 Alshimaa A. Y. Ahmed1
Alshimaa A. Y. Ahmed1 Anber F. Mohammed
Anber F. Mohammed Zainab M. Almarhoon
Zainab M. Almarhoon Stefan Bräse
Stefan Bräse Bahaa G. M. Youssif
Bahaa G. M. Youssif- 1Pharmaceutical Organic Chemistry Department, Faculty of Pharmacy, Assiut University, Assiut, Egypt
- 2Department of Chemistry, College of Science, King Saud University, Riyadh, Saudi Arabia
- 3Institute of Biological and Chemical Systems, Institute of Biological and Chemical Systems—Functional Molecular Systems, Karlsruhe Institute of Technology, Karlsruhe, Germany
Introduction: This work outlines the design, synthesis, and biological evaluation of a new series of benzimidazole/1,2,3-triazole hybrids as apoptotic antiproliferative agents that inhibit the EGFR pathway.
Methods: The research assesses the antiproliferative efficacy of compounds 6a-i and 10a-i against various cancer cell lines.
Results and Discussion: The research emphasizing hybrids 6i and 10e for their remarkable activity, with GI50 values of 29 nM and 25 nM, respectively. The inhibitory effects of the most potent hybrids 6e, 6i, 10d, 10e, and 10g on EGFR were assessed. Compounds 6i and 10e exhibited greater potency than erlotinib as EGFR inhibitors. Compounds 6i and 10e were also examined for their apoptotic potential, revealing that these compounds promote apoptosis by activating caspase-3, caspase-8, and Bax, while down-regulating the anti-apoptotic protein Bcl-2. Molecular docking experiments are thoroughly examined to validate the binding interactions of the most active hybrids, 6i and 10e, with the EGFR active site. Furthermore, our new study examined the ADME properties of the new hybrids.
1 Introduction
Cancer remains one of the most severe diseases globally. The World Health Organization (WHO) states that cancer is the second leading cause of death globally (Workie et al., 2023). By 2030, 21.6 million new cancer cases will be annually (Gona et al., 2024). The epidermal growth factor receptor (EGFR) is a member of the ErbB family of receptor tyrosine kinases. Various malignancies, such as breast cancer and head and neck squamous cell carcinoma, increase its expression. Furthermore, EGFR overexpression occurs in approximately fifty percent of patients with non-small cell lung cancer (NSCLC) (Palumbo et al., 2023; Hatil et al., 2020). Consequently, EGFR represents a compelling target for anticancer treatment, leading to the development of many EGFR tyrosine kinase inhibitors (TKIs) (Metibemu et al., 2019; Abourehab et al., 2021; Levantini et al., 2022). They function by competitive inhibition of adenosine triphosphate (ATP) binding in the tyrosine kinase domain.
Benzimidazole is well-known in pharmacology, as its derivatives are often linked to diverse biological activities. The scaffold is a structural isostere of indole and purine; hence, its derivatives are anticipated to have a favorable affinity with diverse receptor types (Nardi et al., 2023; Acar Çevik et al., 2024). Benzimidazole derivatives demonstrate anticancer action by several mechanisms, including the inhibition of topoisomerase I and II, DNA intercalation, PARP-poly inhibition, and the inhibition of dihydrofolate reductase (DHFR) and aromatase (Çevik et al., 2022; Karadayi et al., 2020; Mostafa et al., 2019). For example, derivatives are used in Veliparib and Nocodazole, two well-known cancer medications that inhibit poly (ADP ribose) polymerase (PARP) and disrupt microtubule function, respectively (Yanaihara et al., 2023). Benzimidazole interacts with Met769 of EGFR in a binding manner similar to quinazoline, with the nitrogen atoms in the nucleus acting as hydrogen bond acceptors (Abdullah et al., 2022). Because benzimidazole is structurally similar to quinazoline, which is the building block for first- and second-generation drugs, this chemical could be a good starting point for future EGFR antagonists (Peerzada et al., 2023). However, more studies are needed before the benzimidazole-based drug is clinically approved for EGFR inhibition applications. Most research that examined the structures of benzimidazole derivatives produced conflicting results on the type and placement of substituents on the primary structure, as well as how well adding electron-withdrawing and electron-donating groups increased the capacity to bind to EGFR (JALIL and ABD HAMID, 2023).
We recently (Youssif et al., 2024) disclosed compound I (Figure 1), a benzimidazole-based anticancer agent selected by NCI for five-dose evaluation against 60 human carcinoma cell lines. Compound I exhibited significant selectivity towards the leukemia subpanel, with a selectivity ratio 5.96 at the GI50 level. Compound I was evaluated for its inhibitory effect on EGFR as a possible target for antiproliferative activity. The findings indicated that I exhibited a substantial antiproliferative effect as an EGFR inhibitor. Furthermore, compound I triggered apoptosis by elevating caspase-3, caspase-8, and Bax levels while reducing the anti-apoptotic protein Bcl2.
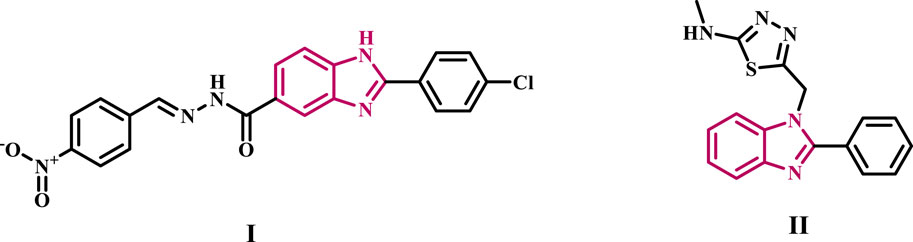
Figure 1. Structure of some benzimidazole-based EGFR inhibitors I and II.
Celik et al. (2019) identified compound II, a benzimidazole derivative, as a strong inhibitor of EGFR with antiproliferative properties. The docking investigation revealed that compound II had two hydrogen bonding interactions with the residues Lys721 and Thr830 within the binding pocket of EGFR.
On the other hand, 1,2,3-triazoles are nitrogen-containing heterocycles with three nitrogen atoms per ring. 1,2,3-Triazoles are stable molecules that form hydrogen bonds with biological targets. This makes them important building blocks for finding new drugs (Mahmoud et al., 2024; Maghraby et al., 2023). 1,2,3-Triazole compounds have diverse pharmacological properties, with anticancer action being the most prominent (Mahmoud et al., 2023). Researchers have documented the anticancer properties of 1,2,3-triazoles through various mechanisms. 1,2,3-Triazoles inhibit enzymes implicated in the advancement of this lethal disease, such as carbonic anhydrases (CAs) (Fatima et al., 2024), aromatase (Mishra and Upadhyay, 2022), vascular endothelial growth factor receptor (VEGFR) (Nguyen et al., 2022), and epidermal growth factor receptor (EGFR) (Kumar et al., 2023).
The phthalimide scaffold hybridized with the 1,2,3-triazole moiety (III, Figure 2), with an IC50 value of 0.22 μM, was particularly effective as an antiproliferative agent against MCF-7 cells. It also demonstrated strong EGFR inhibition, with an IC50 value of 79 nM, slightly higher than that of erlotinib. Compound III caused MCF-7 cells to undergo apoptosis, cell cycle arrest in the S/pre-G1 stages, and DNA fragmentation. The docking of III revealed hydrogen bonding interactions between the nitrogen of the 1,2,3-triazole ring and the Met769 residue, identical to the reference medication erlotinib. This demonstrates the role of the 1,2,3-triazole fragment in blocking the EGFR for anticancer therapy (Ihmaid et al., 2021).

Figure 2. Structure of some 1,2,3-triazole-based EGFR inhibitors III and IV.
We recently (Mahmoud et al., 2024) reported on the design and synthesis of a novel class of 1,2,3-triazole/1,2,4-oxadiazole hybrids that act as dual inhibitors of EGFR/VEGFR-2. The newly synthesized compounds were tested as antiproliferative agents using erlotinib as the reference medication. The results showed that most of the compounds tested had strong antiproliferative effects, with GI50 values ranging from 28 to 104 nM, whereas erlotinib’s GI50 value was 33 nM. The finding’s showed compound IV was the best derivative as an EGFR inhibitor, with an IC50 value of 76 nM, which is lower (more potent) than the reference drug erlotinib’s value of 80 nM. The docking analysis of IV within the EGFR active site demonstrated that the phenyl triazole moiety was deeply embedded in the hydrophobic pocket, corresponding with the phenylacetylene moiety of erlotinib. Furthermore, the 1,2,3-triazole molecule establishes a hydrogen connection with the Lys721 amino acid residue.
In our continuous pursuit of anticancer drugs targeting EGFR (El-Sherief et al., 2018; Abou-Zied et al., 2019; Mahmoud et al., 2022; Al-Wahaibi et al., 2023; Mostafa et al., 2024), we synthesized and evaluated a series of benzimidazole/1,2,3-triazole hybrids (6a-i and 10a-i, Figure 3) for their efficacy against EGFR. The recently synthesized compounds are classified into two categories (Figure 3): compounds of scaffold A are 2 (1-aryl-1,2,3-triazole-4-methylthio)benzimidazoles 6a-i, while scaffold B comprises 2-benzylthio-1-(1-aryl-1,2,3-triazole-4-methyl)benzimidazoles 10a-i. The newly synthesized compounds were tested in vitro against a panel of four cancer cell lines as antiproliferative agents. The most effective compounds were subsequently evaluated for EGFR inhibitory activity. Furthermore, the apoptotic efficacy of the most potent derivatives was assessed. Ultimately, docking analysis and ADMET evaluations were performed to determine the most effective variants.
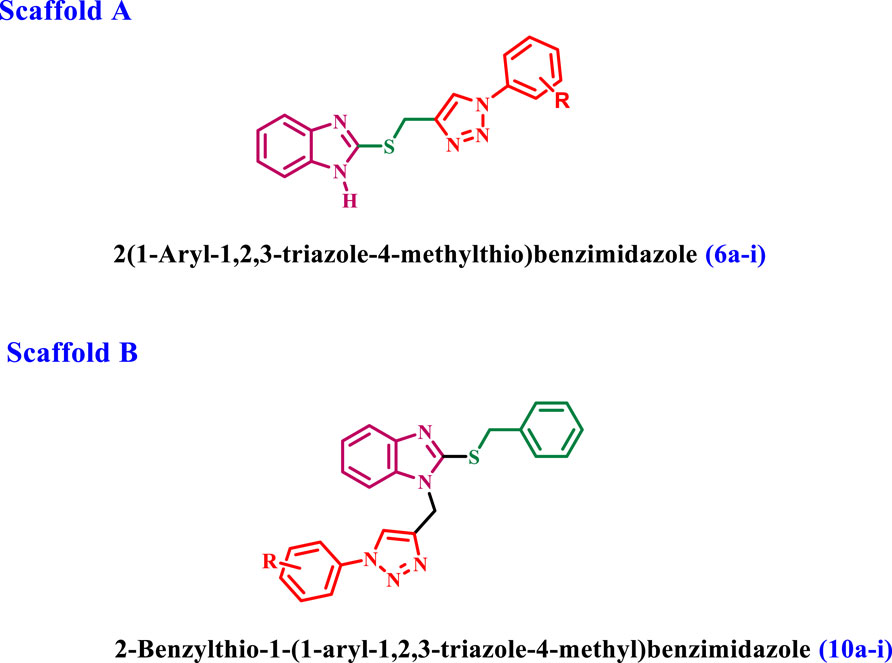
Figure 3. Structure of new benzimidazole/1,2,3-triazole hybrids 6a-i and 10a-i.
2 Results and discussion
2.1 Chemistry
Scheme 1 outlines the synthetic routes of the key intermediates and novel compounds 6a-i. Benzimidazole-2-thione (2) was synthesized through the reaction of o-phenylenediamine (1) with carbon disulfide in the presence of potassium hydroxide in an ethanol/water mixture in 82% yield (Latif et al., 2021). Compound 3, 2-(prop-2-yn-1-ylthio)-1H-benzimidazole was synthesized via the alkylation of compound 2 using propargyl bromide in the presence of anhydrous potassium carbonate in dry acetone (Dunga et al., 2022). Furthermore, we synthesized the substituted azide derivatives 5a-i from aryl amines through arene-diazonium salts using a documented method (Kutonova et al., 2013).
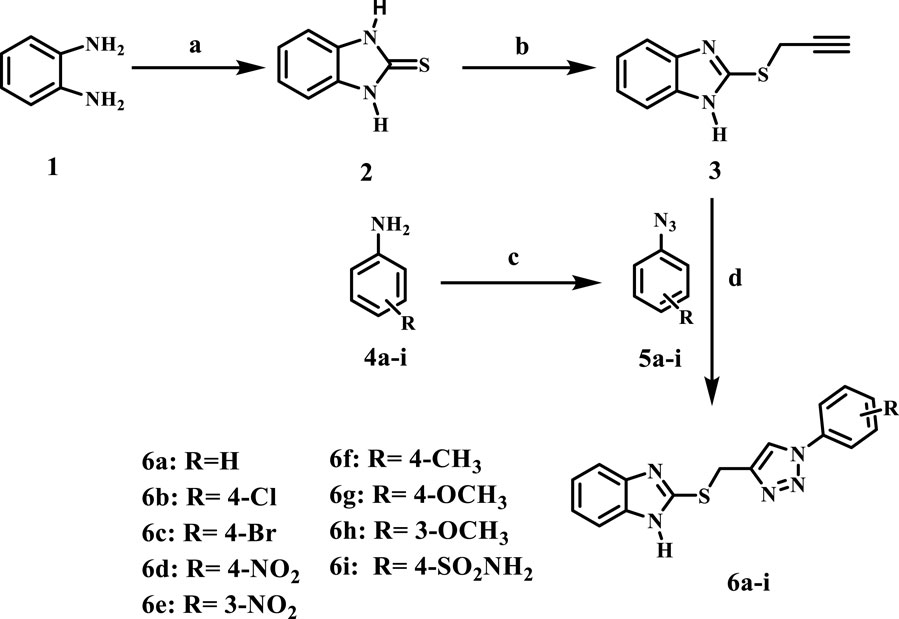
Scheme 1. Synthesis of the new compounds 6a-i. Reagents and reaction conditions: (A) CS2, KOH, EtOH: H2O (1.5:1), reflux, 30 h, yield, 90% (B) propargyl bromide, K2CO3, dry acetone, 7 h, yield, 70% (C) NaNO2, NaN3, HCl, H2O, stirring, 1 h, yield, 53% (D) CuSO4, Na ascorbate, THF: H2O (1:1), stirring, 24 h, yield, 50%–60%.
The final target compounds 6a-i was prepared through the click reaction of 2-propargayl-thiobenzimidazole 3 with the appropriate azides 5a-i in the presence of CuSO4 and sodium ascorbate with a THF: H2O (1:1) mixture, Scheme 1. 1H NMR, 13C NMR, elemental microanalyses, and representative IR spectral analysis established the structure elucidation of the new compounds. In general, the 1H NMR spectra of 6a-i verified the appearance of a broad singlet signal at 12.62–13.04 ppm corresponding to benzimidazole NH. In addition, two singlet signals appeared at 8.63–8.99 ppm and 4.69–4.81 ppm for the triazole-CH and SCH2 groups, respectively. Also, 13C NMR spectra of 6a-i showed signals at 25.8–27.4 ppm for the SCH2 group, and the aromatic carbon signals at δ = 110.9–140 ppm correspond to the benzimidazole carbons. The spectra also revealed the existence of additional signals in the aromatic region for the introduced phenyl moiety. As a representative example, the IR spectrum of compound 6d displayed a broad band at 3,400 cm-1 for the benzimidazole NH as well as two stretching bands at 1,340 cm-1 and 1,523 cm-1 for the NO2 group, along with a bending band at 854 cm-1 that confirms the para-disubstituted pattern.
Scheme 2 outlines the synthetic routes of the key intermediates and novel compounds 10a-i. Compound 8, 2-benzylthiobenzimidazole, was synthesized via the alkylation of compound 2 using benzyl bromide in the presence of anhydrous potassium carbonate in dry acetone (Dunga et al., 2022). Subsequently, 2-benzylthiobenzimidazole 8 was alkylated with propargyl bromide at the benzimidazole NH according to the compound 3 method of synthesis using K2CO3 and acetone to afford compound 9 (Kalyani and Manikyamba, 2004), Scheme 2.
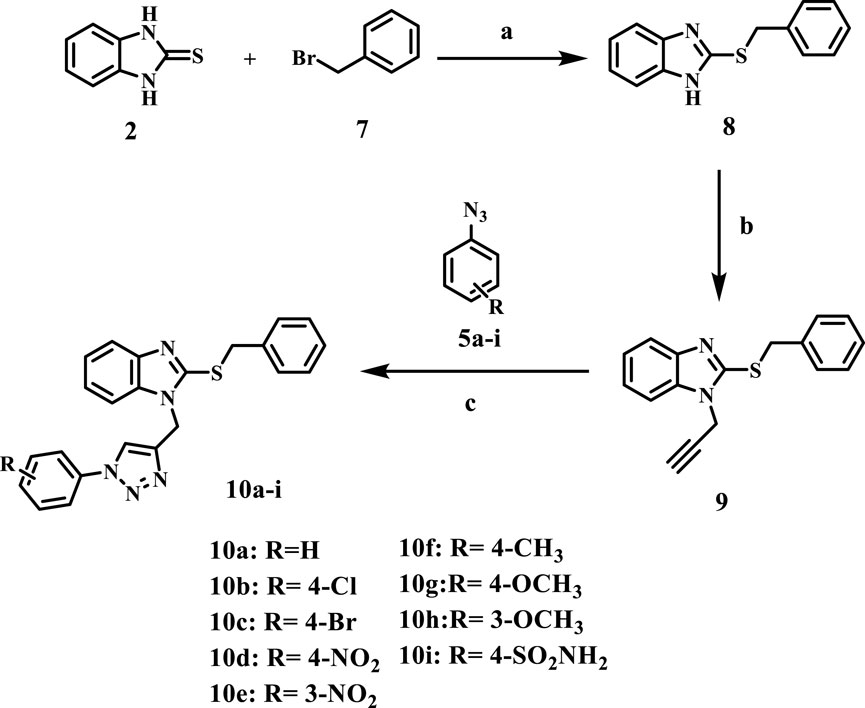
Scheme 2. Synthesis of the new compounds (10a-i). Reagents and reaction conditions: (A) benzyl bromide, K2CO3, dry acetone, 15 h, yield, 72% (B) propargyl bromide, K2CO3, dry acetone, 10 h, yield, 71% (C) CuSO4, Na ascorbate, THF: H2O (1:1), stirring, 24 h, yield, 30%–40%.
Like 6a-i, compounds 10a-i were synthesized by a click reaction between 9 and the appropriate azide derivatives 5a-i. Their structures were elucidated using IR, NMR, and elemental microanalyses. The 1H NMR spectra of 10a-i generally confirmed the appearance of two singlet signals at 4.72–4.79 ppm, 5.47–5.49 ppm, and 8.76–8.90 ppm for the SCH2, NCH2, and triazole CH groups, respectively. The spectra also revealed the existence of additional signals in the aromatic region for the introduced phenyl moiety along with the aromatic protons of the benzimidazole ring and the 2-thiobenzyl ring. In addition, 13C NMR spectra of 10a-i displayed signals at 36.57–36.61 ppm and 39.11–39.34 ppm for the SCH2 and NCH2 groups, respectively. Benzimidazole carbon signals appear in the δ = 110.5–137.8 ppm range. As a representative example, the IR spectrum of compound 10d displayed two stretching bands at 1,340 cm-1 and 1,523 cm-1 for the NO2 group, along with a bending band at 854 cm-1 that confirms the para-disubstituted pattern.
2.2 Biology
2.2.1 Cell viability assay
The MCF-10A normal human mammary gland epithelial cell line was used to test the effects of compounds 6a-i and 10a-i on cell viability. The MTT assay was used to check the cell viability effect of compounds 6a-i and 10a-i after 4 days of treatment with MCF-10A cells (Mekheimer et al., 2022; Hisham et al., 2022). Table 1 results indicate that none of the examined compounds exhibited cytotoxicity, as all hybrids maintained cell viability above 84% at a concentration of 50 µM.
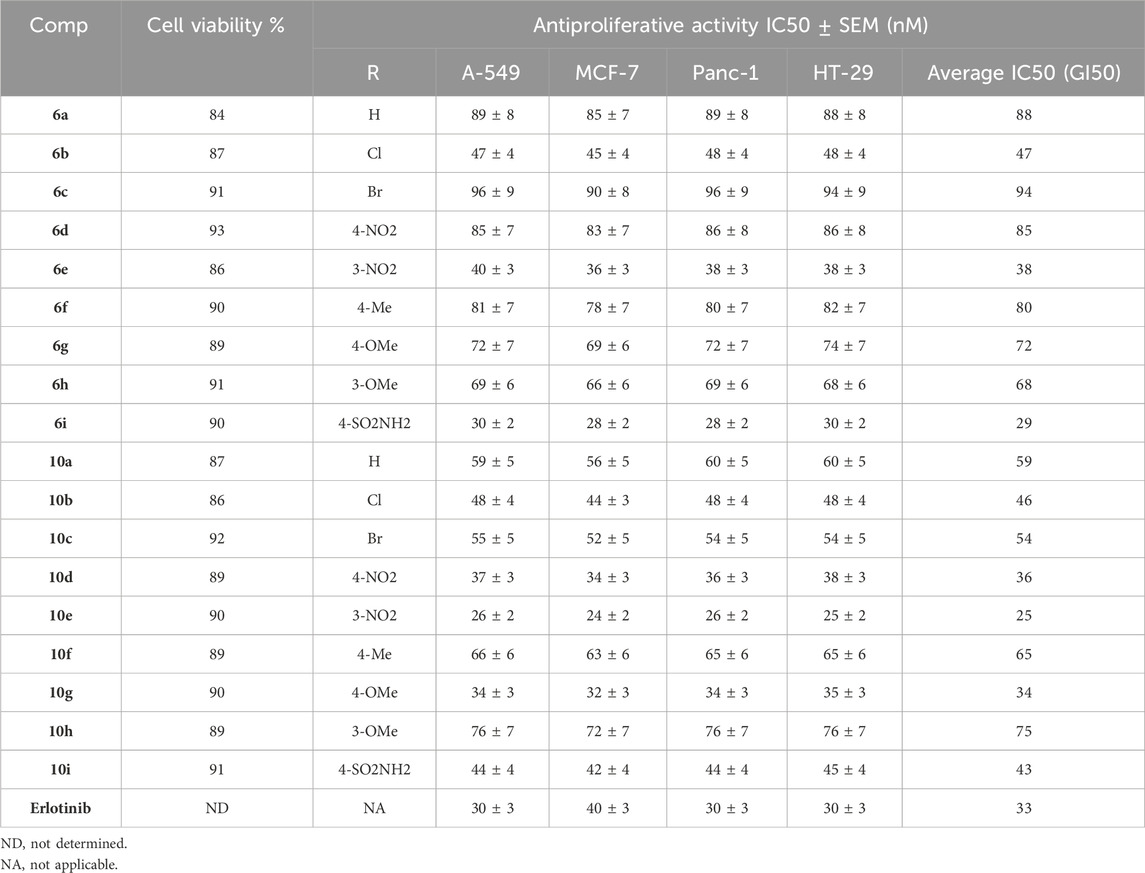
Table 1. IC50 values of compounds 6a-i and 10a-i against four cancer cell lines.
2.2.2 Antiproliferative assay
We assessed the antiproliferative efficacy of novel compounds 6a-i (Scaffold A) and 10a-i (Scaffold B) against four human cancer cell lines (colon: HT-29, lung: A-549, breast: MCF-7, and pancreatic: Panc-1) (El-Sherief et al., 2019; Al-Wahaibi et al., 2022). Erlotinib served as the control in this investigation. Table 1 displays the median inhibitory concentration (IC50) and GI50 (mean IC50) values for the four cancer cell lines.
The tested compounds 6a-i and 10a-i had strong antiproliferative activity, with GI50 values ranging from 25 nM to 94 nM against the four cancer cell lines that were tested. This is in comparison to the standard erlotinib, which had a GI50 value of 33 nM. The five most potent derivatives were compounds 6e, 6i, 10d, 10e, and 10g, with GI50 values ranging from 25 nM to 38 nM. Compounds 6i and 10e, with GI50 values of 25 and 29 nM, demonstrated greater potency than erlotinib, which had a GI50 of 33 nM.
With a GI50 value of 25 nM, compound 10e (R = 3-NO2, Scaffold B) was the most effective of the newly synthesized derivatives 6a-i and 10a-i. It was 1.4 times stronger than erlotinib (GI50 = 33 nM). Compound 10e demonstrated greater potency than the reference erlotinib against all tested cancer cell lines. Compound 6i (R = 4-SO2NH2, Scaffold A) had the second-highest activity, with a GI50 value of 29 nM. It was slightly more effective than the standard erlotinib, whose GI50 value was 33 nM. Compound 6i exhibited greater potency than erlotinib against breast (MCF-7) and pancreatic (Panc-1) cancer cell lines.
The findings show that the type and/or the position of substitutions found on the phenyl ring at position one of the 1,2,3-triazole moiety in both scaffold A and B compounds are essential for antiproliferative action. Compound 10d (R = 4-NO2, Scaffold B), possessing an identical backbone to compound 10e but featuring a nitro group at the 4-position on the phenyl ring, exhibited a GI50 of 36 nM (1.5-fold less potent than 10e), indicating that the nitro group at the 3-position is more conducive to antiproliferative activity than at the 4-position. Unfortunately, this requirement does not apply to all derivatives of scaffold B compounds. Compound 10g (R = 4-OMe, Scaffold B) demonstrated the third greatest activity, with a GI50 of 34 nM. Shifting the methoxy group from position 4 on the phenyl ring to position 3, as in compound 10h (R = 3-OMe, Scaffold B), resulted in a considerable drop in antiproliferative activity. Compound 10h had a GI50 of 75 nM, two times less potent than the 4-methoxy derivative, compound 10g.
Compounds 10a (R = H, Scaffold B), 10b (R = 4-Cl, Scaffold B), 10c (R = 4-Br, Scaffold B), 10f (R = 4-Me, Scaffold B), and 10i (R = 4-SO2NH2, Scaffold B) demonstrated GI50 values of 59, 46, 54, 65, and 43 nM, respectively. All these compounds exhibited lower potency than 10e (R = 3-NO2, Scaffold B) and even 10d (R = 4-NO2, Scaffold B). These data demonstrate that in scaffold B compounds, the nature and/or position of the substitutions significantly influences activity, with activity increasing in the following order: 3-NO2 > 4-OMe > 4-NO2 > 4-SO2NH2 > Cl > Br > H > Me. The same is true for scaffold A compounds: the most active derivatives are those with NO2, SO2NH2, and OMe groups, followed by those with halogen substituents, and the least active are those with methyl substituting or non-substituting. Finally, a future goal for this research is to synthesize and evaluate more triazole and/or benzimidazole moiety derivatives to achieve an accurate SAR.
2.2.3 EGFR inhibitory assay
The most effective antiproliferative derivatives, 6e, 6i, 10d, 10e, and 10g, were evaluated for their ability to inhibit EGFR using the EGFR-TK test (Mahmoud et al., 2023; Alshammari et al., 2022). The results are presented in Table 2. Erlotinib served as the reference compound. The assay results align with those of the antiproliferative assay, indicating that compounds 6i (R = 4-SO2NH2, Scaffold A) and 10e (R = 3-NO2, Scaffold B), identified as the most potent antiproliferative agents, are the most efficacious derivatives of EGFR inhibitors, exhibiting IC50 values of 78 ± 5 and 73 ± 4, respectively. Compounds 6i and 10e demonstrated more potency than erlotinib as EGFR inhibitors, with an IC50 value of 80 nM. Compounds 6e, 10d, and 10g exhibited substantial inhibition of EGFR, with IC50 values of 89, 86, and 82 nM, respectively. These molecules exhibited marginally reduced potency compared to erlotinib. The data suggest that compounds 6e, 6i, 10d, 10e, and 10g are extremely effective antiproliferative candidates that may function as EGFR inhibitors.
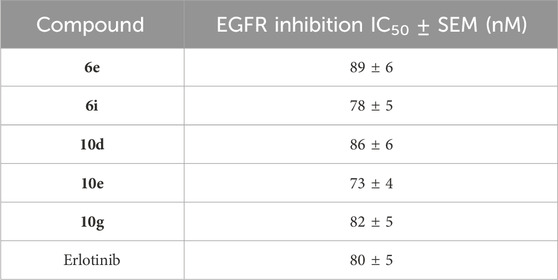
Table 2. IC50 values of compounds 6e, 6i, 10d, 10e, 10g, and erlotinib against EGFR.
2.2.4 Apoptotic markers assays
Apoptosis is an essential cellular process in animal growth, tissue homeostasis, and immune responses. In a healthy body, a vital equilibrium exists between apoptotic and anti-apoptotic mediators during normal physiological processes. Nonetheless, an imbalance may occur in some circumstances, potentially resulting in illnesses (Al-Mahmoudy et al., 2022). Excessive activation or suppression of apoptotic mediators frequently results in this imbalance. Pathological disorders, such as cancer, can disrupt this equilibrium. Compounds 6i and 10e, which demonstrated the greatest potency in all laboratory assays, were examined to assess their ability to initiate the apoptosis cascade and display proapoptotic activity.
2.2.4.1 Assays for caspases 3 and 8
Cells experience apoptosis in reaction to specific signaling cues, resulting in significant modifications. Caspases are considered the primary mediators of apoptosis, initiating the process at an early stage (Sahoo et al., 2023). They decompose vital cellular components, including nuclear proteins, such as DNA repair enzymes and structural proteins within the cytoskeleton, essential for optimal cellular function. Caspases can activate DNases, enzymes that damage nuclear DNA (Larsen and Sørensen, 2017). Compounds 6i and 10e were assessed as activators of caspase-3/8 in the MCF-7 breast cancer cell line (Abdelbaset et al., 2019). The results of this experiment are presented in Table 3.
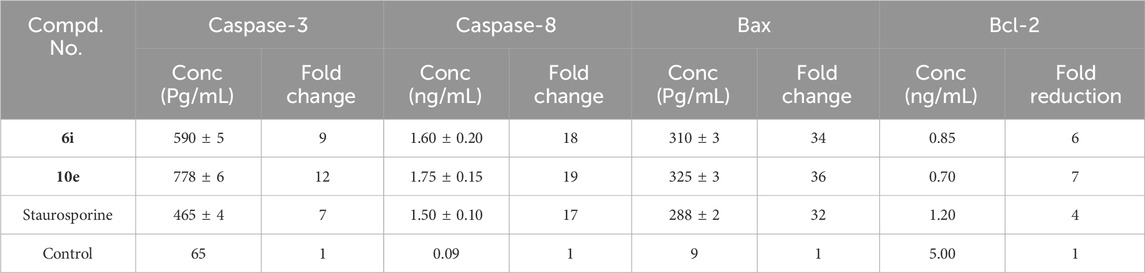
Table 3. Apoptotic capabilities of compounds 6i and 10e.
When MCF-7 cells were treated with compound 10e at its IC50 concentration, it greatly increased the levels of activated caspases 3 and 8. Table 3 shows a 12-fold increase in active caspase-3 expression and a 19-fold increase in active caspase-8 expression. Upon treatment with compound 6i, the levels of caspase-3 and caspase-8 increase significantly—by 9 and 18 times, respectively, compared to untreated cells. In every instance, compounds 6i and 10e demonstrated superior efficacy as activators of caspase-3 and caspase-8 compared to the reference staurosporine.
2.2.4.2 Assays for the proapoptotic Bax and anti-apoptotic Bcl-2
The current study supplied compounds 6i and 10e to breast (MCF-7) cancer cell lines at their respective IC50 values. This led to a notable elevation of pro-apoptotic Bax expression, with a fold increase of 34 for compound 6i and 36 for compound 10e. Also, the treatment significantly decreased anti-apoptotic Bcl-2 expression levels, with a reduction of approximately 6-fold for compound 6i and 7-fold for compound 10e. The results are given in Table 3. Compounds 6i and 10e significantly increased the Bax/Bcl-2 ratio relative to the control untreated cells. These data suggest that apoptosis may be one of the factors contributing to the antiproliferative activity of these compounds.
2.3 Molecular modelling
Docking simulations of the new compounds 6a-i and 10a-i were performed at the ATP-binding site of EGFR to explore their potential binding modes and rationalize the biological results. The crystal structure of the EGFR in complex with the aniline-quinazoline inhibitor erlotinib (PDB: 1M17) (Stamos et al., 2002) was used in the present investigation. All minimizations were performed using the MOE force field (OPLS-AA) and the Born solvation model (Belal et al., 2022). Accuracy of the docking protocol was achieved by redocking the co-crystallized ligand into the EGFR binding site from where the docked ligand displayed an RMSD value of 0.96 Å (Figure 4). The new compounds exhibited good docking scores (−5.70 to −8.82 kcal/mol) relative to the erlotinib docking score of −11.80 kcal/mol. The docking results of the compounds were compared with erlotinib, and the results are shown in Table 4. Regarding Scaffold A (6a-i), the ligand 2-thiobenzimidazole inserts into the hydrophobic pocket in alignment with the erlotinib phenylacetylene moiety, forming stacking between Lys721 and Thr766. Also, the sulfur atom forms a similar water-bridged H-bond with Asp831 at the DFG motif as the NH spacer of the reference erlotinib. In addition, the ligand triazole methylene moiety occupies the location of erlotinib quinazoline, forming pi-H contacts with Leu694 and Gly772. The triazole nitrogen of the most active derivatives (6e and 6i) accepts an additional H bond from the backbone NH of the key amino acid Met769, similar to the quinazoline N-1 of erlotinib. The substituted-phenyl tail projects past an ether chain of the erlotinib, forming pi-H interactions with Leu694 at the gate of the binding site. Interestingly, the amino sulfonyl group of the utmost active derivative 6i donates unique H-bond contact to Leu694 (Figure 5). Meanwhile, compound 6e probes an opposite orientation within the binding site, where the ligand 3-NO2-phenyl moiety is inserted into the hydrophobic pocket and accepts an H-bond contact from Thr766. The 2-thiobenzimidazole alternatively forms an H-bond with the amide carbonyl of Met769 and a pi-H contact between the aryl moiety and the Leu694 residue. On the other hand, scaffold B derivatives (10a-i) are better at probing the space of the binding site than scaffold A derivatives. The ligand benzyl group was inserted into the hydrophobic pocket in alignment with the erlotinib phenylacetylene moiety. Furthermore, the ligand 2-thiobenzimidazole ring is lying in the location of the erlotinib quinazoline moiety close to the amino acid residue Met769. In the complexes of the unsubstituted compound 10a and the 4-methoxy compound 10g, the benzimidazole nitrogen accepts an H-bond from Met769 amide nitrogen compared to the H-bond with the quinazoline N-1 of erlotinib. At the same time, the nitro-containing derivatives (10d and 10e) form a water-bridged H-bond with Thr766, similar to the erlotinib quinazoline N-3. Also, the sulfur atom of these derivatives forms an additional H-bond interaction with Leu820. The substituted-phenyl triazole tail projects past the erlotinib ether chains, forming pi-H interactions with Leu694 at the gate of the binding site. Besides, the 3-NO2 moiety of the utmost active derivative 10e forms a water-bridged H-bond with Pro770, similar to the ether chain of erlotinib at the gate of the binding site (Figure 6). By shifting the methoxy group to position 4 at the phenyl ring in the 10d protein complex, the interaction with the amino acid residue Pro770 at the binding gate is lost. Moreover, the triazole ring in the 10c, 10g, and 10i complexes forms a water-bridged pi-H contact with Cys773 at the binding gate. According to the docking simulation results, EGFR might be a plausible target for the antiproliferative action of novel scaffolds.
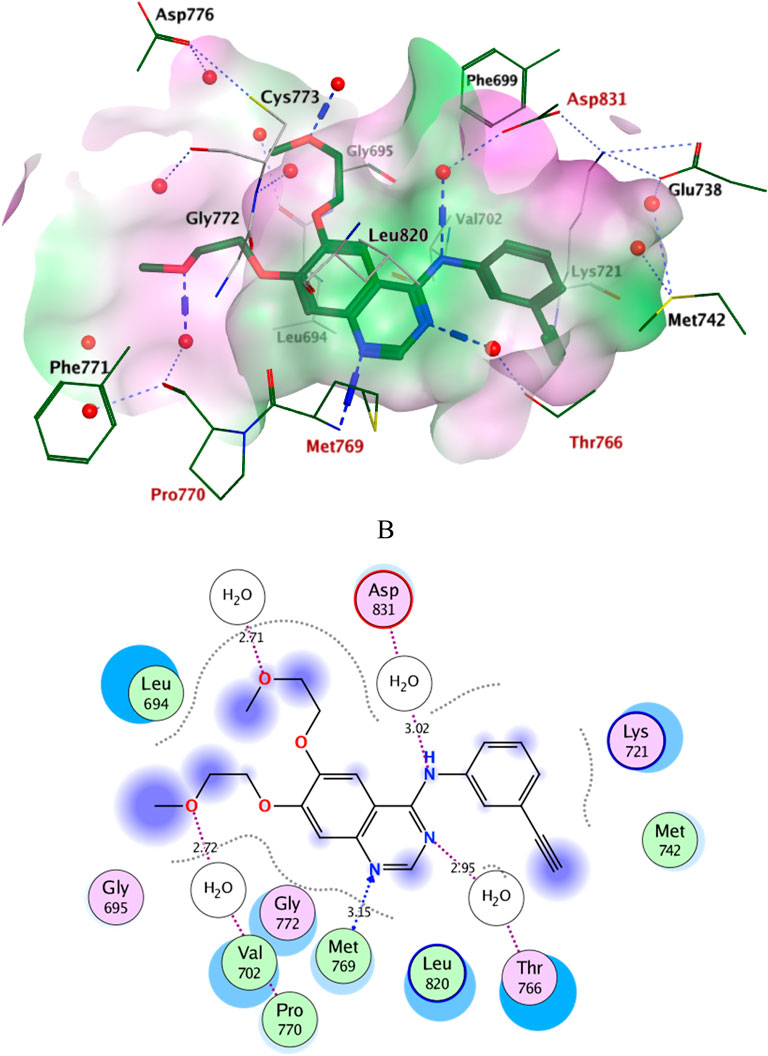
Figure 4. Ligplots at ATP-binding site of EGFR; (A) 3D-docked model of erlotinib (dark green) showing the protein lipophilicity surface (purple: hydrophilic, white: neutral, Green: lipophilic), (B) 2D-docked model of erlotinib. A.
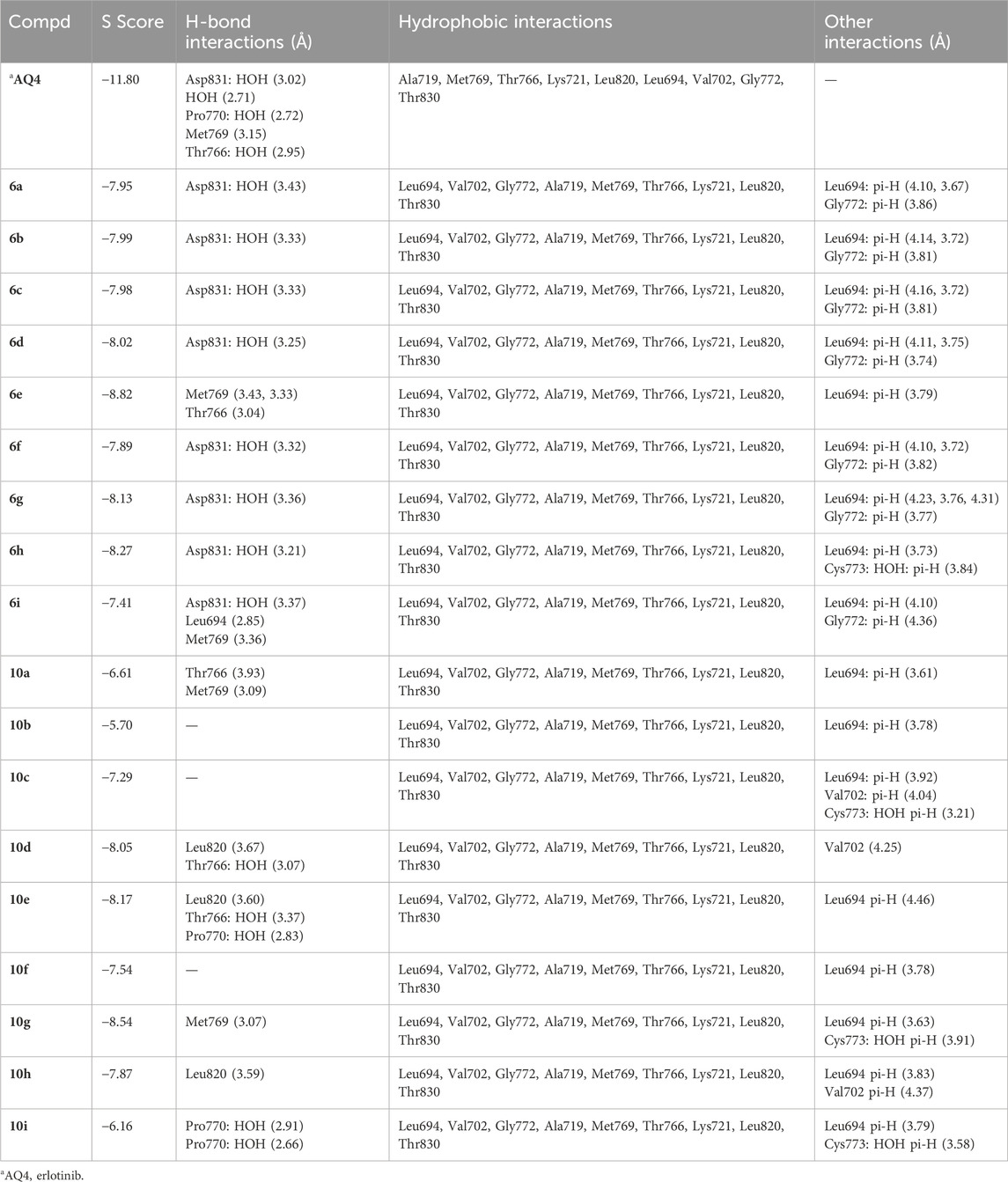
Table 4. Ligand-protein complex interactions of the tested compounds 6a-i and 10a-i within the ATP-binding site of EGFR.
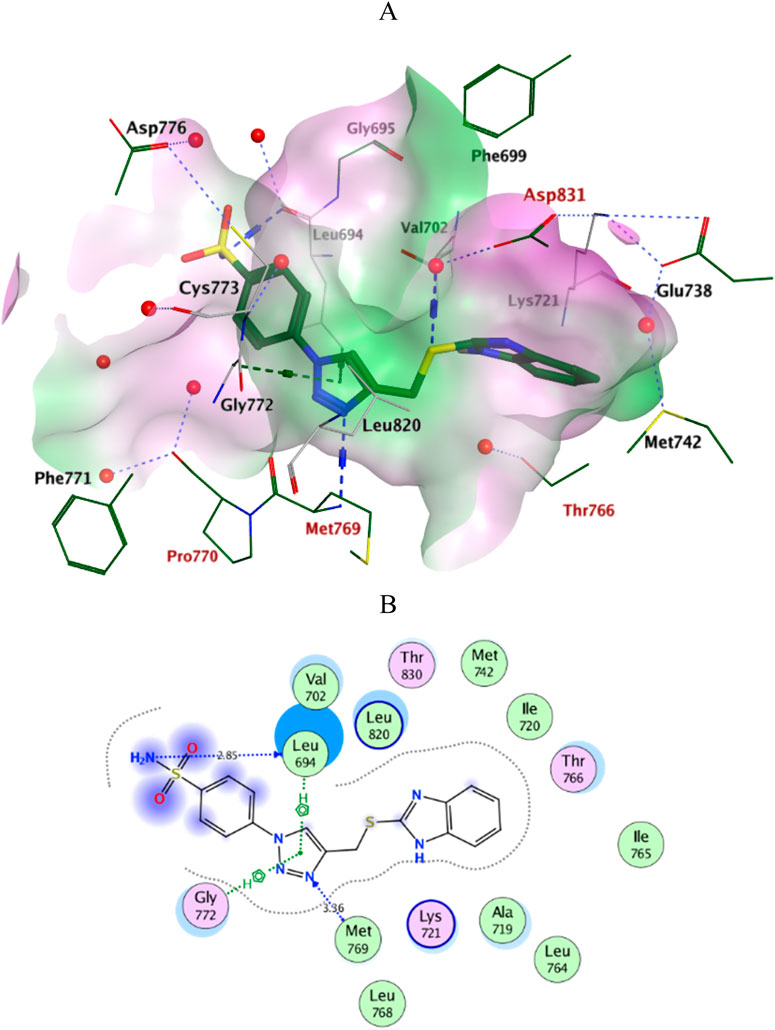
Figure 5. Ligplots at ATP-binding site of EGFR; (A) 3D-docked model of Scaffold A 6i (dark green) showing the protein lipophilicity surface (purple: hydrophilic, white: neutral, Green: lipophilic), (B) 2D-docked model of 6i. A.
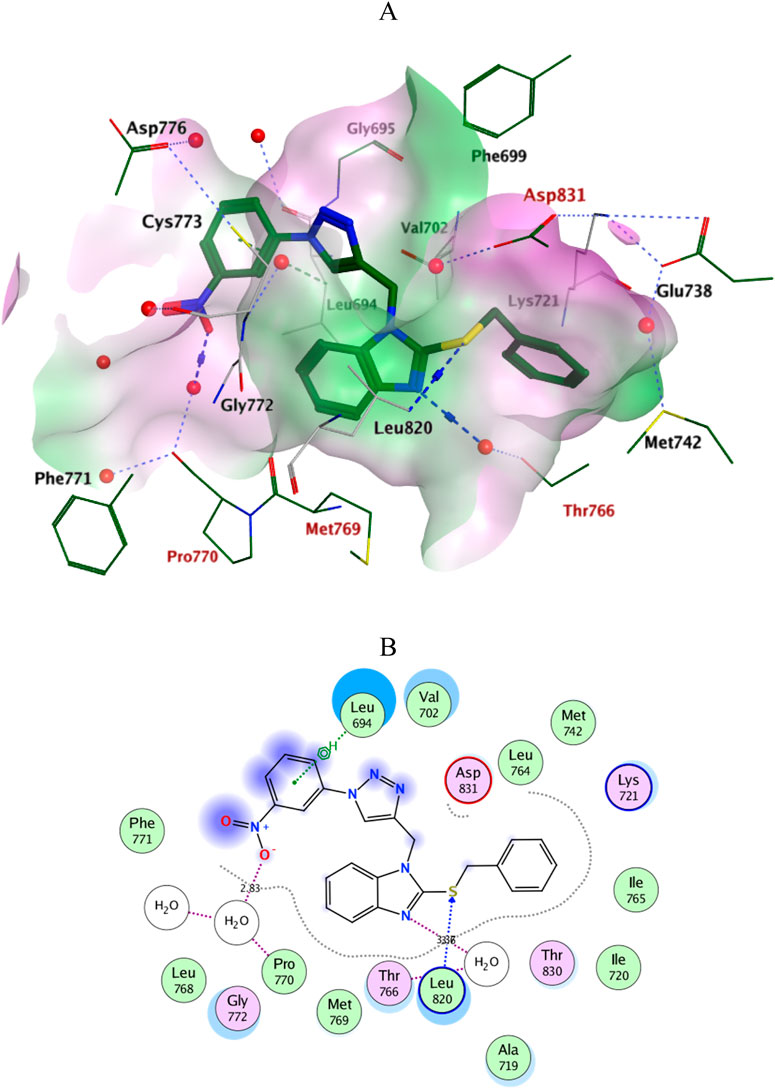
Figure 6. Ligplots at ATP-binding site of EGFR; (A) 3D-docked model of Scaffold B 10e (dark green) showing the protein lipophilicity surface (purple: hydrophilic, white: neutral, Green: lipophilic), (B) 2D-docked model of 10e.
2.4 In silico ADMET/pharmacokinetics studies
The ADMET properties of compounds 6i and 10e were predicted using the pkCSM-pharmacokinetics server (http://biosig.unimelb.edu.au/pkcsm/)by generating SMILES (Simplified Molecule Input Line Entry Specification) of the compounds using ChemDraw software (Pires et al., 2015). The compound’s efficacy as an orally active drug is determined using Caco2 permeability and intestinal absorption models. Both compounds obey Lipinski’s rules of five with zero violation, Table 5. The scaffold B derivative 10e displays a higher Caco2 permeability value and demonstrates superior intestinal absorption ability with 100% than the scaffold A compound 6i. The second variable in absorption is skin permeability, and both compounds have permeability values of less than −2.5 log Kp, suggesting poor permeability. P-glycoprotein is a factor of the ATP-binding cassette (ABC) transporter, essential for the active molecular transport across cell membranes. Both compounds are predicted to be P-glycoprotein substrates, suggesting they can move through the cell membrane via the ABC transporter. Besides this, both compounds were effective as inhibitors for P-glycoprotein II transporters. In contrast, only 10e was effective as an inhibitor for the P-glycoprotein I transporters, implying that 6i cannot inhibit type II drug efflux pumps. The VDss assay estimates the total amount of drug needed for uniform drug distribution in the blood. However, both compounds show low VDss values, indicating slower diffusion in blood. The compound’s ability to move to the brain can be determined via the permeability of the Blood-Brain Barrier (BBB). They will be poorly distributed to the brain if the logBB values are less than −1. Thus, both compounds might be unable to cross BBB. The blood-brain permeability surface area product (logPS) has a more direct and accurate estimation. Both compounds have logPS > −3 and can penetrate the CNS. The metabolism of the compounds in the body was predicted using seven different cytochrome models. Both Compounds are likely to be metabolized by CYP3A4 and also going to be CYP1A2 and CYP2C9 inhibitors. The predicted total clearance rates for the compounds are shown in Table 6, and only 10e appeared as a substrate for the organic cation transporter 2 (OCT2). Furthermore, only 10e predicted AMES toxicity, suggesting this compound might have carcinogenicity or mutagenicity. Also, both compounds will likely be hERG II inhibitors without effect on hERG I. Skin sensitization and hepatotoxicity were not seen in compound 10e, while 6i might show hepatotoxicity. The toxic effects of compounds are shown in Table 6, along with other ADMET properties.

Table 5. Physicochemical properties of compounds 6i and 10e.
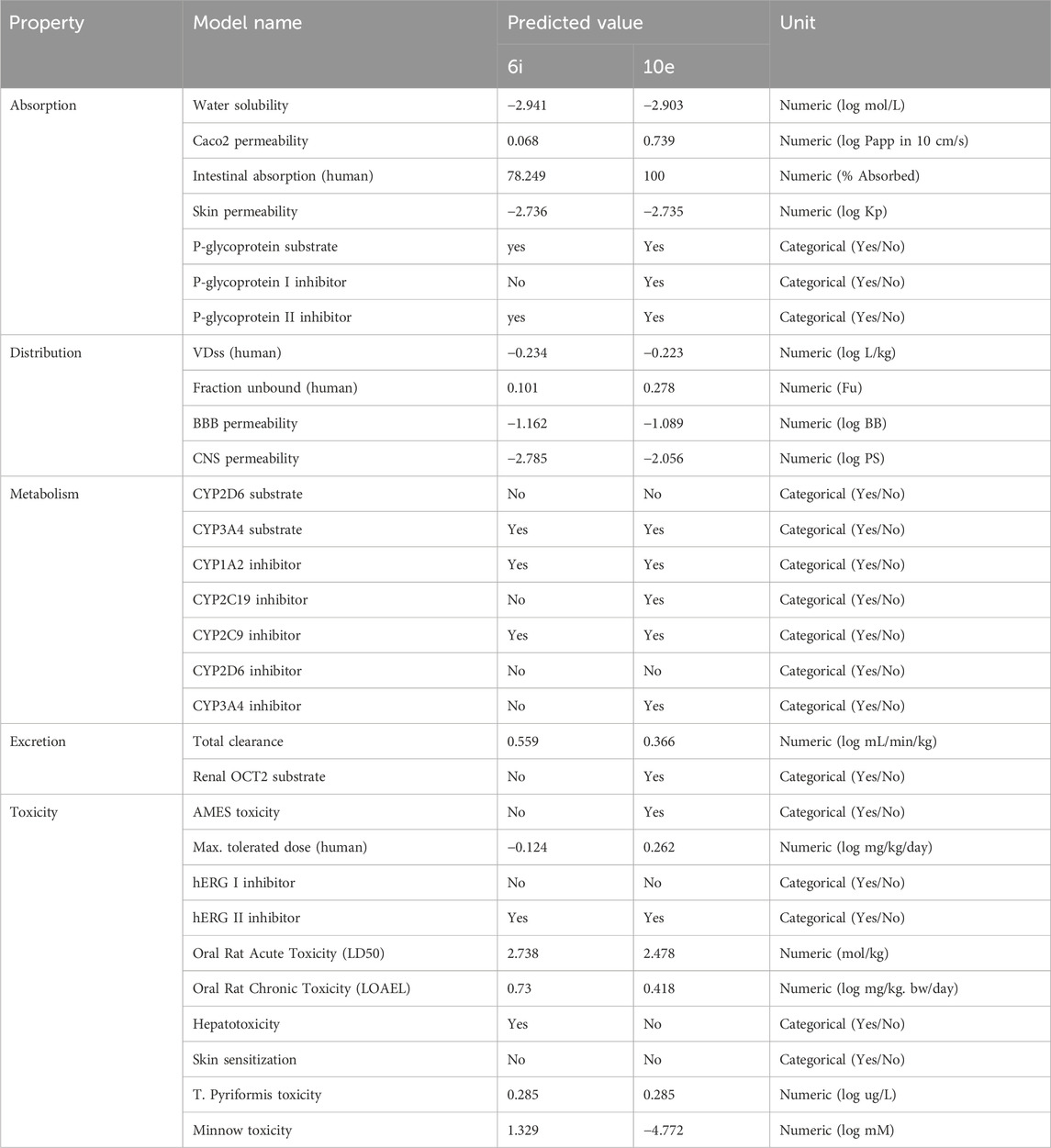
Table 6. ADMET properties of compounds 6i and 10e.
3 Conclusion
This study presents the design and synthesis of novel benzimidazole/1,2,3-triazole hybrids that can block the EGFR enzyme. Compounds 6i and 10e were identified as the most compelling due to their potent EGFR inhibition, with IC50 values of 78 and 73 nM, respectively. Furthermore, they exhibited possible antiproliferative properties against the MCF-7 breast cancer cell line (IC50 = 28 and 24 nM, respectively). At a dose of 50 μM, compounds 6i and 10e exhibited no impact on non-tumor cells MCF-10A, suggesting the potential tumor-cell selectivity of these derivatives. Molecular docking experiments have effectively illustrated the unique binding interactions of compounds 6i and 10e with the EGFR active site. This comprehensive examination is essential for comprehending their mode of action as EGFR inhibitors. The thorough evaluation of these hybrids’ absorption, distribution, metabolism, and excretion (ADME) characteristics highlights their potential as therapeutic agents. The findings indicate that 6i and 10e are attractive candidates for the development of novel medicines for cancer therapy. Furthermore, further exploration of the mechanism of action, in vivo carcinogenic animal models, and lead optimization is underway in our laboratory.
4 Experimental
4.1 Chemistry
4.1.1 Materials and methods
See Supplementary Appendix A Compounds 2, 3, 5a-i, 8, and 9 were prepared according to reported procedures (Latif et al., 2021; Dunga et al., 2022; Kutonova et al., 2013; Kalyani and Manikyamba, 2004).
4.1.2 General method for the synthesis of compounds 6a-i and 10a-i
To a stirred solution of the compound 3 or 9 (1 eq.) in a mixture of 10 mL THF and 10 mL H2O, the appropriate azide derivatives 5a-i (1.5 eq.) were added. The mixture was stirred for 45 min. Then sodium ascorbate (0.05 g, 0.2 eq.) was added initially, followed by the addition of CuSO4 (0.08 g, 0.1 eq.) to the reaction mixture and continued stirring till the completion of the reaction (monitored by TLC). Compounds 6a-i were purified by column chromatography using silica gel eluted gradually with hexane: EtOAc (100:0 to 30:70, v/v). Compounds 10a-i were purified by recrystallization using DMF: H2O (1:2).
4.1.2.1 2-(((1-Phenyl-1H-1,2,3-triazol-4-yl)methyl)thio)-1H-benzimidazole (6a)
Yield: 0.48 g (56%), Yellow powder, m. p: 137°C–138°C, Rf: 0.3 (hexane: ethyl acetate, 1:1, v/v); 1H NMR (400 MHz, DMSO-d6): δ = 12.95 (br s, 1H, NH), 8.79 (s, 1H; triazole CH), 7.82 (t, J = 8.6 Hz, 2H, Ar-H), 7.68 (d, J = 7.7 Hz, 1H, Ar-H), 7.61–7.52 (m, 2H, Ar-H), 7.48 (d, J = 7.3 Hz, 2H, Ar-H), 7.28–7.05 (m, 2H, ArH-5.6), 4.79 (s, 2H, SCH2). 13C NMR (101 MHz, DMSO-d6) δ 143.0, 136.3, 129.8, 128.7, 122.0, 121.8, 120.0, 117.8, 109.2, 26.8. Anal. Calc. (%) for C16H13N5S: C, 62.52; H, 4.26; N, 22.78; S, 10.43. Found: C, 62.35; H, 4.47; N, 23.05; S, 10.59.
4.1.2.2 2-(((1-(4-Chlorophenyl)-1H-1,2,3-triazol-4-yl)methyl)thio)-1H-benzimidazole (6b)
Yield: 0.24 g (89%), Yellow powder, m. p: 145°C–147°C, Rf: 0.35 (hexane: ethyl acetate, 1:1, v/v); 1H NMR (400 MHz, DMSO-d6): δ = 12.71 (br s, 1H, NH), 8.78 (s, 1H; triazole CH), 7.91 (d, J = 8.8 Hz, 2H, ArH-3′,5′), 7.65 (d, J = 8.8 Hz, 2H, Ar-H-2′,6′), 7.51–7.44 (m, 2H, ArH-4.7), 7.24–7.10 (m, 2H, ArH-5.6), 4.71 (s, 2H; SCH2). 13C NMR (101 MHz, DMSO-d6) δ 144.6, 135.3, 132.9, 129.8, 127.1, 121.9, 121.7, 121.5, 117.9, 25.8. Anal Calc. (%) for: C16H12ClN5S C,56.22; H, 3.54; N, 20.49; S,9.38 Found: C,56.49; H,3.66; N,20.73; S,9.41.
4.1.2.3 2-(((1-(4-Bromophenyl)-1H-1,2,3-triazol-4-yl)methyl)thio)-1H-benzimidazole (6c)
Yield: 0.52 g (51%), Yellow powder, m. p: 168°C–170°C, Rf: 0.41 (hexane: ethyl acetate, 1:1, v/v); 1H NMR (400 MHz, DMSO-d6): δ = 12.64 (br s, 1H, NH), 8.78 (s, 1H, triazole CH), 7.85 (d, J = 9.0 Hz, 2H, Ar-H -3′,5′), 7.78 (d, J = 9.0 Hz, 2H, Ar-H-2′,6′), 7.54–7.43 (m, 2H, Ar-H-4.7), 7.14 (dd, J = 6.0, 3.2 Hz, 2H, Ar-H-5.6), 4.72 (s, 2H, SCH2). 13C NMR (101 MHz, DMSO-d6) δ 149.2, 144.6, 143.3, 142.9, 135.7, 127.3, 121.9, 121.8, 121.3, 110.1, 25.8. Anal. Calc. (%) for: C16H12BrN5S C, 49.75; H, 3.13 N, 18.13; S, 8.30, Found: C,49.93; H,3.25; N, 18.40; S, 8.25.
4.1.2.4 2-(((1-(4-Nitrophenyl)-1H-1,2,3-triazol-4-yl)methyl)thio)-1H-benzimidazole (6d)
Yield: 0.52 (59%), Yellow powder, m. p:162°C–164°C, Rf: 0.28 (hexane: ethyl acetate, 1:1, v/v); IR (KBr, ύ cm-1): 3,400 (NH), 3,084 (=CH), 2,962 (CH2), 1,640, 1,598 (C=N, C=C), 1,523, 1,340 (NO2), 854 (p-bending). 1H NMR (400 MHz, DMSO-d6): δ = 12.64 (br s, 1H, NH), 8.97 (s, 1H, triazole CH), 8.43 (d, J = 9.2 Hz, 2H, Ar-H-3′,5′), 8.20 (d, J = 9.2 Hz, 2H, Ar-H-2′,6′), 7.61–7.52 (m, 1H, Ar-H), 7.44–7.35 (m, 1H, Ar-H), 7.21–7.08 (m, 2H, Ar-H-5.6), 4.74 (s, 2H, SCH2). 13C NMR (101 MHz, DMSO-d6) δ 149, 146.6, 145.2, 140.7, 125.5, 122.2, 121.2, 120.6, 117.5, 110.4, 25.7. Anal Calc. (%) for C16H12N6O2S: C, 54.54; H, 3.43; N, 23.85; S, 9.10 Found: C,54.71; H,3.54; N,24.01; S,9.23.
4.1.2.5 2-(((1-(3-Nitrophenyl)-1H-1,2,3-triazol-4-yl)methyl)thio)-1H-benzimidazole (6e)
Yield: 0.1 g (56%) Yellow powder, m. p: 123°C–125°C, Rf: 0.32 (hexane: ethyl acetate, 1:1, v/v). 1H NMR (400 MHz, DMSO-d6): δ = 12.64 (br s, 1H, NH), 8.99 (s, 1H, triazole CH), 8.68 (s, 1H, Ar-H), 8.35 (dd, J = 8.2, 2.1 Hz, 1H, Ar-H), 8.29 (dd, J = 8.2, 2.2 Hz, 1H, Ar-H), 7.86 (t, J = 8.1 Hz, 1H, Ar-H), 7.66–7.50 (m, 1H, Ar-H), 7.43–7.32 (m, 1H, Ar-H), 7.12 (dd, J = 6.2, 3.0 Hz, 2H, Ar-H), 4.74 (s, 2H, SCH2). 13C NMR (101 MHz, DMSO-d6): δ 148.9, 145.2, 144.0, 137.5, 131.9, 126.5, 123.6, 122.7, 122.3, 118.4, 115.2, 110.6, 27.3. Anal. Calc. (%) for C16H12N6O2S: C,54.54; H, 3.43; N, 23.85; S,9.10, Found: C,54.70; H,3.52; N,24.09; S,9.17.
4.1.2.6 2-(((1-(p-Tolyl)-1H-1,2,3-triazol-4-yl)methyl)thio)-1H-benzimidazole (6f)
Yield: 0.8 g (94%), Yellow powder, m. p: 134–136̊C, Rf: 0.21 (hexane: ethyl acetate, 1:1, v/v). 1H NMR (400 MHz, DMSO-d6): δ = 12.64 (br s, 1H, NH), 8.69 (s, 1H, triazole CH), 7.73 (d, J = 8.3 Hz, 2H, Ar-H-3′,5′), 7.48 (brs, 2H, Ar-H), 7.37 (d, J = 8.1 Hz, 2H, Ar-H), 7.18–7.08 (m, 2H, Ar-H-5.6), 4.71 (s, 2H, SCH2), 2.37 (s, 3H, CH3). 13C NMR (101 MHz, DMSO-d6) δ 149.2, 144.2, 138.5, 134.3, 130.3, 121.7, 121.3, 120.0, 117.5, 110.5, 26.9, 21.1. Anal. Calc. (%) for C17H15N5S: C, 63.53; H, 4.70 N, 21.79; S, 9.98, Found: C, 63.42; H, 4.88; N, 22.06; S, 10.05.
4.1.2.7 2-(((1-(4-Methoxyphenyl)-1H-1,2,3-triazol-4-yl)methyl)thio)-1H-benzimidazole (6g)
Yield: 0.5 g (56%), Yellow powder, m. p: 131–135̊C, Rf: 0.2 (hexane: ethyl acetate, 1:1, v/v). 1H NMR (400 MHz, DMSO-d6): δ = 12.62 (brs, 1H, NH), 8.63 (s, 1H, triazole CH), 7.75 (d, J = 9.0 Hz, 2H, Ar-H-3′,5′), 7.72–7.65 (m, 1H,Ar-H), 7.55 (brs, 1H, Ar-H), 7.37 (bs, 1H,Ar-H), 7.18–7.01 (m, 3H, Ar-H), 4.69 (s, 2H, SCH2), 2.51 (s, 3H, OCH3). 13C NMR (101 MHz, DMSO-d6) δ 162, 159.2, 158.9, 155.0, 147.1, 144.0, 128.8, 125.6, 121.6, 114.6, 55.2, 26.7. Anal. Calc. (%) for C17H15N5OS: C, 60.52; H, 4.48 N, 20.76; S, 9.50, Found: C,60.41; H,4.30; N, 21.03; S, 9.61.
4.1.2.8 2-(((1-(3-Methoxyphenyl)-1H-1,2,3-triazol-4-yl)methyl)thio)-1H-benzimidazole (6h)
Yield: 0.3 g (60%), Yellow powder, m. p:131-132̊C, Rf: 0.37 (hexane: ethyl acetate, 1:1, v/v); 1H NMR (400 MHz, DMSO-d6): δ = 13.09 (br s,1H, NH), 8.82 (s, 1H, triazole CH), 7.68 (d, J = 7.4 Hz, 1H, Ar-H), 7.49–7.27 (m, 4H, Ar-H), 7.23–7.13 (m, 1H, Ar-H), 7.10–7.00 (m, 2H, Ar-H), 4.81 (s, 2H, SCH2), 3.82 (s, 3H, OCH3). 13C NMR (101 MHz, DMSO-d6) δ 160.0, 142.9, 137.3, 130.7, 122.0, 121.9, 117.8, 114.4, 111.9, 110.2, 105.6, 103.6, 55.5, 26.8. Anal. Calc. (%) for C17H15N5OS: C, 60.52; H, 4.48 N, 20.76; S,9.50 Found: C,60.34; H,4.59; N,20.94; S, 9.61.
4.1.2.9 4-(4-(((1H-benzimidazol-2-yl)thio)methyl)-1H-1,2,3-triazol-1-yl)benzene-sulfonamide (6i)
Yield: 0.1 g (32%), Yellow powder, m. p: 233°C–234°C, Rf: 0.075 (hexane: ethyl acetate, 1:1, v/v); 1H NMR (400 MHz, DMSO-d6): δ = 12.64 (brs, 1H, NH), 8.85 (s, 1H, triazole CH),8.09 (d, J = 8.4 Hz, 2H, Ar-H-3′,5′), 7.99 (d, J = 8.4 Hz, 2H; Ar-H-2′,6′), 7.62–7.53 (m, 1H, Ar-H), 7.51 (s, 2H, NH2), 7.42–7.34 (m, 1H, Ar-H), 7.18–7.09 (m, 2H, Ar-H-5.6), 4.73 (s, 2H, SCH2). 13C NMR (101 MHz, DMSO-d6) δ 145.3, 144.3, 138.9, 127.9, 122.5, 122.2, 121.6, 120.8, 118.0, 110.9, 26.3. Anal. Calc. (%) for C16H14N6O2S2: C, 49.73; H, 3.65; N, 21.75; S,16.59 Found: C,50.02; H,3.74; N,21.97; S,16.45.
4.1.2.10 2-(Benzylthio)-1-((1-phenyl-1H-1,2,3-triazol-5-yl)methyl)-1H-benzimidazole (10a)
Yield: 0.06 g (43%), Yellow powder, m. p: 139°C–140°C, Rf: 0.575 (hexane: ethyl acetate, 1:1, v/v); 1H NMR (400 MHz, DMSO-d6): δ = 8.78 (s, 1H, triazole CH), 7.83 (d, J = 7.7 Hz, 2H, Ar-H), 7.64 (d, J = 7.0 Hz, 1H, Ar-H), 7.62–7.54 (m, 3H, Ar-H), 7.52–7.41 (m, 3H, Ar-H)), 7.36–7.13 (m, 5H, Ar-H), 5.49 (s, 2H, N-CH2), 4.62 (s, 2H, S-CH2). 13C NMR (101 MHz, DMSO-d6) δ 143.5, 143.3, 137.7, 136.8, 136.3, 130.3, 129.4, 129.2, 128.9, 127.9, 122.2, 120.6, 118.2, 110.5, 39.1, 36.5. Anal. Calc. (%) for: C23H19N5S: C, 69.50; H, 4.82; N,17.62; S,8.07, Found: C,69.67; H,5.01; N,17.54; S,7.98.
4.1.2.11 2-(Benzylthio)-1-((1-(4-chlorophenyl)-1H-1,2,3-triazol-5-yl)methyl)-1H-benzimidazole (10b)
Yield 0.25 g (33%), Yellow powder, m. p: 149°C–150°C, Rf: 0.78 (hexane: ethyl acetate, 1:1, v/v); 1H NMR (400 MHz, DMSO-d6): δ = 8.80 (s, 1H, triazole CH), 7.88 (d, J = 8.4 Hz, 2H, Ar-H-4.7), 7.64 (d, J = 8.5 Hz, 4H, Ar-H), 7.45 (d, J = 7.3 Hz, 2H, Ar-H), 7.33–7.18 (m, 5H, Ar-H), 5.49 (s, 2H, N-CH2 (, 4.62 (s, 2H, S-CH2). 13C NMR (101 MHz, DMSO-d6) δ 143.7, 143.3, 137.6, 135.6, 133.5, 130.3, 129.4, 128.9, 127.9, 122.4, 122.2, 118.2, 110.5, 39.1, 36.5. Anal. Calc. (%) for: C23H18ClN5S: C,63.96; H, 4.20 N,16.21; S,7.42 Found: C,64.15; H,4.37; N,16.49; S,7.55.
4.1.2.12 2-(Benzylthio)-1-((1-(4-bromophenyl)-1H-1,2,3-triazol-5-yl)methyl)-1H-benzimidazole (10c)
Yield 0.25 g (30%), Yellow powder, m. p:158°C–159°C, Rf: 0.81 (hexane: ethyl acetate, 1:1, v/v); 1H NMR (400 MHz, DMSO-d6): δ = 8.80 (s, 1H, triazole CH), 7.87 (d, J = 8.4 Hz, 2H, Ar-H-3 ̏,5 ̏), 7.68–7.57 (m, 4H, Ar-H), 7.45 (d, J = 7.3 Hz, 2H, Ar-H), 7.40–7.14 (m, 5H, Ar-H), 5.49 (s, 2H, N-CH2(, 4.62 (s, 2H, S-CH2). 13C NMR (101 MHz, DMSO-d6) δ 143.7, 137.7, 135.6, 133.5, 131, 130.2, 129.4, 128.9, 127.9, 122.3, 122.2, 118.2, 110.5, 39.3, 31.1. Anal. Calc. (%) for: C23H18BrN5S: C, 57.99; H, 3.81; N, 14.70; S, 6.73 Found: C,58.21; H,3.92; N,14.93; S,6.80.
4.1.2.13 2-(Benzylthio)-1-((1-(4-nitrophenyl)-1H-1,2,3-triazol-5-yl)methyl)-1H-benzimidazole (10d)
Yield 0.25 g (32%), Yellow powder, m. p: 160°C–161°C, Rf: 0.71 (hexane: ethyl acetate, 1:1, v/v); IR (KBr, ύ cm-1): IR (KBr, ύ cm-1): 3,085 (=CH), 2,925 (CH2), 1,597, 1,507 (C=N, C=C), 1,523, 1,340 (NO2), 854 (p-bending), 1H NMR (400 MHz, DMSO-d6): δ = 9 (s, 1H, triazole CH), 8.42 (d, J = 9.0 Hz, 2H, Ar-H-3 ̏,5 ̏), 8.17 (d, J = 8.9 Hz, 2H, Ar-H 2 ̏,6 ̏), 7.75–7.58 (m, 2H, Ar-H-4.7), 7.44 (d, J = 7.4 Hz, 2H, Ar-H), 7.33–7.18 (m, 5H, Ar-H), 5.53 (s, 2H, NCH2(,4.63 (s, 2H, S-CH2). 13C NMR (101 MHz, DMSO-d6) δ 147.2, 144.2, 143.4, 141.1, 137.7, 129.4, 128.9, 127.9, 126, 122.7, 122.4, 121.1, 118.2, 110.5, 39.1, 36.6. Anal. Calc. (%) for: C23H18N6O2S: C, 62.43; H, 4.10 N, 18.99; S,7.25 Found: C,62.31; H,4.28; N,19.05; S,7.32.
4.1.2.14 2-(Benzylthio)-1-((1-(3-nitrophenyl)-1H-1,2,3-triazol-5-yl)methyl)-1H-benzimidazole (10e)
Yield 0.06 g (20%), Yellow powder, m. p:123°C–124°C, Rf: 0.68 (hexane: ethyl acetate, 1:1, v/v); 1H NMR (400 MHz, DMSO-d6): δ = 8.93 (s, 1H, triazole CH), 8.62 (d, J = 7.9 Hz, 1H, Ar-H), 8.29 (d, J = 7.9 Hz, 2H, Ar-H), 7.86 (t, J = 7.9 Hz, 2H, Ar-H), 7.62 (bs, 2H, Ar-H), 7.41 (d, J = 7.4 Hz, 2H, Ar-H), 7.25 (m, 4H, Ar-H), 5.49 (s, 2H, NCH2), 4.58 (s, 2H, SCH2). 13C NMR (101 MHz, DMSO-d6) δ 148.8, 143.8, 137.4, 137.2, 131.9, 129.2, 128.8, 127.8, 126.5, 123.6, 122.6, 122.6, 122.4, 118.1, 115.2, 110.5, 36.6, 29.2. Anal. Calc. (%) for: C23H18N6O2S: C, 62.43; H, 4.10; N,18.99; S,7.25, Found: C,62.60; H, 4.24; N, 19.17; S, 7.39.
4.1.2.15 2-(Benzylthio)-1-((1-(p-tolyl)-1H-1,2,3-triazol-5-yl)methyl)-1H-benzimidazole (10f)
Yield 0.3 g (42%), Yellow powder, m. p:138°C–140°C, Rf: 0.62 (hexane: ethyl acetate, 1:1, v/v), 1H NMR (400 MHz, DMSO-d6): δ = 8.71 (s, 1H, triazole CH), 7.70 (d, J = 8.4 Hz, 2H, Ar-H), 7.67–7.56 (m, 2H, Ar-H), 7.45 (d, J = 6.8 Hz, 2H, Ar-H), 7.37 (d, J = 8.2 Hz, 2H, Ar-H), 7.31–7.19 (m, 5H, Ar-H), 5.47 (s, 2H, NCH2(,4.62 (s, 2H, SCH2), 2.36 (s, 3H, CH3), 13C NMR (101 MHz, DMSO-d6) δ 143.4, 138.9, 137.7, 134.6, 130.6, 129.4, 128.9, 127.9, 122.3, 122.2, 122.1, 120.5, 118.2, 110.5, 36.5, 31.1, 21.0; Anal. Calc. (%) for: C24H21N5S: C, 70.05; H, 5.14; N,17.02; S,7.79 Found: C, 70.24; H, 5.22; N, 17.29; S, 8.07.
4.1.2.16 2-(Benzylthio)-1-((1-(4-methoxyphenyl)-1H-1,2,3-triazol-5-yl)methyl)-1H-benzimidazole (10g)
Yield 0.1 g (34%), Yellow powder, 139°C–140°C Rf: 0.525 (hexane: ethyl acetate, 1:1, v/v), 1H NMR (400 MHz, DMSO-d6): δ = 8.66 (s, 1H, triazole CH), 7.73 (d, J = 9.0 Hz, 2H, Ar-H), 7.65–7.56 (m, 2H, Ar-H-4.7), 7.49–7.42 (m, 2H, Ar-H), 7.34–7.15 (m, 5H, Ar-H), 7.10 (d, J = 9.0 Hz, 2H, Ar-H), 5.47 (s, 2H, NCH2), 4.62 (s, 2H, SCH2), 3.81 (s, 3H, OCH3). 13C NMR (101 MHz, DMSO-d6) δ 159.8, 143.3, 137.7, 131, 130.2, 129.4, 128.9, 127.9, 122.3, 122.3, 122.2, 118.2, 115.3, 110.5, 56.0, 39.3, 36.5. Anal. Calc. (%) for: C24H21N5OS: C, 67.43; H, 4.95; N,16.38; S,7.50 Found: C,67.61; H, 5.12; N,16.65; S,7.62.
4.1.2.17 2-(Benzylthio)-1-((1-(3-methoxyphenyl)-1H-1,2,3-triazol-5-yl)methyl)-1H-benzimidazole (10h)
Yield 0.09 g (30%), Yellow powder, 118°C–119°C, Rf: 0.24 (hexane: ethyl acetate, 1:1, v/v), 1H NMR (400 MHz, DMSO-d6): δ = 8.81 (s, 1H, triazole CH), 7.67–7.58 (m, 2H,Ar-H), 7.51–7.38 (m, 5H, Ar-H), 7.33–7.13 (m, 5H, Ar-H), 7.04 (d, J = 8.2 Hz, 1H, Ar-H), 5.49 (s, 2H, NCH2(, 4.63 (s, 2H, SCH2), 3.82 (s, 3H, OCH3). 13C NMR (101 MHz, DMSO-d6) δ 160.6, 143.5, 137.8, 137.6, 131.3, 129.4, 128.9, 127.9, 126.5, 122.4, 122.3, 118.2, 112.6, 110.6, 56.1, 39.3, 36.6. Anal. Calc. (%) for: C24H21N5OS: C, 67.43; H, 4.95; N, 16.38; S, 7.50, Found: C, 67.29; H, 5.06; N, 16.60; S, 7.61.
4.1.2.18 4-(5-((2-(Benzylthio)-1H-benzo [d]imidazole-1-yl)methyl)-1H-1,2,3-triazol-1-yl) benzene-sulfonamide (10i)
Yield 0.3 g (38%), Yellow powder, m. p: 139°C–140°C, Rf: 0.075 (hexane: ethyl acetate, 1:1, v/v); 1H NMR (400 MHz, DMSO-d6): δ = 8.88 (s, 1H, triazole CH), 8.07 (d, J = 8.7 Hz, 2H, Ar-H-2 ̏, 6 ̏), 7.99 (d, 2H, Ar-H-3 ̏,5 ̏), 7.69–7.58 (m, 2H, Ar-H), 7.52 (s, 2H, NH2), 7.45 (d, J = 6.9 Hz, 2H, Ar-H), 7.33–7.16 (m, 5H, Ar-H), 5.51 (s, 2H, NCH2(, 4.63 (s, 2H, S-CH2). 13C NMR (101 MHz, DMSO-d6) δ 144.4, 143.9, 138.8, 137.7, 129.4, 128.9, 127.9, 122.4, 122.4, 122.2, 120.8, 118.2, 110.5, 36.2, 31.1. Anal. Calc. (%) for: C23H20N6O2S2: C, 57.97; H, 4.23; N, 17.63; S, 13.45. Found: C, 58.24; H, 4.51; N, 17.89; S, 13.34.
4.2 Biology
4.2.1 Cell viability assay
The normal human mammary gland epithelial (MCF-10A) cell line was employed to assess the viability of the evaluated substances. Cell viability was assessed using the MTT test following 4 days of incubation of MCF-10A cells with 50 µM of each examined compound (Mekheimer et al., 2022; Hisham et al., 2022). See Supplementary Appendix A for more details.
4.2.2 Antiproliferative assay
The MTT assay was employed to examine the antiproliferative efficacy of 6a-i and 10a-i against four human cancer cell lines, utilizing erlotinib as a control (El-Sherief et al., 2019; Al-Wahaibi et al., 2022). Refer to Supplementary Appendix A for more information.
4.2.3 EGFR inhibitory assay
The EGFR-TK assay evaluated the inhibitory efficacy of the most potent antiproliferative derivatives 6e, 6i, 10d, 10e, and 10g against EGFR (Alshammari et al., 2022). Refer to Supplementary Appendix A for additional information.
4.2.4 Apoptotic marker assays
Compounds 6i and 10e were assessed for their ability to activate caspase-3, caspase-8, and Bax, as well as to downregulate the anti-apoptotic protein Bcl2 in the MCF-7 breast cancer cell line (Abdelbaset et al., 2019). Supplementary Appendix A provides more details.
4.2.5 Docking study
All the molecular modeling calculations and docking simulation studies were performed on a Processor Intel(R) Pentium(R) CPU N3510@ 1.99GHz and 4 GB Memory with Microsoft Windows 8.1 pro (64 Bit) operating system using Molecular Operating Environment (MOE 2019.0102, 2020; Chemical Computing Group, Canada) as the computational software (Stamos et al., 2002). Refer to Supplementary Appendix A for additional information.
4.2.6 Calculations of ADMET
Pharmacokinetics and drug-likeness predictions for all newly synthesized compounds were conducted using the pkCSM-pharmacokinetics server (http://biosig.unimelb.edu.au/pkcsm/) developed by the Bio21 Institute University of Melbourne (Pires et al., 2015).
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author contributions
AA: Formal Analysis, Investigation, Methodology, Software, Writing–original draft. AM: Conceptualization, Formal Analysis, Investigation, Methodology, Software, Supervision, Validation, Visualization, Writing–original draft, Writing–review and editing. ZA: Funding acquisition, Project administration, Software, Writing–original draft. SB: Investigation, Visualization, Writing–original draft, Writing–review and editing. BY: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The authors acknowledge the support form The Researchers Supporting Project Number (RSPD2025R603) King Saud University, Riyadh, Saudi Arabia. The authors also acknowledge support from the KIT-Publication Fund of the Karlsruhe Institute of Technology.
Acknowledgments
The authors acknowledge the support form The Researchers Supporting Project Number (RSPD2025R603) King Saud University, Riyadh, Saudi Arabia. The authors also acknowledge support from the KIT-Publication Fund of the Karlsruhe Institute of Technology.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2024.1541846/full#supplementary-material
References
Abdelbaset, M. S., Abdel-Aziz, M., Abuo-Rahma, G. E. D. A., Abdelrahman, M. H., Ramadan, M., and Youssif, B. G. (2019). Novel quinoline derivatives carrying nitrones/oximes nitric oxide donors: design, synthesis, antiproliferative and caspase-3 activation activities. Arch. Pharm. 352 (1), 1800270. doi:10.1002/ardp.201800270
Abdullah, M. i.N., Ali, Y., and Abd Hamid, S. (2022). Insights into the structure and drug design of benzimidazole derivatives targeting the epidermal growth factor receptor (EGFR). Chem. Biol. and Drug Des. 100 (6), 921–934. doi:10.1111/cbdd.13974
Abourehab, M. A., Alqahtani, A. M., Youssif, B. G., and Gouda, A. M. (2021). Globally approved EGFR inhibitors: insights into their syntheses, target kinases, biological activities, receptor interactions, and metabolism. Molecules 26 (21), 6677. doi:10.3390/molecules26216677
Abou-Zied, H. A., Youssif, B. G., Mohamed, M. F., Hayallah, A. M., and Abdel-Aziz, M. (2019). EGFR inhibitors and apoptotic inducers: design, synthesis, anticancer activity and docking studies of novel xanthine derivatives carrying chalcone moiety as hybrid molecules. Bioorg. Chem. 89, 102997. doi:10.1016/j.bioorg.2019.102997
Acar Çevik, U., Işik, A., Kaya, B., Kapavarapu, R., Rudrapal, M., Halimi, G., et al. (2024). Benzimidazole-containing compounds as anticancer agents. ChemistrySelect 9 (32), e202401566. doi:10.1002/slct.202401566
Al-Mahmoudy, A., Hassan, A., Ibrahim, T., Youssif, B. G., Taher, E., Tantawy, M., et al. (2022). Novel benzyloxyphenyl pyrimidine-5-carbonitrile derivatives as potential apoptotic antiproliferative agents. Anti-Cancer Agents Med. Chem. Former. Curr. Med. Chemistry-Anti-Cancer Agents 22 (5), 978–990. doi:10.2174/1871520621666210612043812
Alshammari, M. B., Aly, A. A., Youssif, B. G., Bräse, S., Ahmad, A., Brown, A. B., et al. (2022). Design and synthesis of new thiazolidinone/uracil derivatives as antiproliferative agents targeting EGFR and/or BRAFV600E. Front. Chem. 10, 1076383. doi:10.3389/fchem.2022.1076383
Al-Wahaibi, L. H., Mahmoud, M. A., Mostafa, Y. A., Raslan, A. E., and Youssif, B. G. (2023). Novel piperine-carboximidamide hybrids: design, synthesis, and antiproliferative activity via a multi-targeted inhibitory pathway. J. Enzyme Inhibition Med. Chem. 38 (1), 376–386. doi:10.1080/14756366.2022.2151593
Al-Wahaibi, L. H., Mostafa, Y. A., Abdelrahman, M. H., El-Bahrawy, A. H., Trembleau, L., and Youssif, B. G. (2022). Synthesis and biological evaluation of indole-2-carboxamides with potent apoptotic antiproliferative activity as egfr/cdk2 dual inhibitors. Pharmaceuticals 15 (8), 1006. doi:10.3390/ph15081006
Belal, A., Elanany, M. A., Raafat, M., Hamza, H. T., and Mehany, A. B. (2022). Calendula officinalis phytochemicals for the treatment of wounds through matrix metalloproteinases-8 and 9 (MMP-8 and MMP-9): in silico approach. Nat. Product. Commun. 17 (5), 1934578X221098848. doi:10.1177/1934578x221098848
Celik, I., Ayhan-Kılcıgil, G., Guven, B., Kara, Z., Gurkan-Alp, A. S., Karayel, A., et al. (2019). Design, synthesis and docking studies of benzimidazole derivatives as potential EGFR inhibitors. Eur. J. Med. Chem. 173, 240–249. doi:10.1016/j.ejmech.2019.04.012
Çevik, U. A., Celik, I., Mella, J., Mellado, M., Özkay, Y., and Kaplancıklı, Z. A. (2022). Design, synthesis, and molecular modeling studies of a novel benzimidazole as an aromatase inhibitor. ACS omega 7 (18), 16152–16163. doi:10.1021/acsomega.2c01497
Dunga, A. K., Allaka, T. R., Kethavarapu, Y., Nechipadappu, S. K., Pothana, P., Ravada, K., et al. (2022). Design, synthesis and biological evaluation of novel substituted indazole-1, 2, 3-triazolyl-1, 3, 4-oxadiazoles: antimicrobial activity evaluation and docking study. Results Chem. 4, 100605. doi:10.1016/j.rechem.2022.100605
El-Sherief, H. A., Youssif, B. G., Abdelazeem, A. H., Abdel-Aziz, M., and Abdel-Rahman, H. M. (2019). Design, synthesis and antiproliferative evaluation of novel 1, 2, 4-triazole/schiff base hybrids with EGFR and B-RAF inhibitory activities. Anti-Cancer Agents Med. Chem. Former. Curr. Med. Chemistry-Anti-Cancer Agents 19 (5), 697–706. doi:10.2174/1871520619666181224115346
El-Sherief, H. A., Youssif, B. G., Bukhari, S. N. A., Abdelazeem, A. H., Abdel-Aziz, M., and Abdel-Rahman, H. M. (2018). Synthesis, anticancer activity and molecular modeling studies of 1, 2, 4-triazole derivatives as EGFR inhibitors. Eur. J. Med. Chem. 156, 774–789. doi:10.1016/j.ejmech.2018.07.024
Fatima, N., Saeed, A., Ullah, S., Halim, S. A., Khan, A., Yaseen, M., et al. (2024). Synthesis of novel 1, 2, 3-triazole-tethered N-acyl hydrazones as a new class of carbonic anhydrase II inhibitors: in vitro and in silico potentials. Bioorg. Chem. 153, 107822. doi:10.1016/j.bioorg.2024.107822
Gona, P., Gona, C., Ballout, S., Mapoma, C., Rao, S., Mokdad, A., et al. (2024). Trends in the burden of most common obesity-related cancers in 16 Southern Africa development community countries, 1990–2019. Findings from the global burden of disease study. Obes. Sci. and Pract. 10 (1), e715. doi:10.1002/osp4.715
Hatil, A. C., Cicek, E., Oyken, M., and Erson-Bensan, A. E., EGFR epidermal growth factor receptor, atlas of genetics and cytogenetics in oncology and haematology 24(9) (2020).
Hisham, M., Hassan, H. A., Gomaa, H. A., Youssif, B. G., Hayallah, A. M., and Abdel-Aziz, M. (2022). Structure-based design, synthesis and antiproliferative action of new quinazoline-4-one/chalcone hybrids as EGFR inhibitors. J. Mol. Struct. 1254, 132422. doi:10.1016/j.molstruc.2022.132422
Ihmaid, S. K., Alraqa, S. Y., Aouad, M. R., Aljuhani, A., Elbadawy, H. M., Salama, S. A., et al. (2021). Design of molecular hybrids of phthalimide-triazole agents with potent selective MCF-7/HepG2 cytotoxicity: synthesis, EGFR inhibitory effect, and metabolic stability. Bioorg. Chem. 111, 104835. doi:10.1016/j.bioorg.2021.104835
Jalil, N. A. S., and Abd Hamid, S. (2023). Molecular docking analysis on the designed benzimidazole derivatives as EGFR inhibitors: comparison between EGFR wild-type (EGFRWT) and T790M mutant. Sains Malays. 52 (4), 1203–1215. doi:10.17576/jsm-2023-5204-13
Kalyani, P., and Manikyamba, P., Solvation models in the reaction between benzyl bromide and 2-mercapto benzimidazole—a kinetic study, (2004).
Karadayi, F. Z., Yaman, M., Kisla, M. M., Keskus, A. G., Konu, O., and Ates-Alagoz, Z. (2020). Design, synthesis and anticancer/antiestrogenic activities of novel indole-benzimidazoles. Bioorg. Chem. 100, 103929. doi:10.1016/j.bioorg.2020.103929
Kumar, S., Ali, I., Abbas, F., Khan, N., Gupta, M. K., Garg, M., et al. (2023). In-silico identification of small molecule benzofuran-1, 2, 3-triazole hybrids as potential inhibitors targeting EGFR in lung cancer via ligand-based pharmacophore modeling and molecular docking studies. Silico Pharmacol. 11 (1), 20. doi:10.1007/s40203-023-00157-1
Kutonova, K. V., Trusova, M. E., Postnikov, P. S., Filimonov, V. D., and Parello, J. (2013). A simple and effective synthesis of aryl azides via arenediazonium tosylates. Synthesis 45 (19), 2706–2710. doi:10.1055/s-0033-1339648
Larsen, B. D., and Sørensen, C. S. (2017). The caspase-activated DN ase: apoptosis and beyond. FEBS J. 284 (8), 1160–1170. doi:10.1111/febs.13970
Latif, A., Bibi, S., Ali, S., Ammara, A., Ahmad, M., Khan, A., et al. (2021). New multitarget directed benzimidazole-2-thiol-based heterocycles as prospective anti-radical and anti-Alzheimer's agents. Drug Dev. Res. 82 (2), 207–216. doi:10.1002/ddr.21740
Levantini, E., Maroni, G., Del Re, M., and Tenen, D. G. (2022). “EGFR signaling pathway as therapeutic target in human cancers,” in Seminars in cancer biology. Elsevier, 253–275.
Maghraby, M. T. E., Salem, O. I., Youssif, B. G., and Sheha, M. M. (2023). Design, synthesis, and modelling study of new 1, 2, 3-triazole/chalcone hybrids with antiproliferative action as epidermal growth factor receptor inhibitors. Chem. Biol. and Drug Des. 101 (3), 749–759. doi:10.1111/cbdd.14178
Mahmoud, M. A., Mohammed, A. F., Salem, O. I., Almutairi, T. M., Bräse, S., and Youssif, B. G. (2024). Design, synthesis, and apoptotic antiproliferative action of new 1, 2, 3-triazole/1, 2, 4-oxadiazole hybrids as dual EGFR/VEGFR-2 inhibitors. J. Enzyme Inhibition Med. Chem. 39 (1), 2305856. doi:10.1080/14756366.2024.2305856
Mahmoud, M. A., Mohammed, A. F., Salem, O. I., Gomaa, H. A., and Youssif, B. G. (2022). New 1, 3, 4-oxadiazoles linked with the 1, 2, 3-triazole moiety as antiproliferative agents targeting the EGFR tyrosine kinase. Arch. Pharm. 355 (6), 2200009. doi:10.1002/ardp.202200009
Mahmoud, M. A., Mohammed, A. F., Salem, O. I., Rabea, S. M., and Youssif, B. G. (2023). Design, synthesis, and antiproliferative properties of new 1, 2, 3-triazole-carboximidamide derivatives as dual EGFR/VEGFR-2 inhibitors. J. Mol. Struct. 1282, 135165. doi:10.1016/j.molstruc.2023.135165
Mekheimer, R. A., Allam, S. M., Al-Sheikh, M. A., Moustafa, M. S., Al-Mousawi, S. M., Mostafa, Y. A., et al. (2022). Discovery of new pyrimido [5, 4-c] quinolines as potential antiproliferative agents with multitarget actions: rapid synthesis, docking, and ADME studies. Bioorg. Chem. 121, 105693. doi:10.1016/j.bioorg.2022.105693
Metibemu, D. S., Akinloye, O. A., Akamo, A. J., Ojo, D. A., Okeowo, O. T., and Omotuyi, I. O. (2019). Exploring receptor tyrosine kinases-inhibitors in Cancer treatments. Egypt. J. Med. Hum. Genet. 20 (1), 35–16. doi:10.1186/s43042-019-0035-0
Mishra, K. N., and Upadhyay, H. C. (2022). Coumarin-1, 2, 3-triazole hybrids as leading-edge anticancer agents. Front. Drug Discov. 2, 1072448. doi:10.3389/fddsv.2022.1072448
Mostafa, A. S., Gomaa, R. M., and Elmorsy, M. A. (2019). Design and synthesis of 2-phenyl benzimidazole derivatives as VEGFR-2 inhibitors with anti-breast cancer activity. Chem. Biol. and Drug Des. 93 (4), 454–463. doi:10.1111/cbdd.13433
Mostafa, Y. A., Assoud, J. A., Desoky, A. Y., Mohamady, S., Mohamed, N. M., Salem, O. I., et al. (2024). New series of 4, 6-diaryl pyrimidines: facile synthesis and antiproliferative activity as dual EGFR/VEGFR-2 inhibitors. Front. Chem. 12, 1498104. doi:10.3389/fchem.2024.1498104
Nardi, M., Cano, N. C. H., Simeonov, S., Bence, R., Kurutos, A., Scarpelli, R., et al. (2023). A review on the green synthesis of benzimidazole derivatives and their pharmacological activities. Catalysts 13 (2), 392. doi:10.3390/catal13020392
Nguyen, D. T., Do, S. H., Le, T. H., Nguyen, T. H., Nguyen, M. H., Vu, T. N. B., et al. (2022). Synthesis and antiproliferative activity of 1 H-1, 2, 3-triazole-4 H-chromene-d-glucose hybrid compounds with dual inhibitory activity against EGFR/VEGFR-2 and molecular docking studies. New J. Chem. 46 (48), 23179–23197. doi:10.1039/d2nj04373d
Palumbo, C., Benvenuto, M., Focaccetti, C., Albonici, L., Cifaldi, L., Rufini, A., et al. (2023). Recent findings on the impact of ErbB receptors status on prognosis and therapy of head and neck squamous cell carcinoma. Front. Med. 10, 1066021. doi:10.3389/fmed.2023.1066021
Peerzada, M. N., Hamdy, R., Rizvi, M. A., and Verma, S. (2023). Privileged scaffolds in drug discovery against human epidermal growth factor receptor 2 for cancer treatment. Curr. Pharm. Des. 29 (44), 3563–3578. doi:10.2174/0113816128283615231218094706
Pires, D. E., Blundell, T. L., and Ascher, D. B. (2015). pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 58 (9), 4066–4072. doi:10.1021/acs.jmedchem.5b00104
Sahoo, G., Samal, D., Khandayataray, P., and Murthy, M. K. (2023). A review on caspases: key regulators of biological activities and apoptosis. Mol. Neurobiol. 60 (10), 5805–5837. doi:10.1007/s12035-023-03433-5
Stamos, J., Sliwkowski, M. X., and Eigenbrot, C. (2002). Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem. 277 (48), 46265–46272. doi:10.1074/jbc.m207135200
Workie, M. S., Jalali, R., and Cheng, Z. (2023). “Background: cancer is a leading cause of death in the world, and the estimated new cancer cases were 19 million and the estimated cancer deaths were around 10 million worldwide in 2020,” in Proton therapy (PT) is a promising treatment for cancers, 17.
Yanaihara, N., Yoshino, Y., Noguchi, D., Tabata, J., Takenaka, M., Iida, Y., et al. (2023). Paclitaxel sensitizes homologous recombination-proficient ovarian cancer cells to PARP inhibitor via the CDK1/BRCA1 pathway. Gynecol. Oncol. 168, 83–91. doi:10.1016/j.ygyno.2022.11.006
Keywords: benzimidazole, triazole, EGFR, apoptosis, caspases
Citation: Ahmed AAY, Mohammed AF, Almarhoon ZM, Bräse S and Youssif BGM (2025) Design, synthesis, and apoptotic antiproliferative action of new benzimidazole/1,2,3-triazole hybrids as EGFR inhibitors. Front. Chem. 12:1541846. doi: 10.3389/fchem.2024.1541846
Received: 08 December 2024; Accepted: 23 December 2024;
Published: 13 January 2025.
Edited by:
Ilkay Erdogan Orhan, Lokman Hekim University, TürkiyeReviewed by:
Sergio Hernan Szajnman, University of Buenos Aires, ArgentinaPenke Vijaya Babu, Tikvah Pharma Solutions Pvt. Ltd., India
Copyright © 2025 Ahmed, Mohammed, Almarhoon, Bräse and Youssif. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bahaa G. M. Youssif, Ymd5b3Vzc2lmMkBnbWFpbC5jb20=; Stefan Bräse, YnJhZXNlQGtpdC5lZHU=