


95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Infect. Microbiol. , 11 April 2012
Sec. Molecular Bacterial Pathogenesis
Volume 2 - 2012 | https://doi.org/10.3389/fcimb.2012.00029
This article is part of the Research Topic Research Advances in the Study of Campylobacter, Helicobacter & Related Organisms View all 29 articles
The bacterial pathogen Helicobacter pylori is capable of colonizing the gastric mucosa of the human stomach using a variety of factors associated with or secreted from its outer membrane (OM). Lipopolysaccharide (LPS) and numerous OM proteins have been shown to be involved in adhesion and immune stimulation/evasion. Many of these factors are essential for colonization and/or pathogenesis in a variety of animal models. Despite this wide array of potential targets present on the bacterial surface, the ability of H. pylori to vary its OM profile limits the effectiveness of vaccines or therapeutics that target any single one of these components. However, it has become evident that the proteins comprising the complexes that transport the majority of these molecules to the OM are highly conserved and often essential. The field of membrane biogenesis has progressed remarkably in the last few years, and the possibility now exists for targeting the mechanisms by which β-barrel proteins, lipoproteins, and LPS are transported to the OM, resulting in loss of bacterial fitness and significant altering of membrane permeability. In this review, the OM transport machinery for LPS, lipoproteins, and outer membrane proteins (OMPs) are discussed. While the principal investigations of these transport mechanisms have been conducted in Escherichia coli and Neisseria meningitidis, here these systems will be presented in the genetic context of ε proteobacteria. Bioinformatic analysis reveals that minimalist genomes, such as that of Helicobacter pylori, offer insight into the smallest number of components required for these essential pathways to function. Interestingly, in the majority of ε proteobacteria, while the inner and OM associated apparatus of LPS, lipoprotein, and OMP transport pathways appear to all be intact, most of the components associated with the periplasmic compartment are either missing or are almost unrecognizable when compared to their E. coli counterparts. Eventual targeting of these pathways would have the net effect of severely limiting the delivery/transport of components to the OM and preventing the bacterium's ability to infect its human host.
Membrane permeability has long been understood as a critical factor of bacterial survival (Hall et al., 1980; Ruiz et al., 2005). A growing field of knowledge regarding the trafficking and insertion of bacterial membrane components has succeeded in characterizing the key factors involved in membrane biogenesis in a variety of model systems. The eventual targeting of these essential barriers by molecular inhibitors or using them as vaccine candidates has the potential for inducing either bactericidal effects outright, or increased susceptibility to more established antibiotics (Chiu et al., 2007, 2009; Ghanei et al., 2007). Enzymes/proteins found in the cytoplasm, periplasmic space, and outside the bacterial membrane have all evolved under the confines and conditions of each of these unique microenvironments. While localization of these proteins to their ultimate destination within the cell is often essential, the controlled interaction and movement of signaling molecules/nutrients/metabolites between these regions and spaces is also critical for basic microbial metabolism. More recently, studies characterizing bacterial mutants with membrane permeability defects have begun to establish that nearly all permeability deficiencies arise from mutations in the protein machinery that make up the transport apparatus for the basic components of bacterial membranes: lipoproteins, lipopolysaccharide, and outer membrane proteins (OMPs). These fundamental transport pathways are for the most part well-conserved throughout Gram-negative bacteria (Nikaido, 2003; Ruiz et al., 2006). One notable exception to this exists in the bacterial class of ε proteobacteria. Consisting of genera spanning digestive tract colonizing symbionts/pathogens (Marshall and Warren, 1984) to deep-sea thermal vent chemolithotrophs (Takai et al., 2005), this bacterial class contains numerous extremophiles for whom membrane barrier function should arguably play an indispensible role. However, upon closer bioinformatic examination, the transport pathways for each of the three major outer membrane (OM) components in ε proteobacteria appear to have significantly diverged when compared with most other Gram-negative bacteria (Cummings et al., 2002).
One member of this class of ε proteobacteria with significant medical relevance is the gastric pathogen Helicobacter pylori. Able to survive and colonize the mucosal layer in close proximity to the human gastric epithelium (Blaser, 1992), H. pylori is capable of inhabiting an environment with an acidic pH. While adaptations to this gastric environment include motility/chemotaxis (Foynes et al., 2000; Ottemann and Lowenthal, 2002; Croxen et al., 2006) and the production of urease, which can substantially buffer the pH around the bacterium (Scott et al., 2000; Weeks et al., 2000), maintaining a level of barrier function from the OM remains an essential element in allowing this chronic colonizer to survive in this extreme environment. In addition, each of the previously mentioned transport pathways play key roles in permitting H. pylori to chronically colonize its human host.
This review highlights what is known of the three pathways involved in membrane biogenesis, specifically the transport of three major components/transport systems of the OM in the majority of Gram-negative bacteria: lipoprotein, LPS, and OMPs. The importance of each of these three membrane components in H. pylori will be discussed in terms of relevance to infection. Each transport pathway will be examined bioinformatically and the implications for potential targets for future small molecule inhibitors and candidates for vaccine development analyzed. Special attention will be given to the identification of the periplasmic components for these transport pathways, as it appears that many are significantly divergent from those found in other model bacteria or perhaps are even absent in H. pylori entirely (Figure 1). Given the continued prominence of this bacterium in the developing world (Frenck and Clemens, 2003), the current state of vaccine development for this pathogen (Czinn and Blanchard, 2011), and an unsettling rise in the number of reports of antibiotic resistant strains (Boyanova and Mitov, 2010; De Francesco et al., 2010), identifying novel targets for future antimicrobials is of paramount importance.
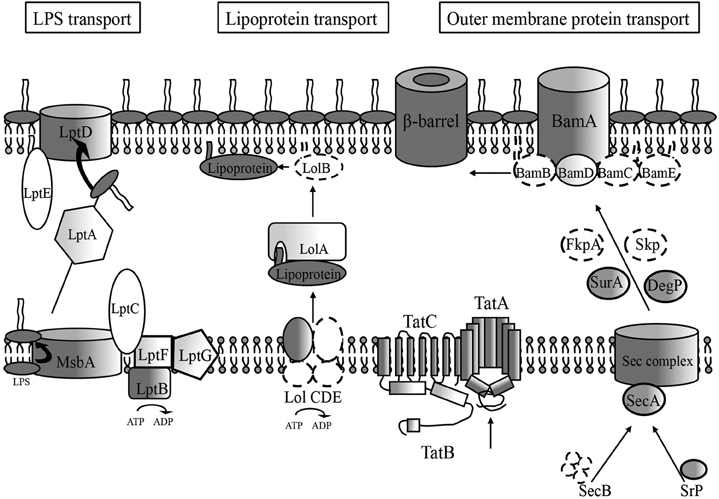
Figure 1. Membrane biogenesis pathway conserved features in H. pylori. Dark features indicate high similarity scores (≤e4) for H. pylori genes compared to E. coli outer membrane biogenesis pathway components, which generally consist of the IM and OM spanning portions of the pathway. Lighter shaded components indicate lower similarity scores requiring comparison of numerous bacterial homologs before attaining a significant similarity score (≤e4) for an H. pylori locus. An absence of shading indicates no homolog for that component was found through sequence similarity scores, but the existence of a potentially functional protein exists, given the presence of a conserved genetic orientation at the particular locus and/or structural similarity as determined by crystal structure analysis software (RaptorX). Dotted lines indicate a complete genetic absence of homology of a pathway component. Those proteins with low sequence similarity, low structural similarity, and apparent absence of sequence and structural similarity make up most of the components found in the periplasmic regions for each of the three OM transport pathways.
A large number of putative lipoproteins have been found in the H. pylori genome (Tomb et al., 1997). Their abundance and potential for facilitating extensive linkages between inner and OMs has been hypothesized to explain the difficulty reported in experimentally separating inner and OM layers in most Helicobacter and Campylobacter species (O'Toole and Clyne, 2001). Few lipoproteins have been well-characterized in H. pylori, but those that have been studied play important roles in infection and the host immune response. One such lipoprotein is the highly conserved H. pylori adhesin A (HpaA), which was originally characterized as a neuraminyllactose-binding hemagglutinin (Evans et al., 1988, 1993), and was shown later to be a lipoprotein (O'Toole et al., 1995). HpaA has since been studied in depth, due to its conserved nature in most strains and its role in bacterial adhesion. Localization of HpaA has been highly disputed in the field, with some studies pointing to flagellar sheath localization (Luke and Penn, 1995), others showing cytoplasmic localization (O'Toole et al., 1995), and others simply showing membrane association (Bolin et al., 1995; Lundstrom et al., 2001). Due to its putative adhesin classification and putative membrane association, HpaA has been examined for use as a potential vaccine candidate (Flach et al., 2011). Similarly, another membrane-associated lipoprotein in H. pylori is Lpp20, first characterized as an 18-kilodalton OM antigen (Keenan et al., 2000), has been shown to be immunodominant in several studies (Keenan et al., 2002; Bakos et al., 2003). While these two lipoproteins are currently the only well-examined examples in H. pylori to date, the presence of several other recognized lipoproteins involved in the transport pathways of both LPS and OMPs (which will be discussed subsequently) adds additional importance to this class of membrane components.
The mechanism for lipoprotein trafficking has been well-characterized in E. coli (Matsuyama et al., 1995, 1997; Yakushi et al., 2000). In this system, lipoproteins are transported to the inner membrane via the Sec pathway, embedded into the membrane, and are subsequently sorted according to the presence or absence of an inner membrane (IM) retention signal (Masuda et al., 2002). Lipoproteins lacking the signal are transported to the OM. Early lipoprotein transport studies utilized spheroplasts that were generated by EDTA-lysozyme treatment, making them devoid of periplasmic proteins (Matsuyama et al., 1995). While these spheroplasts continued to secrete proteins from the cytoplasm that are normally destined for the periplasm and OM, the major OM lipoprotein (Lpp) was not secreted and remained in the IM, which suggested that a periplasmic factor was required for OM lipoprotein release from the IM (Matsuyama et al., 1995). When periplasmic-derived materials were added externally to the spheroplasts, Lpp was released from the IM, and purification of the periplasmic components identified the periplasmic chaperone LolA (Masuda et al., 2002). Further research involving the generation of various proteoliposomes went on to characterize the OM component of the trafficking pathway, LolB (Matsuyama et al., 1997), whose role is the insertion of lipoproteins into the OM. Further analysis identified integral membrane proteins LolC and LolE, as well as a soluble, cytoplasmic ATP-binding protein (LolD) that is recruited by the membrane-associated heterodimer LolCE to form a complex that acts as an ATP-binding cassette (ABC) transporter in the IM (Yakushi et al., 2000; Ruiz et al., 2006). Lipoproteins are taken up by this complex and their lipid moiety bound to LolA, which is believed to protect their fatty-acid chains from the aqueous environment of the periplasm. LolA then transports them to an OM-associated lipoprotein, LolB, which stabilizes their internalization into the OM. Exactly how LolB inserts lipoproteins into the OM has yet to be elucidated. LolA and LolB share similarity of structure, both possessing hydrophobic cavities believed to be the binding sites for the lipid moieties of lipoproteins (Takeda et al., 2003). The cavities are comprised of unclosed β-barrels with α helical lid domains thought to close when not in complex with any lipoprotein (Takeda et al., 2003). It is worth noting that LolC and LolE are highly similar in sequence, and that unlike the Lol complex found in E. coli, the integral membrane portion of the complex in N. meningitidis appears to consists of only a single LolCE homolog (encoded by NMB1235) instead of two. This can potentially be explained in that the single N. meningitidis protein homolog may simply form a homodimer to supplement for the missing protein component (Bos et al., 2007). Given the essential nature of most of these Lol complex components in E. coli (Narita et al., 2002) and the large number of lipoproteins encoded in the H. pylori genome previously mentioned (Alm and Trust, 1999; Baltrus et al., 2009), their use as potential antimicrobial targets is promising.
A single, IM component of the Lol complex from E. coli appears to be conserved in H. pylori (HP0787; Table 1), with highest similarity to LolE. As mentioned previously, LolE and LolC are highly similar in E. coli and it has been postulated that possessing only one of these proteins may be adequate for Lol complex formation (Bos et al., 2007), as in the N. meningitidis model. Insertion mutations in HP0787 exist (Salama et al., 2004), however this result indicating a potential lack of essentiality of this gene should be interpreted with caution, as the generation of deletion mutants for this gene have not been possible to date (Liechti and Goldberg, unpublished results). A few genome sequencing projects have assigned lolD designations to a putative H. pylori homolog (strain B8, HPB8_1387/HP0179) (Farnbacher et al., 2010), however, this characterization is based solely on the ABC transporter domain, and upon closer inspection, the N-terminus of the LolD protein (approximately 100 amino acids) is missing from this H. pylori gene (similar dubious gene designations have been assigned in C. jejuni). While no definitive LolD homolog appears to exist in H. pylori as such, ABC transport domains are notoriously divergent in this bacteria compared with other microbes (Tomb et al., 1997), and dozens of potential candidates with similar domain structure exist in the genome whose functions have not yet been identified (Tomb et al., 1997; Marais et al., 1999). In addition, LolB (the lipoprotein involved in inserting new lipoproteins into the OM in E. coli and N. meningitidis) appears to be absent in H. pylori, and in fact is missing in all ε proteobacteria sequenced to date. A protein with poor genetic similarity to LolA, the periplasmic chaperone component, does exist in H. pylori (HP0785, E value = 0.03) and has been shown to be non-essential (Chalker et al., 2001). Whether this protein acts similarly to the LolA characterized in E. coli has yet to be discerned, however amino acid similarity alignments between HP0785 and LolA show significant similarity for the core functional regions of the chaperone, with greatest similarity existing on one of the two sides of the open β-barrel domain (Figure 2). When examining the H. pylori amino acid sequence for the less well-conserved side of the β-barrel domain, amino acids that make up two of the β strands in this region in the E. coli structure are absent in the H. pylori sequence (Figure 2, shown in blue) potentially indicating a smaller, more tightly packed β sheet on that side of the open β-barrel domain.
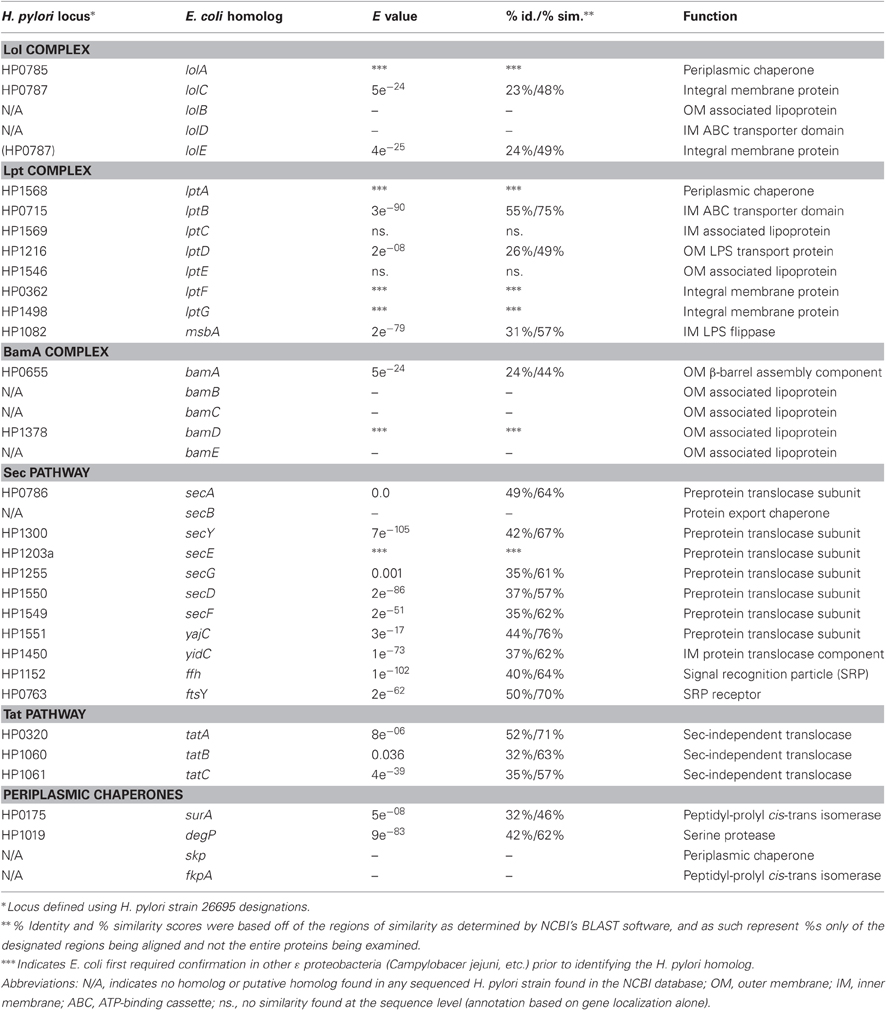
Table 1. H. pylori putative outer membrane biogenesis complex components.

Figure 2. LolA periplasmic lipoprotein chaperone from E. coli and H. pylori display structural homology at N-terminal region. Similarity protein alignments were constructed between E. coli and H. pylori LolA (HP0785) amino acid sequences and a “heat map” color scheme of protein homology based on similarity scores was generated. Crystal structures of E. coli LolA (PDB ID: 3KSN) were then used as a template for mapping the similarity scores. Red indicates very high confidence whereas green/blue indicate lower confidence in similarity comparisons. Views presented are an overhead of the putative, lipoprotein binding domain consisting of the open β-barrel with α helix lid (A), the well-conserved, N-terminal β-sheet region (B), and the less conserved, C-terminal β-sheet region (C) (the two β-strands missing in H. pylori LolA are indicated in blue). Protein sequence alignments and similarity scores were generated using T-Coffee analysis software (Poirot et al., 2003), the “heat map” CORE index for the alignment color gradient used algorithms described in the literature (Notredame and Abergel, 2003), and crystal structures were obtained from the Protein Data Bank (PDB) and viewed using ICM Brower Pro (MolSoft). The dotted red line in model represents an region in the amino acid protein sequence not present in the solved crystal structure.
The genetic orientation of the Lol complex-associated genes in ε proteobacteria is, in a word, perplexing. In E. coli, lolCDE (and in Neisseria, lolCD) are found in a single operon (Figure 3A), with lolB and lolA each existing separately, elsewhere in the genome (Yakushi et al., 2000). In most ε proteobacteria however, the single conserved homolog of lolC appears in an operon with secA, the cytoplasmic-localized membrane translocase. While this proximity to an essential gene may explain problems in targeting lolC for deletion in H. pylori, an added caveat exists. The H. pylorilolA homolog is, coincidentally, also located proximal to secA, albeit transcribed in the opposite direction (Figure 3B), potentially hinting at some selective element as of yet undetermined in either protein or lipoprotein trafficking in ε proteobacteria. Given the apparent absence of a lolD homolog at this locus combined with the presence of the membrane protein translocation ATPase (SecA) in its place, overlap in the Sec export pathway and the lipoprotein transport pathway in ε proteobacteria may be a distinct possibility.
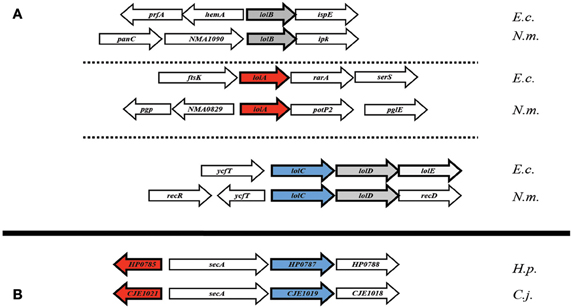
Figure 3. Genetic orientation of Lol complex-homologs. (A) Lol complex associated loci (lolA and lolB, specifically) from E. coli and N. meningitidis appear spread throughout the genome (B) while complex-homologs from H. pylori and C. jejuni appear to be clustered around the cytoplasmic chaperone encoding gene, secA. Abbreviations: E.c., Escherichia coli (K12 substr. W3110); N.m., Neisseria meningitidis (strain Z2491); H.p., Helicobacter pylori (strain 26695); C.j., Campylobacter jejuni (strain RM1221).
It has previously been shown that a lipoprotein signal peptidase gene (HP0074) is essential in H. pylori, indicating that lipoproteins in general are important for H. pylori survival (Chalker et al., 2001). The presence of a potentially essential lolE homolog in the genome would support this hypothesis. The apparent absence of a lolB homolog in addition to lolA proving to not be essential however, indicates that if this transport pathway is still functional in H. pylori, that it may possess redundant/alternate protein components for lipoprotein periplasmic transport and OM insertion. Taken together, despite initial observations indicating a defunct pathway, the lipoprotein transport pathway in H. pylori is potentially viable, and given the prominence of lipoproteins encoded in the genome, this pathway represents a potential molecular target worthy of full characterization and further pursuit.
In most Gram-negative bacteria, the major target for the immune system during a bacterial infection is lipopolysaccharide (LPS) (Chow et al., 1999; Miyake, 2004). LPS is a key component of a Gram-negative OM, contributing to the structural integrity of the bacteria as well as providing a barrier against the host immune system. A number of cellular receptors recognize bacterial LPS and upon binding they induce an innate immune response (Caroff et al., 2002). The primary activator of the host response to the lipid A portion of LPS from E. coli is Toll-like receptor 4 (TLR4) (Chow et al., 1999). In many cases, the immune response resulting from the presence of LPS (also referred to as endotoxin) is so severe, that a dysregulated immune response to infection develops, known as sepsis, which can be life threatening. Studies are currently underway to develop molecular inhibitors of LPS, specifically targeting its ability to induce monocyte activation (Bosshart and Heinzelmann, 2007).
In examining the two predominant model microbes in the field of OM biogenesis, it is worth noting that differences in their LPS exist at a structural basis. While E. coli possesses true LPS, Neiserria expresses lipooligosaccharides (LOS) lacking the repeating O-antigen of most enteric bacteria; this distinction is thought to account for many of the dissimilarities in the immune response to the different bacteria (Zhang et al., 2007). H. pylori does produce a defined length O-chain attached to its core region whose length is variable between strains. Similar to LPS biogenesis in other bacteria, the O-chain of H. pylori is constructed in the cytoplasm and flipped to the periplasm where it is transferred to the core oligosaccharide by the O-antigen ligase (WaaL). Because the host's immune response to LPS is so robust, many pathogenic bacteria express an LPS structure that avoids detection. H. pylori is a well-studied example, as it can express the Lewis Blood Group (LBG) antigens and present them on the O side chain of its LPS (Appelmelk et al., 1996; Monteiro, 2000). LBG antigens are commonly associated with host monocytes, macrophages, granulocytes, and gastric epithelial cells. They are important for low-affinity adhesion to host gastric epithelial cells as well as in phagocytosis (Kirkland et al., 1997; Lepper et al., 2005) and it has been widely speculated that H. pylori uses this key feature to evade the host immune system (Appelmelk et al., 1996; Moran et al., 1999). The genes responsible for the creation of these bacterial LBG antigens are present throughout the genome, and many of them undergo phase variation (Wang et al., 2000). The variation generated in the bacterial population, whether due to phase variation or other mechanisms, results in the production of a wide array of differing LPS profiles within a given population. This is believed to be a “fine-tuning” of the bacterium's defenses, further enhancing the evasion of the immune response (Wang et al., 2000; Khamri et al., 2005; Salaun et al., 2005; Salaun and Saunders, 2006)
In addition, H. pylori modifies the lipid A portion of its LPS. When first characterized, the lipid A from H. pylori was found to be unique and distinguishable from that of enterobacteria in that there is an absence of a phosphoryl group at the 4′-position and fatty acyl groups at the 3- and 3′-position, as well as the presence of 2-aminoethyl phosphate at the 1-position (Suda et al., 1997). Further studies went on to show that H. pylori actually synthesizes two lipid A species, a minor one resembling that of E. coli (bis-phosphorylated and hexa-acylated) and a major species that contains fewer acyl chains, fewer phosphate groups, and a single 2-keto-3-deoxyoctonate (Kdo) sugar attached to the disaccharide backbone (Moran et al., 1997; Tran et al., 2005). This led to the discovery that H. pylori modifies its lipid A and that the result is a molecule with much lower immunobiological activity (Tran et al., 2005). Additional studies identified that while the lipid A from H. pylori is initially synthesized in this hexa-acylated form, a series of modifications after synthesis can result in a tetra-acylated molecule (Stead et al., 2005, 2008).
Lipid A is the “hydrophobic anchor” of the LPS, and is the biological initiator of septic shock (Raetz and Whitfield, 2002). Modifications of the lipid A portion of H. pylori LPS results in reduced immuno-reactivity by as much as 1000 times compared to the lipid A of other bacterial species (Ogawa et al., 1997, 2003a,b; Stead et al., 2005). The exact mechanism by which H. pylori LPS interacts with the immune system is currently in dispute. While it was initially characterized that the host receptor for H. pylori LPS was TLR4 (Kawahara et al., 2001) subsequent papers have shown that H. pylori interacts through TLR2 and not TLR4 (Smith et al., 2003). This controversial finding is apparently not limited to H. pylori, and the same phenomenon has been shown for other Helicobacters as well (Mandell et al., 2004). While another study exists, showing that chemically synthesized lipid A from H. pylori signaled via TLR4 (Ogawa et al., 2003a,b), highly purified H. pylori LPS in an alternate study showed activation only through the TLR2 complex (Yokota et al., 2007). TLR4 expression in gastric cell lines has also been used as evidence of H. pylori LPS activation (Kawahara et al., 2001; Su et al., 2003; Schmausser et al., 2004), however in a recent paper, it was shown that H. pylori LPS first signals via TLR2, which then acts through nuclear factor Y (NF-Y) to activate the transcription of TLR4 (Yokota et al., 2010). Whatever the mechanism, the presentation of LPS on the OM of H. pylori is essential for colonization and pathogenesis partially for to its role in membrane barrier function, but also for its unique ability to mask this pathogen from the host immune system.
The majority of the work on the LPS transport (LPT) pathway has been conducted in E. coli and N. meningitidis, and the pathways are summarized visually in Figure 1. Each model bacterial system provides differing tools that allow the system to be analyzed in a slightly different way. For example, LPS is not essential for N. meningitidis, allowing for comprehensive mutant studies to be conducted, while the E. coli system provides a high degree of technical genetic manipulation to artificially deplete components and analyze the effects. Both systems have shown that the transport pathway consists of two major membrane proteins, the flippase MsbA in the IM and the β-barrel IMP/LptD in the OM. MsbA allows for LPS in the cytoplasmic IM leaflet to be “flipped” onto the periplasmic leaflet. Once there, LPS is extracted from the IM in a process believed to be powered by an ABC transporter consisting of two integral membrane proteins (LptF and LptG) as well as an additional IM protein located solely on the cytoplasmic face of the membrane (LptB). LPS moves from the IM LptBCFG complex to the OM LptDE complex, traversing the periplasm via LptA (Ma et al., 2008; Tran et al., 2008). LptA recognizes the lipid moiety of LPS and is thought to protect the highly hydrophobic lipid element from the aqueous periplasm during transport. The energy source that powers this movement of LPS across the periplasm has yet to be elucidated.
MsbA was initially identified as a multi-copy suppressor of lpxL (htrB) null mutants (Karow and Georgopoulos, 1993), originally characterized as having growth and viability defects due to the accumulation of phospholipids (Karow et al., 1991). Both htrB null mutants and temperature sensitive msbA conditional mutants accumulated a precursor for LPS (N-acetyl [3H]-glucosamine) in the IM (Polissi and Georgopoulos, 1996). Upon subsequent analysis, msbA showed high homology to various ABC transporters; one study using a temperature-sensitive msbA allelle constructed in a polymyxin-resistant background strain of E. coli showed that LPS recovered from the inner membrane did not contain modifications to the lipid A required for polymyxin resistance (Doerrler et al., 2004). These modifications (the covalent modification of lipid A with cationic substituents 4-amino-4-deoxy-L-arabinose and phosphoethanolamine) are believed to occur at the periplasmic leaflet of the IM, and their absence strongly indicated that the LPS in these mutants was, therefore, localized in the cytoplasmic-facing, lipid leaflet (Zhou et al., 1998; Doerrler, 2006).
Attempted mutant studies determined that msbA is essential for in E. coli (Zhou et al., 1998), and this was independently confirmed in Pseudomonas aeruginosa (Ghanei et al., 2007). Similar studies were conducted in N. meningitidis, with somewhat differing results: total LPS levels for the mutant msbA strain were shown to be lower in the parental strain, indicating the potential presence of a feedback inhibition pathway not seen in E. coli (Tefsen et al., 2005). It has been speculated that because the LPS appears to localize on the cytoplasmic side of the IM in E. coli, that MsbA plays a role in the flipping of the lipid A-core oligosaccharide onto the periplasmic side of the membrane. This flippase activity has since been confirmed in a reconstituted system (Eckford and Sharom, 2010). It is believed that MsbA forms a homo-dimer with a chamber oriented toward the cytoplasm capable of accommodating lipid molecules, a finding supported by crystal structure analysis (Ghanei et al., 2007; Ward et al., 2007; Eckford and Sharom, 2008).
The identification of the OM protein LptD (Imp/OstA) was revealed through screening E. coli mutant libraries for strains with alterations in their membrane permeability (Sampson et al., 1989; Aono et al., 1994). lptD mutants resulted in an increased permeability of OMs to maltodextrins, dyes, as well as antibiotics (Sampson et al., 1989). In addition, it has been established that lptD is regulated by sigma factor E, a stress response regulator that reacts to disruptions of LPS structure and found to up-regulate various proteins involved in membrane biogenesis, such as periplasmic and cytoplasmic chaperones (Dartigalongue et al., 2001). Overexpression of LptD was only possible when LptE was also overexpressed simultaneously, and the two proteins were found to form a stable, heterodimeric complex (Chng et al., 2010). In N. meningitidis, inactivating lptD gave results similar to those found in the E. colimsbA conditional mutants: significantly reduced total cellular LPS, further confirming the linkage between LPS synthesis and transport in this model system (Braun and Silhavy, 2002; Bos et al., 2004). By using a PagL induction system (PagL acts by removing an acyl chain on the LPS after it is incorporated into the OM) researchers were able to show that the LPS isolated from N. meningitidis strains with an inactivated lptD showed no signs of this modification, indicating that the LPS was not transported to the outer leaflet of the OM (Bos et al., 2004). Studies in E. coli identified a complex partner of LptD, a lipoprotein originally named RlpB and now denoted as LptE. Conditional mutants for LptE were similar to LptD mutants in that they resulted in an increase in total cellular LPS (Wu et al., 2006).
Screening for essential genes in E. coli lead to the classification of other proteins involved in envelope biogenesis. The proteins LptA and LptB were identified, and when these proteins were down regulated, phenotypes similar to lptD and msbA mutants were observed. In addition, the genes encoding these two proteins were found together in an operon (Sperandeo et al., 2007). In contrast with E. coli, in the N. meningitidis model, these genes are not essential, and when deletion mutants were generated, they were found to have severely reduced LPS production, similar to lptD and msbA mutants. Upon sequence and structural analysis, LptA was found to have no membrane spanning domains while also sharing a conserved N-terminal domain with Imp, indicating a common but as yet un-described function. The operon partner (lptB) encodes a protein that possesses the general features of a typical ABC transporter but also lacks any membrane spanning regions (Sperandeo et al., 2007). LptB exists as a 27-kDa protein that is found in a 140-kDa complex (Stenberg et al., 2005) and biochemical evidence of this LptB-containing complex has also been reported (Narita and Tokuda, 2009). LptB works in conjunction with LptC (previously YrbK) in the complex with LptBFG, an ABC transporter involved in the transport of LPS. LptA is believed to act as a periplasmic chaperone, delivering LPS to the OM, where LptD acts to orient it (Sperandeo et al., 2007; Tran et al., 2008).
While the components of the transport system are characterized, the exact mechanism of transport through the periplasm is still in dispute. The two competing hypotheses differ not in what proteins are involved but rather their orientation to each other during transport. The chaperone hypothesis proposes that LptA acts similarly to LolA of the lipoprotein transport pathway or Skp, DegP, and SurA of the membrane protein transport pathway (Sperandeo et al., 2007). The competing hypothesis is the “periplasmic bridge” model (Bayer, 1991; Tefsen et al., 2005; Sperandeo et al., 2007) in which all components together make up a protein bridge that effectively connect both membranes and simply pass LPS between themselves until it arrives in the OM. Characterized crystal structures of long, fiber-like structures formed by multiple LptA molecules arranged in head-to-tail fashion while in the presence of LPS (Suits et al., 2008; Merten et al., 2012) seem to support the periplasmic bridge model.
A number of other studies seem to favor this “periplasmic bridge” or “trans-envelope” model. Lpt proteins have been shown to colocalize and can be found in both OM and IM preparations (Chng et al., 2010). LptA was first believed to be soluble and localized only in the periplasm (Sperandeo et al., 2009) however, subsequent experimentation has shown that LptA preferentially interacts with membranes and that polyhistidine tags that were used to purify the protein are potentially culprits in effecting the localization that was observed in previous studies (Chng et al., 2010). Subsequent experimentation has also shown that defects in components of the Lpt pathway in either the OM or IM result in LptA degradation, potentially indicating that LptA levels can act as a marker for proper formation of the transmembrane bridge (Sperandeo et al., 2011). The evidence of the existence of these bridges appears definitive, however, whether these bridges are transient or always present remains to be elucidated.
The roles of the stabilizing proteins LptC and LptE have only more recently been clarified. Studies have found that while LptC interacts directly with LptA (Sperandeo et al., 2011; Bowyer et al., 2011), LptE does not (Bowyer et al., 2011). The structure of LptC is highly similar to that of LptA, and it has been shown that like LptA, LptC binds LPS in vitro (Tran et al., 2010). Additionally, LptA can displace LPS from LptC in vitro but LptC cannot displace LPS from LptA, indicating a unidirectional export pathway (Tran et al., 2010). LptE has been shown in E. coli to interact directly with LPS (Chng et al., 2010) and it is theorized to act as a substrate recognition site at the OM. However, when LptE was deleted from N. meningitidis, LPS transport to the cell surface was not noticeably affected and only levels of LptD were altered (Bos and Tommassen, 2011) indicating a potential role in trafficking of LptD to the membrane. One study has shown that several residues of LptE interact directly with LptD in vivo, specifically in a predicted extracellular loop structure, and a “plug-and-barrel” architecture in which LptE resides within the LptD β-barrel has been postulated (Freinkman et al., 2011). The interaction between LptD and LptE has also been shown to occur while LptD is being assembled into the membrane by the Bam complex (Chimalakonda et al., 2011).
It has been proposed that LPS itself may play a role in membrane biogenesis, specifically in OMP transport (Bulieris et al., 2003; Qu et al., 2009). This appears to be species-specific however, as in N. meningitidis, OMP biogenesis is independent of LPS. Predicted binding domains for LPS appear to exist on the periplasmic chaperone Skp in both E. coli (Walton and Sousa, 2004) and N. meningitidis, indicating the potential for at least some overlap between the LPS and OMP transport pathways, however Skp-LPS interactions have to date only been detected in vitro (de Cock et al., 1999). A less direct and more plausible effect of LPS on OMP biogenesis has been described, in which LPS acts merely to stabilize membrane proteins that have already been transported to the membrane via mediating rearrangements of the proteins' surface-exposed loops (de Cock and Tommassen, 1996). Porin trimers have been shown to be stabilized in this fashion (de Cock and Tommassen, 1996).
From a genetic standpoint, the LPS transport pathway in H. pylori appears to be conserved in regard to the integral membrane proteins MsbA, LptB, and LptD (Table 1). The encoding genes (HP1082, HP0715, and HP1215/1216, respectively) are present in all sequenced H. pylori isolates available (strains 26695, J99, HPAG1) as well as Helicobacter acinonychis and Campylobacter jejuni and are highly conserved when compared with their E. coli homologs. Transposon mutagenesis studies have generated mutations in all three genes (Salama et al., 2004), however, whether the proteins are still functional in these mutants is not known.
Both msbA and lptD were found to be significantly up-regulated when H. pylori strain NTUH-C1 was treated with glutaraldehyde (Chiu et al., 2009) and ΔlptD and ΔmsbA mutants have been characterized in the same H. pylori strain (Chiu et al., 2007, 2009). These researchers determined that their mutants, derived from the clinical isolate NTUH-C1, were more susceptible to both n-hexane as well as several antibiotics (novobiocin, erythromycin, tetracycline, and penicillin) when compared to the parental strain, suggesting that MsbA and LptD play an important role in maintaining membrane integrity in H. pylori. Silver staining of proteinase-K digested whole cell lysates indicate lower amounts of total LPS present in ΔlptD, ΔmsbA, and ΔlptDΔmsbA mutants. Immunoblots using antibodies for Lewis antigens A and B on H. pylori LPS preparations were used to show the amounts of O-antigen in the preparations, and both antibodies showed significantly reduced amounts of O-antigen in all three mutants (Chiu et al., 2009). While these studies raise the potential that H. pylori can exist without LPS, as of yet, no examination of the lipid A of these mutants has been conducted. LptA and LptD from E. coli have been shown to interact with the lipid A moiety of LPS, indicating that it is lipid A that is actually the key component that is being transported by the Lpt transport pathway. There are examples in the literature of alterations in H. pylori lipid A resulting in a “truncation of the core oligosaccharide and loss of the attachment of the O sidechain containing Lewis X and Y” (Moran et al., 2004) and the expression of Lewis antigens in a cholesterol-dependent manner (Hildebrandt and McGee, 2009), however, no studies to date have shown that H. pylori can exist without lipid A in its OM, or have shown an alternate pathway for lipid A integration into the membrane. Strain specificity potentially plays a role with regard to the generation of membrane biogenesis mutants in H. pylori, as at present, generating directed insertion or deletion or mutants in lptD, msbA, or any of the other components of the Lpt complex in strains 26695, G27, 1061, or SS1 have proven unsuccessful (Liechti and Goldberg, unpublished results).
In searching for possible homologs for all the components of the LPS trafficking from E. coli in H. pylori, an interesting phenomenon found in all of H. pylori membrane trafficking pathways again presents itself. While the membrane spanning pathway components appear quite similar to their E. coli counterparts (Table 1), the components localizing to the periplasmic region of the pathway appear barely conserved and potentially non-existent in any recognizable form. Putative H. pylori homologs to LptF and LptG (HP0362 and HP1498, respectively) were only found after identifying homologs with either E. coli or N. meningitidis proteins in Helicobacter hepaticus (HH0602 and HH1496, respectively). The purported periplasmic LPS chaperone, LptA, found in both E. coli and N. meningitidis is present in H. pylori, albeit at a very low similarity, whereas the membrane-associated ABC transporter component LptB is highly conserved. Analysis of the putative H. pylori LptA shows high structural similarity at the central region of the protein, and interestingly the majority of divergence from the E. coli homolog exists at both the N- and C-termini (Figure 4). These regions of lower similarity comprise the putative points of interaction between LptA molecules when they form polymers, resulting in a twisted, β-jellyroll structure, in the presence of LPS (Suits et al., 2008). No obvious homologs exist for either LptC and LptE in H. pylori or any ε proteobacteria at the level of amino acid identity/similarity, however one report from the literature indicates that lptE is always present in a common locus in most bacterial genomes, and when it does not appear to exist on the level of genetic similarity, a putative lipoprotein can be found in its same genetic locale (Bos and Tommassen, 2011). In the case of H. pylori, a putative lipoprotein that shares common domain characteristics of LptE orthologs exists at this locus, directly downstream of leuS (Figure 5) and this appears to be the case for all other ε proteobacteria as well (Bos and Tommassen, 2011). When protein structural analysis software [RaptorX, (Peng and Xu, 2011)] is used to compare the putative H. pylori protein at the structural level to all solved crystal structures in the Protein Data Bank, the two most structurally similar proteins are characterized as “rare lipoprotein B” (early nomenclature for LptE) from Nitrosomonas europaea and Shewanella oneidensis (PBD designations 2JXP and 2R76), respectively. Similarly, a conserved protein in ε proteobacteria appears to exist in the same genetic locus as lptC (directly upstream of the lptA homolog) (Figure 6). Similarly, the H. pylori homolog to lptB (HP0715) is found directly upstream of rpoN, similar to its genetic localization in E. coli and N. meningitidis. Amino acid alignments show well-conserved residues in the N-terminus of the putative H. pylori LptC and poorer similarity past ∼ residue 110. The LptC periplasmic domain has been described as a “twisted boat” structure comprised of two β-sheets in opposition to each other (Tran et al., 2010) and the H. pylori putative protein possesses amino acid similarity through the first five β-strands (Figure 7). Unlike LptE, LptC is believed to be the crucial link between LptA and the ABC transporter (LptBFG) and as all localization and protein interaction data to date supports this hypothesis, the presence of a potential LptC-like protein in ε proteobacteria is encouraging.
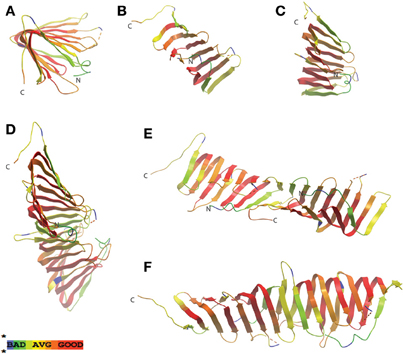
Figure 4. LptA periplasmic LPS chaperone displays greatest structural homology at the central region. Similarity protein alignments were generated between LptA amino acid sequences from E. coli and H. pylori (HP1568) and a color scheme based on similarity scores was generated, described previously in Figure 2. Crystal structures of LptA from E. coli [PDB ID: 2R19, (Suits et al., 2008)] were then used as a template for mapping the similarity scores. Monomeric (A–C), as well as the dimeric (D–F) “head-to-tail” conformation (in which the N-terminal β-strand from each molecule interacts with the C-terminal β-strand of the adjacent LptA molecule) of protein structures are shown. Greatest similarity is found in the central portions of the protein (shown in red) whereas areas believed to be involved in monomer stacking to form filamentous structures, believed to potentially span the periplasm, give significantly lower similarity scores (shown in yellow and green). The dotted yellow and red lines in the model represents regions in the amino acid protein sequence not present in the solved crystal structure.
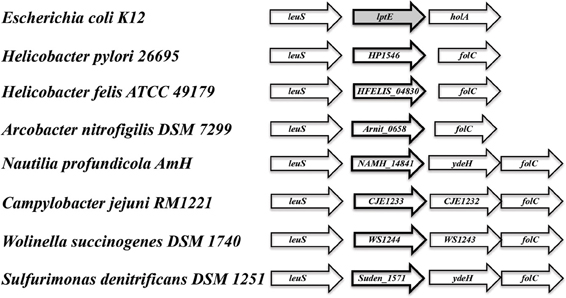
Figure 5. Genes encoding putative lipoproteins of unknown function are found adjacent to leuS (leucyl-tRNA synthetase) in ε proteobacteria. While having limited sequence similarity to lptE, the conserved orientation of the lipoprotein encoding genes downstream of leuS, in addition a high similarity score at the putative protein structural level to LptE from Nitrosomonas europaea and Shewanella oneidensis [Protein DataBase #s 2JXP and 2R76, respectively: analysis conducted utilizing the 3D modeling software RaptorX: (Di Tommaso et al., 2011)] indicates the potential of a functionally equivalent protein in all ε proteobacteria species observed.
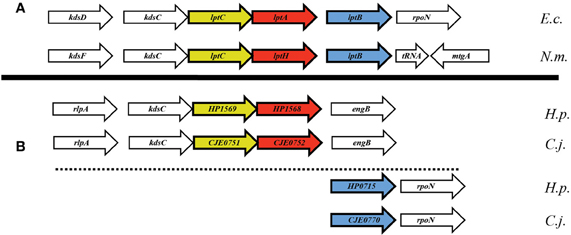
Figure 6. Apparent genetic reorientation of Lpt operon in ε proteobacteria. While the orientation of lptA, lptB, and lptC in most Gram-negative bacteria is that of a single putative operon, the locus in ε proteobacteria appears to have dissociated while still maintaining many of the genes proximal to the Lpt components, namely rpoN and kdsC.
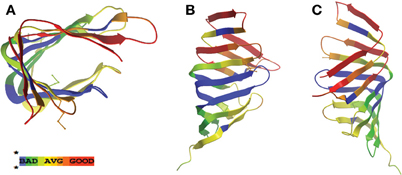
Figure 7. LptC displays greatest structural homology at the N-terminal region. Ribbon structure of LptC protein from E. coli [PDB ID: 3MY2, (Tran et al., 2010)] with areas of similarity to the H. pylori homolog (HP1569) displayed in color code described previously in Figure 2. Views presented are overhead (A) and side (B–C) profiles.
H. pylori contains an OMP family consisting of approximately 33 genes. This family is divided into two subfamilies comprising of the H. pylori outer membrane protein (Hop) and the Hop-related (Hor) families (Alm and Trust, 1999). The vast majority of H. pylori OMPs belong to the Hop sub-family, including almost all known adhesion proteins that allow for the binding of H. pylori to numerous substrates from fucosylated histo-blood group antigens (Ilver et al., 1998) to host laminin (Senkovich et al., 2011). The protein family also contains a substantial number of porins (Exner et al., 1995; Doig et al., 1995; Peck et al., 2001) thought to play roles in nutrient uptake, though to date, no study has closely examined their function with regard to specificity of substrates. It is also worth noting that the flagella apparatus (Eaton et al., 1996) and chemotactic machinery (Foynes et al., 2000; Croxen et al., 2006) of H. pylori are both essential for colonization and both possess OM and IM protein components, as do the transport pathways for LPS and lipoproteins, not to mention the Cag and Com type IV secretion systems, required for injecting the CagA protein into host cells (Censini et al., 1996) and taking up DNA for homologous recombination (Stingl et al., 2010), respectively. It is quite extraordinary how almost everything required for the survival and pathogenesis of H. pylori (DNA and nutrient uptake, adherence, motility, chemotaxis, immune stimulation/evasion, VacA secretion, iron acquisition, etc.) requires β-barrel proteins in the OM; these proteins may all depend on a single mechanism by which they are inserted into the OM.
In attempting to use E. coli as a model to discern proteins involved in OM biogenesis, researchers needed to designate set criteria to judge whether a given protein was likely involved in the transport pathway. They reasoned that defects in membrane transport would result in a loss of barrier function and membrane permeability would significantly increase as a result (Braun and Silhavy, 2002; Ruiz et al., 2006). Therefore, alterations in membrane permeability became a key element to examine when considering potential candidate proteins for OMP transport. It was also shown early on that mis-folding of OMPs resulted in an envelope stress response, in which various chaperones involved in the protein transport pathway were up-regulated (Cosma et al., 1995; Raivio and Silhavy, 1997). Genes involved in the OM transport pathways were also assumed to exist in operons with other transport pathway genes in addition to being essential in E. coli and well-conserved in other bacteria. The ultimate test for proteins playing roles in OM biogenesis was depletion experimentation using either temperature-sensitive mutants or inducer-dependent expression systems. In both cases, levels of OMPs could be examined and locations within the cell detected and compared with wild-type bacteria. It was also shown that, in many cases, problems in OMP transport resulted in a down-regulation of membrane proteins, preventing the accumulation of mis-folded proteins at various stages of the transport process (Werner and Misra, 2005; Ruiz et al., 2006; Bos et al., 2007).
This basic understanding of membrane biogenesis became much less universal once similar studies were conducted in the bacterium N. meningitidis. In stark contrast to E. coli, N. meningitidis proved a more complicated model for recognizing new transport pathway components for a number of reasons. For example, N. meningitidis appears to completely lack the majority of proteins involved and essential for a periplasmic stress response (Bos et al., 2007), as characterized by the E. coli system. In addition, numerous genes identified as essential components of the OM biogenesis apparatus in E. coli are not essential in N. meningitidis (Bos et al., 2007). This does allow for the creation of mutants in this model system however, enabling a more straightforward, reductionist approach.
The sec pathway is a well-established system for trafficking proteins from the cytoplasm through the IM to the periplasm. In this transport pathway, chaperones SecA and SecB sequester proteins bound for the IM transport pathway by recognizing leader sequences on the designated protein (Kumamoto and Francetic, 1993; Valent et al., 1998). These chaperones then bring the newly formed protein to the membrane-localized portion of the Sec complex (SecDEFGY) (Van den denBerg et al., 2004), which acts to move the protein through the IM. SecA then acts as the ATPase motor for this protein transport channel, providing energy for the mechanical “pushing” of the preproteins through the SecYEG complex (Tomkiewicz et al., 2006) and in so doing changes localization from cytoplasmic to peripheral IM and finally to integral IM. The leader sequence on the preprotein is removed by a leader peptidase, and the protein is then generally recognized by various periplasmic chaperones. In addition to the SecB-dependent transport of preproteins to the SecYEG complex, an alternate means of cytoplasmic transport exists (Koch et al., 1999) in the signal recognition particle (SRP) that recognizes either signal recognition peptides or the first transmembrane segment of proteins targeted for the IM. Once bound to the preprotein, the SRP complex migrates to the membrane, where a SRP receptor catalyzes the release of the preprotein and its insertion into the SecYEG complex. While initially it was thought that the majority of OMPs were SecB-dependent (Kumamoto and Beckwith, 1985), it has also been shown that OMPs misdirected to the SRP-dependent pathway can be secreted across the IM and are correctly assembled in the OM (Lee and Bernstein, 2001; Bowers et al., 2003).
Another IM transport pathway for proteins has also been characterized. Known as the Twin-Arginine Translocation (Tat) complex, this system involves three IM proteins (TatA, TatB, and TatC) that allow for protein transport independent of the Sec pathway (Lee et al., 2006). The general model for this complex is that TatA, with its multiple membrane spanning domains, forms a pore complex, while TatB and TatC stabilize the complex and allow for interactions with incoming cytoplasmic proteins. The three genes encoding these proteins are found in a single operon in E. coli and mutants have shown defects in IM trafficking for certain proteins (Lee et al., 2006). It has been postulated that the Tat pathway generally is utilized for proteins that undergo the majority of their conformational changes in the cytoplasm, as the transport pathway is relatively fast as compared with the Sec pathway. Studies have shown that mutations of genes encoding proteins involved in this pathway in various bacteria can result in a loss of infectivity resulting from the mislocalization of virulence determinants, which are normally moved through the periplasm by this mechanism (Voulhoux et al., 2001; Lee et al., 2006).
Recent E. coli studies have led to the discovery of an OM component of the OMP trafficking pathway, the Bam (YaeT) complex (Ruiz et al., 2006). Researchers, in an attempt to identify proteins potentially involved in membrane biogenesis, created an E. coli strain with a defective lptD allele. Barrier function of the OM in wild-type E. coli generally results in resistance to a number of antibiotics and various toxic compounds. However, E. coli strains containing mutations in key OM biogenesis genes, such as lptD, display a “leaky phenotype” in that their membranes are more permeable and as a consequence these mutants show increased susceptibility to toxins and antibiotics. By using the lptD mutant as a starter strain, and then selecting for spontaneous suppressor mutations that result in a return to wild-type levels of antibiotic resistance, researchers were able to characterize other proteins involved in the transport pathway (Ruiz et al., 2005). Among the mutations identified was the yfgL (bamB) gene, encoding an OM-associated lipoprotein. Co-immunoprecipitation studies revealed that BamB existed in a complex in the OM with three other lipoproteins; YfiO (BamD), NlpB (BamC), and SmpA (BamE), as well as the β-barrel protein BamA (Wu et al., 2005).
The homolog of BamA in N. meningitidis (Omp85) had previously been linked to OM biogenesis, and it was initially uncertain whether it also had an additional role in transport of LPS (Voulhoux et al., 2003; Voulhoux and Tommassen, 2004). Subsequent experimentation was able to show that LPS transport is affected when the overall levels of BamA are reduced. This effect was later found to be indirect, as the alterations in LPS transport were the result of the OMP of the LPS transport pathway (LptD) not being inserted correctly into the membrane (Bos et al., 2004). In both bacterial systems, bamA is an essential gene and is involved in the assembly of β-barrel proteins. In examining its genetic orientation, it was discovered that bamA is flanked by two other genes involved in membrane trafficking: the periplasmic chaperone skp as well as yaeL, a protease involved in the envelope stress response described earlier (Bos et al., 2007). Lipoprotein BamD was also shown to be essential for viability in E. coli and N. meningitidis, while mutations in the other three lipoproteins result in only mild alterations in membrane permeability (Onufryk et al., 2005; Wu et al., 2005). However, upon comparison of the pathway mechanics of both microbes, significant differences emerged. Depletion studies for BamA in E. coli showed no alteration in LPS trafficking, indicating no role for BamA involving LPS in E. coli (Doerrler and Raetz, 2005). It was also shown that depletion for BamA in N. meningitidis led to the accumulation of unfolded OMPs in the IM (Voulhoux et al., 2003) while in E. coli, depletion resulted in significantly reduced amounts of all OMPs (Wu et al., 2005). It is believed this is due to the absence of the envelope stress response in N. meningitidis, thereby preventing the down-regulation of OMP synthesis (Bos et al., 2007). Subsequent studies have also shown that BamA also acts as the receptor for contact-dependent growth inhibition (CDI) in E. coli, and that antibodies specific for extracellular epitopes on BamA block inhibitor cell binding (Aoki et al., 2008). The potential for utilization of CDI to target bacterial growth is currently being examined (Aoki et al., 2011).
The four major periplasmic proteins involved in OMP transport in E. coli are SurA (Rouviere and Gross, 1996), DegP (Misra et al., 1991; Krojer et al., 2002), FkpA (Dartigalongue et al., 2001), and Skp (Harms et al., 2001; Bulieris et al., 2003; Walton and Sousa, 2004). Unlike various other members of the OM biogenesis pathway, Skp as well as other periplasmic chaperones, have been shown to be non-singularly essential, and mutants in E. coli show only modest defects in membrane biogenesis (Rizzitello et al., 2001). This has led to the belief that potentially multiple mechanisms exist for periplasmic trafficking, a view supported by greater OM defects seen in sequential knockout studies for other established periplasmic chaperones (Rizzitello et al., 2001), indicating the presence of parallel pathways. To date, SurA is recognized as the main periplasmic chaperone, with Skp/DegP acting as a back-up pathway (Sklar et al., 2007). Evidence does however exist in the literature that some proteins appear to be preferentially transported by SurA (Vertommen et al., 2009). LptD is currently believed to be transported through the periplasm by the SurA chaperone with cooperation from periplasmic disulfide isomerase (DsbC) and thiol oxidase (DsbA) (Denoncin et al., 2010). Further analysis revealed that LptD contains two disulfide bonds that are essential for function that act to stabilize the LptD protein by correctly positioning the N- and C-terminal domains of the protein in the periplasm (Ruiz et al., 2010) and that oxidation is carried out by DsbA. However, this apparent preference for periplasmic chaperones is potentially species-specific, as ΔsurA mutants generated in N. meningitidis did not appear to affect membrane biogenesis, while a Δskp mutant resulted in significantly lower levels of porins PorA and PorB (Volokhina et al., 2011).
A large number of the membrane-associated proteins found on the surface of H. pylori consist of β-barrels, indicating their transport through the membranes from the cytoplasm may be through the Sec and BamA complex (Table 1). The importance of the Sec pathway in H. pylori was made readily apparent via bioinformatic analysis of the H. pylori strain 26695, where it was found that 517 of the 1,590 open reading frames possessed putative signal sequences (Alm et al., 1999). The Sec pathway for IM transport has already been well-characterized in H. pylori and transposon mutagenesis studies have shown that many of the genes encoding components of the Sec-pathway are capable of being mutated (albeit definite conclusions based on these results should be tempered with the knowledge that often insertions in a gene do not always lead to a complete inactivation of that gene), with the exception of secY (Salama et al., 2004). H. pylori does seem to lack the cytoplasmic chaperone SecB, but this is apparently common and has been characterized in other bacterial species (Yamane et al., 2004), indicating SecB acts to supplement the effectiveness of SecA. This may also hint at a more pronounced role for the SRP-dependent pathway in OMP trafficking to the Sec complex in H. pylori, however, to date no studies have tested this potential hypothesis. E. colisecD and secG temperature-sensitive mutants when grown at the non-permissive temperature result in the accumulation of OMPs in the cytoplasm (Fitchen et al., 2003). However, when complementation studies were preformed using plasmids expressing the H. pylori homologues in these mutants, cytoplasmic OMP levels returned to their wild-type levels, indicating a conservation in function for these genes in H. pylori (Fitchen et al., 2003).
Similarly, the Sec-independent IM transport system, the Tat complex, appears to be well-conserved in H. pylori. tatA (HP0320), tatB (HP1060), and tatC (HP1061) are all present (Table 1), with the only difference being the apparent removal of tatA from the original operon and insertion elsewhere in the genome, whereas in E. coli the three genes comprise a single operon (Lee et al., 2006). No characterized mutants for tatB or tatC have been described while a single transposon insertion mutant exists for tatA (Salama et al., 2004) and the low coverage of transposon mutants in these genes suggests that all three maintain an essential function in H. pylori. This would seem to differ with previous studies in E. coli where the Tat system has been shown to be dispensable under laboratory conditions, however, there are examples of bacteria that rely heavily on the Tat system for almost all protein secretion (Dilks et al., 2005).
The periplasmic chaperones DegP and SurA appear to be conserved in H. pylori, while the chaperone Skp does not appear to be well-conserved. N. meningitidis is similar to H. pylori in this respect, as it posseses only surA and skp homologs and is missing a functional degP homolog (Volokhina et al., 2011). The synthetic lethal phenotypes characterized in E. coli double mutants (surA/skp and surA/degP) indicate that SurA and Skp/DegP exist in parallel periplasmic chaperone pathways, and the absence of a Skp homolog in H. pylori is possible due to the presence of SurA. No mutants in degP or surA have been characterized to date in H. pylori however, and insertional mutants in these genes were not generated in the construction of a transposon library in H. pylori of ∼10,000 clones (Salama et al., 2004), indicating that they may be essential.
The integral OMP component of the OMP trafficking pathway, BamA, is well-conserved in H. pylori (Table 1) and all ε proteobacteria. What is striking is that in E. coli and N. meningitidis, this protein is part of a complex along with at least four other conserved lipoproteins (BamB, BamC, BamD, and BamE) while in H. pylori only BamD (ComL) appears to be present. It is worth noting that BamA and BamD appear to form the core of the OMP export machinery, as they are the only two essential components in both E. coli and N. meningitidis. In E. coli BamE has been recently shown to modulate the conformation of BamA, likely through its interactions with BamD, and when Δ bamE mutant cells are treated with proteinase K, BamA is degraded beyond recognition (Rigel et al., 2012). No studies on the Bam complex have been reported in H. pylori to date.
There is currently a relatively large amount of circumstantial genetic evidence suggesting that the OM biogenesis pathways established in E. coli and N. meningitidis are present in some form or derivation in H. pylori and in fact all ε proteobacteria. The OM- and IM-protein components of all of these pathways appear to be conserved (Table 1), but what is striking is that, in these initial genetic and bioinformatic observations, nearly all of the periplasmic components from each of these characterized pathways from E. coli and N. meningitidis are missing or severely altered in all ε proteobacteria (Figure 1). An examination of the divergent residues of putative H. pylori homlogs for LolA, LptA, LptC, and LptE all indicate that in general, the N-terminal domains are conserved, while the more C-terminal domains are not. Divergence at the N- and C-terminal domains of the H. pylori LptA homolog are particularly interesting, as these residues are potentially important for the formation of the long, end-to-end, filamentous structures formed in the presence of LPS. Regions of interaction between filament monomers are generally well-conserved between bacterial species, given the importance of monomer-monomer interaction (Samatey et al., 2001), however, instances of variability at these binding domains are present in the H. pylori literature (Andersen-Nissen et al., 2005). Given that all of the membrane-associated components from each transport system are present and given that in every case, the periplasmic-associated components are highly divergent or potentially missing, there appears to be some strong, and as of yet uncharacterized selection affecting the periplasmic components of all three of the discussed transport pathways in H. pylori and other ε proteobacteria.
Little is known about the conditions inside the periplasm of H. pylori and the field of periplasmic transport has only just begun to expand. Recent studies have examined the transport of urea via UreI for use in buffering the periplasm in H. pylori, maintaining a pH of ∼6.1, compared to a pH as low as ∼2 outside of the OM and pH greater than 7 in the cytoplasm (Scott et al., 2000; Weeks et al., 2000; Krulwich et al., 2011). In this respect “the key to gastric colonization by H. pylori is periplasmic pH homeostasis” (Krulwich et al., 2011). The possibility exists that using the periplasmic space as a pH buffer may be driving this apparent selection affecting the composition of periplasmic-associated lipoproteins as well as potential periplasmic chaperones in all three, membrane biogenesis transport systems. Divergence in sequence perhaps reflects amino acid domains important for survival in harsh environments. However, UreI is found in only a handful of Helicobacters while the divergence in periplasmic-associated lipoproteins is found throughout all ε proteobacteria. Though different from E. coli and N. meningitidis, those lipoprotein components found in H. pylori are highly conserved within ε proteobacteria, indicating the selective element is affecting the entire bacterial family, or at least an early common ancestor. Given the apparent absence of LolB and LolDE, lipoprotein transport in ε proteobacteria may be severely altered and the effect on lipoprotein transport may explain the divergence in lipoprotein components of both the Bam and Lpt transport pathways. Another possibility may be an alteration in membrane lipid composition in ε proteobacteria compared with other bacterial families. This explanation may explain the apparent inability to identify many of the lipoprotein components of all three membrane biogenesis transport systems, as potentially unique alterations in the lipid moiety of these components may be required for insertion into membranes of ε proteobacteria. The fact that the vast majority of sequence divergence in lipoprotein components of trafficking pathways in H. pylori were in domains not involved with interacting with the actual LPS, lipoproteins, or OM proteins indicates that the membrane lipid composition or lipoprotein localization may significantly differ in ε proteobacteria. Phospholipids are known to make up the majority of lipids in H. pylori (Hirai et al., 1995) and cholesterol glucosides, lipids rarely found in animals or bacteria, account for roughly 25% of the lipids in H. pylori (Haque et al., 1996; O'Toole and Clyne, 2001), suggesting an altered membrane composition. As stated previously, inner and OMs in Helicobacters and Campylobacters are notoriously hard to separate, and the alterations seen in both lipid composition and the lipoproteins found in each of these three systems when compared with other bacterial families may offer clues as to the nature of this phenomena.
The field of membrane biogenesis in ε proteobacteria to date is remarkably unexplored. The recent studies undertaken by Chiu et al. have directly shown the importance of the LPS transport pathway components in membrane permeability and that a decrease in membrane integrity is achievable by removing the function of two of these components, resulting in an increased susceptibility of H. pylori to antibiotics (Chiu et al., 2007, 2009). In addition, targeting these pathways not only has the potential to affect membrane permeability, but also prevent virulence determinants essential for the survival of H. pylori from being expressed on the bacterial surface. Despite having shown this correlation between OM biogenesis and membrane integrity, no studies to date have examined the effects of alterations in these pathways on pH resistance, adherence, or colonization in H. pylori. The argument could be made that, given the lack of an effective H. pylori vaccine and the emergence of antibiotic resistant organisms, the time has come to begin exploring therapeutics that target these three essential mechanisms by which all known colonization/virulence determinants are expressed on or secreted from the bacterial surface.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank the reviewers for their helpful comments. The bioinformatic and genetic analysis was funded in part by the University of Virginia Cancer Center through the James and Rebecca Craig Research Scholars Award and the NCI Cancer Center Support Grant (P30 CA44579), and George Liechti was supported in part by a University of Virginia Cancer Training Grant (5T32 CA 009109).
Alm, R. A., Ling, L. S., Moir, D. T., King, B. L., Brown, E. D., Doig, P. C., Smith, D. R., Noonan, B., Guild, B. C., de Jonge, B. L., Carmel, G., Tummino, P. J., Caruso, A., Uria-Nickelsen, M., Mills, D. M., Gibson, R., Merberg, D., Mills, S. D., Jiang, Q., Taylor, D. E., Vovis, G. F., and Trust, T. J. (1999). Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature 397, 176–180.
Alm, R. A., and Trust, T. J. (1999). Analysis of the genetic diversity of Helicobacter pylori: the tale of two genomes. J. Mol. Med. 77, 834–846.
Andersen-Nissen, E., Smith, K. D., Strobe, K. L., Barrett, S. L., Cookson, B. T., Logan, S. M., and Aderem, A. (2005). Evasion of Toll-like receptor 5 by flagellated bacteria. Proc. Natl. Acad. Sci. U.S.A. 102, 9247–9252.
Aoki, S. K., Malinverni, J. C., Jacoby, K., Thomas, B., Pamma, R., Trinh, B. N., Remers, S., Webb, J., Braaten, B. A., Silhavy, T. J., and Low, D. A. (2008). Contact-dependent growth inhibition requires the essential outer membrane protein BamA (YaeT) as the receptor and the inner membrane transport protein AcrB. Mol. Microbiol. 70, 323–340.
Aoki, S. K., Poole, S. J., Hayes, C. S., and Low, D. A. (2011). Toxin on a stick: modular CDI toxin delivery systems play roles in bacterial competition. Virulence 2, 356–359.
Aono, R., Negishi, T., Aibe, K., Inoue, A., and Horikoshi, K. (1994). Mapping of organic solvent tolerance gene ostA in Escherichia coli K-12. Biosci. Biotechnol. Biochem. 58, 1231–1235.
Appelmelk, B. J., Simoons-Smit, I., Negrini, R., Moran, A. P., Aspinall, G. O., Forte, J. G., De Vries, T., Quan, H., Verboom, T., Maaskant, J. J., Ghiara, P., Kuipers, E. J., Bloemena, E., Tadema, T. M., Townsend, R. R., Tyagarajan, K., Crothers, J. M. Jr., Monteiro, M. A., Savio, A., and De Graaff, J. (1996). Potential role of molecular mimicry between Helicobacter pylori lipopolysaccharide and host Lewis blood group antigens in autoimmunity. Infect. Immun. 64, 2031–2040.
Bakos, N., Fekete, B., Prohaszka, Z., Fust, G., and Kalabay, L. (2003). High prevalence of IgG and IgA antibodies to 19-kDa Helicobacter pylori-associated lipoprotein in chronic urticaria. Allergy 58, 663–667.
Baltrus, D. A., Amieva, M. R., Covacci, A., Lowe, T. M., Merrell, D. S., Ottemann, K. M., Stein, M., Salama, N. R., and Guillemin, K. (2009). The complete genome sequence of Helicobacter pylori strain G27. J. Bacteriol. 191, 447–448.
Bayer, M. E. (1991). Zones of membrane adhesion in the cryofixed envelope of Escherichia coli. J. Struct. Biol. 107, 268–280.
Blaser, M. J. (1992). Hypotheses on the pathogenesis and natural history of Helicobacter pylori-induced inflammation. Gastroenterology 102, 720–727.
Bolin, I., Lonroth, H., and Svennerholm, A. M. (1995). Identification of Helicobacter pylori by immunological dot blot method based on reaction of a species-specific monoclonal antibody with a surface-exposed protein. J. Clin. Microbiol. 33, 381–384.
Bos, M. P., Tefsen, B., Geurtsen, J., and Tommassen, J. (2004). Identification of an outer membrane protein required for the transport of lipopolysaccharide to the bacterial cell surface. Proc. Natl. Acad. Sci. U.S.A. 101, 9417–9422.
Bos, M. P., Robert, V., and Tommassen, J. (2007). Biogenesis of the Gram-negative bacterial outer membrane. Annu. Rev. Microbiol. 61, 191–214.
Bos, M. P., and Tommassen, J. (2011). The LptD chaperone LptE is not directly involved in lipopolysaccharide transport in Neisseria meningitidis. J. Biol. Chem. 286, 28688–28696.
Bosshart, H., and Heinzelmann, M. (2007). Targeting bacterial endotoxin: two sides of a coin. Ann. N.Y. Acad. Sci. 1096, 1–17.
Bowyer, A., Baardsnes, J., Ajamian, E., Zhang, L., and Cygler, M. (2011). Characterization of interactions between LPS transport proteins of the Lpt system. Biochem. Biophys. Res. Commun. 404, 1093–1098.
Bowers, C. W., Lau, F., and Silhavy, T. J. (2003). Secretion of LamB-LacZ by the signal recognition particle pathway of Escherichia coli. J. Bacteriol. 185, 5697–5705.
Boyanova, L., and Mitov, I. (2010). Geographic map and evolution of primary Helicobacter pylori resistance to antibacterial agents. Expert Rev. Anti Infect. Ther. 8, 59–70.
Braun, M., and Silhavy, T. J. (2002). Imp/OstA is required for cell envelope biogenesis in Escherichia coli. Mol. Microbiol. 45, 1289–1302.
Bulieris, P. V., Behrens, S., Holst, O., and Kleinschmidt, J. H. (2003). Folding and insertion of the outer membrane protein OmpA is assisted by the chaperone Skp and by lipopolysaccharide. J. Biol. Chem. 278, 9092–9099.
Caroff, M., Karibian, D., Cavaillon, J. M., and Haeffner-Cavaillon, N. (2002). Structural and functional analyses of bacterial lipopolysaccharides. Microbes Infect. 4, 915–926.
Censini, S., Lange, C., Xiang, Z., Crabtree, J. E., Ghiara, P., Borodovsky, M., Rappuoli, R., and Covacci, A. (1996). cag, a pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc. Natl. Acad. Sci. U.S.A. 93, 14648–14653.
Chalker, A. F., Minehart, H. W., Hughes, N. J., Koretke, K. K., Lonetto, M. A., Brinkman, K. K., Warren, P. V., Lupas, A., Stanhope, M. J., Brown, J. R., and Hoffman, P. S. (2001). Systematic identification of selective essential genes in Helicobacter pylori by genome prioritization and allelic replacement mutagenesis. J. Bacteriol. 183, 1259–1268.
Chimalakonda, G., Ruiz, N., Chng, S. S., Garner, R. A., Kahne, D., and Silhavy, T. J. (2011). Lipoprotein LptE is required for the assembly of LptD by the beta-barrel assembly machine in the outer membrane of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 108, 2492–2497.
Chiu, H. C., Lin, T. L., and Wang, J. T. (2007). Identification and characterization of an organic solvent tolerance gene in Helicobacter pylori. Helicobacter 12, 74–81.
Chiu, H. C., Lin, T. L., Yang, J. C., and Wang, J. T. (2009). Synergistic effect of imp/ostA and msbA in hydrophobic drug resistance of Helicobacter pylori. BMC Microbiol. 9, 136.
Chng, S. S., Gronenberg, L. S., and Kahne, D. (2010). Proteins required for lipopolysaccharide assembly in Escherichia coli form a transenvelope complex. Biochemistry 49, 4565–4567.
Chng, S. S., Ruiz, N., Chimalakonda, G., Silhavy, T. J., and Kahne, D. (2010). Characterization of the two-protein complex in Escherichia coli responsible for lipopolysaccharide assembly at the outer membrane. Proc. Natl. Acad. Sci. U.S.A. 107, 5363–5368.
Chow, J. C., Young, D. W., Golenbock, D. T., Christ, W. J., and Gusovsky, F. (1999). Toll-like receptor-4 mediates lipopolysaccharide-induced signal transduction. J. Biol. Chem. 274, 10689–10692.
Cosma, C. L., Danese, P. N., Carlson, J. H., Silhavy, T. J., and Snyder, W. B. (1995). Mutational activation of the Cpx signal transduction pathway of Escherichia coli suppresses the toxicity conferred by certain envelope-associated stresses. Mol. Microbiol. 18, 491–505.
Croxen, M. A., Sisson, G., Melano, R., and Hoffman, P. S. (2006). The Helicobacter pylori chemotaxis receptor TlpB (HP0103) is required for pH taxis and for colonization of the gastric mucosa. J. Bacteriol. 188, 2656–2665.
Cummings, L., Riley, L., Black, L., Souvorov, A., Resenchuk, S., Dondoshansky, I., and Tatusova, T. (2002). Genomic BLAST: custom-defined virtual databases for complete and unfinished genomes. FEMS Microbiol. Lett. 216, 133–138.
Czinn, S. J., and Blanchard, T. (2011). Vaccinating against Helicobacter pylori infection. Nat. Rev. Gastroenterol. Hepatol. 8, 133–140.
Dartigalongue, C., Missiakas, D., and Raina, S. (2001). Characterization of the Escherichia coli sigma E regulon. J. Biol. Chem. 276, 20866–20875.
de Cock, H., and Tommassen, J. (1996). Lipopolysaccharides and divalent cations are involved in the formation of an assembly-competent intermediate of outer-membrane protein PhoE of E. coli. EMBO J. 15, 5567–5573.
de Cock, H., Schafer, U., Potgeter, M., Demel, R., Muller, M., and Tommassen, J. (1999). Affinity of the periplasmic chaperone Skp of Escherichia coli for phospholipids, lipopolysaccharides and non-native outer membrane proteins. Role of Skp in the biogenesis of outer membrane protein. Eur. J. Biochem. 259, 96–103.
De Francesco, V., Giorgio, F., Hassan, C., Manes, G., Vannella, L., Panella, C., Ierardi, E., and Zullo, A. (2010). Worldwide H. pylori antibiotic resistance: a systematic review. J. Gastrointestin. Liver Dis. 19, 409–414.
Denoncin, K., Vertommen, D., Paek, E., and Collet, J. F. (2010). The protein-disulfide isomerase DsbC cooperates with SurA and DsbA in the assembly of the essential beta-barrel protein LptD. J. Biol. Chem. 285, 29425–29433.
Di Tommaso, P., Moretti, S., Xenarios, I., Orobitg, M., Montanyola, A., Chang, J. M., Taly, J. F., and Notredame, C. (2011). T-Coffee: a web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res. 39, W13–W17.
Dilks, K., Gimenez, M. I., and Pohlschroder, M. (2005). Genetic and biochemical analysis of the twin-arginine translocation pathway in halophilic archaea. J. Bacteriol. 187, 8104–8113.
Doerrler, W. T. (2006). Lipid trafficking to the outer membrane of Gram-negative bacteria. Mol. Microbiol. 60, 542–552.
Doerrler, W. T., Gibbons, H. S., and Raetz, C. R. (2004). MsbA-dependent translocation of lipids across the inner membrane of Escherichia coli. J. Biol. Chem. 279, 45102–45109.
Doerrler, W. T., and Raetz, C. R. (2005). Loss of outer membrane proteins without inhibition of lipid export in an Escherichia coli YaeT mutant. J. Biol. Chem. 280, 27679–27687.
Doig, P., Exner, M. M., Hancock, R. E., and Trust, T. J. (1995). Isolation and characterization of a conserved porin protein from Helicobacter pylori. J. Bacteriol. 177, 5447–5452.
Eaton, K. A., Suerbaum, S., Josenhans, C., and Krakowka, S. (1996). Colonization of gnotobiotic piglets by Helicobacter pylori deficient in two flagellin genes. Infect. Immun. 64, 2445–2448.
Eckford, P. D., and Sharom, F. J. (2008). Functional characterization of Escherichia coli MsbA: interaction with nucleotides and substrates. J. Biol. Chem. 283, 12840–12850.
Eckford, P. D., and Sharom, F. J. (2010). The reconstituted Escherichia coli MsbA protein displays lipid flippase activity. Biochem. J. 429, 195–203.
Evans, D. G., Evans, D. J. Jr., Moulds, J. J., and Graham, D. Y. (1988). N-acetylneuraminyllactose-binding fibrillar hemagglutinin of Campylobacter pylori: a putative colonization factor antigen. Infect. Immun. 56, 2896–2906.
Evans, D. G., Karjalainen, T. K., Evans, D. J. Jr., Graham, D. Y., and Lee, C. H. (1993). Cloning, nucleotide sequence, and expression of a gene encoding an adhesin subunit protein of Helicobacter pylori. J. Bacteriol. 175, 674–683.
Exner, M. M., Doig, P., Trust, T. J., and Hancock, R. E. (1995). Isolation and characterization of a family of porin proteins from Helicobacter pylori. Infect. Immun. 63, 1567–1572.
Farnbacher, M., Jahns, T., Willrodt, D., Daniel, R., Haas, R., Goesmann, A., Kurtz, S., and Rieder, G. (2010). Sequencing, annotation, and comparative genome analysis of the gerbil-adapted Helicobacter pylori strain B8. BMC Genomics 11, 335.
Fitchen, N., Williams, P., and Hardie, K. R. (2003). Functional complementation of E. colisecD and secG mutants by Helicobacter pylori homologues. FEMS Microbiol. Lett. 229, 57–63.
Flach, C. F., Svensson, N., Blomquist, M., Ekman, A., Raghavan, S., and Holmgren, J. (2011). A truncated form of HpaA is a promising antigen for use in a vaccine against Helicobacter pylori. Vaccine 29, 1235–1241.
Foynes, S., Dorrell, N., Ward, S. J., Stabler, R. A., McColm, A. A., Rycroft, A. N., and Wren, B. W. (2000). Helicobacter pylori possesses two CheY response regulators and a histidine kinase sensor, CheA, which are essential for chemotaxis and colonization of the gastric mucosa. Infect. Immun. 68, 2016–2023.
Freinkman, E., Chng, S. S., and Kahne, D. (2011). The complex that inserts lipopolysaccharide into the bacterial outer membrane forms a two-protein plug-and-barrel. Proc. Natl. Acad. Sci. U.S.A. 108, 2486–2491.
Frenck, R. W. Jr., and Clemens, J. (2003). Helicobacter in the developing world. Microbes Infect. 5, 705–713.
Ghanei, H., Abeyrathne, P. D., and Lam, J. S. (2007). Biochemical characterization of MsbA from Pseudomonas aeruginosa. J. Biol. Chem. 282, 26939–26947.
Hall, M. N., Emr, S. D., and Silhavy, T. J. (1980). Genetic studies on mechanisms of protein localization in Escherichia coli K-12. J. Supramol. Struct. 13, 147–163.
Haque, M., Hirai, Y., Yokota, K., Mori, N., Jahan, I., Ito, H., Hotta, H., Yano, I., Kanemasa, Y., and Oguma, K. (1996). Lipid profile of Helicobacter spp.: presence of cholesteryl glucoside as a characteristic feature. J. Bacteriol. 178, 2065–2070.
Harms, N., Koningstein, G., Dontje, W., Muller, M., Oudega, B., Luirink, J., and de Cock, H. (2001). The early interaction of the outer membrane protein PhoE with the periplasmic chaperone Skp occurs at the cytoplasmic membrane. J. Biol. Chem. 276, 18804–18811.
Hildebrandt, E., and McGee, D. J. (2009). Helicobacter pylori lipopolysaccharide modification, Lewis antigen expression, and gastric colonization are cholesterol-dependent. BMC Microbiol. 9, 258.
Hirai, Y., Haque, M., Yoshida, T., Yokota, K., Yasuda, T., and Oguma, K. (1995). Unique cholesteryl glucosides in Helicobacter pylori: composition and structural analysis. J. Bacteriol. 177, 5327–5333.
Ilver, D., Arnqvist, A., Ogren, J., Frick, I. M., Kersulyte, D., Incecik, E. T., Berg, D. E., Covacci, A., Engstrand, L., and Boren, T. (1998). Helicobacter pylori adhesin binding fucosylated histo-blood group antigens revealed by retagging. Science 279, 373–377.
Karow, M., Fayet, O., Cegielska, A., Ziegelhoffer, T., and Georgopoulos, C. (1991). Isolation and characterization of the Escherichia coli htrB gene, whose product is essential for bacterial viability above 33 degrees C in rich media. J. Bacteriol. 173, 741–750.
Karow, M., and Georgopoulos, C. (1993). The essential Escherichia colimsbA gene, a multicopy suppressor of null mutations in the htrB gene, is related to the universally conserved family of ATP-dependent translocators. Mol. Microbiol. 7, 69–79.
Kawahara, T., Teshima, S., Oka, A., Sugiyama, T., Kishi, K., and Rokutan, K. (2001). Type I Helicobacter pylori lipopolysaccharide stimulates toll-like receptor 4 and activates mitogen oxidase 1 in gastric pit cells. Infect. Immun. 69, 4382–4389.
Keenan, J., Neal, S., Allardyce, R., and Roake, J. (2002). Serum-derived IgG1-mediated immune exclusion as a mechanism of protection against H. pylori infection. Vaccine 20, 2981–2988.
Keenan, J., Oliaro, J., Domigan, N., Potter, H., Aitken, G., Allardyce, R., and Roake, J. (2000). Immune response to an 18-kilodalton outer membrane antigen identifies lipoprotein 20 as a Helicobacter pylori vaccine candidate. Infect. Immun. 68, 3337–3343.
Khamri, W., Moran, A. P., Worku, M. L., Karim, Q. N., Walker, M. M., Annuk, H., Ferris, J. A., Appelmelk, B. J., Eggleton, P., Reid, K. B., and Thursz, M. R. (2005). Variations in Helicobacter pylori lipopolysaccharide to evade the innate immune component surfactant protein D. Infect. Immun. 73, 7677–7686.
Kirkland, T., Viriyakosol, S., Perez-Perez, G. I., and Blaser, M. J. (1997). Helicobacter pylori lipopolysaccharide can activate 70Z/3 cells via CD14. Infect. Immun. 65, 604–608.
Koch, H. G., Hengelage, T., Neumann-Haefelin, C., MacFarlane, J., Hoffschulte, H. K., Schimz, K. L., Mechler, B., and Muller, M. (1999). In vitro studies with purified components reveal signal recognition particle (SRP) and SecA/SecB as constituents of two independent protein-targeting pathways of Escherichia coli. Mol. Biol. Cell 10, 2163–2173.
Krojer, T., Garrido-Franco, M., Huber, R., Ehrmann, M., and Clausen, T. (2002). Crystal structure of DegP (HtrA) reveals a new protease-chaperone machine. Nature 416, 455–459.
Krulwich, T. A., Sachs, G., and Padan, E. (2011). Molecular aspects of bacterial pH sensing and homeostasis. Nat. Rev. Microbiol. 9, 330–343.
Kumamoto, C. A., and Francetic, O. (1993). Highly selective binding of nascent polypeptides by an Escherichia coli chaperone protein in vivo. J. Bacteriol. 175, 2184–2188.
Kumamoto, C. A., and Beckwith, J. (1985). Evidence for specificity at an early step in protein export in Escherichia coli. J. Bacteriol. 163, 267–274.
Lee, H. C., and Bernstein, H. D. (2001). The targeting pathway of Escherichia coli presecretory and integral membrane proteins is specified by the hydrophobicity of the targeting signal. Proc. Natl. Acad. Sci. U.S.A. 98, 3471–3476.
Lee, P. A., Tullman-Ercek, D., and Georgiou, G. (2006). The bacterial twin-arginine translocation pathway. Annu. Rev. Microbiol. 60, 373–395.
Lepper, P. M., Triantafilou, M., Schumann, C., Schneider, E. M., and Triantafilou, K. (2005). Lipopolysaccharides from Helicobacter pylori can act as antagonists for Toll-like receptor 4. Cell Microbiol. 7, 519–528.
Luke, C. J., and Penn, C. W. (1995). Identification of a 29 kDa flagellar sheath protein in Helicobacter pylori using a murine monoclonal antibody. Microbiology 141, 597–604.
Lundstrom, A. M., Blom, K., Sundaeus, V., and Bolin, I. (2001). HpaA shows variable surface localization but the gene expression is similar in different Helicobacter pylori strains. Microb. Pathog. 31, 243–253.
Ma, B., Reynolds, C. M., and Raetz, C. R. (2008). Periplasmic orientation of nascent lipid A in the inner membrane of an Escherichia coli LptA mutant. Proc. Natl. Acad. Sci. U.S.A. 105, 13823–13828.
Mandell, L., Moran, A. P., Cocchiarella, A., Houghton, J., Taylor, N., Fox, J. G., Wang, T. C., and Kurt-Jones, E. A. (2004). Intact Gram-negative Helicobacter pylori, Helicobacter felis, and Helicobacter hepaticus bacteria activate innate immunity via toll-like receptor 2 but not toll-like receptor 4. Infect. Immun. 72, 6446–6454.
Marais, A., Mendz, G. L., Hazell, S. L., and Megraud, F. (1999). Metabolism and genetics of Helicobacter pylori: the genome era. Microbiol. Mol. Biol. Rev. 63, 642–674.
Marshall, B. J., and Warren, J. R. (1984). Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 1, 1311–1315.
Masuda, K., Matsuyama, S., and Tokuda, H. (2002). Elucidation of the function of lipoprotein-sorting signals that determine membrane localization. Proc. Natl. Acad. Sci. U.S.A. 99, 7390–7395.
Matsuyama, S., Tajima, T., and Tokuda, H. (1995). A novel periplasmic carrier protein involved in the sorting and transport of Escherichia coli lipoproteins destined for the outer membrane. EMBO J. 14, 3365–3372.
Matsuyama, S., Yokota, N., and Tokuda, H. (1997). A novel outer membrane lipoprotein, LolB (HemM), involved in the LolA (p20)-dependent localization of lipoproteins to the outer membrane of Escherichia coli. EMBO J. 16, 6947–6955.
Merten, J. A., Schultz, K. M., and Klug, C. S. (2012). Concentration-dependent oligomerization and oligomeric arrangement of LptA. Protein Sci. 21, 211–218.
Misra, R., Peterson, A., Ferenci, T., and Silhavy, T. J. (1991). A genetic approach for analyzing the pathway of LamB assembly into the outer membrane of Escherichia coli. J. Biol. Chem. 266, 13592–13597.
Miyake, K. (2004). Innate recognition of lipopolysaccharide by Toll-like receptor 4-MD-2. Trends Microbiol. 12, 186–192.
Monteiro, M. A., Zheng, P., Ho, B., Yokota, S., Amano, K., Pan, Z., Berg, D. E., Chan, K. H., MacLean, L. L., and Perry, M. B. (2000). Expression of histo-blood group antigens by lipopolysaccharides of Helicobacter pylori strains from Asian hosts: the propensity to express type 1 blood-group antigens. Glycobiology 10, 701–713.
Moran, A. P., Hynes, S. O., and Heneghan, M. A. (1999). Mimicry of blood group antigen A by Helicobacter mustelae and H. pylori. Gastroenterology 116, 504–505.
Moran, A. P., Lindner, B., and Walsh, E. J. (1997). Structural characterization of the lipid A component of Helicobacter pylori rough- and smooth-form lipopolysaccharides. J. Bacteriol. 179, 6453–6463.
Moran, A. P., Shiberu, B., Ferris, J. A., Knirel, Y. A., Senchenkova, S. N., Perepelov, A. V., Jansson, P. E., and Goldberg, J. B. (2004). Role of Helicobacter pylorirfaJ genes (HP0159 and HP1416) in lipopolysaccharide synthesis. FEMS Microbiol. Lett. 241, 57–65.
Narita, S., Tanaka, K., Matsuyama, S., and Tokuda, H. (2002). Disruption of lolCDE, encoding an ATP-binding cassette transporter, is lethal for Escherichia coli and prevents release of lipoproteins from the inner membrane. J. Bacteriol. 184, 1417–1422.
Narita, S., and Tokuda, H. (2009). Biochemical characterization of an ABC transporter LptBFGC complex required for the outer membrane sorting of lipopolysaccharides. FEBS Lett. 583, 2160–2164.
Nikaido, H. (2003). Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 67, 593–656.
Notredame, C., and Abergel, C. (2003). “Using multiple alignment methods to assess the quality of genomic data analysis,” in Bioinformatics and Genomes: Current Perspectives, ed M. Andrade (Wymondham, UK: Horizon Scientific Press), 30–50.
Ogawa, T., Asai, Y., Sakai, Y., Oikawa, M., Fukase, K., Suda, Y., Kusumoto, S., and Tamura, T. (2003a). Endotoxic and immunobiological activities of a chemically synthesized lipid A of Helicobacter pylori strain 206–1. FEMS Immunol. Med. Microbiol. 36, 1–7.
Ogawa, H., Rafiee, P., Heidemann, J., Fisher, P. J., Johnson, N. A., Otterson, M. F., Kalyanaraman, B., Pritchard, K. A. J., and Binion, D. G. (2003b). Mechanisms of endotoxin tolerance in human intestinal microvascular endothelial cells. J. Immunol. 170, 5956–5964.
Ogawa, T., Suda, Y., Kashihara, W., Hayashi, T., Shimoyama, T., Kusumoto, S., and Tamura, T. (1997). Immunobiological activities of chemically defined lipid A from Helicobacter pylori LPS in comparison with Porphyromonas gingivalis lipid A and Escherichia coli-type synthetic lipid A (compound 506). Vaccine 15, 1598–1605.
Onufryk, C., Crouch, M. L., Fang, F. C., and Gross, C. A. (2005). Characterization of six lipoproteins in the sigma E regulon. J. Bacteriol. 187, 4552–4561.
O'Toole, P. W., and Clyne, M. (2001). “Cell envelope,” in Helicobacter pylori: Physiology and Genetics, eds H. L. T. Mobley, G. L. Mendz, and S. L. Hazell (Washington, DC: ASM Press), 69–80.
O'Toole, P. W., Janzon, L., Doig, P., Huang, J., Kostrzynska, M., and Trust, T. J. (1995). The putative neuraminyllactose-binding hemagglutinin HpaA of Helicobacter pylori CCUG 17874 is a lipoprotein. J. Bacteriol. 177, 6049–6057.
Ottemann, K. M., and Lowenthal, A. C. (2002). Helicobacter pylori uses motility for initial colonization and to attain robust infection. Infect. Immun. 70, 1984–1990.
Peck, B., Ortkamp, M., Nau, U., Niederweis, M., Hundt, E., and Knapp, B. (2001). Characterization of four members of a multigene family encoding outer membrane proteins of Helicobacter pylori and their potential for vaccination. Microbes Infect. 3, 171–179.
Peng, J., and Xu, J. (2011). RaptorX: exploiting structure information for protein alignment by statistical inference. Proteins (Suppl. 10), 161–171.
Poirot, O., O'Toole, E., and Notredame, C. (2003). Tcoffee@igs: a web server for computing, evaluating and combining multiple sequence alignments. Nucleic Acids Res. 31, 3503–3506.
Polissi, A., and Georgopoulos, C. (1996). Mutational analysis and properties of the msbA gene of Escherichia coli, coding for an essential ABC family transporter. Mol. Microbiol. 20, 1221–1233.
Qu, J., Behrens-Kneip, S., Holst, O., and Kleinschmidt, J. H. (2009). Binding regions of outer membrane protein A in complexes with the periplasmic chaperone Skp. A site-directed fluorescence study. Biochemistry 48, 4926–4936.
Raetz, C. R., and Whitfield, C. (2002). Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 71, 635–700.
Raivio, T. L., and Silhavy, T. J. (1997). Transduction of envelope stress in Escherichia coli by the Cpx two-component system. J. Bacteriol. 179, 7724–7733.
Rigel, N. W., Schwalm, J., Ricci, D. P., and Silhavy, T. J. (2012). BamE modulates the Escherichia coli beta-barrel assembly machine component BamA. J. Bacteriol. 194, 1002–1008.
Rizzitello, A. E., Harper, J. R., and Silhavy, T. J. (2001). Genetic evidence for parallel pathways of chaperone activity in the periplasm of Escherichia coli. J. Bacteriol. 183, 6794–6800.
Rouviere, P. E., and Gross, C. A. (1996). SurA, a periplasmic protein with peptidyl-prolyl isomerase activity, participates in the assembly of outer membrane porins. Genes Dev. 10, 3170–3182.
Ruiz, N., Chng, S. S., Hiniker, A., Kahne, D., and Silhavy, T. J. (2010). Nonconsecutive disulfide bond formation in an essential integral outer membrane protein. Proc. Natl. Acad. Sci. U.S.A. 107, 12245–12250.
Ruiz, N., Falcone, B., Kahne, D., and Silhavy, T. J. (2005). Chemical conditionality: a genetic strategy to probe organelle assembly. Cell 121, 307–317.
Ruiz, N., Kahne, D., and Silhavy, T. J. (2006). Advances in understanding bacterial outer-membrane biogenesis. Nat. Rev. Microbiol. 4, 57–66.
Salama, N. R., Shepherd, B., and Falkow, S. (2004). Global transposon mutagenesis and essential gene analysis of Helicobacter pylori. J. Bacteriol. 186, 7926–7935.
Salaun, L., Ayraud, S., and Saunders, N. J. (2005). Phase variation mediated niche adaptation during prolonged experimental murine infection with Helicobacter pylori. Microbiology 151, 917–923.
Salaun, L., and Saunders, N. J. (2006). Population-associated differences between the phase variable LPS biosynthetic genes of Helicobacter pylori. BMC Microbiol. 6, 79.
Samatey, F. A., Imada, K., Nagashima, S., Vonderviszt, F., Kumasaka, T., Yamamoto, M., and Namba, K. (2001). Structure of the bacterial flagellar protofilament and implications for a switch for supercoiling. Nature 410, 331–337.
Sampson, B. A., Misra, R., and Benson, S. A. (1989). Identification and characterization of a new gene of Escherichia coli K-12 involved in outer membrane permeability. Genetics 122, 491–501.
Schmausser, B., Andrulis, M., Endrich, S., Lee, S. K., Josenhans, C., Muller-Hermelink, H. K., and Eck, M. (2004). Expression and subcellular distribution of toll-like receptors TLR4, TLR5 and TLR9 on the gastric epithelium in Helicobacter pylori infection. Clin. Exp. Immunol. 136, 521–526.
Scott, D. R., Marcus, E. A., Weeks, D. L., Lee, A., Melchers, K., and Sachs, G. (2000). Expression of the Helicobacter pyloriureI gene is required for acidic pH activation of cytoplasmic urease. Infect. Immun. 68, 470–477.
Senkovich, O. A., Yin, J., Ekshyyan, V., Conant, C., Traylor, J., Adegboyega, P., McGee, D. J., Rhoads, R. E., Slepenkov, S., and Testerman, T. L. (2011). Helicobacter pylori AlpA and AlpB bind host laminin and influence gastric inflammation in gerbils. Infect. Immun. 79, 3106–3116.
Sklar, J. G., Wu, T., Kahne, D., and Silhavy, T. J. (2007). Defining the roles of the periplasmic chaperones SurA, Skp, and DegP in Escherichia coli. Genes Dev. 21, 2473–2484.
Smith, M. F. Jr., Mitchell, A., Li, G., Ding, S., Fitzmaurice, A. M., Ryan, K., Crowe, S., and Goldberg, J. B. (2003). Toll-like receptor (TLR) 2 and TLR5, but not TLR4, are required for Helicobacter pylori-induced NF-kappa B activation and chemokine expression by epithelial cells. J. Biol. Chem. 278, 32552–32560.
Sperandeo, P., Cescutti, R., Villa, R., Di Benedetto, C., Candia, D., Dehò, G., and Polissi, A. (2007). Characterization of lptA and lptB, two essential genes implicated in lipopolysaccharide transport to the outer membrane of Escherichia coli. J. Bacteriol. 189, 244–253.
Sperandeo, P., Deho, G., and Polissi, A. (2009). The lipopolysaccharide transport system of Gram-negative bacteria. Biochim. Biophys. Acta 1791, 594–602.
Sperandeo, P., Villa, R., Martorana, A. M., Samalikova, M., Grandori, R., Deho, G., and Polissi, A. (2011). New insights into the Lpt machinery for lipopolysaccharide transport to the cell surface: LptA-LptC interaction and LptA stability as sensors of a properly assembled transenvelope complex. J. Bacteriol. 193, 1042–1053.
Stead, C. M., Beasley, A., Cotter, R. J., and Trent, M. S. (2008). Deciphering the unusual acylation pattern of Helicobacter pylori lipid A. J. Bacteriol. 190, 7012–7021.
Stead, C., Tran, A., Ferguson, D. J., McGrath, S., Cotter, R., and Trent, S. (2005). A novel 3-deoxy-D-manno-octulosonic acid (Kdo) hydrolase that removes the outer Kdo sugar of Helicobacter pylori lipopolysaccharide. J. Bacteriol. 187, 3374–3383.
Stenberg, F., Chovanec, P., Maslen, S. L., Robinson, C. V., Ilag, L. L., von Heijne, G., and Daley, D. O. (2005). Protein complexes of the Escherichia coli cell envelope. J. Biol. Chem. 280, 34409–34419.
Stingl, K., Muller, S., Scheidgen-Kleyboldt, G., Clausen, M., and Maier, B. (2010). Composite system mediates two-step DNA uptake into Helicobacter pylori. Proc. Natl. Acad. Sci. U.S.A. 107, 1184–1189.
Su, B., Ceponis, P. J., Lebel, S., Huynh, H., and Sherman, P. M. (2003). Helicobacter pylori activates Toll-like receptor 4 expression in gastrointestinal epithelial cells. Infect. Immun. 71, 3496–3502.
Suda, Y., Ogawa, T., Kashihara, W., Oikawa, M., Shimoyama, T., Hayashi, T., Tamura, T., and Kusumoto, S. (1997). Chemical structure of lipid A from Helicobacter pylori strain 206–1 lipopolysaccharide. J. Biochem. 121, 1129–1133.
Suits, M. D., Sperandeo, P., Deho, G., Polissi, A., and Jia, Z. (2008). Novel structure of the conserved gram-negative lipopolysaccharide transport protein A and mutagenesis analysis. J. Mol. Biol. 380, 476–488.
Takai, K., Campbell, B. J., Cary, S. C., Suzuki, M., Oida, H., Nunoura, T., Hirayama, H., Nakagawa, S., Suzuki, Y., Inagaki, F., and Horikoshi, K. (2005). Enzymatic and genetic characterization of carbon and energy metabolisms by deep-sea hydrothermal chemolithoautotrophic isolates of Epsilonproteobacteria. Appl. Environ. Microbiol. 71, 7310–7320.
Takeda, K., Miyatake, H., Yokota, N., Matsuyama, S., Tokuda, H., and Miki, K. (2003). Crystal structures of bacterial lipoprotein localization factors, LolA and LolB. EMBO J. 22, 3199–3209.
Tefsen, B., Geurtsen, J., Beckers, F., Tommassen, J., and de Cock, H. (2005). Lipopolysaccharide transport to the bacterial outer membrane in spheroplasts. J. Biol. Chem. 280, 4504–4509.
Tomb, J. F., White, O., Kerlavage, A. R., Clayton, R. A., Sutton, G. G., Fleischmann, R. D., Ketchum, K. A., Klenk, H. P., Gill, S., Dougherty, B. A., Nelson, K., Quackenbush, J., Zhou, L., Kirkness, E. F., Peterson, S., Loftus, B., Richardson, D., Dodson, R., Khalak, H. G., Glodek, A., McKenney, K., Fitzegerald, L. M., Lee, N., Adams, M. D., Hickey, E. K., Berg, D. E., Gocayne, J. D., Utterback, T. R., Peterson, J. D., Kelley, J. M., Cotton, M. D., Weidman, J. M., Fujii, C., Bowman, C., Watthey, L., Wallin, E., Hayes, W. S., Borodovsky, M., Karp, P. D., Smith, H. O., Fraser, C. M., and Venter, J. C. (1997). The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388, 539–547.
Tomkiewicz, D., Nouwen, N., van Leeuwen, R., Tans, S., and Driessen, A. J. (2006). SecA supports a constant rate of preprotein translocation. J. Biol. Chem. 281, 15709–15713.
Tran, A. X., Dong, C., and Whitfield, C. (2010). Structure and functional analysis of LptC, a conserved membrane protein involved in the lipopolysaccharide export pathway in Escherichia coli. J. Biol. Chem. 285, 33529–33539.
Tran, A. X., Stead, C. M., and Trent, M. S. (2005). Remodeling of Helicobacter pylori lipopolysaccharide. J. Endotoxin Res. 11, 161–166.
Tran, A. X., Trent, M. S., and Whitfield, C. (2008). The LptA protein of Escherichia coli is a periplasmic lipid A-binding protein involved in the lipopolysaccharide export pathway. J. Biol. Chem. 283, 20342–20349.
Valent, Q. A., Scotti, P. A., High, S., de Gier, J. W., von Heijne, G., Lentzen, G., Wintermeyer, W., Oudega, B., and Luirink, J. (1998). The Escherichia coli SRP and SecB targeting pathways converge at the translocon. EMBO J. 17, 2504–2512.
Van den Berg, B., Clemons, W. M. Jr., Collinson, I., Modis, Y., Hartmann, E., Harrison, S. C., and Rapoport, T. A. (2004). X-ray structure of a protein-conducting channel. Nature 427, 36–44.
Vertommen, D., Ruiz, N., Leverrier, P., Silhavy, T. J., and Collet, J. F. (2009). Characterization of the role of the Escherichia coli periplasmic chaperone SurA using differential proteomics. Proteomics 9, 2432–2443.
Volokhina, E. B., Grijpstra, J., Stork, M., Schilders, I., Tommassen, J., and Bos, M. P. (2011). Role of the periplasmic chaperones Skp, SurA, and DegQ in outer membrane protein biogenesis in Neisseria meningitidis. J. Bacteriol. 193, 1612–1621.
Voulhoux, R., Ball, G., Ize, B., Vasil, M. L., Lazdunski, A., Wu, L. F., and Filloux, A. (2001). Involvement of the twin-arginine translocation system in protein secretion via the type II pathway. EMBO J. 20, 6735–6741.
Voulhoux, R., Bos, M. P., Geurtsen, J., Mols, M., and Tommassen, J. (2003). Role of a highly conserved bacterial protein in outer membrane protein assembly. Science 299, 262–265.
Voulhoux, R., and Tommassen, J. (2004). Omp85, an evolutionarily conserved bacterial protein involved in outer-membrane-protein assembly. Res. Microbiol. 155, 129–135.
Walton, T. A., and Sousa, M. C. (2004). Crystal structure of Skp, a prefoldin-like chaperone that protects soluble and membrane proteins from aggregation. Mol. Cell 15, 367–374.
Wang, G., Ge, Z., Rasko, D. A., and Taylor, D. E. (2000). Lewis antigens in Helicobacter pylori: biosynthesis and phase variation. Mol. Microbiol. 36, 1187–1196.
Ward, A., Reyes, C. L., Yu, J., Roth, C. B., and Chang, G. (2007). Flexibility in the ABC transporter MsbA: alternating access with a twist. Proc. Natl. Acad. Sci. U.S.A. 104, 19005–19010.
Weeks, D. L., Eskandari, S., Scott, D. R., and Sachs, G. (2000). A H+-gated urea channel: the link between Helicobacter pylori urease and gastric colonization. Science 287, 482–485.
Werner, J., and Misra, R. (2005). YaeT (Omp85) affects the assembly of lipid-dependent and lipid-independent outer membrane proteins of Escherichia coli. Mol. Microbiol. 57, 1450–1459.
Wu, T., Malinverni, J., Ruiz, N., Kim, S., Silhavy, T. J., and Kahne, D. (2005). Identification of a multicomponent complex required for outer membrane biogenesis in Escherichia coli. Cell 121, 235–245.
Wu, T., McCandlish, A. C., Gronenberg, L. S., Chng, S. S., Silhavy, T. J., and Kahne, D. (2006). Identification of a protein complex that assembles lipopolysaccharide in the outer membrane of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 103, 11754–11759.
Yakushi, T., Masuda, K., Narita, S., Matsuyama, S., and Tokuda, H. (2000). A new ABC transporter mediating the detachment of lipid-modified proteins from membranes. Nat. Cell Biol. 2, 212–218.
Yamane, K., Bunai, K., and Kakeshita, H. (2004). Protein traffic for secretion and related machinery of Bacillus subtilis. Biosci. Biotechnol. Biochem. 68, 2007–2023.
Yokota, S., Ohnishi, T., Muroi, M., Tanamoto, K., Fujii, N., and Amano, K. (2007). Highly-purified Helicobacter pylori LPS preparations induce weak inflammatory reactions and utilize Toll-like receptor 2 complex but not Toll-like receptor 4 complex. FEMS Immunol. Med. Microbiol. 51, 140–148.
Yokota, S., Okabayashi, T., Rehli, M., Fujii, N., and Amano, K. (2010). Helicobacter pylori lipopolysaccharides upregulate toll-like receptor 4 expression and proliferation of gastric epithelial cells via the MEK1/2-ERK1/2 mitogen-activated protein kinase pathway. Infect. Immun. 78, 468–476.
Zhang, X., Kimura, Y., Fang, C., Zhou, L., Sfyroera, G., Lambris, J. D., Wetsel, R. A., Miwa, T., and Song, W. C. (2007). Regulation of Toll-like receptor-mediated inflammatory response by complement in vivo. Blood 110, 228–236.
Keywords: membrane biogenesis, H. pylori, Bam complex, Lol complex, Lpt complex
Citation: Liechti G and Goldberg JB (2012) Outer membrane biogenesis in Escherichia coli, Neisseria meningitidis, and Helicobacter pylori: paradigm deviations in H. pylori. Front. Cell. Inf. Microbio. 2:29. doi: 10.3389/fcimb.2012.00029
Received: 15 December 2011; Accepted: 28 February 2012;
Published online: 11 April 2012.
Edited by:
D. Scott Merrell, Uniformed Services University, USAReviewed by:
David J. McGee, LSU Health Sciences Center - Shreveport, USACopyright: © 2012 Liechti and Goldberg. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Joanna B. Goldberg, Department of Microbiology, Immunology, and Cancer Biology, University of Virginia Health System, 1300 Jefferson Park Avenue, 7230 Jordan Hall, Charlottesville, VA 22908-0734, USA. e-mail:amJnMmJAdmlyZ2luaWEuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.