
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
METHODS article
Front. Cell. Infect. Microbiol. , 02 April 2012
Sec. Molecular Bacterial Pathogenesis
Volume 2 - 2012 | https://doi.org/10.3389/fcimb.2012.00045
This article is part of the Research Topic Research Advances in the Study of Campylobacter, Helicobacter & Related Organisms View all 29 articles
 William G. Miller1*
William G. Miller1* Mary H. Chapman1
Mary H. Chapman1 Emma Yee1
Emma Yee1 Stephen L. W. On2
Stephen L. W. On2 Desmond K. McNulty3†
Desmond K. McNulty3† Albert J. Lastovica4 Anne M. Carroll3 Eleanor B. McNamara3 Geraldine Duffy5
Albert J. Lastovica4 Anne M. Carroll3 Eleanor B. McNamara3 Geraldine Duffy5 Robert E. Mandrell1
Robert E. Mandrell1
Multilocus sequence typing (MLST) systems have been reported previously for multiple food- and food animal-associated Campylobacter species (e.g., C. jejuni, C. coli, C. lari, and C. fetus) to both differentiate strains and identify clonal lineages. These MLST methods focused primarily on campylobacters of human clinical (e.g., C. jejuni) or veterinary (e.g., C. fetus) relevance. However, other, emerging, Campylobacter species have been isolated increasingly from environmental, food animal, or human clinical samples. We describe herein four MLST methods for five emerging Campylobacter species: C. hyointestinalis, C. lanienae, C. sputorum, C. concisus, and C. curvus. The concisus/curvus method uses the loci aspA, atpA, glnA, gltA, glyA, ilvD, and pgm, whereas the other methods use the seven loci defined for C. jejuni (i.e., aspA, atpA, glnA, gltA, glyA, pgm, and tkt). Multiple food animal and human clinical C. hyointestinalis (n = 48), C. lanienae (n = 34), and C. sputorum (n = 24) isolates were typed, along with 86 human clinical C. concisus and C. curvus isolates. A large number of sequence types were identified using all four MLST methods. Additionally, these methods speciated unequivocally isolates that had been typed ambiguously using other molecular-based speciation methods, such as 16S rDNA sequencing. Finally, the design of degenerate primer pairs for some methods permitted the typing of related species; for example, the C. hyointestinalis primer pairs could be used to type C. fetus strains. Therefore, these novel Campylobacter MLST methods will prove useful in differentiating strains of multiple, emerging Campylobacter species.
Campylobacters are a major cause of human bacterial gastrointestinal illness in the industrialized world (Mølbak and Havelaar, 2008; Olson et al., 2008); campylobacterioses (12.68 cases per 100,000) were second only to Salmonella infections (16.2/100,000) in the United States in 2008 (Anonymous, 2009). The majority of Campylobacter strains isolated from human clinical samples have been identified as C. jejuni subsp. jejuni or, to a lesser extent, C. coli (Lastovica and Allos, 2008). Recently, pathogenic campylobacters outside of the C. jejuni/C. coli group, termed here as emerging Campylobacter species, have been isolated more frequently from food and/or food animals. Recovery of these more fastidious, emerging Campylobacter species from food has not been reported often; isolation of such strains is likely limited by the culture conditions employed, conditions that favor Campylobacter species such as C. jejuni and C. coli. However, Lynch et al. (2011) using novel culture conditions, reported the isolation of multiple emerging Campylobacter spp., e.g., C. concisus, C. curvus, and C. sputorum, from chicken, beef, and pork samples. Emerging campylobacters isolated from food animals are often strains of species associated typically with livestock, such as C. hyointestinalis in sheep, cattle, and swine (Hakkinen et al., 2007; Salihu et al., 2009; Oporto and Hurtado, 2011), C. lanienae in cattle and swine (Sasaki et al., 2003; Inglis et al., 2004; Oporto and Hurtado, 2011), and C. sputorum in cattle and sheep (Terzolo, 1988; On et al., 1998).
The clinical relevance of the emerging Campylobacter spp. is as yet undetermined. Many of the emerging campylobacters are isolated infrequently from human clinical samples, although, as with isolation from food, recovery of these strains from clinical samples is probably limited by the isolation methods and media used. Nevertheless, emerging Campylobacter species are isolated from human clinical samples (Edmonds et al., 1987; Gorkiewicz et al., 2002; Lastovica and Allos, 2008; Bullman et al., 2011). Although the frequency of human illness associated with emerging Campylobacter spp. might be quite low, especially when compared to C. jejuni-associated gastroenteritis, it is possible that some emerging species could be associated with more severe illness. One such example is C. concisus, for which a strong association with Crohn’s disease and ulcerative colitis, has been reported recently (Man et al., 2010; Mahendran et al., 2011; Mukhopadhya et al., 2011).
Although molecular detection methods exist for many of the emerging campylobacters, population analyses, epidemiology, and source tracking of these organisms are limited by the strain typing methods available for these taxa. Molecular typing methods such as amplified fragment length polymorphism (AFLP) analysis and pulsed field gel electrophoresis (PFGE) methods have been employed on emerging Campylobacter strains (reviewed in On et al., 2008); however, sequence-based typing methods are not available for many species. One such sequence-based typing method is multilocus sequence typing (MLST). MLST methods amplify and sequence defined regions of moderately conserved housekeeping loci. At each locus, regions with distinct sequences receive arbitrary but unique allele numbers; similarly, each different allelic profile is assigned a unique sequence type (ST). The first Campylobacter MLST method was developed for C. jejuni (Dingle et al., 2001). This method sequences portions of seven genes: aspA, atpA (uncA), glnA, gltA, glyA, pgm (glmM), and tkt. The C. jejuni MLST method has been used in multiple typing studies and has been used successfully for strain typing and characterization, identification of clonal complexes and lineages, epidemiology, and investigation of host/source-associations (reviewed in Maiden and Dingle, 2008). Since the description of the C. jejuni MLST method, other Campylobacter MLST methods have been constructed that type C. coli (Dingle et al., 2005; Miller et al., 2005), C. lari (Miller et al., 2005), C. upsaliensis (Miller et al., 2005), C. helveticus (Miller et al., 2005), C. fetus (van Bergen et al., 2005), and C. insulaenigrae (Stoddard et al., 2007). Besides the primary use of Campylobacter MLST data for strain typing, MLST data for multiple taxa within Campylobacter are a valuable resource for studies on lateral gene transfer and evolution. MLST data can be used also to identify putative and perhaps clinically relevant taxonomic subdivisions within a species (Miller et al., 2005); additionally, MLST can provide genotypic information for novel species that are diverse phenotypically (Stoddard et al., 2007), especially those for which the only molecular speciation method is 16S rDNA sequencing.
Development of several Campylobacter MLST methods was assisted by the availability of draft genome sequences (Miller et al., 2005). Development of the novel Campylobacter MLST methods described in this study utilized recent draft genomes of C. hyointestinalis, C. lanienae, and C. sputorum (Miller et al., unpublished data), in addition to the closed C. concisus and C. curvus genomes available in the NCBI Microbial Genomes database. We anticipated that the draft genomes would contain some sequencing errors; however, enough reliable sequencing data was available to design MLST primers that could be used to type these five Campylobacter species. Therefore, in this study we describe four novel MLST methods that can be used to type: (1) C. concisus and C. curvus; (2) both subspecies of C. hyointestinalis (subspp. hyointestinalis and lawsonii) and C. fetus (subspp. fetus and venerealis); (3) C. lanienae; and (4) all three biovars of C. sputorum (bvs. fecalis, paraureolyticus, and sputorum). All four MLST gene sets are identical to the C. jejuni gene set [i.e., aspA, atpA(uncA), glnA, gltA, glyA, pgm, and tkt], with the exception of the C. concisus/C. curvus MLST method in which ilvD replaces tkt. A sample set of 213 isolates of diverse geographic origin and source was typed in this study. For all four methods, a total of 163 STs and 729 alleles were identified, indicating that these new MLST methods provide resolution similar to the previous MLST methods described.
All Campylobacter strains were cultured routinely under microaerobic conditions (1.5% O2, 10% H2, 10% CO2, and 78.5% N2) at 37°C on Brain Heart Infusion agar (Becton Dickinson, Sparks, MD, USA) or Anaerobe Basal Agar (ABA; Oxoid, Lenexa, KS, USA) supplemented with 5% (v/v) laked horse blood (Hema Resource and Supply, Aurora, OR, USA). PCR enzymes and reagents were purchased from New England Biolabs (Beverly, MA, USA) or Epicentre (Madison, WI, USA). All chemicals were purchased from Sigma-Aldrich Chemicals (St. Louis, MO, USA) or Fisher Scientific (Pittsburgh, PA, USA). DNA sequencing chemicals and capillaries were purchased from Applied Biosystems (Foster City, CA, USA). PCR and sequencing oligonucleotides were purchased from Eurofins MWG Operon (Huntsville, AL, USA).
Cattle feces were inoculated into wells of a six-well microtiter plate containing 6 ml 1× Anaerobe Basal Broth (Oxoid) amended with Preston supplement (Oxoid), using a sterile cotton swab. Plates were placed inside plastic ZipLoc bags and incubated under microaerobic conditions (as above) for 24 h at 37°C, while shaking at 40 rpm. After incubation, a 10-μl loop of each enrichment culture was plated onto ABA amended with 5% laked horse blood and CAT supplement (Oxoid). Feral swine feces were plated directly, using a sterile cotton swab, onto ABA amended with 5% laked horse blood and CAT supplement. All plates were then incubated under microaerobic conditions at 37°C for 24 h. Bacterial cultures were than filtered through 0.2 μm mixed cellulose ester filters onto ABA plates and incubated at 37°C under microaerobic conditions. After 24 h, single colonies were streaked onto new ABA plates and incubated 24–48 h at 37°C for purification.
Campylobacter strains isolated from the feces of California feral swine or cattle were speciated initially by 16S rDNA sequencing, using the primer pairs 27F (5′ AGA GTT TGA TCC TGG CTC AG 3′) and 1392R (5′ GAC GGG CGG TGT GTA C 3′; Lane, 1991). However, the 27F/1392R primers were not able to type C. hyointestinalis strains past the species level and several strains could not be typed unequivocally. To improve speciation, the atpAF/atpAR primer pairs from Miller et al. (2005) were used. These primers can amplify all Campylobacter taxa described currently, with the exception of C. avium (data not shown). Using these atpA primers, campylobacters of uncertain type were amplified and sequenced; the sequences were then compared by alignments and phylogenetic analyses to strains of known species/subspecies identification, especially those whose genomes had been sequenced. Unlike the 16S primers, the atpA primers could speciate unambiguously all of the strains isolated in this study and could identify clearly both subspecies of C. hyointestinalis. The atpA primer pairs, however, could not differentiate the three known biovars of C. sputorum (i.e., sputorum, fecalis, and paraureolyticus; On et al., 1998). The MLST results provided further confirmatory speciation data; atpA speciation agreed completely with subsequent MLST speciation.
The biovar paraureolyticus can be distinguished from the other two biovars of C. sputorum by the production of urease (On et al., 1998). Therefore, to identify putative bv. paraureolyticus strains, C. sputorum isolates were assayed for urease activity, as follows: a 10-μl loop of an overnight C. sputorum culture was resuspended in 2 ml urease reagent (3 mM NaH2PO4, 110 mM urea, 7 μg/ml phenol red, pH 6.8) and incubated for 1 h at RT. C. sputorum cultures were typed as bv. paraureolyticus based on a positive reaction (solution turning from yellow/orange to magenta). Genome-sequenced strains of biovars sputorum (strain RM3237) and paraureolyticus [strain RM4120 (LMG 11764)] were used as negative and positive controls, respectively. All tests were performed independently at least twice.
The C. sputorum biovars are distinguished also by the production of catalase: bv. fecalis is catalase-positive while the other two biovars are catalase negative (On et al., 1998). To test for catalase activity, a 10-μl loop of an overnight C. sputorum culture was resuspended in 200 μl 3% H2O2 on a glass slide. Presence of bubbles indicated a positive reaction. Genome-sequenced strains of biovars sputorum (strain RM3237) and fecalis [strain RM4121 (CCUG 20703)] were used as negative and positive controls, respectively. All tests were performed independently at least twice.
Each MLST amplification mixture contained: 1× MasterAmp PCR buffer (Epicentre, Madison, WI, USA), 1× MasterAmp PCR enhancer (Epicentre), 2.5 mM MgCl2, 250 μM (each) dNTPs, 50 pmol each primer, and 1 U Taq polymerase (New England Biolabs). For strains where genomic DNA was extracted using kits or standard isolation protocols, 50 ng purified genomic DNA was added to each reaction tube. Otherwise, 2 μl of a boilate was added. Boilates were prepared by resuspending a 1-μl loop of a pure culture or a single Microbank bacterial storage bead (Pro-Lab, Austin, TX, USA) in 100 μl TE and heating at 80°C for 5 min, then 100°C for 20 min, and cooling to 4°C. MLST amplifications were performed on a Tetrad thermocycler (Bio-Rad, Hercules, CA, USA) with the following settings: 94°C for 30 s, 53°C for 30 s, and 72°C for 2 min (30 cycles). Amplicons were purified on a BioRobot 8000 workstation (Qiagen, Valencia, CA, USA). Cycle sequencing reactions were performed on a Tetrad thermocycler, using the ABI PRISM BigDye terminator cycle sequencing kit (version 3.1; Applied Biosystems) and standard protocols. Cycle sequencing extension products were purified using BigDye XTerminator (Applied Biosystems). DNA sequencing was performed on an ABI PRISM 3730 DNA Analyzer (Applied Biosystems), using POP-7 polymer and ABI PRISM Genetic Analyzer Data Collection and ABI PRISM Genetic Analyzer Sequencing Analysis software. Sequences were trimmed, assembled, and analyzed in SeqMan (v 9.1; DNASTAR, Madison, WI, USA).
The Perl program MLSTparser3 (Miller et al., 2009) was modified to include the novel MLST methods for C. concisus, C. curvus, C. hyointestinalis, C. lanienae, and C. sputorum. The expanded MLSTparser3 was used to identify the MLST alleles and ST of each Campylobacter strain typed in this study. New Campylobacter MLST databases were created1; allele and ST data generated in this study were deposited in this database and are available online. The allelic profiles for all 213 strains are listed in Table S1 in Supplementary Material.
A dendrogram of unique Campylobacter STs was constructed by concatenating the allele sequences comprising each ST. Allele sequences for each strain were concatenated in the order aspA-atpA-glnA-gltA-glyA-pgm-tkt with the exception of C. concisus and C. curvus allele sequences, that were concatenated in the order aspA-atpA-glnA-gltA-glyA-ilvD-pgm. Composite concatenate lengths were 3345 bp (C. concisus/C. curvus), 3312 bp (C. fetus, C. hyointestinalis, and C. lanienae), or 3321 bp (C. sputorum). Sequence alignments were performed using CLUSTALX (ver. 2.1)2, and dendrograms were constructed using the neighbor-joining method with the Kimura two-parameter distance estimation method (Kimura, 1980). Phylogenetic analyses were performed using MEGA version 5.1 (Tamura et al., 2011). Polymorphic sites and dn/ds ratios were calculated using START2 (Jolley et al., 2001)
Construction of the novel Campylobacter MLST methods was facilitated by the availability of genome sequences for all of the taxa typed in this study. The genome-sequenced strains were: the completed genomes of C. concisus strain 13826 (NC_009802.1), C. curvus strain 525.92 (NC_009715.1), and C. fetus subsp. fetus strain 82–40 (NC_008599.1), and the draft genomes of C. hyointestinalis subsp. hyointestinalis (Chh) strain RM4092 (LMG 9260), C. hyointestinalis subsp. lawsonii (Chl) strain RM4096 (CCUG 27631), C. lanienae strain RM3663 (NCTC 13004), C. sputorum bv. sputorum strain RM3237, C. sputorum bv. fecalis strain RM4121 (CCUG 20703), C. sputorum bv. paraureolyticus strain RM4120 (LMG 11764), and strain RM6914, exemplar of a novel C. concisus-like clade (Mandrell et al., manuscript in preparation).
Primer design based on a sequence from a single strain might not lead to a successful MLST method if the sequence variation within that taxon prevents the design of primer pairs that efficiently amplify all strains. Therefore, MLST gene sequences from related species would be aligned. Based on this alignment, primers would be designed to bind to regions, 100–200 bp upstream and downstream of the allelic endpoints, that demonstrate a high degree of conservation among the aligned taxa. One to four degenerate bases would be incorporated into the MLST primers, if necessary, to optimize primer binding. This approach was used previously to construct successfully other Campylobacter MLST methods (Miller et al., 2005). Therefore, the full aspA, atpA, glnA, gltA, glyA, pgm, and tkt gene sequences were extracted from the completed and draft genomes and various alignments were performed. Based on sequence similarity between the various Campylobacter taxa, we developed four novel MLST methods to type the strains in this study: Method 1 for typing both C. concisus and C. curvus; Method 2 for typing C. fetus and both subspecies of C. hyointestinalis; Method 3 for typing C. lanienae strains; and Method 4 for typing all three biovars of C. sputorum.
The final MLST primer sets are listed in Table 1. Methods 2, 3, and 4 use the same seven loci and allelic endpoints of the C. jejuni MLST method, i.e., aspA, atpA, glnA, gltA, glyA, pgm, and tkt. However, for the C. concisus/C. curvus MLST method, the sequence diversity at the tkt locus was too great for the construction of suitable primers. Therefore, tkt was replaced by ilvD in Method 1; ilvD was used in a C. jejuni MLST method described previously (Manning et al., 2003). The Method 1 aspA, atpA, glnA, gltA, glyA, and pgm alleles also use the same endpoints of their C. jejuni counterparts; the ilvD allelic endpoints are unique to this study.
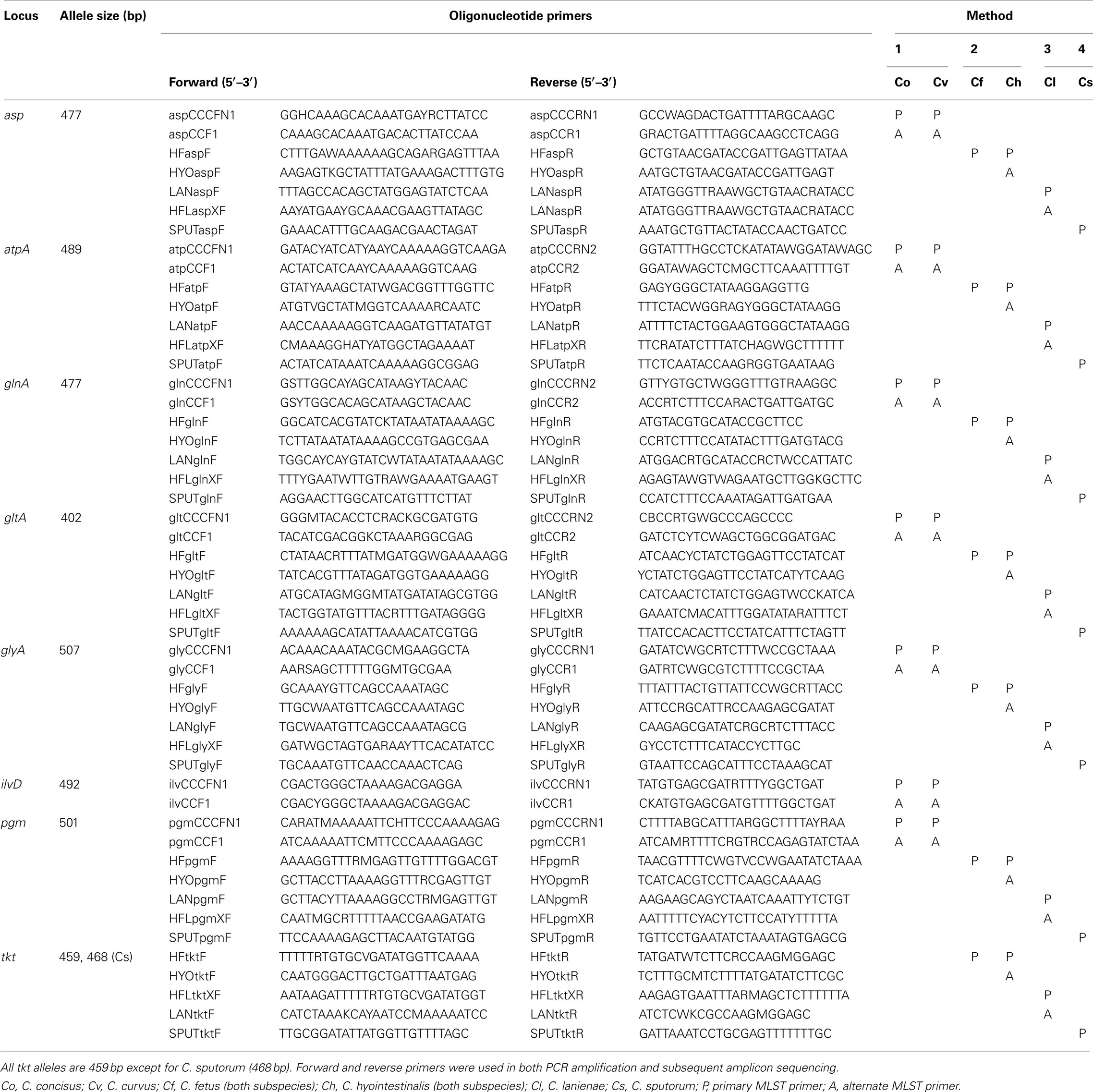
Table 1. Campylobacter MLST primers.
A total of 213 strains were typed in this study. Complete descriptions of each strain, including isolation source, date, and location (if known), allelic profiles and STs are listed in Table S1 in Supplementary Material. Strains typed in this study were isolated over a 30-year period (1981–2010) and were also geographically diverse: strains from each species were isolated on two to three continents (Table S1 in Supplementary Material). All of the C. concisus and C. curvus strains typed were isolated from human clinical samples, whereas nearly all of the Chl and C. lanienae strains typed were isolated from pigs and feral swine; strains from the other three taxa were a mixture of isolates from humans, cattle, and swine (Table 2).
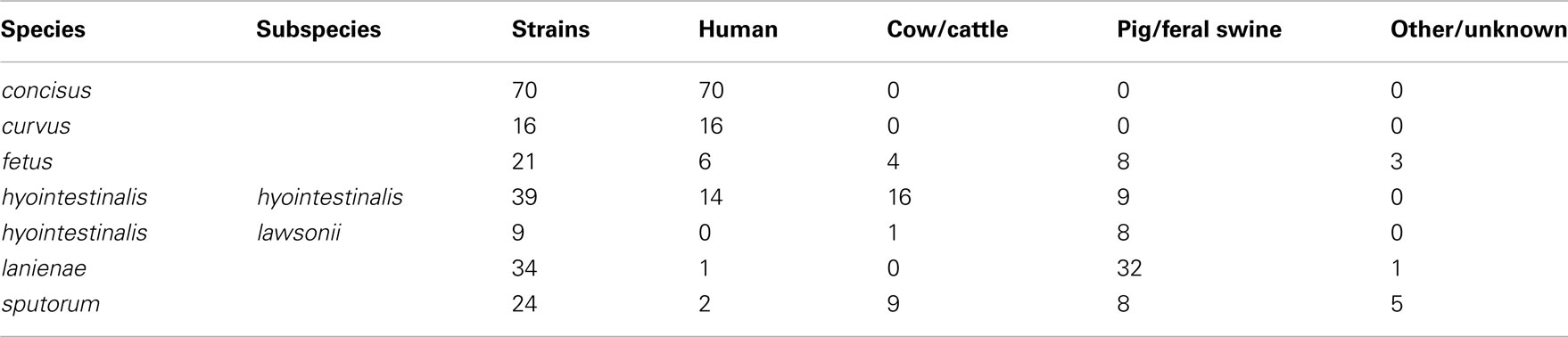
Table 2. Source of the Campylobacter strains typed in this study.
With a few exceptions, the primary MLST primers listed in Table 1 were able to amplify successfully all seven loci and provide high quality sequence data for all 213 strains to generate unambiguous ST. However, despite our best efforts to overcome sequence variation in the initial primer design, in a few instances (∼1–2%), the main primer pairs did not provide sequence quality high enough for an unambiguous ST. Thus, alternate primer pairs (annotated as “A” in Table 1) were used to amplify and sequence these alleles. No strain was excluded from the final strain list because a ST could not be obtained.
Phylogenetic analysis of the MLST STs validated the taxonomic relationships observed previously (Debruyne et al., 2008) for the six Campylobacter species typed in this study. Although the use of different gene sets comprising Method 1 and Methods 2, 3, and 4 prevented the simultaneous analysis of all six species, a clear delineation between C. concisus and C. curvus strains was observed (Figure 1), as well as unambiguous segregation of the other four species (Figure 2), that included the related C. hyointestinalis, C. fetus, and C. lanienae taxa. Moreover, the two C. hyointestinalis subspecies, Chh and Chl, formed distinct clusters (Figure 2). However, the two C. fetus subspecies could not be discriminated by phylogenetic analysis, consistent with previous observations (van Bergen et al., 2005). Divergent STs (C. lanienae STs 1 and 4; C. sputorum STs 8, 13, 14, and 15) were identified within some strains (see below and Figure 2). In C. sputorum, these divergent STs formed a cluster (termed Csp2) distinct from a cluster (termed Csp1) containing the other C. sputorum strains.
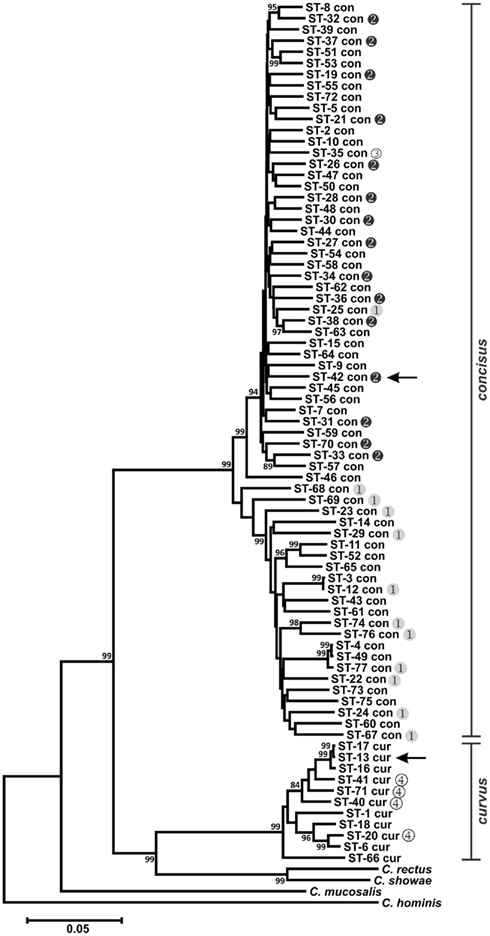
Figure 1. Dendrogram of C. concisus and C. curvus STs. Allele sequences for each strain were concatenated in the order aspA-atpA-glnA-gltA-glyA-ilvD-pgm and aligned using CLUSTALX. The dendrogram was constructed using the neighbor-joining algorithm and the Kimura two-parameter distance estimation method (Kimura, 1980). Bootstrap values >75%, generated from 1000 replicates, are shown at the nodes. Scale bar represents substitutions per site. Genomospecies 1–4 designations, as assigned by Aabenhus et al. (2005), were placed to the right of STs representing strains from that study. Arrows indicate the STs of the C. concisus and C. curvus genome-sequenced strains. The dendrogram contains also the concatenated aspA, atpA, glnA, gltA, glyA, ilvD, and pgm allele sequences of Campylobacter rectus, Campylobacter showae, Campylobacter mucosalis, and Campylobacter hominis. These allele sequences were extracted from draft (C. rectus, C. showae, and C. mucosalis) and completed (C. hominis) genome sequences.

Figure 2. Dendrogram of C. hyointestinalis, C. fetus, C. lanienae, and C. sputorum STs. Allele sequences for each strain were concatenated in the order aspA-atpA-glnA-gltA-glyA-pgm-tkt and aligned using CLUSTALX. The dendrogram was constructed using the neighbor-joining algorithm and the Kimura two-parameter distance estimation method (Kimura, 1980). Bootstrap values >75%, generated from 1000 replicates, are shown at the nodes. Scale bar represents substitutions per site. ST labels indicate taxon: hh, C. hyointestinalis subsp. hyointestinalis; hl, C. hyointestinalis subsp. lawsonii; ff, C. fetus subsp. fetus; fv, C. fetus subsp. venerealis; lan, C. lanienae; sp, C. sputorum; sp (sp), C. sputorum bv. sputorum; sp (f), C. sputorum bv. fecalis; sp (p), C. sputorum bv. paraureolyticus. Arrows indicate the STs of the genome-sequenced strains for each taxon. *Urease-positive strains. Values in parentheses at the nodes represent the average %nt similarity of the STs split at each node, following pairwise comparisons of the concatenated allele sequences.
Many MLST STs and alleles were identified in this study (Table 3). With the exception of C. fetus, for which a previous study identified also a relatively small number of highly clonal STs (van Bergen et al., 2005), the majority of strains within each of the remaining five species possess unique STs. Phylogenetic analysis of the STs of these five species indicated that the least amount of variation exists within Chh and each of the two C. sputorum clades, whereas the greatest amount of variation detected here resides clearly within the C. concisus and C. curvus strain sets (Figures 1–3). Even with the inclusion of the divergent C. lanienae and C. sputorum STs, the average number of base substitutions per site was less in each case (0.0330, C. lanienae; 0.0265, C. sputorum; Figure 3) than that calculated for the C. concisus STs (0.0641; Figure 3).
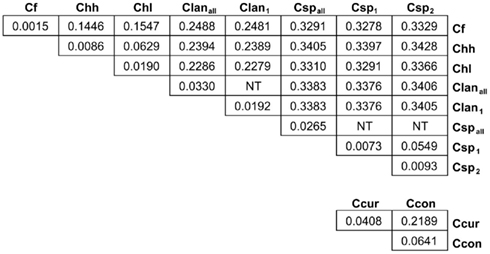
Figure 3. Genetic distance between and within the Campylobacter taxa. Each value represents the average number of base substitutions per site between concatenated allele sequences. Analyses were conducted using the Kimura two-parameter model (Kimura, 1980). The analysis involved 86 nucleotide sequences. Codon positions included were first + second + third+ non-coding. All positions containing gaps and missing data were eliminated. There were a total of 3312 positions in the final dataset. Evolutionary analyses were conducted in MEGA5 (Tamura et al., 2011). Branch lengths used to calculate the averages here were used to construct the dendrograms of Figures 1 and 2. NT, not tested. The C. lanienae and C. sputorum strain sets contained divergent STs. The STs ST-1lan and ST-4lan were removed from the C. lanienae strain set Clanall to create Clan1. Similarly, ST-8sp, ST-13sp, ST-14sp, and ST-15sp were removed from the C. sputorum strain set Cspall to create Csp2; the remaining STs formed Csp1.
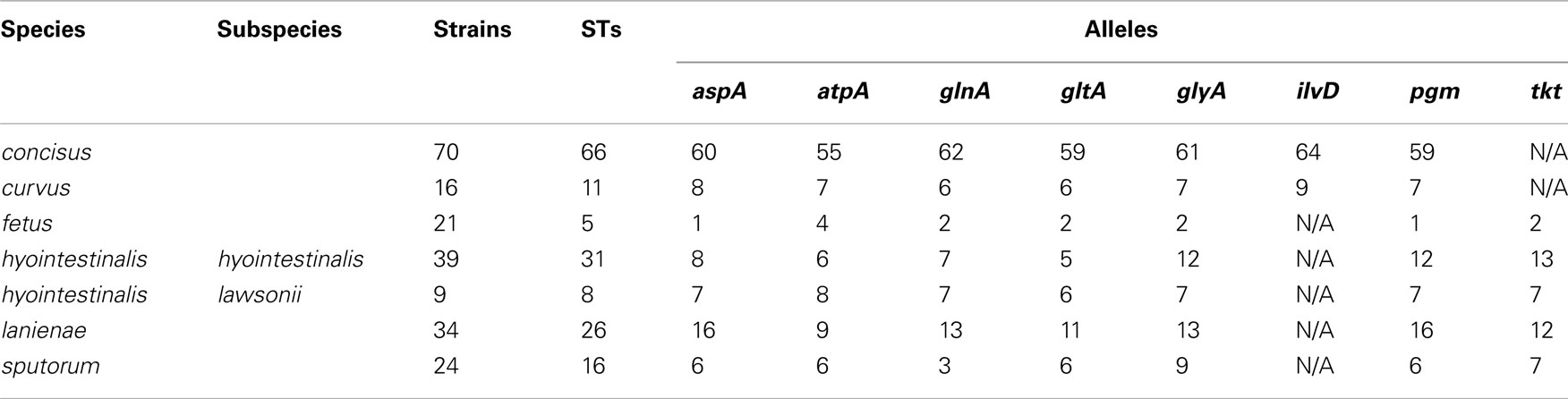
Table 3. Sequence types and alleles identified by the novel MLST methods.
The high degree of variation across the C. concisus STs is reflected by the large number of alleles and polymorphic sites identified within this strain set: for the 70 C. concisus strains, the number of alleles detected at any locus ranged from 55 (atpA) to 64 (ilvD; Table 3). This high density of alleles translated into the large number of polymorphic sites identified at each of the seven C. concisus loci (Table 4). Over 100 polymorphic sites were present within the alleles of each of the C. concisus MLST loci (Table 4), a large number when compared to the relatively few polymorphic sites detected within the Chh alleles, even when the relative sizes of the strain sets were factored into the comparison. For some species, the numbers of polymorphic sites were inflated by the presence of divergent alleles or strains within the strain set. When these alleles and strains were removed from the appropriate strain sets, the number of polymorphic sites decreased substantially. For example, removal of the divergent C. sputorum strains present within C. sputorum clade Csp2 (Figure 3) eliminated 23 of 24 polymorphic sites at the glnA locus (Table 4).
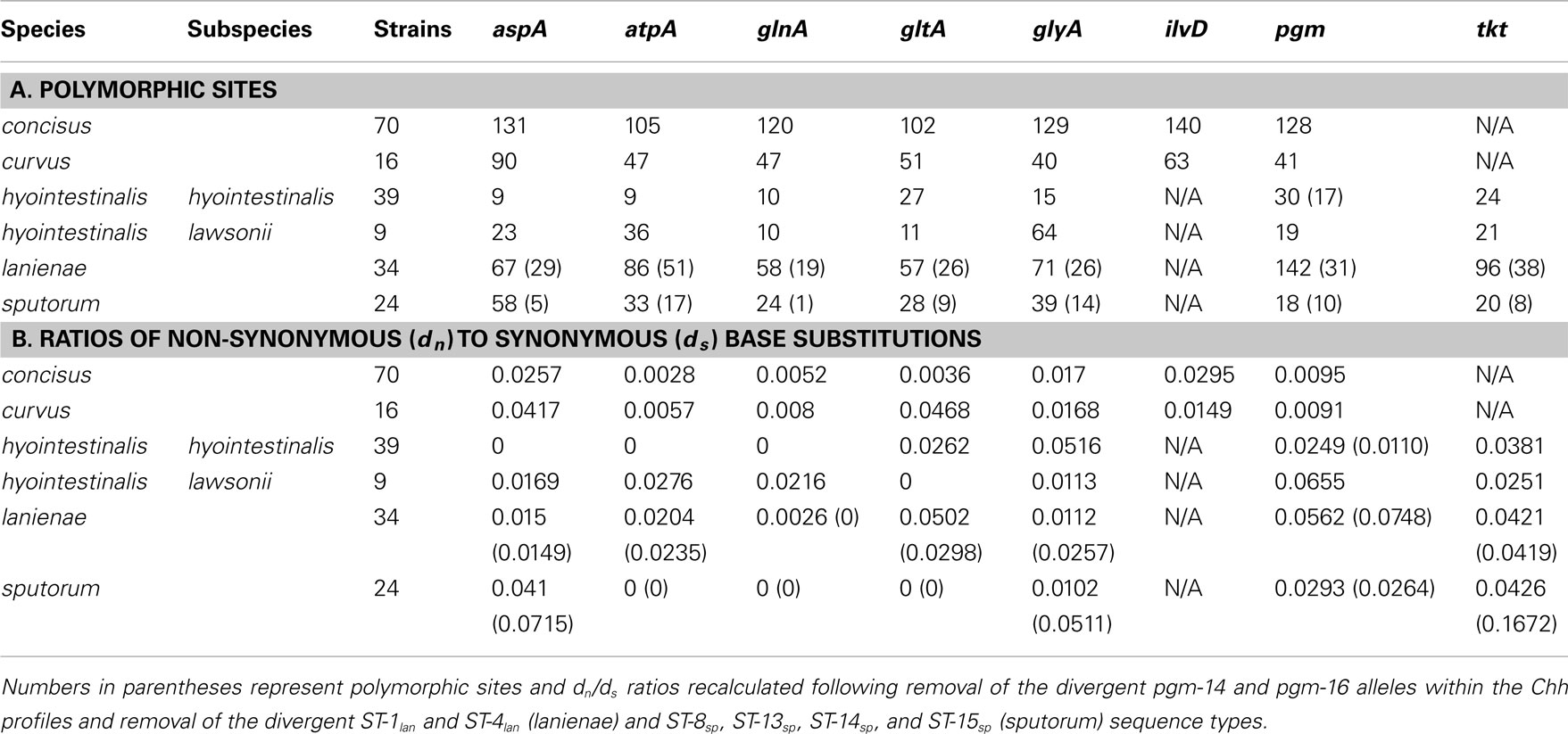
Table 4. Diversity within the Campylobacter MLST loci.
While MLST is often used as a strain typing tool, it can be used also to investigate the population structure of an organism, to identify lineages, for example, that demonstrate a higher association with disease or a particular host. Genes used for MLST methods, typically core housekeeping genes, are usually under purifying or neutral selection. Positively selected genes would be influenced by external/environmental pressures and would evolve more through recombination rather than through the accumulation of point mutations; thus, such genes are not generally used in MLST methods, since they may not provide an accurate representation of the clonal structure of a bacterial population (Maiden, 2006; Perez-Losada et al., 2011). One method of determining the level of selective pressure on a gene is by calculating the ratio between non-synonymous (dn) and synonymous (ds) base substitutions. The rate of synonymous base substitution in genes should equal the neutral substitution rate, in the absence of codon usage bias. Non-synonymous base substitutions (that result in an amino acid change) would be the result of positive selection. Thus, the ratio of non-synonymous to synonymous base substitutions (dn/ds) would be an indicator of potential positive selection: ratios > 1 would be evidence of possible positive selection, whereas ratios < 1 would be more indicative of purifying or stabilizing selection. The dn/ds values for previous Campylobacter methods were quite low: the highest dn/ds values for C. jejuni (0.093), C. coli (0.173), C. lari (0.047), C. upsaliensis (0.097), and C. insulaenigrae (0.110) were substantially <1 (Colles et al., 2003; Miller et al., 2005; Stoddard et al., 2007). Similar ratios (highest dn/ds values) calculated in this study for C. concisus (0.0295), C. curvus (0.0468), Chh (0.0516), Chl (0.0655), C. lanienae (0.0562), and C. sputorum (0.0426; Table 4) are consistent with the previous methods, indicating that these MLST loci are also not subject to positive selection.
Characterization of the Campylobacter MLST methods also identified putative lateral gene transfer events. An allele that was nearly identical to, and clustered phylogenetically with, alleles from another taxon was determined to represent a putative lateral gene transfer event. For example, strains RM14410 and RM14403 in the Chh strain set contain alleles pgm-14 and pgm-16, respectively (Table S1 in Supplementary Material), that are 97.8–99.6% similar at the nt level to pgm alleles from Chl but only 95.2–96.2% similar at the nt level to pgm alleles from Chh (data not shown); these two alleles are also clearly related to other Chl pgm alleles, based on phylogenetic analysis (Figure 4A). Strains RM14410 and RM14403 also contain the atpA alleles atpA-2 and atpA-13, respectively, that cluster with other Chh atpA alleles (Figure 4B). Alleles at the other five loci for these two strains cluster also with other Chh alleles (data not shown), indicating that RM14403 and RM14410 are Chh strains in which a putative lateral transfer event has occurred at the pgm locus. The extent of gene transfer in these two strains could not be determined by MLST and will require further genome sequence analyses. It is not surprising that Chl alleles were discovered in Chh strains: these two taxa are highly related, being subspecies of the same species, and are isolated often from the same food animals (i.e., cattle and swine). No putative lateral transfer events were observed within C. concisus or C. curvus and no alleles were identified that originated tentatively in another species.
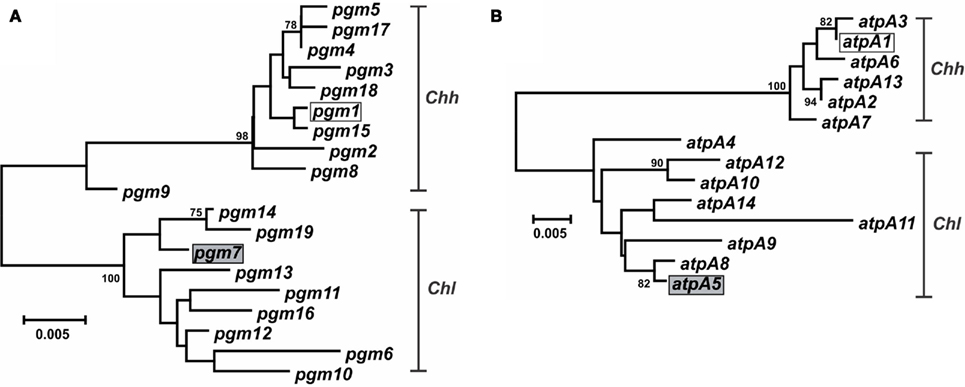
Figure 4. Dendrogram of C. hyointestinalis pgm and atpA alleles. C. hyointestinalis pgm(A) and atpA (B) allele sequences were aligned using CLUSTALX. The dendrogram was constructed using the neighbor-joining algorithm and the Kimura two-parameter distance estimation method (Kimura, 1980). Bootstrap values >75%, generated from 1000 replicates, are shown at the nodes. Scale bar represents substitutions per site. The alleles of the Chh (open boxes) and Chl (gray boxes) genome sequence strains are indicated.
In some instances, phylogenetically divergent alleles within a strain set were indicative of either highly divergent strains or perhaps novel taxa. Here, as in other MLST studies (Miller et al., 2005, 2009), putative lateral gene transfer events were identified at only one of the seven MLST loci. For example, Chh STs containing pgm-14 or pgm-16 (ST-26hh and ST-28hh, respectively; Figure 2) were divergent at only the pgm locus. The alleles for each of the remaining six loci within each ST were of likely Chh origin, and phylogenetic analysis of the concatenated allele sequences clearly placed these two STs within the Chh clade. However, some STs (ST-1lan, ST-4lan, ST-8sp, ST-13sp, ST-14sp, and ST-15sp) contain three to seven variant alleles (Table S1 in Supplementary Material). The C. lanienae STs ST-1lan and ST-4lan differ substantially from the other typed C. lanienae strains: concatenated nucleotide sequences across all seven loci for ST-1lan and ST-4lan are on average only approx. 92 and 88% similar, respectively, to the concatenated sequences of the other C. lanienae STs (Figure 2), which display an average 98% cross-similarity (data not shown). Therefore, these two C. lanienae STs may be exemplars of novel C. lanienae-related taxa (for comparison, the concatenated Chh nucleotide sequences are approx. 94% similar to those of the other C. hyointestinalis subspecies and 87% similar to those of the related species C. fetus; Figure 2). Additionally, six phylogenetically related, urease-negative strains of C. sputorum, all isolated from cattle over a 19-month time period, may be members of a novel taxon. Within these six strains, four divergent ST were identified (ST-8sp, ST-13sp, ST-14sp, and ST-15sp). The concatenated allele sequences of these four STs are 95% similar to STs from the three established C. sputorum biovars, that display only 1% sequence divergence across the 3321-bp (Figure 2). Thus, it is possible that these six strains are members of a C. sputorum-like taxon, perhaps a novel C. sputorum subspecies or biovar. Nevertheless, for both the divergent C. lanienae and C. sputorum strains, additional biochemical and molecular tests will need to be performed to definitively establish their taxonomic position within Campylobacter.
Previous studies investigating the diversity of C. concisus organized strains from this species into two major genetically diverse clusters or genomospecies (GS), based on strain typing using 23S rRNA PCR (Engberg et al., 2005; Kalischuk and Inglis, 2011) or AFLP (Aabenhus et al., 2005; Kalischuk and Inglis, 2011). Included in the C. concisus strain set here were several strains characterized previously by AFLP (Aabenhus et al., 2005). In agreement with these previous studies, phylogenetic analysis of the C. concisus STs identified two clusters: each cluster contained almost exclusively GS1 or GS2 strains (Figure 1). Within C. concisus, two to eight alleles at each of the seven MLST loci were identified in more than one ST (Table S1 in Supplementary Material). It is noteworthy perhaps that of these 33 “common” MLST alleles, only one allele (glyA-31) was identified in both GS1 and GS2 strains, suggesting that minimal genetic exchange occurs between the two genomospecies.
Division of C. concisus strains into two primary genogroups is not merely an academic exercise. In a study analyzing C. concisus strains isolated from diarrheic and non-diarrheic individuals, Kalischuk and Inglis reported that GS1 strains were isolated predominantly from healthy individuals while the GS2 cluster contained isolates primarily from diarrheic individuals (Kalischuk and Inglis, 2011); this correlation between diarrheal disease and GS2 C. concisus species was also observed by Aabenhus et al. (2005). Moreover, GS2 strains were reported to exhibit higher levels of epithelial invasion (Kalischuk and Inglis, 2011). Therefore, GS1 or GS2 strains would be predicted to lead possibly to different clinical outcomes, and the proper placement of C. concisus isolates into these two genomospecies would be critical not only clinically but also for epidemiological studies. The MLST method described here provides another accurate tool for C. concisus genomotyping.
Campylobacter concisus GS4 strains were isolated from severely immunodeficient patients and identified initially by limited phenotyping; comparative AFLP analysis and other DNA-based testing indicated their taxonomic position required clarification (Aabenhus et al., 2005). Given these data, and our MLST results, it is likely that these strains are C. curvus and not C. concisus. These species share many phenotypic traits (On et al., 1996) and are difficult to distinguish with limited phenotypic testing.
At present, C. sputorum biovars are identified by their catalase or urease phenotypes (On et al., 1998). No PCR/sequencing methods exist that differentiate the three described biovars, although whole-genome macro-restriction profiling has been used to suggest biovar clonality (On et al., 1999). However, putative biovar-associated alleles were observed at three of the C. sputorum MLST loci (Table S1 in Supplementary Material). For example, at the atpA locus, bv. sputorum strains are atpA-1, bv. fecalis strains are atpA-3, and bv. paraureolyticus strains are either atpA-2 or atpA-4; strains of the cow-associated C. sputorum clade described above are either atpA-5 or atpA-6. Similar associations exist at the glyA and pgm loci. Indeed, the three biovars segregate also when all seven loci are examined phylogenetically (Figure 2), although the differences are quite small. Obviously, the size of the C. sputorum strain set typed here is too small to reach any definitive conclusions, although the associations are intriguing.
The four MLST methods described in this study typed successfully all 213 Campylobacter strains, representing at least ten Campylobacter taxa that included both subspecies of C. fetus and C. hyointestinalis and all three biovars of C. sputorum. Each method identified multiple novel STs; the small number of STs present in some taxa were more likely due to the limited size of the sample sets for those taxa than a limitation of the method itself. These methods were successful despite the high degree of variation in some species, e.g., C. concisus. Also, the concatenated C. concisus and C. curvus ST sequences were only 81% similar at the nt level (Figure 1); likewise, the C. hyointestinalis and C. fetus ST sequences were 87% similar (Figure 2), yet MLST Methods 1 and 2 could type either set of strains unequivocally. Indeed, the ability to sequence such variable strains provided an unexpected bonus to these MLST methods. The methods described here identified putative novel C. lanienae- and C. sputorum-related taxa and it is likely that these methods could further characterize and type as-yet-undescribed Campylobacter species or subspecies. For example, MLST Method 2 has been used to type reptile-associated C. fetus-like organisms (data not shown).
For many campylobacters, sequence data is restricted currently to ribosomal rRNA loci. While these rDNA sequences can provide crucial speciation data for many taxa, some groups of campylobacters cannot be differentiated readily by 16S rDNA sequencing. One such example includes C. hyointestinalis and C. lanienae strains. Some of the strains in this study from these species could not be typed unequivocally by 16S rDNA sequencing; however, MLST could readily place all strains in their proper taxonomic positions. MLST has been shown also to be of value in identifying strains of species with multiple phenogroups, such as C. insulaenigrae (Stoddard et al., 2007). In this study, a C. sputorum clade was typed that, based on established phenotypic characterization, would likely have been classified as bv. fecalis. While additional tests need to be performed, MLST cast some doubt that these strains were C. sputorum bv. fecalis.
Eighteen of the thirty validly described Campylobacter taxa can now be typed by MLST. This number is likely an underestimate, as some of the C. lari-like species (e.g., C. peloridis) described recently can be typed also using the C. lari MLST method (data not shown). The ability of MLST to type and speciate campylobacters, as well as identify putative horizontal gene transfer, indicates that the multiple Campylobacter MLST methods now available provide a valuable tool in the epidemiology, typing, and evolution of emerging campylobacters.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Nate Simon for technical assistance in the C. hyointestinalis strain typing. We thank Dr. Birgitta Duim for critical reading of this manuscript. We thank also S. Chandler, D. Orthmeyer, G. Baird-Kavanaugh, and S. Hee for providing some of the wildlife and environmental samples; and E. R. Atwill, M. Jay-Russell for their support in projects that supported partially this research and funded by National Research Initiative Competitive Grant nos. 2006-55212-16927 and 2007-35212-18239 from the USDA National Institute of Food and Agriculture. This work was supported mostly by USDA Agricultural Research Service CRIS projects 5325-42000-044 and -045. This publication made use of the Campylobacter Multilocus Sequence Typing website (http://pubmlst.org/campylobacter/) developed by Keith Jolley at the University of Oxford (Jolley and Maiden, 2010).
The Supplementary Material for this article can be found online at http://www.frontiersin.org/Cellular_and_Infection_Microbiology/10.3389/fcimb.2012.00045/abstract
Table S1. Campylobacter strains typed in this study.
Aabenhus, R., On, S. L., Siemer, B. L., Permin, H., and Andersen, L. P. (2005). Delineation of Campylobacter concisus genomospecies by amplified fragment length polymorphism analysis and correlation of results with clinical data. J. Clin. Microbiol. 43, 5091–5096.
Anonymous. (2009). Preliminary FoodNet Data on the incidence of infection with pathogens transmitted commonly through food – 10 States, 2008. MMWR Morb. Mortal. Wkly. Rep. 58, 333–337.
Bullman, S., O’Leary, J., Corcoran, D., Sleator, R. D., and Lucey, B. (2011). Molecular-based detection of non-culturable and emerging campylobacteria in patients presenting with gastroenteritis. Epidemiol. Infect. 140, 684–688.
Colles, F. M., Jones, K., Harding, R. M., and Maiden, M. C. (2003). Genetic diversity of Campylobacter jejuni isolates from farm animals and the farm environment. Appl. Environ. Microbiol. 69, 7409–7413.
Debruyne, L., Gevers, D., and Vandamme, P. (2008). “Taxonomy of the family Campylobacteraceae,” in Campylobacter, 3rd Edn, eds I. Nachamkin, C. M. Szymanski, and M. J. Blaser (Washington, DC: ASM Press), 3–25.
Dingle, K. E., Colles, F. M., Falush, D., and Maiden, M. C. (2005). Sequence typing and comparison of population biology of Campylobacter coli and Campylobacter jejuni. J. Clin. Microbiol. 43, 340–347.
Dingle, K. E., Colles, F. M., Wareing, D. R. A., Ure, R., Fox, A. J., Bolton, F. E., Bootsma, H. J., Willems, R. J. L., Urwin, R., and Maiden, M. C. J. (2001). Multilocus sequence typing system for Campylobacter jejuni. J. Clin. Microbiol. 39, 14–23.
Edmonds, P., Patton, C. M., Griffin, P. M., Barrett, T. J., Schmid, G. P., Baker, C. N., Lambert, M. A., and Brenner, D. J. (1987). Campylobacter hyointestinalis associated with human gastrointestinal disease in the United States. J. Clin. Microbiol. 25, 685–691.
Engberg, J., Bang, D. D., Aabenhus, R., Aarestrup, F. M., Fussing, V., and Gerner-Smidt, P. (2005). Campylobacter concisus: an evaluation of certain phenotypic and genotypic characteristics. Clin. Microbiol. Infect. 11, 288–295.
Gorkiewicz, G., Feierl, G., Zechner, R., and Zechner, E. L. (2002). Transmission of Campylobacter hyointestinalis from a pig to a human. J. Clin. Microbiol. 40, 2601–2605.
Hakkinen, M., Heiska, H., and Hanninen, M. L. (2007). Prevalence of Campylobacter spp. in cattle in Finland and antimicrobial susceptibilities of bovine Campylobacter jejuni strains. Appl. Environ. Microbiol. 73, 3232–3238.
Inglis, G. D., Kalischuk, L. D., and Busz, H. W. (2004). Chronic shedding of Campylobacter species in beef cattle. J. Appl. Microbiol. 97, 410–420.
Jolley, K. A., Feil, E. J., Chan, M. S., and Maiden, M. C. (2001). Sequence type analysis and recombinational tests (START). Bioinformatics 17, 1230–1231.
Jolley, K. A., and Maiden, M. C. (2010). BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics 11, 595. doi:10.1186/1471-2105-11-595
Kalischuk, L. D., and Inglis, G. D. (2011). Comparative genotypic and pathogenic examination of Campylobacter concisus isolates from diarrheic and non-diarrheic humans. BMC Microbiol. 11, 53. doi:10.1186/1471-2180-11-53
Kimura, M. (1980). A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16, 111–120.
Lane, D. J. (1991). “16S/23S rRNA sequencing,” in Nucleic Acid Sequencing Techniques in Bacterial Systematics, eds E. Stakebrandt and M. Goodfellow (New York, NY: Wiley and Sons), 115–175.
Lastovica, A. J., and Allos, B. M. (2008). “Clinical significance of Campylobacter and related species other than Campylobacter jejuni and Campylobacter coli,” in Campylobacter, 3rd Edn, eds I. Nachamkin, C. M. Szymanski, and M. J. Blaser (Washington, DC: ASM Press), 123–149.
Lynch, O. A., Cagney, C., McDowell, D. A., and Duffy, G. (2011). Occurrence of fastidious Campylobacter spp. in fresh meat and poultry using an adapted cultural protocol. Int. J. Food Microbiol. 150, 171–177.
Mahendran, V., Riordan, S. M., Grimm, M. C., Tran, T. A., Major, J., Kaakoush, N. O., Mitchell, H., and Zhang, L. (2011). Prevalence of Campylobacter species in adult Crohn’s disease and the preferential colonization sites of Campylobacter species in the human intestine. PLoS ONE 6, e25417. doi:10.1371/journal.pone.0025417
Maiden, M. C., and Dingle, K. E. (2008). “Population biology of Campylobacter jejuni and related organisms,” in Campylobacter, 3rd Edn, eds I. Nachamkin, C. Szymanski, and M. J. Blaser (Washington, DC: ASM Press), 27–40.
Man, S. M., Zhang, L., Day, A. S., Leach, S. T., Lemberg, D. A., and Mitchell, H. (2010). Campylobacter concisus and other Campylobacter species in children with newly diagnosed Crohn’s disease. Inflamm. Bowel Dis. 16, 1008–1016.
Manning, G., Dowson, C. G., Bagnall, M. C., Ahmed, I. H., West, M., and Newell, D. G. (2003). Multilocus sequence typing for comparison of veterinary and human isolates of Campylobacter jejuni. Appl. Environ. Microbiol. 69, 6370–6379.
Miller, W. G., On, S. L., Wang, G., Fontanoz, S., Lastovica, A. J., and Mandrell, R. E. (2005). Extended multilocus sequence typing system for Campylobacter coli, C. lari, C. upsaliensis, and C. helveticus. J. Clin. Microbiol. 43, 2315–2329.
Miller, W. G., Wesley, I. V., On, S. L., Houf, K., Megraud, F., Wang, G., Yee, E., Srijan, A., and Mason, C. J. (2009). First multi-locus sequence typing scheme for Arcobacter spp. BMC Microbiol. 9, 196. doi:10.1186/1471-2180-9-196
Mølbak, K., and Havelaar, A. (2008). “Burden of illness of campylobacteriosis and sequelae,” in Campylobacter, 3rd Edn, eds I. Nachamkin, C. M. Szymanski, and M. J. Blaser (Washington, DC: ASM Press), 151–162.
Mukhopadhya, I., Thomson, J. M., Hansen, R., Berry, S. H., El-Omar, E. M., and Hold, G. L. (2011). Detection of Campylobacter concisus and other Campylobacter species in colonic biopsies from adults with ulcerative colitis. PLoS ONE 6, e21490. doi:10.1371/journal.pone.0021490
Olson, C. K., Ethelberg, S., Van Pelt, W., and Tauxe, R. V. (2008). “Epidemiology of Campylobacter jejuni infections in industrialized nations,” in Campylobacter, 3rd Edn, eds I. Nachamkin, C. M. Szymanski, and M. J. Blaser (Washington, DC: ASM Press), 163–189.
On, S. L., Atabay, H. I., and Corry, J. E. (1999). Clonality of Campylobacter sputorum bv. paraureolyticus determined by macrorestriction profiling and biotyping, and evidence for long-term persistent infection in cattle. Epidemiol. Infect. 122, 175–182.
On, S. L., Atabay, H. I., Corry, J. E., Harrington, C. S., and Vandamme, P. (1998). Emended description of Campylobacter sputorum and revision of its infrasubspecific (biovar) divisions, including C. sputorum biovar paraureolyticus, a urease-producing variant from cattle and humans. Int. J. Syst. Bacteriol. 48(Pt 1), 195–206.
On, S. L., Holmes, B., and Sackin, M. J. (1996). A probability matrix for the identification of campylobacters, helicobacters and allied taxa. J. Appl. Bacteriol. 81, 425–432.
On, S. L. W., Mccarthy, N., Miller, W. G., and Gilpin, B. J. (2008). “Molecular epidemiology of Campylobacter species,” in Campylobacter, 3rd Edn, eds I. Nachamkin, C. Szymanski, and M. J. Blaser (Washington, DC: ASM Press), 191–212.
Oporto, B., and Hurtado, A. (2011). Emerging thermotolerant Campylobacter species in healthy ruminants and swine. Foodborne Pathog. Dis. 8, 807–813.
Perez-Losada, M., Porter, M. L., Viscidi, R. P., and Crandall, K. A. (2011). “Multilocus sequence typing of pathogens,” in Genetics and Evolution of Infectious Diseases, ed. M. Tibayrenc (Burlington, MA: Elsevier), 503–522.
Salihu, M. D., Abdulkadir, J. U., Oboegbulem, S. I., Egwu, G. O., Magaji, A. A., Lawal, M., and Hassan, Y. (2009). Isolation and prevalence of Campylobacter species in cattle from Sokoto state, Nigeria. Vet. Ital. 45, 501–505.
Sasaki, Y., Fujisawa, T., Ogikubo, K., Ohzono, T., Ishihara, K., and Takahashi, T. (2003). Characterization of Campylobacter lanienae from pig feces. J. Vet. Med. Sci. 65, 129–131.
Stoddard, R. A., Miller, W. G., Foley, J. E., Lawrence, J., Gulland, F. M., Conrad, P. A., and Byrne, B. A. (2007). Campylobacter insulaenigrae isolates from northern elephant seals (Mirounga angustirostris) in California. Appl. Environ. Microbiol. 73, 1729–1735.
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739.
Terzolo, H. R. (1988). Identification of campylobacters from bovine and ovine faeces. Rev. Argent. Microbiol. 20, 53–68.
Keywords: MLST, emerging, Campylobacter hyointestinalis, Campylobacter lanienae, Campylobacter concisus, Campylobacter curvus, Campylobacter sputorum
Citation: Miller WG, Chapman MH, Yee E, On SLW, McNulty DK, Lastovica AJ, Carroll AM, McNamara EB, Duffy G and Mandrell RE (2012) Multilocus sequence typing methods for the emerging Campylobacter species C. hyointestinalis, C. lanienae, C. sputorum, C. concisus, and C. curvus. Front. Cell. Inf. Microbio. 2:45. doi: 10.3389/fcimb.2012.00045
Received: 05 December 2011; Accepted: 16 March 2012;
Published online: 02 April 2012.
Edited by:
Alain Stintzi, Ottawa Institute of Systems Biology, CanadaReviewed by:
Qijing Zhang, Iowa State University, USACopyright: © 2012 Miller, Chapman, Yee, On, McNulty, Lastovica, Carroll, McNamara, Duffy and Mandrell. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: William G. Miller, USDA, ARS, WRRC, Produce Safety and Microbiology Research Unit, 800 Buchanan Street, Albany, CA 94710, USA. e-mail:d2lsbGlhbS5taWxsZXJAYXJzLnVzZGEuZ292
†Present address: Desmond K. McNulty, ICON Central Laboratories, South County Business Park, Leopardstown, Dublin, Ireland.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.