 Dong Liu
Dong Liu Jude E. Uzonna*
Jude E. Uzonna*- Department of Immunology, University of Manitoba, Winnipeg, MB, Canada
The complicated interactions between Leishmania and the host antigen-presenting cells (APCs) have fundamental effects on the final outcome of the disease. Two major APCs, macrophages and dendritic cells (DCs), play critical roles in mediating resistance and susceptibility during Leishmania infection. Macrophages are the primary resident cell for Leishmania: they phagocytose and permit parasite proliferation. However, these cells are also the major effector cells to eliminate infection. The effective clearance of parasites by macrophages depends on activation of appropriate immune response, which is usually initiated by DCs. Here, we review the early interaction of APCs with Leishmania parasites and how these interactions profoundly impact on the ensuing adaptive immune response. We also discuss how the current knowledge will allow further refinement of our understanding of the interplay between Leishmania and its hosts that leads to resistance or susceptibility.
Introduction
Several species of the protozoan parasites belonging to the genus Leishmania are the causative agent of a spectrum of zoonotic disease collectively called leishmaniasis. The disease is considered endemic in over 80 countries and about 2 million new cases occur every year, of which approximately 50% of these new cases are children (Bhattacharya et al., 2006). More than 12 million people are believed to be presently infected and a population of over 350 million people are at risk and around 7000 death annually are due to the disease (Bern et al., 2008). All types of leishmaniasis are transmitted by the female phlebotomine Sandflies, which infect a range of mammals including humans, rodents, and canids (Ashford, 1996). During their relatively simple life cycle, Leishmania parasites alternate between two distinct developmental stages: the flagellated, motile “promastigote” form residing in the midgut of Sandfly vectors, and the non-motile “amastigote” form that reside within phagolysosomal vesicles of the vertebrate host macrophages.
The life cycle of Leishmania is initiated when the infected female Sandfly injects metacyclic promastigotes into the host during a regular blood meal. Within minutes, the promastigotes are rapidly taken up by phagocytic cells, including macrophages and neutrophils. Since neutrophils have a very short life span, macrophages are the final host cells for proliferation of Leishmania parasites (Antoine et al., 1998). After internalization into the phagolysosome of macrophages, the promastigotes start to differentiate into small, non-motile amastigote forms, which divide many times by binary fission, eventually rupturing the macrophages to infect other surrounding macrophages. When the infected host is bitten by another Sandfly, these amastigotes are taken up with the blood meal, undergo series of morphological and developmental changes in the gut of the Sandfly and differentiate into mature or infectious promastigote forms. Fully differentiated infective organisms migrate to the mouthpart of the sandflies and are injected into another host during a blood meal.
Interaction of Leishmania with Host Cells
Although Leishmania parasites interact and infect a variety of host cell types, macrophages and dendritic cells (DCs) are arguably the most important cells that regulate the outcome of infection. After the initial uptake and internalization of promastigotes by macrophages into the phagosome, subsequent fusion with lysosomes occurs, and the parasites must survive/live within this hostile host phagolysosomal environment. Although this is perhaps one of the most challenging environments for most pathogens, Leishmania are among the few protozoa that can survival and multiply in such a harsh environment. Understanding how these organisms are able to survival and manipulate host cells to favor their replication and transmission is critically important for designing new drugs or therapeutic strategies against the disease. In the following discussion, we present an overview of the Leishmania–macrophage/DC interaction and the subsequent impact of this interaction on host immune response and disease pathogenesis. Although we will mention the various subversion strategies employed by the parasite to successfully proliferate in infected cells, we will not go into details of these evasion strategies. For more comprehensive information on these, we refer the reader to these recent and excellent reviews (Gregory and Olivier, 2005; Olivier et al., 2005; Soong, 2008; Xin et al., 2008; Shio et al., 2012).
The Interaction of Leishmania with Macrophages
Because Leishmania parasites are obligatory intracellular pathogens, macrophages are indispensable for parasite survival, replication, and differentiation. After initial infection, both neutrophils and macrophages are recruited to the infection site and their interaction with the parasites significantly influence the outcome of infection (Ribeiro-Gomes et al., 2004, 2007). However, recent studies suggest that more neutrophils are recruited to the infection site (particularly when infection is initiated by Sandfly bite) and these cells are very efficient in parasite uptake (Peters et al., 2008; Peters and Sacks, 2009). Since neutrophils are short-lived phagocytes, they are believed to serve as intermediate host cells, and have been proposed to act as “Trojan horses” used by parasites to hitchhike a ride in order to silently enter macrophages thereby avoiding cell activation (Laufs et al., 2002; van Zandbergen et al., 2004). In fact, L. major infection of polymorphonuclear neutrophils (PMNs) leads to significant delay in their programmed death and secretion of high levels of MIP-1β, which is known to attract macrophages to the site of infection (van Zandbergen et al., 2004). Once macrophages are recruited, they phagocytose free parasites and infected/apoptotic PMNs and become the final definitive host cells for parasite replication as well as effector cells responsible for the destruction of parasites.
The uptake of Leishmania promastigotes by host cells is a classical receptor-mediated process that initiates phagocytosis. A number of parasite and macrophage surface molecules have been implicated in the interaction between Leishmania and macrophages. In the case of promastigotes, the complement receptors (CR)1, CR3 (Mac-1), fibronectin receptor, and the mannose-fucose receptor (MR) on the surface of macrophages play import roles in promastigote binding/attachment (Kane and Mosser, 2000). Interestingly, an earlier study found that individual ligation of these receptors does not trigger the activation of macrophages, suggesting multiple receptor ligation may be important for initiating the appropriate protective response (Aderem and Underhill, 1999). In a recently study Ueno et al. attempted to address the conflicting reports regarding the importance of MR in Leishmania entry into macrophages. They found that metacyclic promastigotes of L. infantum chagasi use CR3 but not MR to enter macrophages, in contrast to avirulent promastigotes, which use both receptors to enter the cells. It is already known that CR3 ligation by itself does not trigger NADPH oxidase activation and subsequent respiratory burst at the phagosome membrane (Sehgal et al., 1993), while MR ligation has been shown to promote inflammatory responses (Linehan et al., 2000). These results may explain why metacyclic promastigotes avoid using detrimental MR during their invasion of macrophages in order to enhance their intracellular survival.
The surface lipophosphoglycan (LPG), GP63, and proteophophoglycans (PPG) on L. major promastigotes are also important determinants in the initiation phagocytosis and subsequent intracellular survival of parasites (Yao et al., 2003; Naderer and McConville, 2008), as they are the target molecules for various opsonins such as the degradation fragments of complement component C3 (C3b/iC3b) (Brittingham and Mosser, 1996), mannose-binding protein (Green et al., 1994), and galectins (Pelletier et al., 2003; Kamhawi et al., 2004). It is important to remember that deficiency of a single molecule (such as LPG) is not detrimental for attachment, because phagocytosis of LPG mutant parasites is not compromised and may be even better when compared to their wild type controls (McNeely and Turco, 1990). However, while deficiency of LPG does not affect phagocytosis, it significantly impacts on the intracellular survival of these mutants (McNeely and Turco, 1990), suggesting these molecules are still critical for inhibiting and subverting the host killing machinery. In the case of amastigote stage of Leishmania, it has been demonstrated that host IgG-coated parasites could bind to Fc receptors (FcγR) on macrophages to facilitate entry into these cells. This engagement and subsequent entry activate downstream signaling pathways that prevent killing and promote intracellular parasite growth (Miles et al., 2005).
Macrophages are also the major effector cells responsible for destruction of the parasites. Macrophages can be activated by different signals leading to their development into functionally distinct subsets with different disease outcomes. Thus, appropriate activation of macrophages is crucial for eliminating this intracellular pathogen. Macrophage activation is generally divided into two functionally distinct spectra: classical and alternative activation (Figure 1). Classical activation is mediated by the products of Th1 and NK cells—in particular, IFN-γ, which stimulates macrophages to produce inducible nitric oxide synthase (iNOS, also known as NOS2), an enzyme which catalyzes L-arginine to generate nitric oxide (NO) (Liew et al., 1990). NO is a toxic molecule that plays a major role in killing intracellular parasites, including Leishmania. In the absence of IFN-γ or microbial products such as LPS, iNOS mRNA, and protein are undetectable in macrophages (Santos et al., 2006), suggesting a critical role for IFN-γ in macrophage activation. The importance of iNOS in facilitating parasite control is further supported by the finding that iNOS deficient mice on a normally resistant background are highly susceptible to L. major infection despite mounting a strong Th1 (IFN-γ) response (Seger and Krebs, 1995). Interestingly, an IFN-γ independent immune-protective mechanism has also been recently proposed. Spath et al. (2009) demonstrated that in vitro infection of Stat1-deficient macrophages with LPG deficient (lpg1-) L. major promastigotes resulted in a two-fold increase in intracellular growth, which was independent of IFN-γ but was associated with a substantial increase in phagosomal pH. Besides IFN-γ, a number of other inflammatory cytokines, such as IL-1, TNF, IFN-α, and IFN-β are also involved in macrophage activation and upregulation of iNOS expression leading to NO production (Bronte and Zanovello, 2005).
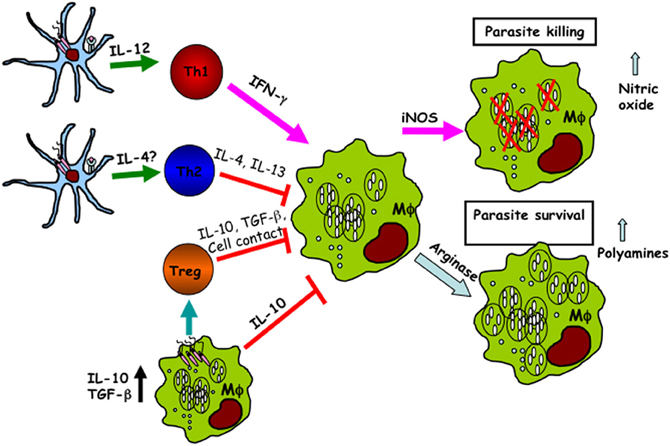
Figure 1. Dendritic cells and macrophages regulate the outcome of Leishmania infection. Following infection, both macrophages and dendritic cells phagocytose Leishmania leading to different functional outcomes. Infected dendritic cells produce IL-12, which is critical for the development of IFN-γ-producing CD4+ Th1 cells. IFN-γ acts on infected macrophages leading to their activation (classical activation), upregulation of iNOS, and production of nitric oxide and other free radicals that are important for intracellular parasite killing. In contrast, the production of IL-4 by other cell types (including keratinocytes and Vβ4 T cells) supports CD4+ Th2 development. Th2 cells produce IL-4 and IL-13, which leads to upregulation of arginase activity, alternative macrophage activation and the production polyamines that favor intracellular parasite proliferation. In addition, naturally occurring regulatory T cells (Treg) and infected macrophages also produce some immunoregulatory cytokines including IL-10 and TGF-β, which further deactivate infected cells leading to impaired parasite killing.
In contrast classical activation, alternative macrophage activation is induced by Th2 cytokines, such as IL-4 and IL-13 (Gordon, 2003). It has been shown that IL-4-induced polyamine biosynthesis (via upregulation of arginase) favors L. major parasite survival in macrophages (Kropf et al., 2005). This finding reinforced the notion that Leishmania encoded arginase is a virulent factor and its expression functions to preferentially enhance alternative macrophage activation leading to parasite survival (Iniesta et al., 2002, 2005; Kropf et al., 2005). Using arginase deficient L. major, we found that the influence of parasite-derived arginase on disease severity is not related to its modulatory effect on the host immune response (Muleme et al., 2009). Rather, Leishmania-encoded arginase enhances disease pathogenesis by augmenting host cellular arginase activities leading to parasite survival and proliferation in infected macrophages. Thus, contrary to popular belief, the host cellular arginase activity in L. major-infected mice may not be regulated by cytokines. Another cytokine that negatively affects classical macrophage activation is IL-10. It inhibits the respiratory burst and inflammatory cytokine productions, particularly TNF, by macrophages (Gordon, 2003), thereby negatively affect parasiticidal activity of infected cells.
To evade killing by activated macrophages, Leishmania must manipulate macrophage activation pathways in a way that favor their survival and proliferation. Interleukin 12 (IL-12) is a critical cytokine required for CD4+ Th1 development and production of IFN-γ (Mosmann and Coffman, 1989). IL-12 is mainly produced by antigen-presenting cells (APCs), and macrophages were initially proposed as the major source of IL-12 in L. major-infected mice. Although macrophages phagocytose Leishmania efficiently, there is evidence that their ability to produce IL-12 is selectively impaired by the parasites (Reiner, 1994; Carrera et al., 1996; Desjardins and Descoteaux, 1998). This impairment in IL-12 production is not strain specific because macrophages from both the susceptible and resistant strains of mice display similar degree of impairment in IL-12 production after L. major infection although other proinflammatory cytokines were only slightly affected (Reiner, 1994). In addition, infection of macrophages by Leishmania also leads to the production of immuno-regulatory cytokines such as IL-10 and TGF-β, cytokines that are known for their inhibition/deactivation of macrophage functions (von der Weid et al., 1996; Kane and Mosser, 2000, 2001).
The balance between the host and parasite factors that control the activation/deactivation of macrophages determines the final outcome (or fate) of the parasites within infected macrophage. In line with this, certain parasite-derived molecules have been shown to regulate the outcome of L. major infection in mice. Recently, we demonstrated that in the absence of phosphoglycan (PG) containing molecules (as seen in lpg2- L. major), Leishmania parasites are unable to induce early IL-4 production in BALB/c mice (Liu et al., 2009), suggesting that parasite-derived molecules can modulate host adaptive immune response to favor their survival. Similarly, Leishmania parasites can also induce the expression of immunomodulatory molecules, such as CD200, to inhibit macrophage activation (Cortez et al., 2011). In this report, it was further demonstrated that CD200-dependent iNOS inhibition was responsible for the increased virulence of L. amazonensis (Cortez et al., 2011).
The Interaction of Leishmania with Dendritic Cells
DCs are hematopoietic bone marrow progenitor-derived leukocytes that are highly heterogeneous and widely distributed all over the body (Banchereau and Steinman, 1998; Steinman, 2007). As professional and most potent APCs, DCs are specialized in antigen uptake, processing, and presentation to T cells. Although macrophages are also professional APCs and the main host for harboring Leishmania parasites as well as effector cells for parasite killing, Leishmania-infected macrophages do not secrete IL-12 (von Stebut et al., 1998) and hence unable to stimulate antigen-specific CD4+ Th1 cell response (Kima et al., 1996). Indeed, several reports show a central role for DCs in orchestrating immune responses in leishmaniasis (Gorak et al., 1998; von Stebut et al., 1998; Leon et al., 2007). The skin contains at least three DC populations that consist of epidermal Langerhan cells and two migratory dermal DC subsets. Early studies demonstrated that epidermal Langerhan cells (MHC IIhigh, CD11c+, CD11blow, CD8αinter, CD205high, and Langerin+) phagocytose L. major in vivo and migrate to draining lymph nodes (dLNs) for presentation to antigen-specific T cells (Moll et al., 1993). Surprisingly, however, later studies showed that DCs harboring parasites in dLNs are Langerin negative, but rather express dermal DC markers (MHC IIhigh, CD11c+, CD11b+, CD8α−, and CD205low) (Ritter et al., 2004), which is the major DC population that primes CD4+ T cell response. Also, mice genetically modified to show MHC class II deficiency exclusively in LCs (but not in dermal DCs) control L. major infection similar to wild type animals (Lemos et al., 2004), suggesting that LCs are dispensable for triggering T cell response during Leishmania infection. Moreover, a more recent paper showed that LCs may even play a pathogenic role during low dose infection via the induction and expansion of regulatory T cells (Kautz-Neu et al., 2011). Some studies showed that parasite harboring dermal DCs most likely migrate out of the skin and transport the antigens to the dLNs (Ritter et al., 2004; Ng et al., 2008). Other studies suggested that blood monocyte-derived DC may differentiate within the inflamed skin following a Sandfly bite, phagocytose parasites, and transport them to the dLN where they present parasite-derived antigen to T cells (Leon et al., 2007).
The production of IL-12 by APCs is critically important for polarization of naive T cells toward Th1 subset and subsequent IFN-γ production (von Stebut et al., 1998). Infection of DC with Leishmania results in functional IL-12p70 production (Marovich et al., 2000). Interestingly, different DC subsets are differentially permissive to Leishmania parasites and this differential infectivity seems to be inversely correlated with the ability of infected cells to produce IL-12p70 (Henri et al., 2002). CD8α+ DCs are the least permissive to L. major amastigotes compared to CD8α− DCs and CD8α− DCs. However, CD8α+ DCs are the most powerful IL-12p70 producers in response to infection (Henri et al., 2002). The mechanism(s) that control the induction of IL-12 from DCs and the functional differences between the IL-12-producing DCs and the non-producers are still not yet known.
It has been speculated that the different outcome of Leishmania infection in the resistant and susceptible mice may be related to differences in their DC function, particularly in promoting the differentiation of naïve CD4+ helper T cells into effector cells (Suzue et al., 2008). However, von Stebut et al. (1998) demonstrated that L. major-infected fetal skin-derived DCs from both BALB/c and C57BL/6 mice upregulate their constimulatory molecules and produce comparable levels proinflammatory cytokines, including IL-12p70. In contrast, Moll et al. (2002) showed that epidermal-derived DCs (Langerhans cells) from BALB/c mice up-regulate IL-4 receptor expression and down-regulate IL-12p40 production in response to L. major infection, suggesting that Leishmania parasites have evolved strategies to inhibit DC Th1 polarizing functions. Baldwin et al. (2004) found that L. major-infected BALB/c mice have an increased number of plasmacytoid DCs in their dLNs and this was associated with higher cell recruitment early after infection compared to infected C57BL/6 mice. However, it is not clear whether the observed differences in DCs between susceptible and resistant mice are relevant to the pathogenesis of disease. At present, there is still limited information on initial DC responses to other species of Leishmania at the site of infection and their contribution to priming of protective or pathogenic T cell responses.
Toll like receptors (TLRs) are important pattern recognition receptors (PRRs) that are not only involved in self/non-self discrimination by the innate arm of host immunity, but also at initiating and directing subsequent adaptive immunity. TLRs are expressed abundantly on macrophages, DCs, and NK cells. Upon recognition of respective pathogen associated molecular patterns (PAMPs), TLRs recruit adaptor molecules, such as MyD88 and TRIF, and initiate a series but highly coordinated downstream signaling events that lead to the production of pro-inflammatory cytokines, chemokines, and antimicrobial peptides (Kawai and Akira, 2010). MyD88 participates in most TLRs's signal transduction by binding to the cytoplasmic domain of Toll/IL-1 like receptor (Takeda and Akira, 2004). de Veer et al. (2003) found that MyD88 deficient C57BL/6 mice were more suscepitble to L. major infection, suggesting a critical role of TLR signaling in initiating anti-Leishmania immunity. They further demonstrated that LPG, the major and most abundant surface molecule of Leishmania, is a TLR2 ligand, and is responsible for the generation of protective immunity against leishmaniasis. To date, three distinct TLRs, TLR2, TLR4, and TLR9, have been suggested to be involved in the recognition of L. major (de Veer et al., 2003; Kropf et al., 2004; Liese et al., 2007; Schleicher et al., 2007; Abou Fakher et al., 2009). However, a recent study (Abou Fakher et al., 2009), using C57BL/6 mice deficient in either TLR2, 4, or 9, showed that only TLR9-deficient mice are more susceptible to L. major infection, indicating that the reported role of TLR2 and TLR4 in immunity to murine leishmaniasis needs to be re-evaluated. It is also not clear whether TLR deficiency specific to DCs or macrophages will likely have any effect on the outcome the disease.
Subversion of Host Macrophage and DC Intracellular Signaling Pathways by Leishmania
Intracellular signaling events leading to the activation of genes involved in production of effector molecules are important for efficient and effective control of pathogens by host infected cells. A number of pathogens are able to manipulate the signal transduction pathways of their host cells to favor their survival (Harnett et al., 1999; Tachado et al., 1999; Sibley, 2011). Since Leishmania are obligate intracellular parasites, their survival inside its mamalian host cells is critically dependent on their ability to successfully disrupt the host cell signaling events that lead to the generation of effector molecules. To evade killing by the host, the parasites are actively involved in almost every aspects of the manipulation/inhibition of host cell signalling, ranging from preventing the production of microbicidal molecules, and protective cytokines to interferring with effective antigen presentation.
In response to cytokines such as IFN-γ and TNF, macrophages become activated and produce microbicidal free radical molecules such as NO and reactive oxgen intermediates (Figure 1). In addition, activated macrophages also produce TNF, which acts in an autocrine manner to further enhance the production of microbicidal free radicals by macrophages. The critical role of NO is confirmed in iNOS deficient mice whose macrophages are unable to eliminate parasites in vitro and these mice are also highly susceptible to Leishmania (Seger and Krebs, 1995). Protein kinase C (PKC) is known to play a key role in the regulation of macrophge functions including production of proinflammatory cytokines, NO, and reactive oxygen intermediates (ROI). By using murine macrophage cell line overexpressing a dominant-negative (DN) mutant of PKC-α, St. Denis et al. (1999) found that although phagocytosis of L. donovani promastigotes was not altered by DN PKC-α overexpression, intracellular survival of the parasites was significantly enhanced. This observation suggests that PKC signaling is critical for macrophage activation and parasite killing by infected cells. In line with this, early studies showed that the abundant promastigote surface protein, LPG, is able to block the bio-activity of PKC (Severn et al., 1992). However, another study (Olivier et al., 1992) questioned the role of LPG in the blockage of PKC function, by using amastigotes which lack or have highly reduced levels of LPG on their surface. In this report, amastigotes were still capable of efficiently inhibiting PKC activity in monocytes, suggesting that other parasite molecules or factors may be involved in modulating PKC activity. Another parasite molecule that has been implicated in the downregulation of intracellular signaling cascade in macrophages resulting in the suppression of macrophage microbicidal activities is gp63 (Sorensen et al., 1994; Descoteaux and Turco, 1999). GP63 is a 63 kDa zinc-dependent metalloproteinase that is abundantly expressed on the cell surface of all Leishmania promastigotes (Yao et al., 2003), and is about 10 times less abundant than LPG (McConville and Blackwell, 1991; Pimenta et al., 1991). Recent studies show that a number of host cell signaling molecules, particularly protein tyrosine kinases, are either proteolytically processed or modulated by gp63 (Gomez et al., 2009; Halle et al., 2009). Thus, the expression of gp63 activity is potentially another possible evasion strategy of Leishmania parasites.
Leishmania has also been shown to downregulate the activation of mitogen-activated protein kinases (MAPKs), a group of enzymes that regulate macrophage functions including the production of proinflammatory cytokines and NO (Seger and Krebs, 1995). Infection of macrophages with L. donovani leads to alteration of MAPK activities resulting in active downregulation of proinflammatory cytokine production (Nandan et al., 1999). This effect was shown to be mediated by L. donovani-dependent induction of protein tyrosine phosphatases (PTPs) (Blanchette et al., 1999; Forget et al., 2006). In line with this observation, the activation of MAPK and production of proinflammatory cytokines was restored to normal levels in L. donovani-infected SHP-1 deficient macrophages (Forget et al., 2006). The induction of PTPs and inhibition of MAPK in macrophages has been associated with the promastigote-specific surface molecules because infection of macrophages with LPG deficient Leishmania leads to robust activation of ERK, although this has minimal effect on p38 and JNK activation (Dermine et al., 2000; Prive and Descoteaux, 2000). In contrast, Balaraman et al. (2005) using immortalized macrophage cell line, found that LPG has a stimulatory effect on macrophages leading to increased activation of MAPKs. These differences might be related to the use of different cells in these studies; primary versus immortalized cell lines.
In addition to downregulation of MAPK, Leishmania also inhibits the JAK/STAT signaling pathways. JAK2/STAT1 signaling pathway is critically important for proper and optimal macrophage response to IFN-γ, which leads to the production of cytotoxic molecules for killing intracelular pathogens, including Leishmania. Nandan and Reiner (1995) showed that infection of human monocytic cell line and peripheral blood monocytes with L. donovani leads to dramatic impairment in IFN-γ-induced tyrosine phosphorylation. This effect was mediated via selective inhibition of JAK/STAT1 signaling pathway (Nandan and Reiner, 1995).
Like macrophages, Leishmania also interferes with intracellular signaling in DCs, which affects their antigen-presenting functions and hence their ability to induce optimal cell-mediated immunity against the parasite (Soong, 2008; Xin et al., 2008). Studies show that L. amazonensis amastigotes can suppress cytokine- and TLR-4-mediated DC activation and IL-12 production via rapid degradation of intracellular signaling proteins, particularly those associated with the JAK/STAT, NFκB, and interferon regulatory factor (IRF) pathways (Xin et al., 2008). The cathepsin-like cysteine proteinases in the parasite are believed to be responsible for the downregulation of NFκB signaling and hence IL-12 production in macrophages (Cameron et al., 2004). Whether these molecules are also involved in modulating NFκB signaling and IL-12 production in DCs is still not known. Interestingly, it has been shown that the alteration of DC response to L. amazonensis infection (including CD40 expression and IL-12 production) was mediated by the activation of ERK, a member of MAPK (Boggiatto et al., 2009). Thus, these reports suggest that the mechanisms through which Leishmania impair signaling in macrophages and DCs may be different.
Concluding Remarks
Although multiple cellular components are involved in the immune response against leishmaniasis, there is no question that macrophages and DCs play critical roles in the initiation, development, and maintenance of a protective immunity (Figure 1). It is quite clear that the production of IL-12 by DCs initiates Th1 response and protective immunity by promoting early NK cell activities (including IFN-γ production and cytotoxicity) (Liese et al., 2007). However, the exact mechanisms driving differentiation of naive CD4+ T cells into Th1 or Th2 phenotypes are still not very clear but are likely to be dictated by the cytokine environments during the early hours after infection (Sokol et al., 2008). On the other hand, the parasite also uses a variety of strategies to manipulate macrophage and DC functions to favor their survival and replication. One of the major mechanisms used by Leishmania is to inhibit the production of Th1-polarizing cytokine, IL-12, as well as preventing DCs from successfully presenting parasite antigens to T cells resulting in impaired cell-mediated immunity. Thus, the interplay between APCs (macrophages and DCs) and Leishmania parasites is complex. The parasite must infect these cells in order to proliferate and survive, but these infected cells are also critical for presenting parasite-derived antigens to T cells to initiate cell-mediated immunity, which is detrimental for the parasite. Given the fact that the disease pathology is highly variable depending on the species of Leishmania, it is very hard to generalize specific modulatory mechanism(s) to all strains and in all hosts. This is important because most of the studies investigating the role of DCs/macrophages (and their modulation thereof) during Leishmania infection usually conducted with a single specie of the parasite in one strain of mice, which precludes multi-species/strain comparison. A more comprehensive study would be very helpful for a better understanding of the role of these cells, the mechanisms that regulate their antigen presentation functions and pathogen factors that influence antigen presentation and subsequent activation of the adaptive immune system. This would be beneficial in the design of new drugs and therapies to control this disease.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
Funding for this study was provided by the Canadian Institutes of Health Research and the Manitoba Health Research Council.
Abbreviations
DC, dendritic cell; dLN, draining lymph node; LPG, lipophosphoglycan; PG, phosphoglycan.
References
Abou Fakher, F. H., Rachinel, N., Klimczak, M., Louis, J., and Doyen, N. (2009). TLR9-dependent activation of dendritic cells by DNA from Leishmania major favors Th1 cell development and the resolution of lesions. J. Immunol. 182, 1386–1396.
Aderem, A., and Underhill, D. M. (1999). Mechanisms of phagocytosis in macrophages. Annu. Rev. Immunol. 17, 593–623.
Antoine, J. C., Prina, E., Lang, T., and Courret, N. (1998). The biogenesis and properties of the parasitophorous vacuoles that harbour Leishmania in murine macrophages. Trends Microbiol. 6, 392–401.
Ashford, R. W. (1996). Leishmaniasis reservoirs and their significance in control. Clin. Dermatol. 14, 523–532.
Balaraman, S., Singh, V. K., Tewary, P., and Madhubala, R. (2005). Leishmania lipophosphoglycan activates the transcription factor activating protein 1 in J774A.1 macrophages through the extracellular signal-related kinase (ERK) and p38 mitogen-activated protein kinase. Mol. Biochem. Parasitol. 139, 117–127.
Baldwin, T., Henri, S., Curtis, J., O'Keeffe, M., Vremec, D., Shortman, K., and Handman, E. (2004). Dendritic cell populations in Leishmania major-infected skin and draining lymph nodes. Infect. Immun. 72, 1991–2001.
Banchereau, J., and Steinman, R. M. (1998). Dendritic cells and the control of immunity. Nature 392, 245–252.
Bern, C., Maguire, J. H., and Alvar, J. (2008). Complexities of assessing the disease burden attributable to leishmaniasis. PLoS Negl. Trop. Dis. 2:e313. doi: 10.1371/journal.pntd.0000313
Bhattacharya, S. K., Sur, D., and Karbwang, J. (2006). Childhood visceral leishmaniasis. Indian J. Med. Res. 123, 353–356.
Blanchette, J., Racette, N., Faure, R., Siminovitch, K. A., and Olivier, M. (1999). Leishmania-induced increases in activation of macrophage SHP-1 tyrosine phosphatase are associated with impaired IFN-gamma-triggered JAK2 activation. Eur. J. Immunol. 29, 3737–3744.
Boggiatto, P. M., Jie, F., Ghosh, M., Gibson-Corley, K. N., Ramer-Tait, A. E., Jones, D. E., and Petersen, C. A. (2009). Altered dendritic cell phenotype in response to Leishmania amazonensis amastigote infection is mediated by MAP kinase, ERK. Am. J. Pathol. 174, 1818–1826.
Brittingham, A., and Mosser, D. M. (1996). Exploitation of the complement system by Leishmania promastigotes. Parasitol. Today 12, 444–447.
Bronte, V., and Zanovello, P. (2005). Regulation of immune responses by L-arginine metabolism. Nat. Rev. Immunol. 5, 641–654.
Cameron, P., McGachy, A., Anderson, M., Paul, A., Coombs, G. H., Mottram, J. C., Alexander, J., and Plevin, R. (2004). Inhibition of lipopolysaccharide-induced macrophage IL-12 production by Leishmania mexicana amastigotes: the role of cysteine peptidases and the NF-kappaB signaling pathway. J. Immunol. 173, 3297–3304.
Carrera, L., Gazzinelli, R. T., Badolato, R., Hieny, S., Muller, W., Kuhn, R., and Sacks, D. L. (1996). Leishmania promastigotes selectively inhibit interleukin 12 induction in bone marrow-derived macrophages from susceptible and resistant mice. J. Exp. Med. 183, 515–526.
Cortez, M., Huynh, C., Fernandes, M. C., Kennedy, K. A., Aderem, A., and Andrews, N. W. (2011). Leishmania promotes its own virulence by inducing expression of the host immune inhibitory ligand CD200. Cell Host Microbe 9, 463–471.
de Veer, M. J., Curtis, J. M., Baldwin, T. M., Didonato, J. A., Sexton, A., McConville, M. J., Handman, E., and Schofield, L. (2003). MyD88 is essential for clearance of Leishmania major: possible role for lipophosphoglycan and Toll-like receptor 2 signaling. Eur. J. Immunol. 33, 2822–2831.
Dermine, J. F., Scianimanico, S., Prive, C., Descoteaux, A., and Desjardins, M. (2000). Leishmania promastigotes require lipophosphoglycan to actively modulate the fusion properties of phagosomes at an early step of phagocytosis. Cell. Microbiol. 2, 115–126.
Descoteaux, A., and Turco, S. J. (1999). Glycoconjugates in Leishmania infectivity. Biochim. Biophys. Acta 1455, 341–352.
Desjardins, M., and Descoteaux, A. (1998). Survival strategies of Leishmania donovani in mammalian host macrophages. Res. Immunol. 149, 689–692.
Forget, G., Gregory, D. J., Whitcombe, L. A., and Olivier, M. (2006). Role of host protein tyrosine phosphatase SHP-1 in Leishmania donovani-induced inhibition of nitric oxide production. Infect. Immun. 74, 6272–6279.
Gomez, M. A., Contreras, I., Halle, M., Tremblay, M. L., McMaster, R. W., and Olivier, M. (2009). Leishmania GP63 alters host signaling through cleavage-activated protein tyrosine phosphatases. Sci. Signal 2, ra58.
Gorak, P. M., Engwerda, C. R., and Kaye, P. M. (1998). Dendritic cells, but not macrophages, produce IL-12 immediately following Leishmania donovani infection. Eur. J. Immunol. 28, 687–695.
Green, P. J., Feizi, T., Stoll, M. S., Thiel, S., Prescott, A., and McConville, M. J. (1994). Recognition of the major cell surface glycoconjugates of Leishmania parasites by the human serum mannan-binding protein. Mol. Biochem. Parasitol. 66, 319–328.
Gregory, D. J., and Olivier, M. (2005). Subversion of host cell signalling by the protozoan parasite Leishmania. Parasitology 130(Suppl.), S27–S35.
Halle, M., Gomez, M. A., Stuible, M., Shimizu, H., McMaster, W. R., Olivier, M., and Tremblay, M. L. (2009). The Leishmania surface protease GP63 cleaves multiple intracellular proteins and actively participates in p38 mitogen-activated protein kinase inactivation. J. Biol. Chem. 284, 6893–6908.
Harnett, W., Deehan, M. R., Houston, K. M., and Harnett, M. M. (1999). Immunomodulatory properties of a phosphorylcholine-containing secreted filarial glycoprotein. Parasite Immunol. 21, 601–608.
Henri, S., Curtis, J., Hochrein, H., Vremec, D., Shortman, K., and Handman, E. (2002). Hierarchy of susceptibility of dendritic cell subsets to infection by Leishmania major: inverse relationship to interleukin-12 production. Infect. Immun. 70, 3874–3880.
Iniesta, V., Carcelen, J., Molano, I., Peixoto, P. M., Redondo, E., Parra, P., Mangas, M., Monroy, I., Campo, M. L., Nieto, C. G., and Corraliza, I. (2005). Arginase I induction during Leishmania major infection mediates the development of disease. Infect. Immun. 73, 6085–6090.
Iniesta, V., Gomez-Nieto, L. C., Molano, I., Mohedano, A., Carcelen, J., Miron, C., Alonso, C., and Corraliza, I. (2002). Arginase I induction in macrophages, triggered by Th2-type cytokines, supports the growth of intracellular Leishmania parasites. Parasite Immunol. 24, 113–118.
Kamhawi, S., Ramalho-Ortigao, M., Pham, V. M., Kumar, S., Lawyer, P. G., Turco, S. J., Barillas-Mury, C., Sacks, D. L., and Valenzuela, J. G. (2004). A role for insect galectins in parasite survival. Cell 119, 329–341.
Kane, M. M., and Mosser, D. M. (2000). Leishmania parasites and their ploys to disrupt macrophage activation. Curr. Opin. Hematol. 7, 26–31.
Kane, M. M., and Mosser, D. M. (2001). The role of IL-10 in promoting disease progression in leishmaniasis. J. Immunol. 166, 1141–1147.
Kautz-Neu, K., Noordegraaf, M., Dinges, S., Bennett, C. L., John, D., Clausen, B. E., and von Stebut, E. (2011). Langerhans cells are negative regulators of the anti-Leishmania response. J. Exp. Med. 208, 885–891.
Kawai, T., and Akira, S. (2010). The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11, 373–384.
Kima, P. E., Soong, L., Chicharro, C., Ruddle, N. H., and McMahon-Pratt, D. (1996). Leishmania-infected macrophages sequester endogenously synthesized parasite antigens from presentation to CD4+ T cells. Eur. J. Immunol. 26, 3163–3169.
Kropf, P., Freudenberg, M. A., Modolell, M., Price, H. P., Herath, S., Antoniazi, S., Galanos, C., Smith, D. F., and Muller, I. (2004). Toll-like receptor 4 contributes to efficient control of infection with the protozoan parasite Leishmania major. Infect. Immun. 72, 1920–1928.
Kropf, P., Fuentes, J. M., Fahnrich, E., Arpa, L., Herath, S., Weber, V., Soler, G., Celada, A., Modolell, M., and Muller, I. (2005). Arginase and polyamine synthesis are key factors in the regulation of experimental leishmaniasis in vivo. FASEB J. 19, 1000–1002.
Laufs, H., Muller, K., Fleischer, J., Reiling, N., Jahnke, N., Jensenius, J. C., Solbach, W., and Laskay, T. (2002). Intracellular survival of Leishmania major in neutrophil granulocytes after uptake in the absence of heat-labile serum factors. Infect. Immun. 70, 826–835.
Lemos, M. P., Esquivel, F., Scott, P., and Laufer, T. M. (2004). MHC class II expression restricted to CD8alpha+ and CD11b+ dendritic cells is sufficient for control of Leishmania major. J. Exp. Med. 199, 725–730.
Leon, B., Lopez-Bravo, M., and Ardavin, C. (2007). Monocyte-derived dendritic cells formed at the infection site control the induction of protective T helper 1 responses against Leishmania. Immunity 26, 519–531.
Liese, J., Schleicher, U., and Bogdan, C. (2007). TLR9 signaling is essential for the innate NK cell response in murine cutaneous leishmaniasis. Eur. J. Immunol. 37, 3424–3434.
Liew, F. Y., Li, Y., and Millott, S. (1990). Tumour necrosis factor (TNF-alpha) in leishmaniasis. II. TNF-alpha-induced macrophage leishmanicidal activity is mediated by nitric oxide from L-arginine. Immunology 71, 556–559.
Linehan, S. A., Martinez-Pomares, L., and Gordon, S. (2000). Mannose receptor and scavenger receptor: two macrophage pattern recognition receptors with diverse functions in tissue homeostasis and host defense. Adv. Exp. Med. Biol. 479, 1–14.
Liu, D., Kebaier, C., Pakpour, N., Capul, A. A., Beverley, S. M., Scott, P., and Uzonna, J. E. (2009). Leishmania major phosphoglycans influence the host early immune response by modulating dendritic cell functions. Infect. Immun. 77, 3272–3283.
Marovich, M. A., McDowell, M. A., Thomas, E. K., and Nutman, T. B. (2000). IL-12p70 production by Leishmania major-harboring human dendritic cells is a CD40/CD40 ligand-dependent process. J. Immunol. 164, 5858–5865.
McConville, M. J., and Blackwell, J. M. (1991). Developmental changes in the glycosylated phosphatidylinositols of Leishmania donovani. Characterization of the promastigote and amastigote glycolipids. J. Biol. Chem. 266, 15170–15179.
McNeely, T. B., and Turco, S. J. (1990). Requirement of lipophosphoglycan for intracellular survival of Leishmania donovani within human monocytes. J. Immunol. 144, 2745–2750.
Miles, S. A., Conrad, S. M., Alves, R. G., Jeronimo, S. M., and Mosser, D. M. (2005). A role for IgG immune complexes during infection with the intracellular pathogen Leishmania. J. Exp. Med. 201, 747–754.
Moll, H., Fuchs, H., Blank, C., and Rollinghoff, M. (1993). Langerhans cells transport Leishmania major from the infected skin to the draining lymph node for presentation to antigen-specific T cells. Eur. J. Immunol. 23, 1595–1601.
Moll, H., Scharner, A., and Kampgen, E. (2002). Increased interleukin 4 (IL-4) receptor expression and IL-4-induced decrease in IL-12 production by Langerhans cells infected with Leishmania major. Infect. Immun. 70, 1627–1630.
Mosmann, T. R., and Coffman, R. L. (1989). TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu. Rev. Immunol. 7, 145–173.
Muleme, H. M., Reguera, R. M., Berard, A., Azinwi, R., Jia, P., Okwor, I. B., Beverley, S., and Uzonna, J. E. (2009). Infection with arginase-deficient Leishmania major reveals a parasite number-dependent and cytokine-independent regulation of host cellular arginase activity and disease pathogenesis. J. Immunol. 183, 8068–8076.
Naderer, T., and McConville, M. J. (2008). The Leishmania-macrophage interaction: a metabolic perspective. Cell. Microbiol. 10, 301–308.
Nandan, D., Lo, R., and Reiner, N. E. (1999). Activation of phosphotyrosine phosphatase activity attenuates mitogen-activated protein kinase signaling and inhibits c-FOS and nitric oxide synthase expression in macrophages infected with Leishmania donovani. Infect. Immun. 67, 4055–4063.
Nandan, D., and Reiner, N. E. (1995). Attenuation of gamma interferon-induced tyrosine phosphorylation in mononuclear phagocytes infected with Leishmania donovani: selective inhibition of signaling through Janus kinases and Stat1. Infect. Immun. 63, 4495–4500.
Ng, L. G., Hsu, A., Mandell, M. A., Roediger, B., Hoeller, C., Mrass, P., Iparraguirre, A., Cavanagh, L. L., Triccas, J. A., Beverley, S. M., Scott, P., and Weninger, W. (2008). Migratory dermal dendritic cells act as rapid sensors of protozoan parasites. PLoS Pathog. 4:e1000222. doi: 10.1371/journal.ppat.1000222
Olivier, M., Brownsey, R. W., and Reiner, N. E. (1992). Defective stimulus-response coupling in human monocytes infected with Leishmania donovani is associated with altered activation and translocation of protein kinase C. Proc. Natl. Acad. Sci. U.S.A. 89, 7481–7485.
Olivier, M., Gregory, D. J., and Forget, G. (2005). Subversion mechanisms by which Leishmania parasites can escape the host immune response: a signaling point of view. Clin. Microbiol. Rev. 18, 293–305.
Pelletier, I., Hashidate, T., Urashima, T., Nishi, N., Nakamura, T., Futai, M., Arata, Y., Kasai, K., Hirashima, M., Hirabayashi, J., and Sato, S. (2003). Specific recognition of Leishmania major poly-beta-galactosyl epitopes by galectin-9, possible implication of galectin-9 in interaction between L. major and host cells. J. Biol. Chem. 278, 22223–22230.
Peters, N. C., Egen, J. G., Secundino, N., Debrabant, A., Kimblin, N., Kamhawi, S., Lawyer, P., Fay, M. P., Germain, R. N., and Sacks, D. (2008). In vivo imaging reveals an essential role for neutrophils in leishmaniasis transmitted by sand flies. Science 321, 970–974.
Peters, N. C., and Sacks, D. L. (2009). The impact of vector-mediated neutrophil recruitment on cutaneous leishmaniasis. Cell. Microbiol. 11, 1290–1296.
Pimenta, P. F., Saraiva, E. M., and Sacks, D. L. (1991). The comparative fine structure and surface glycoconjugate expression of three life stages of Leishmania major. Exp. Parasitol. 72, 191–204.
Prive, C., and Descoteaux, A. (2000). Leishmania donovani promastigotes evade the activation of mitogen-activated protein kinases p38, c-Jun N-terminal kinase, and extracellular signal-regulated kinase-1/2 during infection of naive macrophages. Eur. J. Immunol. 30, 2235–2244.
Reiner, N. E. (1994). Altered cell signaling and mononuclear phagocyte deactivation during intracellular infection. Immunol. Today 15, 374–381.
Ribeiro-Gomes, F. L., Moniz-De-Souza, M. C., Alexandre-Moreira, M. S., Dias, W. B., Lopes, M. F., Nunes, M. P., Lungarella, G., and Dosreis, G. A. (2007). Neutrophils activate macrophages for intracellular killing of Leishmania major through recruitment of TLR4 by neutrophil elastase. J. Immunol. 179, 3988–3994.
Ribeiro-Gomes, F. L., Otero, A. C., Gomes, N. A., Moniz-De-Souza, M. C., Cysne-Finkelstein, L., Arnholdt, A. C., Calich, V. L., Coutinho, S. G., Lopes, M. F., and Dosreis, G. A. (2004). Macrophage interactions with neutrophils regulate Leishmania major infection. J. Immunol. 172, 4454–4462.
Ritter, U., Meissner, A., Scheidig, C., and Korner, H. (2004). CD8 alpha- and Langerin-negative dendritic cells, but not Langerhans cells, act as principal antigen-presenting cells in leishmaniasis. Eur. J. Immunol. 34, 1542–1550.
Santos, J. L., Andrade, A. A., Dias, A. A., Bonjardim, C. A., Reis, L. F., Teixeira, S. M., and Horta, M. F. (2006). Differential sensitivity of C57BL/6 (M-1) and BALB/c (M-2) macrophages to the stimuli of IFN-gamma/LPS for the production of NO: correlation with iNOS mRNA and protein expression. J. Interferon Cytokine Res. 26, 682–688.
Schleicher, U., Liese, J., Knippertz, I., Kurzmann, C., Hesse, A., Heit, A., Fischer, J. A., Weiss, S., Kalinke, U., Kunz, S., and Bogdan, C. (2007). NK cell activation in visceral leishmaniasis requires TLR9, myeloid DCs, and IL-12, but is independent of plasmacytoid DCs. J. Exp. Med. 204, 893–906.
Sehgal, G., Zhang, K., Todd, R. F. 3rd., Boxer, L. A., and Petty, H. R. (1993). Lectin-like inhibition of immune complex receptor-mediated stimulation of neutrophils. Effects on cytosolic calcium release and superoxide production. J. Immunol. 150, 4571–4580.
Severn, A., Wakelam, M. J., and Liew, F. Y. (1992). The role of protein kinase C in the induction of nitric oxide synthesis by murine macrophages. Biochem. Biophys. Res. Commun. 188, 997–1002.
Shio, M. T., Hassani, K., Isnard, A., Ralph, B., Contreras, I., Gomez, M. A., Abu-Dayyeh, I., and Olivier, M. (2012). Host cell signalling and Leishmania mechanisms of evasion. J. Trop. Med. 2012, 819512.
Sibley, L. D. (2011). Invasion and intracellular survival by protozoan parasites. Immunol. Rev. 240, 72–91.
Sokol, C. L., Barton, G. M., Farr, A. G., and Medzhitov, R. (2008). A mechanism for the initiation of allergen-induced T helper type 2 responses. Nat. Immunol. 9, 310–318.
Soong, L. (2008). Modulation of dendritic cell function by Leishmania parasites. J. Immunol. 180, 4355–4360.
Sorensen, A. L., Hey, A. S., and Kharazmi, A. (1994). Leishmania major surface protease Gp63 interferes with the function of human monocytes and neutrophils in vitro. APMIS 102, 265–271.
Spath, G. F., Schlesinger, P., Schreiber, R., and Beverley, S. M. (2009). A novel role for Stat1 in phagosome acidification and natural host resistance to intracellular infection by Leishmania major. PLoS Pathog. 5:e1000381. doi: 10.1371/journal.ppat.1000381
St. Denis, A., Caouras, V., Gervais, F., and Descoteaux, A. (1999). Role of protein kinase C-alpha in the control of infection by intracellular pathogens in macrophages. J. Immunol. 163, 5505–5511.
Steinman, R. M. (2007). Lasker Basic Medical Research Award. Dendritic cells: versatile controllers of the immune system. Nat. Med. 13, 1155–1159.
Suzue, K., Kobayashi, S., Takeuchi, T., Suzuki, M., and Koyasu, S. (2008). Critical role of dendritic cells in determining the Th1/Th2 balance upon Leishmania major infection. Int. Immunol. 20, 337–343.
Tachado, S. D., Mazhari-Tabrizi, R., and Schofield, L. (1999). Specificity in signal transduction among glycosylphosphatidylinositols of Plasmodium falciparum, Trypanosoma brucei, Trypanosoma cruzi and Leishmania spp. Parasite Immunol. 21, 609–617.
van Zandbergen, G., Klinger, M., Mueller, A., Dannenberg, S., Gebert, A., Solbach, W., and Laskay, T. (2004). Cutting edge: neutrophil granulocyte serves as a vector for Leishmania entry into macrophages. J. Immunol. 173, 6521–6525.
von der Weid, T., Beebe, A. M., Roopenian, D. C., and Coffman, R. L. (1996). Early production of IL-4 and induction of Th2 responses in the lymph node originate from an MHC class I-independent CD4+NK1.1- T cell population. J. Immunol. 157, 4421–4427.
von Stebut, E., Belkaid, Y., Jakob, T., Sacks, D. L., and Udey, M. C. (1998). Uptake of Leishmania major amastigotes results in activation and interleukin 12 release from murine skin-derived dendritic cells: implications for the initiation of anti-Leishmania immunity. J. Exp. Med. 188, 1547–1552.
Xin, L., Li, K., and Soong, L. (2008). Down-regulation of dendritic cell signaling pathways by Leishmania amazonensis amastigotes. Mol. Immunol. 45, 3371–3382.
Keywords: rodents, parasitic-protozoan, dendritic cells, macrophages, cytokines, innate immunity
Citation: Liu D and Uzonna JE (2012) The early interaction of Leishmania with macrophages and dendritic cells and its influence on the host immune response. Front. Cell. Inf. Microbio. 2:83. doi: 10.3389/fcimb.2012.00083
Received: 02 March 2012; Paper pending published: 30 March 2012;
Accepted: 27 May 2012; Published online: 12 June 2012.
Edited by:
Albert Descoteaux, INRS-Institut Armand-Frappier, CanadaReviewed by:
Lee-Ann H. Allen, University of Iowa, USAJose A. Bengoechea, Fundacion Caubet-CIMERA Illes Balears, Spain
Copyright: © 2012 Liu and Uzonna. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Jude E. Uzonna, Parasite Vaccines Development Laboratory, Department of Immunology, University of Manitoba, 750 McDermot Avenue, Winnipeg, MB R3E 0T5, Canada. e-mail:dXpvbm5hQGNjLnVtYW5pdG9iYS5jYQ==