia pestis with macrophages: limitations in YopJ-dependent apoptosis. Infect. Immun. 74, 3239–3250.
Zauberman, A., Tidhar, A., Levy, Y., Bar-Haim, E., Halperin, G., Flashner, Y., et al. (2009). Yersinia pestis endowed with increased cytotoxicity is avirulent in a bubonic plague model and induces rapid protection against pneumonic plague. PLoS ONE 4:e5938. doi: 10.1371/journal.pone.0005938
Zhang, Y., and Bliska, J. B. (2003). Role of Toll-like receptor signaling in the apoptotic response of macrophages to Yersinia infection. Infect. Immun. 71, 1513–1519.
Zhang, D. W., Shao, J., Lin, J., Zhang, N., Lu, B. J., Lin, S. C., et al. (2009). RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325, 332–336.
Zhang, Y., Ting, A. T., Marcu, K. B., and Bliska, J. B. (2005). Inhibition of MAPK and NF-kappa B pathways is necessary for rapid apoptosis in macrophages infected with Yersinia. J. Immunol. 174, 7939–7949.
Zheng, Y., Lilo, S., Brodsky, I. E., Zhang, Y., Medzhitov, R., Marcu, K. B., et al. (2011). A Yersinia effector with enhanced inhibitory activity on the NF-kappaB pathway activates the NLRP3/ASC/caspase-1 inflammasome in macrophages. PLoS Pathog. 7:e1002026. doi: 10.1371/journal.ppat.1002026
Zheng, Y., Lilo, S., Mena, P., and Bliska, J. B. (2012). YopJ-induced caspase-1 activation in Yersinia-infected macrophages: independent of apoptosis, linked to necrosis, dispensable for innate host defense. PloS ONE 7:e36019. doi: 10.1371/journal.pone.0036019
Zhou, H., Monack, D. M., Kayagaki, N., Wertz, I., Yin, J., Wolf, B., et al. (2005). Yersinia virulence factor YopJ acts as a deubiquitinase to inhibit NF-kappa B activation. J. Exp. Med. 202, 1327–1332.
Keywords: Yersinia, cell death, YopJ, inflammasome, caspase-1, apoptosis, pyroptosis
Citation: Philip NH and Brodsky IE (2012) Cell death programs in Yersinia immunity and pathogenesis. Front. Cell. Inf. Microbio. 2:149. doi: 10.3389/fcimb.2012.00149
Received: 29 September 2012; Accepted: 13 November 2012;
Published online: 30 November 2012.
Edited by:
Matthew Francis, Umeå University, SwedenCopyright © 2012 Philip and Brodsky. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Igor E. Brodsky, Department of Pathobiology, School of Veterinary Medicine, University of Pennsylvania, 3800 Spruce St., Rosenthal 390EE, Philadelphia, PA 19104, USA. e-mail:aWJyb2Rza3lAdmV0LnVwZW5uLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.