
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Infect. Microbiol. , 20 April 2012
Sec. Molecular Bacterial Pathogenesis
Volume 2 - 2012 | https://doi.org/10.3389/fcimb.2012.00046
This article is part of the Research Topic Research Advances in the Study of Campylobacter, Helicobacter & Related Organisms View all 29 articles
 Linda H. Ta1 Lori M. Hansen1 William E. Sause2 Olga Shiva3 Aram Millstein1 Karen M. Ottemann2
Linda H. Ta1 Lori M. Hansen1 William E. Sause2 Olga Shiva3 Aram Millstein1 Karen M. Ottemann2 Andrea R. Castillo2*
Andrea R. Castillo2* Jay V. Solnick1*
Jay V. Solnick1*The Helicobacter pylori cag pathogenicity island (cag PAI) encodes a type IV secretion system that is more commonly found in strains isolated from patients with gastroduodenal disease than from those with asymptomatic gastritis. Genome-wide organization of the transcriptional units in H. pylori strain 26695 was recently established using RNA sequence analysis (Sharma et al., 2010). Here we used quantitative reverse-transcription polymerase chain reaction of open reading frames and intergenic regions to identify putative cag PAI operons in H. pylori; these operons were analyzed further by transcript profiling after deletion of selected promoter regions. Additionally, we used a promoter-trap system to identify functional cag PAI promoters. The results demonstrated that expression of genes on the H. pylori cag PAI varies by nearly five orders of magnitude and that the organization of cag PAI genes into transcriptional units is conserved among several H. pylori strains, including, 26695, J99, G27, and J166. We found evidence for 20 transcripts within the cag PAI, many of which likely overlap. Our data suggests that there are at least 11 operons: cag1-4, cag3-4, cag10-9, cag8-7, cag6-5, cag11-12, cag16-17, cag19-18, cag21-20, cag23-22, and cag25-24, as well as five monocistronic genes (cag4, cag13, cag14, cag15, and cag26). Additionally, the location of four of our functionally identified promoters suggests they are directing expression of, in one case, a truncated version of cag26 and in the other three, transcripts that are antisense to cag7, cag17, and cag23. We verified expression of two of these antisense transcripts, those antisense to cag17 and cag23, by reverse-transcription polymerase chain reaction. Taken together, our results suggest that the cag PAI transcriptional profile is generally conserved among H. pylori strains, 26695, J99, G27, and J166, and is likely complex.
Helicobacter pylori is a Gram-negative bacterium that infects the stomachs of approximately half the human population. Although infection is typically asymptomatic throughout the lifetime of the host, it causes peptic ulcer disease in about 10% of those infected and gastric adenocarcinoma in about 1–3% (Kusters et al., 2006). The best-studied bacterial factor associated with clinical sequelae of H. pylori infection is the cytotoxin associated gene pathogenicity island (cag PAI). Patients infected with H. pylori strains that contain the cag PAI are at increased risk for both peptic ulcer and gastric cancer (Kusters et al., 2006). Experimental studies in gerbils (Rieder et al., 2005), mice (Arnold et al., 2011), and rhesus macaques (Hornsby et al., 2008) have also demonstrated the pro-inflammatory effects of the cag PAI.
The 40-kb cag PAI contains on average 27 genes, several of which encode a type IV secretion apparatus that is required for translocation of the effector molecules CagA (cag26) and peptidoglycan into host epithelial cells (Segal et al., 1997; Odenbreit et al., 2000; Rohde et al., 2003; Viala et al., 2004). Of the 27 genes on the cag PAI, 18 are required for the translocation of CagA into host cells and 15 are required to induce transcription of the pro-inflammatory cytokine IL-8 (Fischer et al., 2001; Shaffer et al., 2011). CagA is reliant on the secretion chaperone protein CagF (cag22) for recruitment to the type IV translocation channel (Pattis et al., 2007). Upon translocation into the cell, CagA is phosphorylated at C-terminal tyrosine residues by c-Src and other kinases, which results in the activation of receptor tyrosine kinase (RTK)-like signaling pathways (Segal et al., 1997; Selbach et al., 2002). Both phosphorylated and unphosphorylated CagA contribute to H. pylori pathogenesis via multiple mechanisms, including the disruption of the cytoskeleton, interruption of cellular signaling, and interference with adhesion between adjacent cells (Backert and Selbach, 2008).
Several studies have provided a glimpse of the cag PAI transcriptional unit organization. One initial study employed a urease transcription fusion to check for promoters in nine cag PAI DNA regions that were upstream of groups of co-directional genes (Joyce et al., 2001). This analysis determined that there were at least five promoters on the cag PAI. Another early study identified the promoters responsible for regulating cagA and cagB (Spohn et al., 1997). A more recent genome-wide transcriptional unit analysis that used RNA sequencing identified 14 transcriptional units within the cag PAI. Additionally, they found many potential small regulatory RNAs (Sharma et al., 2010). Other studies have suggested that several cag PAI genes are differentially regulated in vivo compared to in vitro (Joyce et al., 2001; Boonjakuakul et al., 2005; Castillo et al., 2008b). In one such study, an in vivo induced promoter called Pivi66, was internal to the cag7 gene (Castillo et al., 2008b), which suggested that promoters may not always be within intergenic regions.
Here we sought to determine the conservation of operon structure in the cag PAI among H. pylori strains, and to identify promoters responsible for the transcription of cag PAI genes in strains 26695, J99, and G27, whose genomes are sequenced (Tomb et al., 1997; Alm et al., 1999), and in strain J166 that we and others have used to infect rhesus macaques (Hornsby et al., 2008). Operon structure was first predicted by a gene expression analysis that used quantitative reverse-transcriptase polymerase chain reaction (qRT-PCR) for both open reading frames (ORFs) and intergenic regions. The predicted putative operons were further defined by qRT-PCR after deletion of selected promoter regions. Since our transcription analyses suggested a potentially complex operon structure, we augmented these studies with a non-biased promoter-trap study that identified cag PAI promoters as DNA regions capable of directing expression of a heterologous reporter. Our results demonstrate that there is remarkable consistency across strains in the expression of genes in the cag PAI, which is organized into at least 20 transcriptional units.
Helicobacter pylori strains 26695 (Tomb et al., 1997), J99 (Alm et al., 1999), J166 (Hornsby et al., 2008), and ACHP17 (mG27 HP0294/295::res1-aphA3-res1; Castillo et al., 2008a) were used for these studies. DNA and RNA for qRT-PCR were prepared from strains cultured on Brucella agar (Difco Laboratories, Detroit, MI, USA) containing 5% bovine calf serum (Invitrogen Life Technologies, Carlsbad, CA, USA) supplemented with 5 μg/mL trimethoprim, 10 μg/mL vancomycin, 2.5 IU/mL polymixin B, 2.5 μg/mL amphotericin B (TVPA, all from Sigma, St. Louis, MO, USA) and incubated at 37°C with an atmosphere that contained 5% CO2. Plate grown bacteria were then transferred to Brucella broth containing bovine calf serum with TVPA and incubated at 37°C in 5% CO2 with gentle rotation at 60 rpm. The OD600 was determined for each culture 18–24 h after inoculation. The promoter reporter H. pylori strain ACHP17 and strain G27 from which RNA was isolated for RT-PCR were grown under microaerobic conditions (10% CO2, 5% O2, and 85% N2) at 37°C on columbia blood agar plates with 4% (w/v) columbia agar base, 5% (w/v) defibrinated horse blood (Hemostat labs), 0.2% (w/v) β-cyclodextrin, 10 μg/mL vancomycin, 50 μg/mL cycloheximide, 5 μg/mL cefsulodin, 8 μg/mL amphotericin B, 2.5 IU/mL polymyxin and 5 μg/mL trimethoprim. H. pylori strains were stored at −80°C in brain heart infusion media supplemented with 10% fetal bovine serum, 1% (w/v) β-cyclodextrin, 25% glycerol, and 5% dimethyl sulfoxide.
Escherichia coli strain DH10B (Grant et al., 1990) was grown at 37°C in Luria–Bertani (LB) broth (1% w/v tryptone, 0.5% w/v yeast extract, 0.5% w/v NaCl), with 100 μg/mL ampicillin. E. coli was also grown on solid LB media consisting of LB broth with 1.5% (w/v) agar. All antibiotics were purchased from Sigma-Aldrich, Fisher, or ISC BioExpress. All culture media were purchased from Remel, Fisher, or Difco unless otherwise indicated.
At OD600 0.4–0.5 (early exponential growth phase) 2 mL aliquots were taken from H. pylori liquid cultures and centrifuged at 16,000 × g for 30 s at room temperature. Supernatants were removed and 1 mL of TriZol (Invitrogen) was immediately added. Samples were vortexed and RNA was extracted according to the manufacturer’s directions. RNA was treated with DNase I (Roche Applied Sciences, Mannheim, Germany), purified using an RNeasy clean up kit (QIAGEN, Inc., Valencia, CA, USA), and suspended in ultra pure water (Invitrogen) at a concentration of 20 ng/μL.
DNA was extracted from plate grown bacteria using a DNeasy Tissue Kit (QIAGEN). DNA samples were diluted in ultra pure water to a concentration of 5 ng/μL and stored at −20°C.
Reverse-transcriptase polymerase chain reactions were carried out using the Super Script One-step RT-PCR kit with Platinum Taq (Invitrogen). One hundred or 250 ng of RNA was used as a template for each RT-PCR reaction. For the reverse-transcription step (55°C for 30 min), only the oligonucleotide that was complementary to the putative transcript was included in the reaction, PIIIR (5′-cctagcgaccaaaagcgatgaa-3′), PIXR (5′-gaaactgctaagaatatcagtg-3′), and PXIIR (5′-cgtcattaatcaaatagaacaaagc-3′). The reverse-transcriptase in these reactions was then inactivated by incubation at 94°C for 5 min. Prior to starting the PCR program (35 cycles, 94°C/30 s, 55°C/30 s, 72°C/30 s) the reactions were briefly incubated on ice (∼1 min) while the second oligonucleotides, PIIIF (5′-cattgtggtctttcccgaaagc-3′), PIXF (5′-cactcttgcctataaaggcc-3′), and PXIIF (5′-ctgagacgacaagctatgatttc-3′) were added. Oligonucleotides for our positive control were HP188F (5′-ccactataaaagagatctttcaagcggaagg-3′) and HP187R (5′-gcttgccctcggtgtctgcatc-3′); HP187R was present in the RT reaction and both HP187R and HP188F were present in the PCR reaction. As a control for amplification, each set of oligonucleotides was used in a PCR reaction with DNA as the template. Additionally, each set of oligonucleotides was used in an RT-PCR reaction with the RNA template and Platinum Taq only. This control was done to verify our RNA samples were DNA free.
Quantitative real time RT-PCR was performed with primer pairs specific for each cag gene (Table A1 in Appendix) and for each intergenic region (Table A2 in Appendix), using methods essentially as described (Boonjakuakul et al., 2004, 2005). In brief, RT and PCR were performed in a single 20 μL reaction mixture using the thermostable recombinant Tth (rTth) DNA polymerase (Applied Biosystems) with 100 ng RNA extracted as described above. In the presence of Mn(OAc)2, rTth has reverse-transcriptase activity and DNA polymerase activity. Two-step amplification was performed with 45 cycles at 95°C for 20 s followed by 59.5°C for 1 min. Accumulation of PCR product was detected during each cycle by excitation of SYBR green at 490 nm. Relative fluorescence was characterized by a cycle threshold (Ct) value, which was defined as the crossover point of the kinetic curve with an arbitrary fluorescence level set at 150 relative fluorescence units. The absence of contaminating DNA was examined by performing the RT-PCR with MgCl2, in which rTth has DNA polymerase but no RT activity. All qRT-PCR products were electrophoresed on a 2% agarose (Invitrogen) gel to verify correct product size. Transcript abundance was calculated only if the observed Ct with RNA template was less than that of the no-template control, and there was a band of the appropriate size on an agarose gel. Otherwise, transcript was considered absent. All transcript copy numbers were normalized to 16S RNA and the data presented represents the average of duplicate wells.
The chloramphenicol resistance conferring cat gene from plasmid pNR9589 (Wang and Taylor, 1990) and 1–2 kb DNA fragments of the genes directly flanking the region targeted for deletion were PCR amplified (oligonucleotides in Table A3 in Appendix) with compatible restriction sites. All three fragments were digested with the appropriate enzymes and ligated with compatibly digested pBluescript SK− (Stratagene, La Jolla, CA, USA) to generate a shuttle plasmid with fragments of the cag PAI flanking the cat gene. The shuttle plasmid was amplified in E. coli Top10 (Invitrogen, Carlsbad, CA, USA), sequence verified, and then used to transform H. pylori strain J166 by a standard natural transformation procedure (Salama et al., 2001). H. pylori transformants were selected on Brucella agar plates with TVPA and 4 μg/mL chloramphenicol. Correct replacement of cag PAI DNA regions with the cat gene was verified using PCR and DNA sequence analyses.
Genomic DNA was isolated from H. pylori J166 and mG27 (Wizard genomic prep kit, Promega). The DNA region representing the cag PAI was amplified from each strain as a set of 13 PCR products of ∼2.5 kb in length with 600 bp of overlap between adjacent PCR products (oligonucleotides in Table A4 in Appendix). For each strain, the PCR products were pooled, partially digested with Sau3A, and ligated to BglII digested pcat-T-tnpR (Castillo et al., 2008a) to generate recombinant plasmids, pcat-T-caglibmG27-tnpR and pcat-T-caglibJ166-tnpR. After ligation, the recombinant plasmids were transformed into E. coli DH10B and the E. coli were plated on LB agar with ampicillin. For these strains, ∼2193 (pcat-T-caglibmG27-tnpR) or 5000 (pcat-T-caglibmJ166-tnpR) individual ampicillin resistant (AmpR) colonies were pooled, grown overnight, and treated (Qiagen miniprep extraction kit, Qiagen) to extract the recombinant plasmids. For a subset of colonies from each library, individual recombinant plasmids were analyzed for the presence and size of a H. pylori cag PAI insert. All recombinant plasmids analyzed contained inserts and had an average insert size of 469 bp for pcat-T-caglibmG27-tnpR and 96 bp for pcat-T-caglibmJ166-tnpR.
To isolate putative promoters, H. pylori strain ACHP17 was transformed using natural transformation (Salama et al., 2001) with either pcat-T-caglibmG27-tnpR or pcat-T-caglibJ166-tnpR, and transformants were selected based on their resistance to chloramphenicol (Cm) on CBA plus 13 μg/mL Cm. Cm resistant (CmR) transformants were passed twice on Cm prior to being analyzed for kanamycin sensitivity (KmS) on CBA plus 15 μg/mL kanamycin.
To examine the diversity of the cag PAI library clones in H. pylori, 10–30 CmR clones were selected from each library and the region upstream of tnpR was sequenced using primers rrnB1 and tnpRbk75 (Castillo et al., 2008a). The average insert size was 232 bp for pcat-T-caglibmG27-tnpR and 100 bp for pcat-T-caglibJ166-tnpR. PCR amplicons were sequenced and compared to the 26695 and G27 genomes to assess randomness of the cloned regions. The number of transformants needed to obtain 100% coverage of the cag PAI for each library was determined using the formula N = ln[1 − P/ln(1 − I/G)] (N = number of independent clones, I = size of averaged cloned fragment, G = size of target genome, and P = probability). These calculations suggested that 791 pcat-T-caglibmG27-tnpR and 1840 pcat-T-caglibJ166-tnpR transformants would be required for complete coverage of the cag PAI.
We used both transcription profiling and a functional genetic approach to define cag PAI operons and the putative promoters that regulate them. First, we performed qRT-PCR to determine the mRNA copy number within each ORF and each intergenic region on the cag PAI. Our assumption was that contiguous genes transcribed in the same direction, with the presence of intergenic message and similar mRNA copy number, would likely form an operon. Selected putative operons were then further analyzed by deletion of the promoter region and reanalysis of mRNA copy number of downstream genes. We then augmented these analyses by using a non-biased promoter-trap system to find active promoters within the cag PAI.
We first calculated the transcript copy number for each gene and intergenic region within the cag PAI of H. pylori J99, 26695, and J166, using methods described previously (Boonjakuakul et al., 2004, 2005). Briefly, three factors were used to calculate copies per cell: (a) a 10-fold change in starting template concentration corresponds to a 3.3-cycles change in Ct (23.3 = 10); (b) 100 ng of RNA equals 106H. pylori cells, and (c) the empirically derived observation that a Ct of 19 corresponds to 1 × 105 copies of starting DNA template (assuming 1 copy per bacterial chromosome). We have previously shown that calculation of mRNA copies/cell using Ct corrected for primer efficiency yields values that are essentially identical to those obtained by the more conventional method using standard curves (Boonjakuakul et al., 2004).
Transcript levels for all genes on the cag PAI for each H. pylori strain are shown in Figure 1. For clarity, intergenic transcript is shown only as present (adjacent bars representing gene transcript levels are shaded identically) or absent (adjacent bars are shaded differently). For example, intergenic transcript was detected between cag1 and cag2 (both black bars) and between cag10 and cag9 (both gray bars), but not between cag15 (gray bar) and cag14 (black bar). Transcript levels varied within each strain by as much as five orders of magnitude, ranging from about 10 copies/cell to as low as 1 copy per 10,000 cells. These estimates are consistent with our previous studies (Boonjakuakul et al., 2004, 2005) and with estimates of gene expression levels in Saccharomyces cerevisiae (Kang et al., 2000) and E. coli (Young and Bremer, 1975). The highest transcript abundance was found for cag26 and for cag25. Since cag26 encodes an effector protein, CagA, secreted via the type IV secretion system, and cag25 encodes a virB2 ortholog that is thought to encode a pilin protein that forms a multimeric structure (Andrzejewska et al., 2006), it is not surprising that these genes are highly expressed. Although in general, the expression level of genes on the cag PAI was similar across the three strains analyzed, there is some variation that appears to occur within the operons predicted by these experiments (Figure 2).
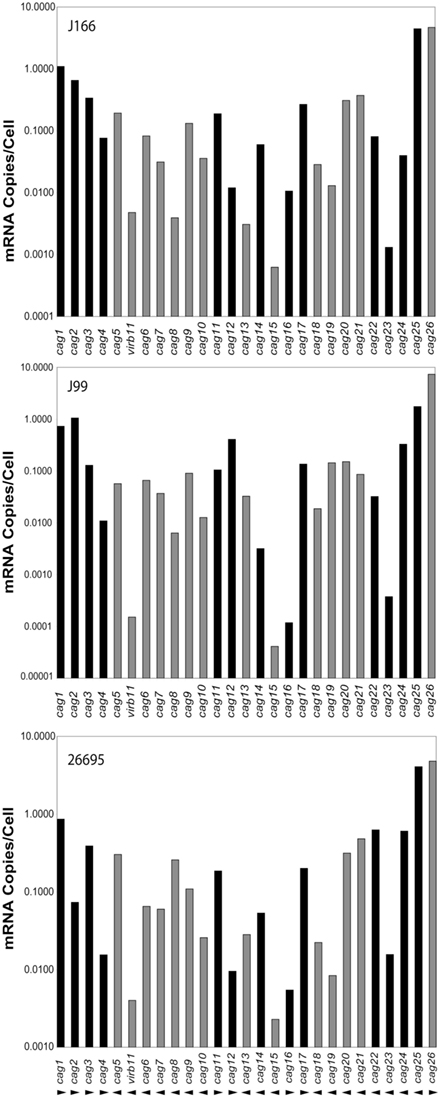
Figure 1. Expression level of each gene on the cag PAI (mRNA copies/cell; normalized to 16S rRNA) for H. pylori strains J166 (top panel), J99 (middle panel), and 26695 (bottom panel). Our data represents the average of duplicate reactions. Adjacent genes for which intergenic transcript was detected are indicated with the same shading (black or gray). Direction of transcription is shown by arrowheads below the bottom panel. Since cag26 (cagA) is not contiguous with the PAI in H. pylori J166, the cag25-26 intergenic message was not measured, but presumed to be absent because cag25 and cag26 transcription is in opposite directions.
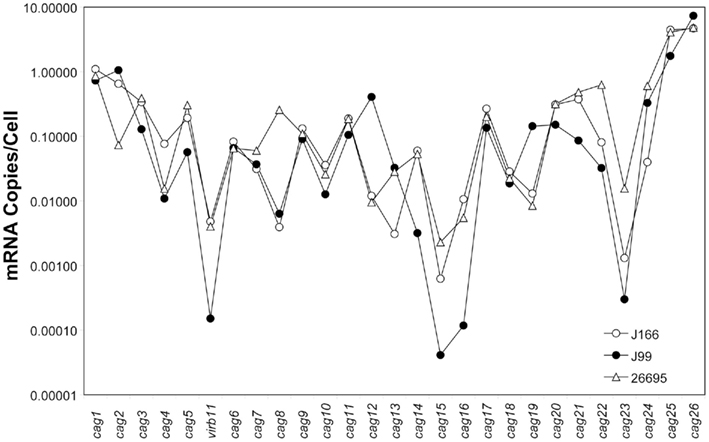
Figure 2. Composite gene expression (mRNA copies/cell, normalized to 16S rRNA) for each gene on the cag PAI of H. pylori strains J166 (open circles), J99 (closed circles), and 26695 (open triangles). Our data represents the average of duplicate reactions.
We reasoned that adjacent genes transcribed with ORFs in the same direction, with the presence of intergenic transcript, might represent a single transcriptional unit, particularly if the transcript abundance was similar across genes. Therefore, we initially considered the possibility that the following may represent cag PAI operons (numbered in the direction of transcription): cag1-4, cag10-5, cag11-12, cag16-17, cag21-18, and cag25-22 (Figure 1). However, there were sometimes marked differences in transcript abundance of genes within these putative operons (e.g., cag25-22, Figures 1 and 2). This might occur due to differential decay of the transcript or possibly because the gene is part of more than one transcriptional unit. To address these possibilities, we deleted the genomic region immediately upstream of the translational start of the first gene in each of six putative operons in H. pylori strain J166, a region likely to contain the promoter, and then measured cag PAI gene transcript abundance. We reasoned that deletion of this region should decrease the expression level of all genes in the transcriptional unit, and leave others unchanged.
Deletion of the putative promoter regions upstream of cag1, cag10, cag11, cag16, cag21, and cag25 had differential effects on the expression of downstream genes when compared to the isogenic wild type H. pylori J166 strain (Figure 3). Deletion of the region upstream of cag1 reduced expression of cag1-3 by three orders of magnitude and cag4 by only 1.5 orders of magnitude. By contrast, expression of cag5, a gene transcribed in the opposite direction of this putative operon, remained essentially unchanged. Deletion of the region upstream of cag10 reduced expression of both cag10 and cag9 by similar levels and had no effect on expression of cag8-7. Deletion of the putative promoters upstream of cag11, cag16, and cag21 reduced expression of the downstream genes, cag11-12, cag16-17, and cag21-18, but in each case to different levels, ranging from 1 to 3 orders of magnitude (Figure 3). Finally, deletion of the region upstream of cag25 reduced expression of the downstream genes cag25-23 to different levels and had no effect on the expression of cag22. In some cases, these results make clear predictions about operon structure. For example, our original prediction of cag10-5 and cag25-22 as operons was incorrect, since in each case one or more downstream genes did not change appreciably in the promoter knockouts. Thus, cag10-5 consists of at least two operons, cag10-9 and cag8-7, which also may be organized into one or more transcriptional units. Similarly, cag25-22 appears to have only cag25-24 on one transcriptional unit, with cag22 and perhaps cag23 on separate transcripts. The variable change we observed in cag PAI gene expression after deletion of the predicted upstream promoter again suggests that either the transcripts are being degraded or that there are additional promoters controlling expression of these genes. To identify additional promoters that may contribute to the more complex expression pattern we observed here, we undertook a non-biased promoter-trap approach.
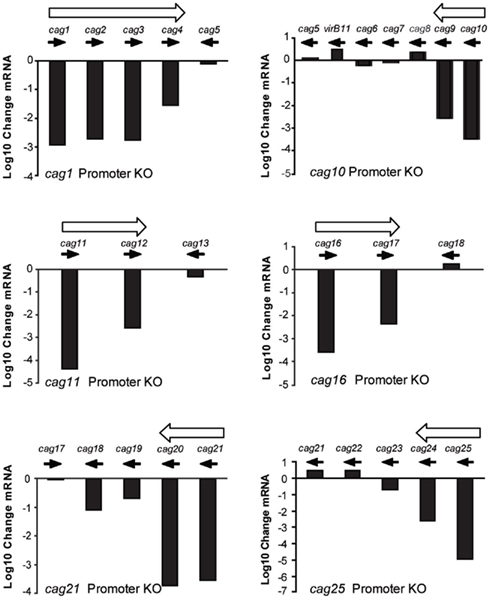
Figure 3. Change in cag PAI gene expression (log 10 change mRNA) between wild type H. pylori J166 and isogenic deletions of DNA upstream of the translational start codon in cag1 (416 bp, top left), cag10 (498 bp, top right), cag11 (498 bp, middle left), cag16 (532 bp, middle right), cag21 (615 bp, bottom left), and cag25 (706 bp, bottom right). Filled arrows indicate direction of transcription and open arrows indicate putative operons.
We next employed a functional identification of cag PAI promoters strategy based on the ability of short cloned regions of the cag PAI to direct expression of a heterologous promoter. We used a tnpR transcriptional reporter developed for Vibrio cholerae (Camilli et al., 1994) that had been previously modified to function in H. pylori (Camilli et al., 1994; Castillo et al., 2008b). We constructed libraries of putative cag PAI promoters using both H. pylori strains J166 and G27 as template for PCR; we cloned the Sau3A-digested fragments upstream of the promoterless tnpR gene in pCT-tnpR. If the cloned cag PAI region contained a promoter, we predicted it would direct tnpR expression and the creation of the TnpR protein. TnpR in turn would catalyze the removal of an unlinked kanamycin resistance (KmR) cassette and convert the H. pylori reporter strain ACHP17 from KmR to KmS.
For promoter identification, H. pylori strain ACHP17 bearing the res1-kan-res1 marker was transformed with pcat-T-caglibmG27-tnpR or pcat-T-caglibJ166-tnpR to CmR, followed by screening for retention or loss of the res1-kan-res1 cassette. We screened 1060 H. pylori pcat-T-caglibmG27-tnpR transformants and 1274 H. pylori pcat-T-caglibmJ166-tnpR transformants, representing 100 and 71% coverage, respectively. This analysis identified 34 and 27 transformants, respectively, that were sensitive to kanamycin and thus had expressed tnpR. After removing redundant clones, we determined that the DNA sequences upstream of tnpR in these KmS transformants correspond to 14 unique loci (Table 1). Eleven and four promoters were identified through the screening of our pcat-T-caglibmG27-tnpR and pcat-T-caglibmJ166-tnpR libraries, respectively; one promoter, PIII (Table 1; Figure 4), was isolated from both libraries.
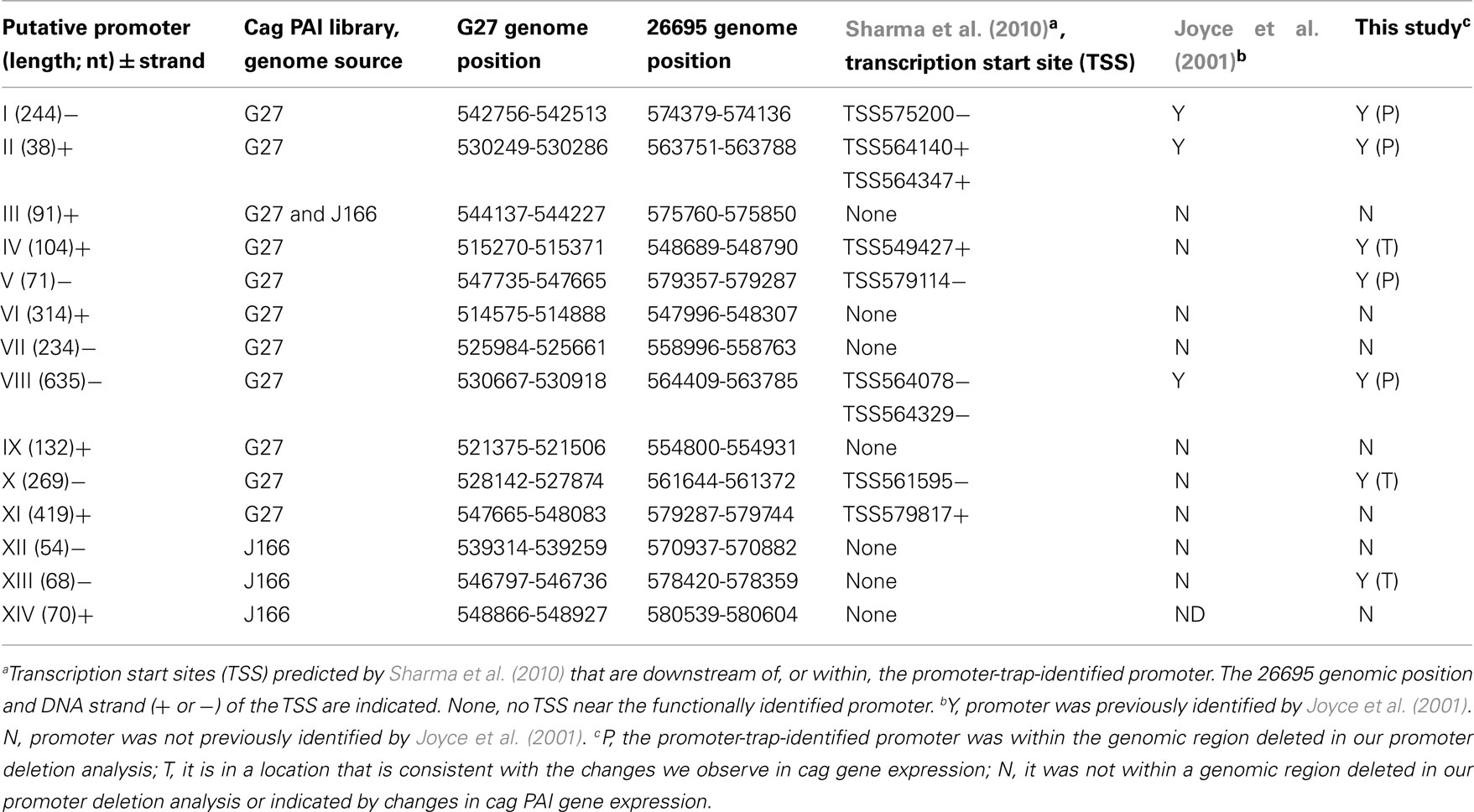
Table 1. Chromosomal location of putative cag PAI promoters.
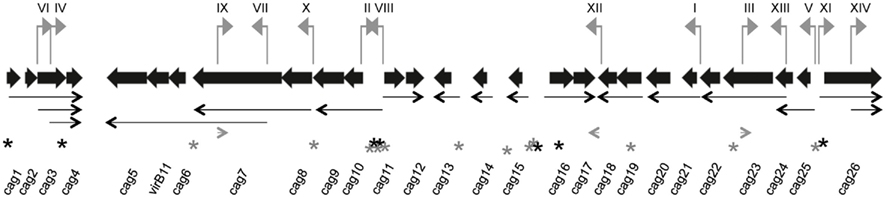
Figure 4. Promoter-trap-identified promoters and proposed transcript map on the H. pylori cag PAI. Each gene on the cag PAI is represented by a thick black arrow, oriented in the direction of transcription, whose length and spacing are approximately proportional to the annotated gene length and intergenic spacing. DNA segments represented in the promoter library that contained functional promoters (numbered I–XIV) to correspond with Table 1 and sequences in Table A5 in Appendix) are shown as small gray flags pointing in the direction they direct transcription and positioned in their cag PAI location. Thin black arrows represent 17 of the 20 proposed transcripts and gray arrowheads represent the three potential antisense transcript start points. All arrows point in the direction of transcription. Transcription start sites identified in Sharma et al. (2010) are indicated by an asterisk (*); black asterisks indicate the transcription start site is on the plus strand and gray asterisks indicate the minus strand.
We next mapped our promoter-trap-identified promoters onto the cag PAI map and compared these promoters to those found in by our initial qRT-PCR analysis and also to the work of others. Several of the promoter-trap-identified promoters were located in cag PAI regions that were either predicted by the qRT-PCR or promoter deletion analyses (Table 1). These include the promoters upstream of cag10 (PVIII), cag11 (PII), cag 21 (PI), and cag25 (PV; Figures 3 and 4; Table 1). The promoter-trap approach also identified several possible promoters that were located within operons that might account for the variable gene expression observed after deleting the main promoter (Figures 3 and 4). These include PIV, PX, and PXIII that are located in genomic positions to suggest they contribute to the expression of cag4, cag8-7, and cag23-22 (Figures 3 and 4).
The other putative promoters identified in our promoter-trap study are either consistent with cag PAI transcripts predicted by other groups, or as of yet, unique. We identified a promoter that is upstream of cag26 (PXI) and one that is within, and in the same direction as, cag26 (PXIV). The promoter upstream of cag26 was identified in work done by Spohn et al. (1997) and more recently by Sharma et al. (2010) as a promoter that drives expression of cag26 (Figure 4; Spohn et al., 1997; Sharma et al., 2010). We also identified a unique putative promoter (PVI) that overlaps cag3 and the adjacent upstream region and is in the correct direction to promote expression of a polycistronic mRNA including cag3 and cag4 (Figure 4). Two of our putative promoters were located within cag7, one in the same direction (PVII) and one antisense (PIX) to cag7 (Figure 4). We hypothesize the promoter located within cag7 contributes to expression of cag6-5 and the promoter that is antisense to cag7 may direct expression of a regulatory sRNA. Neither of these promoters has been identified by other studies. Finally, the last two putative promoters we identified, PIII and PXII, were within and antisense to cag23 and in the 3′ end of cag18 and may direct expression of sRNAs that are antisense to cag23 and cag17, respectively. These promoters are also unique to this study.
Although our transcription, promoter deletion and promoter-trap analyses do not completely overlap, they show reasonable agreement in predicting transcripts and operon structure and are generally consistent with operon structure predicted by others (Table 1, discussion). Taken together our data suggests the existence of at least 20 cag PAI transcripts (Figure 4).
To determine if the promoters PIII, PIX, and PXII direct expression of transcripts that are antisense to cag23, cag7, and cag17, respectively, we carried out additional RT-PCR reactions on RNA isolated from H. pylori strain G27. The oligonucleotides (PIIIR, PIXR, and PXIIR) used in the reverse-transcription reactions were located ∼100–150 nt downstream of PIII, PIX, and PXII and were antisense to the putative transcripts. For the subsequent PCR reactions in which reverse-transcriptase had been inactivated, the sense oligonucleotides, PIIIF, PIXF, and PXII were added. Amplicons were detected downstream of PIII and PXII in the RT-PCR reactions and were absent in the corresponding polymerase only controls, suggesting that these promoters do in fact direct expression of transcripts (Figure 5). We did not detect a transcript downstream of PIX in our experiments; while it is possible that PIX is not a promoter, it is more likely that the transcript is regulated or is in very low abundance. The promoter-trap system by which PIX was identified was designed to capture low abundant and transient expression events.
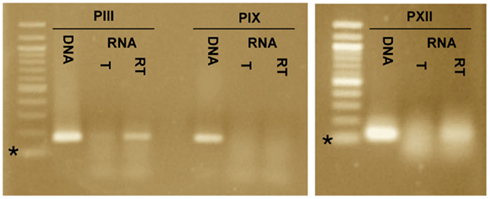
Figure 5. Reverse-transcriptase polymerase chain reaction (RT-PCR) identifies transcripts downstream of promoter-trap-identified promoters PIII and PXII. RT-PCR was carried out for the three promoters that potentially directed expression of antisense transcripts, PIII, PIX, and PXII. Three reactions were included for each promoter, a DNA template + DNA polymerase (DNA), an RNA template + DNA polymerase (T) and an RNA template + reverse-transcriptase and DNA polymerase (RT). Amplicons of the correct size were detected for PIII and PXII in the RT reactions but not in the T reactions. An amplicon was not detected for PIX in the RT or T reactions. This supports expression of transcripts downstream of PIII and PXII, but not PIX. The * indicates the 100-bp marker of the 100-bp ladder.
In this study we used transcript profiling coupled with putative promoter deletion and a non-biased promoter-trap system to analyze expression of cag PAI genes and their organization into transcriptional units across several H. pylori strains. We found that cag PAI gene expression varies by nearly five orders of magnitude across the cag PAI, and that expression of cag PAI genes is similar across strains 26695, J99, and J166. Based on transcript profiling of cag PAI ORFs and intergenic regions, we initially placed cag PAI genes into six polycistrons and four monocistrons. However, subsequent promoter deletions coupled with transcript profiling and promoter-trap promoter identification studies suggested cag PAI operon structure was much more complex. Our data suggests that there are at least 11 operons: cag1-4, cag3-4, cag10-9, cag8-7, cag6-5, cag11-12, cag16-17, cag19-18, cag21-20, cag23-22, and cag25-24, as well as five monocistronic genes (cag4, cag13, cag14, cag15, cag26). Additionally, the location of four of our promoter-trap-identified promoters suggests they direct expression of, in one case, a truncated version of cag26 and in the other three, transcripts that are antisense to cag7, cag17, and cag23. Using RT-PCR we verified the presence of transcripts that are antisense to cag17 and cag23.
Our transcript profiling of cag PAI ORFs and intergenic regions of three H. pylori strains, 26695, J99, and J166, suggested that cag PAI expression is generally conserved among strains. There were some genes, however, whose expression showed appreciable differences across strains. Potential reasons for these differences may be attributed to one or a combination of the following: (1) difficulty in accurate quantitation of low abundance transcripts, (2) differential stability of the transcripts, and (3) differential strength of the promoters. We suspect that the differences in cag15 expression between strains may be due to its very low expression in vitro (Joyce et al., 2001). The reduced expression of cag12, cag13, and cag19 in H. pylori strain 26695 compared to that of J99 and J166 is more likely attributed to transcript instability and differences in promoter strength. Our expression findings should allow researchers to more confidently apply our and other cag PAI expression data to unique clinically isolated H. pylori strains.
Our findings are generally consistent with previous predictions of cag PAI promoters, expression and operon structure. First, our promoter-trap and promoter deletion studies identified four of the five cag PAI promoters, upstream of cag1, cag10, cag21, and cag25 (not cag15), that were predicted by Joyce et al. (2001) in the H. pylori Alston strain. However our transcript profiling of cag PAI ORFs and intergenic regions did predict the promoter upstream of the cag15 (Figure 1). The failure of our promoter-trap to identify the promoter upstream of cag15 was not surprising, as Joyce et al. (2001) found that this promoter was only induced in co-culture with epithelial cells or in mice. A similar profile of promoters between the clinically isolated Alston strain and 26695, J99, J166, and G27 again supports conservation of cag PAI operon structure and expression between H. pylori strains.
Our promoter analyses also identified promoters upstream of cag25 (cagB) and cag26 (cagA) that were in positions similar to those previously reported by Spohn et al. (1997) for H. pylori strain G27, and by Sharma et al. (2010) for H. pylori strain 26695. Spohn et al. (1997) identified two transcription start points upstream of cag25 that are ∼200 bp upstream of what we and Sharma et al. (2010) found for cag25. All three studies predicted the same start point that is upstream of cag26, but we found an additional promoter that is located within cag26. The significance of multiple start sites upstream of cag25 and within cag26 are, as of yet, unclear. However, a recent study suggests discreet roles for the amino- and carboxy-terminus of Cag26 (CagA) and it is interesting to speculate this promoter could separate Cag26 function by creating a truncated protein (Pelz et al., 2011).
Our transcript profiles obtained from our work were also consistent with many of the 14 cag PAI operons identified in the H. pylori genome-wide transcript analysis conducted by Sharma et al. (2010). In common, we predicted five polycistrons: cag1-4, cag6-5, cag8-7, cag11-12, and cag16-17, and the three monocistrons: cag4, cag13, and cag26. Our promoter locations are consistent with their transcripts that start at cag10, cag14, and cag25, but our data did not predict that the transcripts extended to cag7, cag13, and cag18, respectively. We also did not find functional promoters upstream of cag17 and cag18 that would suggest they were also expressed as monocistrons. However, in addition to the truncated cag26 transcript mentioned above, we also identified the following set of transcripts that were not identified by Sharma et al. (2010), including the polycistrons cag3-4, cag21-20, cag19-18 and three transcripts that were antisense to cag7, cag17, and cag23. A transcript for cag15 was also not identified by Sharma et al. (2010), likely due to its very low abundance in vitro (Joyce et al., 2001). We speculate that these discrepancies are due to potential issues with transcript abundance and stability here and in Sharma et al. (2010) and incomplete screening of our cag PAI promoter libraries.
Outstanding observations in our screening of the pcat-T-caglibJ166-tnpR and pcat-T-caglibG27-tnpR libraries in H. pylori ACHP17 were that we only identified four promoters grouped at the 3′-end of the cag PAI from pcat-T-caglibJ166-tnpR and that we did not identify promoters from the central region of the cag PAI from pcat-T-caglibG27-tnpR. We hypothesize that this was due to a combination of two things: (1) incomplete representation of the cag PAI region in both of our libraries and then (2) restriction modification system differences that were apparent in transforming our G27 based reporter strain ACHP17 with J166 cag PAI DNA. Although our library screening calculations (see Materials and Methods) suggested that we had screened 100% of the H. pylori G27 cag PAI and 71% of the H. pylori J166 cag PAI, our control experiments with 10 or 30 randomly selected H. pylori transformants, respectively, suggested that our libraries were biased; the H. pylori caglibG27-tnpR library was biased toward the left and right ends of the cag PAI and the H. pylori caglibJ166 library was biased toward the right side of the cag PAI. Nonetheless, this methodology was very effective at identifying promoters in positions where we observed slight differences in expression of adjacent genes. Specific amplification of cag PAI regions (e.g., cag12-17) that were underrepresented in our cag PAI libraries will ensure better representation of the G27 cag PAI region in our library for future in vivo analyses.
This and previous studies have contributed to building a more complete expression profile of the clinically important cag PAI of H. pylori grown in vitro (Spohn et al., 1997; Joyce et al., 2001; Sharma et al., 2010). The promoters identified by these in vitro studies can now be analyzed for their potential regulation during H. pylori infection of a host. It is clear in at least two cases that in vitro predicted promoters, those upstream of cag15 and cag21, are expressed at higher levels when co-cultured with an epithelial cell monolayer and in mice (Joyce et al., 2001). While we anticipate that a subset of our in vitro identified promoters will be regulated in vivo and may contribute to virulence, other studies suggest that there is a set of promoters or transcripts uniquely expressed in vivo (Scott et al., 2007; Castillo et al., 2008b); analysis of H. pylori transcripts isolated from gerbil stomachs predicted that cag25 is expressed as a monocistron in vivo (Scott et al., 2007) and a promoter-trap study identified a unique promoter, Pivi66, within cag7 (Castillo et al., 2008b). Analysis of our H. pylori cat-T-caglibmG27-tnpR library in rodents has the potential to identify additional in vivo induced cag PAI promoters.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by Public Health Service Grant (AI42081) from the NIH to Jay V. Solnick, a Research Scholar Grant (RSG-05-249-01-MBC) from the American Cancer Society to Karen M. Ottemann, and an Eastern Washington University Foundation Grant to Andrea R. Castillo. We thank Michael Hornsby, Wendy Axsen, and Thanh Vo for their technical assistance with experiments.
Alm, R. A., Ling, L. S., Moir, D. T., King, B. L., Brown, E. D., Doig, P. C., Smith, D. R., Noonan, B., Guild, B. C., Dejonge, B. L., Carmel, G., Tummino, P. J., Caruso, A., Uria-Nickelsen, M., Mills, D. M., Ives, C., Gibson, R., Merberg, D., Mills, S. D., Jiang, Q., Taylor, D. E., Vovis, G. F., and Trust, T. J. (1999). Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature 397, 176–180.
Andrzejewska, J., Lee, S. K., Olbermann, P., Lotzing, N., Katzowitsch, E., Linz, B., Achtman, M., Kado, C. I., Suerbaum, S., and Josenhans, C. (2006). Characterization of the pilin ortholog of the Helicobacter pylori type IV cag pathogenicity apparatus, a surface-associated protein expressed during infection. J. Bacteriol. 188, 5865–5877.
Arnold, I. C., Lee, J. Y., Amieva, M. R., Roers, A., Flavell, R. A., Sparwasser, T., and Muller, A. (2011). Tolerance rather than immunity protects from Helicobacter pylori-induced gastric preneoplasia. Gastroenterology 140, 199–209.
Backert, S., and Selbach, M. (2008). Role of type IV secretion in Helicobacter pylori pathogenesis. Cell. Microbiol. 10, 1573–1581.
Boonjakuakul, J. K., Canfield, D. R., and Solnick, J. V. (2005). Comparison of Helicobacter pylori virulence gene expression in vitro and in the rhesus macaque. Infect. Immun. 73, 4895–4904.
Boonjakuakul, J. K., Syvanen, M., Suryaprasad, A., Bowlus, C. L., and Solnick, J. V. (2004). Transcription profile of Helicobacter pylori in the human stomach reflects its physiology in vivo. J. Infect. Dis. 190, 946–956.
Camilli, A., Beattie, D. T., and Mekalanos, J. J. (1994). Use of genetic recombination as a reporter of gene expression. Proc. Natl. Acad. Sci. U.S.A. 91, 2634–2638.
Castillo, A. R., Arevalo, S. S., Woodruff, A. J., and Ottemann, K. M. (2008a). Experimental analysis of Helicobacter pylori transcriptional terminators suggests this microbe uses both intrinsic and factor-dependent termination. Mol. Microbiol. 67, 155–170.
Castillo, A. R., Woodruff, A. J., Connolly, L. E., Sause, W. E., and Ottemann, K. M. (2008b). Recombination-based in vivo expression technology identifies Helicobacter pylori genes important for host colonization. Infect. Immun. 76, 5632–5644.
Fischer, W., Puls, J., Buhrdorf, R., Gebert, B., Odenbreit, S., and Haas, R. (2001). Systematic mutagenesis of the Helicobacter pylori cag pathogenicity island: essential genes for CagA translocation in host cells and induction of interleukin-8. Mol. Microbiol. 42, 1337–1348.
Grant, S. G., Jessee, J., Bloom, F. R., and Hanahan, D. (1990). Differential plasmid rescue from transgenic mouse DNAs into Escherichia coli methylation-restriction mutants. Proc. Natl. Acad. Sci. U.S.A. 87, 4645–4649.
Hornsby, M. J., Huff, J. L., Kays, R. J., Canfield, D. R., Bevins, C. L., and Solnick, J. V. (2008). Helicobacter pylori induces an antimicrobial response in rhesus macaques in a Cag pathogenicity island-dependent manner. Gastroenterology 134, 1049–1057.
Joyce, E. A., Gilbert, J. V., Eaton, K. A., Plaut, A., and Wright, A. (2001). Differential gene expression from two transcriptional units in the cag pathogenicity island of Helicobacter pylori. Infect. Immun. 69, 4202–4209.
Kang, J. J., Watson, R. M., Fisher, M. E., Higuchi, R., Gelfand, D. H., and Holland, M. J. (2000). Transcript quantitation in total yeast cellular RNA using kinetic PCR. Nucleic Acids Res. 28, e2.
Kusters, J. G., Van Vliet, A. H., and Kuipers, E. J. (2006). Pathogenesis of Helicobacter pylori infection. Clin. Microbiol. Rev. 19, 449–490.
Odenbreit, S., Puls, J., Sedlmaier, B., Gerland, E., Fischer, W., and Haas, R. (2000). Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science 287, 1497–1500.
Pattis, I., Weiss, E., Laugks, R., Haas, R., and Fischer, W. (2007). The Helicobacter pylori CagF protein is a type IV secretion chaperone-like molecule that binds close to the C-terminal secretion signal of the CagA effector protein. Microbiology 153, 2896–2909.
Pelz, C., Steininger, S., Weiss, C., Coscia, F., and Vogelmann, R. (2011). A novel inhibitory domain of Helicobacter pylori protein CagA reduces CagA effects on host cell biology. J. Biol. Chem. 286, 8999–9008.
Rieder, G., Merchant, J. L., and Haas, R. (2005). Helicobacter pylori cag-type IV secretion system facilitates corpus colonization to induce precancerous conditions in Mongolian gerbils. Gastroenterology 128, 1229–1242.
Rohde, M., Puls, J., Buhrdorf, R., Fischer, W., and Haas, R. (2003). A novel sheathed surface organelle of the Helicobacter pylori cag type IV secretion system. Mol. Microbiol. 49, 219–234.
Salama, N. R., Otto, G., Tompkins, L., and Falkow, S. (2001). Vacuolating cytotoxin of Helicobacter pylori plays a role during colonization in a mouse model of infection. Infect. Immun. 69, 730–736.
Scott, D. R., Marcus, E. A., Wen, Y., Oh, J., and Sachs, G. (2007). Gene expression in vivo shows that Helicobacter pylori colonizes an acidic niche on the gastric surface. Proc. Natl. Acad. Sci. U.S.A. 104, 7235–7240.
Segal, E. D., Lange, C., Covacci, A., Tompkins, L. S., and Falkow, S. (1997). Induction of host signal transduction pathways by Helicobacter pylori. Proc. Natl. Acad. Sci. U.S.A. 94, 7595–7599.
Selbach, M., Moese, S., Meyer, T. F., and Backert, S. (2002). Functional analysis of the Helicobacter pylori cag pathogenicity island reveals both VirD4-CagA-dependent and VirD4-CagA-independent mechanisms. Infect. Immun. 70, 665–671.
Shaffer, C. L., Gaddy, J. A., Loh, J. T., Johnson, E. M., Hill, S., Hennig, E. E., Mcclain, M. S., Mcdonald, W. H., and Cover, T. L. (2011). Helicobacter pylori exploits a unique repertoire of type IV secretion system components for pilus assembly at the bacteria-host cell interface. PLoS Pathog. 7, e1002237. doi:10.1371/journal.ppat.1002237
Sharma, C. M., Hoffmann, S., Darfeuille, F., Reignier, J., Findeiss, S., Sittka, A., Chabas, S., Reiche, K., Hackermuller, J., Reinhardt, R., Stadler, P. F., and Vogel, J. (2010). The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 464, 250–255.
Spohn, G., Beier, D., Rappuoli, R., and Scarlato, V. (1997). Transcriptional analysis of the divergent CagAB genes encoded by the pathogenicity island of Helicobacter pylori. Mol. Microbiol. 26, 361–372.
Tomb, J. F., White, O., Kerlavage, A. R., Clayton, R. A., Sutton, G. G., Fleischmann, R. D., Ketchum, K. A., Klenk, H. P., Gill, S., Dougherty, B. A., Nelson, K., Quackenbush, J., Zhou, L., Kirkness, E. F., Peterson, S., Loftus, B., Richardson, D., Dodson, R., Khalak, H. G., Glodek, A., Mckenney, K., Fitzegerald, L. M., Lee, N., Adams, M. D., Hickey, E. K., Berg, D. E., Gocayne, J. D., Utterback, T. R., Peterson, J. D., Kelley, J. M., Cotton, M. D., Weidman, J. M., Fujii, C., Bowman, C., Watthey, L., Wallin, E., Hayes, W. S., Borodovsky, M., Karp, P. D., Smith, H. O., Fraser, C. M., and Venter, J. C. (1997). The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388, 539–547.
Viala, J., Chaput, C., Boneca, I. G., Cardona, A., Girardin, S. E., Moran, A. P., Athman, R., Memet, S., Huerre, M. R., Coyle, A. J., Distefano, P. S., Sansonetti, P. J., Labigne, A., Bertin, J., Philpott, D. J., and Ferrero, R. L. (2004). Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat. Immunol. 5, 1166–1174.
Wang, Y., and Taylor, D. E. (1990). Chloramphenicol resistance in Campylobacter coli: nucleotide sequence, expression, and cloning vector construction. Gene 94, 23–28.
Young, R., and Bremer, H. (1975). Analysis of enzyme induction in bacteria. Biochem. J. 152, 243–254.
Table A1. Open reading frame primer pairs selected for real time RT-PCR.
Table A2. Intergenic primer pairs selected for real time RT-PCR.
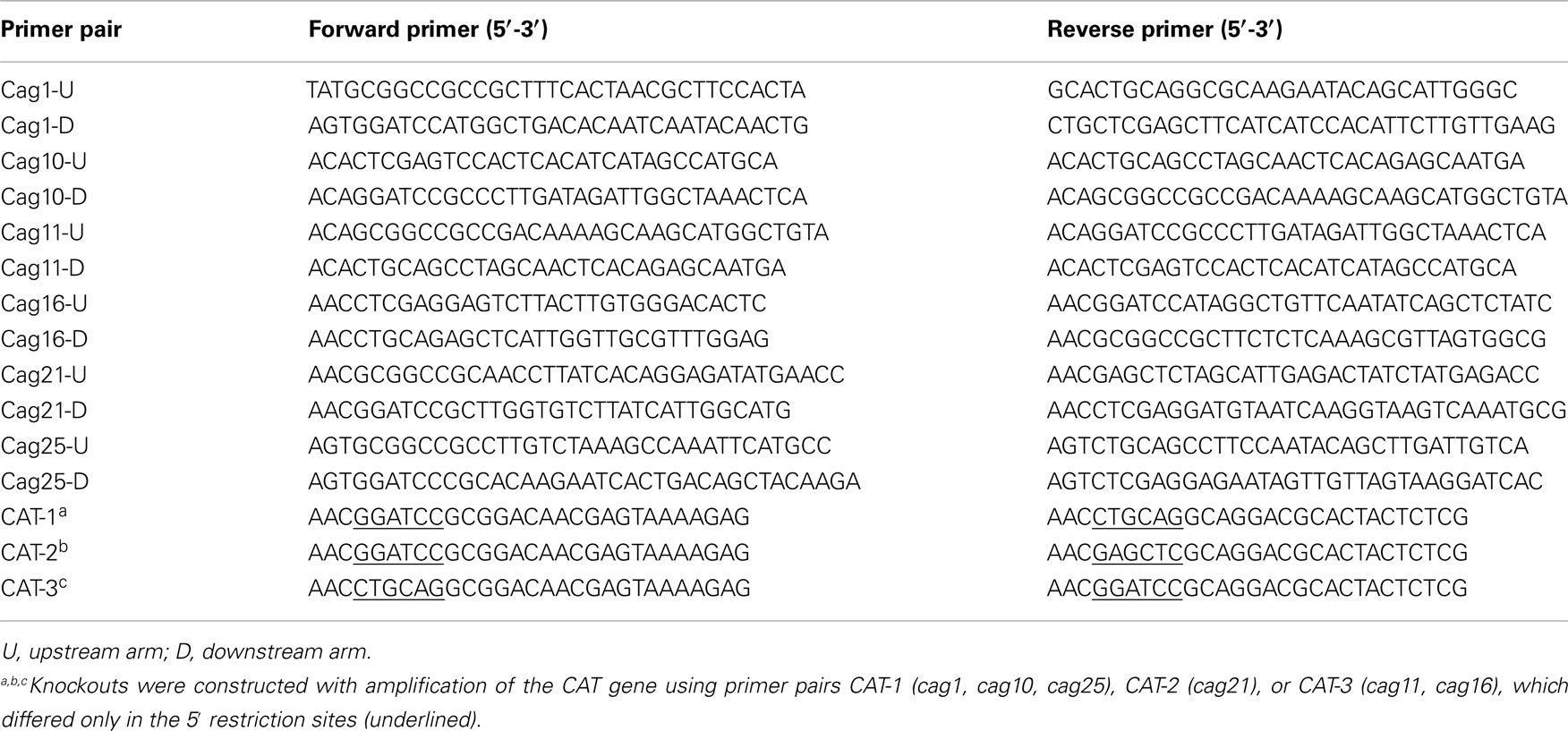
Table A3. Primer pairs used to construct promoter knockouts.
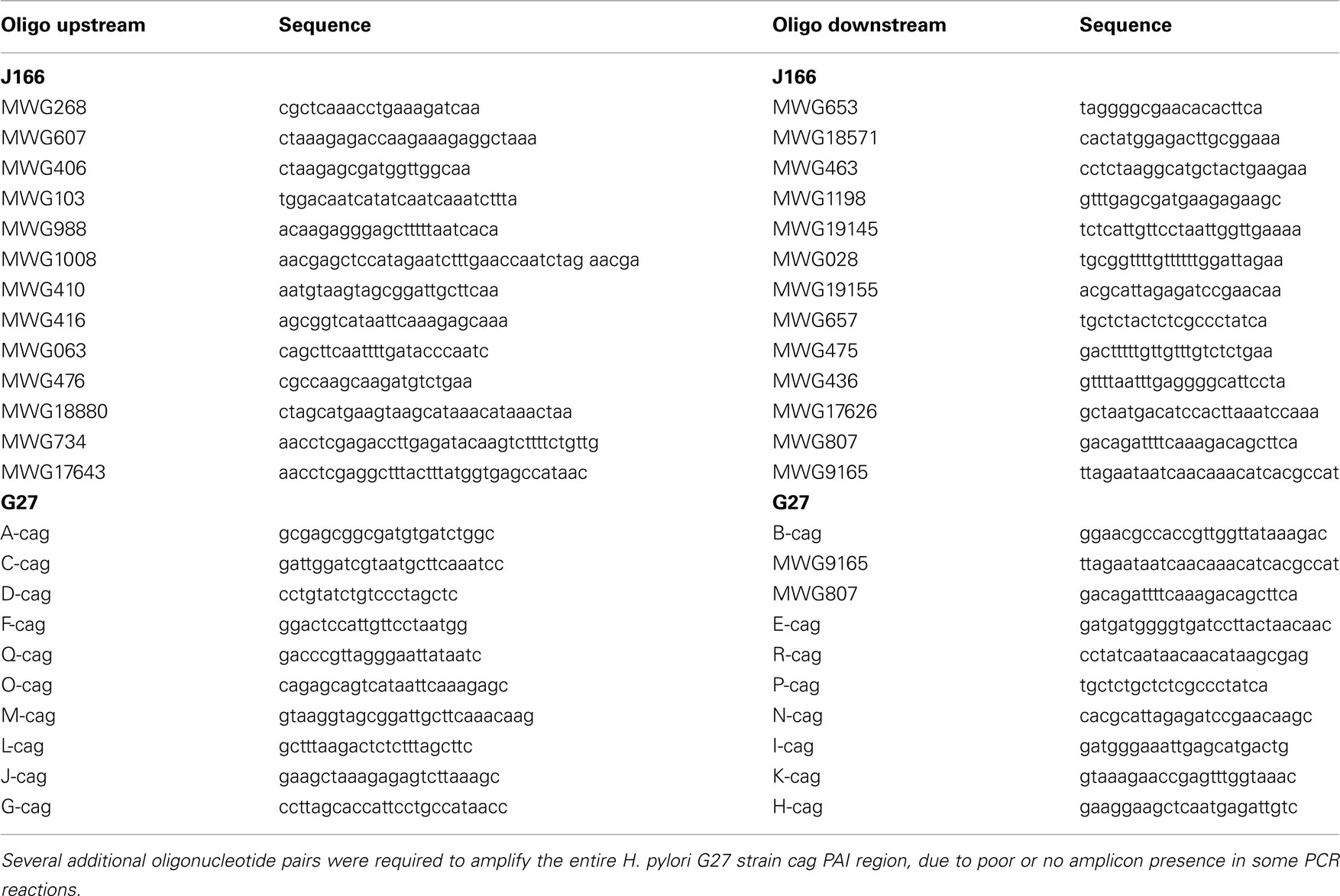
Table A4. Primer pairs used to generate amplicons for cag PAI libraries.

Table A5. Putative promoter sequences that direct expression of the reporter tnpR.
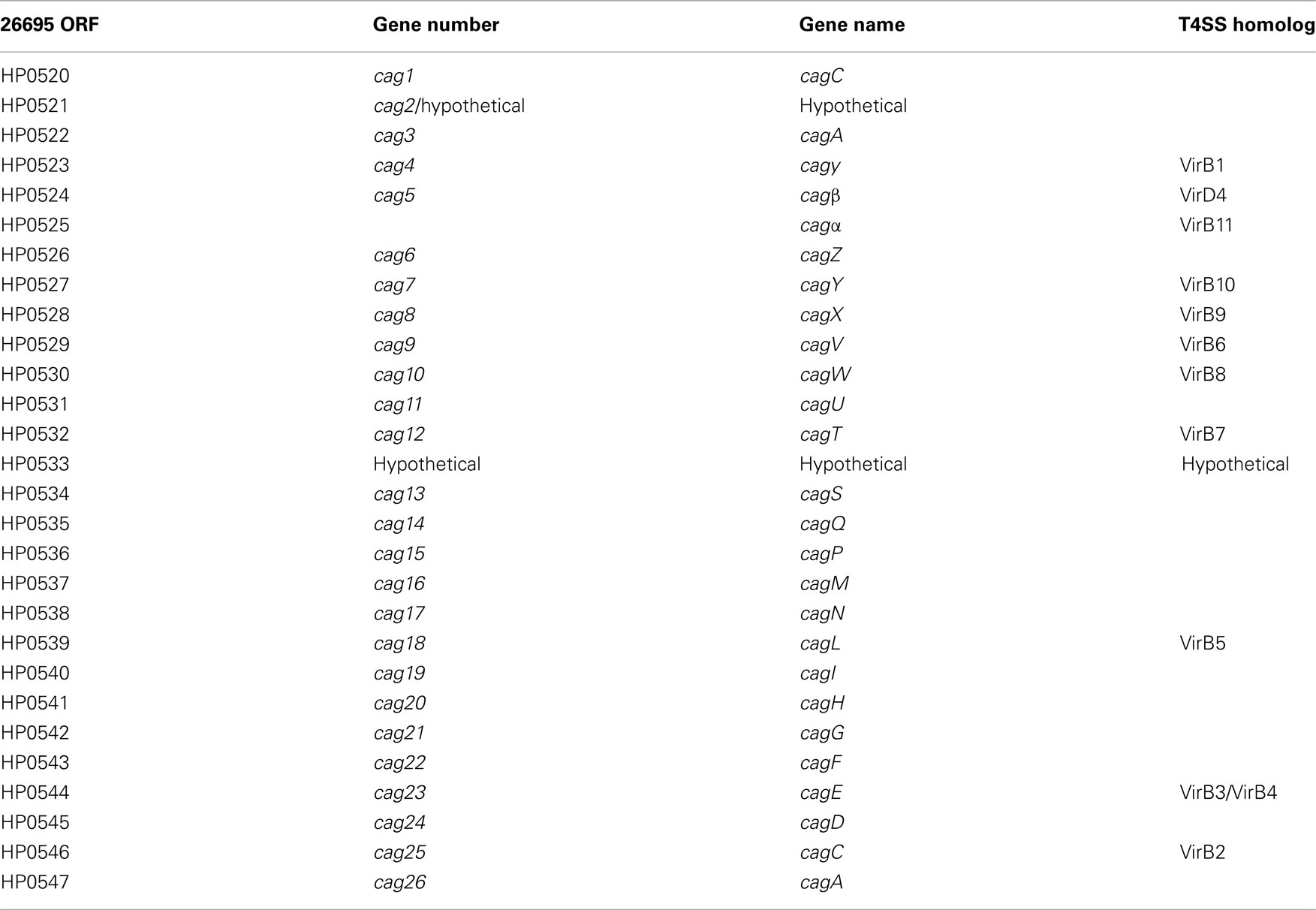
Table A6. Key to cag PAI gene names.
Keywords: cag PAI, operon structure, expression
Citation: Ta LH, Hansen LM, Sause WE, Shiva O, Millstein A, Ottemann KM, Castillo AR and Solnick JV (2012) Conserved transcriptional unit organization of the cag pathogenicity island among Helicobacter pylori strains. Front. Cell. Inf. Microbio. 2:46. doi: 10.3389/fcimb.2012.00046
Received: 02 November 2011; Accepted: 17 March 2012;
Published online: 20 April 2012.
Edited by:
D. Scott Merrell, Uniformed Services University, USAReviewed by:
Jeong-Heon Cha, Yonsei University, South KoreaCopyright: © 2012 Ta, Hansen, Sause, Shiva, Millstein, Ottemann, Castillo and Solnick. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Andrea R. Castillo, Department of Biology, Eastern Washington University, Cheney, WA 99004, USA. e-mail:YWNhc3RpbGxvQGV3dS5lZHU=; Jay V. Solnick, Center for Comparative Medicine, University of California Davis, Davis, CA 95616, USA. e-mail:anZzb2xuaWNrQHVjZGF2aXMuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.