 Michele J. Anderson1
Michele J. Anderson1 Ying-Chi Lin2
Ying-Chi Lin2 Aaron N. Gillman1 Patrick J. Parks1,3
Aaron N. Gillman1 Patrick J. Parks1,3 Patrick M. Schlievert4
Patrick M. Schlievert4 Marnie L. Peterson1*
Marnie L. Peterson1*- 1 Department of Experimental and Clinical Pharmacology, University of Minnesota, Minneapolis, MN, USA
- 2 School of Pharmacy, College of Pharmacy, Kaohsiung Medical University, Kaohsiung, Taiwan
- 3 3M Skin and Wound Care Division, 3M Company, St. Paul, MN, USA
- 4 Department of Microbiology, Carver College of Medicine, University of Iowa, Iowa City, IA, USA
Staphylococcus aureus causes many diseases in humans, ranging from mild skin infections to serious, life-threatening, superantigen-mediated Toxic Shock Syndrome (TSS). S. aureus may be asymptomatically carried in the anterior nares or vagina or on the skin, serving as a reservoir for infection. Pulsed-field gel electrophoresis clonal type USA200 is the most widely disseminated colonizer and the leading cause of TSS. The cytolysin α-toxin (also known as α-hemolysin or Hla) is the major epithelial proinflammatory exotoxin produced by TSS S. aureus USA200 isolates. The current study aims to characterize the differences between TSS USA200 strains [high (hla+) and low (hla−) α-toxin producers] in their ability to disrupt vaginal mucosal tissue and to characterize the subsequent infection. Tissue viability post-infection and biofilm formation of TSS USA200 isolates CDC587 and MN8, which contain the α-toxin pseudogene (hla−), MNPE (hla+), and MNPE isogenic hla knockout (hlaKO), were observed via LIVE/DEAD® staining and confocal microscopy. All TSS strains grew to similar bacterial densities (1–5 × 108 CFU) on the mucosa and were proinflammatory over 3 days. However, MNPE formed biofilms with significant reductions in the mucosal viability whereas neither CDC587 (hla−), MN8 (hla−), nor MNPE hlaKO formed biofilms. The latter strains were also less cytotoxic than wild-type MNPE. The addition of exogenous, purified α-toxin to MNPE hlaKO restored the biofilm phenotype. We speculate that α-toxin affects S. aureus phenotypic growth on vaginal mucosa by promoting tissue disruption and biofilm formation. Further, α-toxin mutants (hla−) are not benign colonizers, but rather form a different type of infection, which we have termed high density pathogenic variants (HDPV).
Introduction
Biofilms are a community of micro-organisms that are embedded in an extracellular matrix (ECM) composed of lipids, proteins, polysaccharides, and DNA. They are formed in a multi-stage process which includes initial attachment, accumulation, maturation, and dispersion (Gotz, 2002; Hall-Stoodley et al., 2004; Latasa et al., 2006; Heilmann, 2011). The members of a biofilm are protected from environmental factors such as ultraviolet light and dehydration, as well as from host immune factors such as neutrophils and macrophages (Hall-Stoodley et al., 2004). Biofilm-associated bacteria are also much more resistant to antibiotics and antimicrobial agents (Mah and O’Toole, 2001; Stewart and Costerton, 2001). This resistance is on the order of 100- to 1000-fold higher than the planktonic form and can be attributed to genetic factors, growth rate, metabolic factors, and the physical properties of the biofilm ECM (Gilbert et al., 2002).
Recently, there has been a great deal of interest in the role of biofilms in infectious diseases. The National Institutes of Health has estimated that ∼80% of human infections are caused by pathogenic biofilms (biofilms). Biofilm-mediated infections affect wounds, blood, mucosal surfaces (sinuses, respiratory, and genito-urinary tracts), and medical devices (endotracheal tubes, intravascular and urinary catheters, orthopedic implants, and arterial stents; Towne et al., 1994; Donlan, 2001; Bakaletz, 2007; Wolcott et al., 2008; Rohrich et al., 2010). Device-related biofilm infections were the first to be described in the 1980s (Marrie et al., 1982). Staphylococcus epidermidis and Staphylococcus aureus are the most frequent causes of medically associated biofilms (Arciola et al., 2001; Lentino, 2003).
Staphylococcus aureus is an opportunistic pathogen which causes a wide variety of diseases in humans, ranging from relatively mild skin and soft tissue infections to severe, life-threatening necrotizing pneumonia, bacteremia, and Toxic Shock Syndrome (TSS; Lowy, 1998). This organism is the most common cause of ventilator-associated pneumonia, surgical site infections, and catheter-associated bloodstream infections in the healthcare setting (Kuehnert et al., 2005). S. aureus is also a common cause of skin and pulmonary infections in the community (Zetola et al., 2005). Infections caused by S. aureus are often preceded by colonization. In fact, carriers have a threefold higher risk for S. aureus infections (Wertheim et al., 2005). S. aureus is carried asymptomatically in the anterior nares, skin, and vaginal mucosa by up to 30% of the population (Kuehnert et al., 2006; Gorwitz et al., 2008). USA200 methicillin-susceptible S. aureus (MSSA) are the most common pulsed-field gel electrophoresis (PFGE) type isolated from the nose and are a frequent cause of healthcare associated infections (Patel et al., 2008; Tenover et al., 2008). USA200 MSSA are also a major cause of TSS, as they are often associated with expression of the TSS toxin-1 gene (tst; Peacock et al., 2002; Diep et al., 2006; Tenover et al., 2008).
Staphylococcus aureus illnesses are usually initiated at mucosal surfaces or skin. Bacteremia is not typically associated with menstrual TSS; however, toxin penetration across the mucosa can be facilitated by the activity of other exotoxins, including the cytotoxin, α-toxin (Peterson et al., 2005; Brosnahan et al., 2009), which is an exotoxin known to play a role in the pathogenesis of S. aureus (Patel et al., 1987; Bramley et al., 1989). This cytotoxin is secreted as a water soluble monomer which forms a heptameric β-barrel pore in host cell membranes (Song et al., 1996). Targets of the toxin include lymphocytes, macrophages, epithelium, endothelium, and erythrocytes (Bhakdi and Tranum-Jensen, 1991).
Recently, the exoprotein profiles of two USA200 TSS S. aureus isolates, MNPE (α-toxin positive, hla+) and CDC587 (α-toxin mutant, hla−) were biochemically characterized (Lin et al., 2011). The MNPE strain is a pulmonary post-influenza TSS isolate, whereas CDC587 is a menstrual, vaginal TSS isolate which contains a pseudogene for α-toxin (Schlievert et al., 1982; MacDonald et al., 1987). The exoproteins from MNPE were significantly more proinflammatory and cytotoxic, in vitro, to immortalized human vaginal epithelial cells (HVECs) than those from the vaginal isolate CDC587 (Lin et al., 2011). This was due to the presence of large amounts of α-toxin in supernatants from MNPE.
An ex vivo porcine vaginal mucosa infection model was used to further characterize the differences between TSS USA200 S. aureus hla+ and low α-toxin-producing hla− strains with regard to their abilities to infect and disrupt vaginal mucosal tissue. All TSS strains grow to similar bacterial densities on the vaginal mucosa. MNPE (hla+) proved to be highly cytotoxic to the vaginal epithelium as early as 24 h post-infection. Cytotoxic effects of CDC587 (hla−) and MN8 (hla−) are much lower and do not appear until 48 h post-infection. Of particular interest, MNPE forms mature biofilms by 72 h post-infection, whereas CDC587, MN8, and MNPE hlaKO isolates infect the tissue but do not form biofilms. Further, exogenous α-toxin restores the mature biofilm phenotype of the MNPE hlaKO strain. These data suggest that α-toxin contributes to TSS S. aureus mucosal biofilm formation while causing significant disruption to the mucosa. Additionally, the absence of α-toxin did not inhibit TSS S. aureus isolates from infecting the vaginal mucosa, but growth occurred with a different, non-biofilm phenotype. As these non-biofilm-forming strains grow to a high density on the tissue and are proinflammatory, we have termed them high density pathogenic variants (HDPV).
Materials and Methods
Bacteria
USA200 isolates MNPE, CDC587, and MN8 were used extensively in this study (Table 1). All three strains are methicillin sensitive S. aureus (MSSA). MNPE was isolated from a post-influenza pulmonary TSS case in 1987 and most likely originated from a skin source (MacDonald et al., 1987). CDC587 and MN8 are typical menstrual vaginal TSS strains, isolated from patients in the 1980s (Schlievert et al., 1982). Previously described isogenic α-toxin deletion mutants, MNPE hlaKO, CDC587 hlaKO, were used in some studies (Lin et al., 2011). All bacteria are maintained in the laboratory as frozen glycerol stocks. Prior to experimentation, Tryptic Soy Agar containing 5% sheep’s blood (TSB, Becton–Dickinson, Franklin Lakes, NJ, USA) is inoculated from frozen stocks. On the afternoon prior to initiation of experiment, Todd Hewitt broth (Becton–Dickinson) is inoculated with colonies from the fresh TSB plates. Stationary phase (overnight) cultures of each strain are washed in RMPI 1640 (Invitrogen, Carlsbad, CA, USA) and resuspended to a concentration of approximately 5 × 108 colony forming units (CFU/ml). Two microliters of this suspension are used to inoculate explants on the mucosal surface (1 × 106 CFU/explants). Explants are returned to 37°C and incubated for up to 4 days. All studies are performed in a biosafety level 2 (BSL2) laboratory.

Table 1. Staphylococcus aureu s strains used in this study.
Ex vivo Porcine Vaginal Mucosa Preparation and Infection
Porcine vaginal mucosa has been reported to be histologically and structurally similar to human vaginal mucosa (Squier et al., 2008). Therefore, we developed an ex vivo porcine vaginal mucosa infection model to characterize the role of α-toxin in S. aureus mucosal biofilm formation. Normal healthy porcine vaginal tissue is excised from animals at slaughter and transported to the laboratory in RPMI 1640 media supplemented with 10% fetal calf serum, penicillin (50 IU/ml), streptomycin (50 mg/ml), and amphotericin B (2.5 μg/ml). Antibiotics are included to decolonize vaginal normal flora which may interfere with biofilm formation (Iwase et al., 2010). Tissue is utilized within 3 h of excision. Explants of uniform size are obtained from the porcine vagina using a 5-mm biopsy punch. Excess muscle tissue is trimmed away with a scalpel. Tissue explants are washed in antibiotic-free RPMI 1640 media three times. The explants are then placed mucosal side up on a PET track-etched 0.4 μm cell culture insert (BD Bioscience, Franklin Lakes, NJ, USA) in 6-well plates containing fresh serum- and antibiotic-free RPMI 1640 and incubated at 37°C. The mucosal surface is continually exposed to air. The mucosal surface of each is inoculated with ∼1 × 106 CFU of bacteria suspended in antibiotic-free RPMI 1640. Infected explants are incubated at 37°C with 7% CO2. At various times post-infection, samples are removed from the incubator and processed for bacterial recovery and IL-8 and α-toxin analysis. Some samples were stained and imaged by confocal laser scanning microscopy (CLSM) after removal from the incubator.
Bacterial Biofilm and Mucosal Viability Determinations
The physical properties of the LIVE/DEAD® staining kit from Invitrogen and CLSM are utilized to simultaneously determine microbial biofilm formation and corresponding toxicity to mucosal tissue. The FilmTracer™ LIVE/DEAD® Biofilm Viability kit provides a two-color fluorescence assay of bacterial and mucosal viability based on membrane integrity. This kit utilizes mixtures of SYTO® 9 green fluorescent nucleic acid stain and the red-fluorescent nucleic acid stain, propidium iodide. These stains differ both in their spectral characteristics and in their ability to penetrate healthy cells. When used alone, the SYTO® 9 stain generally labels all mucosal epithelial cells and/or bacteria in a population – those with intact membranes and those with damaged membranes. In contrast, propidium iodide penetrates only mucosal cells or bacteria with damaged membranes, causing a reduction in the SYTO® 9 stain fluorescence when both dyes are present. Thus, with the mixture of the SYTO® 9 and propidium iodide stains, mucosal cells or bacteria with intact cell membranes stain fluorescent green, while bacteria or mucosal cells with damaged membranes stain fluorescent red.
Bacterial Recovery from Porcine Vaginal Explants
Bacteria are enumerated from unwashed, infected explants by vortex mixing (highest setting for 2 min or medium high for 4 min) in 250 μl sterile phosphate buffered saline (Sigma, St. Louis, MO, USA). Supernatants are then serially diluted in PBS (or plated neat) and spread on Tryptic Soy Agar containing 5% sheep’s blood (Becton Dickinson, Franklin Lakes, NJ, USA) using a spiral plater (Biotek, Microbiology International, MD, USA).
Quantification of Vaginal Mucosa IL-8
Interleukin-8 (IL-8) is a cytokine which is used as an indicator of inflammation or irritation. The amount of IL-8 produced in the vaginal mucosal explants is determined by homogenizing the explants in 250 μl sterile phosphate buffered saline. The homogenate is clarified by centrifugation and stored at −20°C until assayed. We use a porcine specific IL-8 ELISA (R&D Systems, Minneapolis, MN, USA) to quantify the amount of IL-8 present in each explant.
Alpha-Toxin Detection by Western Immunoblotting
Clarified homogenates are concentrated by ethanol precipitation (five-fold) and resuspended in sterile water. Samples are mixed 1:1 with sample loading buffer and electrophoresed on a 4–20% gradient acrylamide gel (BioRad, Hercules, CA, USA) under reducing conditions, transferred onto polyvinylidene fluoride (PVDF) membranes (BioRad, Hercules, CA, USA) and immunoblotted with rabbit α-toxin anti-sera (obtained as described in Lin et al., 2011). Proteins are detected by chemiluminescence using SuperSignal West Dura Extended Duration substrate (Pierce, Rockford, IL, USA).
Staining and Examination by CLSM
Infected and control explants are stained using FilmTracer™ LIVE/DEAD® Biofilm Viability kit (Invitrogen, Carlsbad, CA, USA) according to manufacturer’s instructions. After staining, specimens are gently washed three times in Hank’s balanced salt solution (Invitrogen, Carlsbad, CA, USA) and transferred to glass slides. A coverslip with 1 mm spacer (Electron Microscopy Sciences, Hatfield, PA, USA) is then applied and specimens are imaged on an Olympus FluoView 1000 BX2 or Zeiss Axio Scope II. Images are captured using either FluoView or LaserSharp software and processed using either FluoView or ImageJ software. All images are 60× original magnification.
Purification of α-Toxin
The cytolysin α-toxin is purified as previously described (Brosnahan et al., 2009). Briefly, α-toxin is isolated from S. aureus strain MNJA grown in beef heart medium. The culture is precipitated with ethanol at 4°C. The precipitate is resolubilized in water, and α-toxin is purified by isoelectric focusing (IEF). IEF is conducted in two phases: the first phase uses a pH gradient of 3.5–10, followed by a second phase using a pH gradient from 7 to 9. The isoelectric point of α-toxin is 8.5. Purity is confirmed by SDS-PAGE and quantified using the Bio-Rad protein assay (Bio-Rad, Hercules, CA, USA).
Alpha-Toxin Treatment
In some experiments, exogenous α-toxin is added to explants before or after infection with MNPE hlaKO. As a pre-treatment, purified α-toxin (5 μg/explant) is added and explants are incubated for 6 h at 37°C. Explants are then infected with MNPE hlaKO and further incubated for 3 day. Alternatively, explants are infected with MNPE hlaKO and incubated at 37°C for 24 h, then 5 μg of purified α-toxin is added and explants are further incubated for two more days. Following incubation, explants are stained and imaged as described above.
Biofilm Formation on Plastic
The role of α-toxin in biofilm formation on plastic is evaluated using two different kinds of 96-well tissue culture plates (TDD 92096, MidSci, St. Louis, MO, USA and Corning 3595, Fisher Scientific, Pittsburgh, PA, USA) according to the procedure described previously (Christensen et al., 1985). Briefly, overnight cultures of S. aureus are diluted 1:20 in Tryptic Soy Broth (BD Bacto, Sparks, MD, USA) supplemented with 0.2% sterile glucose (Sigma Aldrich, St. Louis, MO, USA). Ninety-six well plates are inoculated with 200 μl of diluted bacteria and allowed to incubate for 24 or 72 h at 37°C; care was taken to prevent shaking during incubation. Following incubation, wells are aspirated and gently washed with sterile saline three times. Adherent bacteria are fixed overnight at room temperature with 200 μl of Bouin’s fixative (Ricca Chemicals, Arlington, TX, USA). The fixative is then aspirated and bacteria are stained for 5 min at room temperature with Gentian Violet (Ricca Chemicals, Arlington, TX, USA). Plates are washed under running water. Stain is resolubilized with 30% glacial acetic acid (Fisher Scientific, Pittsburgh, PA, USA) and optical density is read at 570 nm.
Statistical Analysis
The data in Figures 2A,B were analyzed by one-way ANOVA followed by Bonferroni’s post-test using GraphPad Prism 5 Software (GraphPad Software Inc., La Jolla, CA, USA) and are representative of two experiments (pooled) with n = 3–4 per group, per experiment. The data in Figure 5 were analyzed by one-way ANOVA followed by Bonferroni’s post-test using GraphPad Prism 5 software. The results are representative of one experiment with n = 5–8 per strain.
Results
hla+ and hla− Carrying Strains Produce Different Infection Phenotypes on Porcine Vaginal Mucosa
Previous results indicated that secreted exotoxins from MNPE were highly proinflammatory and cytotoxic to immortalized HVECs when compared to secreted exotoxins from a mTSS strain, CDC587 (Lin et al., 2011); thus, it was hypothesized that an α-toxinhigh strain of S. aureus (MNPE, hla+) would be more cytotoxic to full thickness porcine vaginal mucosal epithelium compared to hla− carrying menstrual TSS isolates. To test this hypothesis, a LIVE/DEAD® staining kit from Invitrogen (green = live cells, red = dead cells) and CLSM were employed. Throughout the course of the experiment (up to 72 h), uninfected mucosal tissue remains alive and intact as demonstrated by a predominance of green color (Figures 1A,E,I). In contrast, at 24 h post-infection with MNPE, all of the epithelial cells are stained red and rounded, demonstrating disrupted membranes and loss of cell-to-cell adhesion (Figure 1B). The mucosal epithelium of explants infected with hla− strains (either CDC587 or MN8) remains highly viable (green staining) at 24 h post-infection (Figures 1C,D), although there is evidence of some cell rounding in CDC587-infected explants (Figure 1C), suggesting a loss of cell-to-cell adhesion. At the 48-h timepoint, there is still significant red staining in MNPE-infected explants (Figure 1F). Cytotoxicity and cell rounding are evident in CDC587-infected explants at 48 h post-infection (Figure 1G). The epithelium of explants infected with MN8 also shows signs of disruption at this intermediate time point, although there are still many viable cells and areas of intact cell junctions (Figure 1H). After 72 h, the only evidence of epithelium in MNPE-infected explants are faint red spots in random patterns (Figure 1J, lower right quadrant). In contrast, bright green live epithelial cells remain (although they are sparse) in explants infected with CDC587 (Figure 1K). MN8 appears to be less cytotoxic to the mucosal tissue than either CDC587 or MNPE, as evidenced by larger areas of bright green stained epithelial cells (Figure 1L). These results indicate that strains-producing α-toxin are more cytotoxic to the mucosal epithelium than strains producing low amounts or no α-toxin.
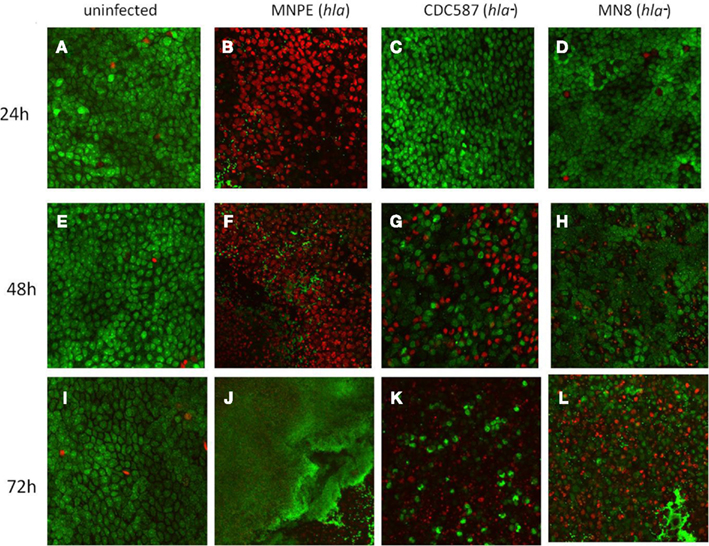
Figure 1. hla+ and hla− carrying strains produce different infection phenotypes on porcine vaginal mucosa. Explants of normal, healthy porcine vaginal mucosa were infected with ∼1 × 106 CFU of live S. aureus strains MNPE, CDC587, or MN8. Uninfected or infected porcine vaginal mucosal epithelium explants were stained with FilmTracer™ LIVE/DEAD stain, then gently washed to remove excess stain. CLSM was used to image explants at the indicated times post-infection. (A,E,I) Uninfected tissue remains live (green) and intact (asymmetrical) throughout the course of the experiment. (B) MNPE-infected tissue is dead (red) by 24 h. Some bacteria are attached to the tissue (small, green staining). (C,D) In contrast, the tissue infected with CDC587 or MN8 resembles uninfected tissue at 24 h. (F) At 48 h, more bacteria are evident in the MNPE-infected tissue. (G,H) Some of the tissue infected with CDC587 or MN8 has died (red) and sloughed away (black). Small, individual cocci (small, bright green punctuate staining) are beginning to appear. (J) At 72 h, MNPE biofilm (diffuse green) has overtaken the tissue. (K) At 72 h post-infection, most of the tissue has been killed by CDC587, very few cocci are observed. (L) At 72 h post-infection with MN8 results in ∼50% cell death (red/black areas). Some macro-colonies are beginning to form in the lower right quadrant. Original magnification was 60×.
The dyes used for CLSM also allowed imaging of the bacteria growing on the porcine vaginal mucosa over time. At 24 h, in MNPE-infected explants, some bacteria are observed (small green, Figure 1B). In contrast, the hla− bacteria are not visualized at this time point, based on a lack of small bright green punctate staining in explants (Figures 1C,D). At the intermediate time point (48 h post-infection), MNPE has entered the accumulation phase of biofilm development (Figure 1F). Microcolonies are observed to be interspersed between dead epithelial cells. Both hla− strains are beginning to be visible on the tissue (small, green, punctate staining, Figures 1G,H). Further, it appears that at least a portion of the MN8 bacteria are intracellular, as evidenced by the small, bright green punctate staining within the more diffuse green staining (Figure 1H). By 72 h post-infection, biofilm has overtaken most of the MNPE-infected explants (large green mass, Figure 1J), which is in stark contrast to explants infected with strains carrying the hla pseudogene (hla−). Only sparse, individual colonies are observed in CDC587-infected explants (Figure 1K). S. aureus growth on MN8-infected explants was intermediate, with microcolonies beginning to form by 72 h (Figure 1L). These data suggest a role for α-toxin in the formation of mucosal biofilms.
S. aureus (hla+ and hla−) Grow to Similar Densities on the Mucosal Surface
The CFU recovered from infected, unwashed tissue were enumerated to determine if there were intrinsic differences in the ability of hla+ or hla− strains to grow on porcine vaginal mucosa. No significant differences across strains were observed in the number of bacteria recovered from the tissue at any time point examined (Figure 2A, n = 6, p > 0.05). CFU values across strains were then pooled and evaluated for an effect of time. There is an increase in the amount of bacteria recovered from 24 to 48 h. Twenty-four hours after infection of the tissue with ∼6.0 Log10 CFU/explant, the bacterial densities are 7.83 ± 0.07 Log10 CFU/explant. By 48 h, the bacterial density is significantly increased to 8.41 ± 0.04 Log10 CFU/explant (n = 18, p < 0.0001). No further increase is observed between 48 and 72 h (8.41 ± 0.02 Log10 CFU/explants, n = 18, p = 0.9543). These data demonstrate that all of the strains grew to a density at which cytolysins would be expressed.
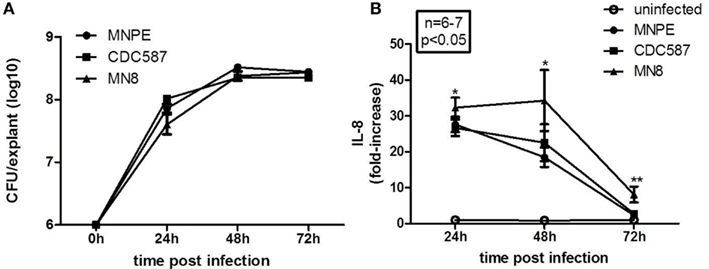
Figure 2. Staphylococcus aureus (hla+ and hla−) grow to similar densities on the mucosal tissue and are proinflammatory. (A) Quantitation of bacteria growth on tissue. Bacteria were recovered from unwashed explants infected with MNPE (circles), CDC587 (squares), or MN8 (triangles). The initial inoculum was ∼1 × 106 CFU/explant. After the indicated time, explants were transferred to tubes containing 250 μl PBS and vortex mixed for 4 min to recover bacteria. Supernatants were serially diluted in PBS and plated on TSB agar. Over the first 24–48 h, the bacteria grew logarithmically. There was no change in density from 48 to 72 h. Data are the combined results of two experiments, with n = 3 each. (B) Quantitation of IL-8 from the tissue during the course of infection. Explants of porcine vaginal mucosa were infected with ∼1 × 106 CFU of MNPE, CDC587, or MN8 and incubated at 37°C for the indicated times. Explants were then placed in tube containing 250 μl PBS and homogenized for 30 s. Homogenates were clarified by centrifugation and IL-8 was detected by porcine specific ELISA. Data were normalized to uninfected controls at each time point. n = 3–4 explants/experiment, results reflect pooled data from two experiments. IL-8 from infected explants was significantly higher than control (uninfected) at 24 and 48 h post-infection (p < 0.05). By 72 h, the amount of IL-8 detected in homogenates of explants from MNPE or CDC587 was reduced compared to earlier time points and is no longer statistically significant. IL-8 detected in MN8-infected explant homogenates was reduced compared to earlier time points, but remains significantly higher than controls.
S. aureus Strains (hla+ or hla−) are Proinflammatory to the Vaginal Mucosa
The cytokine interleukin-8 (IL-8) is chemotactic for neutrophils and is considered a marker of inflammation. A significant increase in the amount of IL-8 produced by vaginal explants at 24 and 48 h post-infection is observed with all strains of S. aureus tested (Figure 2B). At 24 h, explants infected with MNPE, CDC587, or MN8 produce 27.7 ± 2.16-fold, 26.7 ± 2.28-fold, and 32.3 ± 2.79-fold higher levels of IL-8 compared to uninfected explants, respectively. At 48 h post-infection, MNPE-infected explants contain the lowest amount of IL-8, with an 18.5 ± 2.70-fold increase, followed by CDC587-infected explants, which is increased 22.54 ± 5.16-fold. MN8-infected explants contain 34.3 ± 8.52-fold higher IL-8 than uninfected control tissue. By 3 days (72 h) post-infection, the differences in IL-8 production by MNPE and CDC587 compared to that of uninfected control are less than threefold and no longer significantly different. MN8-infected explants produce significantly higher IL-8, 8.09 ± 2.21-fold more than uninfected explants do. The decline in IL-8 detected in MNPE- and CDC587-infected mucosal tissue over time is likely due to a combination of increased cell death and proteolytic degradation. As time increases, the proportion of live cells decreases, as does the concentration of IL-8 detected. These data show that although CDC587 and MN8 are less cytotoxic than MNPE, they are equally proinflammatory to the mucosa.
α-Toxin is Detected in the Mucosa after Infection
The post-influenza TSS isolate (MNPE) and the mTSS isolates (CDC587 and MN8) are known to differ in their ability to produce α-toxin. MNPE, which contains the wild-type gene, expresses large amounts of α-toxin. CDC587 and MN8, which carry the pseudogene, is hypothesized to produce less α-toxin. To test this hypothesis, the accumulation of this toxin in the porcine vaginal mucosa at 72 h was examined by performing western blotting on tissue homogenates (Figure 3A). As expected, no toxin is detected in uninfected tissue, and α-toxin accumulates in the tissue infected with MNPE. Alpha-toxin accumulates in the tissue infected with CDC587 to a far lesser extent and appears to be slightly smaller in size (likely a truncated form of α-toxin). It was a bit surprising that no α-toxin bands are identified with S. aureus MN8 because in a previous exoprotein characterization (Lin et al., 2011), a small band was identified. Homogenates from tissue infected with wild-type MNPE, MNPE hlaKO, wild-type CDC587 or CDC587 hlaKO were analyzed for α-toxin expression to confirm α-toxin specificity of the immunoblotting antibody (Figure 3B). As expected, in the wt MNPE-infected tissue there is an abundance of α-toxin, but a lack of α-toxin in the homogenate from MNPE hlaKO. Again, a small amount of slightly (1–2 kDa) truncated α-toxin in wt CDC587-infected homogenates is observed. This band is not detected in the CDC587 hlaKO-infected homogenates, demonstrating a specific interaction of the anti-sera with α-toxin and indicates that CDC587 indeed expresses low amounts of α-toxin. These data demonstrate that α-toxin accumulates in the vaginal mucosa infected with strains carrying wild-type (hla+) and in contrast is minimally detected in mutant (hla−) strains.
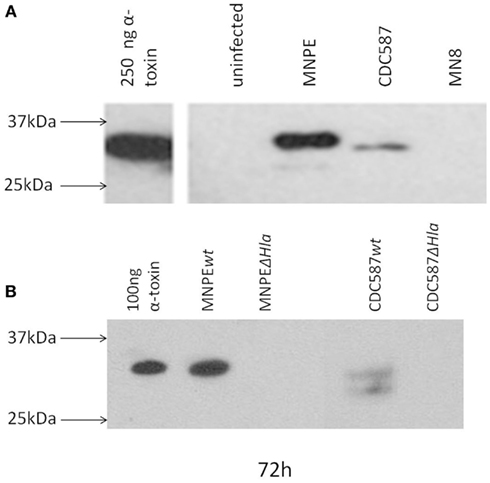
Figure 3. α-Toxin is detected in the mucosa after infection. Detection of α-toxin in explants of porcine vaginal mucosa by western immunoblotting. At 72 h post-infection with different strains of S. aureus, explants were transferred to tubes containing 250 μl PBS and homogenized for 30 s. Homogenates were clarified by centrifugation and concentrated fivefold by ethanol precipitation. Proteins were separated by SDS-PAGE on a 4–20% gradient gel, transferred and probed with rabbit anti-α-toxin serum. (A) Based on the position of purified α-toxin, full-length α-toxin is only detected in tissue infected with MNPE. Truncated forms were detected in CDC587 and MNPE-infected tissue. No bands were observed in tissue infected with MN8 or in uninfected tissue. (B) No bands were detected in hlaKO strains.
α-Toxin Promotes S. aureus Mucosal Biofilm Formation
Rescue experiments with exogenous α-toxin were performed to determine if α-toxin is both necessary and sufficient for biofilm formation. As expected with MNPE, large areas of biofilm (green) over ECM (black) are observed 72 h post-infection (Figure 4B). In contrast, the epithelium of the MNPE hlaKO-infected explants contain many live cells (large green ovals) interspersed with dead cells (large red circles/ovals; Figure 4C). Live bacteria are also visible (small, green, punctate) as individual cocci or small aggregates. Treatment with 5 μg exogenous α-toxin for 72 h resulted in an increase in epithelial cytotoxicity as evidenced by more red staining (Figure 4D) compared to untreated, unifected control (Figure 4A). Treatment with 5 μg exogenous α-toxin for 6 h prior to infection (Figure 4E) or 24 h after infection with the MNPE hlaKO restored the biofilm phenotype (Figure 4F). These data demonstrate that α-toxin is both necessary and sufficient for biofilm formation on a mucosal surface.
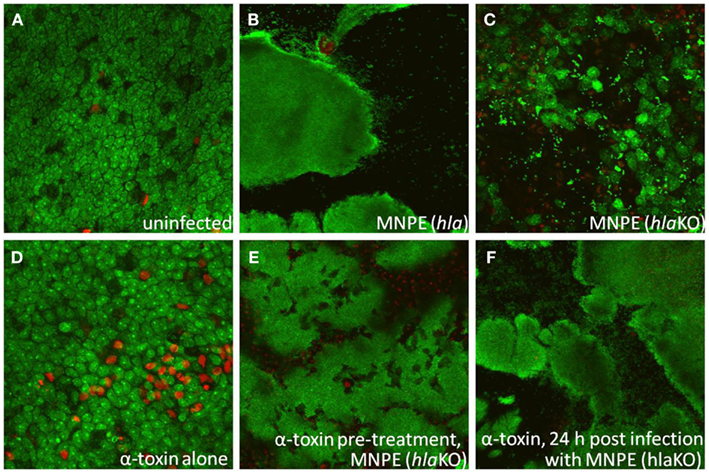
Figure 4. α-Toxin promotes S. aureus biofilm formation. Explants of normal, healthy porcine vaginal mucosa were infected with ∼1 × 106 CFU of live S. aureus strain MNPE or an isogenic α-toxin deletion mutant (MNPE hlaKO). FilmTracer™ LIVE/DEAD stain was used to stain infected porcine vaginal mucosal epithelium explants. CLSM was used to image explants 3 days post-infection. (A) Mucosa is largely viable (green staining) and intact (asymmetry). (B) Large masses of biofilm are observed on mucosa infected with wt MNPE. (C) No biofilm is observed on mucosa infected with MNPE hlaKO. Live individual cocci and small aggregates (small, bright punctuate, and irregular shaped green staining) are evident. Many cells of the mucosa remain alive (green ovals/circles). (D) Treatment of mucosa with 5 μg purified α-toxin alone for 72 h results in a slight increase in cytotoxicity (red staining). (E) Treatment of mucosa with 5 μg purified α-toxin for 6 h prior to infection with MNPE hlaKO restores biofilm formation. (F) Treatment with 5 μg α-toxin 24 h post-infection with MNPE hlaKO restores biofilm formation. Original magnification was 60×.
α-Toxin is Not Required for S. aureus Biofilm Formation on Plastic
As the vast majority of biofilm experiments are done using a plastic substrate and optimal growth media, experiments were performed to determine if α-toxin plays a critical role in that model. Biofilms formed in all S. aureus strains tested by 24 h post-inoculation of 96-well tissue culture plates (Figure 5A). No significant differences were observed between strains. This was also the case at 72 h post-inoculation of the tissue culture plates (Figure 5B). These data suggest that under optimum conditions, α-toxin is not required to form biofilm on plastic.
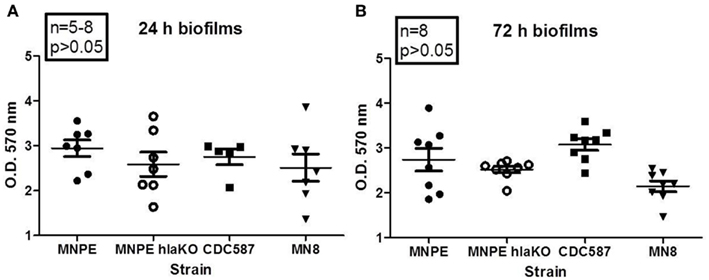
Figure 5. α-Toxin is not required for S. aureus biofilm formation on plastic. Overnight cultures of various strains of S. aureus were diluted 1:20 in TSB + 0.2% glucose and inoculated into 96-well plates. Cultures were returned to 37°C incubator, without shaking, for the indicated times. After incubation, wells were washed, fixed and stained. O.D. at 570 nm was read after solubilizing dye with 30% glacial acetic acid. Robust biofilm forms at 24 h (A) and remains through 72 h (B). No differences were observed between strains, regardless of α-toxin presence.
Discussion
Since the 1980s, biofilms have increasingly been recognized as an important component of human infectious diseases. Biofilm formation by S. aureus is a multi-stage process involving many gene products (Gotz, 2002; Heilmann, 2011). The majority of genes involved in biofilm formation have been identified using in vitro culture techniques. The classic in vitro microtiter plate assay for biofilm formation on abiotic surfaces has been a valuable tool; however, it does not represent the characteristics of biofilm-associated infections in vivo and may have led to an overestimation of the impact of some molecules in clinical biofilm formation (Otto, 2008). In vivo experiments are the gold standard in pathogenesis research, but they are highly labor intensive, expensive, and subject to regulation. Recognizing these limitations, we developed an ex vivo porcine mucosal infection model to study microbial colonization, infection, and biofilm formation on mucosal surfaces. The histological and permeability characteristics of porcine vaginal mucosa are very similar to human vaginal mucosa (Squier et al., 2008).
Staphylococcus aureus initiates diseases at skin and mucosal surfaces, which are also frequent sites of S. aureus colonization. A previously published study identified differences in the secreted proteins between two clonally related USA200 TSS S. aureus isolates, MNPE (hla+) and CDC587 (hla−; Lin et al., 2011). Stationary phase supernatants from these two isolates differed in their epithelial cytotoxicity and proinflammatory potential, which was proportional to the amount of α-toxin produced by each strain. The current study investigates the ability of these strains to infect vaginal mucosa and form biofilms on the mucosal surface. The cytotoxicity and proinflammatory effects of the strains on vaginal mucosal tissue are also characterized. Using an ex vivo porcine vaginal mucosa model, a dramatic difference in infection phenotypes and mucosal vaginal epithelial cytotoxicity in hla+ strains compared to hla− strains was observed. Specifically, the hla+ strain (MNPE) developed a mature biofilm over 3 days and was significantly more cytotoxic over the first 24 h. While the hla− strains CDC587 and MN8 did not form biofilm phenotypes, despite growing to similar bacterial cell densities and being proinflammatory, and were less cytotoxic. This observation is further supported by our findings that disruption of the S. aureus α-toxin gene (MNPE hlaKO) abrogated biofilm formation on mucosal tissue, while the addition of exogenous α-toxin restored the biofilm phenotype. Our proposed mechanism is that S. aureus α-toxin contributes to the formation of biofilms on mucosal surfaces by disrupting the host epithelium, which exposes the ECM for attachment, provides nutrients for bacterial survival, and facilitates intercellular bacterial adhesion.
The western blot analysis revealed an abundance of α-toxin in porcine vaginal mucosa infected with the biofilm-forming MNPE but none in MNPE hlaKO-infected tissue. This is the first study to examine α-toxin production by MNPE in a biofilm state on mucosal tissue and these data are consistent with previous findings of α-toxin in stationary phase supernatants (Lin et al., 2011). In this previous study by Lin et al. western blot analysis also revealed very low amounts of α-toxin secreted by hla− strains of S. aureus (CDC587 and MN8). Additionally, peptide reads on both sides of the stop codon at position 113 were observed via mass spectrometry. Furthermore, a rabbit red blood cell hemolytic assay demonstrated that CDC587 and MN8 had more biological activity (hemolysis) compared to their isogenic α-toxin knockouts. In the present study, tissue homogenates from CDC587 (hla−) contained small amounts of α-toxin and the protein appears to be slightly truncated compared to MNPE and purified α-toxin (Figure 3A). Several truncated forms of α-toxin have been reported in the literature (Kwak et al., 2010; Vecsey-Semjen et al., 2010). These fragments demonstrate significantly lower biological activity than the full-length protein, requiring much higher concentrations and longer incubation times to achieve similar pathological effects (Kwak et al., 2010). Alpha-toxin is not detected from tissue homogenates infected with MN8 (hla−) despite of this strain being very similar to CDC587. This may be a function of the concentration being below the limit of detection for this assay. Regardless, this finding is consistent with a role for α-toxin in biofilm formation in that MN8 and CDC587 strains failed to form biofilms over 3 days.
Classically, α-toxin is thought of as a cytolysin. The toxin binds to host cells, forms a pore and causes leakage of intracellular contents. A recent study reported that α-toxin not only causes irreversible pathological changes to the epithelial membrane by its insertion, but also activates a disintigrin and metalloproteinase 10 (ADAM10). This activation results in the shedding of E-cadherin and severing of homotypic, adherens junction-based linkages between adjacent cells, which causes disruption at the tissue level (Inoshima et al., 2011). Disruption of the tissue caused by α-toxinhigh producing strains likely contributes to the penetration of TSST-1 and other superantigens (SAgs) across vaginal epithelial tissue as has been reported previously (Brosnahan et al., 2009). Low levels of α-toxin or other secreted exotoxins may also contribute to SAg accumulation within the tissue. TSST-1 accumulated within the tissue may then serve as a reservoir for toxin-mediated diseases.
Alpha-toxin has been proposed to play a role in the formation of biofilms on abiotic surfaces. A mutant defective in α-toxin failed to form biofilms in vitro under static conditions in microtiter plate assays and in flow cell chambers (Caiazza and O’Toole, 2003). The proposed mechanism for α-toxin promoting biofilm formation was to facilitate bacterial cell-to-cell interactions, which in turn facilitates macrocolony formation. In contrast to these findings, we observed no significant differences in the ability of the USA200 strains (hla+ and hla−) to form biofilms on plastic microtiter plates. This was not entirely surprising as other factors are known to be important for biofilm formation on abiotic surfaces when organisms are grown under optimal conditions, such as in nutrient-rich media on plastic substrates (Otto, 2008). Unlike plastic microtiter biofilm assays, the vaginal mucosa is a comparatively harsh environment, where nutrients are scarce and bacteria are subject to innate immune defenses, necessitating a role for α-toxin on the vaginal mucosa in biofilm formation. This is supported by our current observation of the hla+ strain having a biofilm phenotype whereas hla− and hlaKO do not.
In contrast to this study, S. aureus strain (UAMS-1), which carries the α-toxin pseudogene (hla−), causes osteomyelitis, a disease considered to be biofilm-associated (Smeltzer et al., 1997). The reasons for this apparent discrepancy are unclear, but perhaps hla− carrying strains of S. aureus eventually form biofilms on mucosal surfaces beyond our tested 3 days. Due to limitations in ex vivo tissue viability, our experiments were conducted for only 3 or 4 days, whereas osteomyelitis takes weeks to develop. Alternatively, α-toxin may not play a role in colonization and subsequent infection of bone, especially since the biofilm substrate of bone is very different from the biofilm substrate of epithelial tissue. Together, in vitro and in vivo studies suggest that regulation of α-toxin by quorum sensing or by the presence of a pseudogene (hla−) and the role of α-toxin in S. aureus biofilm formation on host tissues is complicated and not completely understood. This underscores the need for anatomical site specificity for accurate and clinically predictive models of staphylococcal diseases.
Although the ex vivo porcine mucosal tissue infection model is similar to human vaginal mucosa and was developed to more closely mimic in vivo conditions, the vaginal mucosa was not subjected to vaginal secretions or menstrual fluids, which may cause the outer mucosa to slough and/or impede the ability of the organisms to infect and/or produce exotoxins such as α-toxin. Further, due to the lack of a blood supply, there is no neutrophil influx, which is critical for controlling early bacterial growth. Taking these limitations into consideration, we still believe that our model is an improvement over in vitro abiotic biofilm models.
Our biofilm model is of much shorter experimental duration than some existing models due to tissue viability limitations. However, menstrual TSS commonly occurs within 2–3 days post initiation of menses, which is within the timeframe of our study. In addition, this study only describes one hla+USA200 MSSA TSST-1+ isolate compared to its isogenic hlaKO and two hla− containing organisms. Therefore, whether all hla+ TSST-1+S. aureus isolates would form a biofilm over 72 h on mucosal tissue is unknown. However, we have determined that a methicillin-resistant S. aureus α-toxinhigh, TSST-1+ isolate (I6) is also highly toxic to vaginal mucosal tissue and forms a biofilm by day 3 post-infection (unpublished data).
The role of α-toxin in S. aureus mucosal biofilm formation in vivo has not been characterized. However, this cytolysin is known to play a role in the pathogenesis of S. aureus, based on studies of mutants lacking α-toxin (Bramley et al., 1989). For example, in a murine model of peritonitis, α-toxin was shown to be of critical importance when only mice injected with wild-type hla+ died (Patel et al., 1987). More recently, α-toxin has been reported to play a significant role in lung tissue damage during murine staphylococcal pneumonia (Bubeck Wardenburg and Schneewind, 2008). Researchers determined that virulence correlated with expression levels of α-toxin and further demonstrated that vaccination with a non-pore forming α-toxin (hlaH35L) protected mice from S. aureus pneumonia (Kennedy et al., 2010). Vaccines targeting α-toxin may prevent S. aureus vaginal mucosal biofilm formation, but not S. aureus infection or toxin (SAg)-mediated diseases. This is because α-toxin-deficient strains (MN8 and CDC587) were not avirulent, but rather caused a different type of infection.
We have termed these non-biofilm-forming isolates HDPV because they grow to a similar density on tissue, are proinflammatory to the tissue and can secrete other virulence factors, such as SAgs, which can lead to toxin-mediated diseases. These data also suggest that α-toxin may be a potent therapeutic target for some diseases. More specifically, neutralizing α-toxin may reduce the organism’s ability to form biofilms, rendering it more sensitive to antimicrobial agents and host immune responses. Ultimately, ex vivo tissue models, like the one described, and follow-up in vivo studies are needed to determine the bacterial and host factors which contribute to S. aureus colonization, infection and biofilm formation in humans so optimal therapeutics to prevent and treat staphylococcal infections can be developed.
Conflict of Interest Statement
This work was funded in part by a research grant from 3M, Skin and Wound Care Division, St. Paul, MN, USA (Marnie L. Peterson and Michele J. Anderson); and Patrick J. Parks is the Medical Director and employe of 3M Skin and Wound Care Division and Adjunct Associate Professor in Experimental and Clinical Pharmacology, College of Pharmacy, University of Minnesota.
Acknowledgments
This work was supported by National Institutes of Health, National Institute of Allergy and Infectious Diseases AI-73366 (Marnie L. Peterson) and AI-74283 (Patrick M. Schlievert), 3M (Marnie L. Peterson) and National Science Council, Taiwan 101-2320-B-037-021 (Ying-Chi Lin). We thank Liz Horn and Laurie Hovde for their technical assistance. We would also like to thank Alexander Horswill, University of Iowa, for his assistance in constructing α-toxin mutant strains described previously (Lin et al., 2011) and in this report.
References
Arciola, C. R., Baldassarri, L., and Montanaro, L. (2001). Presence of icaA and icaD genes and slime production in a collection of staphylococcal strains from catheter-associated infections. J. Clin. Microbiol. 39, 2151–2156.
Bakaletz, L. O. (2007). Bacterial biofilms in otitis media: evidence and relevance. Pediatr. Infect. Dis. J. 26(Suppl. 10), S17–S19.
Bhakdi, S., and Tranum-Jensen, J. (1991). Alpha-toxin of Staphylococcus aureus. Microbiol. Rev. 55, 733–751.
Bramley, A. J., Patel, A. H., O’Reilly, M., Foster, R., and Foster, T. J. (1989). Roles of alpha-toxin and beta-toxin in virulence of Staphylococcus aureus for the mouse mammary gland. Infect. Immun. 57, 2489–2494.
Brosnahan, A. J., Mantz, M. J., Squier, C. A., Peterson, M. L., and Schlievert, P. M. (2009). Cytolysins augment superantigen penetration of stratified mucosa. J. Immunol. 182, 2364–2373.
Bubeck Wardenburg, J., and Schneewind, O. (2008). Vaccine protection against Staphylococcus aureus pneumonia. J. Exp. Med. 205, 287–294.
Caiazza, N. C., and O’Toole, G. A. (2003). Alpha-toxin is required for biofilm formation by Staphylococcus aureus. J. Bacteriol. 185, 3214–3217.
Christensen, G. D., Simpson, W. A., Younger, J. J., Baddour, L. M., Barrett, F. F., Melton, D. M., and Beachey, E. H. (1985). Adherence of coagulase-negative staphylococci to plastic tissue culture plates: a quantitative model for the adherence of staphylococci to medical devices. J. Clin. Microbiol. 22, 996–1006.
Diep, B. A., Carleton, H. A., Chang, R. F., Sensabaugh, G. F., and Perdreau-Remington, F. (2006). Roles of 34 virulence genes in the evolution of hospital- and community-associated strains of methicillin-resistant Staphylococcus aureus. J. Infect. Dis. 193, 1495–1503.
Gilbert, P., Allison, D. G., and McBain, A. J. (2002). Biofilms in vitro and in vivo: do singular mechanisms imply cross-resistance? J. Appl. Microbiol. 92(Suppl.), 98S–110S.
Gorwitz, R. J., Kruszon-Moran, D., McAllister, S. K., McQuillan, G., McDougal, L. K., Fosheim, G. E., Jensen, B. J., Killgore, G., Tenover, F. C., and Kuehnert, M. J. (2008). Changes in the prevalence of nasal colonization with Staphylococcus aureus in the United States, 2001–2004. J. Infect. Dis. 197, 1226–1234.
Hall-Stoodley, L., Costerton, J. W., and Stoodley, P. (2004). Bacterial biofilms: from the natural environment to infectious diseases. Nat. Rev. Microbiol. 2, 95–108.
Inoshima, I., Inoshima, N., Wilke, G. A., Powers, M. E., Frank, K. M., Wang, Y., and Bubeck Wardenburg, J. (2011). A Staphylococcus aureus pore-forming toxin subverts the activity of ADAM10 to cause lethal infection in mice. Nat. Med. 17, 1310–1314.
Iwase, T., Uehara, Y., Shinji, H., Tajima, A., Seo, H., Takada, K., Agata, T., and Mizunoe, Y. (2010). Staphylococcus epidermidis Esp inhibits Staphylococcus aureus biofilm formation and nasal colonization. Nature 465, 346–349.
Kennedy, A. D., Bubeck Wardenburg, J., Gardner, D. J., Long, D., Whitney, A. R., Braughton, K. R., Schneewind, O., and DeLeo, F. R. (2010). Targeting of alpha-hemolysin by active or passive immunization decreases severity of USA300 skin infection in a mouse model. J. Infect. Dis. 202, 1050–1058.
Kuehnert, M. J., Hill, H. A., Kupronis, B. A., Tokars, J. I., Solomon, S. L., and Jernigan, D. B. (2005). Methicillin-resistant-Staphylococcus aureus hospitalizations, United States. Emerging Infect. Dis. 11, 868–872.
Kuehnert, M. J., Kruszon-Moran, D., Hill, H. A., McQuillan, G., McAllister, S. K., Fosheim, G., McDougal, L. K., Chaitram, J., Jensen, B., Fridkin, S. K., Killgore, G., and Tenover, F. C. (2006). Prevalence of Staphylococcus aureus nasal colonization in the United States, 2001–2002. J. Infect. Dis. 193, 172–179.
Kwak, Y. K., Hogbom, M., Colque-Navarro, P., Möllby, R., and Vécsey-Semjén, B. (2010). Biological relevance of natural alpha-toxin fragments from Staphylococcus aureus. J. Membr. Biol. 233, 93–103.
Latasa, C., Solano, C., Penadés, J. R., and Lasa, I. (2006). Biofilm-associated proteins. C. R. Biol. 329, 849–857.
Lentino, J. R. (2003). Prosthetic joint infections: bane of orthopedists, challenge for infectious disease specialists. Clin. Infect. Dis. 36, 1157–1161.
Lin, Y. C., Anderson, M. J., Kohler, P. L., Strandberg, K. L., Olson, M. E., Horswill, A. R., Schlievert, P. M., and Peterson, M. L. (2011). Proinflammatory exoprotein characterization of toxic shock syndrome Staphylococcus aureus. Biochemistry 50, 7157–7167.
MacDonald, K. L., Osterholm, M. T., Hedberg, C. W., Schrock, C. G., Peterson, G. F., Jentzen, J. M., Leonard, S. A., and Schlievert, P. M. (1987). Toxic shock syndrome. A newly recognized complication of influenza and influenzalike illness. JAMA 257, 1053–1058.
Mah, T. F., and O’Toole, G. A. (2001). Mechanisms of biofilm resistance to antimicrobial agents. Trends Microbiol. 9, 34–39.
Marrie, T. J., Nelligan, J., and Costerton, J. W. (1982). A scanning and transmission electron microscopic study of an infected endocardial pacemaker lead. Circulation 66, 1339–1341.
Patel, A. H., Nowlan, P., Weavers, E. D., and Foster, T. (1987). Virulence of protein A-deficient and alpha-toxin-deficient mutants of Staphylococcus aureus isolated by allele replacement. Infect. Immun. 55, 3103–3110.
Patel, M., Waites, K. B., Hoesley, C. J., Stamm, A. M., Canupp, K. C., and Moser, S. A. (2008). Emergence of USA300 MRSA in a tertiary medical centre: implications for epidemiological studies. J. Hosp. Infect. 68, 208–213.
Peacock, S. J., Moore, C. E., Justice, A., Kantzanou, M., Story, L., Mackie, K., O’Neill, G., and Day, N. P. (2002). Virulent combinations of adhesin and toxin genes in natural populations of Staphylococcus aureus. Infect. Immun. 70, 4987–4996.
Peterson, M. L., Ault, K., Kremer, M. J., Klingelhutz, A. J., Davis, C. C., Squier, C. A., and Schlievert, P. M. (2005). The innate immune system is activated by stimulation of vaginal epithelial cells with Staphylococcus aureus and toxic shock syndrome toxin 1. Infect. Immun. 73, 2164–2174.
Rohrich, R. J., Monheit, G., Nguyen, A. T., Brown, S. A., and Fagien, S. (2010). Soft-tissue filler complications: the important role of biofilms. Plast. Reconstr. Surg. 125, 1250–1256.
Schlievert, P. M., Osterholm, M. T., Kelly, J. A., and Nishimura, R. D. (1982). Toxin and enzyme characterization of Staphylococcus aureus isolates from patients with and without toxic shock syndrome. Ann. Intern. Med. 96(6 Pt 2), 937–940.
Smeltzer, M. S., Thomas, J. R., Hickmon, S. G., Skinner, R. A., Nelson, C. L., Griffith, D., Parr, T. R. Jr., and Evans, R. P. (1997). Characterization of a rabbit model of staphylococcal osteomyelitis. J. Orthop. Res. 15, 414–421.
Song, L., Hobaugh, M. R., Shustak, C., Cheley, S., Bayley, H., and Gouaux, J. E. (1996). Structure of staphylococcal alpha-hemolysin, a heptameric transmembrane pore. Science 274, 1859–1866.
Squier, C. A., Mantz, M. J., Schlievert, P. M., and Davis, C. C. (2008). Porcine vagina ex vivo as a model for studying permeability and pathogenesis in mucosa. J. Pharm. Sci. 97, 9–21.
Stewart, P. S., and Costerton, J. W. (2001). Antibiotic resistance of bacteria in biofilms. Lancet 358, 135–138.
Tenover, F. C., McAllister, S., Fosheim, G., Mcdougal, L. K., Carey, R. B., Limbago, B., Lonsway, D., Patel, J. B., Kuehnert, M. J., and Gorwitz, R. (2008). Characterization of Staphylococcus aureus isolates from nasal cultures collected from individuals in the United States in 2001 to 2004. J. Clin. Microbiol. 46, 2837–2841.
Towne, J. B., Seabrook, G. R., Bandyk, D., Freischlag, J. A., and Edmiston, C. E. (1994). In situ replacement of arterial prosthesis infected by bacterial biofilms: long-term follow-up. J. Vasc. Surg. 19, 226–233; discussion 233–225.
Vecsey-Semjen, B., Kwak, Y. K., Högbom, M., and Möllby, R. (2010). Channel-forming abilities of spontaneously occurring alpha-toxin fragments from Staphylococcus aureus. J. Membr. Biol. 234, 171–181.
Wertheim, H. F., Melles, D. C., Vos, M. C., van Leeuwen, W., van Belkum, A., Verbrugh, H. A., and Nouwen, J. L. (2005). The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect. Dis. 5, 751–762.
Wolcott, R. D., Rhoads, D. D., and Dowd, S. E. (2008). Biofilms and chronic wound inflammation. J. Wound Care 17, 333–341.
Keywords: alpha-toxin, Staphylococcus aureus, biofilm, epithelium, vaginal mucosa, toxic shock syndrome
Citation: Anderson MJ, Lin Y-C, Gillman AN, Parks PJ, Schlievert PM and Peterson ML (2012) Alpha-toxin promotes Staphylococcus aureus mucosal biofilm formation. Front. Cell. Inf. Microbio. 2:64. doi: 10.3389/fcimb.2012.00064
Received: 28 January 2012; Accepted: 20 April 2012;
Published online: 09 May 2012.
Edited by:
Martin John McGavin, University of Western Ontario, CanadaReviewed by:
Sang S. Yoon, Yonsei University College of Medicine, South KoreaChristiane Wolz, University Tübingen, Germany
Daniel O. Sordelli, University of Buenos Aires, Argentina
Copyright: © 2012 Anderson, Lin, Gillman, Parks, Schlievert and Peterson. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Marnie L. Peterson, Department of Experimental and Clinical Pharmacology, College of Pharmacy, University of Minnesota, 4-212 McGuire Translational Research Facility, 2001 6th Street SE, Minneapolis, MN 55455, USA. e-mail:cGV0ZXIzNzdAdW1uLmVkdQ==