
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. , 04 April 2025
Sec. Medicinal and Pharmaceutical Chemistry
Volume 13 - 2025 | https://doi.org/10.3389/fchem.2025.1571646
This article is part of the Research Topic Recent Advances in Synthetic Organic Chemistry at the Biomedical Interface: Honoring Professor Iwao Ojima on the Occasion of his 80th Birthday View all 8 articles
 Hamada Hashem1
Hamada Hashem1 Shadwa Abdelfattah2
Shadwa Abdelfattah2 Hesham M. Hassan3
Hesham M. Hassan3 Ahmed Al-Emam3
Ahmed Al-Emam3 Mohammed Alqarni4
Mohammed Alqarni4 Ghallab Alotaibi5Ibrahim Taha Radwan6Kirandeep Kaur7
Ghallab Alotaibi5Ibrahim Taha Radwan6Kirandeep Kaur7 Devendra Pratap Rao8*
Devendra Pratap Rao8* Stefan Bräse9*Abdullah Alkhammash5
Stefan Bräse9*Abdullah Alkhammash5Introduction: Carbonic anhydrase IX (CA IX) is a tumor-associated enzyme involved in cancer progression and survival. Targeting CA IX with selective inhibitors like SLC-0111 has shown therapeutic potential. This study aimed to develop a novel 4-pyridyl analog (Pyr) of SLC-0111 with enhanced anticancer activity.
Methods: Pyr was synthesized using a tail-based design and characterized by NMR. Its cytotoxicity was tested against cancer and normal cell lines. CA inhibition, cell cycle effects, apoptosis induction, and protein expression changes were evaluated. Molecular docking and ADMET predictions assessed binding and drug-like properties.
Results and Discussion: Pyr showed selective cytotoxicity toward cancer cells and potent CA IX inhibition. It induced G0/G1 arrest, apoptosis, and modulated p53, Bax, and Bcl-2 levels. Docking confirmed strong CA IX binding, and ADMET analysis indicated good oral bioavailability. These results support Pyr as a promising anticancer candidate.
Cancer remains one of the most challenging health conditions worldwide, characterized by uncontrolled cell growth, invasion into surrounding tissues, and potential metastasis to distant organs (Wikipedia, 2024). It affects millions of lives annually, highlighting the urgent need for effective therapeutic strategies (WHO, 2024). Advances in cancer biology have paved the way for developing anticancer agents targeting various cellular and molecular mechanisms involved in tumor initiation, progression, and metastasis (Doostmohammadi et al., 2024). Current treatments, including chemotherapy, targeted therapy, and immunotherapy, often aim to disrupt specific pathways critical to cancer survival and growth (Yang et al., 2024; P. Inc, 2024; Tilsed et al., 2022). Despite significant progress, the emergence of drug resistance and adverse side effects highlight the need for novel therapeutic targets and agents with improved efficacy and safety profiles (Targeted Cancer Therapy, 2024; Tian et al., 2024).
Among the emerging molecular targets in cancer therapy, carbonic anhydrases (CAs) have gained attention due to their critical role in maintaining the pH balance within tumor cells and their microenvironment (Pastorekova and Gillies, 2019). Carbonic anhydrases are zinc metalloenzymes with 15 known isoforms in humans, of which CA IX and CA XII are overexpressed in hypoxic tumor cells (Baranauskienė et al., 2019). These isoforms play a pivotal role in regulating intracellular and extracellular pH by catalyzing the reversible hydration of carbon dioxide to bicarbonate and protons, enabling cancer cells to thrive in acidic and hypoxic conditions (Sedlakova et al., 2014). By supporting an acidic extracellular environment, CA IX and CA XII facilitate tumor progression, invasion, and immune evasion. Consequently, carbonic anhydrase inhibitors (CAIs) have emerged as promising anticancer agents, selectively targeting tumor-associated isoforms to disrupt these critical processes (Merkx et al., 2021). Recent studies have demonstrated the potential of CAI to inhibit tumor growth, enhance the effectiveness of conventional chemotherapeutics, and overcome resistance mechanisms (Kalinin et al., 2021).
SLC-0111, a ureido-substituted benzenesulfonamide, is a potent inhibitor of CA IX, disrupting pH regulation in tumor cells and impairing their survival and invasiveness (MedChemExpress, 2024). Preclinical studies have demonstrated that SLC-0111 enhances the efficacy of chemotherapeutic agents such as cisplatin in head and neck squamous carcinoma models, leading to reduced tumor growth and invasion (Sarnella et al., 2022). Additionally, SLC-0111 has progressed to Phase I clinical trials, where it was evaluated for safety and tolerability in patients with advanced solid tumors (McDonald et al., 2020).
Building upon the success of SLC-0111, researchers have developed various analogs to enhance selectivity and potency against tumor-associated carbonic anhydrase (CA) isoforms (Angeli et al., 2023). For instance, benzofuran-based derivatives have been synthesized, exhibiting selective inhibition of CA IX and CA XII (Figure 1) and demonstrating promising anticancer activity, with compound 1 displaying significant inhibitory activity against CA IX with a Ki value of 7.1 nM (Shaldam et al., 2021). Similarly, thiazole and thiadiazole-based analogs have shown substantial inhibitory activity against tumor-associated CA isoforms with thiadiazole analog 2 exhibited remarkable Ki values of 7.9 and 9.9 nM for CA IX and CA XII inhibition, respectively (Abo-Ashour et al., 2019). These derivatives have effectively suppressed tumor cell proliferation and migration, underscoring their promise as therapeutic agents in cancer treatment by targeting carbonic anhydrases.
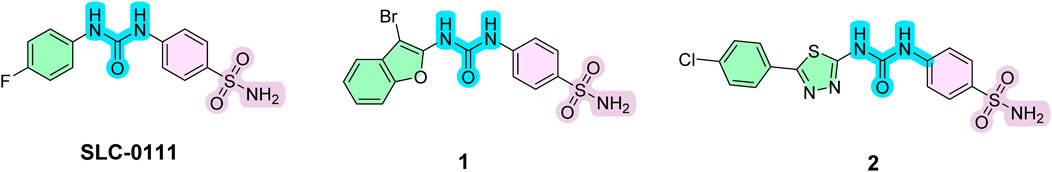
Figure 1. Examples of important inhibitors of tumor-associated carbonic anhydrase isoforms.
In parallel with the development of small molecule inhibitors targeting CA IX, immunotherapeutic strategies have also emerged as promising approaches for the treatment of CA IX-expressing tumors. Notably, bi-specific adapter molecules capable of engaging universal chimeric antigen receptor T (CAR-T) cells have been designed to facilitate the selective targeting of CA IX-positive tumor cells (Low et al., 2024). These adapters act as molecular bridges that redirect CAR-T cells to tumor sites while providing an additional layer of control, potentially reducing off-tumor toxicity and enhancing safety profiles. By enabling the specific recognition and elimination of hypoxic, CA IX-expressing tumor cells, these bi-specific adapters offer a novel immunotherapeutic avenue that could complement the effects of CA IX inhibitors and broaden the therapeutic landscape for solid tumors characterized by hypoxia and acidosis.
Inspired by these findings on SLC-0111 and its related analogs, we employed a tail approach to design and synthesize a new analog of SLC-0111, incorporating a 4-pyridyl moiety linked with sulfanilamide as the zinc-binding group. This analog was evaluated for its anticancer properties and inhibitory activity against four CA isoforms: CA I, II, IX, and XII. The design retained the zinc-binding sulfonamide motif and urea linker from SLC-0111 while introducing a 4-pyridyl moiety as a tail to enhance interactions with the hydrophobic region of the CA active site (Figure 2).
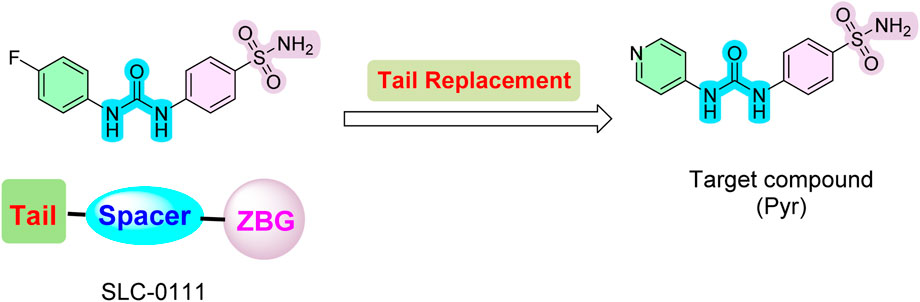
Figure 2. Design of novel SLC-0111 analog (Pyr).
Scheme 1 illustrates the stepwise synthesis of a sulfonamide-based urea derivative starting from pyridine-4-carboxylic acid (isonicotinic acid). The synthesis begins with the Fischer esterification of isonicotinic acid, converting its carboxylic acid group into an ethyl ester using absolute ethanol in the presence of concentrated sulfuric acid as a catalyst. In the second step, the ethyl ester reacts with hydrazine hydrate, replacing the ethoxy group with a hydrazino group to form the corresponding hydrazide (Isoniazid). The hydrazide is then subjected to diazotization using sodium nitrite in the presence of hydrochloric acid, leading to the formation of the acyl azide intermediate. This reactive intermediate is key to the next transformation, the Curtius rearrangement, which is induced by heating the acyl azide in toluene. During this rearrangement, the azide undergoes nitrogen gas evolution (N2) and acyl migration, forming an isocyanate intermediate. The highly reactive isocyanate is then reacted with sulfanilamide under heating in toluene to yield the final sulfonamide-based urea derivative Pyr. This step involves a nucleophilic attack by the amine group of sulfanilamide on the electrophilic carbon of the isocyanate group, forming the desired product.
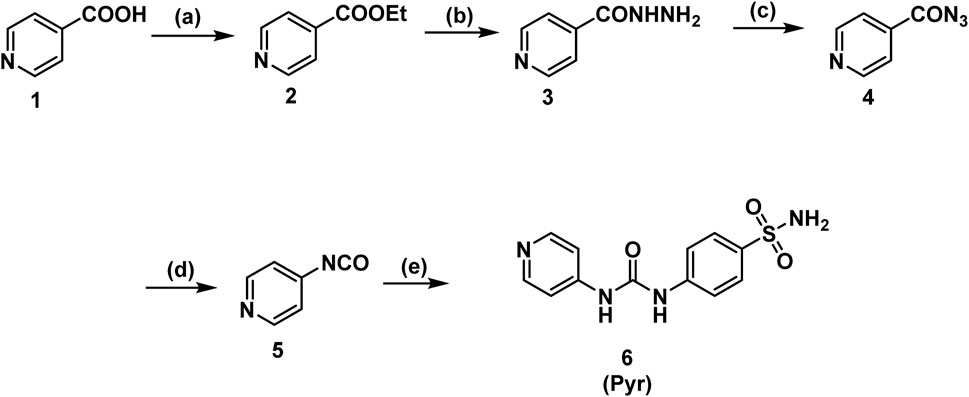
Scheme 1. Synthesis of the target compound (Pyr). Reagents and Conditions: (a) EtOH, H2SO4, reflux, 8 h; (b) hydrazine hydrate, EtOH, reflux, 18 h; (c) NaNO2, HCl, 0°C, 1 h; (d) toluene, reflux, 1 h; (e) sulfanilamide, toluene, reflux, 2h.
The final compound, Pyr, was characterized using 1H-NMR, 13C-NMR, and elemental analysis. The 1H-NMR spectrum confirms the structure of the compound with distinct peaks corresponding to its key protons. The urea moiety is represented by two singlets at 8.97 ppm and 9.10 ppm, confirming the presence of the urea hydrogens. The protons of the pyridine ring appear as two doublets at 8.34 ppm and 7.44 ppm, consistent with the expected splitting for this system. Similarly, the protons of the para-substituted benzene ring are represented by two doublets at 7.62 ppm and 7.73 ppm. The NH2 group of the sulfonamide appears as a singlet at 7.24 ppm. In the 13C-NMR spectrum, eight peaks are observed, consistent with the distinct carbon atoms in the molecule. Four peaks are assigned to the benzene ring carbons, while three peaks correspond to the pyridine ring carbons. The carbonyl carbon of the urea group is confirmed by a characteristic peak at 150.40 ppm, supporting the successful formation of the urea moiety. These findings from the NMR data align well with the proposed structure of the target compound.
Many sulfonamide-based compounds have shown promising cytotoxic activities (Güleç et al., 2024a; Güleç et al., 2024b) against various cancer cell lines. Accordingly, the cytotoxic activity of Pyr was evaluated against HT-29, MCF7, and PC3 cancer cell lines, as well as the normal CCD-986sk cell line. Based on the cell viability assay results shown in Figure 3, Pyr exhibited moderate cytotoxicity against the tested cancer cell lines (HT-29, MCF7, and PC3) with IC50 values of 27.74, 11.20, and 8.36 µg/mL, respectively, while showing significantly lower toxicity toward the normal cell line CCD-986sk (IC50 = 50.32 µg/mL), indicating good selectivity. Compared to the reference SLC-0111, Pyr demonstrated superior cytotoxicity against the MCF7 cell line (IC50 = 11.20 µg/mL vs. 18.15 µg/mL for SLC-0111) and comparable activity against the PC3 cell line (IC50 = 8.36 µg/mL vs. 8.71 µg/mL), whereas SLC-0111 exhibited better potency against the HT-29 cell line (IC50 = 13.53 µg/mL vs. 27.74 µg/mL for Pyr). Regarding safety, Pyr also showed slightly lower toxicity toward normal CCD-986sk cells (IC50 = 50.32 µg/mL) compared to SLC-0111 (IC50 = 45.70 µg/mL), highlighting its favorable selectivity profile. In contrast, the reference drug Staurosporine is significantly more potent across all cell lines, with much lower IC50 values for HT-29 (4.07 µg/mL), MCF7 (2.93 µg/mL), and PC3 (1.20 µg/mL). Still, it also exhibits considerable toxicity to normal cells (IC50 = 18.66 µg/mL), reflecting poor selectivity. These findings suggest that while Staurosporine is highly effective, its lack of selectivity limits its therapeutic potential. By contrast, with its selective cytotoxicity, superior or comparable efficacy to SLC-0111 in certain cancer cell lines, and reduced toxicity to normal cells, Pyr presents a more favorable profile for further development as an anti-cancer agent.
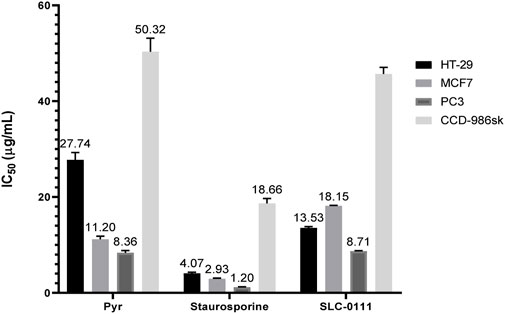
Figure 3. IC50 values (µg/mL) for Pyr and Staurosporine against HT-29, MCF7, PC3 cancer cells, and CCD-986sk normal cells. Pyr exhibited moderate cytotoxicity with better selectivity for cancer cells, while Staurosporine showed higher potency but less selectivity. Data are mean ± SEM.
Sulfonamides are well-known inhibitors of carbonic anhydrase (CA) enzymes (Buza et al., 2024; Buza et al., 2023; Kakakhan et al., 2023) and have been widely studied for their potential therapeutic applications, particularly in cancer and other diseases. Given their significance as CA inhibitors, we evaluated Pyr’s inhibitory activity against various human carbonic anhydrase isoforms, specifically CA I, CA II, CA IX, and CA XII. The results in Table 1 present the IC50 values (µg/mL) of Pyr and the reference drugs acetazolamide (AAZ) and SLC-0111 against carbonic anhydrase (CA) isoforms I, II, IX, and XII. Pyr showed moderate inhibition of CA I (20.29 ± 0.92 µg/mL), CA II (0.569 ± 0.03 µg/mL) and CA XII (2.97 ± 0.17 µg/mL) with higher potency against the cancer-related isoform CA IX (0.399 ± 0.02 µg/mL). In comparison, AAZ, a standard carbonic anhydrase inhibitor, exhibited significantly lower IC50 values across all isoforms, with exceptional potency against CA II (0.153 ± 0.01 µg/mL), CA IX (0.105 ± 0.01 µg/mL), and CA XII (0.029 ± 0.001 µg/mL). Similarly, SLC-0111, a selective inhibitor of tumor-associated carbonic anhydrases, showed strong inhibition of CA IX (0.048 ± 0.006 µg/mL) and CA XII (0.096 ± 0.008 µg/mL). These results suggest that while Pyr is less potent than AAZ and SLC-0111 overall, it demonstrated selective inhibition of CA IX, a tumor-associated isoform, indicating its potential as a targeted inhibitor for cancer-related applications.
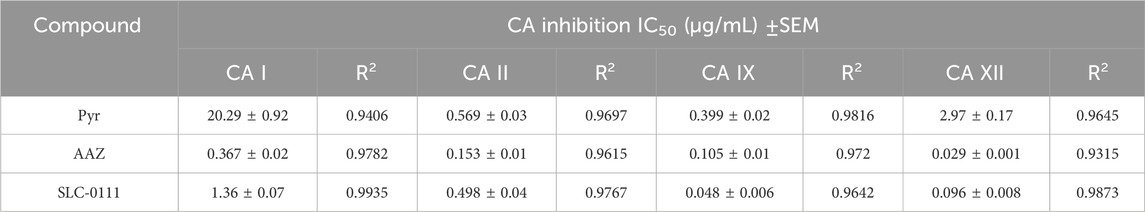
Table 1. IC50 values (µg/mL) of Pyr and acetazolamide (AAZ) against CA isoforms I, II, IX, and XII. Data are expressed as mean ± SEM.
The data in Table 2 and Figure 4 illustrate the impact of Pyr on the cell cycle distribution of PC3 cells compared to untreated cells (treated with DMSO as a control). Treatment of PC3 cells with Pyr significantly increased the percentage of cells in the G0-G1 phase (76.59%) compared to the control (53.84%), indicating a G0-G1 phase arrest. Correspondingly, the percentage of cells in the S phase decreased notably in Pyr-treated cells (16.23%) compared to the control (28.66%), suggesting inhibition of DNA synthesis and progression to the S phase. Furthermore, the percentage of cells in the G2/M phase was reduced in Pyr-treated cells (7.18%) compared to the control (17.50%), indicating impaired transition to mitosis. These results suggest that Pyr effectively induces cell cycle arrest at the G0-G1 phase in PC3 cells, thereby inhibiting cell proliferation.
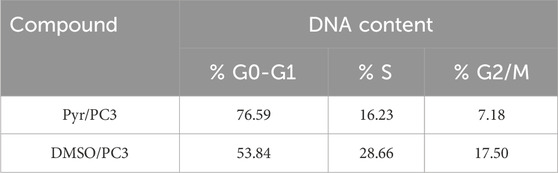
Table 2. Effect of Pyr derivative on PC3 cell cycle distribution compared to DMSO-treated cells.
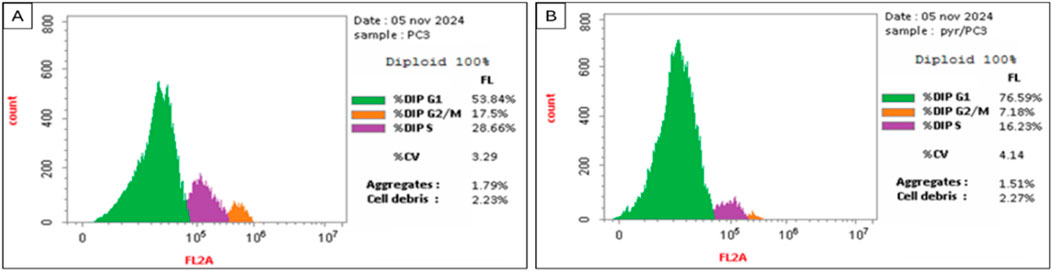
Figure 4. Effect of DMSO (Control, (A)) and Pyr (IC50, 8.63 μg/mL, (B)) on cell accumulation across various phases of the cell cycle of prostate PC3 cancer cells.
The data in Table 3 and Figure 5 highlight the effect of Pyr on apoptosis and necrosis in PC3 cells compared to untreated cells (DMSO-treated cells). Treatment of PC3 cells significantly increased total apoptosis to 29.46% compared to 2.39% in the untreated cells. This increase is driven by both early apoptosis (9.33%) and late apoptosis (15.74%), indicating that Pyr effectively triggers the apoptotic pathway in PC3 cells. Necrosis levels were also slightly elevated in Pyr-treated cells (4.39%) compared to the control (1.82%), though apoptosis remains the predominant mode of cell death. These findings suggest that Pyr induces substantial apoptotic cell death in PC3 cells, with minimal necrotic activity, highlighting its potential as an anti-cancer agent targeting apoptosis.
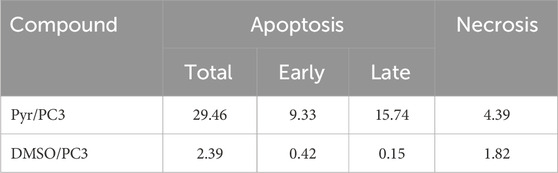
Table 3. Effect of Pyr derivative on apoptosis and necrosis in PC3 cells compared to DMSO-treated cells.
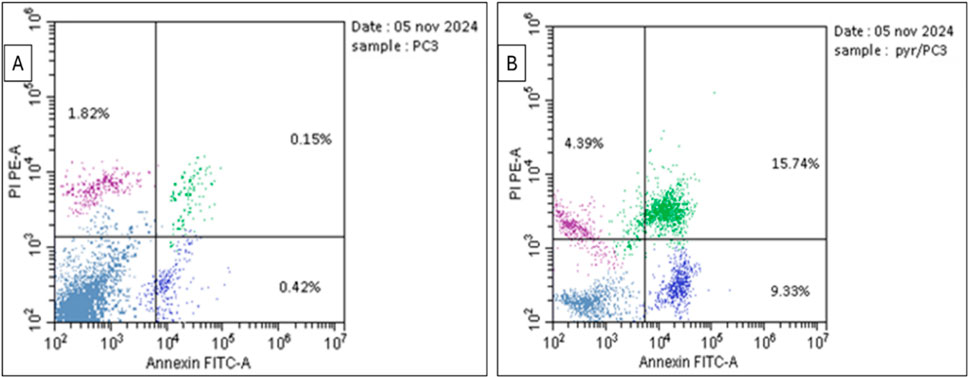
Figure 5. Flow cytometric dot plot for prostate PC3 cancer cells treated with Pyr and untreated cells following Annexin V-FITC/PI staining. (A) shows untreated cells, while (B) shows PC3 cells treated with Pyr at IC50 (8.63 μg/mL). The four quadrants are labeled as follows: Lower Left (LL) for viable cells, Lower Right (LR) for early apoptotic cells, Upper Left (UL) for necrotic cells, and Upper Right (UR) for late apoptotic cells.
The effect of Pyr on the expression of pro-apoptotic (p53, Bax) and anti-apoptotic (Bcl-2) proteins in PC3 cells compared to untreated cells was studied. As shown in Table 4, Pyr treatment markedly increased the expression levels of p53 and Bax, while simultaneously reducing Bcl-2 levels when compared to the DMSO-treated control group. Specifically, Pyr treatment elevated p53 protein concentration to 1461.33 ± 10.00 pg/mL, corresponding to a 9.66-fold increase relative to control cells (151.33 ± 3.33 pg/mL). This substantial upregulation of p53 suggests the activation of a p53-dependent apoptotic pathway, which is known to play a critical role in tumor suppression through cell cycle arrest and apoptosis induction. Concurrently, Bax, a key pro-apoptotic member of the Bcl-2 family, was significantly increased to 477.27 ± 18.54 pg/mL (5.85-fold), supporting the pro-apoptotic effect of Pyr by promoting mitochondrial outer membrane permeabilization and the subsequent release of apoptogenic factors. In contrast, Bcl-2, an anti-apoptotic protein that antagonizes Bax and preserves mitochondrial integrity, was notably decreased following Pyr treatment, reaching 8.88 ± 0.30 pg/mL, which represents a 0.409-fold reduction compared to control (21.68 ± 0.70 pg/mL). This decline in Bcl-2 expression further emphasizes the potential of Pyr to disrupt cellular survival pathways, favoring apoptosis over proliferation in PC3 cells.
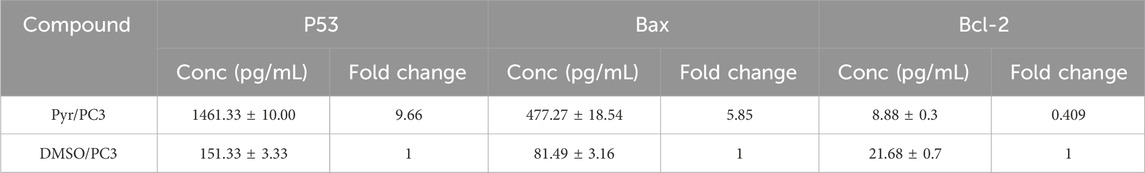
Table 4. Effects of Pyr on p53, Bax, and Bcl-2 protein expression levels in PC3 cells.
To further explore the apoptotic mechanism induced by the synthesized compound Pyr, the activities of caspase-3 and caspase-9—two critical executioners of the intrinsic apoptosis pathway—were assessed in PC3 cells. As presented in Table 5, Pyr treatment led to a significant increase in the activities of both caspase-3 and caspase-9 when compared to the DMSO-treated control. The concentration of caspase-3 in Pyr-treated cells reached 573.19 ± 22.27 pg/mL, reflecting an 8.55-fold elevation relative to the control (67.02 ± 2.60 pg/mL). This notable increase underscores the activation of downstream apoptotic processes, as caspase-3 is a key effector protease responsible for the cleavage of vital cellular substrates during apoptosis. Similarly, the concentration of caspase-9, an essential initiator of the mitochondrial apoptosis pathway, was significantly enhanced following Pyr treatment, rising to 41.82 ± 0.95 ng/mL, which corresponds to an 8.97-fold increase compared to control cells (4.66 ± 0.41 ng/mL). The activation of caspase-9 indicates the involvement of the intrinsic (mitochondrial) pathway, consistent with the observed modulation of upstream regulators, including p53, Bax, and Bcl-2.
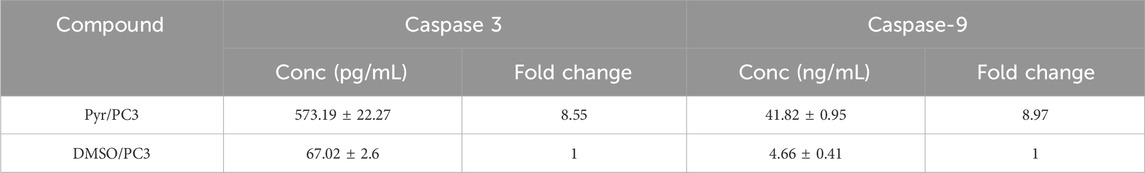
Table 5. Effects of Pyr on caspase-3 and caspase-9 activities in PC3 cells.
Figure 6 illustrates the studied geometrical structural convergence of Pyr in the ground state using the same theoretical level. To better understand the conformational behavior of the structure under consideration, it is feasible to talk about two essential structural properties: bond lengths and bond angles. The compound’s difference in planarity is explained by the face direction of the two aromatic rings, directing the sulphonyl amino group (SO2NH2) away from the plane of the molecule. Furthermore, an important hint to the bent structure found during optimization is provided by the CO group that considered the center of molecule bending and forms two H-bonds with the N1H5 and N2H6 with estimated bond distances of 2.367 Å and 2.370 Å, respectively. This feature may contribute to the structural stability of Pyr. Given that the covalent link between N and H is 1.012 Å longer than the usual bond length (Sayed and Abdelrehim, 2022), the observed N5-H and N6-H bond length validates the strength of the H-bond formation. The optimized structure’s bond angle values allow for elaborating the atoms’ planarity behavior. Bond angle values evaluated the similarity in the environment surrounding the molecule, as in the case of C5-N1-C7 and C7-N2-C8 with 130.77°.
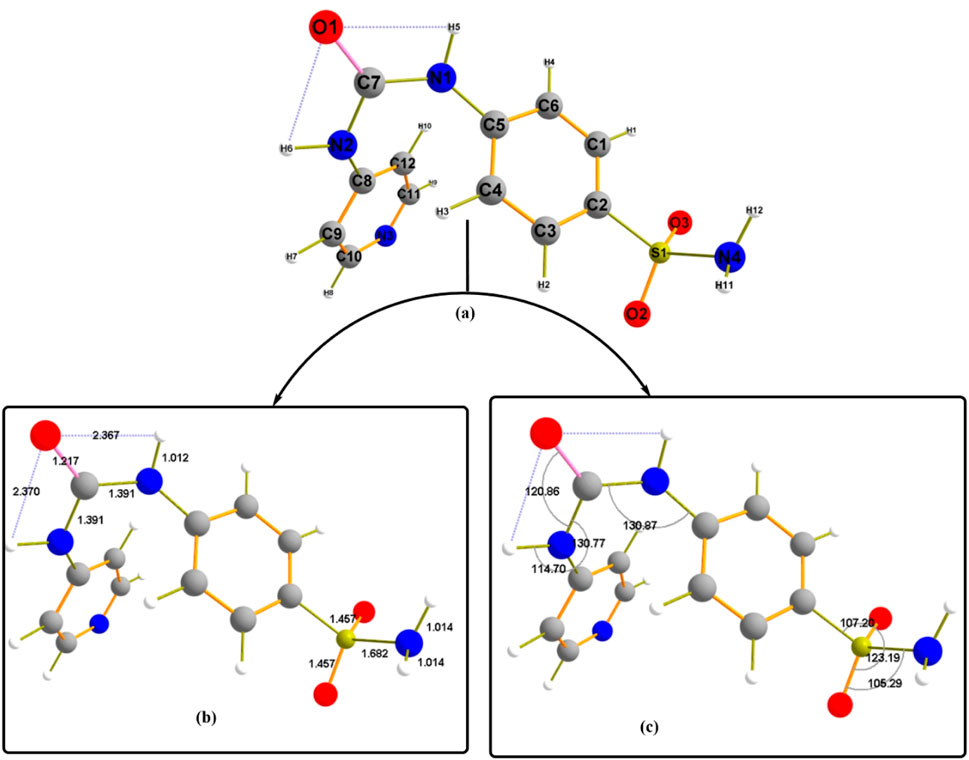
Figure 6. The geometrical structure of the designed compound Pyr (a) labeled with (b) bond lengths, (c) bond angles.
To predict the stability and reactivity of different molecules, it is essential to study the features of compounds’ frontier molecular orbitals (FMOs) (Abdelrehim and El-Sayed, 2020). Figure 7 displays the energy distribution of the most significant molecular orbitals for the gaseous optimized structures (HOMO-2, HOMO-1, HOMO, LUMO, LUMO+1, and LUMO+2). The stability of the current heterocyclic complex can be predicted using the amplitude of the energy gap (∆E = 2,558 eV) between HOMO and LUMO levels, a crucial parameter in the study of stability and reactivity (Chen et al., 2021). The orbital contribution at all examined levels was mostly observed throughout the proposed molecule, providing additional evidence for the stability of both ground and excited states. The orbital contribution in the FMOs mostly occurs around SO2 and CONH groups, leading to the concept of successive donor-acceptor interactions in the excited states.
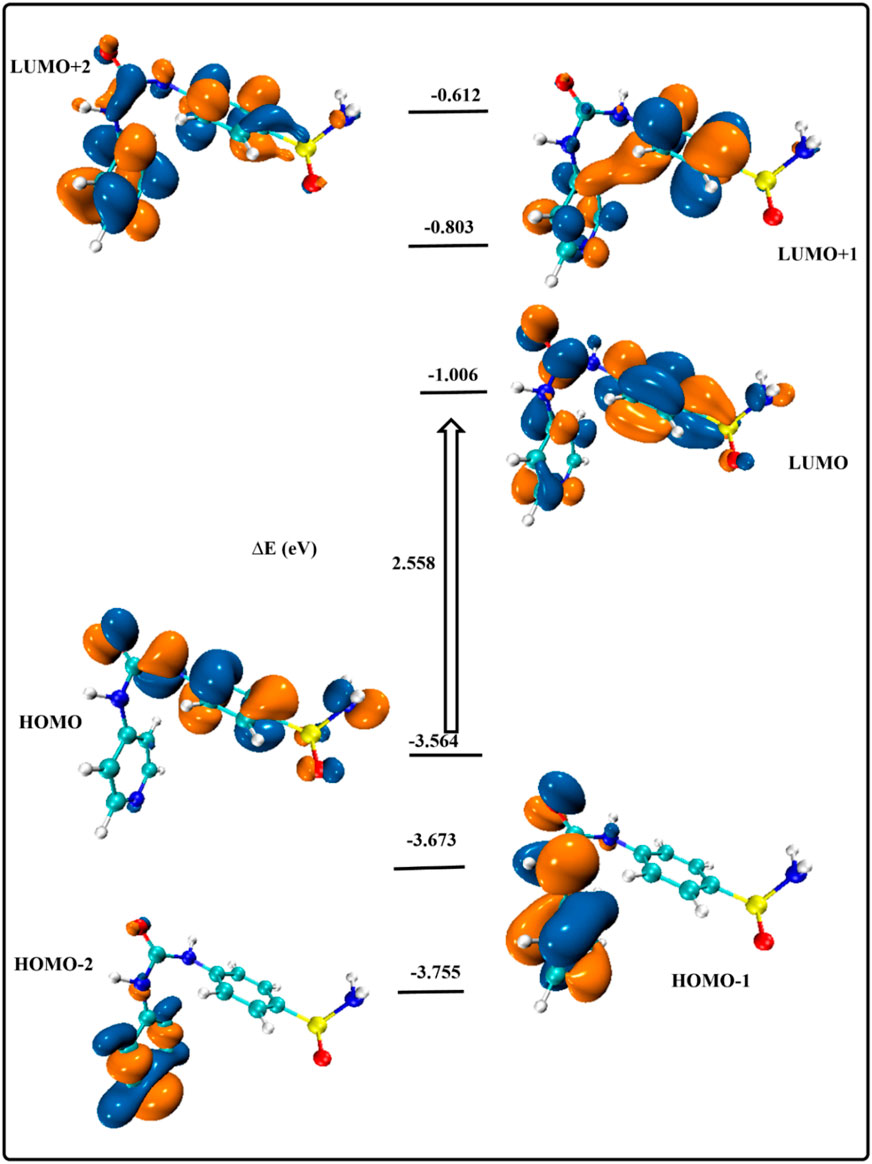
Figure 7. Energy excitation levels with energy values (eV).
The TDDFT and CPCM solvation model was used to characterize the electrical behavior of Pyr. The default Gaussian 09 parameters were used for the TD-DFT calculations. The software settings were adjusted for Nstate = 6 to study six states. In Figure 8, three transition bands are shown as lines. The LOG file of the Gaussian calculation shows a singlet strong absorption band for the first transition, which is the n-π* transition. With a 64.3% transition contribution, an excitation energy of 4.505 eV, and a maximum wavelength (λmax) of 275 nm, the orbital contribution comprises HOMO→LUMO excitation with n-π type of electronic transition. The oscillator strength (F) revealed the strong ability of electronic achievement to LUMO excited state, where its value is higher in the first transition (0.347). Table 6 shows that while the other transitions were anticipated from HOMO→LUMO+1and from HOMO-1→LUMO with comparable contribution %, their F values differ based on the ability of electron transition from one level to another.
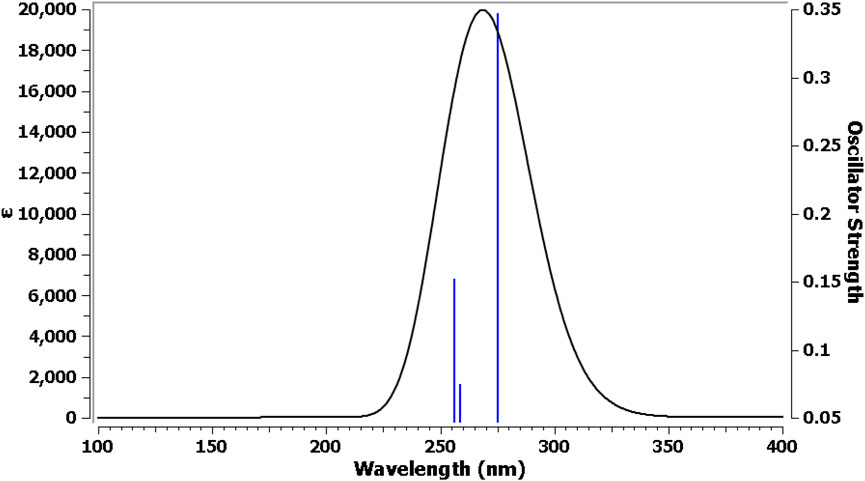
Figure 8. UV–Vis electronic absorption spectra for Pyr.
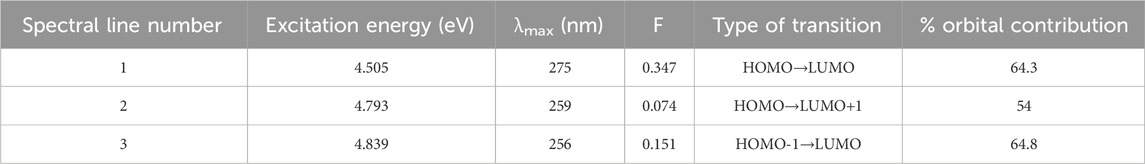
Table 6. Excitation energies, maximum wavelengths, oscillator strengths, and % orbital contribution for Pyr.
The electron localization function (ELF) explores the empirical concepts of electron localization, especially the localization of electron pairs, in the spirit of Lewis structures. An Electron Localization Function (ELF) in atomic space designates a point where electron confinement and bond type are known (Emara et al., 2023). One of the most important two-dimensional planes offers details on the type of bond in three distinct planes: H11-N4-H12, N1-C7-N2, N1-C7-O1, and O2-S1-O3. All the atoms of interest are exhibited at the same plane, as shown in Figure 9. The selected planes estimate a localized electronic area (red color scale) between C7 and N atoms (both N1 and N2) and the same environment (appear in the same plane). At the same time, the plane of the amino group exhibits unusual behavior as no additional atoms are observed in this plane with slightly localized electronic density in the bond of interaction between N4 and S1 (not of the same plane). So, the electron localization between N and the second coordinated atom located in the other plane is predicted by this finding, completing the geometrical structure with the optimum conditions. The map of electronic deformation around O1 decreases the electronic localization between it and C7, as displayed in Figure 9c. Another reason for this deformation is a prediction of H-bond formation between O and the other surrounded H atoms. The bond nature between S and O atoms in the O2-S1-O3 is exhibited to be less strong due to the delocalization of electrons around the two O atoms.
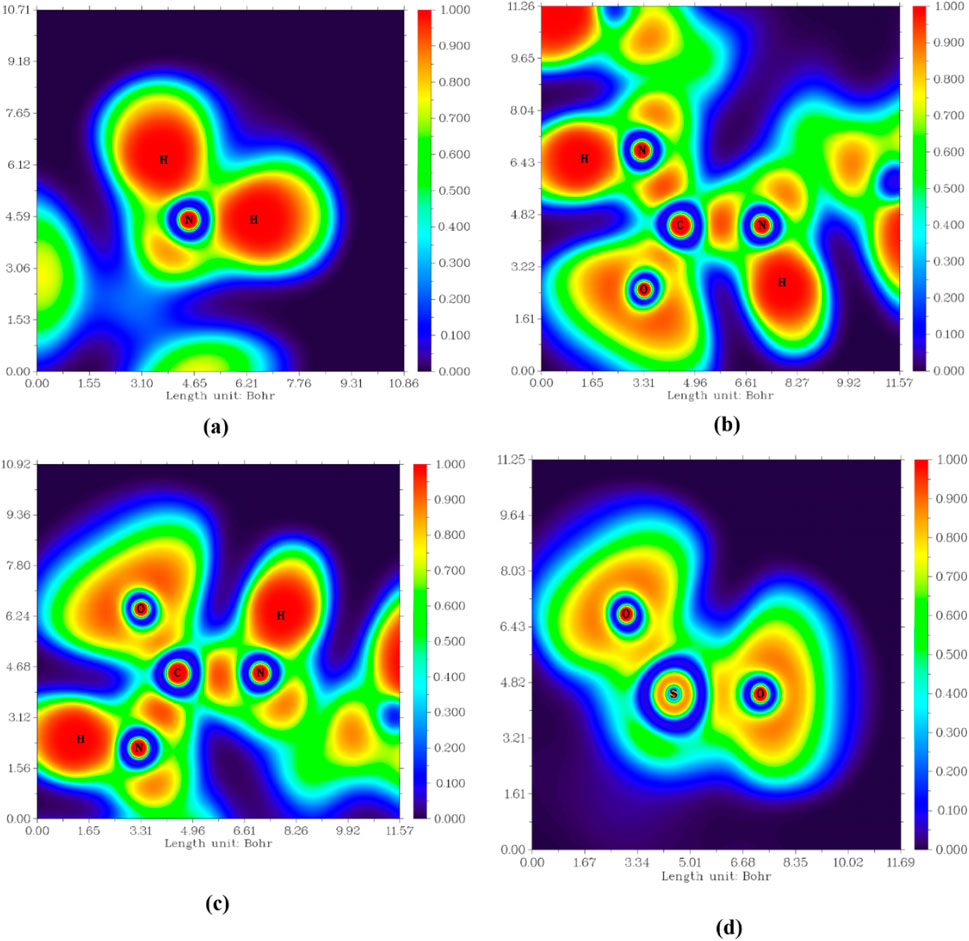
Figure 9. Electron localization function (ELF) colored map of Pyr (a) H11-N4-H12, (b) N1-C7-N2, (c) N1-C7-O1, and (d) O2-S1-O3.
The molecular electrostatic potential (MEP) 3D map is used to analyze the electronegativities of atomic locations on molecules. This topological index helps explain molecular contacts and recognition processes since long-range interactions are mostly caused by electrostatic forces (Issa et al., 2024). The structures of the studied compounds were visually evaluated using colors such as red, orange, yellow, green, and blue to identify potential sites of electrophilic or nucleophilic assaults (Figure 10). The following order of colors represents the decreasing potential for each atomic site: orange, red, green, yellow, and blue. As a result, a red zone indicates an electron-rich site and a blue zone indicates an electron-deficient site. More precisely, it was predicted that all oxygen atom sites in Pyr would have the maximum electronic richness, with the remaining portion of the molecule striving to reach a neutral state and only having a small density of π-electrons of the phenyl ring (yellow color scale). This discrepancy is believed to direct a significant electron donation from the nucleophilic O site to the electron acceptor of electrophilic sites, mainly including amino group hydrogen atoms (primary and secondary groups).
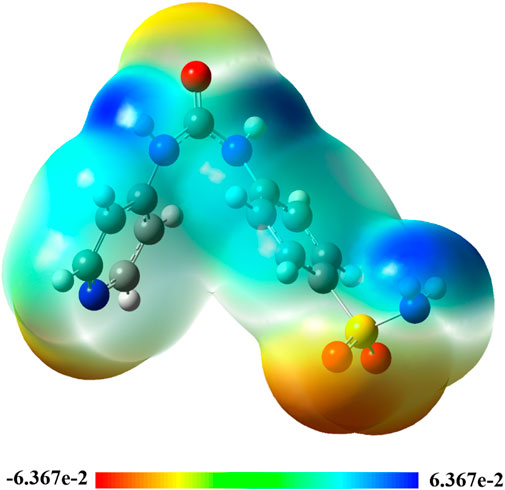
Figure 10. 3D-colored map of the molecular electrostatic potential of Pyr.
Utilizing reduced density gradient (RDG) research, noncovalent bond interactions (NCIs) between several molecular sites were identified. Different color codes were used to illustrate noncovalent interactions (El-Sayed et al., 2023). As illustrated in Figure 11, the interactions readily discernible on the surface of each molecule are vdW interactions with a green color scale and repulsion (steric) interactions with a red color scale. The sign (λ2)ρ, obtained by multiplying the electron density by the sign of the second Hessian eigenvalue, indicates the strength of the HB interaction in compounds. In this study, vdW is visible in the cage of C1-C2-S1 and C3-C2-S1. Other electrostatic interactions appear between the two phenyl rings. In this map, H-bond spikes do not appear in the chart; the high distance between H atoms and O atoms (around 2.4 Å) may lead to less sensitivity to be estimated with reduced density parameters. The unfavorable repulsion forces in the molecule were attributed to the steric effect of phenyl rings that export closed 6-atomic system constrain behavior. This type of steric interaction can be vanished by favorable electrostatic interactions.
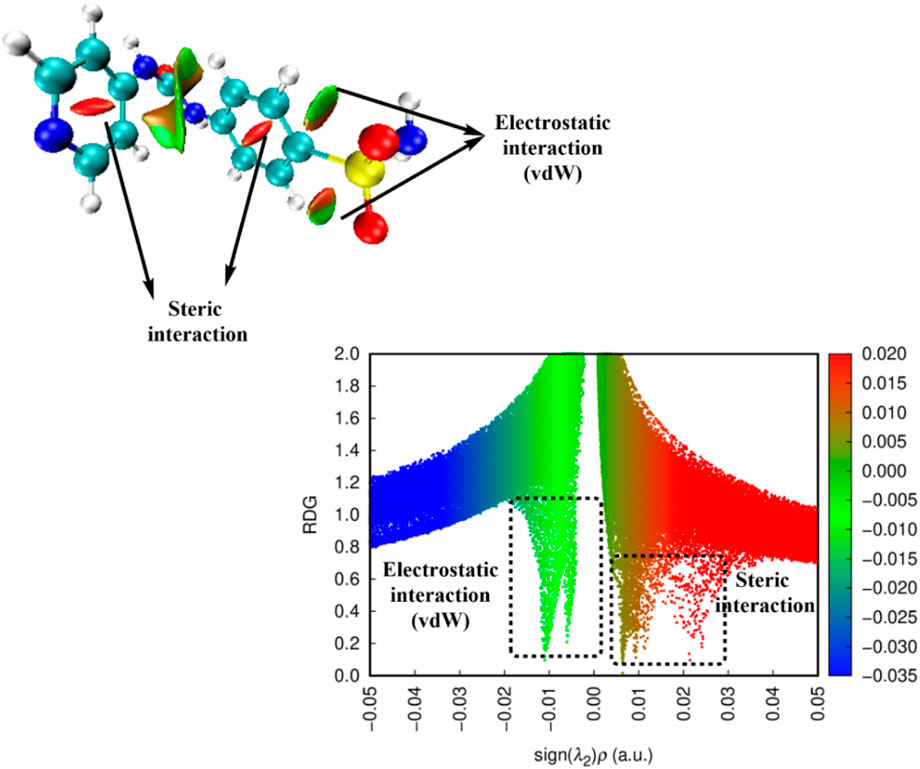
Figure 11. 3D-NCI map and RDG plot of Pyr.
Molecular docking is a computational method used to predict the preferred orientation of a small molecule when bound to a receptor, such as a protein, and to evaluate the strength and stability of the interaction (Chang et al., 2023). For this study, the synthesized compound Pyr was docked into the active site of the carbonic anhydrase IX using PDB entry 5FL4, which contains the structure of CAIX bound to its cocrystallized ligand 9FK. Autodock Vina, a widely used docking program, was employed to perform the docking simulations, and the results were visualized using Discovery Studio Visualizer, a tool that allows for detailed analysis of molecular interactions.
To ensure the accuracy of the docking procedure, the cocrystallized ligand 9FK was redocked into the CAIX structure. The root-mean-square deviation (RMSD) between the original and redocked ligand poses was calculated to validate the results. An RMSD of 0.9458 Å, which is well below the typical threshold of 2 Å for reliable docking, indicated that the docking procedure and the employed parameters were accurate and capable of reproducing the experimental binding mode of the ligand. The binding affinity of the redocked ligand was found to be −8.6 kcal/mol, demonstrating a stable interaction with the CAIX enzyme. The superimposition of the redocked ligand with its original conformation is shown in Figure 12.
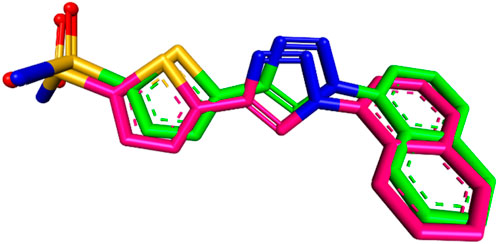
Figure 12. Superimposition of the redocked (green) and co-crystallized (magenta) poses of 9FK inside the CAIX active site (RMSD = 0.9458 Å).
Following the validation of the docking protocol, Pyr was docked into the CAIX active site, and a comparative analysis was performed with acetazolamide, the reference compound used in the in-vitro CA inhibition assay. The binding affinity of Pyr was calculated to be −7.4 kcal/mol, indicating a moderately stable interaction with the CAIX enzyme, while acetazolamide showed a binding affinity of −7.7 kcal/mol, slightly higher than Pyr, which suggests a similar but slightly stronger binding interaction.
Pyr exhibited several key interactions that were similar to those of acetazolamide. Specifically, the NH2 group of Pyr’s sulfonamide interacted with the zinc ion in the active site, mirroring the interaction of acetazolamide, where the sulfonamide group serves as the zinc-binding moiety. Additionally, both the oxygen and NH2 groups of Pyr’s sulfonamide formed classical hydrogen bonds with THR200, as seen with acetazolamide. The sulfur atom of Pyr’s sulfonamide also engaged in Pi-sulfur interactions with Trp210 and His94, akin to acetazolamide’s interactions. The 2D and 3D interactions of Pyr are shown in Figure 13, illustrating these interactions in detail.
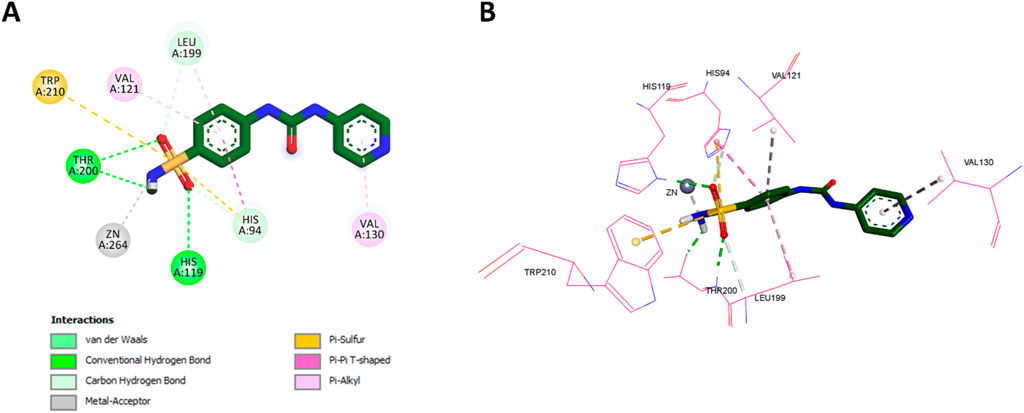
Figure 13. (A) 2D interaction diagram and (B) 3D binding mode of Pyr within the CAIX active site.
However, Pyr exhibited some unique interactions not observed with acetazolamide. The oxygen of Pyr’s sulfonamide formed a classical hydrogen bond with His119, a distinct interaction that was absent in acetazolamide. Moreover, Pyr formed non-classical hydrogen bonds with both Leu199 and His94, which were not seen in the acetazolamide docking. Pyr’s benzene ring demonstrated hydrophobic interactions with Leu199, a feature similar to the thiadiazole group of acetazolamide, but also formed additional hydrophobic interactions with both Val121 and His94, which were not present in acetazolamide. Finally, the pyridine ring (tail region) of Pyr exhibited Pi-alkyl (hydrophobic) interactions with Val130, a unique feature not observed in acetazolamide. The 2D and 3D interactions of acetazolamide are shown in Figure 14 for comparison.
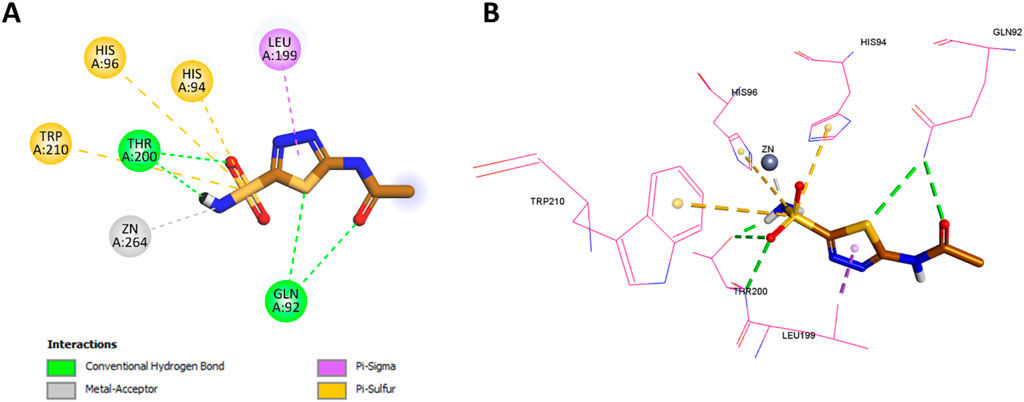
Figure 14. (A) 2D interaction diagram and (B) 3D binding mode of Pyr within the CAIX active site.
The Lipinski rule can assess the validity of the therapeutic properties in the compounds under investigation based on five determinants: molecular weight <500 Da, strong lipophilic qualities, such as LogP value <5, H-bond donors <5, H-bond acceptors <10 (Baell et al., 2013). The ligand under investigation may show signs of a drug when it meets more than two of these requirements. The drug-likeness features of PYR are shown in Table 7. It was calculated that the molecular weight of the ligand is 292.31 g/mol. It has five hydrogen bond acceptors and three hydrogen bond donors, where the investigated ligand meets these specifications. Excellent permeability across the cell membrane is indicated by the iLog P value of the molecule, which is determined to be within the range of less than 5, as predicted theoretically. It was found that the title ligand’s computed TPSA value was less than 140 Å2, which indicates good intestinal absorption values. Based on Lipinski’s rule, these findings support the hypothesis that PYR will absorb well. Significant bioavailability is predicted based on the TPSA and number of rotatable bond values. The primary focus of medical chemistry’s drug design, discovery, and contemporary drug development methods is on tiny molecules that resemble drugs and have high biological activity while being lowly harmful (Rautio et al., 2018). In silico prediction techniques, which enhance activity and toxicity study time and resource efficiency, can be used to identify (ADMET) traits (Ferreira and Andricopulo, 2019). The lipophilicity and pharmacokinetic parameters (ADMET features) are shown in Table 8. Drawing from the identified result, it can be shown that PYR exhibited a penetration capability that manifests as BBB permeant and GI absorption, indicating a reduced ability to cross the BBB and enter the central nervous system (Mani et al., 2023). The permeability value is −7.88 cm/s in human skin, showing that they cannot be absorbed within human skin. The propensity of a substance to inhibit five isoenzymes related to cytochrome P450, a major drug-metabolizing enzymes (CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4) can be evaluated using metabolic factors. CPY is utilized to develop medicinal effects and inhibit their capability. This trait is important for toxicity and other unfavorable medication interactions (Zhao et al., 2021). The predicted drug-potent ligand cannot affect these types of drug-metabolizing enzymes. Pyr exported a potential physicochemical characteristic that qualifies it for oral bioavailability. A suitable way to visualize this behavior is with the bioavailability radar, as shown in Figure 15. The pink zone in the bioavailability radar plot shows the optimal range for each of the six physicochemical parameters—size, solubility, lipophilicity, polarity, saturation, and flexibility. These characteristics are thought to be ideal for reaching the best oral bioavailability. A dependable model that accurately forecasts drug candidates’ absorption in the gastrointestinal system and their accessibility via the blood-brain barrier is the BOILED Egg method. Pyr was in a white zone, suggesting a high gastrointestinal absorption, as shown in Figure 16.
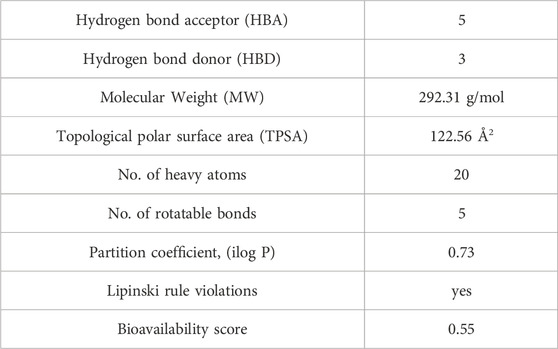
Table 7. Drug-likeness descriptors of the studied ligand Pyr.
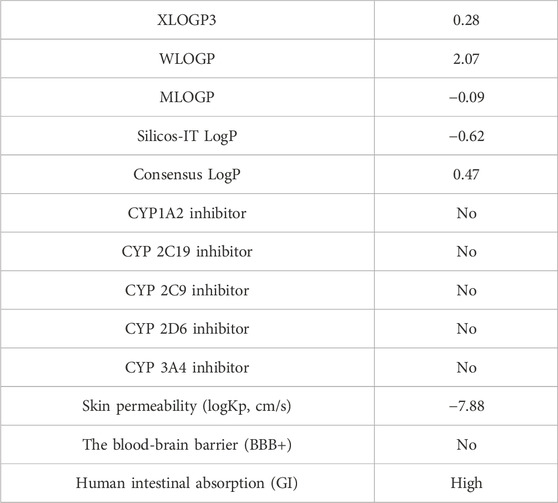
Table 8. Lipophilicity and pharmacokinetic parameters for Pyr.
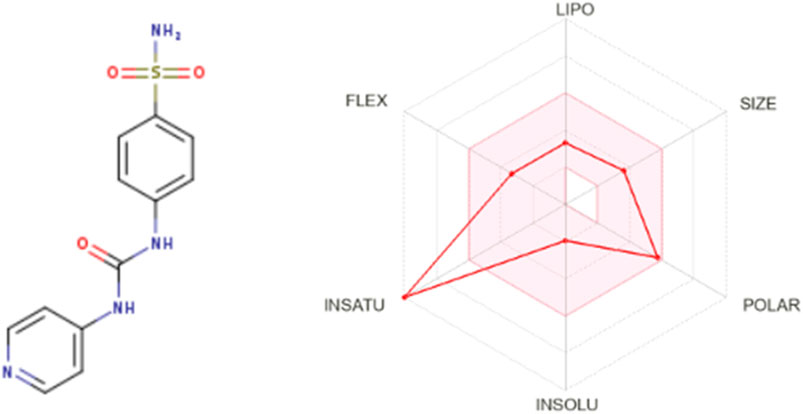
Figure 15. Bioavailability Radar model for Pyr.
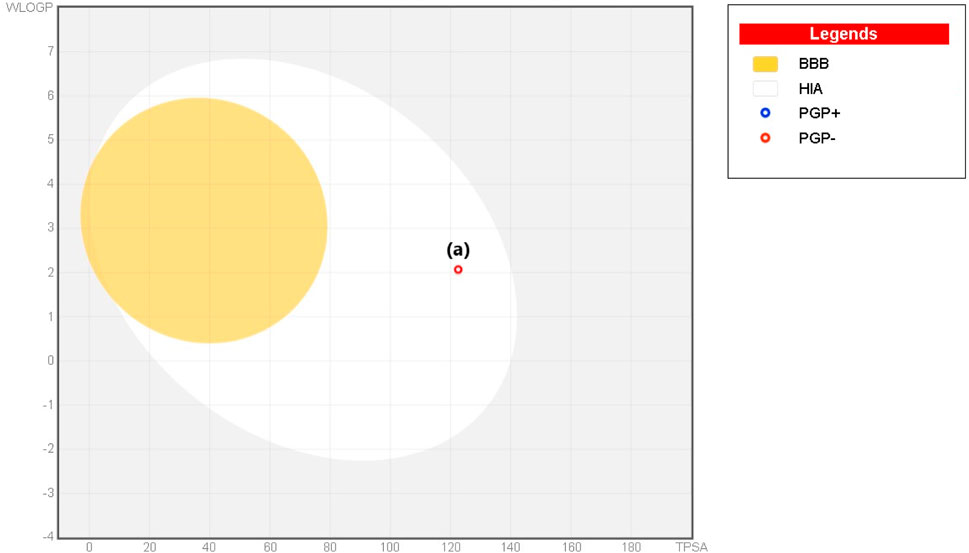
Figure 16. BOILED Egg plot of Pyr.
All reactions were monitored using thin-layer chromatography (TLC) on Merck (Boston, MA, United States) 9385 pre-coated aluminum silica gel plates (Kieselgel 60) measuring 5 cm × 20 cm with a 0.2 mm layer thickness. Spots were visualized under UV light at a wavelength of 254 nm. Melting points were determined using Stuart’s electrothermal melting point apparatus and were uncorrected. Nuclear magnetic resonance (NMR) spectra for protons (^1H, 400 MHz) were recorded in DMSO-d6 on a Bruker AM400 spectrometer, with tetramethylsilane (TMS) as the internal standard. Chemical shift values are reported in parts per million (ppm), using DMSO-d6 as the solvent, and coupling constants (J) are expressed in hertz (Hz). Signal splitting patterns are described as follows: s (singlet), d (doublet), dd (doublet of doublets), t (triplet), q (quartet), m (multiplet), and brs (broad singlet). Intermediates 1-5 were synthesized according to the reported procedures in the literature (Maconi et al., 2002).
Freshly prepared azide derivative (4) (1.5 mmol, 0.178) was suspended in 10 mL of toluene, and the mixture was heated under reflux for 1 h until the evolution of N2 gas stopped. Then sulfanilamide (1 mmol, 0.172 g) was added portionwise to the solution, and the reaction mixture was heated under reflux for 2 h. The formed fluffy precipitate was filtered off and washed extensively with boiling toluene to remove unreacted isocyanate. The crude urea product was recrystallized from absolute ethanol.
White powder; 0.216 g, 74% yield; mp: 232°C–234°C; 1H NMR (400 MHz, DMSO-d6) δ = 9.10 (1H, s, urea-NH), 8.97 (1H, s, urea-NH), 8.34 (2H, d, pyridyl-2CH), 7.73 (2H, d, phenyl-2CH), 7.62 (2H, d, phenyl-2CH), 7.44 (2H, d, pyridyl-2CH), 7.24 (2H, s, pyridyl-NH2); 13C NMR (100 MHz, DMSO-d6) δ = 152.90, 150.40, 146.76, 143.78, 137.16, 126.92, 117.84, 112.15. Anal. Calcd for C12H12N4O3S (292.31): C, 49.31; H, 4.14; N, 19.17. Found: C, 49.43; H, 3.99; N, 19.40.
The cell viability of colon HT29, breast MCF7, and prostate PC3 cancer cells, along with CCD-986sk normal cells, was evaluated for Pyr using MTT assay protocols as described in the literature (Ali et al., 2024). All cell lines were obtained from the Vacsera Cell Culture Library, Tissue Culture Unit, Cairo, Egypt, with ATCC certification. For further details, See Supplementary Appendix A in the supplementary data.
According to the assay protocol, Pyr was incubated at room temperature for 10 min and analyzed in triplicate. Absorbance measurements were recorded at 405 nm in kinetic mode for 1 h at room temperature. The resulting data were plotted linearly to derive absorbance values, from which the IC50 value was determined using the slope of the plot (Abdelhakeem et al., 2024).
The impact of Pyr on the cell cycle progression of PC3 cells was examined by analyzing DNA content with a flow cytometer, following the procedure outlined in the literature (Ali et al., 2024). For further details, See Supplementary Appendix A in the supplementary data.
To detect apoptosis in PC3 cancer cells, the Annexin V FITC assay protocol was employed as described in the literature (Ali et al., 2024). For further details, See Supplementary Appendix A in the supplementary data.
The impact of Pyr on caspase-3 protein levels, Bax, and Bcl-2 was evaluated following the protocols provided in the manufacturer’s kit instructions.
Computerized Prism 5 program was used to statistically analyzed data using one-way ANOVA test followed by Tukey’s as post ANOVA for multiple comparison at P ≤ 0.05. Data were presented as mean ± SEM.
The target compound Pyr was subjected to a computational analysis using density functional theory (DFT) in order to provide fully optimized geometrical and electronic calculations using the hybrid B3LYP technique (Parr et al., 1980; Fayed et al., 2020). For further details, See Supplementary Appendix A in the supplementary data.
The docking procedure essentially follows a crucial path for extremely successful results. Consequently, the docking of the investigated complexes with control comparison was simulated using the AutoDock 4.2 program (Morris et al., 2009). Additionally, Discovery Studio was used to analyze and visualize docking data. For further details, See Supplementary Appendix A in the supplementary data.
The ADMET parameters were calculated and in silico tests of the drug-like properties of Pyr were carried out using the open-source SwissADME server (Daina et al., 2017).
In conclusion, this study highlights the potential of a novel 4-pyridyl analog of SLC-0111 (Pyr) as a promising candidate for cancer therapy. Pyr exhibited selective cytotoxicity against various cancer cell lines (HT-29, MCF7, PC3) with reduced toxicity toward normal cells, demonstrating a favorable therapeutic index. Moreover, Pyr demonstrated potent and selective inhibition of tumor-associated carbonic anhydrase IX (CA IX) with an IC50 of 0.399 µg/mL with moderate inhibition of other CA isoforms I, II and XII. Further mechanistic investigations revealed its ability to induce G0/G1 phase cell cycle arrest and promote apoptosis in PC3 cells, supported by increased caspase-3 and caspase-9 activities and modulation of Bax/Bcl-2 and p53 protein levels. Molecular docking studies further validated Pyr’s strong binding affinity and selective inhibition of tumor-associated carbonic anhydrase IX. Additionally, ADMET predictions confirmed its drug-like properties and oral bioavailability. These findings establish Pyr as a viable candidate for further development in anticancer drug discovery and targeted therapy applications.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Ethical approval was not required for the studies on humans in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used.
HH: Methodology, Writing – original draft. SA: Resources, Writing – original draft. HMH: Software, Visualization, Writing – review and editing. AA-E: Software, Visualization, Writing – review and editing. MA: Data curation, Formal Analysis, Writing – review and editing. GA: Investigation, Methodology, Writing – review and editing. IR: Conceptualization, Data curation, Writing – original draft. KK: Formal Analysis, Writing – review and editing. DR: Writing – original draft. SB: Conceptualization, Project administration, Writing – original draft. AA: Investigation, Methodology, Writing – review and editing.
The author(s) declare that financial support was received for the research and/or publication of this article. This study was funded by Deanship of Research and Graduate Studies at King Khalid University through a Large Research Project under grant number RGP2/417/45.
The authors extend their appreciation to the Deanship of Research and Graduate Studies at King Khalid University for funding this work through a Large Research Project under grant number RGP2/417/45.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2025.1571646/full#supplementary-material
Abdelhakeem, M. M., Morcoss, M. M., Hanna, D. A., and Lamie, P. F. (2024). Design, synthesis and in silico insights of novel 1,2,3-triazole benzenesulfonamide derivatives as potential carbonic anhydrase IX and XII inhibitors with promising anticancer activity. Bioorg. Chem. 144, 107154. doi:10.1016/j.bioorg.2024.107154
Abdelrehim, E.-S. M., and El-Sayed, D. S. (2020). A new synthesis of poly heterocyclic compounds containing [1,2,4]triazolo and [1,2,3,4]tetrazolo moieties and their DFT study as expected anti-cancer reagents. Curr. Org. Synth. 17, 211–223. doi:10.2174/1570179417666200226092516
Abo-Ashour, M. F., Eldehna, W. M., Nocentini, A., Ibrahim, H. S., Bua, S., Abdel-Aziz, H. A., et al. (2019). Novel synthesized SLC-0111 thiazole and thiadiazole analogues: determination of their carbonic anhydrase inhibitory activity and molecular modeling studies. Bioorg Chem. 87, 794–802. doi:10.1016/j.bioorg.2019.04.002
Ali, D. M. E., Aziz, H. A., Bräse, S., Al Bahir, A., Alkhammash, A., Abuo-Rahma, G.E.-D. A., et al. (2024). Unveiling the anticancer potential of a new ciprofloxacin-chalcone hybrid as an inhibitor of topoisomerases I and II and apoptotic inducer. Molecules 29, 5382. doi:10.3390/molecules29225382
Angeli, A., Paoletti, N., and Supuran, C. T. (2023). Five-membered heterocyclic sulfonamides as carbonic anhydrase inhibitors. Molecules 28, 3220. doi:10.3390/molecules28073220
Baell, J., Congreve, M., Leeson, P., and Abad-Zapatero, C. (2013). Ask the experts: past, present and future of the rule of five. Future Med. Chem. 5, 745–752. doi:10.4155/fmc.13.61
Baranauskienė, L., and Matulis, D. (2019). “Overview of human carbonic anhydrases,” in Carbonic anhydrase as drug target: thermodynamics and structure of inhibitor binding. Editor D. Matulis (Cham: Springer International Publishing), 3–14.
Buza, A., Türkeş, C., Arslan, M., Demir, Y., Dincer, B., Nixha, A. R., et al. (2023). Discovery of novel benzenesulfonamides incorporating 1,2,3-triazole scaffold as carbonic anhydrase I, II, IX, and XII inhibitors. Int. J. Biol. Macromol. 239, 124232. doi:10.1016/j.ijbiomac.2023.124232
Buza, A., Türkeş, C., Arslan, M., Demir, Y., Dincer, B., Nixha, A. R., et al. (2024). Novel benzenesulfonamides containing a dual triazole moiety with selective carbonic anhydrase inhibition and anticancer activity. RSC Med. Chem. 16, 324–345. doi:10.1039/d4md00617h
Chang, Y., Hawkins, B. A., Du, J. J., Groundwater, P. W., Hibbs, D. E., and Lai, F. (2023). A guide to in silico drug design. Pharmaceutics 15, 49. doi:10.3390/pharmaceutics15010049
Chen, S., An, R., Li, Y., Zhu, Y., Zhu, X., Liu, R., et al. (2021). Strategy of regulating the electrophilic/nucleophilic ability by ionic ratio in poly(ionic liquid)s to control the coupling reaction of epoxide. Catal. Sci. Technol. 11, 6498–6506. doi:10.1039/D1CY01024G
Daina, A., Michielin, O., and Zoete, V. (2017). SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 7, 42717. doi:10.1038/srep42717
Doostmohammadi, A., Jooya, H., Ghorbanian, K., Gohari, S., and Dadashpour, M. (2024). Potentials and future perspectives of multi-target drugs in cancer treatment: the next generation anti-cancer agents. Cell Commun. Signal. 22, 228. doi:10.1186/s12964-024-01607-9
El-Sayed, D. S., Tawfik, E. M., Elhusseiny, A. F., and El-Dissouky, A. (2023). A perception into binary and ternary copper (II) complexes: synthesis, characterization, DFT modeling, antimicrobial activity, protein binding screen, and amino acid interaction. BMC Chem. 17, 55. doi:10.1186/s13065-023-00962-x
Emara, M. M., Hafez, S. T., Khalil, T. E., Kashyout, A.E.-H. B., El-Dissouky, A., and El-Sayed, D. S. (2023). Electronic and structural perturbations of microporous ZIF-67 nanoparticles and Cr(VI) molecule during adsorptive water decontamination unveiled by experimental and quantum computational investigations. J. Mol. Liq. 390, 123042. doi:10.1016/j.molliq.2023.123042
Fayed, T. A., Gaber, M., Abu El-Reash, G. M., and El-Gamil, M. M. (2020). Structural, DFT/B3LYP and molecular docking studies of binuclear thiosemicarbazide Copper (II) complexes and their biological investigations. Appl. Organomet. Chem. 34, e5800. doi:10.1002/aoc.5800
Ferreira, L. L. G., and Andricopulo, A. D. (2019). ADMET modeling approaches in drug discovery. Drug Discov. Today 24, 1157–1165. doi:10.1016/j.drudis.2019.03.015
Güleç, Ö., Türkeş, C., Arslan, M., Demir, Y., Dincer, B., Ece, A., et al. (2024a). Novel beta-lactam substituted benzenesulfonamides: in vitro enzyme inhibition, cytotoxic activity and in silico interactions. J. Biomol. Struct. Dyn. 42, 6359–6377. doi:10.1080/07391102.2023.2240889
Güleç, Ö., Türkeş, C., Arslan, M., Demir, Y., Dincer, B., Ece, A., et al. (2024b). Bioactivity, cytotoxicity, and molecular modeling studies of novel sulfonamides as dual inhibitors of carbonic anhydrases and acetylcholinesterase. J. Mol. Liq. 410, 125558. doi:10.1016/j.molliq.2024.125558
Issa, A. A., Kamel, M. D., and El-Sayed, D. S. (2024). Depicted simulation model for removal of second-generation antipsychotic drugs adsorbed on Zn-MOF: adsorption locator assessment. J. Mol. Model 30, 106. doi:10.1007/s00894-024-05896-2
Kakakhan, C., Türkeş, C., Güleç, Ö., Demir, Y., Arslan, M., Özkemahlı, G., et al. (2023). Exploration of 1,2,3-triazole linked benzenesulfonamide derivatives as isoform selective inhibitors of human carbonic anhydrase. Bioorg Med. Chem. 77, 117111. doi:10.1016/j.bmc.2022.117111
Kalinin, S., Malkova, A., Sharonova, T., Sharoyko, V., Bunev, A., Supuran, C. T., et al. (2021). Carbonic anhydrase IX inhibitors as candidates for combination therapy of solid tumors. Int. J. Mol. Sci. 22, 13405. doi:10.3390/ijms222413405
Low, P., Chu, H. A., Huang, B., Kasireddy, S., Singh, Y., Zheng, S., et al. (2024). Bi-specific adapters and their use with universal car-t cells in the treatment of caix-expressing tumors. Available online at: https://patents.google.com/patent/WO2024191887A2/en?oq=WO2024191887A2 (Accessed March 3, 2025).
Maconi, A., Pastorin, G., Da Ros, T., Spalluto, G., Gao, Z., Jacobson, K. A., et al. (2002). Synthesis, biological properties, and molecular modeling investigation of the first potent, selective, and water-soluble human A3 adenosine receptor antagonist. J. Med. Chem. 45, 3579–3582. doi:10.1021/jm020974x
Mani, N., Nicksonsebastin, D., and Prasath, M. (2023). Quantum computational, spectroscopic, ADMET, molecular docking and dynamics simulation revealing the inhibition of psoralidin against anti-tuberculosis. Chem. Phys. Impact 7, 100292. doi:10.1016/j.chphi.2023.100292
McDonald, P. C., Chia, S., Bedard, P. L., Chu, Q., Lyle, M., Tang, L., et al. (2020). A phase 1 study of SLC-0111, a novel inhibitor of carbonic anhydrase IX, in patients with advanced solid tumors. Am. J. Clin. Oncol. 43, 484–490. doi:10.1097/coc.0000000000000691
MedChemExpress (2024). MedchemExpress.Com, CA IX/XII inhibitor. U-104 (SLC-0111). Available online at: https://www.medchemexpress.com/U-104.html (Accessed November 24, 2024).
Merkx, R. I. J., Mulders, P. F. A., and Oosterwijk, E. (2021). “Carbonic anhydrase IX: current and emerging therapies,” in The carbonic anhydrases: current and emerging therapeutic targets. Editors W. R. Chegwidden, and N. D. Carter (Cham: Springer International Publishing), 205–219.
Morris, G. M., Huey, R., Lindstrom, W., Sanner, M. F., Belew, R. K., Goodsell, D. S., et al. (2009). AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791. doi:10.1002/jcc.21256
Parr, R. G. (1980). “Density functional theory of atoms and molecules,” in Horizons of quantum chemistry. Editors K. Fukui, and B. Pullman (Dordrecht: Springer Netherlands), 5–15.
Pastorekova, S., and Gillies, R. J. (2019). The role of carbonic anhydrase IX in cancer development: links to hypoxia, acidosis, and beyond. Cancer Metastasis Rev. 38, 65–77. doi:10.1007/s10555-019-09799-0
P. Inc (2024). Targeted therapy vs immunotherapy: which is Right for you, key differences and similarities - OncoDaily. Available online at: https://oncodaily.com/oncolibrary/immune-oncology/targeted-therapy-vs-immunotherapy (Accessed November 24, 2024).
Rautio, J., Meanwell, N. A., Di, L., and Hageman, M. J. (2018). The expanding role of prodrugs in contemporary drug design and development. Nat. Rev. Drug Discov. 17, 559–587. doi:10.1038/nrd.2018.46
Sarnella, A., Ferrara, Y., Auletta, L., Albanese, S., Cerchia, L., Alterio, V., et al. (2022). Inhibition of carbonic anhydrases IX/XII by SLC-0111 boosts cisplatin effects in hampering head and neck squamous carcinoma cell growth and invasion. J. Exp. and Clin. Cancer Res. 41, 122. doi:10.1186/s13046-022-02345-x
Sayed, D. S. E., and Abdelrehim, E.-S. M. (2022). Spectroscopic details on the molecular structure of pyrimidine‑2‑thiones heterocyclic compounds: computational and antiviral activity against the main protease enzyme of SARS-CoV-2. BMC Chem. 16, 82. doi:10.1186/s13065-022-00881-3
Sedlakova, O., Svastova, E., Takacova, M., Kopacek, J., Pastorek, J., and Pastorekova, S. (2014). Carbonic anhydrase IX, a hypoxia-induced catalytic component of the pH regulating machinery in tumors. Front. Physiol. 4, 400. doi:10.3389/fphys.2013.00400
Shaldam, M., Eldehna, W. M., Nocentini, A., Elsayed, Z. M., Ibrahim, T. M., Salem, R., et al. (2021). Development of novel benzofuran-based SLC-0111 analogs as selective cancer-associated carbonic anhydrase isoform IX inhibitors. Eur. J. Med. Chem. 216, 113283. doi:10.1016/j.ejmech.2021.113283
Targeted Cancer Therapy (2024). Targeted drug therapy for cancer. Available online at: https://www.cancer.org/cancer/managing-cancer/treatment-types/targeted-therapy/what-is.html (accessed November 24, 2024).
Tian, Y., Wang, X., Wu, C., Qiao, J., Jin, H., and Li, H. (2024). A protracted war against cancer drug resistance. Cancer Cell Int. 24, 326. doi:10.1186/s12935-024-03510-2
Tilsed, C. M., Fisher, S. A., Nowak, A. K., Lake, R. A., and Lesterhuis, W. J. (2022). Cancer chemotherapy: insights into cellular and tumor microenvironmental mechanisms of action. Front. Oncol. 12, 960317. doi:10.3389/fonc.2022.960317
WHO (2024). Cancer. Available online at: https://www.who.int/news-room/fact-sheets/detail/cancer (Accessed November 24, 2024).
Wikipedia (2024). The hallmarks of cancer. Wikipedia. Available online at: https://en.wikipedia.org/w/index.php?title=The_Hallmarks_of_Cancer&oldid=1259244236 (Accessed November 24, 2024).
Yang, M., Cui, M., Sun, Y., Liu, S., and Jiang, W. (2024). Mechanisms, combination therapy, and biomarkers in cancer immunotherapy resistance. Cell Commun. Signal. 22, 338. doi:10.1186/s12964-024-01711-w
Zhao, M., Ma, J., Li, M., Zhang, Y., Jiang, B., Zhao, X., et al. (2021). Cytochrome P450 enzymes and drug metabolism in humans. Int. J. Mol. Sci. 22, 12808. doi:10.3390/ijms222312808
ADMET Absorption, Distribution, Metabolism, Excretion, and Toxicity
AAZ Acetazolamide
BBB Blood-Brain Barrier
Bcl-2 B-cell lymphoma 2
CA Carbonic Anhydrase
CA I Carbonic Anhydrase Isoform I
CA II Carbonic Anhydrase Isoform II
CA IX Carbonic Anhydrase Isoform IX
CA XII Carbonic Anhydrase Isoform XII
CAR-T Chimeric antigen receptor T
CAIs Carbonic Anhydrase Inhibitors
CCD-986sk Human normal fibroblast cell line
DFT Density Functional Theory
DMSO Dimethyl Sulfoxide
ELF Electron Localization Function
FMOs Frontier Molecular Orbitals
GI Gastrointestinal
HOMO Highest Occupied Molecular Orbital
IC50 Half-maximal Inhibitory Concentration
Ki Inhibition Constant
LUMO Lowest Unoccupied Molecular Orbital
MCF7 Human breast cancer cell line
MEP Molecular Electrostatic Potential
MTT 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
NCI Non-Covalent Interaction
NMR Nuclear Magnetic Resonance
PC3 Human prostate cancer cell line
RMSD Root-Mean-Square Deviation
TD-DFT Time-Dependent Density Functional Theory
TLC Thin-Layer Chromatography
TPSA Topological Polar Surface Area
UV–Vis Ultraviolet–Visible Spectroscopy
vdW van der Waals
Keywords: sulphonamide, SLC-0111, carbonic anhydrase, cytotoxicity, apoptosis
Citation: Hashem H, Abdelfattah S, Hassan HM, Al-Emam A, Alqarni M, Alotaibi G, Radwan IT, Kaur K, Rao DP, Bräse S and Alkhammash A (2025) Discovery of a novel 4-pyridyl SLC-0111 analog targeting tumor-associated carbonic anhydrase isoform IX through tail-based design approach with potent anticancer activity. Front. Chem. 13:1571646. doi: 10.3389/fchem.2025.1571646
Received: 05 February 2025; Accepted: 11 March 2025;
Published: 04 April 2025.
Edited by:
Naohiko Yoshikai, Tohoku University, JapanReviewed by:
Cüneyt Türkeş, Erzincan Binali Yıldırım University, TürkiyeCopyright © 2025 Hashem, Abdelfattah, Hassan, Al-Emam, Alqarni, Alotaibi, Radwan, Kaur, Rao, Bräse and Alkhammash. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Devendra Pratap Rao, ZGV2ZW5kcmFwcmF0YXByYW9AeWFob28uY29t; Stefan Bräse, c3RlZmFuLmJyYWVzZUBraXQuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.