 Ramsha Munir1
Ramsha Munir1 Ameer Fawad Zahoor1*
Ameer Fawad Zahoor1* Muhammad Naveed Anjum2
Muhammad Naveed Anjum2 Asim Mansha1
Asim Mansha1 Ali Irfan1Aijaz Rasool Chaudhry3
Ali Irfan1Aijaz Rasool Chaudhry3 Ahmad Irfan4
Ahmad Irfan4 Katarzyna Kotwica-Mojzych5
Katarzyna Kotwica-Mojzych5 Mariola Glowacka6
Mariola Glowacka6 Mariusz Mojzych6*
Mariusz Mojzych6*- 1Department of Chemistry, Government College University Faisalabad, Faisalabad, Pakistan
- 2Department of Applied Chemistry, Government College University Faisalabad, Faisalabad, Pakistan
- 3Department of Physics, College of Science, University of Bisha, Bisha, Saudi Arabia
- 4Department of Chemistry, College of Science, King Khalid University, Abha, Saudi Arabia
- 5Department of Basic Sciences, Department of Histology, Embriology and Cytophysiology, Medical University of Lublin, Lublin, Poland
- 6Faculty of Health Sciences Collegium Medicum, The Mazovian Academy in Plock, Płock, Poland
The Yamaguchi reagent, based on 2,4,6-trichlorobenzoyl chloride (TCBC) and 4-dimethylaminopyridine (DMAP), is an efficient tool for conducting the intermolecular (esterification) reaction between an acid and an alcohol in the presence of a suitable base (Et3N or iPr2NEt) and solvent (THF, DCM, or toluene). The Yamaguchi protocol is renowned for its ability to efficiently produce a diverse array of functionalized esters, promoting high yields, regioselectivity, and easy handling under mild conditions with short reaction times. Here, the recent utilization of the Yamaguchi reagent was reviewed in the synthesis of various natural products such as macrolides, terpenoids, polyketides, peptides, and metabolites.
1 Introduction
Ester linkage is the cornerstone of modern synthetic chemistry for containing carbonyl functionality and the structural part of most of the precursors in the synthesis of medicinally important natural and synthetic compounds (Haslam, 1980). Apart from the pharmaceutical industry, other industries (such as textile, cosmetics, fragrance, pesticides, fungicides, and coatings) are also dependent on the ester linkage-based synthetic intermediates. Therefore, esterification is an eminent conversion reaction that is usually performed between acid chloride and alcohol, acid anhydride and alcohol, or carboxylic acid and alcohol (Khan et al., 2021). With the profound interest in the ester linkage, several methodologies have been developed, and the most common methodologies are the Mitsunobu reaction (Munawar et al., 2022), Fischer esterification (involving a Lewis acid as the catalyst) (Joseph et al., 2005), Steglich esterification [usually takes place in the presence of DCC, 4-dimethylaminopyridine (DMAP), and DCM) (Munawar et al., 2024), and Yamaguchi protocol (Inanaga et al., 1979). Each of these methods have their limitations; for example, Fischer esterification is a slower reaction and provides a low yield of product (Khan et al., 2021), whereas Steglich esterification utilizes toxic carbodiimide (Jordan et al., 2021). Steglich and Yamaguchi’s methods are distinguished for the use of DMAP as a strong nucleophilic base. Among others, Yamaguchi esterification is a leading and beneficial tool for esterification and has gained significance in the regioisomeric synthesis of macrolides and many other natural products (Majhi, 2021).
The Yamaguchi coupling protocol was first developed by Masaru Yamaguchi et al. (in 1979) during the synthesis of ester (Inanaga et al., 1979). This methodology was mainly based on the reaction between acids and alcohols in the presence of 2,4,6-trichlorobenzoyl chloride (TCBC), with DMAP as the coupling agent and Et3N as the base, providing corresponding esters in moderate-to-good yield. Later, this procedure was extended for the synthesis of a variety of macrolactones. The most commonly used solvents are THF, toluene, and DCM to smoothly furnish both primary and secondary esters. Yamaguchi esterification ensures wide substrate scope, mild conditions, and the formation of a regioselective product in moderate-to-good yield. Side by side, Yamaguchi esterification also has disadvantages of less reactivity of TCBC (due to its steric environment), decomposition of the substrates, or poor yield in the case of the total synthesis of very few compounds. Still, this reaction has a wide scope and has been extended to the synthesis of a variety of macrolactones with no epimerization of stereochemistry. The reaction usually takes place either in a single step (direct reaction of carboxylic acid and alcohol) or in two steps (via the formation of acid anhydride from carboxylic acid and 2,4,6-trichlorobenzoyl chloride, followed by the attack of alcohol). The two-step method has previously been reported for the synthesis of various lactones of a large ring size, such as 2,4,6-tridemethyl-3-deoymethynolide. The original Yamaguchi procedure was based on a two-step methodology that was later modified as a one-pot (single-step) reaction by Hikota et al. (1990). The detailed mechanistic pathway of this reaction was studied by Dhimitruka and SantaLucia, and the methodology was successfully used for the formation of a Lux-S aspartic acid suppressant. In their investigative studies, benzoyl chloride, p-tolyl chloride, and TCBC as an electrophile were used, and the formation of the regioselective product was confirmed only with 2,4,6-trichlorobenzoyl chloride (Yamaguchi reagent), which is the key feature of this esterification reaction (Dhimitruka and SantaLucia, 2006). As shown in Figure 1, the carboxylic acid after deprotonation (via the involvement of a base) provides the carboxylate. The coupling of this carboxylate with 2,4,6-trichlorobenzoyl chloride leads to the formation of an anhydride, which, after further coupling with another carboxylate, results in acid anhydride (Park et al., 2022). In the next step, the addition of DMAP results in the formation of pyridinium salt, followed by the nucleophilic attack of the base, which leads toward the formation of the desired ester.
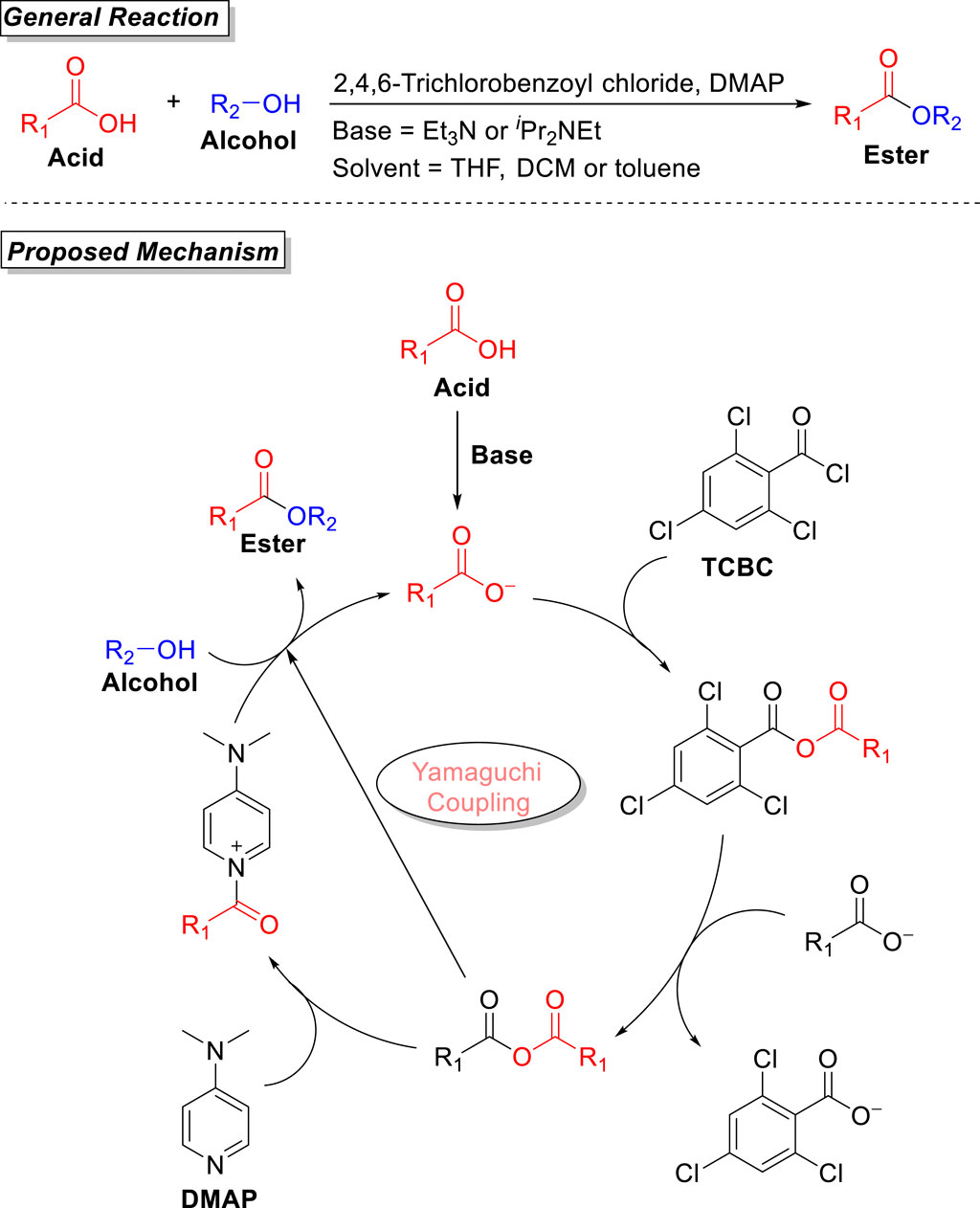
Figure 1. General reaction and proposed mechanism of the Yamaguchi coupling reaction.
In 2014, a commendable strategy for esterification was introduced by Okuno et al. via the use of 2,4,6-trichlorobenzoyl-4-dimethylaminopyridinium chloride as a modified Yamaguchi reagent (Okuno et al., 2014). This TCB-DMAP reagent was prepared (in 90% yield) simply by a reaction between TCBC and DMAP in the presence of THF. It facilitates esterification for broad substrate groups, avoiding the formation of anhydride and can be stored for many years (Yamamoto and Muramatsu, 2019). In 2016, Nishio et al. synthesized a recoverable fluorous Yamaguchi reagent for the efficient esterification of several benzoic acids with alcohols (Nishio et al., 2016) (Supplementary Figure 1).
In addition to esterification and macro-lactonization, the Yamaguchi reagent is much significant for many other organic reactions, especially for the synthesis of carboxylic acid derivatives (Radha Krishna et al., 2022; Mukhopadhyay and Trauner, 2022). For instance, Chandra et al. (2018) utilized their own modified Yamaguchi reagent for amidation, thioesterification, and peptide synthesis. Zulquranain et al. (2020) synthesized pyrazine-2-carboxylic acid derivatives (via the use of the Yamaguchi reagent) to be a cytotoxic agent against Mycobacterium tuberculosis (Zulqurnain et al., 2023). In natural product synthesis, the Yamaguchi reagent has been involved in the synthesis of biologically active compounds (Kotammagari, 2014; Wu et al., 2012; Valeev et al., 2019; Molawi et al., 2010), such as stagonolide C (a herbicide isolated from Cirsium arvense) (Wu et al., 2012), amphidinolide W (a marine dinoflagellate and a cytotoxic agent against the murine lymphoma cell line with IC50 = 3.9 μg/mL) (Shimbo et al., 2002), palmerolide A (exhibits cytotoxicity against melanoma cell line UACC-62 and renal cancer cell line RXF 393) (Pujari et al., 2011), and xyolide (bioactive against Pythium ultimum, a plant pathogen) (Maram and Das, 2015) (Supplementary Figure 2). Fascinated by the synthetic utility of the Yamaguchi reagent, Majhi et al. published a review article on the application of Yamaguchi’s method in the synthesis of biologically potent natural products in 2021 (Majhi, 2021). However, an updated compilation of its recent application (2021–2023) in the synthesis of natural products has been presented here.
2 Review of the literature
2.1 Synthesis of natural macrolides
Esterification and macrolactonization are the commonly involved reactions in the construction of macrolides consisting of simpler to complex frameworks. Here, we present various examples, demonstrating the strong potential of the Yamaguchi reagent in the synthesis of 10–30 (ring size)-membered macrolides.
2.1.1 Synthesis of 10-membered macrolides
2.1.1.1 Reddy’s total synthesis of sumalactone A
Sumalactone A 10 is a 10-membered macrolactone that was isolated from Penicillium sumatrense, a marine fungus (Wu et al., 2017). This benzannulated macrolactone is famous for its numerous biological activities, such as anti-fungal, anti-cancer, and anti-inflammatory effects (Scheme 1) (Allu et al., 2019; Shahzadi et al., 2022). As an attractive target of various organic chemists, Reddy et al. (2022) performed the stereoselective synthesis of sumalactone A 10 using easily available inexpensive starting materials 1 and 2 (Reddy et al., 2022). The key steps in their synthetic part involve Yamaguchi esterification with the proper maintenance of stereochemistry of the reacting substrates. As illustrated in the scheme, acid 3 and alcohol 4 were successfully achieved from acid 1 and alcohol 2, respectively. Both the synthesized compounds, in hand, were subjected to esterification using a Yamaguchi reagent (2,4,6-trichlorobenzoyl chloride) in the presence of Et3N, DMAP, and toluene, resulting in high (78%)-yielding ester 5. Furthermore, Grubb’s second-generation catalyst was used for the RCM reaction of ester 5, followed by its palladium-catalyzed reduction and deprotection in the presence of BBr3 and DCM to finally afford sumalactone A 6 with 77% yield.
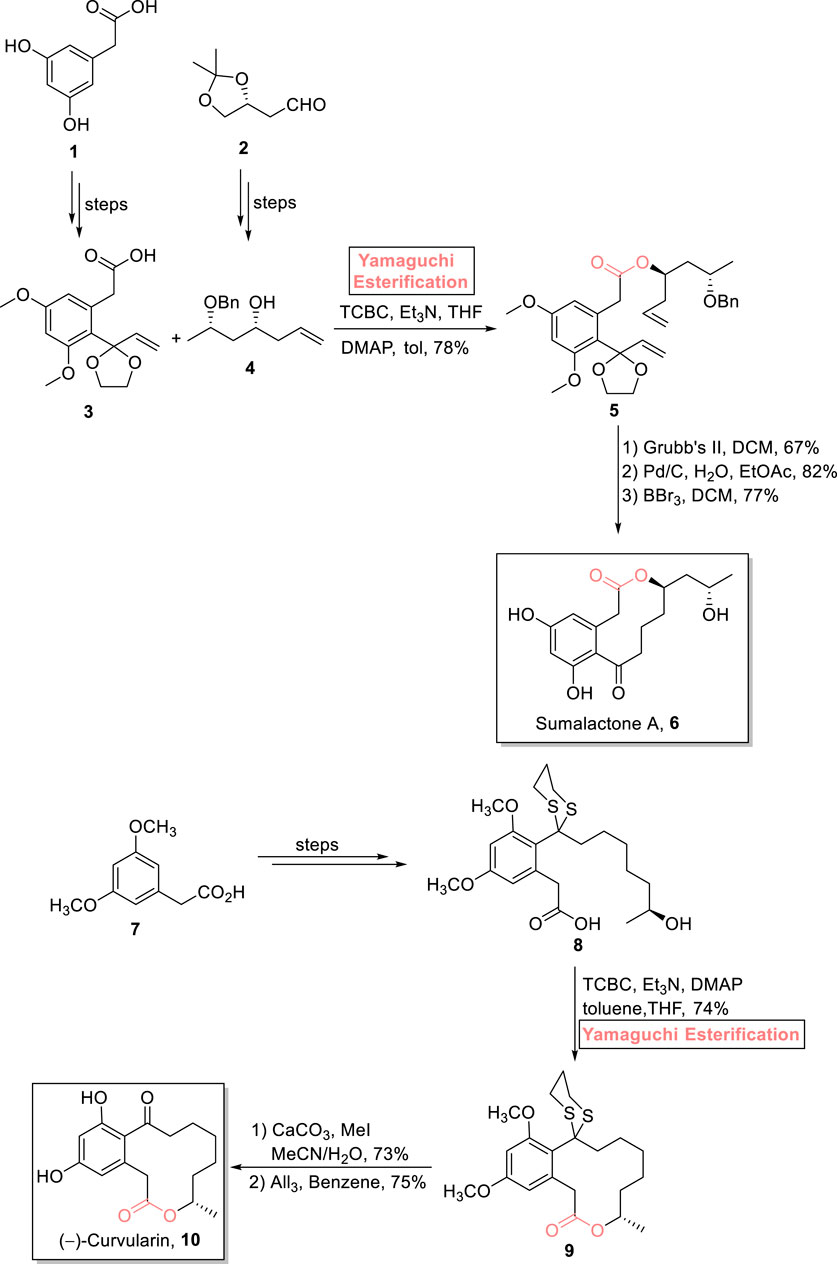
Scheme 1. Synthesis of sumalactone A 10 and (−)-curvularin 11.
2.1.2 Synthesis of 12-membered macrolides
2.1.2.1 Radha Krishna’s total synthesis of (−)-curvularin
One of the 12-membered macrolides, curvularin, is a resorcylic acid lactone that is produced by various fungal sources, that is, Alternaria, Penicillium, and Curvularia. It exhibits diverse biological activities such as cell division prohibition and cytotoxicity against sea urchin embryogenesis (Zhan and Gunatilaka, 2005). With a great deal of interest, Radha Krishna et al. (2022) accomplished its total synthesis using Yamaguchi macrolactonization as a crucial step. For achieving the targeted product, (3,5-dimethoxyphenyl)acetic acid 7 was used as an easily available starting material to build compound 8. Next, compound 8 was subjected to a well-suited Yamaguchi reagent, leading to the formation of compound 9 with 74% yield. After this, the removal of the 1,3 dithiane group and deprotection of methyl ester groups in ester 9 resulted in the synthesis of (−)-curvularin 10 with 75% yield (Scheme 1).
2.1.2.2 Chambers’s total synthesis of 10-deoxymethynolide
Enones are unique synthetic intermediates that have widespread applications in the synthesis of many biologically active natural products. An enone, 10-deoxymethynolide contains a popular polyketide macrolide, having four stereogenic centers, and is expected to be a medicinally important natural product (Wei and Shi, 2013). Chambers et al. (2023) accomplished the efficient 14-step total synthesis of 10-deoxymethynolide 15 by using ester 11 as an easily available enantioenriched starting material (Scheme 2) (Chambers et al., 2023). The settlement of four stereocenters in the target compound was no doubt a challenging task that was successfully made possible using the Yamaguchi protocol. To set the stage for the Yamaguchi esterification protocol, the modification of compound 11 (over a few steps) into compound 12, followed by treatment with compound 13 in the presence of TCBC, triethyl amine (Et3N), DMAP, and toluene, successfully furnished ester 14 with 56% yield. Next, the ring closure reaction of compound 14 using Grubb’s second-generation catalyst and subsequent desilylation completed the total synthesis of 10-deoxymethynolide 15 with 34% yield.
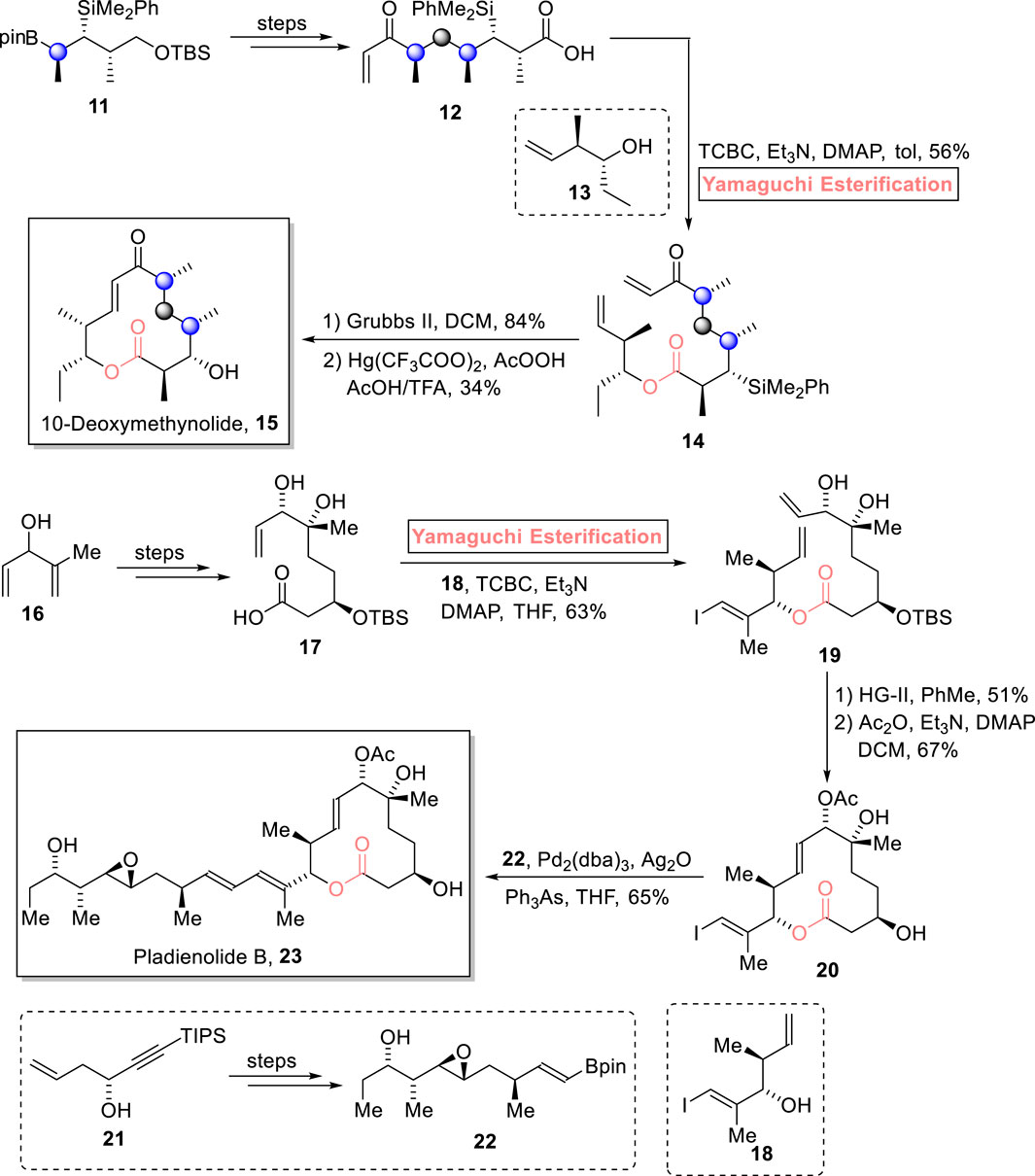
Scheme 2. Synthesis of 10-deoxymethynolide 15 and pladienolide B 23.
2.1.2.3 Yoo and Krische’s total synthesis of pladienolide B
Pladienolides are 12-membered macrolides isolated from Streptomyces platensis in 2004. Owing to their anti-proliferative activity in multi-resistant human tumor cells, this unique family of natural products is gaining prominence as anti-cancer pharmacodynamics in medicinal chemistry (Sakai et al., 2004; Villa et al., 2012). To date, various synthetic reports on the synthesis of pladienolide derivatives have been published. Pioneering this groundbreaking endeavor, Yoo and Krische (2021) devised a robust and economical synthetic route for the synthesis of pladienolide B 23 (consisting of 10 stereogenic centers) in just 10 steps (Scheme 2) (Yoo and Krische, 2021). The salient feature of their synthesis lies in the strategic use of the Yamaguchi method as the leading tool for esterification. To set the stage for Yamaguchi esterification, compound 16 was turned into acid 17 over a few steps. Compound 17, in hand, was allowed to get esterified with fragment 18 in the presence of TCBC, Et3N, DMAP, and THF to result in ester 19 with 63% yield. In the next step, the ring-closing metathesis of compound 19, followed by acetylation, resulted in compound 20 with 67% yield. Moving toward the final step, compound 20 was made to couple with compound 22 (from compound 21) under Suzuki conditions to successfully accomplish the target pladienolide B 23 with 65% yield.
2.1.3 Synthesis of 14-membered macrolides
2.1.3.1 Meyer’s total synthesis of amphidinolide R
Amphidinolides are cytotoxic macrolides, and these were isolated from Amphidinium sp. marine dinoflagellates by Kobayashi et al.. Structurally, amphidinolide R is 14-membered, while amphidinolide J and amphidinolide S are 15-membered macrolides (Ishibashi and Kobayashi, 1997; Shotwell and Roush, 2004). Meyer et al. (2023) accomplished the diastereoselective and enantioselective synthesis of amphidinolide R 28 (9 steps) and amphidinolide J 29 (9 steps), along with the first total synthesis of amphidinolide S 30 (10 steps). The achievement of the desired stereochemistry in the products was assured via the use of Yamaguchi esterification as a powerful step (Scheme 3) (Meyer et al., 2023). Their methodology involved the independent synthesis of fragments 25 and 27 from compounds 24 and 26, respectively. Then, fragments 25 and 27 were subjected to esterification using the Yamaguchi protocol in the presence of diethyl amine, TCBC, Et3N, and THF. The esterified intermediate was then subjected to Grubb’s second-generation catalyst and DDQ, followed by corresponding work procedures under given conditions to furnish amphidinolide R 28 in 78% yield.
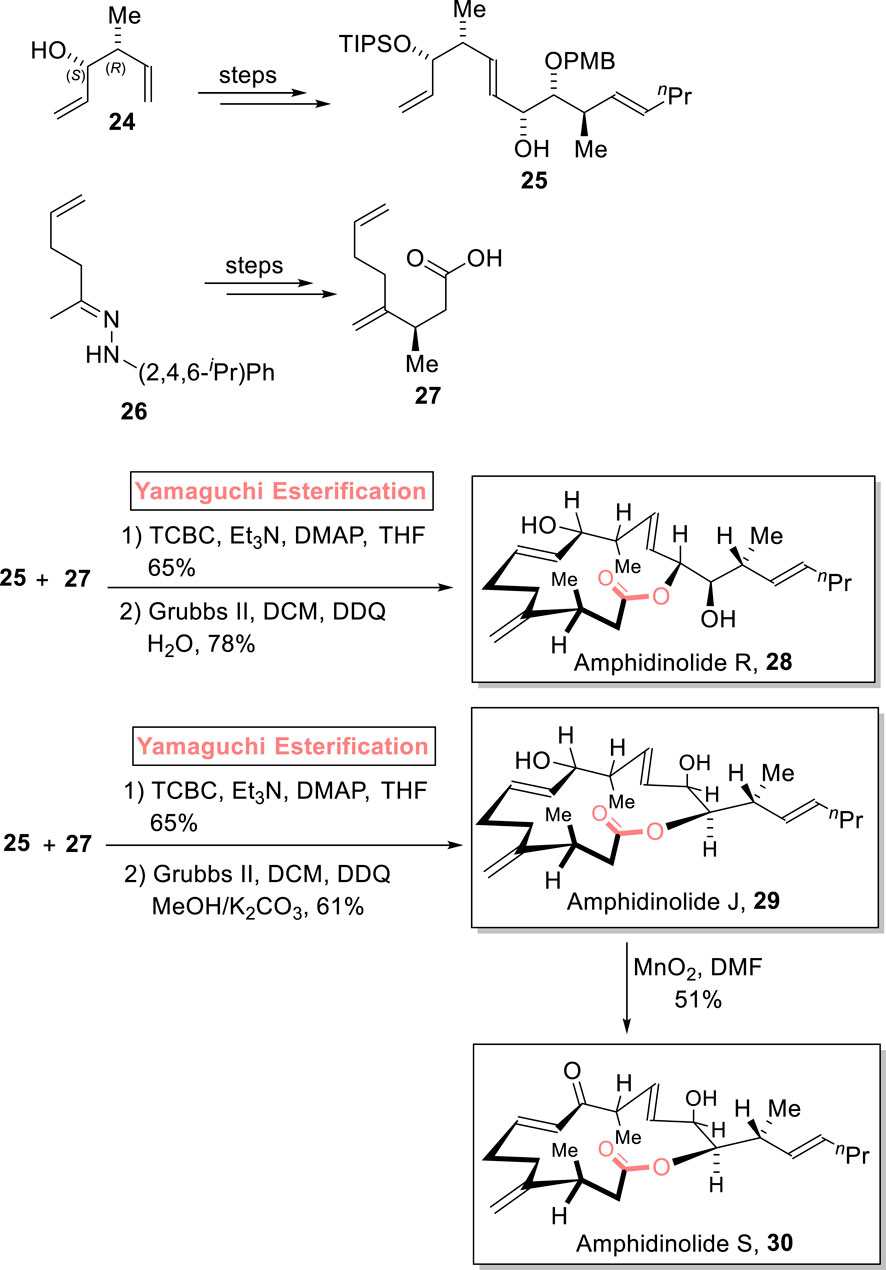
Scheme 3. Synthesis of amphidinolide R 28, amphidinolide J 29, and amphidinolide S 30.
2.1.3.2 Nakazato’s total synthesis of (+)-neopeltolide
The tetrahydropyran ring is present in most biologically important natural products. One of the tetrahydropyran rings containing a natural product, (+)-neopeltolide 40, is a 14-membered macrolide, consisting of a lactone ring associated with a 2,4,6-trisubstituted tetrahydropyran scaffold. It was isolated from a deep-water sponge in Jamaica by Wright et al. (2007). It exhibits cytotoxic activity against A549 (human lung adenocarcinoma cells) and NCI-ADR-RES (human ovarian sarcoma cells) with IC50 values of 1.2 nM and 5.1 nM, respectively. Furthermore, it also exhibits growth inhibition against Candida albicans with a MIC value of 0.62 μg/mL (Ulanovskaya et al., 2008; Bai and Dai, 2015). With these distinctive medicinal features, this scaffold has been the focus of various researchers, and more than 20 reports on its synthesis have been documented. Continuing with the ongoing effort, Nakazato et al. (2022) performed the 11-step total synthesis of (+)-neopeltolide 40 (with 12% overall yield) via Yamaguchi esterification of intermediates 32 and 34 as the key step (Scheme 4) (Nakazato et al., 2022). In their synthetic methodology, compounds 32, 34, and 37 were prepared from starting materials 31, 33, and 36, respectively. After this, the Yamaguchi esterification of compounds 32 and 34 in the presence of TCBC, Et3N, DMAP, and TsOH successfully furnished ester 38 (with 90% yield) as a precursor for the construction of the 14-membered anti-cancer macrolide. Ester 38 then underwent a sequence of Meyer–Schuster rearrangement, Zhan-catalyzed RCM reaction, and Michael addition to result in tetrahydropyran 39 with 69% yield. Next, compound 39 was subjected to Zn-mediated methylenation, hydrogenolysis, and subsequent Mitsunobu coupling with compound 37 to afford the desired natural product 40 with 94% yield.
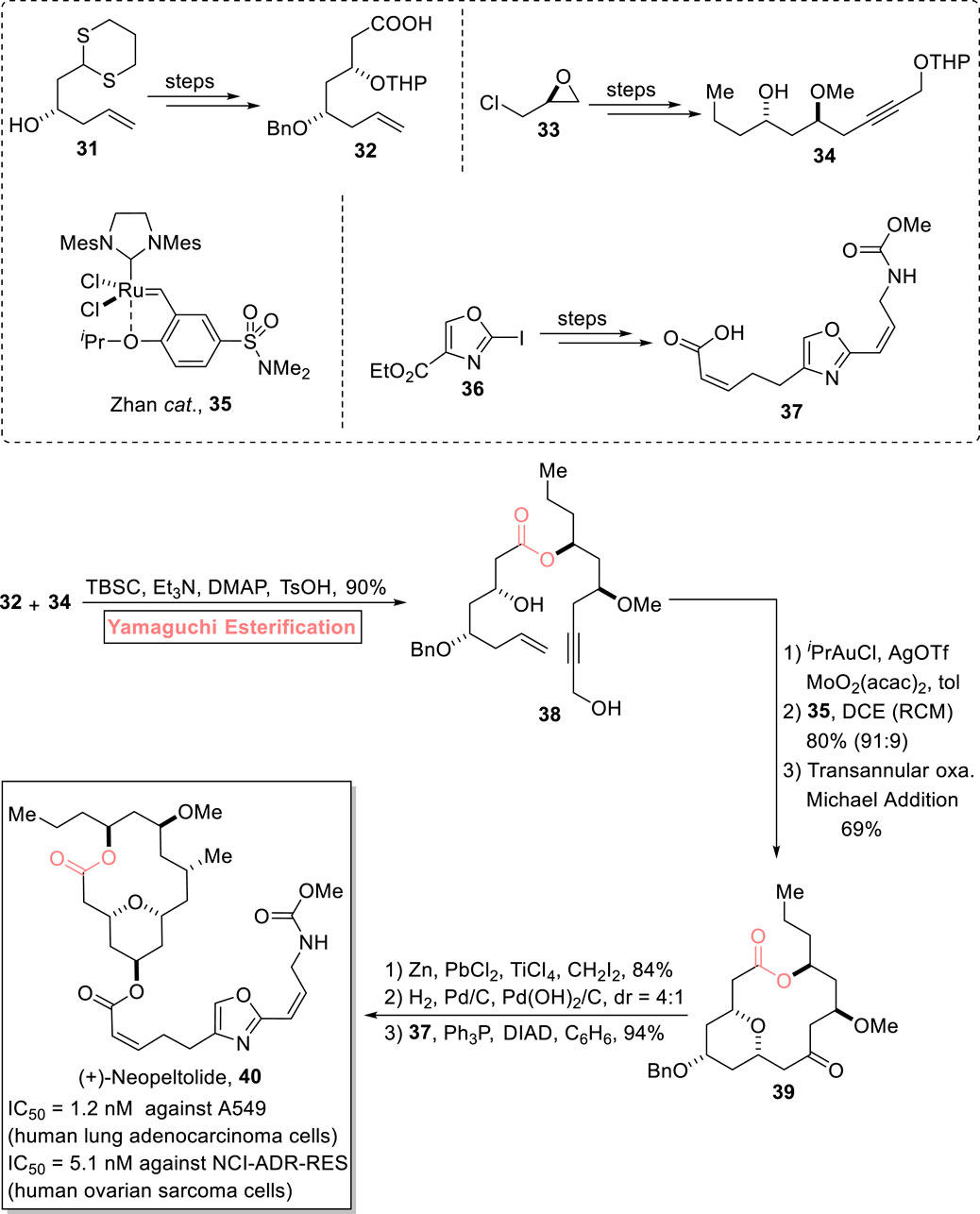
Scheme 4. Synthesis of (+)-neopeltolide 40.
2.1.3.3 Depa’s total synthesis of neocosmosin A
Resorcyclic acid lactones are well known in the medicinal world for their remarkable biological profile as they exhibit estrogenic, cytotoxic, nematocidal, anti-viral, and anti-fungal biological activities (Patocka et al., 2013; Hellwig et al., 2003; Abid-Essefi et al., 2004). The 14-membered macrolides, neocosmosin A 47 and neocosmosin B 51, are resorcyclic acid lactones according to their structural composition. These were isolated in 2012 from Neocosmospora sp. of a fungal strain. Neocosmosin A 47 possesses an affinity for binding with human cannabinoid and opioid receptors (Gao et al., 2013). Depa et al. (2021) performed the efficient (14-step) total synthesis of this natural product using propylene oxide 41 and 4-methoxy salicylic acid 43 (as easily available starting materials) with a 4.45% overall yield (Scheme 5) (Depa et al., 2021). The construction of this targeted natural product with the desired stereochemistry entails Yamaguchi macrolactonization as a crucial step. The methodology involved the coupling of bromide 42 and dithiane 44, followed by hydrolysis and desilylation to furnish hydroxy acid 45 with 91% yield. Next, the Yamaguchi protocol was used for the esterification of acid 45 by treating it with TCBC, Et3N, THF, DMAP, and toluene, which resulted in compound 46 with 66% yield. Proceeding toward the last stage of the total synthesis, the dithiane group was removed via the treatment of lactone 46 with calcium carbonate and methyl iodide, followed by TiCl4-mediated deprotection to successfully afford neocosmosin A 47 with 78% yield.
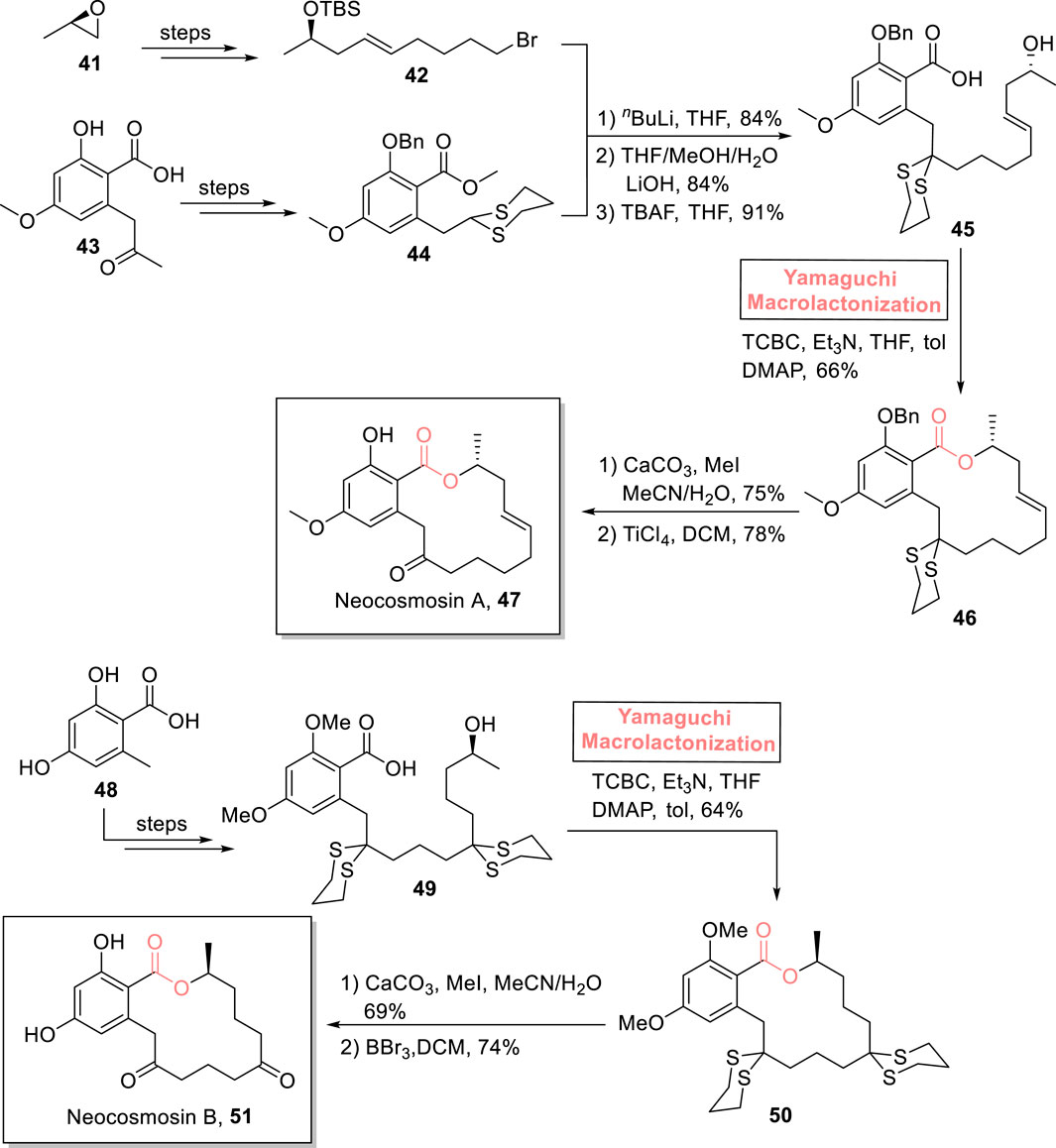
Scheme 5. Synthesis of neocosmosin A 47 and neocosmosin B 51.
2.1.3.4 Kumari’s total synthesis of neocosmosin B
Kumari et al. (2022) reported the first total synthesis of neocosmosin B 51 in 12 steps by using Yamaguchi macrolactonization as a key step (Scheme 5) (Kumari et al., 2022). Their synthesis commenced with easily available orsellinic acid 48, which, over a few steps, provided compound 49. In the following step, the Yamaguchi reagent was used for the macrolactonization of hydroxyl acid 49 by treating it with TCBC, Et3N, THF, and then with DMF and toluene to finally furnish lactone 50 with 64% yield. Finally, the removal of dithiane groups and demethoxylation of lactone 50 successfully furnished the desired neocosmosin B 51 with 74% yield.
2.1.3.5 Dissanayake’s total synthesis of sanctolide A
Sanctolide A 57 is a 14-membered polyketide and peptide-based macrolide. It was isolated in 2012 by Orjala et al. from Oscillatoria sancta, a cyanobacterium (Kang et al., 2012). The structural framework of this hybrid scaffold comprises an N-methyl-substituted macrocyclic diester attached with a lipophilic side chain. With these significant structural features, this natural product is expected to show promising pharmaceutical effects. Dissanayake et al. (2023) performed both the total and formal syntheses of sanctolide A 57 by using Yamaguchi esterification as the main step (Scheme 6) (Dissanayake et al., 2023). As shown in Scheme 6, alcohol 53 (synthesized from compound 52 in several steps) was made to react with 2,4,6-trichlorobenzoyl chloride and diisopropylethylamine in THF. Then, carboxylic acid 55 (prepared from the reaction of isovaleric acid 54 and acryloyl chloride) was added to complete the esterification process in the presence of DMAP and toluene, resulting in ester (R)-56 with 78% yield. Hence, the coherence of stereochemistry in the reactants (alcohol 53 and acid 55) and the product (ester 56) highlights the success of choosing the Yamaguchi reagent for the esterification process. In the following step, the RCM reaction of ester (R)-56 and the subsequent treatment with (PPh3)3RuH(CO)Cl furnished the desired sanctolide A (R)-57 with 51% yield.
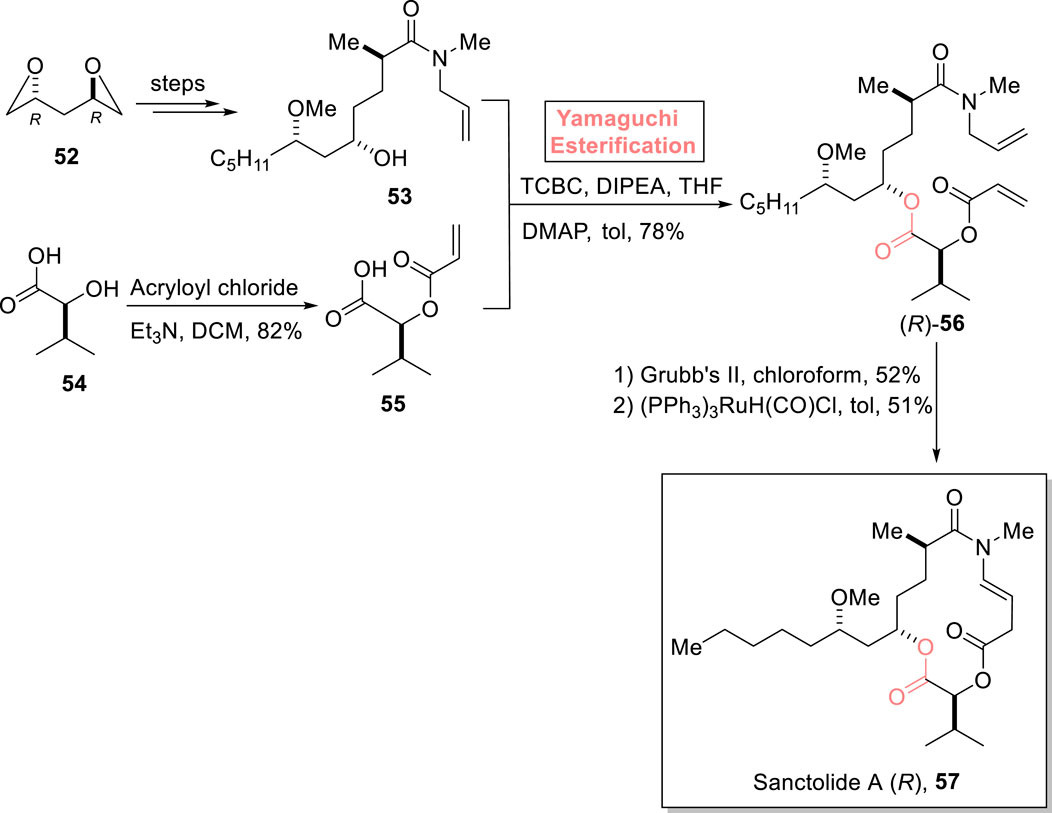
Scheme 6. Synthesis of sanctolide A 57.
2.1.4 Synthesis of 15-membered macrolides
2.1.4.1 Meyer’s total synthesis of amphidinolide J and amphidinolide S
As mentioned previously, Meyer et al. (2023) accomplished the synthesis of amphidinolide R 28 and amphidinolide J 29 and the first total synthesis of amphidinolide S 30. Their simple and facile methodology involved the Yamaguchi esterification of compounds 25 and 27 and subsequent treatment with Grubb’s second-generation catalyst to furnish amphidinolide J 29 with 61% yield. Next, the MnO2-induced oxidation of amphidinolide J 29 led to the formation of amphidinolide S 30 with 51% yield (Scheme 3).
2.1.4.2 Lai and Dai’s total synthesis of palmyrolide A
One of the neuroactive 15-membered macrolides, palmyrolide A, was isolated from an assembly of marine cyanobacteria consisting of Oscillatoria spp. and Leptolyngbya cf. It exhibits Ca influx suppression in cerebrocortical neurons (with an IC value of 3.70 μm) and Na channel suppression (with an IC value of 5.2 μm) (Pereira et al., 2010; Tan, 2007). Based on these captivating aspects, Lai and Dai (2021) enclosed the total synthesis of (−)-palmyrolide A 64a and (+)-5,7-epi-palmyrolide A 64b (Scheme 7) (Lai and Dai, 2021). Their multi-module strategic route toward synthesizing diasteroisomeric macrolides used Yamaguchi esterification as the main step. First, compounds 60 and 61 were subjected to the Negishi coupling reaction (in the presence of Pd(OAc)2, Aphos-Y, and THF), followed by hydrolysis to provide acid 62 with 60% yield. In the next steps, acid 62 was esterified with alcohol 59 (from starting material 58) using a well-suited Yamaguchi protocol (TBSCl, iPr2Net, DMAP, and THF) to obtain an inseparable mixture of esters 63a and 63b with a combined yield of 71%. Furthermore, the ring-closing metathesis of compounds 63a and 63b (in the presence of Grubb’s second-generation catalyst) and subsequent treatment with RuH(PPh3)3(CO)Cl successfully provided (−)-palmyrolide A 64a and (+)-5,7-epi-palmyrolide A 64b with a combined yield of 42%.
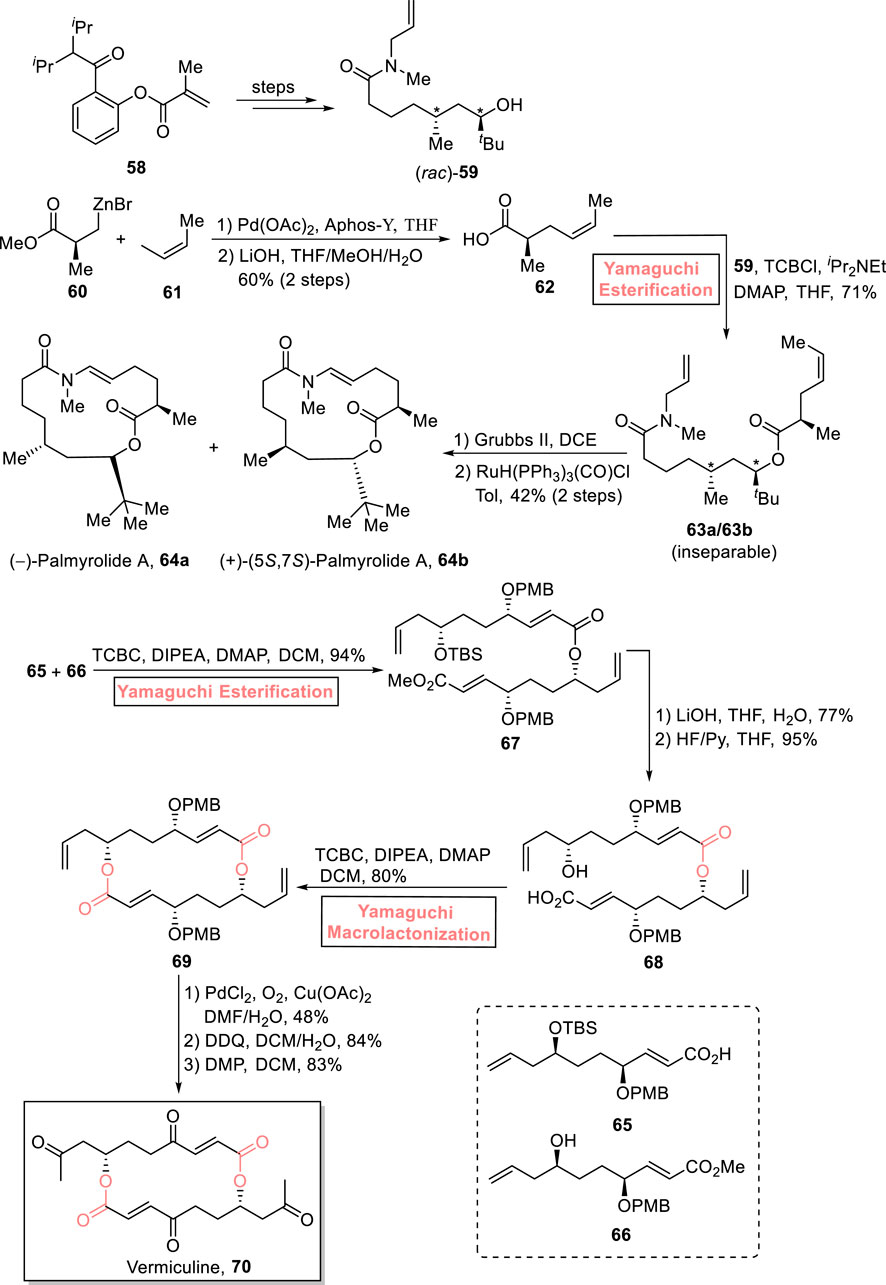
Scheme 7. Synthesis of (−)-palmyrolide A 64a, (+)-5,7-epi-palmyrolide A 64b, and (−)-vermiculine 70.
2.1.5 Synthesis of 16-membered macrolides
2.1.5.1 Liu’s total synthesis of vermiculine
(−)-Vermiculine 70 is a 16-membered macrodiolide and was first isolated from Penicillium vermiculatum in 1972 by Kuhr et al. (Fuska et al., 1972). It holds significant medicinal importance owing to its anti-cancer, anti-protozoal, and immunomodulatory biological effects (Fuska et al., 1972; Fuska et al., 1974; Horáková et al., 1976). Liu et al. (2021) devised a highly flexible and efficient synthetic route for the synthesis of (−)-vermiculine 70 (as well as its analogs) in 14 steps and 9% overall yield (Scheme 7) (Liu et al., 2021). The cornerstone of their synthetic scheme lies in the utilization of Yamaguchi esterification (for the efficient ligation of compounds 65 and 66) and Yamaguchi macrolactonization (for cyclization toward the construction of the 16-membered macrolide) as key steps. In their synthetic path, compounds 65 and 66 were subjected to Yamaguchi esterification in the presence of TCBC, diisopropyleyhylamine (DIPEA), DMAP, and DCM to achieve compound 67 with 94% yield. Dimer 67 was subjected to hydrolysis and TBS group removal to yield acid 68 (in 95% yield), which was proceeded further for macrolactonization under the Yamaguchi conditions, resulting in macrodiolide 69 with 80% yield. The oxidation of macrodiolide 69 in the presence of PdCl2, O2, and Cu(OAc)2, followed by PMB group removal and its Dess–Martin oxidation, resulted in the desired vermiculine 70 with 83% yield.
2.1.5.2 Schmidt’s total synthesis of berkeleylactone A
One of the 16-membered macrolides, berkeleylactone A 77, was isolated from a fungal strain. It is a famous anti-biotic that exhibits a strong anti-microbial effect against multi-drug resistant S. aureus (Stierle et al., 2017; Michel et al., 1977; Vakiti et al., 2022). Schmidt et al. (2023) disclosed a novel synthetic route toward the synthesis of berkeleylactone A 77 and its analog 78 via easily accessible intermediate 76 (Scheme 8) (Schmidt et al., 2023). The whole methodology was started from alkyne 71, which was reacted with propylene epoxide (under the given conditions) and then subjected to an alkyne zipper reaction, followed by treatment with potassium tertiary butoxide to result in alcohol 72 with 98% yield. In the next step, Yamaguchi esterification took precedence over Steglich esterification for furnishing ester in high yield as the substrate for te RCM reaction. Thus, the esterification of alcohol 72 with acid 73 was well managed with the exposure of the Yamaguchi reagent (TBSC, Et3N, and DMAP) in toluene as the solvent provided access to ester 74 with 94% yield. The dihydroxylation of compound 74 using AD-mix-α and subsequent acetonide protection resulted in compound 75 (in 99% yield), which was transformed into compound 76 over a few steps. Compound 76 was exposed to Rosenmund’s catalyst for hydrogenation and deprotection using TFA, resulting in berkeleylactone A 77 with 87% yield, while its analog was acquired by the direct deprotection of compound 76. After the successful synthesis of both natural product 77 and its analog 78, both of these were subjected to biological analysis against various bacterial and fungal strains, that is, S. aureus, Candida glabrata, C. albicans, Enterococcus faecalis, and Enterococcus faecium. Both compounds exhibited average-to-good anti-microbial effects against these strains.
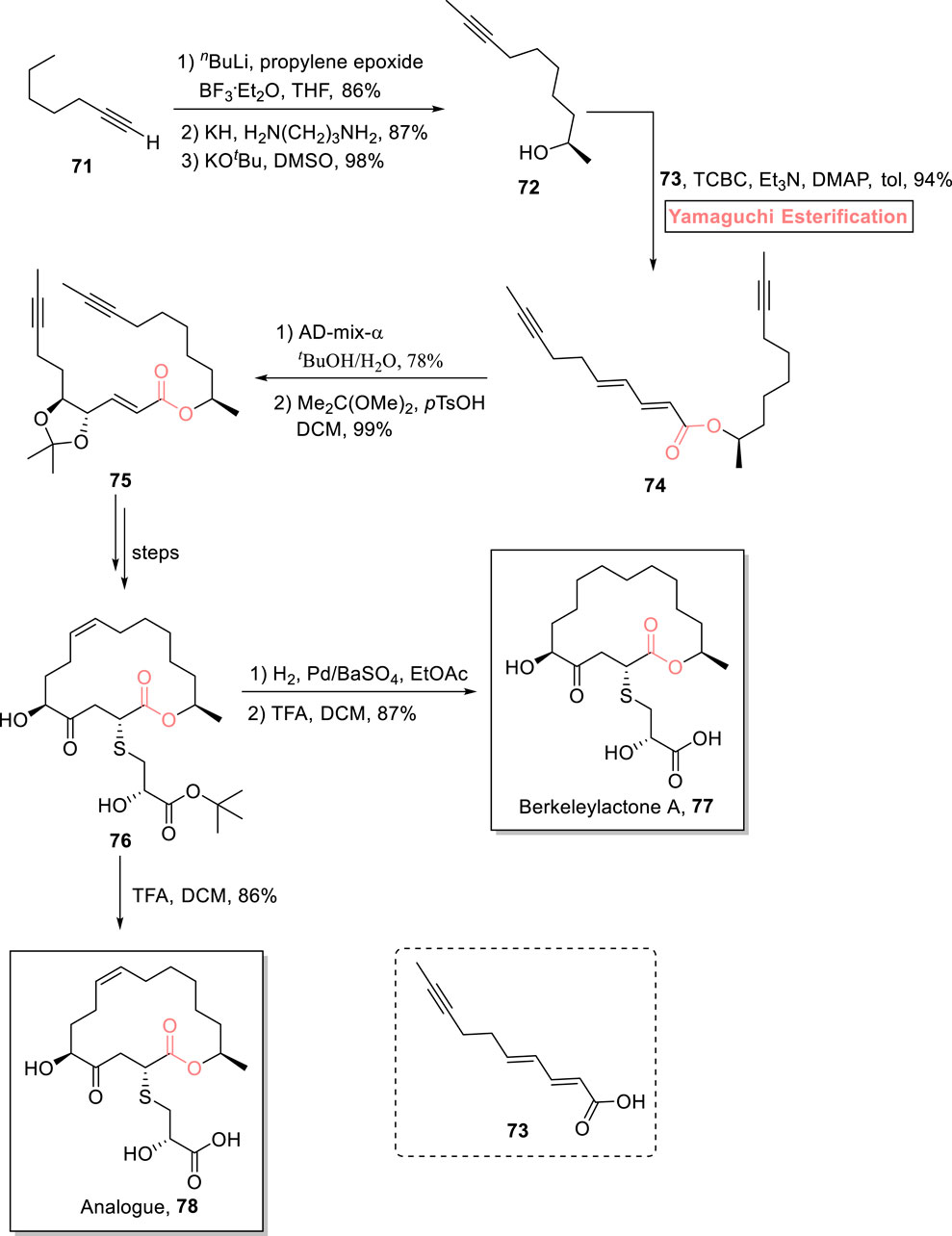
Scheme 8. Synthesis of berkeleylactone A 77 and its analog 78.
2.1.5.3 Kumari’s total synthesis of aspergillide D
Aspergillide D 82 is a 16-membered macrolide that was isolated from a fungal strain of Aspergillus sp. SCSGAF 0076. Its first total synthesis was performed by Mohapatra et al. in 2017 (Jena et al., 2017). After that, many reports on the total synthesis of this natural product have been published, with a common issue of low yield. Kumari et al. (2023) presented a high-yielding 15-step synthetic scheme for the synthesis of aspergillide D 82 via an epoxide ring-opening reaction (Ahmad et al., 2018) and Yamaguchi macrolactonization as the main steps (Scheme 9) (Kumari et al., 2023). In their synthesis, 3-butene-1-ol 79, as a readily available compound, was utilized to produce hydroxy acid 80. In order to perform macrolactonization, hydroxy acid 80 was treated with TCBC, Et3N, and DMAP in toluene to furnish highly regioselective lactone 81 with 67% yield. In the last step, deprotection was conducted in the presence of DDQ and DCM to produce the aspired aspergillide D 82 with 86% yield.
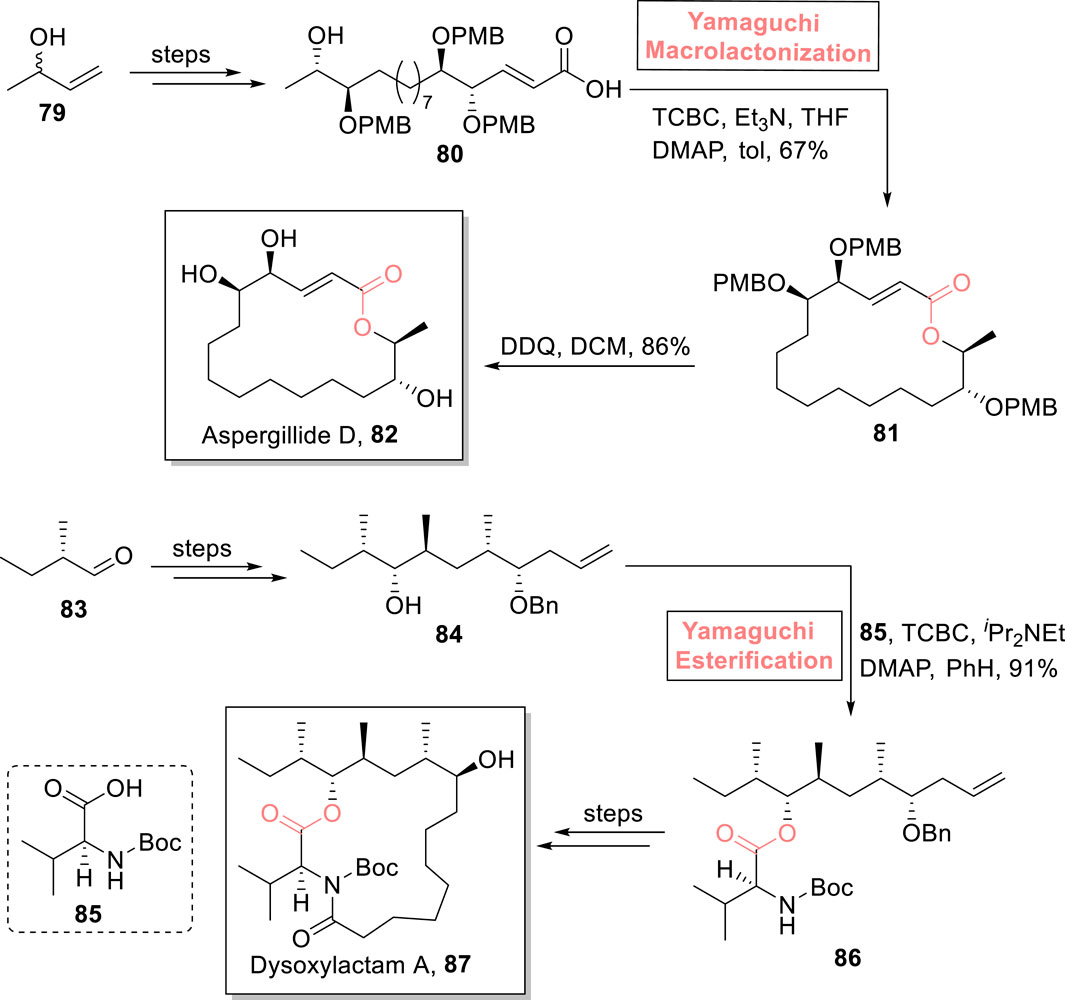
Scheme 9. Synthesis of aspergillide D 82 and dysoxylactam A 87.
2.1.6 Synthesis of 17-membered macrolides
2.1.6.1 Yang’s total synthesis of dysoxylactam A
One of the 17-membered macrolide, dysoxylactam A 87, was isolated from Dysoxylum hongkongense. It is a macrocyclic lipopeptide, consisting of a C19-branched fatty acid (Liu et al., 2019). Owing to its potential to reverse the P-glycoprotein-mediated multidrug resistance in tumor cells, Chandankar et al. performed its first total synthesis in 2020, and after that, various researchers have performed its total synthesis (Chandankar and Raghavan, 2020). In the continuation of these studies, Yang et al. (2022) designed an easy and concise synthetic route toward the total synthesis of this attractive unprecedented natural product (Scheme 9) (Yang et al., 2022). Their synthesis was based on 12 steps, starting from 2-methylbutanal 83, and a 23.2% overall yield was acquired. After achieving compound 84 (from compound 83), the Yamaguchi reagent (TCBC), iPr2NEt, and DMAP played their role in its esterification with compound 85 to successfully produce ester 86 (in 91% yield) with no epimerization. After several steps, dysoxylactam A 87 was easily obtained from ester 86. The synthesized compound exhibited the cytotoxic activity in combination with vinorelbine (anti-cancer drug) with an IC50 value of 3.5 nmol. L‒1.
2.1.7 Synthesis of 18-membered macrolactones
2.1.7.1 Goda and Fuwa’s total synthesis of (−) enigmazole B
Among cytotoxic marine macrolides, enigmazoles were isolated from Cinachyrella enigmatica (Oku et al., 2010). (−)-Enigmazole A exhibits cytotoxic activity against a human cancer cell line, with GT50 = 1.7 μm. The intriguing anti-cancer biological profile of the enigmazole family gained the attention of many organic chemists (Takada et al., 2023). With profound interest, Goda and Fuwa, (2023) performed the first total synthesis of (−)-enigmazole B 91 in 20 consecutive steps using Yamaguchi macrolactonization as a key step (Scheme 10) (Goda and Fuwa, 2023). Their methodology was commenced with the modification of compound 88 over a few steps to provide compound 89, which underwent macrocyclization via treatment with a well-suited Yamaguchi reagent in the presence of Et3N, THF, DMAP, and toluene, resulting in macrolactone 90 with 93% yield. Next, macrolactone 90 underwent a sequence of DDQ-mediated deprotection (of the PMB group), phosphorylation, and K2CO3-mediated deprotection to successfully yield the desired natural product (−)-enigmazole B 91 with 89% yield.
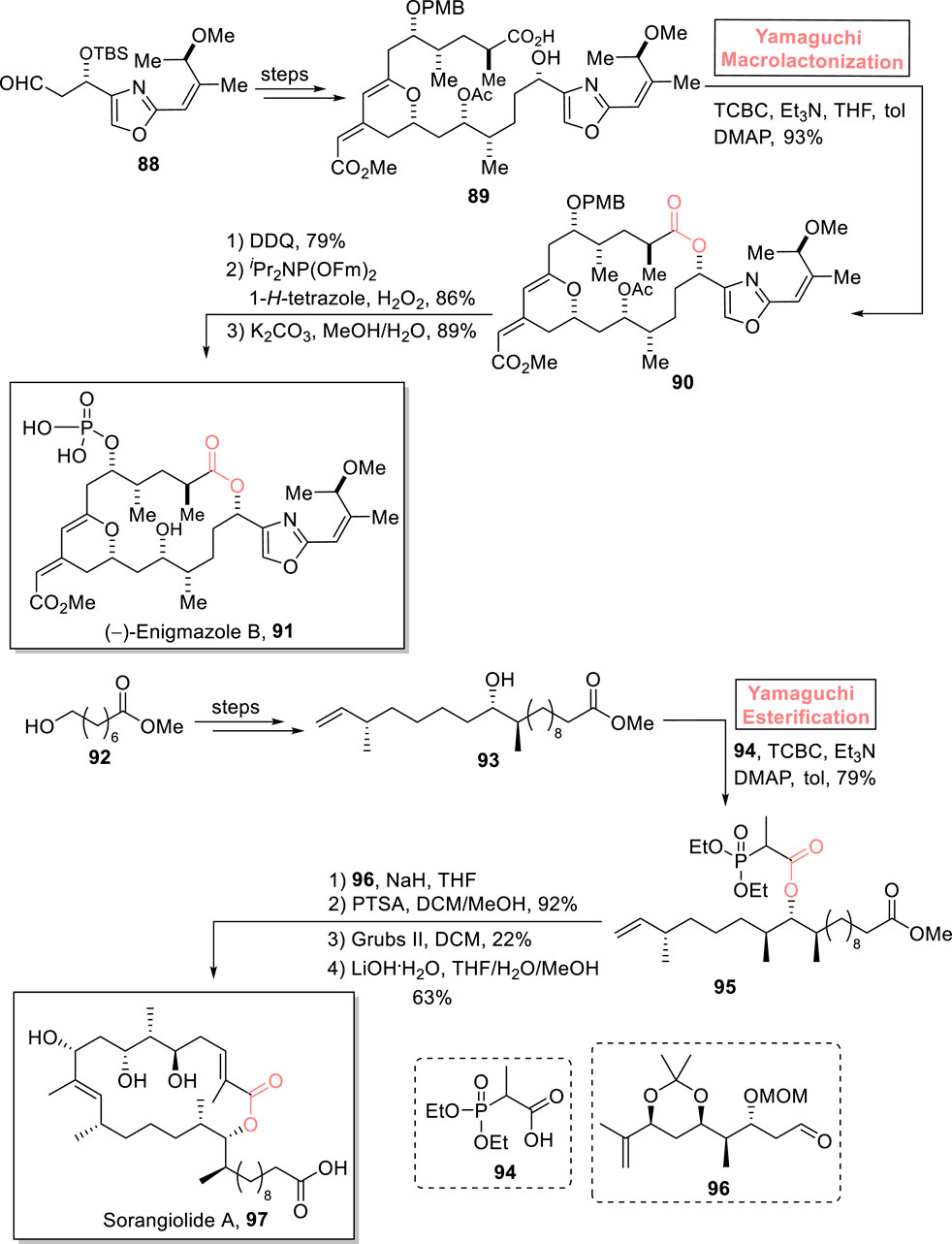
Scheme 10. Synthesis of (−)-enigmazole B 91 and sorangiolide A 97.
2.1.7.2 Sahana’s total synthesis of sorangiolide A
Sorangiolide A 97 is an 18-membered macrolide and a polyketide that was isolated by Jansen et al. from Sorangium cellulosum, a myxobacterial strain. Structurally, the heterocyclic cage of sorangiolide A 97 consists of four methylated centers, two trisubstituted olefins, and four hydroxylated centers. This natural product is renowned for its anti-bacterial activity against Staphylococcus aureus (a Gram-positive bacterium with a MIC value of 5–10 μg/mL) (Jansen et al., 1995; Irschik et al., 1995). Sahana et al. (2022) designed a convergent asymmetric synthetic tool for the first successful total synthesis of sorangiolide A 97 (0.9% overall yield) by using Yamaguchi esterification as a key step (Scheme 10) (Sahana et al., 2023). In their synthetic route, alcohol 93 (prepared from compound 92) was treated with triethyl amine, 2,4,6-trichlorobenzoyl chloride, DMAP, and toluene with the addition of acid 94. This Yamaguchi protocol successfully produced ester 95 with 79% yield. Compound 95 was then made to couple with aldehyde 96 (in the presence of sodium hydride and THF), followed by acetonoid group deprotection (by using PTSA) to provide the intermediate with 92% yield. After that, it was subjected to RCM and subsequent treatment with LiOH.H2O to successfully produce the target product 97 with 63% yield.
2.1.7.3 Salituro’s total synthesis of strasseriolides (A and B)
Strasseriolides (A and B) were first isolated in 2020 by Rayes et al. from the fungal strain of Strasseria geniculata (CF-247251) in New Zealand (Annang et al., 2020). Structurally, they are 18-membered macrolides having 2 trisubstituted alkenes, 5 methyl centers, and 1 free carboxylic acid group (Saha et al., 2020). Strasseriolide B shows anti-malarial potential against Plasmodium falciparum (the most virulent parasite) (Pérez-Moreno et al., 2016). Inspired by this fact, Salituro et al. (2022) reported a robust synthetic route for the first total synthesis of strasseriolide A 102 and strasseriolide B 103 in 15-step and 16-step sequences, respectively (Scheme 11) (Salituro et al., 2022). The methodology entails Yamaguchi esterification and the Nozaki–Hiyama–Kishi (NHK) reaction as key steps commencing from readily available starting materials, i.e., acid 98 and alcohol 99; both were explored for esterification under various protocols (EDCI, DCC, HBTU, and Shiina), but only the Yamaguchi method (TCBC, triethyl amine, and DMAP) was successful in the synthesis of the desired ester with 39% yield. The coupling intermediate underwent the NHK reaction to furnish compounds 100 and 101 with 73% combined yield (dr = 1.1:1). Compound 101 was oxidized in the presence of DMP, followed by hydrolysis, to produce strasseriolide A 102 with 64% yield. Furthermore, strasseriolide B 103 was easily acquired via the direct hydrolysis of compound 101, with 45% yield.

Scheme 11. Synthesis of strasseriolide A 102 and strasseriolide B 103.
2.1.7.4 Sahana’s total synthesis of strasseriolide A
Pioneering the synthetic work on strasseriolides, Sahana et al. (2022) performed the total synthesis of strasseriolide A 102 in 22 steps with 1.0% overall yield (Scheme 11) (Sahana et al., 2022). In their synthesis, S-Roche ester 104 was used as the starting material, and Yamaguchi macrolactonization was used as a key step. After achieving compound 105 (from ester 104), it was allowed to undergo macrolactonization following the Yamaguchi protocol (as the leading tool) by using TCBC, Et3N, DMAP, and toluene to obtain compound 106 (in 30% yield) with the desired stereochemistry. The whole synthetic scheme ended with the successful synthesis of strasseriolide A 102 (in 52% yield) by treating compound 106 with LiOH·H2O.
2.1.8 Synthesis of 19-membered macrolides
2.1.8.1 Zhang’s total synthesis of 27-deoxylyngbyabellin A
Lyngbyabellins are two thiazole rings containing marine metabolites, which are famous for their anti-cancer pharmaceutical effects (Pirovani et al., 2015). A 19-membered macrolide, 27-deoxylyngbyabellin A 116 was first isolated from Lyngbya bouillonii (a marine cyanobacterium) (Matthew et al., 2010). It exhibits cytotoxic effects against HeLa cervical cancer cells and HT-29 colorectal adeno cancer cells with IC50 values of 12 nM and 7.3 nM, respectively. Zhang et al. (2021) accomplished the total synthesis of 27-deoxylyngbyabellin A 116 in 10 consecutive steps with 9.7% overall yield (Scheme 12) (Zhang et al., 2021). Their efficient synthetic scheme began with the starting compounds 107 and 108, which were allowed to react in the presence of pentafluorophenyl diphenylphosphinate, PPh3, and triethyl amine, followed by HCl-mediated Boc group removal and coupling with Boc-Gly-OH, which resulted in compound 109 with 68% yield. In the next step, compound 109 (after hydrolysis) was coupled with compound 111 (using DCC and DMAP), and the subsequent removal of allyl ester in the presence of morpholine and Pd(PPh3)4 produced acid 112. In the next step of esterification of acid 112 with alcohol 114, Keck esterification was attempted, but after its failure after a few attempts, the well-suited Yamaguchi reagent (2,4,6-trichlorobenzoyl chloride) was used in the presence of diisopropylethylamine as the base and THF as a solvent to result in the successful synthesis of ester 115 with 65% yield. After that, the TMSe and Boc groups were removed by treating ester 115 with TBAF and p-TsOH in a sequence, followed by its macrocyclization in the presence of diphenyl phosphorazidate and DMF to result in the successful synthesis of 27-deoxylyngbyabellin A 116 with 45% yield.
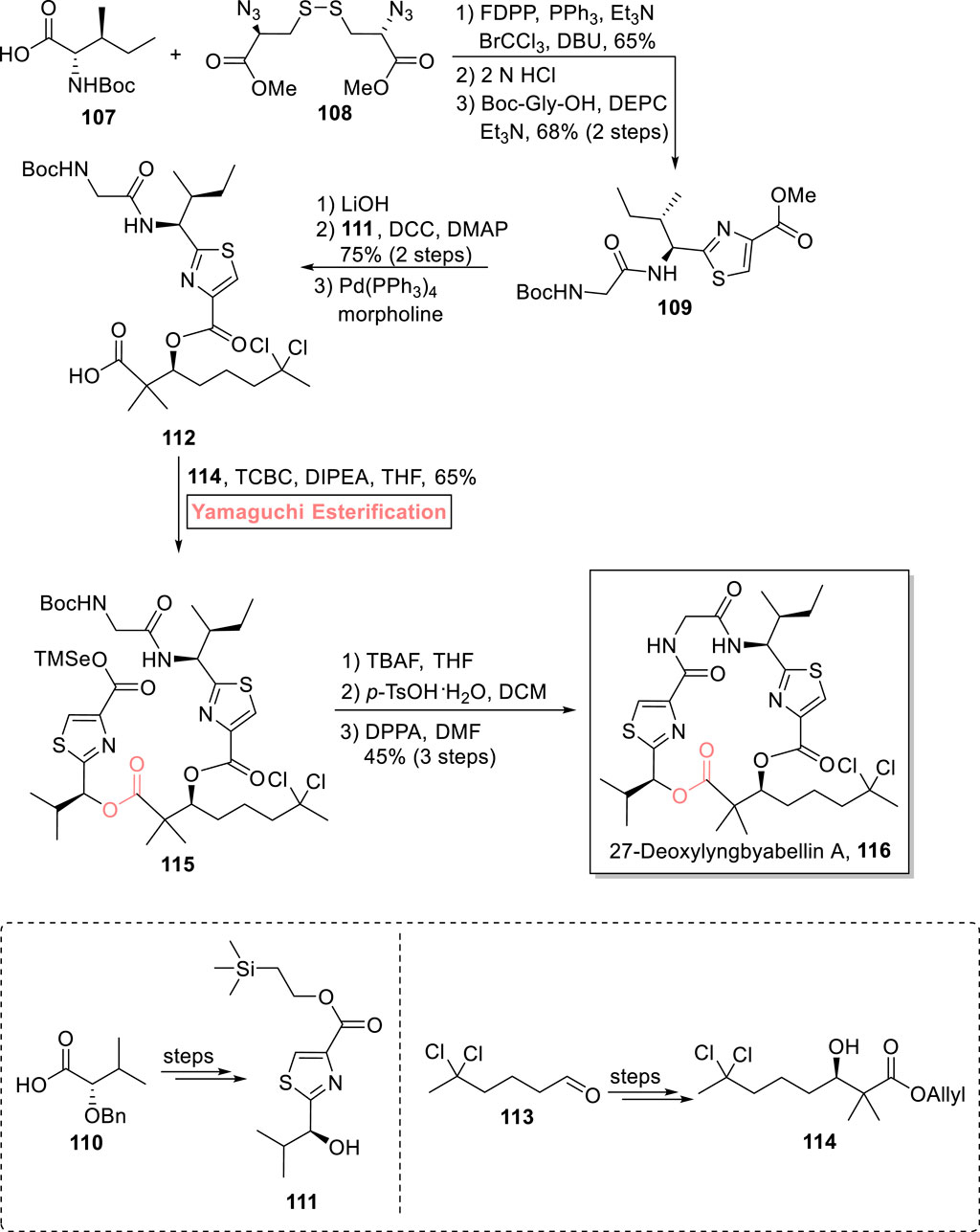
Scheme 12. Synthesis of 27-deoxylyngbyabellin A 116.
2.1.9 Synthesis of 20-membered macrolides
2.1.9.1 Gosh’s total synthesis of iriomoteolide-1a and iriomoteolide-1b
Iriomoteolide-1a 126 and iriomoteolide-1b 127 are 20-membered macrolides that were isolated independently from the HY A024 strain of Amphidinium sp. (a dinoflagellate found in Japan) by Tsuda et al. (2007a) and Tsuda et al. (2007b). Both these natural marine products are structurally related to each other and are famous for their intriguing biological potential. In particular, iriomoteolide-1a exhibits cytotoxic activity against various human cell lines, that is, lymphocyte DG-75 and EBC-infected lymphocyte Raji cells with IC50 values of 2 ng/mL and 3 ng/mL, respectively (Munir et al., 2023). Considering these interesting facts, Gosh et al. (2022) devised a robust synthetic route for the synthesis of (the proposed structures) these natural products by using Yamaguchi macrolactonization as the key step (Scheme 13) (Ghosh and Yuan, 2022). Their synthesis was initiated with the synthesis of key fragments 118 and 120 (from starting materials 117 and 119), followed by their coupling via the Julia–Kocienski reaction to result in olefin 121 with 83% yield. Olefin 121 (after benzyl group deprotection) was oxidized in the presence of DMP and NaHCO3. As a result, the oxidized product was then coupled with sulfone 123 (from alcohol 122) via the Julia–Kocienski reaction, followed by PMB group and TBS group removal in sequence using DDQ and NaClO2, yielding alcohol 124 with 72% yield. Next, the oxidation of alcohol 124 first with MnO2 and second with NaClO2 adjusted the stage for Yamaguchi macrolactonization as the resulting carboxylic acid was treated with TCBC, DIPEA, and DMAP to successfully furnish 20-membered macrolactone 125 with 61% yield. The next few steps involved the replacement of the TBS group with the TES group, bromocatecholborane-induced deprotection of the MOM group, TES-protected oxidation of the free hydroxyl group (by using DMP), and a final deprotection in the presence of HF/Py, which resulted in the desired natural products iriomoteolide-1a 126 (in 56% yield) and iriomoteolide-1b 127 (in 17% yield).
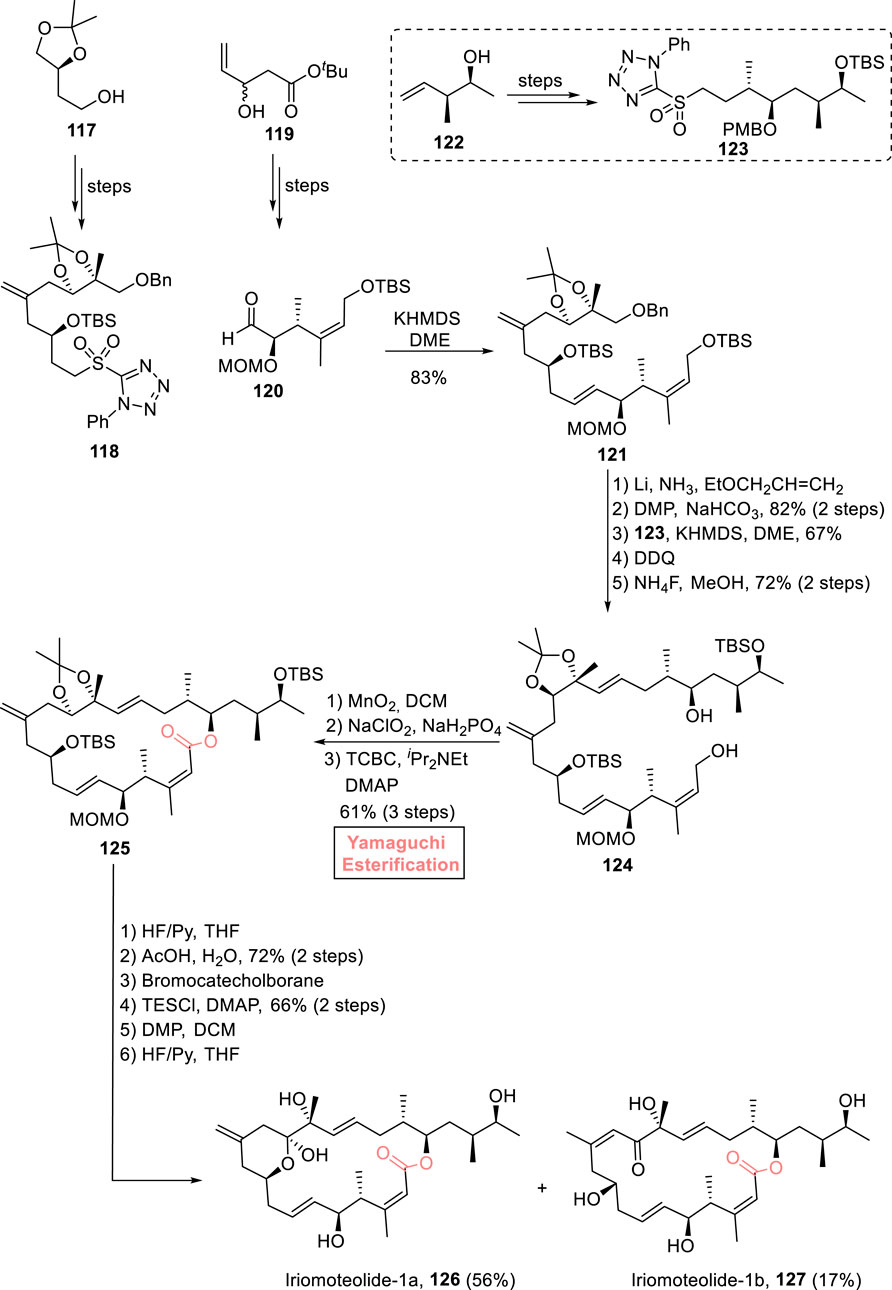
Scheme 13. Synthesis of iriomoteolide-1a 126 and iriomoteolide-1b 127.
2.1.9.2 Bold’s synthesis of zampanolide analogs
The famous anti-cancer (−)-zampanolide 139 is a 20-membered macrolide, which was isolated from Fasciospongia rimosa by Tanaka and Higa (1996). This natural product was re-isolated by Field et al. from F. rimosa in 1996 (Field et al., 2009). The structural activity studies of this unique pharmacophore underscore the necessity of developing analogs to maintain a broad-spectrum medicinal library (Chen and Kingston, 2014). In 2021, Bold et al. synthesized morpholine analogs of (−)-zampanolide with the desired stereochemistry that was assured via the Yamaguchi reagent (Scheme 15) (Bold et al., 2021). The synthetic scheme was initiated from alcohol 128 that was subjected to tosylation and treatment with a base (KOH), followed by Jacobsen epoxidation and copper catalyst-induced hydrolysis to yield epoxide 130 with 65% yield and 99.5% ee. After the transformation of epoxide 130 into alcohol 131, it was reacted with epoxide 132 in the presence of butyl lithium and BF3·OEt2, providing compound 133 in 76% yield. In the next step, the tosyl group of compound 133 was removed via treatment with magnesium, which led to the formation of compound 134 with 85% yield. Compound 135 (with 96% yield) was achieved from the acylation of compound 134 in the presence of isopropenyl acetate. Meanwhile, the reaction of compound 134 with benzoyl chloride in the presence of triethyl amine and DCM provided benzamide 136 with 98% yield (Scheme 14). The Yamaguchi esterification of the synthesized compounds 135, 133, and 136 with alcohol 137 (in the presence of TCBC, TEA, DMAP, and THF) and the subsequent deprotection step resulted in the independent synthesis of compounds 138a–c with 78%–89% yields, which, after modification in some required steps, completed the synthesis of analogs 139a–c.
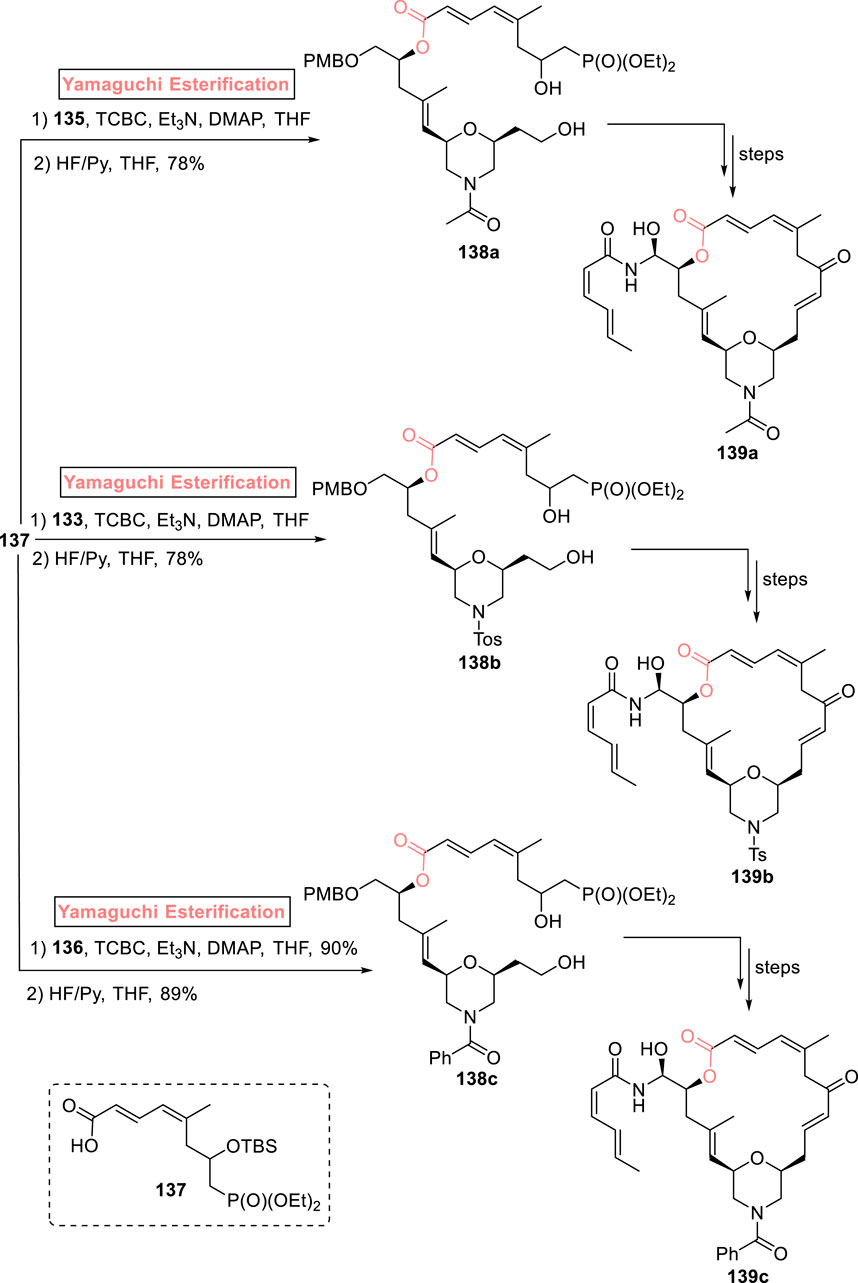
Scheme 15. Synthesis of (−)-zampanolide analogs 139a–c.
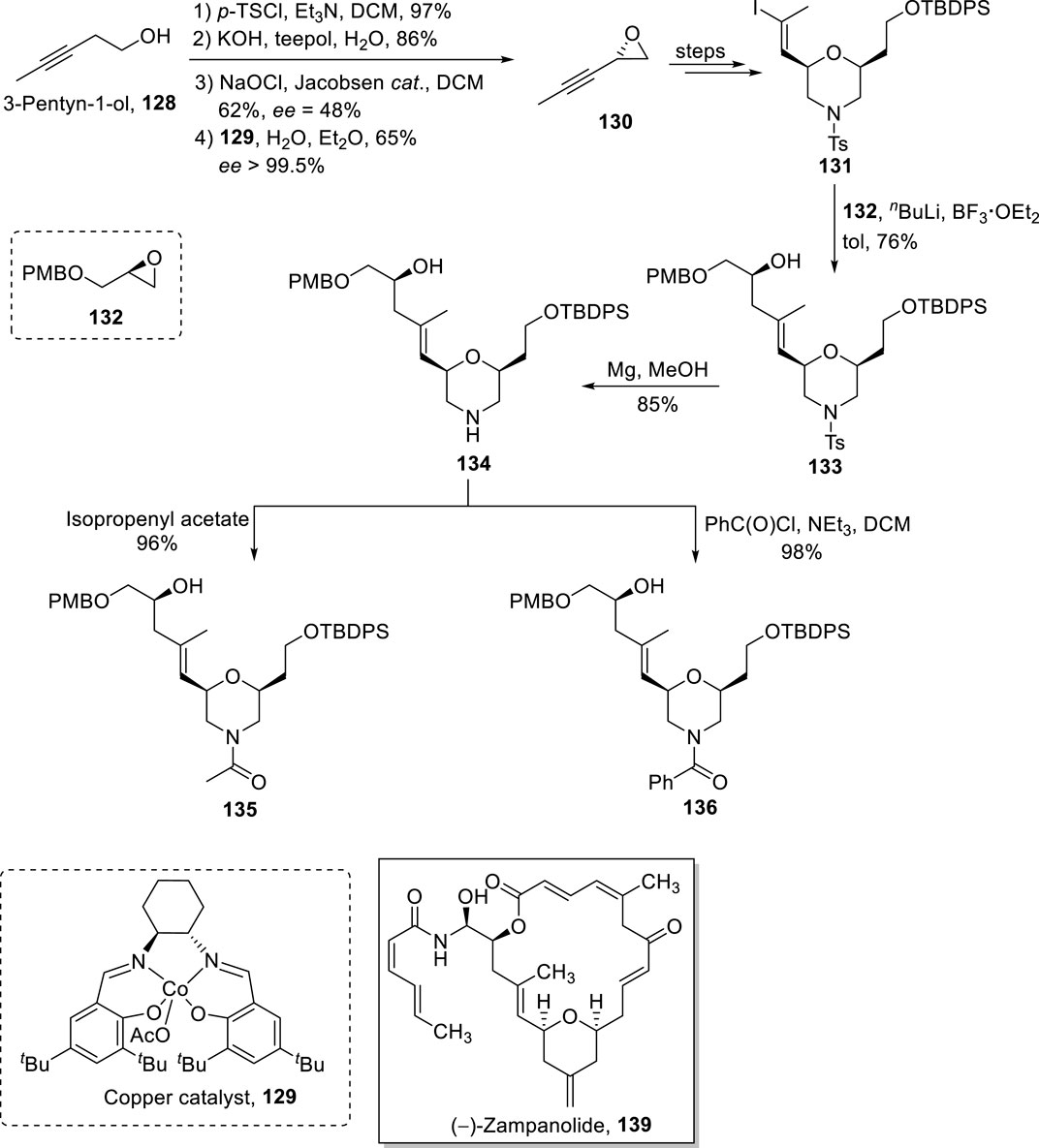
Scheme 14. Synthesis of compounds 135 and 136 toward the synthesis of (−)-zampanolide analogs.
2.1.9.3 Umana’s synthesis of (−)-zampanolide analog
In the continuation of work on (−)-zampanolide 139 analogs, Umana et al. (2023) developed a linear analog 139d of (−)-zampanolide as a potential anti-cancer agent (Scheme 16). The synthesis was commenced from alcohol 140, which was converted into compound 141 over a few steps. To set the stage for the incorporation of the side chain hemiaminal group, a challenging task with concern to attain the required configuration in the desired product, Yamaguchi esterification was preferred over any other step. Thus, alcohol 141 was made to undergo Yamaguchi esterification (with acid 142) by treating it with 2,4,6-trichlorobenzoyl chloride, triethyl amine, and DMAP to achieve ester 143 with 60% yield. Compound 143 was treated with methanol and HCl (for silyl group deprotection), followed by DMP-induced oxidation and installation of a hemiaminal side group (via reaction with compound 144), successfully producing the desired compound 139d with 40% yield.
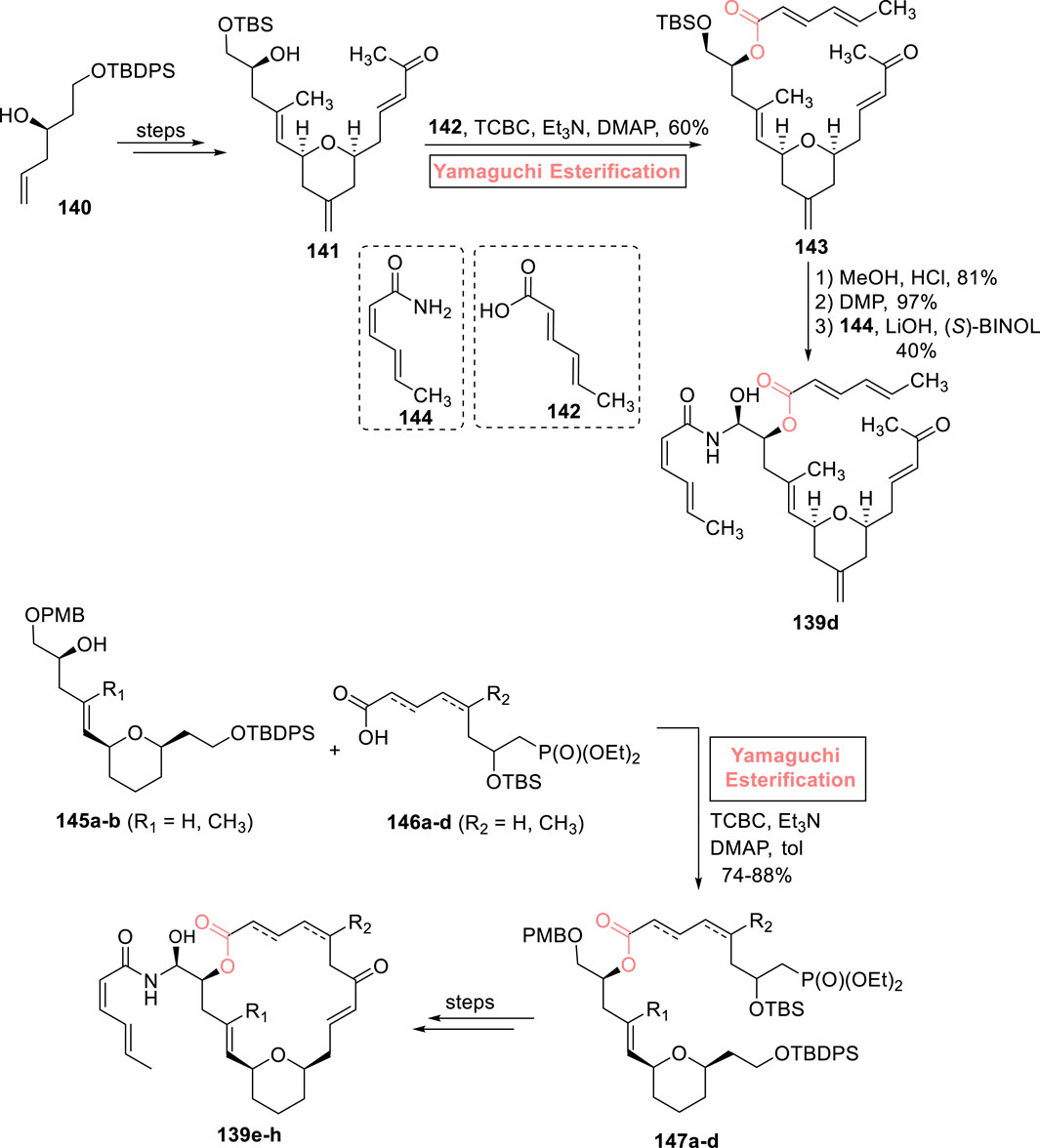
Scheme 16. Synthesis of (−)-zampanolide analog 139d–h.
2.1.9.4 Brutsch’s synthesis of (−)-zampanolide analogs
With the profound interest in the field of natural product synthesis, Brütsch et al. (2023) contributed their efforts to the synthesis of four desmethylene analogs of (−)-zampanolide 139d–h (Scheme 16). Among these, the synthesis of three analogs involves the use of the efficient Yamaguchi reagent. The compounds 145a–b were made to couple with compounds 146a–d (macrocyclization) under the presented Yamaguchi conditions (TCBC, Et3N, DMAP, and toluene) to achieve the compounds 147a–d within the yield range of 74%–88%. Over a few steps, (−)-zampanolide analogs 139e–h were successfully attained from compounds 147a–d (Supplementary Figure 3).
2.1.9.5 Wender’s synthesis of bryostatin analogs
Bryostatin is a 20-membered polyketidic macrolide, which was isolated from Bugula neritina. It has various medicinal applications as it is used in the treatment of cancer, AIDS, Alzheimer’s disease, and many other degenerative diseases (Farlow et al., 2019; Gutiérrez et al., 2016). Considering the remarkable pharmacophore of bryostatin, Wender et al. (2022) introduced a novel strategy for the synthesis of its analog 152 (Scheme 17) (Wender et al., 2022). In their methodology, compound 149 (prepared from compound 148 in a few steps) esterified with alcohol 150 in the presence of a Yamaguchi reagent, Et3N, DMAP, and toluene, to obtain ester 151 with 70% yield. The deprotection of ester 151 in sequential steps using PPTS and HF/Py successfully produced bryostatin analog 152 with 65% yield.
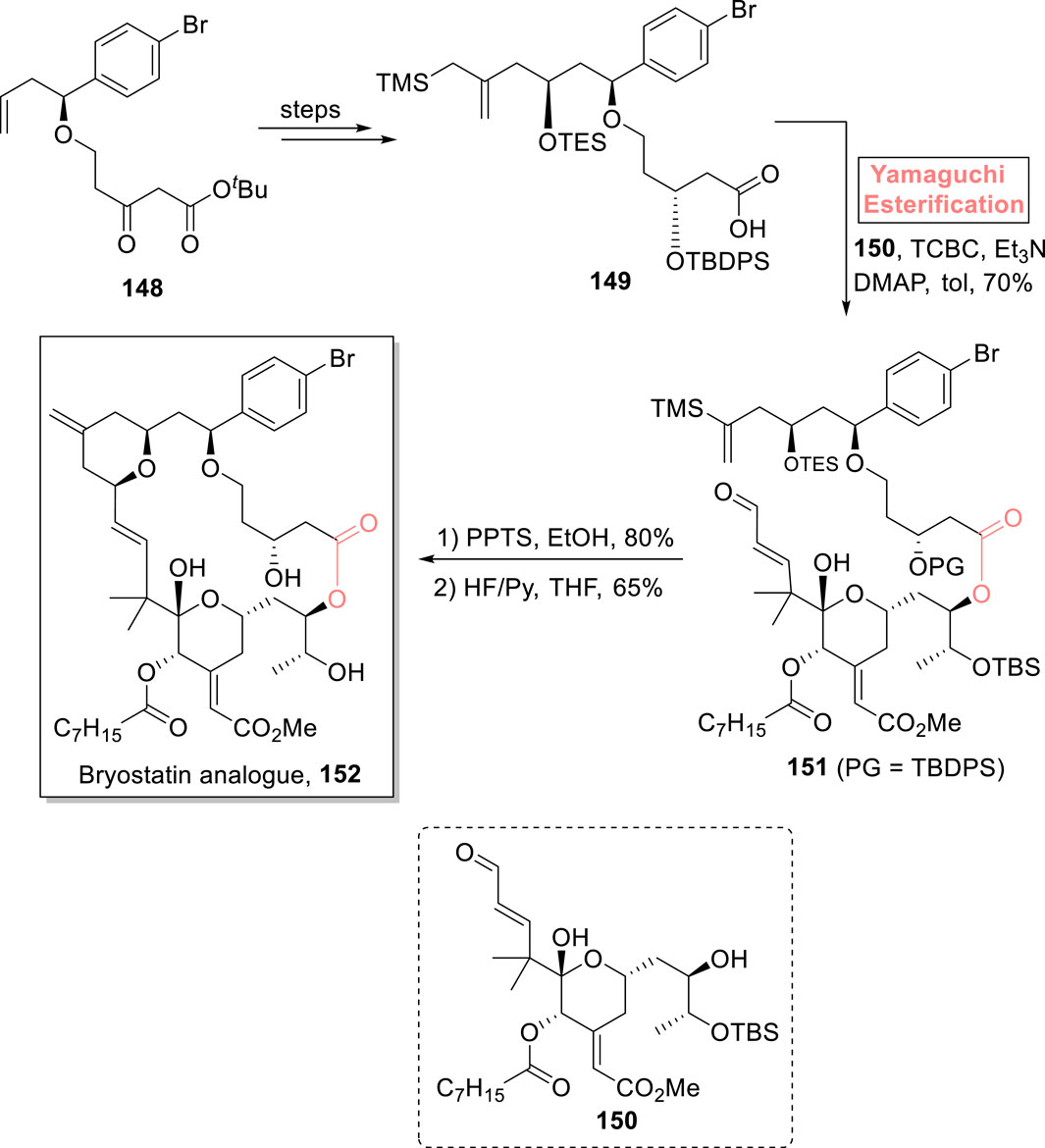
Scheme 17. Synthesis of bryostatin analog 152.
2.1.10 Synthesis of 22-membered natural macrolides
2.1.10.1 Fritz’s total synthesis of pulvomycin D
A famous antibiotic, pulvomycin D, is a 22-membered macrolide that was isolated from Streptomyces sp. by Zief et al. (1957), and its structure was confirmed by Moon et al. (2020). This polyketidic macrolide also shows remarkable anti-cancer effects and has structural stability even under strong acidic and basic conditions (Kim et al., 1990). These interesting facts prompted Fritz et al. to perform its total synthesis in 2021 (Fritz et al., 2022). The synthesis of the precursor was achieved in consecutive steps with 0.23% overall yield via the use of a Yamaguchi reagent for the installation of the C1–C7 fragments (Supplementary Scheme 1).
2.1.11 Synthesis of 23-membered natural macrolides
2.1.11.1 Decultot and Clark’s total synthesis toward amphidinolide F
In 2022, Decultot and Clark performed the facile synthesis of precursor 162 toward the total synthesis of amphidinolide F 163 using Yamaguchi esterification as a key step (Scheme 18) (Decultot and Clark, 2022). In their synthetic path, alcohol 153 was subjected to DMP-promoted oxidation, followed by a reaction with the Grignard reagent, TMS group deprotection, and Sonogashira coupling reaction (with Me2CCHBr) to afford compound 154 with 83% yield. After a few steps, ketone 155 was acquired from compound 154. Ketone 155 was allowed to react with aldehyde 157 in the presence of dicyclohexylboron chloride to generate a diastereomeric pair of alcohols 158a and 158b with 55% and 10% yields, respectively. In the next step, compound 158a proceeded for TBS protection and subsequent hydrolysis, producing alcohol 159 with 77% yield. For the esterification of compound 159 with alcohol 161, the well-optimized Yamaguchi protocol (Et3N, DMAP, and TCBC) was used to successfully produce fragment 162 with 69% yield.

Scheme 18. Synthesis of fragment 162 toward the synthesis of amphidinolide F 163.
2.1.12 Synthesis of 28-membered polyketidic macrolides
2.1.12.1 Babczyk and Menche’s total synthesis of pentamycin
Pentamycin 176 belongs to the class of polyene macrolides and polyketides. It was first isolated in 1958 from Streptomyces pentaticus (Hamilton-Miller, 1973). The unique structural framework of this macrolide consists of a 28-membered core with an adjacent polyol fragment, having 12 stereomeric centers. Pentamycin 176 exhibits remarkable biological activities against C. albicans and Trichomonas vaginalis. Furthermore, it can also be used as an anti-cancer agent along with bleomycin (Kranzler et al., 2015; Payero et al., 2015). These fascinating features prompted Babczyk and Menche to perform its total synthesis by using Yamaguchi esterification as the crucial step (Scheme 19) (Babczyk and Menche, 2023). For the accomplishment of this task, fragment 166 (from compounds 164 and 165) and fragment 168 (from compound 167) were subjected to coupling reaction (in the presence of LDA and DMPU), followed by Birch reduction to produce compound 169 in 84% yield. In the four steps for the synthesis of alkyne 171, acetyl group protection and TBS group deprotection of compound 169 provided favorable conditions for subsequent oxidation and Bestmann–Ohira homologation (with compound 170). Alkyne 171 was then subjected to a sequence of stannyl-cupration (under TES protection), methylation, iodination, IBX-induced oxidation, and Pinnick oxidation to furnish compound 174 with 76% yield. To perform the challenging esterification of this sterically hindered carboxylic acid 174 with alcohol 173, the well-suited Yamaguchi protocol was used by using TCBC, Et3N, and DMAP, which yielded ester 175 with 84% yield. After a few steps, the successful synthesis of the targeted pentamycin 176 was achieved gratifyingly.
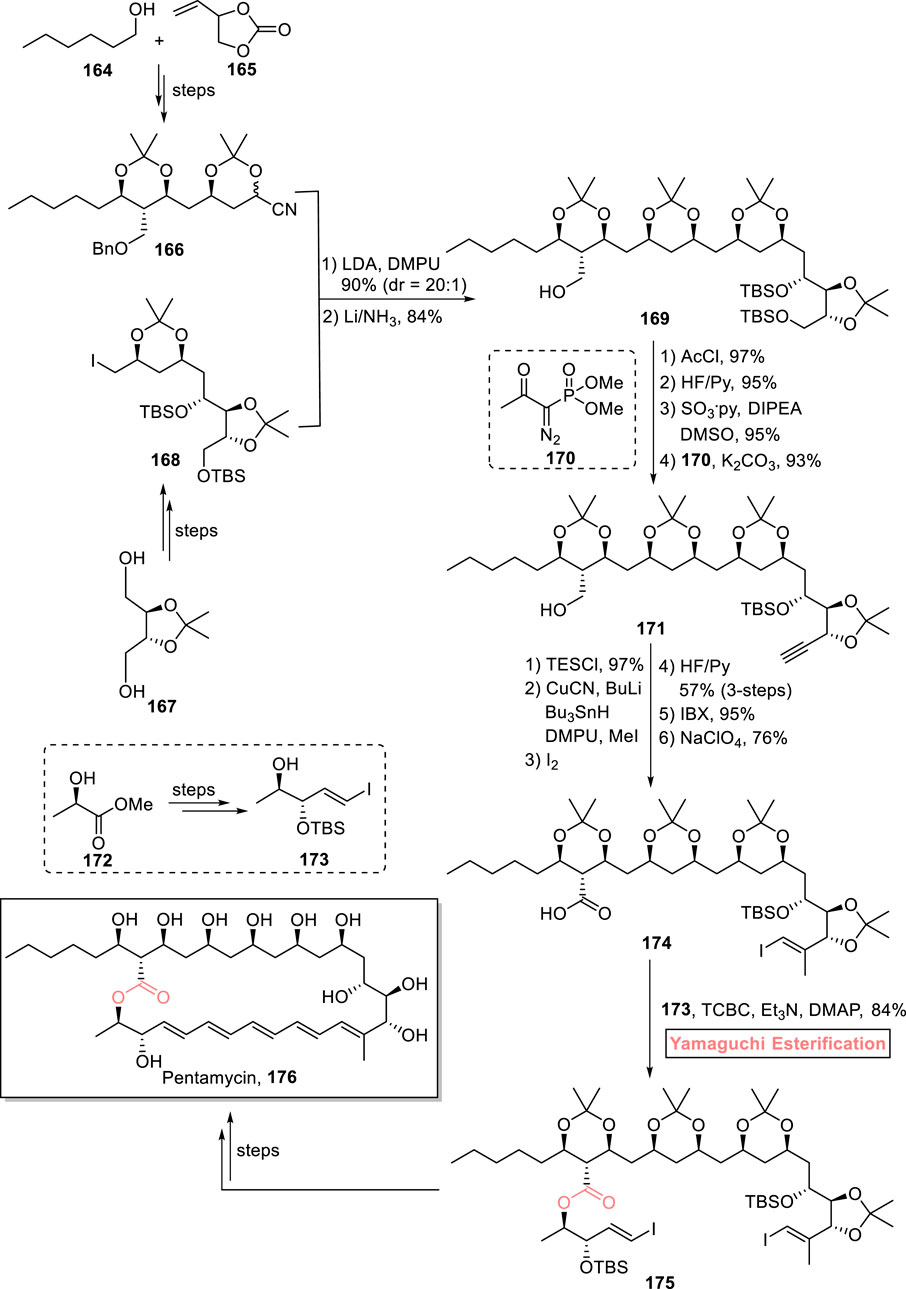
Scheme 19. Synthesis of pentamycin 176.
2.1.13 Synthesis of 30-membered macrolides
2.1.13.1 Lizzadro’s synthesis of disorazole C1 and analogs
Disorazoles are an intriguing class of natural products well known for their anti-tubulin and anti-cancer effects. They were isolated in 1994 from S. cellulosum, a myxobacterium, by Hofle and Reichenbach (Jansen et al., 1994; Jensen, 1994). One of the disorazoles, (−)-disorazole C1 181, is a 30-membered macrolide. It exhibits a highly cytotoxic effect in various mammalian cell lines (Hopkins and Wipf, 2009). Lizzadro et al. (2021) devised an efficient and well-designed strategy for the total synthesis of (−)-disorazole C1 (Scheme 20) (Lizzadro et al., 2021). The interesting features of their synthetic scheme involved both Yamaguchi esterification and Yamaguchi macrolactonization as the main steps to obtain the 30-membered macrolide (Supplementary Scheme 2). The Yamaguchi esterification was made possible by reacting synthesized compounds 177 and 178 in the presence of 2,4,6-trichlorobenzoyl chloride, triethyl amine, DMAP, and toluene, producing ester 179 with 75% yield. For the intramolecular reaction, a sequence of silyl group deprotection and hydrolysis of ester 179, followed by exposure to Yamaguchi conditions for macrolactonization, resulted in cyclic macrocycle 180 with 70% yield. In the end, metal-catalyzed reduction of compound 180, followed by a reaction with HBr in acetonitrile and water, resulted in the synthesis of the targeted natural product 181 with 56% yield. After the successful synthesis of (−)-disorazole C1 181, Lizzadro et al. (2022), extended their methodology for the synthesis of three novel analogs of disorazole as potent cytotoxic agents against cancer (Lizzadro et al., 2022) (Supplementary Figure 4).
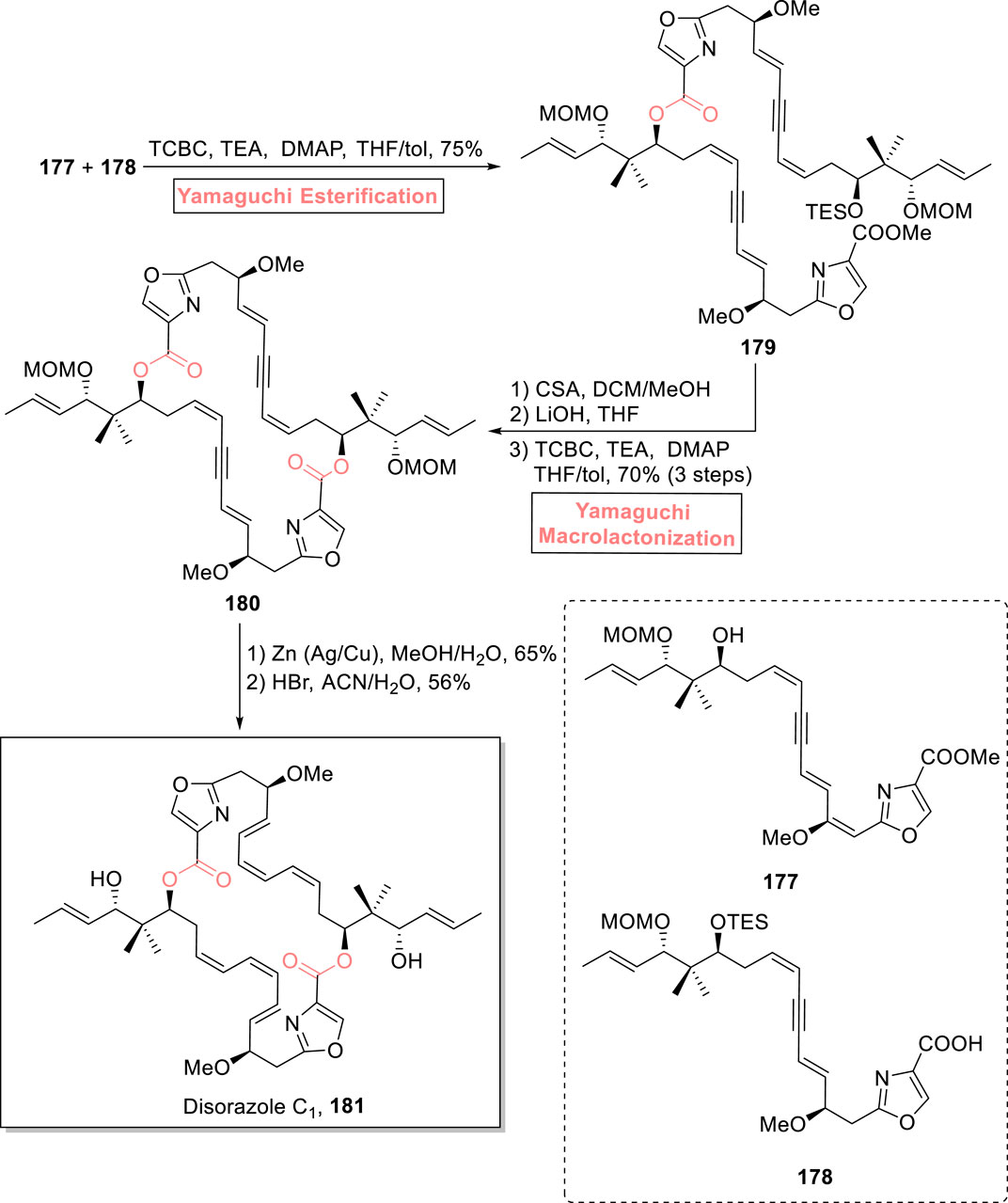
Scheme 20. Synthesis of disorazole C1 181.
2.2 Synthesis of natural metabolites
2.2.1 Gillsch’s total synthesis of ophiofuranones (A and B)
Ophiofuranone A and ophiofuranone B, belonging to the class of fungal metabolites, were isolated from Ophiosphaerella korrae by Lou et al. (2019) (Li et al., 2019). In 2022, Gillsch et al. disclosed the first total synthesis and microbial analysis of ophiofuranone A and ophiofuranone B by using cheap starting materials, i.e., tiglic acid and methallyl alcohol (Gillsch et al., 2022b). The challenging molecular architecture (with 4 stereogenic centers) of these 2 natural products was easily built in 16 steps by using Yamaguchi esterification as a powerful step (Supplementary Scheme 3).
2.2.2 Gillsch’s total synthesis of thiocarboxylic acid and analogs
A fungal metabolite, thiocarboxylic acid, was isolated from Penicillium sp. Sb62. This unique natural product exhibits anti-microbial activity against S. aureus, Escherichia coli, and C. albicans with MIC values of 1.7–3.0 μg/mL (Chang et al., 2020). Gillsch et al. (2022) devised an efficient 14-step synthetic route toward the synthesis of thiocarboxylic acid and its three analogs (Gillsch et al., 2022a). The key step entails the Yamaguchi esterification, which assisted in the separation of E and Z isomers with no scrambling (Supplementary Scheme 3).
2.2.3 Wittman’s synthesis of the JBIR-141 analog
Cancer is the second leading cause of death, and it has numerous causes. So, the search for new anti-cancer drugs with improved cytotoxicity has always remained a significant interest of scientists. Among some anti-cancer compounds, JBIR-141 inhibits the transcription of Foxo3a with an IC50 value of 23.1 nM. It is a tetramic acid metabolite that was isolated from the 4587H4S strain of Streptomyces sp. Furthermore, this natural product possesses significant structural features for having N-nitrosohydroxylamine, 3-acyltetramic acid, and oxazoline-4-carboxamide adorned with six stereogenic centers (Kawahara et al., 2015; Yasoshima et al., 2022). With great interest, Wittman et al. (2022) devised an efficient strategy for the synthesis of a close analog of JBIR-141245 using readily available starting materials L-threonine 189, L-alanine 187, and L-glutamic acid 182 (Scheme 21) (Wittmann et al., 2022). The synthesized compounds 184 and 185 (from starting compounds 182 and 183), in hand, were subjected to Yamaguchi conditions (TCBC, Et3N, DMAP, and toluene), facilitating a smooth esterification process. The resulting ester was then treated with HCl and ethyl acetate to yield compound 186. For the synthesis of key fragment 188, compound 187 was protected by using carbazole chloride. On the other hand, compound 189 was turned into ester 190 via its treatment with SO2 and methanol. The resulting ester (in 100% yield) was then made to react with protected compound 188 under the given conditions for condensation, followed by cyclization in the presence of (NH4)6Mo7O24.4H2O, to provide dipeptide 191 with 82% yield. Next, the removal of the carbazole group of compound 191 was made possible by hydrogenolysis in the presence of formaldehyde. This was followed by CsOH-promoted saponification, coupling of the intermediate with ester 186 (in the presence of EDC, DIPEA, HOBt, and DCM), and a final step of debenzylation, leading to the target analog 192 with 92% yield.
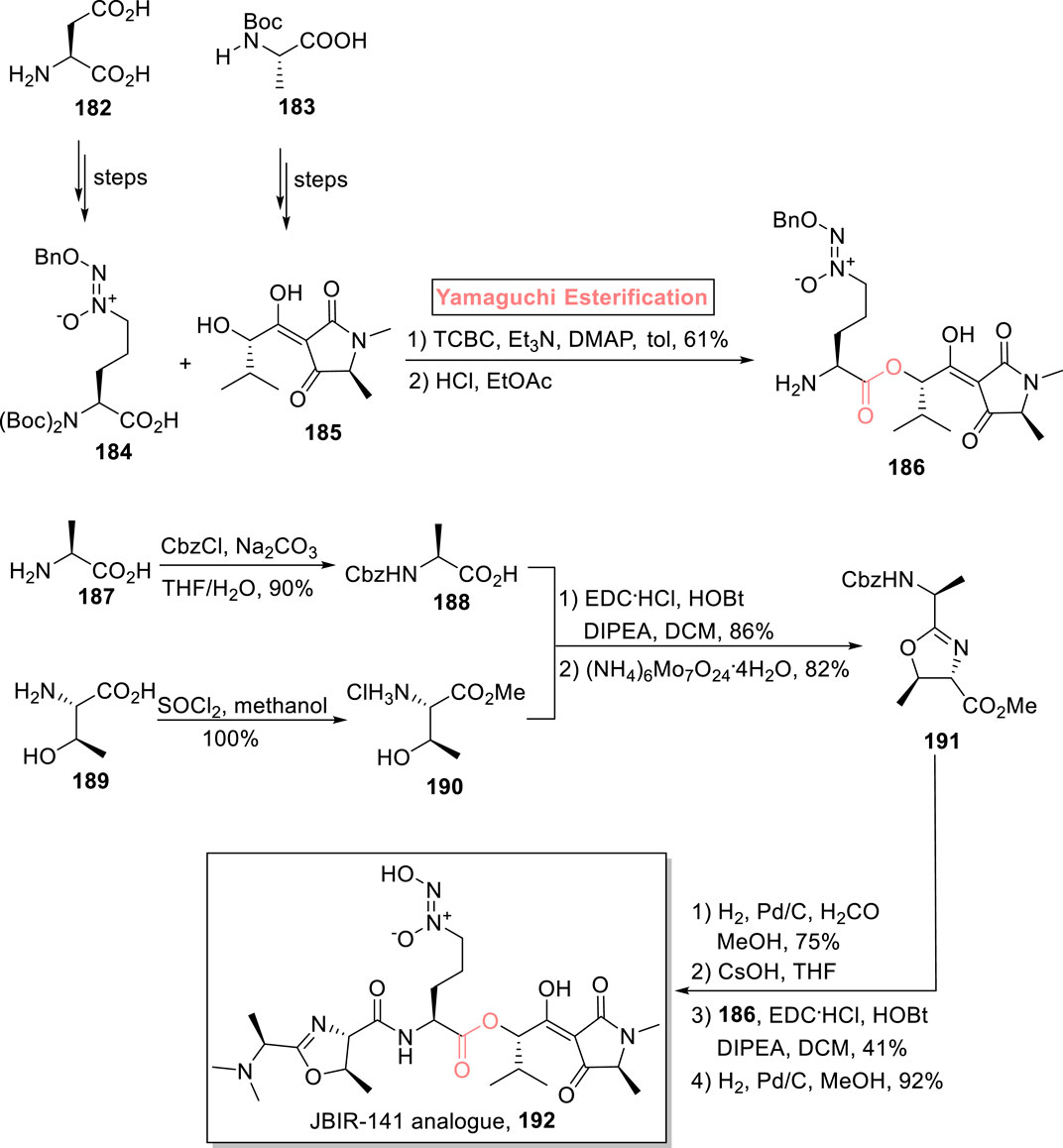
Scheme 21. Synthesis of JBIR-141 192.
2.2.4 Jansen’s total synthesis of desferri-exochelin 772SM
Exochelins are mycobacterial secretions that are used to chelate iron (as is necessary for the replication and energy metabolism of these microorganisms). One of these natural products, desferri-exochelin 772SM 202, was first isolated by Horwitz et al. from M. tuberculosis in 1995 (Horwitz and Horwitz, 2014; Gobin et al., 1995). Their significant Fe-chelating capability prompted Jansen et al. (2023) to devise a convergent and concise synthetic route for the total synthesis of desferri-exochelin 772SM 202 in 11 consecutive steps with an 8.6% overall yield (Scheme 22) (Jansen et al., 2023). The key step in the synthesis entails the coupling of advanced, hugely decorated fragments using Yamaguchi esterification. In their methodology, the easily available starting material 193 was transformed into compound 194 over a few steps. On one hand, compound 194 was used for the synthesis of alcohol 197 with the assistance of Pd(PPh3)4-mediated deprotection and PyBOP-induced coupling with N-ethylmorpholine of compound 194, resulting in intermediate 196 (with 46% yield). Subsequently, the Boc group protection of compound 196 was performed, followed by its PyBOP-induced coupling with 3-hydroxybutanoic acid, resulting in cobactin 197 with 84% yield, while in another route, compound 194 was made to react with monomethyl pimelate bis-(trichloromethyl)carbonate, 2,4,6-collidine, and THF to yield compound 195, proceeded by Boc-group deprotection, PyBOP-assisted amide coupling (with compound 199), and allyl group deprotection, to provide acid 200 (ready to esterify). Next, the essence of the Yamaguchi protocol can be realized as it was applied for the esterification (after the failure of other methods) of compound 200 with already synthesized alcohol 197 in the presence of TCBC, Et3N, DMAP, and toluene to produce ester 201 in 36% yield. In the final step, ester 201 was treated with hexafluoroisopropanol (HFIP) in the presence of sunlight to successfully produce desferri-exochelin 772SM 202 with 62% yield.
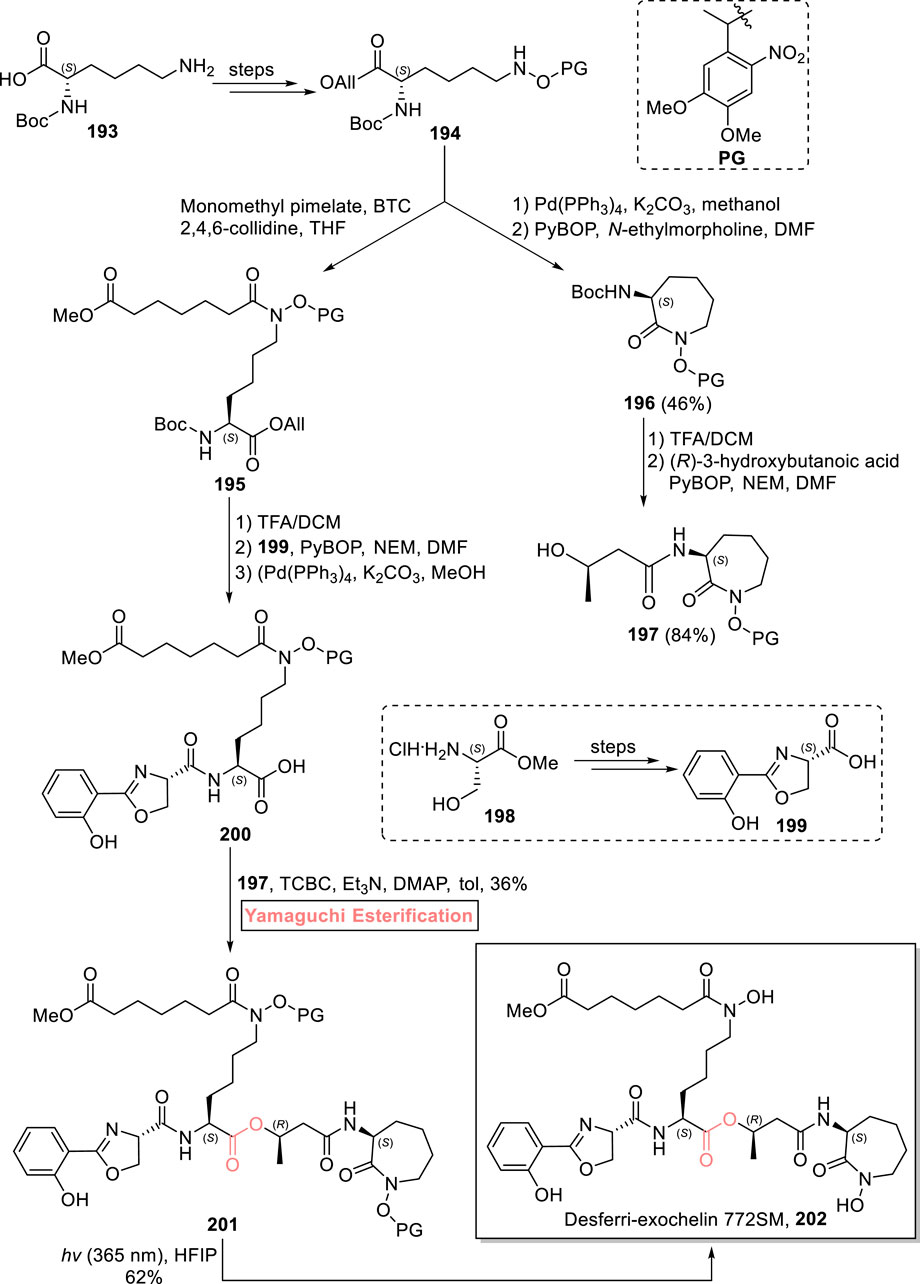
Scheme 22. Synthesis of desferri-exochelin 772SM 202.
2.3 Synthesis of natural polyketides
2.3.1 Morishita’s total synthesis of nhatrangin A
Nhatrangin A 207 belongs to the class of polyketide natural products and is a truncated derivative and a synthetic intermediate of oscillatoxin D and aplysiatoxin. Nhatrangin A 207 was first isolated from Lyngbya majuscula, a marine cyanobacterium, by Orjala et al. in 2010 (Chlipala et al., 2010). More interestingly, aplysiatoxin is renowned for its anti-inflammatory and anti-tumor activities (Fujiki et al., 1984). Being the synthetic intermediate of aplysiatoxin, nhatrangin A 207 gained the attention of various research groups; Morishita et al. (2023) accomplished its total synthesis and confirmed its configuration with a previously reported synthesis (Scheme 23) (Morishita et al., 2023). Their synthesis commenced with the efficient Yamaguchi esterification of compound 203 (after silyl protection) with compound 205 (from compound 204) in the presence of TCBC, Et3N, DMAP, and toluene to yield compound 206 with 90% yield. Subsequently, compound 206 was subjected to ozonolysis (in the presence of ozone and triphenyl phosphine), Pinnick oxidation (with 2-methyl-2-butene), and TBS-group deprotection in sequence to successfully produce the targeted nhatrangin A 207 in 92% yield.
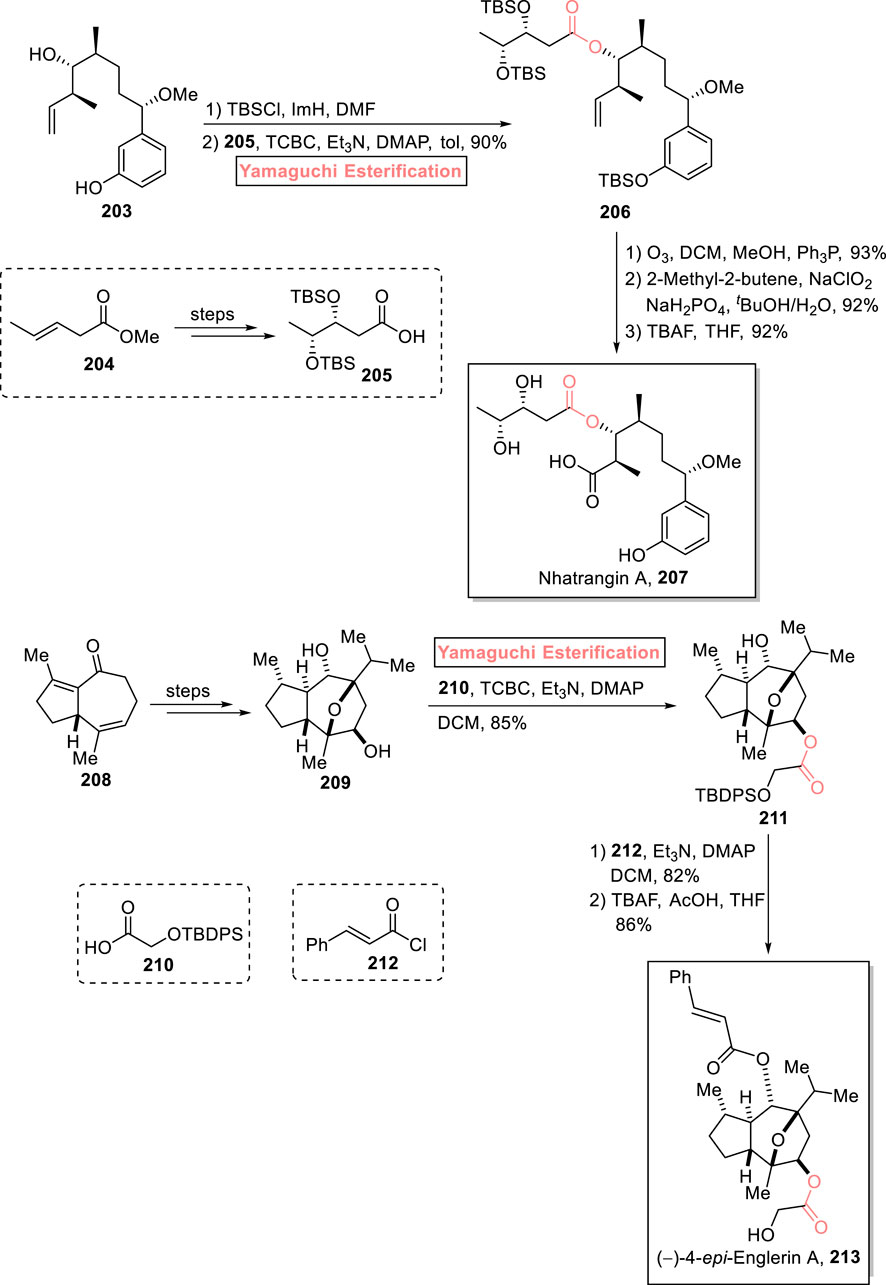
Scheme 23. Synthesis of nhatrangin A 207 and (−)-4-epi-englerin A 213.
2.4 Synthesis of natural terpenoids
2.4.1 Kumar Palli’s total synthesis of (−)-4-epi-englerin A
The natural extracts of the plant Phyllanthus engleri are renowned for their cytotoxic activity against renal cancer cells (Ratnayake et al., 2009). One of these natural products includes (−)-4-epi-englerin A 213, which is a sesquiterpenoid. The first total synthesis of this medicinally important scaffold was performed by Christmann in 2009 (Willot et al., 2010). Kumar Palli et al. (2021) also reported a valuable approach (based on 22 steps) for its total synthesis (with 4% overall yield) by using Yamaguchi esterification as the crucial step (Scheme 23) (Kumar Palli et al., 2021). Their synthesis started with compound 208, which was modified into diol 209 through a few steps. Next, the standard conditions for strategic Yamaguchi esterification were adjusted by reacting diol 209 with protected alcohol 210 in the presence of TCBC, Et3N, DMAP, and CH2Cl2 (solvent) to attain ester 211 with 85% yield. Then, ester 211 was allowed to react with trans-cinnamoyl chloride 212, followed by desilylation to gain the desired (−)-4-epi-englerin A 213 with 86% yield.
2.5 Synthesis of natural peptides
2.5.1 Chen’s total synthesis of colletopeptide A and colletotrichamide A
Colletopeptide A 223 and colletotrichamide A 224 are cyclic depsipeptides that were isolated independently from Colletotrichum gloeosporioides JS419 and Colletotrichum sp. S8 (Feng et al., 2019). These natural products are renowned for their broad range of biological activities. In particular, colletopeptide A 223 exhibits cytotoxic activity against RAW264.7 macrophages with IC50 = 8.3 μM (Oh et al., 2006; Ohno et al., 2004). With a profound interest in these medicinally active peptides, Chen et al. (2023) devised an impressive strategy for the first total synthesis of colletopeptide A 223 (in 15 steps) and colletotrichamide A 224 (in 17 steps) via a common precursor 221 (Scheme 24) (Chen et al., 2023). Their successful stereoselective synthesis entails Yamaguchi esterification as the main step. In the first step, the easily available starting materials, i.e., alkenes 214 and 215, were subjected to cross-metathesis to produce alkene 216 with 81% yield. Next, ester 218 was easily obtained in the desired stereochemistry using the Yamaguchi protocol for esterification. For this, compounds 216 and 217 were treated with TCBC and Et3N, yielding ester 218 with 76% yield. Compound 218 was treated with TMSOTf (for removal of the Boc group), and its amide coupling reaction with compound 219 led to the synthesis of tridepsipeptide 220 with 61% yield. It took a few steps for the achievement of precursor 221, which, after TBS removal, directly furnished colletopeptide A 223 with 67% yield. However, for the synthesis of colletotrichamide A 224, the precursor 221 was allowed to react with mannose derivative 222 in the presence of DTBMP, Tf2O, and DCM, followed by sequential steps involving benzyl group and TBS group removal under given conditions, to successfully achieve the desired natural product 224 with 83% yield.
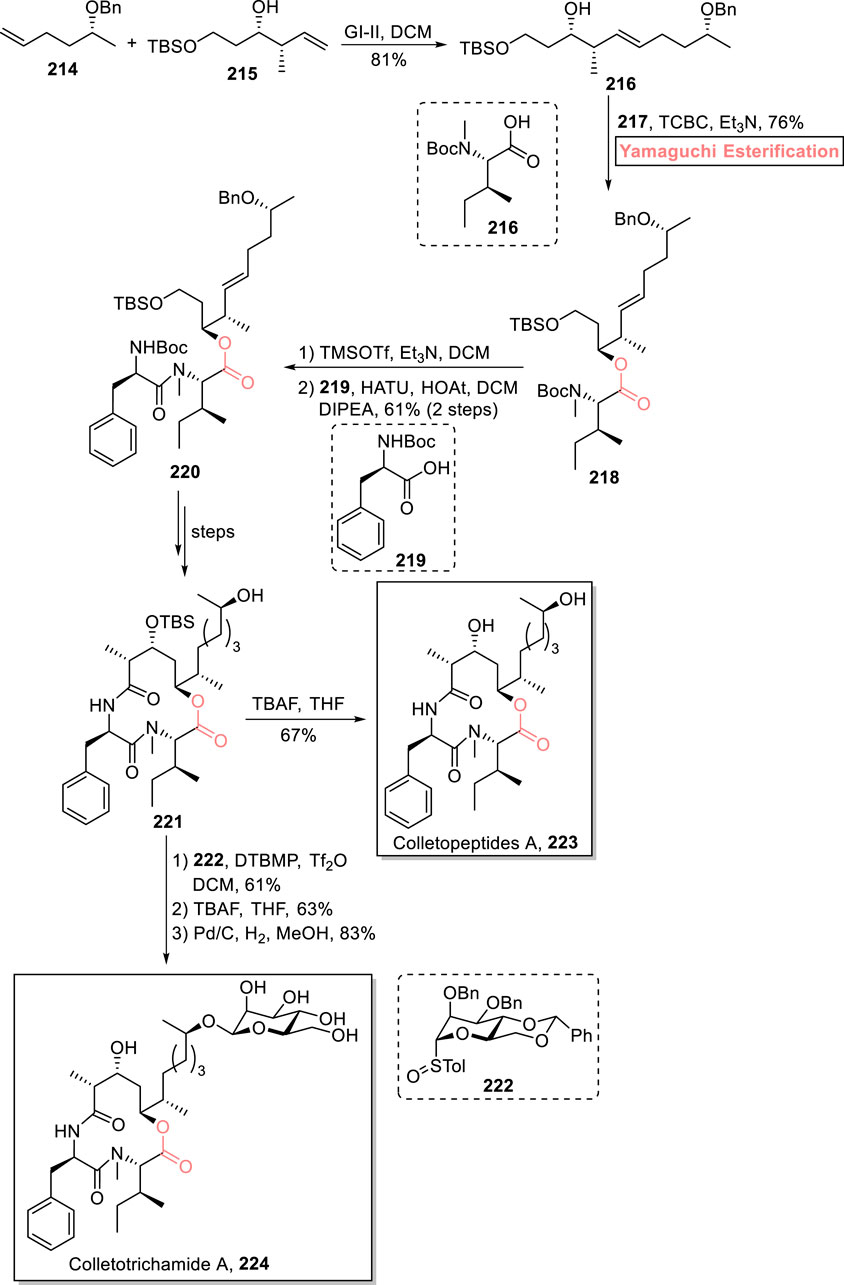
Scheme 24. Synthesis of colletopeptide A 223 and colletotrichamide A 224.
2.5.2 Bauer and Kazmaier’s total synthesis of thiamyxins (A, B, C, and E)
Thiamyxins (A, B, E and C) are depsipeptides in nature, and these were isolated by Muller from a myxobacterial variant of the Myxococcaceae (MCy9487) family (Haack et al., 2022). These thiazoline-containing natural products are popular owing to their anti-viral activities (more particularly against Zika, dengue, and Coronavirus). These interesting features prompted Bauer and Kazmaier (2023) to perform their challenging total synthesis by using Yamaguchi esterification as a key step for macrolactonization with the careful embellishment of stereocenters (Supplementary Scheme 3) (Bauer and Kazmaier, 2023).
2.6 Synthesis of natural α-pyrone
2.6.1 Zhao’s total synthesis of brevipolide H
Brevipolides A–J are α-pyrone-based natural products. Brevipolides G–I were isolated from Lippia alba by Hegde et al. (2004). Structurally, these are dihydro-α-pyrone motifs containing a cyclopropane ring in their side chain. This family of natural products shows anti-cancer activities against various cell lines (Deng et al., 2009). Considering their impressive pharmaceutical profile, Zhao et al. (2023) conducted the total synthesis of brevipolide H 293 in 12 consecutive steps with 8.65% overall yield (Zhao et al., 2023). The methodology was based on the synthesis of allylic alcohol 291 starting from D-galactal 289, covering a few steps. Next, envisaging the Yamaguchi protocol as a leading tool for esterification, compound 291 was subjected to Et3N, TCBC, and DMAP with the addition of p-methoxycinnamic acid to provide ester 292 with 73% yield. In the final step, HCl-mediated acetyl group deprotection of ester 292 produced brevipolide H 293 with 77% yield.
2.7 Synthesis of natural biosurfactants
2.7.1 Mittendorf’s total synthesis of (+)-aureosurfactin 1a and (−)-aureosurfactin 1b
Biosurfactants are important (ecofriendly) natural products, with extensive applications in the textile, cosmetic, food, and pharmaceutical industries (Singh et al., 2007; Mulligan, 2005). One of the biosurfactants, aureosurfactin, was isolated first by Yun et al. from Aureobasidium pullulans in 2016 (Kim et al., 2016). Structurally, aureosurfactin is an ester of acyclic dimer 3,5-dihydroxydecanoic acid. In 2023, Mittendorf et al. devised a concise strategy for the total synthesis of both enantiomers, i.e., (+)-aureosurfactin 299a and (−)-aureosurfactin 299b with 18% and 13.5% overall yields, respectively (Mittendorf et al., 2023). In their synthetic scheme, valeraldehydes 294a and 294b (as starting materials), in a few steps, provided acids 296a and 296b (independently), which further underwent methylation and TES group deprotection to result in alcohols 297a and 297b in 78% and 67% yields, respectively. Next, the key fragments 296a and 297b, as well as 296b and 297b, were subjected to coupling via the use of a well-compatible reagent, i.e., TBSCl, Et3N, and toluene (Yamaguchi esterification) to generate esters 298a and 298b with 92% and 93% yields, respectively. Finally, an additional HF/Py-mediated deprotection step (independently) generated both enantiomers, namely, (+)-aureosurfactin 299a and (−)-aureosurfactin 299b, in 99% and 79% yields, correspondingly.
3 Conclusion
The formal and total syntheses, structural elucidation, and revision of (newly isolated, as well as previously existing) natural products are meticulous steps in the design and development of drugs. In this endeavor, many organic reactions and reagents play prominent roles, as reported in the literature. The Yamaguchi reagent (2,4,6-trichlorobenzoyl chloride) is one among such reagents, and it is responsible for esterification between an acid and alcohol in the presence of DMAP as a coupling reagent. This reagent (despite its limitations including low yield and high cost) has been efficiently used in the synthesis of a wide variety of esters and lactones that are present in various biologically active natural products as key structural motifs. Our article presents an up-to-date compilation of Yamaguchi reagent-based synthetic schemes utilized for the synthesis of natural products and is aimed at helping related research groups in possibly devising further synthetic routes toward the synthesis of a diverse range of natural products (whose total syntheses are yet to be unlocked) using this reagent.
Author contributions
RM: data curation, methodology, and writing–original draft. AZ: resources, writing–original draft, writing–review and editing, project administration, and conceptualization. MA: writing–review and editing, resources, and data curation. AM: data curation, resources, software, and writing–review and editing. AlI: data curation, methodology, resources, software, and writing–review and editing. AC: writing–review and editing, resources, and investigation. AhI: writing–review and editing, resources, and investigation. KK-M: data curation, funding acquisition, resources, software, and writing–review and editing. MG: funding acquisition, resources, and writing–review and editing. MM: data curation, funding acquisition, resources, and writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. The research was partly financed by the following research projects: Collegium Medicum, the Mazovian Academy in Plock, and Medical University of Lublin (DS 730). A. Irfan acknowledges the Deanship of Research and graduate studies at King Khalid University for funding this work through the Large Groups Research Project under grant number RGP2/156/45.
Acknowledgments
Author A. R. Chaudhry acknowledges the Deanship of Graduate Studies and Scientific Research at the University of Bisha for supporting this work through the Fast-Track Research Support Program.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2024.1477764/full#supplementary-material
References
Abid-Essefi, S., Ouanes, Z., Hassen, W., Baudrimont, I., Creppy, E., and Bacha, H. (2004). Cytotoxicity, inhibition of DNA and protein syntheses and oxidative damage in cultured cells exposed to zearalenone. Toxicol. vitro 18 (4), 467–474. doi:10.1016/j.tiv.2003.12.011
Ahmad, S., Zahoor, A. F., Naqvi, S. A. R., and Akash, M. (2018). Recent trends in ring opening of epoxides with sulfur nucleophiles. Mol. Divers. 22 (1), 191–205. doi:10.1007/s11030-017-9796-x
Allu, S. R., Banne, S., Jiang, J., Qi, N., Guo, J., and He, Y. (2019). A unified synthetic approach to optically pure curvularin-type metabolites. J. Org. Chem. 84 (11), 7227–7237. doi:10.1021/acs.joc.9b00776
Annang, F., Pérez-Moreno, G., Gonzalez-Menendez, V., Lacret, R., Pérez-Victoria, I., Martín, J., et al. (2020). Strasseriolides A–D, a family of antiplasmodial macrolides isolated from the fungus Strasseria geniculata CF-247251. Org. Lett. 22 (17), 6709–6713. doi:10.1021/acs.orglett.0c01665
Babczyk, A., and Menche, D. (2023). Total synthesis of pentamycin by a conformationally biased double stille ring closure with a trienyl-bis-stannane. J. Am. Chem. Soc. 145 (20), 10974–10979. doi:10.1021/jacs.3c03011
Bai, Y., and Dai, M. (2015). Strategies and methods for the synthesis of anticancer natural product neopeltolide and its analogs. Curr. Org. Chem. 19 (10), 871–885. doi:10.2174/1385272819666150119225149
Bauer, K., and Kazmaier, U. (2023). Total synthesis of thiamyxins A–C and thiamyxin E, a potent class of RNA-virus-inhibiting (cyclo) depsipeptides. Angew. Chem. Int. Ed. 62, e202305445. doi:10.1002/anie.202305445
Bold, C. P., Gut, M., Schürmann, J., Lucena-Agell, D., Gertsch, J., Diaz, J. F., et al. (2021). Synthesis of morpholine-based analogues of (−)-Zampanolide and their biological activity. Chemistry–A Eur. J. 27 (19), 5936–5943. doi:10.1002/chem.202003996
Brütsch, T. M., Cotter, E., Lucena-Agell, D., Redondo-Horcajo, M., Davies, C., Pfeiffer, B., et al. (2023). Synthesis and structure-activity relationship studies of C (13)-desmethylene-(−)-zampanolide analogs. Chemistry–A Eur. J. 29 (36), e202300703. doi:10.1002/chem.202300703
Chambers, K. J., Sanghong, P., Carter Martos, D., Casoni, G., Mykura, R. C., Prasad Hari, D., et al. (2023). Stereospecific conversion of boronic esters into enones using methoxyallene: application in the total synthesis of 10-deoxymethynolide. Angew. Chem. 135 (50), e202312054. doi:10.1002/anie.202312054
Chandankar, S. S., and Raghavan, S. (2020). Stereoselective synthesis of dysoxylactam A. Org. Lett. 22 (2), 653–655. doi:10.1021/acs.orglett.9b04426
Chandra, J., Manne, S. R., Mondal, S., and Mandal, B. (2018). (E)-Ethyl-2-cyano-2-(((2, 4, 6-trichlorobenzoyl) oxy) imino) acetate: A Modified Yamaguchi Reagent for Enantioselective Esterification, Thioesterification, Amidation, and Peptide Synthesis. ACS omega 3 (6), 6120–6133. doi:10.1021/acsomega.8b00732
Chang, J.-L., Xu, H.-Z., Zhou, J., Zhou, M., Zhang, X., Guo, Y., et al. (2020). Antimicrobial furancarboxylic acids from a Penicillium sp. J. Nat. Prod. 83 (12), 3606–3613. doi:10.1021/acs.jnatprod.0c00758
Chen, J., Jiang, Y., Yan, J., Xu, C., and Ye, T. (2023). Total syntheses of colletopeptide A and colletotrichamide A. Molecules 28 (20), 7194. doi:10.3390/molecules28207194
Chen, Q.-H., and Kingston, D. G. (2014). Zampanolide and dactylolide: cytotoxic tubulin-assembly agents and promising anticancer leads. Nat. Product. Rep. 31 (9), 1202–1226. doi:10.1039/c4np00024b
Chlipala, G. E., Tri, P. H., Hung, N. V., Krunic, A., Shim, S. H., Soejarto, D. D., et al. (2010). Nhatrangins A and B, aplysiatoxin-related metabolites from the marine cyanobacterium Lyngbya majuscula from Vietnam. J. Nat. Prod. 73 (4), 784–787. doi:10.1021/np100002q
Decultot, L., and Clark, J. S. (2022). Synthetic studies on amphidinolide F: exploration of macrocycle construction by intramolecular stille coupling. Org. Lett. 24 (41), 7600–7604. doi:10.1021/acs.orglett.2c03045
Deng, Y., Balunas, M. J., Kim, J.-A., Lantvit, D. D., Chin, Y.-W., Chai, H., et al. (2009). Bioactive 5, 6-dihydro-α-pyrone derivatives from Hyptis brevipes. J. Nat. Prod. 72 (6), 1165–1169. doi:10.1021/np9001724
Depa, M. R., Potla, S., Narkhede, U. C., Jadhav, V. D., Sridhar, G., and Vidavalur, S. (2021). Total synthesis of neocosmosin A. Synth. Commun. 51 (18), 2817–2823. doi:10.1080/00397911.2021.1952435
Dhimitruka, I., and SantaLucia, J. (2006). Investigation of the Yamaguchi esterification mechanism. Synthesis of a Lux-S enzyme inhibitor using an improved esterification method. Org. Lett. 8 (1), 47–50. doi:10.1021/ol0524048
Dissanayake, G. C., Ndi, C. N., Markley, J. L., Martinez, J. B., and Hanson, P. R. (2023). Total synthesis of sanctolide A and formal synthesis of (2 S)-sanctolide A. J. Org. Chem. 88 (2), 805–817. doi:10.1021/acs.joc.2c01922
Farlow, M. R., Thompson, R. E., Wei, L.-J., Tuchman, A. J., Grenier, E., Crockford, D., et al. (2019). A randomized, double-blind, placebo-controlled, phase II study assessing safety, tolerability, and efficacy of bryostatin in the treatment of moderately severe to severe Alzheimer’s disease. J. Alzheimer's Dis. 67 (2), 555–570. doi:10.3233/jad-180759
Feng, L., Wang, J., Liu, S., Zhang, X.-J., Bi, Q.-R., Hu, Y.-Y., et al. (2019). Colletopeptides A–D, anti-inflammatory cyclic tridepsipeptides from the plant endophytic fungus Colletotrichum sp. S8. J. Nat. Prod. 82 (6), 1434–1441. doi:10.1021/acs.jnatprod.8b00829
Field, J. J., Singh, A. J., Kanakkanthara, A., Halafihi, T. i., Northcote, P. T., and Miller, J. H. (2009). Microtubule-stabilizing activity of zampanolide, a potent macrolide isolated from the Tongan marine sponge Cacospongia mycofijiensis. J. Med. Chem. 52 (22), 7328–7332. doi:10.1021/jm901249g
Fritz, L., Wienhold, S., Hackl, S., and Bach, T. (2022). Total synthesis of Pulvomycin D. Chemistry–A Eur. J. 28 (3), e202104064. doi:10.1002/chem.202104064
Fujiki, H., Tanaka, Y., Miyake, R., Kikkawa, U., Nishizuka, Y., and Sugimura, T. (1984). Activation of calcium-activated, phospholipid-dependent protein kinase (protein kinase C) by new classes of tumor promoters: teleocidin and debromoaplysiatoxin. Biochem. biophysical Res. Commun. 120 (2), 339–343. doi:10.1016/0006-291x(84)91259-2
Fuska, J., Ivanitskaya, L., Horakova, K., and Kuhr, I. (1974). The cytotoxic effects of a new antibiotic vermiculine. J. Antibiotics 27 (2), 141–142. doi:10.7164/antibiotics.27.141
Fuska, J., Nemec, P., and Kuhr, I. (1972). Vermiculine, a new antiprotozoal antibiotic from Penicillium vermiculatum. J. Antibiotics 25 (4), 208–211. doi:10.7164/antibiotics.25.208
Gao, J., Radwan, M. M., León, F., Dale, O. R., Husni, A. S., Wu, Y., et al. (2013). 'Neocosmospora sp.-derived resorcylic acid lactones with in vitro binding affinity for human opioid and cannabinoid receptors. J. Nat. Prod. 76 (5), 824–828. doi:10.1021/np300653d
Ghosh, A. K., and Yuan, H. (2022). Total syntheses of the proposed structure of iriomoteolide-1a,-1b and synthesis of three derivatives for structural studies. Mar. Drugs 20 (10), 587. doi:10.3390/md20100587
Gillsch, F., Mbui, F., Bilitewski, U., and Schobert, R. (2022a). Synthesis and bioactivity of thiocarboxylic A and derivatives. J. Nat. Prod. 85 (12), 2828–2835. doi:10.1021/acs.jnatprod.2c00870
Gillsch, F., Zeng, H., Bär, S. I., Schrey, H., and Schobert, R. (2022b). Synthesis and bioactivity of ophiofuranones A and B. J. Org. Chem. 87 (9), 6520–6523. doi:10.1021/acs.joc.2c00521
Gobin, J., Moore, C. H., Reeve Jr, J. R., Wong, D. K., Gibson, B. W., and Horwitz, M. A. (1995). Iron acquisition by Mycobacterium tuberculosis: isolation and characterization of a family of iron-binding exochelins. Proc. Natl. Acad. Sci. 92 (11), 5189–5193. doi:10.1073/pnas.92.11.5189
Goda, Y., and Fuwa, H. (2023). Total synthesis of (−)-Enigmazole B. Org. Lett. 25 (47), 8402–8407. doi:10.1021/acs.orglett.3c03002
Gutiérrez, C., Serrano-Villar, S., Madrid-Elena, N., Pérez-Elías, M. J., Martín, M. E., Barbas, C., et al. (2016). Bryostatin-1 for latent virus reactivation in HIV-infected patients on antiretroviral therapy. Aids 30 (9), 1385–1392. doi:10.1097/qad.0000000000001064
Haack, P. A., Harmrolfs, K., Bader, C. D., Garcia, R., Gunesch, A. P., Haid, S., et al. (2022). 'Thiamyxins: structure and biosynthesis of myxobacterial RNA-virus inhibitors. Angew. Chem. Int. Ed. 61 (52), e202212946. doi:10.1002/anie.202212946
Hamilton-Miller, J. (1973). Chemistry and biology of the polyene macrolide antibiotics. Bacteriol. Rev. 37 (2), 166–196. doi:10.1128/mmbr.37.3.166-196.1973
Haslam, E. (1980). Recent developments in methods for the esterification and protection of the carboxyl group. Tetrahedron 36 (17), 2409–2433. doi:10.1016/0040-4020(80)80219-5
Hegde, V. R., Pu, H., Patel, M., Das, P. R., Strizki, J., Gullo, V. P., et al. (2004). Three new compounds from the plant Lippia alva as inhibitors of chemokine receptor 5 (CCR5). Bioorg. and Med. Chem. Lett. 14 (21), 5339–5342. doi:10.1016/j.bmcl.2004.08.021
Hellwig, V., Mayer-Bartschmid, A., Müller, H., Greif, G., Kleymann, G., Zitzmann, W., et al. (2003). Pochonins A− F, new antiviral and antiparasitic resorcylic acid lactones from Pochonia c hlamydosporia var. c atenulata. J. Nat. Prod. 66 (6), 829–837. doi:10.1021/np020556v
Hikota, M., Sakurai, Y., Horita, K., and Yonemitsu, O. (1990). Synthesis of erythronolide A via a very efficient macrolactonization under usual acylation conditions with the Yamaguchi reagent. Tetrahedron Lett. 31 (44), 6367–6370. doi:10.1016/s0040-4039(00)97066-7
Hopkins, C. D., and Wipf, P. (2009). 'Isolation, biology and chemistry of the disorazoles: new anti-cancer macrodiolides. Nat. Product. Rep. 26 (5), 585–601. doi:10.1039/b813799b
Horáková, K., Kernacova, B., Nemec, P., and Fuska, J. (1976). Characterization of the cytotoxic activity of vermiculine. J. Antibiotics 29 (10), 1109–1111. doi:10.7164/antibiotics.29.1109
Horwitz, L. D., and Horwitz, M. A. (2014). The exochelins of pathogenic mycobacteria: unique, highly potent, lipid-and water-soluble hexadentate iron chelators with multiple potential therapeutic uses. Antioxidants and redox Signal. 21 (16), 2246–2261. doi:10.1089/ars.2013.5789
Inanaga, J., Hirata, K., Saeki, H., Katsuki, T., and Yamaguchi, M. (1979). A rapid esterification by means of mixed anhydride and its application to large-ring lactonization. Bull. Chem. Soc. Jpn. 52 (7), 1989–1993. doi:10.1246/bcsj.52.1989
Irschik, H., Jansen, R., Gerth, K., Höfle, G., and Reichenbach, H. (1995). Antibiotics from gilding bacteria. No.67. Sorangiolid A, a new antibiotic isolated from the myxobacterium Sorangium cellulosumSo ce 12. J. Antibiotics 48 (8), 886–887. doi:10.7164/antibiotics.48.886
Ishibashi, M., and Kobayashi, J. i. (1997). Amphidinolides: unique macrolides from marine dinoflagellates. Heterocycles 1 (44), 543–572. doi:10.3987/rev-96-sr1
Jansen, C. U., Nørskov, A., Mortensen, K. T., and Qvortrup, K. M. (2023). Convergent total synthesis of exochelin 772SM. J. Org. Chem. 88 (13), 8669–8673. doi:10.1021/acs.joc.3c00561
Jansen, R., Irschik, H., Meyer, H., Reichenbach, H., Wray, V., Schomburg, D., et al. (1995). Antibiotics from gliding bacteria, LXIV. Isolation and structure elucidation of sorangiolides A and B, novel macrocyclic lactone carboxylic acids from Sorangium cellulosum. Liebigs Ann. 1995 (5), 867–872. doi:10.1002/jlac.1995199505125
Jansen, R. (1994). Disorazoles, highly cytotoxic metabolites from the sorangicin-producing bacterium Sorangium cellulosum, strain So ce 12. Liebigs Ann. Chem. 759–773. doi:10.1002/jlac.199419940802
Jena, B. K., Reddy, G. S., and Mohapatra, D. K. (2017). First asymmetric total synthesis of aspergillide D. Org. and Biomol. Chem. 15 (8), 1863–1871. doi:10.1039/c6ob02435a
Jordan, A., Whymark, K. D., Sydenham, J., and Sneddon, H. F. (2021). A solvent-reagent selection guide for Steglich-type esterification of carboxylic acids. Green Chem. 23 (17), 6405–6413. doi:10.1039/d1gc02251b
Joseph, T., Sahoo, S., and Halligudi, S. (2005). Brönsted acidic ionic liquids: a green, efficient and reusable catalyst system and reaction medium for Fischer esterification. J. Mol. Catal. A Chem. 234 (1-2), 107–110. doi:10.1016/j.molcata.2005.03.005
Kang, H.-S., Krunic, A., and Orjala, J. (2012). Sanctolide A, a 14-membered PK–NRP hybrid macrolide from the cultured cyanobacterium Oscillatoria sancta (SAG 74.79). Tetrahedron Lett. 53 (28), 3563–3567. doi:10.1016/j.tetlet.2012.04.136
Kawahara, T., Kagaya, N., Masuda, Y., Doi, T., Izumikawa, M., Ohta, K., et al. (2015). Foxo3a inhibitors of microbial origin, JBIR-141 and JBIR-142. Org. Lett. 17 (21), 5476–5479. doi:10.1021/acs.orglett.5b02842
Khan, Z., Javed, F., Shamair, Z., Hafeez, A., Fazal, T., Aslam, A., et al. (2021). Current developments in esterification reaction: a review on process and parameters. J. Industrial Eng. Chem. 103, 80–101. doi:10.1016/j.jiec.2021.07.018
Kim, G., Chu-Moyer, M. Y., and Danishefsky, S. J. (1990). The total synthesis of dl-indolizomycin. J. Am. Chem. Soc. 112 (5), 2003–2005. doi:10.1021/ja00161a059
Kim, J.-S., Lee, I.-K., Kim, D.-W., and Yun, B.-S. (2016). Aureosurfactin and 3-deoxyaureosurfactin, novel biosurfactants produced by Aureobasidium pullulans L3-GPY. J. Antibiotics 69 (10), 759–761. doi:10.1038/ja.2015.141
Kotammagari, T. K. (2014). '2, 4, 6-trichlorobenzoyl chloride (Yamaguchi reagent). Synlett 25 (09), 1335–1336. doi:10.1055/s-0033-1341245
Kranzler, M., Syrowatka, M., Leitsch, D., Winnips, C., and Walochnik, J. (2015). Pentamycin shows high efficacy against Trichomonas vaginalis. Int. J. Antimicrob. agents 45 (4), 434–437. doi:10.1016/j.ijantimicag.2014.12.024
Kumari, K., Syed, T., Krishna, A., Muvvala, S., Nowduri, A., Sridhar, C., et al. (2022). Steroselective total synthesis of Neocosmosin B. Tetrahedron 112, 132723. doi:10.1016/j.tet.2022.132723
Kumari, K., Syed, T., Krishna, A., Muvvala, S., Nowduri, A., Sridhar, C., et al. (2023). Stereoselective total synthesis of Aspergillide D. Nat. Prod. Res. 37 (20), 3402–3408. doi:10.1080/14786419.2022.2078321
Kumar Palli, K., Reddy Anugu, R., and Chandrasekhar, S. (2021). Total synthesis of (−)-4-epi-Englerin A. Eur. J. Org. Chem. 2021 (22), 3190–3196. doi:10.1002/ejoc.202100354
Lai, Y., and Dai, W. M. (2021). Modular total synthesis of (–)-Palmyrolide A and (+)-(S,S)-Palmyrolide A via ring-closing metathesis and alkene isomerization†. Chin. J. Chem. 39 (1), 69–74. doi:10.1002/cjoc.202000458
Li, Y.-L., Zhu, R.-X., Li, G., Wang, N.-N., Liu, C.-Y., Zhao, Z.-T., et al. (2019). Secondary metabolites from the endolichenic fungus Ophiosphaerella korrae. RSC Adv. 9 (8), 4140–4149. doi:10.1039/c8ra10329a
Liu, C.-P., Xie, C.-Y., Zhao, J.-X., Ji, K.-L., Lei, X.-X., Sun, H., et al. (2019). Dysoxylactam a: a macrocyclolipopeptide reverses p-glycoprotein-mediated multidrug resistance in cancer cells. J. Am. Chem. Soc. 141 (17), 6812–6816. doi:10.1021/jacs.9b02259
Liu, H., Ottosen, R. N., Jennet, K. M., Svenningsen, E. B., Kristensen, T. F., Biltoft, M., et al. (2021). Macrodiolide diversification reveals broad immunosuppressive activity that impairs the cGAS-STING pathway. Angew. Chem. Int. Ed. 60 (34), 18734–18741. doi:10.1002/anie.202105793
Lizzadro, L., Spieß, O., Collisi, W., Stadler, M., and Schinzer, D. (2022). Extending the structure-activity relationship of disorazole C1: exchanging the oxazole ring by thiazole and influence of chiral centers within the disorazole core on cytotoxicity. ChemBioChem 23 (20), e202200458. doi:10.1002/cbic.202200458
Lizzadro, L., Spieß, O., and Schinzer, D. (2021). Total synthesis of (−)-disorazole C1. Org. Lett. 23 (12), 4543–4547. doi:10.1021/acs.orglett.1c01123
Majhi, S. (2021). Applications of Yamaguchi method to esterification and macrolactonization in total synthesis of bioactive natural products. ChemistrySelect 6 (17), 4178–4206. doi:10.1002/slct.202100206
Maram, L., and Das, B. (2015). A stereoselective total synthesis of xyolide, a natural bioactive nonenolide. Helvetica Chim. Acta 98 (5), 674–682. doi:10.1002/hlca.201400291
Matthew, S., Salvador, L. A., Schupp, P. J., Paul, V. J., and Luesch, H. (2010). Cytotoxic halogenated macrolides and modified peptides from the apratoxin-producing marine cyanobacterium Lyngbya bouillonii from Guam. J. Nat. Prod. 73 (9), 1544–1552. doi:10.1021/np1004032
Meyer, C. C., Verboom, K. L., Evarts, M. M., Jung, W.-O., and Krische, M. J. (2023). Allyl alcohol as an acrolein equivalent in enantioselective C–C coupling: total synthesis of amphidinolides R, J, and S. J. Am. Chem. Soc. 145 (14), 8242–8247. doi:10.1021/jacs.3c01809
Michel, K., Demarco, P., and Nagarajan, R. (1977). The isolation and structure elucidation of macrocyclic lactone antibiotic, A26771B. J. antibiotics 30 (7), 571–575. doi:10.7164/antibiotics.30.571
Mittendorf, F., Quambusch, M., and Kirsch, S. F. (2023). Total synthesis of both enantiomers of the biosurfactant aureosurfactin via bidirectional synthesis with a chiral Horner–Wittig building block. Org. and Biomol. Chem. 21 (22), 4574–4577. doi:10.1039/d3ob00650f
Molawi, K., Delpont, N., and Echavarren, A. M. (2010). Enantioselective synthesis of (−)-englerins A and B. Angew. Chem. 20 (122), 3595–3597. doi:10.1002/anie.201000890
Moon, K., Cui, J., Kim, E., Riandi, E. S., Park, S. H., Byun, W. S., et al. (2020). Structures and biosynthetic pathway of pulvomycins B–D: 22-membered macrolides from an estuarine Streptomyces sp. Org. Lett. 22 (14), 5358–5362. doi:10.1021/acs.orglett.0c01249
Morishita, M., Hada, K., Kita, M., and Nishikawa, T. (2023). The asymmetric total synthesis and configuration confirmation of aplysiaenal and nhatrangin A, truncated derivatives of aplysiatoxin and oscillatoxin. J. Nat. Prod. 86 (4), 1033–1041. doi:10.1021/acs.jnatprod.3c00077
Mukhopadhyay, T. K., and Trauner, D. (2022). Concise synthesis of glycerophospholipids. J. Org. Chem. 88 (15), 11253–11257. doi:10.1021/acs.joc.2c02096
Mulligan, C. N. (2005). Environmental applications for biosurfactants. Environ. Pollut. 133 (2), 183–198. doi:10.1016/j.envpol.2004.06.009
Munawar, S., Zahoor, A. F., Ali, S., Javed, S., Irfan, M., Irfan, A., et al. (2022). Mitsunobu reaction: a powerful tool for the synthesis of natural products: a review. Molecules 27 (20), 6953. doi:10.3390/molecules27206953
Munawar, S., Zahoor, A. F., Hussain, S. M., Ahmad, S., Mansha, A., Parveen, B., et al. (2024). Steglich esterification: a versatile synthetic approach toward the synthesis of natural products, their analogues/derivatives. Heliyon 10 (1), e23416. doi:10.1016/j.heliyon.2023.e23416
Munir, R., Zahoor, A. F., Nazeer, U., Saeed, M. A., Mansha, A., Irfan, A., et al. (2023). Gilman reagent toward the synthesis of natural products. RSC Adv. 13 (50), 35172–35208. doi:10.1039/d3ra07359a
Nakazato, K., Oda, M., and Fuwa, H. (2022). Total synthesis of (+)-neopeltolide by the macrocyclization/transannular pyran cyclization strategy. Org. Lett. 24 (22), 4003–4008. doi:10.1021/acs.orglett.2c01429
Nishio, Y., Kawazu, A., Hirano, S., and Matsubara, H. (2016). Preparation of fluorous Yamaguchi reagents and evaluation of their reactivity in esterification. Tetrahedron 72 (5), 720–725. doi:10.1016/j.tet.2015.12.026
Oh, D.-C., Jensen, P. R., and Fenical, W. (2006). Zygosporamide, a cytotoxic cyclic depsipeptide from the marine-derived fungus Zygosporium masonii. Tetrahedron Lett. 47 (48), 8625–8628. doi:10.1016/j.tetlet.2006.08.113
Ohno, O., Ikeda, Y., Sawa, R., Igarashi, M., Kinoshita, N., Suzuki, Y., et al. (2004). 'Isolation of heptadepsin, a novel bacterial cyclic depsipeptide that inhibits lipopolysaccharide activity. Chem. and Biol. 11 (8), 1059–1070. doi:10.1016/j.chembiol.2004.05.016
Oku, N., Takada, K., Fuller, R. W., Wilson, J. A., Peach, M. L., Pannell, L. K., et al. (2010). Isolation, structural elucidation, and absolute stereochemistry of enigmazole A, a cytotoxic phosphomacrolide from the Papua New Guinea marine sponge Cinachyrella enigmatica. J. Am. Chem. Soc. 132 (30), 10278–10285. doi:10.1021/ja1016766
Okuno, Y., Isomura, S., Nishibayashi, A., Hosoi, A., Fukuyama, K., Ohba, M., et al. (2014). Modified Yamaguchi reagent: convenient and efficient esterification. Synth. Commun. 44 (19), 2854–2860. doi:10.1080/00397911.2014.919659
Park, J., Ngo, H. V., Jin, H.-E., Lee, K. W., and Lee, B.-J. (2022). Hydroxyl group-targeted conjugate and its self-assembled nanoparticle of peptide drug: effect of degree of saturation of fatty acids and modification of physicochemical properties. Int. J. Nanomedicine Vol. 17, 2243–2260. doi:10.2147/ijn.s356804
Patocka, J., Soukup, O., and Kuca, K. (2013). Resorcylic acid lactones as the protein kinase inhibitors, naturally occuring toxins. Mini Rev. Med. Chem. 13 (13), 1873–1878. doi:10.2174/13895575113136660096
Payero, T. D., Vicente, C. M., Rumbero, Á., Barreales, E. G., Santos-Aberturas, J., de Pedro, A., et al. (2015). Functional analysis of filipin tailoring genes from Streptomyces filipinensis reveals alternative routes in filipin III biosynthesis and yields bioactive derivatives. Microb. Cell Factories 14, 114–14. doi:10.1186/s12934-015-0307-4
Pereira, A. R., Cao, Z., Engene, N., Soria-Mercado, I. E., Murray, T. F., and Gerwick, W. H. (2010). Palmyrolide A, an unusually stabilized neuroactive macrolide from Palmyra Atoll cyanobacteria. Org. Lett. 12 (20), 4490–4493. doi:10.1021/ol101752n
Pérez-Moreno, G., Cantizani, J., Sánchez-Carrasco, P., Ruiz-Pérez, L. M., Martín, J., El Aouad, N., et al. (2016). Discovery of new compounds active against Plasmodium falciparum by high throughput screening of microbial natural products. PLoS One 11 (1), e0145812. doi:10.1371/journal.pone.0145812
Pirovani, R. V., Brito, G. A., Barcelos, R. C., and Pilli, R. A. (2015). Enantioselective total synthesis of (+)-lyngbyabellin M. Mar. drugs 13 (6), 3309–3324. doi:10.3390/md13063309
Pujari, S. A., Gowrisankar, P., and Kaliappan, K. P. (2011). A shimizu non-aldol approach to the formal total synthesis of palmerolide A. Chemistry–An Asian J. 6 (11), 3137–3151. doi:10.1002/asia.201100429
Radha Krishna, M., Sridhar, G., Syed, T., and Jayaprakash, H. V. (2022). Stereoselective total synthesis of (-)-curvularin. Synth. Commun. 52 (1), 37–42. doi:10.1080/00397911.2021.1979043
Ratnayake, R., Covell, D., Ransom, T. T., Gustafson, K. R., and Beutler, J. A. (2009). Englerin A, a selective inhibitor of renal cancer cell growth, from Phyllanthus engleri. Org. Lett. 11 (1), 57–60. doi:10.1021/ol802339w
Reddy, G. N., Gudisela, M. R., Boddiboyena, R., and Prasad, K. (2022). Total synthesis of sumalactone A. Synth. Commun. 52 (17), 1713–1720. doi:10.1080/00397911.2022.2111527
Saha, S., Paul, D., and Goswami, R. K. (2020). Cyclodepsipeptide alveolaride C: total synthesis and structural assignment. Chem. Sci. 11 (41), 11259–11265. doi:10.1039/d0sc04478d
Sahana, M. H., Paul, D., Sharma, H., and Goswami, R. K. (2023). Total synthesis of antibacterial macrolide sorangiolide A. Org. Lett. 25 (43), 7827–7831. doi:10.1021/acs.orglett.3c03066
Sahana, M. H., Saha, D., and Goswami, R. K. (2022). Total synthesis of strasseriolide A. J. Org. Chem. 87 (17), 11805–11815. doi:10.1021/acs.joc.2c01595
Sakai, T., Sameshima, T., Matsufuji, M., Kawamura, N., Dobashi, K., and Mizui, Y. (2004). 'Pladienolides, new substances from culture of Streptomyces platensis Mer-11107 I. Taxonomy, fermentation, isolation and screening. J. antibiotics 57 (3), 173–179. doi:10.7164/antibiotics.57.173
Salituro, L. J., Pazienza, J. E., and Rychnovsky, S. D. (2022). Total syntheses of strasseriolide A and B, antimalarial macrolide natural products. Org. Lett. 24 (5), 1190–1194. doi:10.1021/acs.orglett.1c04340
Schmidt, F., Viswanathan Ammanath, A., Götz, F., and Maier, M. E. (2023). Synthesis of berkeleylactone A by ring-closing alkyne metathesis. Eur. J. Org. Chem. 26 (36), e202300615. doi:10.1002/ejoc.202300615
Shahzadi, I., Zahoor, A. F., Tüzün, B., Mansha, A., Anjum, M. N., Rasul, A., et al. (2022). Repositioning of acefylline as anti-cancer drug: synthesis, anticancer and computational studies of azomethines derived from acefylline tethered 4-amino-3-mercapto-1, 2, 4-triazole. Plos one 17 (12), e0278027. doi:10.1371/journal.pone.0278027
Shimbo, K., Tsuda, M., Izui, N., and Kobayashi, J. i. (2002). Amphidinolide W, a new 12-membered macrolide from dinoflagellate Amphidinium sp. J. Org. Chem. 67 (3), 1020–1023. doi:10.1021/jo016089o
Shotwell, J. B., and Roush, W. R. (2004). Synthesis of the C11− C29 fragment of amphidinolide F. Org. Lett. 6 (21), 3865–3868. doi:10.1021/ol048381z
Singh, A., Van Hamme, J. D., and Ward, O. P. (2007). Surfactants in microbiology and biotechnology: Part 2. Application aspects. Biotechnol. Adv. 25 (1), 99–121. doi:10.1016/j.biotechadv.2006.10.004
Stierle, A. A., Stierle, D. B., Decato, D., Priestley, N. D., Alverson, J. B., Hoody, J., et al. (2017). The berkeleylactones, antibiotic macrolides from fungal coculture. J. Nat. Prod. 80 (4), 1150–1160. doi:10.1021/acs.jnatprod.7b00133
Takada, K., Oku, N., Peach, M. L., Ransom, T. T., Henrich, C. J., and Gustafson, K. R. (2023). Enigmazole phosphomacrolides from the marine sponge Cinachyrella enigmatica. J. Org. Chem. 88 (15), 10996–11002. doi:10.1021/acs.joc.3c00963
Tan, L. T. (2007). Bioactive natural products from marine cyanobacteria for drug discovery. Phytochemistry 68 (7), 954–979. doi:10.1016/j.phytochem.2007.01.012
Tanaka, J.-i., and Higa, T. (1996). 'Zampanolide, a new cytotoxic marcrolide from a marine sponge. Tetrahedron Lett. 37 (31), 5535–5538. doi:10.1016/0040-4039(96)01149-5
Tsuda, M., Oguchi, K., Iwamoto, R., Okamoto, Y., Fukushi, E., Kawabata, J., et al. (2007a). Iriomoteolides-1b and-1c, 20-membered macrolides from a marine dinoflagellate Amphidinium species. J. Nat. Prod. 70 (10), 1661–1663. doi:10.1021/np0702537
Tsuda, M., Oguchi, K., Iwamoto, R., Okamoto, Y., Kobayashi, J. i., Fukushi, E., et al. (2007b). Iriomoteolide-1a, a potent cytotoxic 20-membered macrolide from a benthic dinoflagellate Amphidinium species. J. Org. Chem. 72 (12), 4469–4474. doi:10.1021/jo070414b
Ulanovskaya, O. A., Janjic, J., Suzuki, M., Sabharwal, S. S., Schumacker, P. T., Kron, S. J., et al. (2008). Synthesis enables identification of the cellular target of leucascandrolide A and neopeltolide. Nat. Chem. Biol. 4 (7), 418–424. doi:10.1038/nchembio.94
Umaña, C. A., Henry, J. L., Saltzman, C. T., Sackett, D. L., Jenkins, L. M., and Taylor, R. E. (2023). Linear (−)-Zampanolide: flexibility in conformation–activity relationships. ChemMedChem 18 (19), e202300292. doi:10.1002/cmdc.202300292
Vakiti, A. R., Valluru, K. R., Verma, S. N., Syed, T., Sridhar, G., and Kumar, J. S. (2022). Total synthesis of berkeleylactone F. Synth. Commun. 52 (2), 205–211. doi:10.1080/00397911.2021.2012804
Valeev, R., Sunagatullina, G., and Miftakhov, M. (2019). Yamaguchi esterification in the synthetic approaches to precursors of epothilone D analogs. Russ. J. Org. Chem. 55, 1439–1441. doi:10.1134/s1070428019090264
Villa, R., Mandel, A. L., Jones, B. D., La Clair, J. J., and Burkart, M. D. (2012). Structure of FD-895 revealed through total synthesis. Org. Lett. 14 (21), 5396–5399. doi:10.1021/ol3023006
Wei, Y., and Shi, M. (2013). Recent advances in organocatalytic asymmetric morita–baylis–hillman/aza-morita–baylis–hillman reactions. Chem. Rev. 113 (8), 6659–6690. doi:10.1021/cr300192h
Wender, P. A., Luu-Nguyen, Q. H., Sloane, J. L., and Ranjan, A. (2022). Trimethylene methane dianion equivalent for the asymmetric consecutive allylation of aldehydes: applications to prins-driven macrocyclizations for the synthesis of bryostatin 1 and analogues. J. Org. Chem. 87 (23), 15925–15937. doi:10.1021/acs.joc.2c02047
Willot, M., Radtke, L., Könning, D., Fröhlich, R., Gessner, V., Strohmann, C., et al. (2010). Synthesis of engerlin A. Synfacts 2010 (03), 0262. doi:10.1055/s-0029-1219246
Wittmann, L., Wachter, A. C., Schrey, H., and Schobert, R. (2022). Synthetic approach to the natural N-nitrosohydroxylamino tetramic acid JBIR-141. Org. Lett. 24 (28), 5171–5175. doi:10.1021/acs.orglett.2c02006
Wright, A. E., Botelho, J. C., Guzmán, E., Harmody, D., Linley, P., McCarthy, P. J., et al. (2007). Neopeltolide, a macrolide from a lithistid sponge of the family Neopeltidae. J. Nat. Prod. 70 (3), 412–416. doi:10.1021/np060597h
Wu, J.-Z., Wang, Z., and Qiao, C. (2012). Synthesis of stagonolide C from mulzer epoxide. Tetrahedron Lett. 53 (9), 1153–1155. doi:10.1016/j.tetlet.2011.12.102
Wu, Y.-H., Zhang, Z.-H., Zhong, Y., Huang, J.-J., Li, X.-X., Jiang, J.-Y., et al. (2017). Sumalactones A–D, four new curvularin-type macrolides from a marine deep sea fungus Penicillium Sumatrense. RSC Adv. 7 (63), 40015–40019. doi:10.1039/c7ra06933b
Yamamoto, H., and Muramatsu, W. (2019). Modified Yamaguchi reagent mediated coupling reactions. Synfacts 15 (01), 0100. doi:10.1055/s-0037-1611429
Yang, G. Z., Wang, L., Fan, Y. Y., Lai, Z. W., Yu, X. N., Lou, L. G., et al. (2022). Concise total synthesis of dysoxylactam A and a simplified analog. Chin. J. Chem. 40 (17), 2027–2034. doi:10.1002/cjoc.202200123
Yasoshima, K., Yoshida, M., and Doi, T. (2022). The concise synthesis of a tert-butoxycarbonyl derivative of (3 R, 4 S)-4-Amino-3-hydroxy-7-(N-nitrosohydroxyamino)-2, 2-dimethylheptanoate, a component of JBIR-141. Bull. Chem. Soc. Jpn. 95 (5), 830–832. doi:10.1246/bcsj.20220035
Yoo, M., and Krische, M. J. (2021). Total synthesis of the spliceosome modulator pladienolide B via asymmetric alcohol-mediated syn-and anti-diastereoselective carbonyl crotylation. Angew. Chem. 133 (25), 14042–14047. doi:10.1002/ange.202103845
Zhan, J., and Gunatilaka, A. L. (2005). Microbial transformation of curvularin. J. Nat. Prod. 68 (8), 1271–1273. doi:10.1021/np0580309
Zhang, Y., Liu, Y., and Du, Y. (2021). First total synthesis of 27-deoxylyngbyabellin A. Synthesis 53 (16), 2874–2880. doi:10.1055/a-1478-9088
Zhao, Z., Lv, Q., Geng, J., Liu, Y., Hu, C., Du, Y., et al. (2023). Stereoselective total synthesis of (+)-brevipolide H from D-galactal. Synthesis 55 (02), 341–346. doi:10.1055/a-1700-3520
Zief, M., Woodside, R., and Schmitz, H. (1957). Pulvomycin. Antibiotics and Chemother. 7 (7), 384–386.
Keywords: Yamaguchi esterification, macrolides, terpenoids, polyketides, peptides, metabolites
Citation: Munir R, Zahoor AF, Anjum MN, Mansha A, Irfan A, Chaudhry AR, Irfan A, Kotwica-Mojzych K, Glowacka M and Mojzych M (2024) Yamaguchi esterification: a key step toward the synthesis of natural products and their analogs—a review. Front. Chem. 12:1477764. doi: 10.3389/fchem.2024.1477764
Received: 08 August 2024; Accepted: 09 September 2024;
Published: 11 October 2024.
Edited by:
Essa M. Saied, Humboldt University of Berlin, GermanyReviewed by:
Ramesh Mamidala, Cambrex Corporation, a pharmaceutical company, United StatesSomeshwar Nagamalla, University of Kansas, United States
Pallavi Sharma, University of Lincoln, United Kingdom
Copyright © 2024 Munir, Zahoor, Anjum, Mansha, Irfan, Chaudhry, Irfan, Kotwica-Mojzych, Glowacka and Mojzych. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ameer Fawad Zahoor, ZmF3YWQuemFob29yQGdjdWYuZWR1LnBr; Mariusz Mojzych, bW1vanp5Y2hAeWFob28uY29t