
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Infect. Microbiol., 18 April 2012
Sec. Molecular Bacterial Pathogenesis
Volume 2 - 2012 | https://doi.org/10.3389/fcimb.2012.00044
This article is part of the Research TopicThe Staphylococci and staphylococcal pathogenesisView all 19 articles
 Nouri L. Ben Zakour1,2
Nouri L. Ben Zakour1,2 Scott A. Beatson2 Adri H. M. van den Broek3 Keith L. Thoday3
Scott A. Beatson2 Adri H. M. van den Broek3 Keith L. Thoday3 J. Ross Fitzgerald1*
J. Ross Fitzgerald1*The Staphylococcus intermedius group consists of three closely related coagulase-positive bacterial species including S. intermedius, Staphylococcus pseudintermedius, and Staphylococcus delphini. S. pseudintermedius is a major skin pathogen of dogs, which occasionally causes severe zoonotic infections of humans. S. delphini has been isolated from an array of different animals including horses, mink, and pigeons, whereas S. intermedius has been isolated only from pigeons to date. Here we provide a detailed analysis of the S. pseudintermedius whole genome sequence in comparison to high quality draft S. intermedius and S. delphini genomes, and to other sequenced staphylococcal species. The core genome of the SIG was highly conserved with average nucleotide identity (ANI) between the three species of 93.61%, which is very close to the threshold of species delineation (95% ANI), highlighting the close-relatedness of the SIG species. However, considerable variation was identified in the content of mobile genetic elements, cell wall-associated proteins, and iron and sugar transporters, reflecting the distinct ecological niches inhabited. Of note, S. pseudintermedius ED99 contained a clustered regularly interspaced short palindromic repeat locus of the Nmeni subtype and S. intermedius contained both Nmeni and Mtube subtypes. In contrast to S. intermedius and S. delphini and most other staphylococci examined to date, S. pseudintermedius contained at least nine predicted reverse transcriptase Group II introns. Furthermore, S. pseudintermedius ED99 encoded several transposons which were largely responsible for its multi-resistant phenotype. Overall, the study highlights extensive differences in accessory genome content between closely related staphylococcal species inhabiting distinct host niches, providing new avenues for research into pathogenesis and bacterial host-adaptation.
Population genetic analysis has revealed the existence of a group of closely related coagulase-positive staphylococcal species associated with different host-species, collectively known as the Staphylococcus intermedius group (SIG; Varaldo et al., 1988; Bannhoer et al., 2007; Sasaki et al., 2007b). The SIG consists of Staphylococcus pseudintermedius, an opportunistic pathogen which predominantly colonizes the skin and mucosal surfaces of dogs (Kloos, 1980; Greene and Lammler, 1993), Staphylococcus delphini which has been isolated from a wide array of animals, including minks, horses, cows, and pigeons (Sasaki et al., 2007b), and S. intermedius, which has only been isolated from pigeons to date (Bannoehr et al., 2007; Sasaki et al., 2007b). Although S. pseudintermedius is a component of the canine normal flora, disruption of the normal skin flora or an underlying condition such as atopic dermatitis, can lead to S. pseudintermedius infections such as superficial and deep canine pyoderma, and otitis media or externa (Cole et al., 1998). Recently, strains of S. pseudintermedius, which are refractory to treatment by most commonly used classes of antibiotic including methicillin have emerged and disseminated widely (Perreten et al., 2010; Ruscher et al., 2010). S. pseudintermedius is rarely isolated from humans but can occasionally cause severe zoonotic infections, typically through dog bite wounds (Mahoudeau et al., 1997; Tanner et al., 2000; Pottumarthy et al., 2004). Furthermore, S. pseudintermedius has the capacity to produce enterotoxins related to those made by Staphylococcus aureus and has been implicated in several food poisoning outbreaks (Khambaty et al., 1994; Becker et al., 2001). Our understanding of the molecular pathogenesis of S. pseudintermedius canine pyoderma is very limited (Fitzgerald, 2009). However, the recent announcement of the first genome sequences for S. pseudintermedius strains has revealed the complement of genes encoding putative virulence factors (Ben Zakour et al., 2011; Tse et al., 2011), leading to proteomic studies which have identified novel host–pathogen interactions (Bannoehr et al., 2011a,b). An enhanced understanding of the pathogenesis of S. pseudintermedius is required in order to facilitate the design of novel therapeutics for the control of bacterial pyoderma infection caused by multi-resistant S. pseudintermedius. Furthermore, the distinct host-tropisms of the three members of the SIG suggest that these closely related species would represent an excellent system for investigating evolutionary events underlying bacterial host-adaptation, a fundamental aspect of bacteriology which has not been well examined to date.
Recently, we published an announcement of the S. pseudintermedius ED99 genome, briefly listing several noteworthy features of the genome (Ben Zakour et al., 2011). Here we provide a comprehensive analysis of the ED99 genome in comparison to high quality draft genomes of the closely related species S. delphini, and S. intermedius generated in the current study, and to publicly available genomes for other staphylococcal species. The resulting data represent an excellent framework for investigations into the pathogenesis of canine pyoderma, and the molecular basis for staphylococcal host-specificity.
The previously sequenced S. pseudintermedius ED99 (formerly M732/99) was isolated from a clinical case of canine bacterial pyoderma in 1999 in Scotland and was selected to represent one of the common clones identified in a previous population genetic study of S. pseudintermedius (Bannoehr et al., 2007; Ben Zakour et al., 2011). S. delphini 8086 was isolated from the trachea of a horse in the UK (Bannoehr et al., 2007), and the type strain S. intermedius NCTC11048, from the anterior nares of a pigeon in the Czech Republic (Hajek, 1976).
Genomic DNA was isolated from 1 ml of overnight culture of S. pseudintermedius in BHI (Oxoid) at 37°C with shaking at 200 rpm. Genomic DNA extraction was carried out with a bacterial genomic DNA purification kit (Edge Biosystems) according to the manufacturer’s instructions, except that prior to incubation at 37°C for 10 min, 125 μg/ml lysostaphin (AMBI L) was included.
Whole genome sequencing of S. pseudintermedius ED99 was carried out as previously described (Ben Zakour et al., 2011). Genome sequencing of S. delphini 8086 and S. intermedius NCTC11048 was carried out using the Illumina 3G Genome Analyzer. For each strain, we generated a total of 4,087,613 and 3,879,139 paired-end reads, respectively, with a fixed length of 36 bp and an average insert size of 200 bp, corresponding to more than 58× and 50× genome coverage, respectively. De novo assembly was performed by using the Velvet short reads assembler program (Zerbino and Birney, 2008). For each genome, contigs were mapped against the completed whole genome of S. pseudintermedius ED99 using MauveAligner (Rissman et al., 2009) and manually inspected for potential mis-assemblies. To confirm the reliability of the sequences obtained by this de novo sequencing approach based only on very short reads, re-sequencing of S. pseudintermedius ED99 as an internal control was also performed in parallel to S. delphini 8086 and S. intermedius NCTC11048. An automatic annotation was then performed by the RAST annotation server to predict CDS and their putative functions (Aziz et al., 2008). Functional categories were assigned by searching all predicted proteins against the COG database (www.ncbi.nlm.nih.gov/COG). The software AlienHunter (Vernikos and Parkhill, 2006) was used to detect atypical genome regions corresponding to putative horizontal gene transfer, insertion sequence (IS) elements were identified by interrogation of the IS database (Siguier et al., 2006), and clustered regularly interspaced short palindromic repeat (CRISPR) elements were characterized by the CRISPRFinder web software (Grissa et al., 2007a). The draft genome sequences of S. delphini 8086 and S. intermedius NCTC11048 have been deposited in the GenBank WGS database and have Genome Bioproject ID numbers PRJEA87011 and PRJEA87009, respectively.
Orthologous CDS between S. pseudintermedius ED99 (accession number CP002478), S. aureus Mu50 (accession number BA000017; Kuroda et al., 2001), Staphylococcus epidermidis RP62A (accession number CP000029; Gill et al., 2005), Staphylococcus haemolyticus JCSC1435 (accession number AP006716; Takeuchi et al., 2005), Staphylococcus saprophyticus ATCC15305 (accession number AP008934; Kuroda et al., 2005), Staphylococcus carnosus TM300 (accession number AM295250; Rosenstein et al., 2009), S. delphini 8086, and S. intermedius NCTC11048, were identified by reciprocal best hits using BLASTP, with a maximum e-value of 0.01, a minimum percentage identity of 40% and a minimum percentage sequence coverage of 80%. The average nucleotide identity (ANI) between the complete and draft genome sequences available for all staphylococcal species and the outgroup Macrococcus caseolyticus JCSC5402 (accession number AP009484; Baba et al., 2009), was determined by using the in silico DNA–DNA hybridization method of the JSpecies software using default parameters (Richter and Rossello-Mora, 2009). The distance matrix based on the ANI values obtained was then used in Splitstree to construct a Neighbor-Joining tree (Huson and Bryant, 2006).
We previously sequenced and annotated the complete genome of S. pseudintermedius ED99, used in this study as a reference for comparative genomic analysis with other SIG species (Ben Zakour et al., 2011). As previously reported, the ED99 genome was composed of a single circular chromosome of 2,572,216 bp (Figure 1) with an average G + C content of 37.6%, contained five ribosomal operons, 58 tRNA loci and encoded for 2401 predicted protein-coding sequences (CDSs; Bannoehr et al., 2011a). Wider analysis for the current study revealed that 44 putative CDS are pseudogenes, and functional information could be assigned to 77.6% of all CDS (1863 CDS), with 13.1% encoding conserved hypothetical proteins (315 CDS), and 9.3% encoding hypothetical proteins without significant homology to proteins of known function.
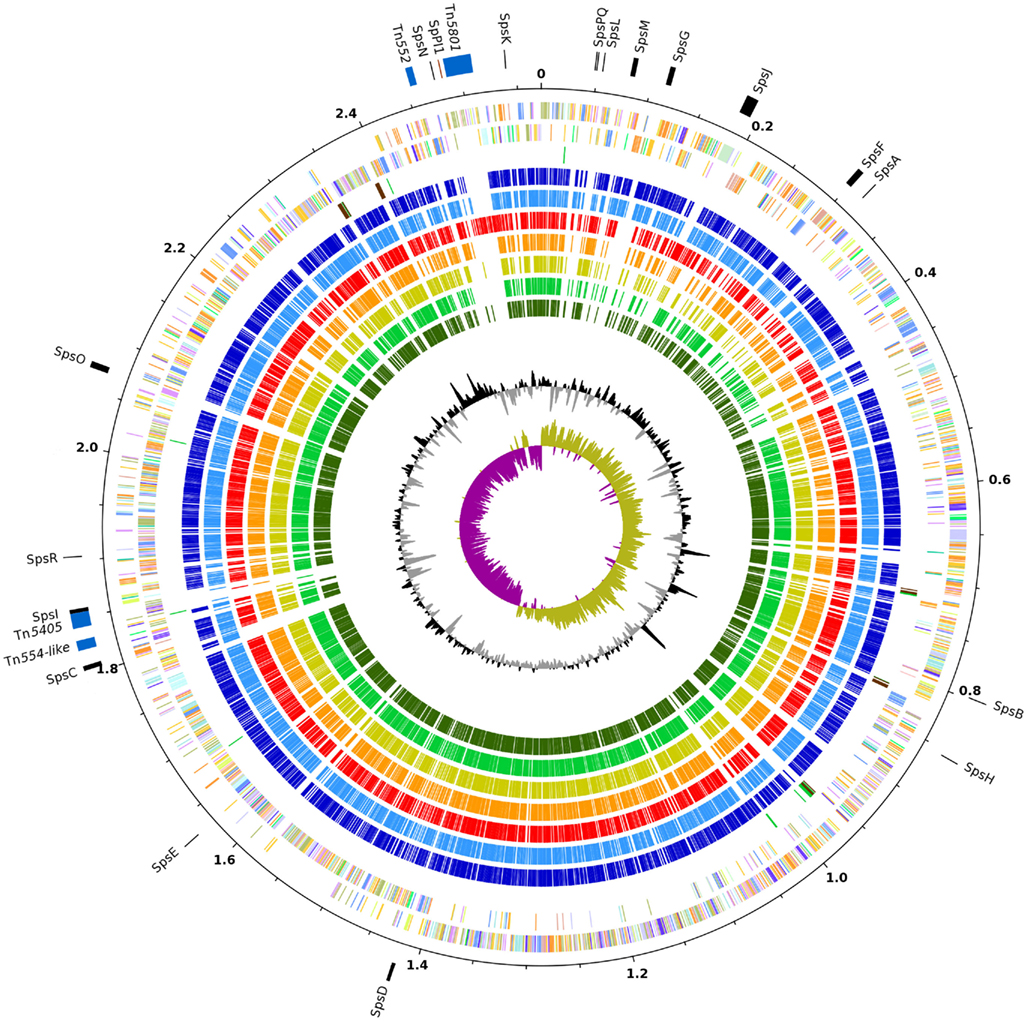
Figure 1. Genome atlas of S. pseudintermedius ED99 compared to other staphylococci. Orthologs depicted in other species are identified by reciprocal best hits using BLASTP. Major features of the S. pseudintermedius ED99 are indicated on the outermost circle, including MGE (blue), CWA proteins (black), and SpPI1 (maroon) with positions on the reference genome indicated in Mb. From the outer to inner circles, circle 2 and 3, represent S. pseudintermedius ED99 genes on plus and minus strands, respectively (colored according to their COG category); circle 4, tRNA (green), rRNA (brown); circle 5, S. delphini 8086 (dark blue); circle 6, S. intermedius NCTC11048 (blue); circle 7, S. aureus Mu50 (red); circle 8, S. epidermidis RP62A (orange); circle 9, S. haemolyticus JCSC1435 (yellow); circle 10, S. saprophyticus ATCC15305 (light green); circle 11, S. carnosus TM300 (dark green); circle 12, G + C content plot (black and gray); and circle 13, GC skew plot (purple and khaki).
The draft genome sequence of S. delphini 8086 was assembled into 133 contigs and had an approximate size of 2.53 Mb including 2369 predicted CDS. The draft genome sequence of S. intermedius NCTC11048 was assembled into 228 contigs, and had an approximate size of 2.78 Mb, significantly larger than the two other members of the SIG, and was predicted to encode 2589 CDS (Table 1). Of note, the average GC content of the three SIG genomes ranges from 37.4 to 38.3%, which is considerably higher than any other staphylococcal species sequenced to date suggesting the existence of distinct selective pressures influencing the SIG genome nucleotide composition.
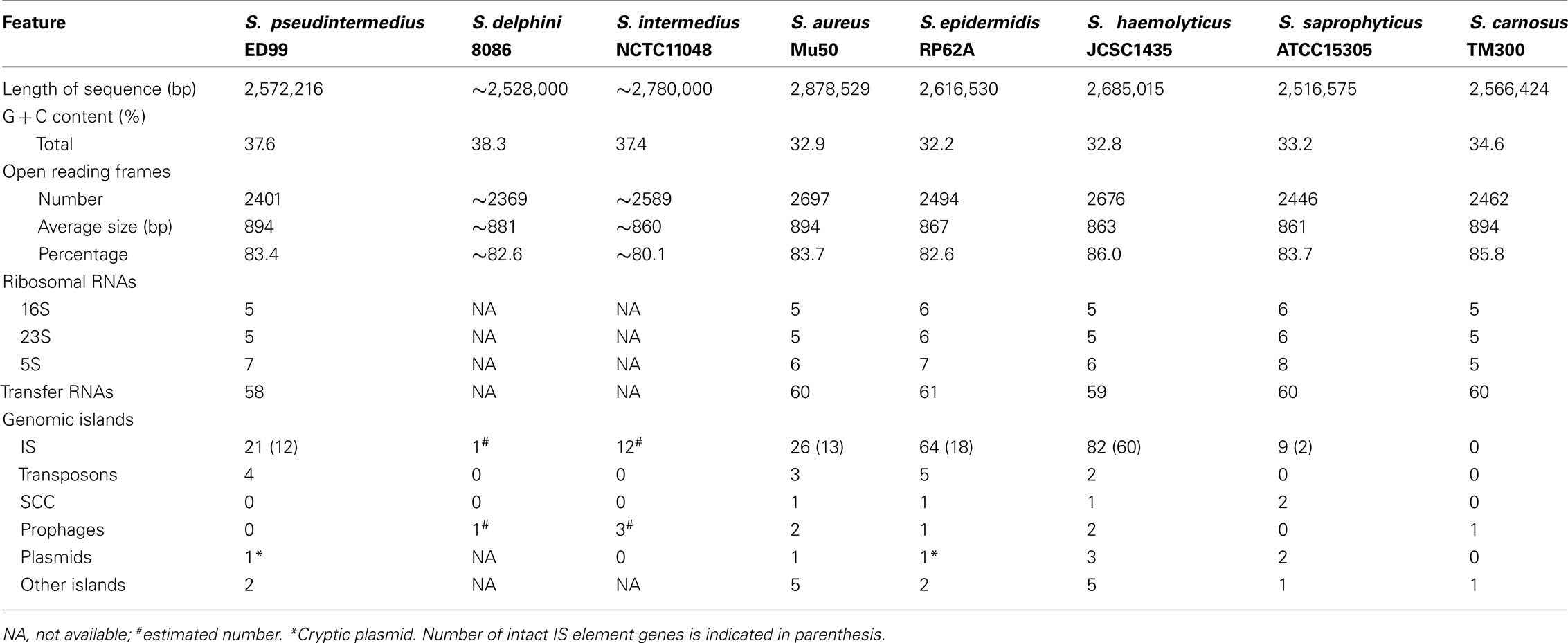
Table 1. General features of the SIG genomes in comparison with other staphylococci.
The genomes of the three SIG species were compared with those of S. aureus Mu50, S. epidermidis RP62A, S. haemolyticus JCSC1435, S. saprophyticus ATCC15305, and S. carnosus TM300 (Table 1). Comparison of the eight selected staphylococcal chromosomes by reciprocal BLASTP revealed a high level of conservation and synteny (Figure 1). However, a large chromosomal region (∼465 kb) of low similarity was identified which corresponds to the oriC environ, a region of staphylococcal genomes containing many species-specific CDS (Takeuchi et al., 2005). Similar to S. saprophyticus ATCC15305 and S. haemolyticus JCSC1435, the genome of S. pseudintermedius ED99 contains a large chromosomal inversion at the beginning of the oriC environ, from the position 33,035 to 2,568,216, and which spans the entire chromosome. Consistent with their close-relatedness, the oriC environ exhibits a high level of gene content conservation among the closely related SIG members.
By performing reciprocal best blast hits analysis, we have determined a set of 1214 genes defining the core genome shared by S. pseudintermedius ED99, S. aureus Mu50, S. epidermidis RP62A, S. haemolyticus JCSC1435, S. saprophyticus ATCC15305, S. carnosus TM300, S. delphini 8086, and S. intermedius NCTC11048 (Table S2 in Supplementary Material). Of the predicted proteins, 12.4% have a general predicted function, 9.1% are of unknown function and 4.3% have no homolog found in the COG database, which is consistent with previous studies (Takeuchi et al., 2005; Rosenstein et al., 2009). Based on the core genome only, the average percentage similarity of S. pseudintermedius ED99 proteins compared to other staphylococcal species ranges from 68.8% with S. carnosus TM300 to 95.2 and 97.7% with S. delphini 8086 and S. intermedius NCTC11048, respectively. The evolutionary relatedness of the staphylococcal species was examined by calculation of their ANI. The phylogenetic tree based on pairwise comparison of the ANI confirms the close-relatedness of the SIG compared to the other staphylococcal species and is consistent with previously determined phylogenies (Figure 2). Of note, despite sharing distinct ecological niches, S. pseudintermedius ED99 and S. delphini 8086 share an ANI of 93.61%, which is very close to the threshold of species delineation of 95% ANI, equivalent to the DNA–DNA re-association threshold of 70% (Goris et al., 2007).
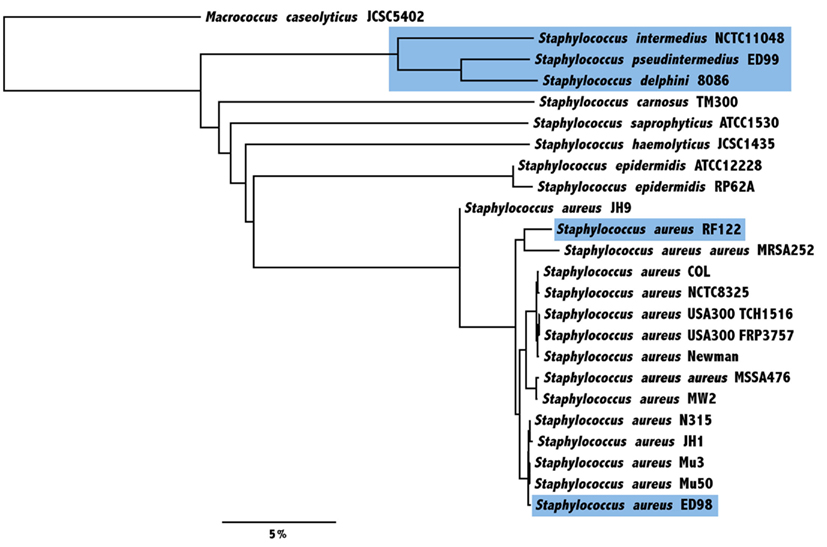
Figure 2. Average nucleotide identity (ANI)-based Neighbor-Joining tree of 23 sequenced strains from eight staphylococcal species in addition to an outgroup Macrococcus caseolyticus. The evolutionary relatedness was examined by calculation of ANI, based on core genes only. Species and strains of animal origin are highlighted in blue (scale indicates the % difference ANI).
Although the S. pseudintermedius ED99 genome demonstrated a large degree of conservation and synteny with the other staphylococcal genomes, numerous regions of differences (RDs) of greater than 5 kb in size were identified including IS elements, genomic islands, and prophage- and plasmid-related sequences (Figure 1). A total of 21 predicted IS elements were identified including 7 identical copies of ISSp1 which had 47% nucleotide identity with ISSha1 of S. haemolyticus and IS1182 integrated in Tn5405 which had 100% nucleotide homology with the same element in S. aureus. In several cases the closest homologs of S. pseudintermedius IS elements were in other genera such as Clostridium tetani E88 (46% identity), and Geobacillus thermodenitrificans NG80-2 (52% identity). In addition, transposase-related sequences indicating the presence of at least one IS element in S. delphini 8086 and 12 IS elements in S. intermedius NCTC11048 were identified.
Reverse transcriptase (RT) Group II introns are self-splicing retro-elements composed of an intron RNA domain and a RT gene which are present in 25% of sequenced bacterial genomes, including members of the Firmicutes such as Bacillus, Streptococcus, and Lactococcus (Matsuura et al., 1997; Granlund et al., 2001; Tourasse and Kolsto, 2008). RT Group II introns are extremely rare in staphylococcal genomes, and generally present in low number with one or two copies, such as in S. aureus JKD6159 (Chua et al., 2011) and S. pseudintermedius HKU-10 (Tse et al., 2011). A total of nine intact copies and one pseudogene of the RT gene were identified in the genome of S. pseudintermedius ED99. All copies had a predicted intergenic insertion site, and no particular trend in their location or the nature of the genes adjacent to these elements was observed. However, it is possible that the integration of RT Group II introns may influence the expression of downstream genes or promote recombinational events (Tourasse and Kolsto, 2008). Of note, RT Group II introns were not identified in the genomes of S. delphini 8086 and S. intermedius NCTC11048.
The S. pseudintermedius ED99 genome does not contain any predicted complete prophages but contains three RDs containing phage-related genes, and a putative integrated 3.5 kb plasmid with 38% overall identity to plasmid pC221 of S. aureus was detected within the oriC environ. The genome also contains a novel member of the staphylococcal pathogenicity island family (SpPI1) adjacent to Tn5801, which is 4.1 kb in size, and includes attachment sites and genes with 55% identity to the terminase orf5, 67% identity to the repressor orf20, and 90% identity to the integrase int of SaPIbov of S. aureus (Figure 3). Of note, these three genes belong to the core minimal set of genes required for a functional mobile SaPI (Ubeda et al., 2008). In theory, SpPI1 may represent an ancestral minimal form of SaPI but orf20 and int contain premature stop codons implying that SpPI1 is no longer mobile. Predicted virulence genes were not identified in SpPI1 but two genes encoding hypothetical proteins of unknown function were present. We examined the distribution of SpPI1 by PCR screening of 13 S. pseudintermedius strains, which represented diverse sequence types, with various geographical, host and clinical origins, and found that it was present in 11 strains (data not shown).
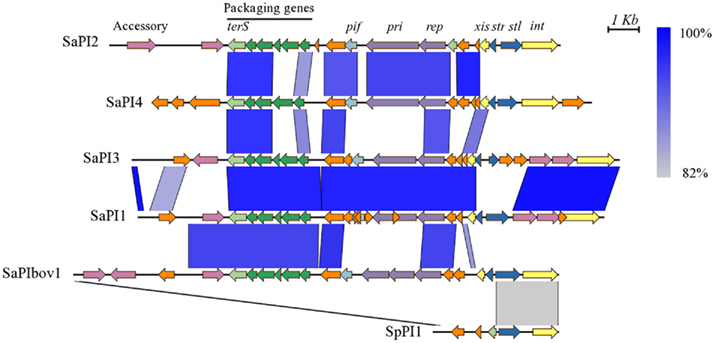
Figure 3. Comparison of SpPI1 with the major staphylococcal pathogenicity islands. The pathogenicity island SpPI1 from S. pseudintermedius ED99 is compared to SaPI2 from S. aureus RN3984, SaPI4 from S. aureus MRSA252, SaPI3 from S. aureus COL, SaPI1 from S. aureus RN4282, and SaPIbov1 from S. aureus RF122. The similarity between regions is indicated by a spectrum of blue to gray, from 100 to 82% similarity. Genes are colored according to their sequence and function, as described previously (Novick et al., 2010): int (integrase) and xis (excisionase) in yellow; transcription regulators in dark blue; replication genes including the primase gene, pri, and the replication initiator gene, rep in purple; encapsidation genes in green; terminase small subunit gene (terS) in light green; superantigen and other accessory genes in pink; pif (related to phage interference) in light blue; and genes encoding hypothetical proteins in orange. Figure produced with EasyFig (Sullivan et al., 2011).
Staphylococcus pseudintermedius ED99 contains a novel 14 kb genomic island located adjacent to a tRNA locus, which is also present in S. delphini 8086 (Figure A1 in Appendix). The island contains two genes with 51 and 32% homology respectively to orf32 and orf33 of ΦMu50B, two genes encoding the bi-component leukotoxin Luk-I identified previously in S. pseudintermedius SD91 (Prevost et al., 1995), and seven genes related to L-ascorbate transport and utilization not found in other staphylococci sequenced to date. The L-ascorbate utilization operon is organized in a novel combination of the ula operon of Escherichia coli (Yew and Gerlt, 2002) and shares similarity ranging from 55 to 68% with the ascorbate PTS system IIABC of Streptococcus sp. and the other members of the operon (ulaG, ulaD, ulaE, and ulaF) found in Enterococcus sp. The operon was identified in all 25 representative strains of S. pseudintermedius, S. intermedius, and S. delphini, with the exception of S. intermedius NCTC11048, which only contains the bi-component leukotoxin Luk-I (data not shown). In addition, despite being located downstream of a tRNA locus, which is a well-known integration site for mobile elements, mobility genes and flanking repeats regions were not identified in the sequence of both islands. Taken together, these characteristics imply an ancient acquisition in a progenitor of the SIG group and active maintenance of the acquired function among the SIG species.
Clustered regularly interspaced short palindromic repeats, recently described as an adaptative bacterial immune system, were shown to provide protection against viruses in Streptococcus thermophilus (Barrangou et al., 2007) and prevent conjugative transfer of plasmids in S. epidermidis (Marraffini and Sontheimer, 2008). CRISPR are widespread and have been identified in ∼40 and 90% of bacterial and archeal genomes sequenced (Grissa et al., 2007b). Occurrences of CRISPR are rare in staphylococcal species and are so far represented by the Mtube subtype (Haft et al., 2005) in the genome of S. epidermidis RP62A (Marraffini and Sontheimer, 2008), S. lugdunensis HKU09-01 (Tse et al., 2010) and on a novel SCCmecV element carried by livestock-associated methicillin-resistant S. aureus (Golding et al., 2010). Both S. pseudintermedius and S. intermedius genomes harbor a CRISPR locus of the Nmeni subtype, exclusively associated with vertebrate pathogens and commensals (Gill et al., 2005), and for which the closest homologs identified were found in Streptococcus M1 GAS and Ruminococcus lactaris ATCC 29176 (Figure 4). The identity between S. pseudintermedius and S. intermedius CRISPR-associated genes ranged from 78.7 to 86.7% while the 36-bp-repeat units were almost identical (97%), suggesting that the locus may have been acquired before speciation by a common ancestor to the SIG and then lost by S. delphini, although independent acquisition by both species cannot be ruled out. The repeat array of the S. pseudintermedius CRISPR locus contains 22 different spacers, with a size ranging from 29 to 31 bp, for which 6 share similarity with sequences of prophages and plasmids such as Φ2638A, pMG1, and pH308197 associated with staphylococcal, enterococcal, and bacillus species, respectively. Of note, eight additional spacers were found to share similarity with prophage-related regions from the S. intermedius genome. In addition, a CRISPR locus of the Mtube subtype was also identified in the genome of S. intermedius, for which the closest homologs were the CRISPR loci found in S. epidermidis RP62A and S. lugdunensis HKU09-01 (Figure 4). The presence of CRISPR loci in S. pseudintermedius and S. intermedius correlates with the absence of plasmids, and prophages as previously described (Barrangou et al., 2007; Marraffini and Sontheimer, 2008). However, the role of CRISPRs in immunity to non-phage genomic islands is unclear, as illustrated by the presence of several recently acquired transposons in the S. pseudintermedius ED99 genome. Furthermore, other mechanisms such as restriction–modification can limit the transfer of mobile genetic elements (MGE) between bacteria (Kobayashi, 2001). Although, we did not mention it specifically in the paper, S. delphini contains several genes related to these functions, which could partly explain the lack of genomic islands in S. delphini. In addition, little is known about the bacterial composition of the environmental niche occupied by S. delphini. Therefore, hypotheses explaining the lack of genomic islands in S. delphini include (i) that less diversity is encountered by this species in its niche, or (ii) most of the other species encountered are distantly related, resulting in less frequent successful horizontal gene transfers.
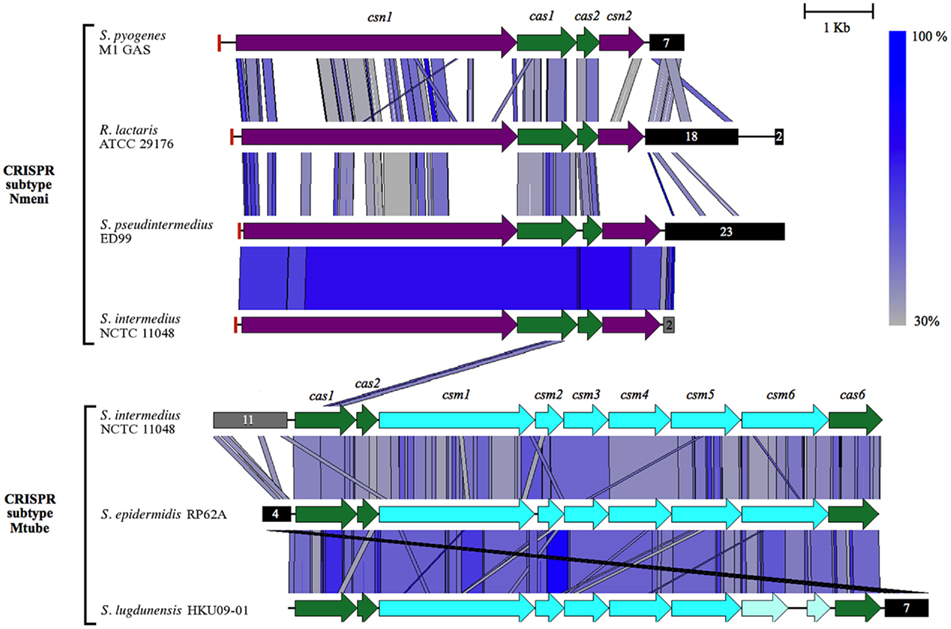
Figure 4. Comparison of the CRISPR loci of S. pseudintermedius ED99 and S. intermedius NCTC 11048 with related Nmeni and Mtube types. The CRISPR locus subtype Nmeni of S. pseudintermedius ED99 and S. intermedius NCTC 11048 are compared to those of Streptococcus M1 GAS and Ruminococcus lactaris ATCC 29176. The CRISPR locus subtype Mtube of S. intermedius NCTC 11048 is compared to those of S. epidermidis RP62A and S. lugdunensis HKU09-01. The similarity between regions is indicated by a spectrum of blue to gray colors depicting a range of 100–30% similarities. Genes of the csn family are shown in purple, the cas family in green, and the csm family in light blue. Of note, the csm6 gene in S. lugdunensis HKU09-01 appears to be a pseudogene. Black boxes represent clusters of direct repeats regions with the number of spacers between the direct repeats indicated. The red box at the beginning of each Nmeni subtype CRISPR locus corresponds to a single copy of the repeats identified in the cluster of downstream direct repeats. The gray boxes indicate the approximate number of direct repeats at the locus in S. intermedius NCTC 11048 (due to the unfinished status of the genome). Figure produced with EasyFig (Sullivan et al., 2011).
All SIG species genomes encode a number of predicted toxins including several previously identified such as the enterotoxin Se-int (Becker et al., 2001; Hendricks et al., 2002; Futagawa-Saito et al., 2004), hemolysin III, the β-hemolysin (Dziewanowska et al., 1996), the bi-component leukotoxin Luk-I (Prevost et al., 1995) and several exfoliative toxins (Terauchi et al., 2003; Futagawa-Saito et al., 2009; Iyori et al., 2010; Table 2). We also identified a putative novel exfoliative toxin specific to S. pseudintermedius ED99 designated SPETA which has 78 and 76% amino acid identity with the exfoliative toxin A SHETA from Staphylococcus hyicus and ETA from S. aureus, respectively. The existence of several exfoliative toxin variants made by different S. pseudintermedius strains is consistent with the skin tropism of S. pseudintermedius and the characteristic skin pathology associated with pyoderma. A wide range of exoenzymes is encoded by all three SIG species, such as the serine protease HtrA, two lipases encoded by the genes lip and geh, a thermonuclease, a protease ClpX, and a thermolysin (Wladyka et al., 2008). In contrast to other staphylococcal genome sequences, S. pseudintermedius ED99 and S. delphini 8086 genomes both contain genes encoding a putative sialidase or neuraminidase. The neuraminidase modification of host sugars may contribute to host colonization by providing a carbon source for growth, contributing to biofilm formation, or by enhancing adherence by exposing receptors on the host cell (Sakarya et al., 2010). Overall, the SIG species share a large number of toxins and exoenzymes reflecting their recent common ancestry and possibly their common skin niche.
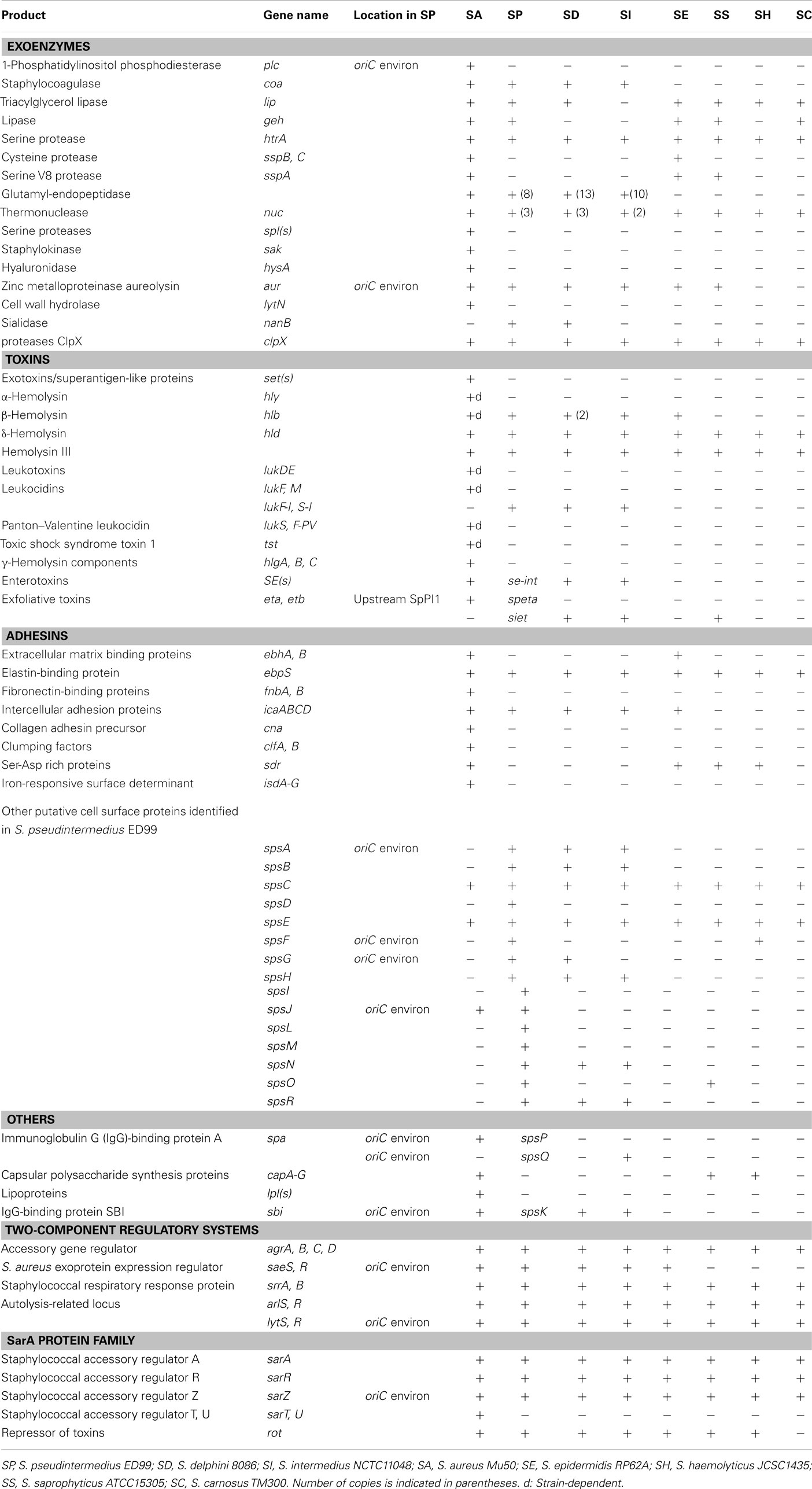
Table 2. Distribution of virulence factors identified in eight staphylococcal species.
All three SIG species contained genes encoding proteins with ∼42% identity to the Von Willebrand-binding protein of S. aureus which is involved in the formation of abscesses (Cheng et al., 2010). S. pseudintermedius ED99 contains a cluster of eight genes encoding predicted glutamyl-endopeptidases which share 56–75% identity with each other, and 29–42% identity with the probable glutamyl-endopeptidase ORF2 encoded by the etd pathogenicity island of S. aureus TY114 (Yamaguchi et al., 2002). This glutamyl-endopeptidases cluster is also present in S. intermedius NCTC 11048 and S. delphini 8086 and is not associated with a predicted MGE implying it is part of the core genome of the SIG species.
A total of 18 genes encoding putative cell wall-associated (CWA) proteins, designated SpsA to SpsR, were previously identified in the genome of S. pseudintermedius ED99 (Bannoehr et al., 2011a). Of these, nine were also encoded in the S. intermedius and S. delphini genomes, revealing a considerable number of S. pseudintermedius-specific CWA proteins which may be important for its canine host tropism. Recently, it was demonstrated that SpsD and SpsL, mediated binding to several extracellular matrix proteins. Of note, there was enhanced affinity of SpsL for canine in comparison to human fibrinogen implying a role in host-specific interactions (Bannoehr et al., 2011a).
Finally, the control of S. aureus virulence gene expression is coordinated by an array of global regulators (Novick, 2003). With the exception of the S. aureus regulators (saeSR) and the staphylococcal accessory regulators encoded by sarSTU, homologs of numerous well-characterized regulators of virulence previously identified in sequenced staphylococcal species were detected in the genomes of the SIG species (Table 2).
Compared to human-specific bacterial species, the SIG species encounter distinct environmental conditions dependent on their host-species with several biophysical parameters influenced, including skin hydration and pH (Montagna, 1967). For instance, in contrast to humans, sweat glands in the skin of animals such as dogs are not involved in thermoregulation (Affolter and Moore, 1994) resulting in lower relative skin hydration (Boelsma et al., 2003; Shimada et al., 2009). In addition, skin pH in dogs has been reported to differ from humans and to vary greatly depending on the anatomical site and breed of dog (Matousek and Campbell, 2002). Furthermore, there are known host-dependent differences in the availability of iron which influence the mechanisms of iron acquisition employed by staphylococci (Pishchany et al., 2010). In relation to these differing environments, the SIG species have a diverse complement of genes encoding proteins potentially involved in osmo-protection and resistance to oxidative stress. For example, S. pseudintermedius ED99 has four putative nitroreductase genes, one more than S. intermedius NCTC11048 and S. delphini 8086. All SIG species harbor several sodium/salt transporters, with the exception of the high-affinity potassium system Kdp which is absent from S. intermedius NCTC11048. S. pseudintermedius ED99 and S. delphini 8086 also contain a second catalase gene known as katB which has to date only been identified among staphylococcal species that inhabit high osmotic and oxidative stress niches including, S. xylosus, S. saprophyticus, and S. equorum (Blaiotta et al., 2010). Another source of genetic variation among the SIG species which could be related to their host niche is the transport and metabolism of small molecules such as carbohydrates. S. pseudintermedius ED99 contains genes encoding proteins predicted to mediate the transport and utilization of lactose/galactose in contrast to S. intermedius NCTC11048 and S. delphini 8086 which encode variant sugar transporters likely to have distinct substrates. S. intermedius NCTC11048 also encodes a novel ribose ABC transporter while S. delphini 8086 harbors two additional PTS systems, predicted to be involved in transport of cellobiose and glucitol/sorbitol, respectively.
In common with other staphylococci, the SIG species have developed several mechanisms for acquisition of iron (Table 3). One such mechanism is the production of low-molecular-weight chelating agents called siderophores. For example, the SIG genomes contain the genes sfaABCD and htsABC involved in the biosynthesis and transport of staphyloferrin A and transport of heme (Hammer and Skaar, 2011). In contrast to other non-S. aureus species sequenced, the SIG species can also produce and transport staphylobactin A mediated by the operons sbnA-I and sirABC. Uptake of ferrous iron is also enabled through the presence of the FeoAB system, which is not shared with S. saprophyticus ATCC15305 or S. haemolyticus JCSC1435. Further differences with other staphylococcal species include the absence of the siderophore–Fe transport system SstABC, the lipoprotein receptor for Fe3+ (FhuD) and the hemoglobin–Fe transport system IsdA-H, found in S. aureus. In addition, variation among the three SIG species were observed with the absence of the ferrous iron transporter EfeUOB in S. pseudintermedius ED99 and the presence of an inactivating mutation in the iron–manganese mntABC operon in S. delphini 8086. Overall, in common with other staphylococci, the SIG species have an extensive array of systems for acquiring iron from the host, which may be important in the context of the skin environment where iron sources are particularly limited.
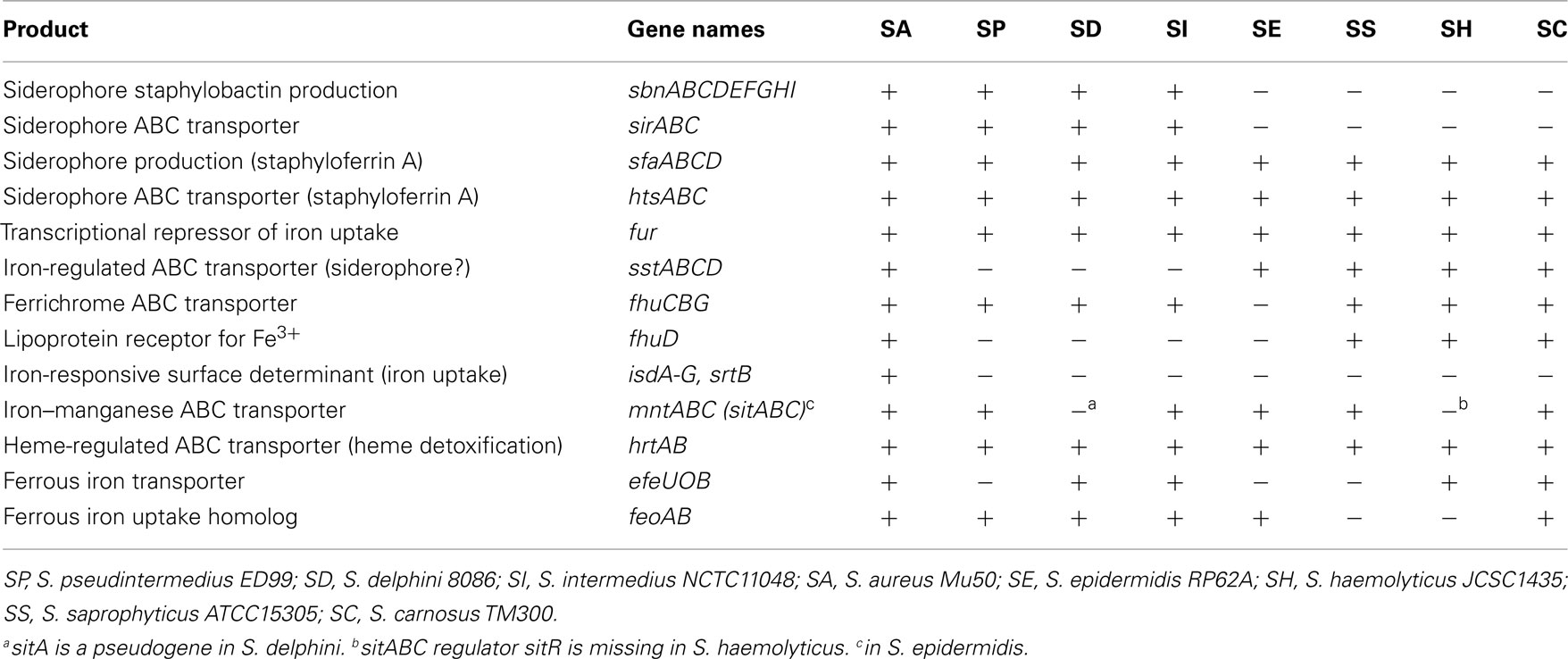
Table 3. Distribution of iron transport related genes identified in eight staphylococcal species.
In common with S. aureus and S. haemolyticus, multi-resistance in S. pseudintermedius strains is frequent and includes resistance to tetracycline (Schwarz et al., 1998), macrolides, lincosamides and streptogramins (Eady et al., 1993; Boerlin et al., 2001), aminoglycosides and aminocyclitols (Noble et al., 1996; Boerlin et al., 2001), fluoroquinolones (Intorre et al., 2007), and methicillin (Piriz et al., 1995; Kania et al., 2004; Sasaki et al., 2007a). We determined that S. pseudintermedius ED99 is resistant to ampicillin, erythromycin, tetracycline, and trimethoprim whereas S. delphini 8086 and S. intermedius NCTC11048 are sensitive to all antibiotics cited above (data not shown). Four transposons containing one or more antibiotic resistance genes were identified in the genome of S. pseudintermedius ED99 (Table 4). Of these, 2 (Tn552 and Tn554-like) found also in S. haemolyticus JCSC1435, and in S. epidermidis and S. aureus strains respectively, encode the bla operon, which confers β-lactam resistance. Tn5405 encodes the aminoglycoside–streptothricin resistance genes aad6–sat4–aphA-3 and is associated with the macrolide–lincosamide–streptogramin resistance gene ermB previously described in Enterococcus faecium (Werner et al., 2003) and S. intermedius (Boerlin et al., 2001), and the putative conjugative transposon Tn5801 (25.8 kb) encodes the tetM gene responsible for resistance to tetracycline. Tn5801 was previously identified in S. aureus Mu50 and is related to the conjugative transposon Tn916 of Enterococcus faecalis (Flannagan et al., 1994). The existence of transposons which are nearly identical to those found in human-associated staphylococcal species indicates a recent inter-species horizontal transfer of antibiotic resistance. In contrast to the closely related species S. delphini and S. intermedius, which have limited clinical importance, S. pseudintermedius encounters considerable antibiotic selective pressures, which have contributed to the spread of MGE encoding antibiotic resistance. Since the 1990s, methicillin-resistant S. pseudintermedius strains have emerged through several independent SCCmec acquisition events and disseminated widely (Piriz et al., 1995; Kania et al., 2004; Bannhoer et al., 2007; Sasaki et al., 2007a). Importantly, S. pseudintermedius strains containing MGE encoding antibiotic resistance could represent a reservoir for the spread of resistance genes to human commensal skin flora (Guardabassi et al., 2004a,b).

Table 4. Mobile genetic elements associated with antibiotic resistance identified in S. pseudintermediu s ED99.
The spread of antibiotic-resistant strains of S. pseudintermedius and the lack of an effective vaccine means that alternative approaches for controlling canine pyoderma are required. The identification of novel virulence determinants in the genome of S. pseudintermedius has provided candidate new targets for therapeutic interventions. In particular, the identification of CWA-associated proteins and toxins which contribute to the pathology associated with pyoderma infection should provide the impetus for detailed characterization of these critical host–pathogen interactions and for investigations into their potential as vaccine components. Furthermore, the discovery that transposons are largely responsible for the multi-resistant phenotype of S. pseudintermedius provides important insights into the evolution of antibiotic resistance within the species. Future genome sequencing projects should include representatives of the widespread methicillin-resistant S. pseudintermedius clones (Bannoehr et al., 2007). Furthermore, comparative genomic analysis also revealed the presence of CRISPR loci, and an unusually high %GC content of the SIG species, which provides intriguing avenues for basic research into bacterial genome evolution. Finally, several additional species-specific features which likely reflect the distinct ecological niches occupied, were identified such as genes involved in carbohydrate metabolism iron acquisition and resistance to oxidative stress. The identification of the genetic events which led to differentiation of the SIG species and the determinants which correlate with their distinct host-tropisms provide avenues for fundamental studies into the molecular basis of bacterial host-adaptation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was funded by The Petplan Charitable Trust, Pfizer Animal Health, and the Australian National Health and Medical Research Council (project 511224). Scott A. Beatson is the recipient of an Australian Research Council Fellowship (project DP0881347). We are grateful to J. Bannoehr for helpful discussions.
The Supplementary Material for this article can be found online at http://www.frontiersin.org/Cellular_and_Infection_Microbiology/10.3389/fcimb.2012.00044/abstract
Table S1. Genes specific to S. pseudintermedius ED99.
Table S2. Core genes identified in S. pseudintermedius ED99.
Affolter, V. K., and Moore, P. F. (1994). Histologic features of normal canine and feline skin. Clin. Dermatol. 12, 491–497.
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., Formsma, K., Gerdes, S., Glass, E. M., Kubal, M., Meyer, F., Olsen, G. J., Olson, R., Osterman, A. L., Overbeek, R. A., McNeil, L. K., Paarmann, D., Paczian, T., Parrello, B., Pusch, G. D., Reich, C., Stevens, R., Vassieva, O., Vonstein, V., Wilke, A., and Zagnitko, O. (2008). The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9, 75. doi:10.1186/1471-2164-9-75
Baba, T., Kuwahara-Arai, K., Uchiyama, I., Takeuchi, F., Ito, T., and Hiramatsu, K. (2009). Complete genome sequence of Macrococcus caseolyticus strain JCSCS5402, [corrected] reflecting the ancestral genome of the human-pathogenic Staphylococci. J. Bacteriol. 191, 1180–1190.
Bannhoer, J., Ben Zakour, N. L., Waller, A. S., Guardabassi, L., Thoday, K. M., van den Broek, A. H. M., and Fitzgerald, J. R. (2007). Population genetic structure of the Staphylococcus intermedius group: insights into agr diversification and the emergence of methicillin-resistant strains. J. Bacteriol. 189, 8685–8692.
Bannoehr, J., Ben Zakour, N. L., Reglinski, M., Inglis, N. F., Prabhakaran, S., Fossum, E., Smith, D. G., Wilson, G. J., Cartwright, R. A., Haas, J., Hook, M., van den Broek, A. H., Thoday, K. L., and Fitzgerald, J. R. (2011a). Genomic and surface proteomic analysis of the canine pathogen Staphylococcus pseudintermedius reveals proteins that mediate adherence to the extracellular matrix. Infect. Immun. 79, 3074–3086.
Bannoehr, J., Brown, J. K., Shaw, D. J., Fitzgerald, R. J., van den Broek, A. H., and Thoday, K. L. (2011b). Staphylococcus pseudintermedius surface proteins SpsD and SpsO mediate adherence to ex vivo canine corneocytes. Vet. Dermatol. 23, 119–e26.
Bannoehr, J., Ben Zakour, N. L., Waller, A. S., Guardabassi, L., Thoday, K. L., van den Broek, A. H., and Fitzgerald, J. R. (2007). Population genetic structure of the Staphylococcus intermedius group: insights into agr diversification and the emergence of methicillin-resistant strains. J. Bacteriol. 189, 8685–8692.
Barrangou, R., Fremaux, C., Deveau, H., Richards, M., Boyaval, P., Moineau, S., Romero, D. A., and Horvath, P. (2007). CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712.
Becker, K., Keller, B., von Eiff, C., Brück, M., Lubritz, G., Etienne, J., and Peters, G. (2001). Enterotoxigenic potential of Staphylococcus intermedius. Appl. Environ. Microbiol. 67, 5551–5557.
Ben Zakour, N. L., Bannoehr, J., van den Broek, A. H., Thoday, K. L., and Fitzgerald, J. R. (2011). Complete genome sequence of the canine pathogen Staphylococcus pseudintermedius. J. Bacteriol. 193, 2363–2364.
Blaiotta, G., Fusco, V., Ercolini, D., Pepe, O., and Coppola, S. (2010). Diversity of Staphylococcus species strains based on partial kat (catalase) gene sequences and design of a PCR-restriction fragment length polymorphism assay for identification and differentiation of coagulase-positive species (S. aureus, S. delphini, S. hyicus, S. intermedius, S. pseudintermedius, and S. schleiferi subsp. coagulans). J. Clin. Microbiol. 48, 192–201.
Boelsma, E., van de Vijver, L. P., Goldbohm, R. A., Klöpping-Ketelaars, I. A., Hendriks, H. F., and Roza, L. (2003). Human skin condition and its associations with nutrient concentrations in serum and diet. Am. J. Clin. Nutr. 77, 348–355.
Boerlin, P., Burnens, A. P., Frey, J., Kuhnert, P., and Nicolet, J. (2001). Molecular epidemiology and genetic linkage of macrolide and aminoglycoside resistance in Staphylococcus intermedius of canine origin. Vet. Microbiol. 79, 155–169.
Cheng, A. G., McAdow, M., Kim, H. K., Bae, T., Missiakas, D. M., and Schneewind, O. (2010). Contribution of coagulases towards Staphylococcus aureus disease and protective immunity. PLoS Pathog. 6, e1001036. doi:10.1371/journal.ppat.1001036
Chua, K. Y., Seemann, T., Harrison, P. F., Monagle, S., Korman, T. M., Johnson, P. D., Coombs, G. W., Howden, B. O., Davies, J. K., Howden, B. P., and Stinear, T. P. (2011). The dominant Australian community-acquired methicillin-resistant Staphylococcus aureus clone ST93-IV [2B] is highly virulent and genetically distinct. PLoS ONE 6, e25887. doi:10.1371/journal.pone.0025887
Cole, L. K., Kwochka, K. W., Kowalski, J. J., and Hillier, A. (1998). Microbial flora and antimicrobial susceptibility patterns of isolated pathogens from the horizontal ear canal and middle ear in dogs with otitis media. J. Am. Vet. Med. Assoc. 212, 534–538.
Dziewanowska, K., Edwards, V. M., Deringer, J. R., Bohach, G. A., and Guerra, D. J. (1996). Comparison of the beta-toxins from Staphylococcus aureus and Staphylococcus intermedius. Arch. Biochem. Biophys. 335, 102–108.
Eady, E. A., Ross, J. I., Tipper, J. L., Walters, C. E., Cove, J. H., and Noble, W. C. (1993). Distribution of genes encoding erythromycin ribosomal methylases and an erythromycin efflux pump in epidemiologically distinct groups of Staphylococci. J. Antimicrob. Chemother. 31, 211–217.
Fitzgerald, J. R. (2009). The Staphylococcus intermedius group of bacterial pathogens: species re-classification, pathogenesis and the emergence of methicillin resistance. Vet. Dermatol. 20, 490–495.
Flannagan, S. E., Zitzow, L. A., Su, Y. A., and Clewell, D. B. (1994). Nucleotide sequence of the 18-kb conjugative transposon Tn916 from Enterococcus faecalis. Plasmid 32, 350–354.
Futagawa-Saito, K., Makino, S., Sunaga, F., Kato, Y., Sakurai-Komada, N., Ba-Thein, W., and Fukuyasu, T. (2009). Identification of first exfoliative toxin in Staphylococcus pseudintermedius. FEMS Microbiol. Lett. 301, 176–180.
Futagawa-Saito, K., Suzuki, M., Ohsawa, M., Ohshima, S., Sakurai, N., Ba-Thein, W., and Fukuyasu, T. (2004). Identification and prevalence of an enterotoxin-related gene, se-int, in Staphylococcus intermedius isolates from dogs and pigeons. J. Appl. Microbiol. 96, 1361–1366.
Gill, S. R., Fouts, D. E., Archer, G. L., Mongodin, E. F., Deboy, R. T., Ravel, J., Paulsen, I. T., Kolonay, J. F., Brinkac, L., Beanan, M., Dodson, R. J., Daugherty, S. C., Madupu, R., Angiuoli, S. V., Durkin, A. S., Haft, D. H., Vamathevan, J., Khouri, H., Utterback, T., Lee, C., Dimitrov, G., Jiang, L., Qin, H., Weidman, J., Tran, K., Kang, K., Hance, I. R., Nelson, K. E., and Fraser, C. M. (2005). Insights on evolution of virulence and resistance from the complete genome analysis of an early methicillin-resistant Staphylococcus aureus strain and a biofilm-producing methicillin-resistant Staphylococcus epidermidis strain. J. Bacteriol. 187, 2426–2438.
Golding, G. R., Bryden, L., Levett, P. N., McDonald, R. R., Wong, A., Wylie, J., Graham, M. R., Tyler, S., Van Domselaar, G., Simor, A. E., Gravel, D., and Mulvey, M. R. (2010). Livestock-associated methicillin-resistant Staphylococcus aureus sequence type 398 in humans, Canada. Emerging Infect. Dis. 16, 587–594.
Goris, J., Konstantinidis, K. T., Klappenbach, J. A., Coenye, T., Vandamme, P., and Tiedje, J. M. (2007). DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 57(Pt 1), 81–91.
Granlund, M., Michel, F., and Norgren, M. (2001). Mutually exclusive distribution of IS1548 and GBSi1, an active group II intron identified in human isolates of group B streptococci. J. Bacteriol. 183, 2560–2569.
Greene, R. T., and Lammler, C. (1993). Staphylococcus intermedius: current knowledge on a pathogen of veterinary importance. Zentralblatt Veterinarmedizin Reihe B 40, 206–214.
Grissa, I., Vergnaud, G., and Pourcel, C. (2007a). CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 35, W52–W57.
Grissa, I., Vergnaud, G., and Pourcel, C. (2007b). The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinformatics 8, 172. doi:10.1186/1471-2105-8–172
Guardabassi, L., Loeber, M. E., and Jacobson, A. (2004a). Transmission of multiple antimicrobial-resistant Staphylococcus intermedius between dogs affected by deep pyoderma and their owners. Vet. Microbiol. 98, 23–27.
Guardabassi, L., Schwarz, S., and Lloyd, D. H. (2004b). Pet animals as reservoirs of antimicrobial-resistant bacteria. J. Antimicrob. Chemother. 54, 321–332.
Haft, D. H., Selengut, J., Mongodin, E. F., and Nelson, K. E. (2005). A guild of 45 CRISPR-associated (Cas) protein families and multiple CRISPR/Cas subtypes exist in prokaryotic genomes. PLoS Comput. Biol. 1, e60. doi:10.1371/journal.pcbi.0010060
Hajek, V. (1976). Staphylococcus intermedius, a new species isolated from animals. Int. J. Syst. Evol. Microbiol. 26, 401–408.
Hammer, N. D., and Skaar, E. P. (2011). Molecular mechanisms of Staphylococcus aureus iron acquisition. Annu. Rev. Microbiol. 65, 129–147.
Hendricks, A., Schuberth, H. J., Schueler, K., and Lloyd, D. H. (2002). Frequency of superantigen-producing Staphylococcus intermedius isolates from canine pyoderma and proliferation-inducing potential of superantigens in dogs. Res. Vet. Sci. 73, 273–277.
Huson, D. H., and Bryant, D. (2006). Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267.
Intorre, L., Vanni, M., Di Bello, D., Pretti, C., Meucci, V., Tognetti, R., Soldani, G., Cardini, G., and Jousson, O. (2007). Antimicrobial susceptibility and mechanism of resistance to fluoroquinolones in Staphylococcus intermedius and Staphylococcus schleiferi. J. Vet. Pharmacol. Ther. 30, 464–469.
Iyori, K., Hisatsune, J., Kawakami, T., Shibata, S., Murayama, N., Ide, K., Nagata, M., Fukata, T., Iwasaki, T., Oshima, K., Hattori, M., Sugai, M., and Nishifuji, K. (2010). Identification of a novel Staphylococcus pseudintermedius exfoliative toxin gene and its prevalence in isolates from canines with pyoderma and healthy dogs. FEMS Microbiol. Lett. 312, 169–175.
Kania, S. A., Williamson, N. L., Frank, L. A., Wilkes, R. P., Jones, R. D., and Bemis, D. A. (2004). Methicillin resistance of Staphylococci isolated from the skin of dogs with pyoderma. Am. J. Vet. Res. 65, 1265–1268.
Khambaty, F. M., Bennett, R. W., and Shah, D. B. (1994). Application of pulsed-field gel electrophoresis to the epidemiological characterization of Staphylococcus intermedius implicated in a food-related outbreak. Epidemiol. Infect. 113, 75–81.
Kloos, W. E. (1980). Natural populations of the genus Staphylococcus. Annu. Rev. Microbiol. 34, 559–592.
Kobayashi, I. (2001). Behavior of restriction-modification systems as selfish mobile elements and their impact on genome evolution. Nucleic Acids Res. 29, 3742–3756.
Kuroda, M., Ohta, T., Uchiyama, I., Baba, T., Yuzawa, H., Kobayashi, I., Cui, L., Oguchi, A., Aoki, K., Nagai, Y., Lian, J., Ito, T., Kanamori, M., Matsumaru, H., Maruyama, A., Murakami, H., Hosoyama, A., Mizutani-Ui, Y., Takahashi, N. K., Sawano, T., Inoue, R., Kaito, C., Sekimizu, K., Hirakawa, H., Kuhara, S., Goto, S., Yabuzaki, J., Kanehisa, M., Yamashita, A., Oshima, K., Furuya, K., Yoshino, C., Shiba, T., Hattori, M., Ogasawara, N., Hayashi, H., and Hiramatsu, K. (2001). Whole genome sequencing of methicillin-resistant Staphylococcus aureus. Lancet 357, 1225–1240.
Kuroda, M., Yamashita, A., Hirakawa, H., Kumano, M., Morikawa, K., Higashide, M., Maruyama, A., Inose, Y., Matoba, K., Toh, H., Kuhara, S., Hattori, M., and Ohta, T. (2005). Whole genome sequence of Staphylococcus saprophyticus reveals the pathogenesis of uncomplicated urinary tract infection. Proc. Natl. Acad. Sci. U.S.A. 102, 13272–13277.
Mahoudeau, I., Delabranche, X., Prevost, G., Monteil, H., and Piemont, Y. (1997). Frequency of isolation of Staphylococcus intermedius from humans. J. Clin. Microbiol. 35, 2153–2154.
Marraffini, L. A., and Sontheimer, E. J. (2008). CRISPR interference limits horizontal gene transfer in Staphylococci by targeting DNA. Science 322, 1843–1845.
Matousek, J. L., and Campbell, K. L. (2002). A comparative review of cutaneous pH. Vet. Dermatol. 13, 293–300.
Matsuura, M., Saldanha, R., Ma, H., Wank, H., Yang, J., Mohr, G., Cavanagh, S., Dunny, G. M., Belfort, M., and Lambowitz, A. M. (1997). A bacterial group II intron encoding reverse transcriptase, maturase, and DNA endonuclease activities: biochemical demonstration of maturase activity and insertion of new genetic information within the intron. Genes Dev. 11, 2910–2924.
Noble, W. C., Rahman, M., Karadec, T., and Schwarz, S. (1996). Gentamicin resistance gene transfer from Enterococcus faecalis and E. faecium to Staphylococcus aureus, S. intermedius and S. hyicus. Vet. Microbiol. 52, 143–152.
Novick, R. P. (2003). Autoinduction and signal transduction in the regulation of staphylococcal virulence. Mol. Microbiol. 48, 1429–1449.
Novick, R. P., Christie, G. E., and Penadés, J. R. (2010). The phage-related chromosomal islands of Gram-positive bacteria. Nat. Rev. Microbiol. 8, 541–551.
Perreten, V., Kadlec, K., Schwarz, S., Grönlund Andersson, U., Finn, M., Greko, C., Moodley, A., Kania, S. A., Frank, L. A., Bemis, D. A., Franco, A., Iurescia, M., Battisti, A., Duim, B., Wagenaar, J. A., van Duijkeren, E., Weese, J. S., Fitzgerald, J. R., Rossano, A., and Guardabassi, L. (2010). Clonal spread of methicillin-resistant Staphylococcus pseudintermedius in Europe and North America: an international multicentre study. J. Antimicrob. Chemother. 65, 1145–1154.
Piriz, S., de la Fuente, R., Valle, J., Mateos, E., Hurtado, M. A., Cid, D., Ruiz-Santaquiteria, J. A., and Vadillo, S. (1995). Comparative in vitro activity of 11 beta-lactam antibiotics against 91 Staphylococcus intermedius strains isolated from staphylococcal dermatitis in dogs. Zentralblatt Veterinarmedizin Reihe B 42, 293–300.
Pishchany, G., McCoy, A. L., Torres, V. J., Krause, J. C., Crowe, J. E. Jr., Fabry, M. E., and Skaar, E. P. (2010). Specificity for human hemoglobin enhances Staphylococcus aureus infection. Cell Host Microbe 8, 544–550.
Pottumarthy, S., Schapiro, J. M., Prentice, J. L., Houze, Y. B., Swanzy, S. R., Fang, F. C., and Cookson, B. T. (2004). Clinical isolates of Staphylococcus intermedius masquerading as methicillin-resistant Staphylococcus aureus. J. Clin. Microbiol. 42, 5881–5884.
Prevost, G., Bouakham, T., Piemont, Y., and Monteil, H. (1995). Characterisation of a synergohymenotropic toxin produced by Staphylococcus intermedius. FEBS Lett. 376, 135–140.
Richter, M., and Rossello-Mora, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U.S.A. 106, 19126–19131.
Rissman, A. I., Mau, B., Biehl, B. S., Darling, A. E., Glasner, J. D., and Perna, N. T. (2009). Reordering contigs of draft genomes using the Mauve aligner. Bioinformatics 25, 2071–2073.
Rosenstein, R., Nerz, C., Biswas, L., Resch, A., Raddatz, G., Schuster, S. C., and Götz, F. (2009). Genome analysis of the meat starter culture bacterium Staphylococcus carnosus TM300. Appl. Environ. Microbiol. 75, 811–822.
Ruscher, C., Lübke-Becker, A., Semmler, T., Wleklinski, C. G., Paasch, A., Soba, A., Stamm, I., Kopp, P., Wieler, L. H., and Walther, B. (2010). Widespread rapid emergence of a distinct methicillin- and multidrug-resistant Staphylococcus pseudintermedius (MRSP) genetic lineage in Europe. Vet. Microbiol. 144, 340–346.
Sakarya, S., Ertugrul, M. B., Oztürk, T., and Gökbulut, C. (2010). Effect of pharynx epithelial cells surface desialylation on receptor-mediated adherence of Staphylococcus aureus. J. Appl. Microbiol. 108, 1313–1322.
Sasaki, T., Kikuchi, K., Tanaka, Y., Takahashi, N., Kamata, S., and Hiramatsu, K. (2007a). Methicillin-resistant Staphylococcus pseudintermedius in a veterinary teaching hospital. J. Clin. Microbiol. 45, 1118–1125.
Sasaki, T., Kikuchi, K., Tanaka, Y., Takahashi, N., Kamata, S., and Hiramatsu, K. (2007b). Reclassification of phenotypically identified Staphylococcus intermedius strains. J. Clin. Microbiol. 45, 2770–2778.
Schwarz, S., Roberts, M. C., Werckenthin, C., Pang, Y., and Lange, C. (1998). Tetracycline resistance in Staphylococcus spp. from domestic animals. Vet. Microbiol. 63, 217–227.
Shimada, K., Yoon, J. S., Yoshihara, T., Iwasaki, T., and Nishifuji, K. (2009). Increased transepidermal water loss and decreased ceramide content in lesional and non-lesional skin of dogs with atopic dermatitis. Vet. Dermatol. 20, 541–546.
Siguier, P., Perochon, J., Lestrade, L., Mahillon, J., and Chandler, M. (2006). ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 34, D32–D36.
Sullivan, M. J., Petty, N. K., and Beatson, S. A. (2011). Easyfig: a genome comparison visualizer. Bioinformatics 27, 1009–1010.
Takeuchi, F., Watanabe, S., Baba, T., Yuzawa, H., Ito, T., Morimoto, Y., Kuroda, M., Cui, L., Takahashi, M., Ankai, A., Baba, S., Fukui, S., Lee, J. C., and Hiramatsu, K. (2005). Whole-genome sequencing of Staphylococcus haemolyticus uncovers the extreme plasticity of its genome and the evolution of human-colonizing staphylococcal species. J. Bacteriol. 187, 7292–7308.
Tanner, M. A., Everett, C. L., and Youvan, D. C. (2000). Molecular phylogenetic evidence for noninvasive zoonotic transmission of Staphylococcus intermedius from a canine pet to a human. J. Clin. Microbiol. 38, 1628–1631.
Terauchi, R., Sato, H., Hasegawa, T., Yamaguchi, T., Aizawa, C., and Maehara, N. (2003). Isolation of exfoliative toxin from Staphylococcus intermedius and its local toxicity in dogs. Vet. Microbiol. 94, 19–29.
Tourasse, N. J., and Kolsto, A. B. (2008). Survey of group I and group II introns in 29 sequenced genomes of the Bacillus cereus group: insights into their spread and evolution. Nucleic Acids Res. 36, 4529–4548.
Tse, H., Tsoi, H. W., Leung, S. P., Lau, S. K., Woo, P. C., and Yuen, K. Y. (2010). Complete genome sequence of Staphylococcus lugdunensis strain HKU09-01. J. Bacteriol. 192, 1471–1472.
Tse, H., Tsoi, H. W., Leung, S. P., Urquhart, I. J., Lau, S. K., Woo, P. C., and Yuen, K. Y. (2011). Complete genome sequence of the veterinary pathogen Staphylococcus pseudintermedius strain HKU10-03, isolated in a case of canine pyoderma. J. Bacteriol. 193, 1783–1784.
Ubeda, C., Maiques, E., Barry, P., Matthews, A., Tormo, M. A., Lasa, I., Novick, R. P., and Penadés, J. R. (2008). SaPI mutations affecting replication and transfer and enabling autonomous replication in the absence of helper phage. Mol. Microbiol. 67, 493–503.
Varaldo, P. E., Kilpper-Balz, R., Biavasco, F., Satta, G., and Schleifer, K. H. (1988). Staphylococcus delphini sp-nov, a coagulase-positive species isolated from dolphins. Int. J. Syst. Bacteriol. 38, 436–439.
Vernikos, G. S., and Parkhill, J. (2006). Interpolated variable order motifs for identification of horizontally acquired DNA: revisiting the Salmonella pathogenicity islands. Bioinformatics 22, 2196–2203.
Werner, G., Hildebrandt, B., and Witte, W. (2003). Linkage of erm(B) and aadE-sat4-aphA-3 in multiple-resistant Enterococcus faecium isolates of different ecological origins. Microb. Drug Resist. 9(Suppl. 1), S9–S16.
Wladyka, B., Bista, M., Sabat, A. J., Bonar, E., Grzeszczuk, S., Hryniewicz, W., and Dubin, A. (2008). A novel member of the thermolysin family, cloning and biochemical characterization of metalloprotease from Staphylococcus pseudintermedius. Acta Biochim. Pol. 55, 525–536.
Yamaguchi, T., Nishifuji, K., Sasaki, M., Fudaba, Y., Aepfelbacher, M., Takata, T., Ohara, M., Komatsuzawa, H., Amagai, M., and Sugai, M. (2002). Identification of the Staphylococcus aureus etd pathogenicity island which encodes a novel exfoliative toxin, ETD, and EDIN-B. Infect. Immun. 70, 5835–5845.
Yew, W. S., and Gerlt, J. A. (2002). Utilization of L-ascorbate by Escherichia coli K-12: assignments of functions to products of the yjf-sga and yia-sgb operons. J. Bacteriol. 184, 302–306.
Zerbino, D. R., and Birney, E. (2008). Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829.
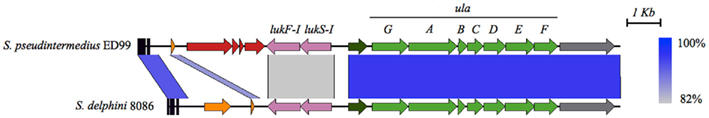
Figure A1. Comparison of the genomic island containing the bi-component leukotoxin Luk-I and the L-ascorbate transport and utilization operon in S. pseudintermedius ED99 and S. delphini 8086. The similarity between regions is indicated by a spectrum of blue to gray, ranging from 100 to 82% similarity. Genes are colored according to their sequence and function as follow: phage-related genes in red; leukotoxin genes in pink; putative phosphoglucomutase in dark green; L-ascorbate transport and utilization genes in green; putative transcriptional regulator in gray; tRNA genes are represented by black boxes; and genes encoding hypothetical proteins in orange.
Keywords: Staphylococcus, genomics, pathogenesis, host-adaptation, animal, antibiotic resistance
Citation: Ben Zakour NL, Beatson SA, van den Broek AHM, Thoday KL and Fitzgerald JR (2012) Comparative genomics of the Staphylococcus intermedius group of animal pathogens. Front. Cell. Inf. Microbio. 2:44. doi: 10.3389/fcimb.2012.00044
Received: 15 December 2011; Paper pending published: 19 January 2012;
Accepted: 15 March 2012; Published online: 18 April 2012.
Edited by:
Martin John McGavin, University of Western Ontario, CanadaReviewed by:
Steven Gill, University of Rochester School of Medicine and Dentistry, USACopyright: © 2012 Ben Zakour, Beatson, van den Broek, Thoday and Fitzgerald. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: J. Ross Fitzgerald, The Roslin Institute and Edinburgh Infectious Diseases, Royal (Dick) School of Veterinary Studies, University of Edinburgh, Easter Bush Campus, Midlothian, Edinburgh EH25 9RG, UK. e-mail:cm9zcy5maXR6Z2VyYWxkQGVkLmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.