1 Introduction
Ionic liquids consist of mixtures of cations and anions, usually of asymmetric size, that can remain in a liquid phase at temperatures typically below 100°C (Hayes et al., 2015). After initially being described as “water-free salts” (Walden, 1914), ionic liquids continue to gain attention due to their unique properties, including low volatility, high thermal stability, and excellent solvating capabilities (Sowmiah et al., 2009; Eyckens and Henderson, 2019). Given the diversity of molecular structures that form ionic liquids (Hayes et al., 2015) and their potential applications in energy conversion and storage (Armand et al., 2009; Wishart, 2009; Torimoto et al., 2010; Watanabe et al., 2017), it is not surprising that optimizing function–structure relationships is both complex and highly rewarding (Silva et al., 2020; Wang et al., 2020; Khlyupin et al., 2023; Nesterova et al., 2025). The challenges extend even to the level of understanding underlying thermodynamic and dynamic properties of ionic liquids (Weingärtner, 2008; Silva et al., 2020; Nordness and Brennecke, 2020). Compared to the ions in ordinary electrolytes, those of ionic liquids are densely packed and thus subject to ion–ion correlations due to electrostatic and non-electrostatic interactions (Del Pópolo and Voth, 2004).
Classical mean-field modeling (Kornyshev, 2007; May, 2019; Goodwin et al., 2023), which is a powerful conceptual tool to characterize ions in a solvent at sufficient dilution, fails for ionic liquids. Consequently, significant efforts have been undertaken in the past to incorporate ion correlations due to electrostatic and non-electrostatic interactions into the modeling of ionic liquids (Kondrat et al., 2023). Examples include the use of field theory (Démery et al., 2012; Nakamura, 2015; Budkov, 2020; Chao and Wang, 2020), density functional theory (Wu et al., 2011; Henderson et al., 2011; Ma et al., 2014), a microscopic cluster expansion model (Avni et al., 2020), molecular dynamics (Wang et al., 2007; Fedorov and Kornyshev, 2008; Coles et al., 2020; Zeman et al., 2021; Ye and Wang, 2022) and Monte Carlo (Kondrat et al., 2011; Bhuiyan et al., 2012) simulations, as well as phenomenological extensions of mean-field approaches (Frydel, 2016; Goodwin et al., 2017; Downing et al., 2018a). To allow for oscillations in ionic concentration profiles, Bazant, Storey, and Kornyshev (BSK) proposed a phenomenological Landau–Ginzburg-like model that leads to a fourth-order modified Poisson–Boltzmann equation with a correlation length that accounts for short-range ion–ion interactions (Bazant et al., 2011). The BSK approach has been used widely (Yochelis, 2014; Shalabi et al., 2019; Varner and Wang, 2022; Nesterova et al., 2025) and linked to microscopic approaches (Blossey et al., 2017; Avni et al., 2020). Yet, understanding of the physical basis of higher-order Poisson–Boltzmann approaches and their relation to even the most basic molecular properties such as charge discreteness, ion polarizability, non-electrostatic interactions, and electrostatic ion–ion correlations remains incomplete.
The present work aims at incorporating both charge discreteness and electrostatic correlations between ion pairs into a lattice-based model of an ionic liquid that is in contact with a single planar electrode. To account for charge discreteness, we separate the short-range and long-range parts of the electrostatic interaction by truncating the Coulomb potential below a distance to the neighboring ions. The operator that results from this truncation involves an infinite number of higher-order derivatives, yet it becomes equivalent to the BSK model in the limit of small . We focus on this limit, which results in an approximate incorporation of electrostatic interactions on the level of continuum electrostatics except for nearest neighbors. In contrast, electrostatic interactions among nearest neighbor ions are modeled explicitly, thereby accounting for their discrete nature. To account for electrostatic correlations among neighboring ions, we adopt the quasi-chemical approximation (QCA) approach (Sher et al., 1987; Davis, 1996) when expressing the configurational entropy of the lattice gas. In principle, QCA can account for a correlation among any number of ions, but in the present work, we only include two-body correlations among nearest neighbor ions. Hence, we propose a method that accounts for both charge discreteness and electrostatic correlations between ion pairs in the modeling of ionic liquids. We recognize that the present work assumes is sufficiently small and neglects ion correlations that involve more than two ions. Yet, we also point out that, in principle, this approach can be extended to account for larger and higher-order correlations. In addition to presenting the method and deriving the self-consistency relationship (a fourth-order differential equation) plus its boundary conditions, we calculate analytic results for the differential capacitance up to quadratic order in the surface charge density of the electrode and numerical results for larger surface charge densities. Our results are compared with limiting cases, such as the absence of electrostatic correlations between ion pairs and the negligence of ion discreteness. We find that charge discreteness enhances the magnitude of the electrostatic potential, and thus also the differential capacitance, whereas electrostatic correlations between ion pairs induce a transition from a bell-shaped to a camel-shaped profile of the differential capacitance.
2 Theory
Consider a compact, solvent-free ionic liquid containing monovalently charged cations and anions of the same size. We represent the ionic liquid by a lattice of coordination number , volume per lattice site, and lattice spacing , with each site being occupied by either a cation or an anion. and denote the local mole fraction of cations (index “1”) and anions (index “2”), respectively, at position . The absence of solvent is expressed by the condition at every position . In the present work, we aim at separating contributions to the electrostatic energy of the ionic liquid that originate from the short-range and long-range parts of the Coulomb potential. To introduce this method, we write for the electrostatic energy due to the long-range part.
where we have defined the difference in mole fractions between the cations and anions. Note that specifies the local volume charge density, where denotes the elementary charge, and that is positive in regions with an excess of cations over anions. Equation 1 accounts for all pairwise interactions among the ions within the ionic liquid: for cation–cation and anion–anion pairs, and for cation–anion and anion–cation pairs, given that the two ions are separated by a distance . We introduce a dimensionless potential through:
where, here and in the following, we express energies in units of , where is the Boltzmann constant and is the absolute temperature. In order to recast Equation 2 into a local equation for the potential , we introduce a yet unknown differential operator such that , where is the Dirac delta function. This indeed implies the local equation:
and corresponding electrostatic energy
Only after specifying a concrete interaction potential , we can determine the corresponding differential operator . This is addressed in the following section.
2.1 Truncated Coulomb potential
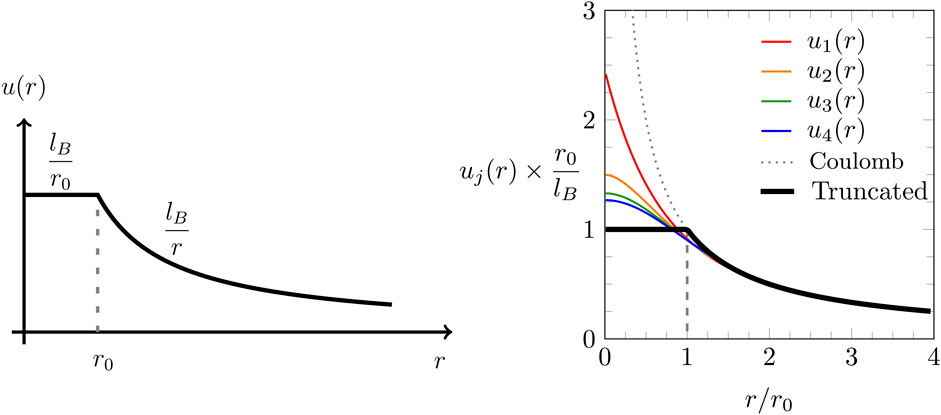
To capture the long-range part of the electrostatic interaction, we choose a truncated Coulomb potential
where is the Bjerrum length and is the truncation length. Truncated Coulomb potentials have been employed in the past to cut off the long-range part with the goal to facilitate efficient computer simulations (Linse and Andersen, 1986; Baker et al., 1999). In contrast, we propose to cut off the short-range part of the Coulomb interaction, as shown in Figure 1: the black solid line in both diagrams of Figure 1 shows the truncated potential according to Equation 5. The operator can be identified conveniently in Fourier space, where the Fourier transformation
together with , gives rise to
In Equation 6, and denote the nabla differential operator and the ith Bernoulli number, respectively. Recall that the set of Bernoulli numbers can be defined via a series expansion of the generating function . If the truncated Coulomb potential is approximated by the -th order operator,
then the corresponding potential is given by the following equation:
The function in Equation 8 is the -th order approximation of the truncated Coulomb potential. For , Equation 8 gives rise to the Coulomb potential . In the limit , the truncated Coulomb potential , as specified in Equation 5, is recovered. As has been pointed out by Lee et al. (2015), the first-order approximation of the truncated Coulomb potential is as follows:
The right diagram in Figure 1 shows the truncated Coulomb potential together with the increasingly more accurate approximations , , , and . In the present work, we will focus on the first-order approximation only, leading to a fourth-order Poisson equation that utilizes the differential operator similar to the BSK model (Bazant et al., 2011). However, Equations 7, 8 provide a method for the incorporation of higher-order approximations in a straightforward manner.
We point out that the BSK model (Bazant et al., 2011) is an effective mean-field approach based on the pair potential . The emergence of from the bare Coulomb potential can be the result of charge discreteness, which we consider in the present work, or of short-range correlations. Our present work considers electrostatic correlations beyond the short-range correlations of the BSK model. Electrostatic correlations between ion pairs are not accounted for by the BSK model.
2.2 Free energy minimization
Before formulating and minimizing a free energy that accounts for charge discreteness and electrostatic correlations between ion pairs, we start with three comments: first, choosing the truncation radius to be equal or slightly larger than the lattice spacing enables us to explicitly include the electrostatic interactions between neighboring ions: between cation–cation and anion–anion pairs and between cation–anion and anion–cation pairs. This introduces the definition and the notation that we use in Equation 10. Second, we account for electrostatic correlations between ions located at neighboring lattice sites through QCA (Davis, 1996). Correlations involving more than two ions can, in principle, be incorporated into QCA (Bossa et al., 2015), but they are ignored in the present work. To account for correlated ion pairs, we consider the local fraction of - pairs, where the indices and adopt the value 1 for a cation and 2 for an anion. Third, we introduce the chemical potentials and of the cations and anions, respectively, and choose them to ensure coexistence with a bulk phase of the ionic liquid, where . We also recall the definition of the lattice coordination number . With that, we express the free energy of a lattice-based ionic liquid as follows (Downing et al., 2018b):
The four contributions to this free energy account for the electrostatic energy due to the long-range part of the Coulomb potential, the mixing entropy of ion pairs according to QCA, the electrostatic energy associated with neighboring ion pairs, and a Legendre transformation that fixes the chemical potentials of the cations and anions in the ionic liquid. The in Equation 10 must fulfill the three conservation relations , , and . This leaves one degree of freedom for the local distribution of ion pairs, which we define conveniently as . The difference in mole fractions constitutes another degree of freedom. We thus have the following equation:
Note that both and depend on the position inside the ionic liquid. Calculation of the first variation in the free energy leads to
We have not included boundary terms in Equation 12 because they will be discussed separately below. By symmetry, the chemical potentials are equal. Thermal equilibrium demands , thus leading to the following two equations:
which must be fulfilled at each position . Equation 13 defines the two relations and , with and as additional parameters. Spatial compositional variations arise from the solutions of Equation 3. However, to simplify calculations, we replace the operator by its first-order approximation , as defined in Equation 7. This renders the level of modeling the long-range part of the electrostatic interactions similar to the BSK model (Bazant et al., 2011) yet with different boundary conditions as discussed below. Equation 3 then reads as follows:
where we have defined the length . For example, and imply . We will use as unit length throughout this work. Equation 14 is a self-consistency relationship that takes the form of a fourth-order differential equation with the function defined through a set of two algebraic equations. As pointed out above, electrostatic interactions are represented by the potential in Equation 9. Unlike interpretations in terms of short-range ion correlations (Storey and Bazant, 2012; Moon et al., 2015; Blossey et al., 2017; Shalabi et al., 2019; de Souza and Bazant, 2020), we associate the use of with an approximation of the truncated Coulomb potential in Equation 5, which originates in our goal to model charge discreteness rather than correlations. A complementary interpretation of would be in terms of excluded volume interactions between adjacent ions, and such an interpretation has been proposed recently (Gupta et al., 2020a).
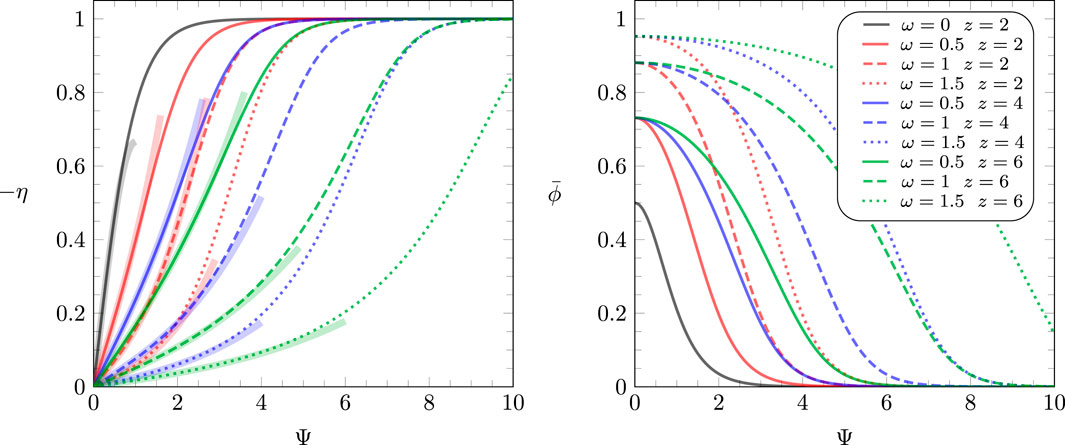
The function that is needed to solve Equation 14 satisfies Equation 13 and is displayed together with in Figure 2 for different parameters and . The solution of Equation 13 for is , irrespective of , and is shown as a black line in the left diagram of Figure 2. Curves of the same color refer to (red), (blue), and (green) and are calculated for different values of , as indicated in the legend. Note that for (the red lines in Figure 2), Equation 13 yields the following simple analytic result:
which corresponds to the solution of the one-dimensional Ising model (Davis, 1996) in an external field .
2.3 Boundary conditions
From this point forward, we consider a single charged planar electrode that carries a uniform surface charge density and is in contact with the ionic liquid. We locate the origin of a Cartesian coordinate system at the electrode surface, with its -axis pointing normal into the ionic liquid. The electrostatic potential then depends only on the distance to the electrode, and Equation 14 reads as follows:
where a prime denotes the derivative with respect to the position . We derive solutions of this fourth-order non-linear differential equation for subject to the boundary conditions as well as
where is a scaled (dimensionless) surface charge density. Different sets of boundary conditions at the electrode surface have been proposed in the past for fourth-order differential equations of the type in Equation 16: and in the original BSK model (Bazant et al., 2011) and and based on variational free energy minimization (Gupta et al., 2020b). In addition, and have been proposed based on the continuity of the Maxwell stress tensor at the charged electrode (de Souza and Bazant, 2020) and on interpreting in Equation 9 as a specific combination of Coulomb and Yukawa potentials (Bossa and May, 2020). We take the point of view that the truncated Coulomb potential applies to all charges in the system, including those within the ionic liquid and those at the electrode surface. In this case, the energy (per unit area ) of the system associated with the long-range part of the electrostatic interactions is as follows:
Its first variation is as follows:
The integral in Equation 19 is , which, when combined with the variation in the non-electrostatic contributions to the free energy, will vanish, as demonstrated in Equation 12. Poisson’s equation for the truncated Coulomb potential, , relates the potential to the local volume charge density . Integrating that equation across the surface of the electrode yields . Because the scaled surface charge density is fixed, the ensuing relation causes the first surface term in Equation 19 to vanish. Vanishing of the second surface term demands . Because of , the term in the third line of Equation 18 vanishes. The remaining two terms, the electrostatic energy due to the surface charges on the electrode and the electrostatic energy due the ions in the ionic liquid, then render Equation 18 identical to Equation 4. Hence, the boundary conditions at the electrode, and , lead to the consistent vanishing of with defined in Equation 10, including all surface terms.
The following three comments complete our discussion of the boundary conditions. First, for , the potential is constant, . Hence, because the region does not contribute to , the integration in Equation 18 may start from instead of also including negative values of . Second, short-range electrostatic interactions of the ionic liquid with the electrode surface contribute another surface term to in Equation 18. Because the variation of this term vanishes, it does not add to the boundary conditions. Third, our approach recovers the boundary condition , used previously to solve the second-order self-consistency differential equation that results from our model for (Downing et al., 2018b).
The solution of Equation 16 reveals how the surface potential depends on the scaled surface charge density , allowing us to compute the differential capacitance as follows:
Calculation of the scaled differential capacitance is a major focus of the present work.
3 Results and discussion
Before computing general results for numerically, we analytically derive quadratic expressions of the form through linearizing our model followed by a perturbation approach to access the nonlinear regime. The determination of the two constants and as functions of , , and provides insights about the role of charge discreteness and electrostatic correlations between ion pairs.
3.1 Linearized theory
For small potentials, , only the linear part of the relationship is significant. We use Equation 13 to perform a series expansion of up to the linear order. The result, , can conveniently be expressed as , where we have defined the length:
This length will be shown to play a role in separating different regimes of the solution for the linear problem. We thus solve the equation
subject to , , . The solution of the linear problem, denoted in the following by , can generally be expressed as the sum of two contributions:
with
The two constants and follow from the boundary conditions in Equation 17,
In the following, we discuss how the structure of the solution changes as is varied. For , the corresponding potential,
is characterized by a single decay length. For , there is a double-exponential decay with the two decay lengths and . At , the decay length diverges, and the potential becomes as follows:
Finally, for , the quantities and , as well as and , adopt complex conjugate values with
and
The potential
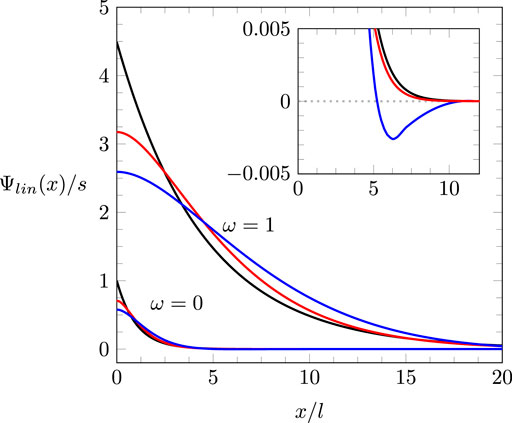
then exhibits exponentially decaying oscillations. Figure 3 shows for (upper three curves) and (lower three curves) and three different ratios of , namely (black), (red), and (blue). Figure 3 suggests the tendency of to increase the magnitude and decay length of the potential. The truncation length only moderately modifies this tendency. Irrespective of the ratio , the same linear relationship,
between surface potential and scaled surface charge density results. Hence, Equation 27 is valid within the linearized model for any choice of , , and . This leads to a prediction for the differential capacitance in the limit of an uncharged electrode, where both and ,
Recall that depends on and through Equation 20. Clearly, increasing lowers , whereas larger increases .
3.2 Non-linear model: a perturbation approach
The linearized model is based on the relationship . In this section, we employ a perturbation approach to analytically model the nonlinear region, where the potential deviates only slightly from . To this end, we expand the relationship up to the third order,
From Equation 13, we find the third-order coefficient as follows:
The left diagram in Figure 2 displays the third-order approximation, as specified in Equations 29, 30 (color-matching thick transparent lines). Hence, we aim to solve the nonlinear differential equation
subject to , , and . We express the potential as the sum of the solution for the linear problem and a small perturbation . Substituting into Equation 31 leads to the linear, inhomogeneous differential equation
for the perturbation contribution of the potential , where is given by Equation 21. The solution of Equation 32 can be expressed as the sum of a specific solution for the inhomogeneous equation (index “inh”):
where , , , and are defined in Equations 22, 23, and a solution for the homogeneous equation (index “hom”):
The two constants and in Equation 33 can be determined such that besides , Equation 32 also satisfies the boundary conditions and . This results in an expression for the surface contribution of the perturbation potential with
The total surface potential can be used to calculate the scaled differential capacitance,
analytically up to quadratic order in . Using the expressions for , , and in Equations 28, 30, 34, respectively, to calculate the quadratic-order coefficient yields the following:
The quadratic expression for in Equation 35 with the analytic results for and given in Equations 28, 36 is the central outcome of this work, accounting (albeit on an approximate level) for both charge discreteness and electrostatic correlations.
3.3 Limiting cases
3.3.1 Continuum mean-field model, and
For and , Equation 16 reduces to the well-known classical mean-field equation that emerges from the lattice-based Poisson–Boltzmann framework of a solvent-free ionic liquid (Borukhov et al., 1997; Kornyshev, 2007). Neither charge discreteness nor ion correlations are accounted for. Subject to the boundary conditions and , we obtain a surface potential for and for , and thus, a scaled differential capacitance . For small , this can be represented by with and . All models discussed in the present work recover this limit if both and .
3.3.2 Vanishing truncation length,
For , the expressions for and in Equations 28, 36 simplify to the following:
This is identical to the results derived previously by Downing et al. (2018b), where an approach analogous to the present one yet without truncating the Coulomb potential was proposed. Not truncating the Coulomb potential while still adding nearest-neighbor electrostatic interactions separately into the model implies a double counting of nearest neighbor ion–ion interactions. The truncation of the Coulomb potential introduced in the present work eliminates this inconsistency.
As pointed out by Downing et al. (2018b), changes sign upon increasing , indicating a transition from bell shape to camel shape of the differential capacitance . This transition is caused by electrostatic correlations between ion pairs, as shown below by comparing the predictions for in the presence and absence of electrostatic correlations.
3.3.3 Vanishing central charge,
The choice eliminates the explicit account of electrostatic interactions between nearest neighbors. Without these interactions, ion correlations between neighboring ions are no longer present. The solution of Equation 13 is , and the corresponding relation implies mean-field electrostatics subject to a truncated Coulomb potential. The resulting fourth-order differential equation is equivalent to the modified Poisson–Boltzmann equation of the solvent-free BSK model (Bazant et al., 2011) yet with different boundary conditions. For , we obtain the following from Equations 28, 36:
The negative sign of signifies a bell shape of the differential capacitance . Hence, for , cannot exhibit a camel shape.
3.3.4 The influence of electrostatic correlations between ion pairs
The usage of the QCA approximation accounts for electrostatic correlations between neighboring ion pairs, whereas three-body and higher-order correlations are neglected. To investigate the role of electrostatic ion pair correlations, we compare QCA to a mean-field model, which is based on a random mixing approximation (RMA) (Davis, 1996). RMA ignores ion pair correlations by modeling the entropy contribution of the free energy through an ideal lattice gas. This is accomplished by imposing , which, when used in Equation 11, leads to the free energy
Electrostatic interactions between neighboring ions are thus described on the level of the familiar Bragg–Williams model (Davis, 1996), whereas the long-range components are accounted for by the truncated Coulomb potential. As above, we use and and note that . Vanishing first variation of the free energy
then implies the relation
This implicitly defines the function to be used in the fourth-order self-consistency differential equation, Equation 14, namely, . Up to the third order in , this function is given by . As above, we solve the linearized model and perform a perturbation approach, yielding again an analytic expression for the scaled differential capacitance up to quadratic order in the scaled surface charge density . We find that
where we have defined . Equation 39 accounts for the discrete nature of the ionic charges, but neglects electrostatic correlations between ion pairs. As for Equation 37, the negative sign of implies a bell shape of . Hence, in the absence of electrostatic correlations between ion pairs, our model does not predict a transition to a camel shape of .
3.4 Numerical results
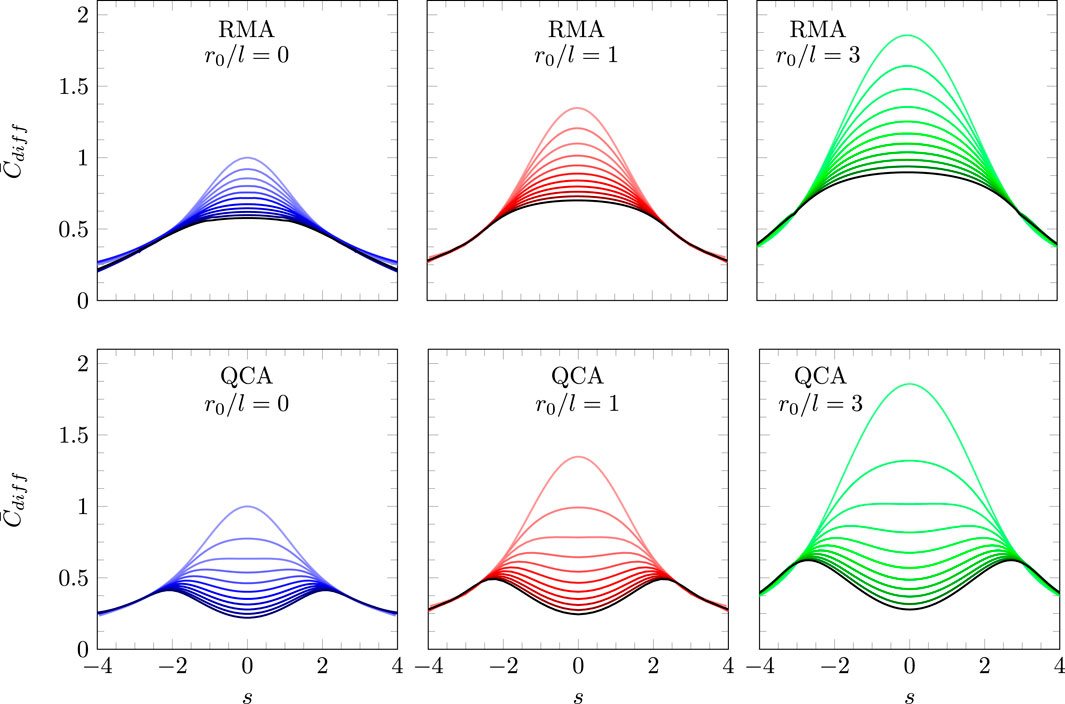
In this section, we present numerical results for , with various parameter choices for , , and , and then contrast the predictions of the model in the presence and absence of electrostatic ion–ion correlations. Solutions of Equation 14 employ the function according to Equation 13 in the presence of electrostatic ion correlations (modeled according to QCA) and according to Equation 38 in the absence of electrostatic ion correlations (modeled according to RMA).
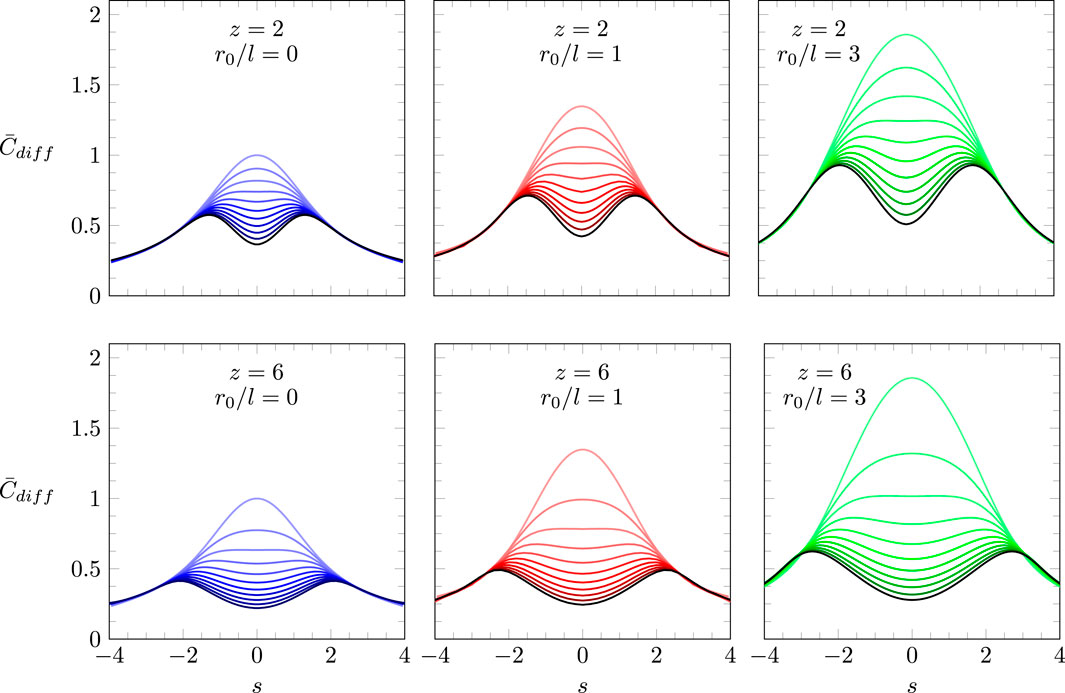
Figure 4 summarizes the predictions of our model according to Equation 16 with specified in Equation 13 and the boundary conditions in Equation 17. Recall that this model incorporates both the truncated Coulomb potential and QCA, thus accounting, approximatively, for both charge discreteness and electrostatic correlations. The scaled differential capacitance is displayed as a function of the scaled surface charge density with (upper three diagrams) and (lower three diagrams), as well as (left, colored purple), (middle, colored red), and (right, colored green). Each graph contains 11 curves, varying from (the light colored curve on the top of each diagram) to (the black curve on the bottom of each diagram) in steps of 0.1.
We point out that in the vicinity of , where can be represented by a quadratic function, each curve reproduces our analytic expression, as specified in Equations 28, 36. Hence, Figure 4 reinforces the validity of our analytic results and provides an extension of beyond the quadratic regime. As already predicted analytically, we observe the transition from a bell-shaped to a camel-shaped profile of the differential capacitance as the strength of the electrostatic interaction is increased. The transition occurs at a value of that satisfies the equation . For and , this amounts to and , respectively, independently of . The truncation length does affect the value of where the bell-to-camel shape transition occurs, but the value of where the transition happens is independent of . The role of is to amplify the magnitudes of . This is most clearly evidenced by the explicit relationship for the dependence of on in Equation 28.
Figure 4 displays the dependence of for the two parameters and (given is fixed). Yet, if we identify the truncation radius with the lattice spacing, there should be a relationship between and . Recall the definitions and . The lattice spacing represents the size of the ions in the ionic liquid, typically on the order of a nanometer. If we identify the truncation radius with the lattice spacing, we obtain . Hence, the choice (implying ) corresponds to , which is displayed by the black curve in the right column of Figure 4. Further increasing the Bjerrum length (implying ) yields larger values for . These cases are not shown in Figure 4 but are easily accessible through our analytic expression for .
Figure 5 shows a comparison of our model with and without accounting for electrostatic correlations between ion pairs. The lower three diagrams in Figure 5 reproduce the lower three diagrams of Figure 4, calculated for and using QCA where ion pair correlations are accounted for. The upper three diagrams show analogous results, yet in the absence of ion pair correlations, employing RMA, as introduced in Section 3.3.4. We have verified the agreement of the numerical results for RMA (upper three diagrams) in the region of small with the analytic expression specified in Equation 39. As already pointed out, RMA does not predict a transition from a bell-shaped to a camel-shaped profile of the differential capacitance as the strength of the electrostatic interaction is increased. Hence, we conclude that electrostatic correlations rather than charge discreteness or excluded volume interactions cause camel-shape profiles of . Starting from the classical mean-field approach, , displayed by the uppermost purple line on the left two diagrams in Figure 5, and introducing charge discreteness without accounting for electrostatic correlations corresponds to transitioning to the black curve on the right top diagram of Figure 5: the curve widens but without qualitatively changing its shape.
Because the presence of the bell-to-camel shape transition is independent of , the reason of its existence for a compact, solvent-free ionic liquid reflects the role of electrostatic ion–ion correlations. Correlated clusters of ions increase the decay length of the potential , as shown in Figure 3. Hence, if the system of electrode and diffuse layer of counterions is represented by an effective parallel-plate capacitor, the distance between the capacitor plates is larger in the presence of electrostatic ion–ion correlations. The correspondingly smaller value of is consistent with the existence of a transition from the bell-shaped to a camel-shaped profile of when electrostatic ion–ion correlations are accounted for.
4 Conclusion
The classical lattice-based mean-field theory for a densely packed, solvent-free ionic liquid employs continuum electrostatics and ignores ion–ion correlations, leading to a bell-shaped profile according to for the scaled differential capacitance as a function of the scaled charge density . The present work is an attempt to analyze how changes if charge discreteness and electrostatic correlations between ion pairs are accounted for. To account for charge discreteness, we employ a truncated Coulomb potential (shown in Figure 1) to model the long-range part of the electrostatic interactions but exclude those of a given ion with its nearest neighbors. These short-range interactions are modeled explicitly as interactions between individual point charges. Electrostatic correlations between pairs of neighboring ions are accounted for using the quasi-chemical approximation (QCA) approach. Downing et al. (2018b) included QCA similarly to the present approach but without truncating the Coulomb potential, which led to a double counting of electrostatic interactions between nearest neighbor ions. The present work is no longer subject to this inconsistency. We emphasize that our approach is still subject to approximations: the truncation of the Coulomb potential is modeled only on the level of fourth-order electrostatics ( in Equation 7), and QCA ignores correlations of higher order and between pairs of ions separated by distances larger than one lattice spacing. Yet, these approximations can, in principle, be overcome by considering sixth- or higher-order electrostatics ( or higher in Equation 7) and by incorporating larger clusters into QCA (Bossa et al., 2015).
Our approximation of the full self-consistency relationship, , by the fourth-order differential equation, , renders our model structurally equivalent to the BSK model (Bazant et al., 2011). An important aspect of our study includes the boundary conditions associated with that equation (Equation 17), which emerge as part of the functional minimization of the free energy (Equations 12, 19) and differ from previously proposed sets of boundary conditions (Bazant et al., 2011; Gupta et al., 2020b; de Souza and Bazant, 2020; Bossa and May, 2020).
A major result of our study is analytic expressions for the coefficients and in an expansion of up to quadratic order in . We have obtained from solving the linearized model and from performing a perturbation approach into the non-linear regime. The quadratic expression of reveals the existence of a transition from a bell-shaped to a camel-shaped profile of the differential capacitance as a function of the electrostatic interaction strength between neighboring ions. However, such a transition exists only in the presence of electrostatic correlations, not in their absence. Hence, we conclude that electrostatic correlations between ion pairs are able to turn the bell-shaped profile predicted by mean-field theory, , into a camel-shaped profile. Ion discreteness, on the other hand, enhances the magnitude but does not qualitatively alter the profile of . Our analytic predictions agree with numerical calculations of the full profiles for , i.e., beyond the quadratic regime, that we have presented in Figures 4, 5.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material; further inquiries can be directed to the corresponding author.
Author contributions
GB: conceptualization, formal analysis, writing–original draft, and writing–review and editing. SM: conceptualization, formal analysis, writing–original draft, and writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. Author G.V.B. thanks Fondecyt (grant no. 11240064) and VIDCA/UACh (grant INS-INV 2022-09) for the support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Armand, M., Endres, F., MacFarlane, D. R., Ohno, H., and Scrosati, B. (2009). Ionic-liquid materials for the electrochemical challenges of the future. Nat. Mater. 8, 621–629. doi:10.1038/nmat2448
PubMed Abstract | CrossRef Full Text | Google Scholar
Baker, N. A., Hünenberger, P. H., and McCammon, J. A. (1999). Polarization around an ion in a dielectric continuum with truncated electrostatic interactions. J. Chem. Phys. 110, 10679–10692. doi:10.1063/1.479013
CrossRef Full Text | Google Scholar
Bazant, M. Z., Storey, B. D., and Kornyshev, A. A. (2011). Double layer in ionic liquids: overscreening versus crowding. Phys. Rev. Lett. 106, 046102. doi:10.1103/physrevlett.106.046102
PubMed Abstract | CrossRef Full Text | Google Scholar
Bhuiyan, L. B., Lamperski, S., Wu, J., and Henderson, D. (2012). Monte Carlo simulation for the double layer structure of an ionic liquid using a dimer model: a comparison with the density functional theory. J. Phys. Chem. B 116, 10364–10370. doi:10.1021/jp304362y
PubMed Abstract | CrossRef Full Text | Google Scholar
Blossey, R., Maggs, A., and Podgornik, R. (2017). Structural interactions in ionic liquids linked to higher-order Poisson-Boltzmann equations. Phys. Rev. E 95, 060602. doi:10.1103/physreve.95.060602
PubMed Abstract | CrossRef Full Text | Google Scholar
Borukhov, I., Andelman, D., and Orland, H. (1997). Steric effects in electrolytes: a modified Poisson-Boltzmann equation. Phys. Rev. Lett. 79, 435–438. doi:10.1103/physrevlett.79.435
CrossRef Full Text | Google Scholar
Bossa, G. V., and May, S. (2020). Stability of ionic liquid modeled by composite Coulomb-Yukawa potentials. Phys. Rev. Res. 2, 032040. doi:10.1103/physrevresearch.2.032040
CrossRef Full Text | Google Scholar
Bossa, G. V., Roth, J., and May, S. (2015). Modeling lipid–lipid correlations across a bilayer membrane using the quasi-chemical approximation. Langmuir 31, 9924–9932. doi:10.1021/acs.langmuir.5b01719
PubMed Abstract | CrossRef Full Text | Google Scholar
Coles, S. W., Park, C., Nikam, R., Kanduc, M., Dzubiella, J., and Rotenberg, B. (2020). Correlation length in concentrated electrolytes: insights from all-atom molecular dynamics simulations. J. Phys. Chem. B 124, 1778–1786. doi:10.1021/acs.jpcb.9b10542
PubMed Abstract | CrossRef Full Text | Google Scholar
Davis, H. T. (1996). “Statistical mechanics of phases, interfaces, and thin films,” in Advances in interfacial engineering series (VCH).
Google Scholar
Del Pópolo, M. G., and Voth, G. A. (2004). On the structure and dynamics of ionic liquids. J. Phys. Chem. B 108, 1744–1752. doi:10.1021/jp0364699
CrossRef Full Text | Google Scholar
Démery, V., Dean, D. S., Hammant, T. C., Horgan, R. R., and Podgornik, R. (2012). The one-dimensional Coulomb lattice fluid capacitor. J. Chem. Phys. 137, 064901. doi:10.1063/1.4740233
PubMed Abstract | CrossRef Full Text | Google Scholar
de Souza, J. P., and Bazant, M. Z. (2020). Continuum theory of electrostatic correlations at charged surfaces. J. Phys. Chem. C 124, 11414–11421. doi:10.1021/acs.jpcc.0c01261
CrossRef Full Text | Google Scholar
Downing, R., Berntson, B. K., Bossa, G. V., and May, S. (2018a). Differential capacitance of ionic liquids according to lattice-gas mean-field model with nearest-neighbor interactions. J. Chem. Phys. 149, 204703. doi:10.1063/1.5047490
PubMed Abstract | CrossRef Full Text | Google Scholar
Downing, R., Bossa, G. V., and May, S. (2018b). The role of ion–ion correlations for the differential capacitance of ionic liquids. J. Phys. Chem. C 122, 28537–28544. doi:10.1021/acs.jpcc.8b09756
CrossRef Full Text | Google Scholar
Fedorov, M. V., and Kornyshev, A. A. (2008). Towards understanding the structure and capacitance of electrical double layer in ionic liquids. Electroch. Acta 53, 6835–6840. doi:10.1016/j.electacta.2008.02.065
CrossRef Full Text | Google Scholar
Frydel, D. (2016). Mean field electrostatics beyond the point charge description. Adv. Chem. Phys. 160, 209–260. doi:10.1002/9781119165156.ch4
CrossRef Full Text | Google Scholar
Goodwin, Z. A., de Souza, J. P., Bazant, M. Z., and Kornyshev, A. A. (2023). “Mean-field theory of the electrical double layer in ionic liquids,” in Encyclopedia of ionic liquids (Springer), 837–850.
CrossRef Full Text | Google Scholar
Goodwin, Z. A., Feng, G., and Kornyshev, A. A. (2017). Mean-field theory of electrical double layer in ionic liquids with account of short-range correlations. Electroch. Acta 225, 190–197. doi:10.1016/j.electacta.2016.12.092
CrossRef Full Text | Google Scholar
Gupta, A., Govind Rajan, A., Carter, E. A., and Stone, H. A. (2020a). Ionic layering and overcharging in electrical double layers in a Poisson-Boltzmann model. Phys. Rev. Lett. 125, 188004. doi:10.1103/physrevlett.125.188004
PubMed Abstract | CrossRef Full Text | Google Scholar
Gupta, A., Govind Rajan, A., Carter, E. A., and Stone, H. A. (2020b). Thermodynamics of electrical double layers with electrostatic correlations. J. Phys. Chem. C 124, 26830–26842. doi:10.1021/acs.jpcc.0c08554
CrossRef Full Text | Google Scholar
Henderson, D., Lamperski, S., Jin, Z., and Wu, J. (2011). Density functional study of the electric double layer formed by a high density electrolyte. J. Phys. Chem. B 115, 12911–12914. doi:10.1021/jp2078105
PubMed Abstract | CrossRef Full Text | Google Scholar
Khlyupin, A., Nesterova, I., and Gerke, K. (2023). Molecular scale roughness effects on electric double layer structure in asymmetric ionic liquids. Electrochim. Acta 450, 142261. doi:10.1016/j.electacta.2023.142261
CrossRef Full Text | Google Scholar
Kondrat, S., Feng, G., Bresme, F., Urbakh, M., and Kornyshev, A. A. (2023). Theory and simulations of ionic liquids in nanoconfinement. Chem. Rev. 123, 6668–6715. doi:10.1021/acs.chemrev.2c00728
PubMed Abstract | CrossRef Full Text | Google Scholar
Kondrat, S., Georgi, N., Fedorov, M. V., and Kornyshev, A. A. (2011). A superionic state in nano-porous double-layer capacitors: insights from Monte Carlo simulations. Phys. Chem. Chem. Phys. 13, 11359–11366. doi:10.1039/c1cp20798a
PubMed Abstract | CrossRef Full Text | Google Scholar
Linse, P., and Andersen, H. C. (1986). Truncation of Coulombic interactions in computer simulations of liquids. J. Chem. Phys. 85, 3027–3041. doi:10.1063/1.451011
CrossRef Full Text | Google Scholar
Ma, K., Woodward, C. E., and Forsman, J. (2014). Classical density functional study on interfacial structure and differential capacitance of ionic liquids near charged surfaces. J. Phys. Chem. C 118, 15825–15834. doi:10.1021/jp504001u
CrossRef Full Text | Google Scholar
May, S. (2019). Differential capacitance of the electric double layer: mean-field modeling approaches. Curr. Opin. Electrochem. 13, 125–131. doi:10.1016/j.coelec.2018.12.002
CrossRef Full Text | Google Scholar
Moon, G. J., Ahn, M. M., and Kang, I. S. (2015). Osmotic pressure of ionic liquids in an electric double layer: prediction based on a continuum model. Phys. Rev. E 92, 063020. doi:10.1103/physreve.92.063020
PubMed Abstract | CrossRef Full Text | Google Scholar
Nakamura, I. (2015). Dipolar self-consistent field theory for ionic liquids: effects of dielectric inhomogeneity in ionic liquids between charged plates. J. Phys. Chem. C 119, 7086–7094. doi:10.1021/jp511770r
CrossRef Full Text | Google Scholar
Nesterova, I., Evstigneev, N. M., Ryabkov, O. I., Gerke, K. M., and Khlyupin, A. (2025). Mechanism of overscreening breakdown by molecular-scale electrode surface morphology in asymmetric ionic liquids. J. Colloid Interface Sci. 677, 396–405. doi:10.1016/j.jcis.2024.08.040
PubMed Abstract | CrossRef Full Text | Google Scholar
Shalabi, A., Daniels, L., Scott, M., and Mišković, Z. (2019). Differential capacitance of ionic liquid interface with graphene: the effects of correlation and finite size of ions. Electrochim. Acta 319, 423–434. doi:10.1016/j.electacta.2019.06.171
CrossRef Full Text | Google Scholar
Silva, W., Zanatta, M., Ferreira, A. S., Corvo, M. C., and Cabrita, E. J. (2020). Revisiting ionic liquid structure-property relationship: a critical analysis. Int. J. Mol. Sci. 21, 7745. doi:10.3390/ijms21207745
PubMed Abstract | CrossRef Full Text | Google Scholar
Walden, P. (1914). Molecular weights and electrical conductivity of several fused salts. Bull. Acad. Imper. Sci. St. Petersbg. 1800.
Google Scholar
Wang, Y., Jiang, W., Yan, T., and Voth, G. A. (2007). Understanding ionic liquids through atomistic and coarse-grained molecular dynamics simulations. Acc. Chem. Res. 40, 1193–1199. doi:10.1021/ar700160p
PubMed Abstract | CrossRef Full Text | Google Scholar
Wang, Y.-L., Li, B., Sarman, S., Mocci, F., Lu, Z.-Y., Yuan, J., et al. (2020). Microstructural and dynamical heterogeneities in ionic liquids. Chem. Rev. 120, 5798–5877. doi:10.1021/acs.chemrev.9b00693
PubMed Abstract | CrossRef Full Text | Google Scholar
Watanabe, M., Thomas, M. L., Zhang, S., Ueno, K., Yasuda, T., and Dokko, K. (2017). Application of ionic liquids to energy storage and conversion materials and devices. Chem. Rev. 117, 7190–7239. doi:10.1021/acs.chemrev.6b00504
PubMed Abstract | CrossRef Full Text | Google Scholar
Weingärtner, H. (2008). Understanding ionic liquids at the molecular level: facts, problems, and controversies. Angew. Chem. Int. Ed. 47, 654–670. doi:10.1002/anie.200604951
CrossRef Full Text | Google Scholar
Wu, J., Jiang, T., Jiang, D., Jin, Z., and Henderson, D. (2011). A classical density functional theory for interfacial layering of ionic liquids. Soft Matter 7, 11222–11231. doi:10.1039/c1sm06089a
CrossRef Full Text | Google Scholar
Ye, B. B., and Wang, Z.-G. (2022). A coarse-grained model of room-temperature ionic liquids between metal electrodes: a molecular dynamics study. Phys. Chem. Chem. Phys. 24, 11573–11584. doi:10.1039/d2cp00166g
PubMed Abstract | CrossRef Full Text | Google Scholar
Yochelis, A. (2014). Transition from non-monotonic to monotonic electrical diffuse layers: impact of confinement on ionic liquids. Phys. Chem. Chem. Phys. 16, 2836–2841. doi:10.1039/c3cp55002h
PubMed Abstract | CrossRef Full Text | Google Scholar
 Guilherme Volpe Bossa
Guilherme Volpe Bossa Sylvio May
Sylvio May