 Hafsa Abdul Ghuffar
Hafsa Abdul Ghuffar Hyunho Noh
Hyunho Noh
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. , 14 June 2024
Sec. Porous Crystalline Networks
Volume 12 - 2024 | https://doi.org/10.3389/fchem.2024.1427536
This article is part of the Research Topic Retroconstruction of Porous Crystalline Networks for A Sustainable Future View all 4 articles
Interfacial charge transfer reactions involving cations and electrons are fundamental to (photo/electro) catalysis, energy storage, and beyond. Lithium-coupled electron transfer (LCET) at the electrode-electrolyte interfaces of lithium-ion batteries (LIBs) is a preeminent example to highlight the importance of charge transfer in modern-day society. The thermodynamics of LCET reactions define the minimal energy for charge/discharge of LIBs, and yet, these parameters are rarely available in the literature. Here, we demonstrate the successful incorporation of tungsten oxides (WOx) within a chemically stable Zr-based metal−organic framework (MOF), MOF-808. Cyclic voltammograms (CVs) of the composite, WOx@MOF-808, in Li+-containing acetonitrile (MeCN)-based electrolytes showed an irreversible, cathodic Faradaic feature that shifted in a Nernstian fashion with respect to the Li+ concentration, i.e., ∼59 mV/log [(Li+)]. The Nernstian dependence established 1:1 stoichiometry of Li+ and e−. Using the standard redox potential of Li+/0, the apparent free energy of lithiation of WOx@MOF-808 (ΔGapp,Li) was calculated to be −36 ± 1 kcal mol−1. ΔGapp,Li is an intrinsic parameter of WOx@MOF-808, and thus by deriving the similar reaction free energies of other metal oxides, their direct comparisons can be achieved. Implications of the reported measurements will be further contrasted to proton-coupled electron transfer (PCET) reactions on metal oxides.
Cation-coupled electron transfer reactions at electrode-electrolyte interfaces are critical to modern-day electrolyzers, (photo) electrocatalysts, batteries, and many others. These serve as the core technologies to shift the energy and chemical sectors away from fossil fuels to those that are more renewable, such as solar and wind energy (Adams et al., 2003; Peper and Mayer, 2019; Prather et al., 2023). Nearly all charge transfer reactions relevant to renewable energy involve interfacial electron transfer reactions that are coupled with cations. Perhaps the most seminal example of coupled charge transfer reaction occurs within lithium-ion batteries (LIBs). Electrons transferred between the anode (graphite) and the cathode (layered metal oxides) are coupled with lithium cations in the electrolyte. Eq. 1 below shows the lithium-coupled electron transfer (LCET) reaction at the metal oxide (Mn+Ox) cathode (Van Noorden, 2014; Kim et al., 2019; Manthiram, 2020).
In surface science, electrocatalysis, and energy storage literature, it is often implicitly assumed that the stoichiometry of the involved cation and the electron is equal, i.e., in Eq. 1, y = z (Poizot et al., 2002; Choi et al., 2003; Kim et al., 2019; Mayur et al., 2019; Manthiram, 2020). This is self-evident from the square scheme of an LCET reaction using metal oxide (Figure 1), as equimolar amounts of cation and electron prevent the formation of charged, and therefore energetically unfavorable species. Computational modeling of TiO2 clusters further elucidated an increase in the equilibrium constant (K) of LCET reaction by 108 upon the addition of a single electron (Zhang et al., 2012). However, for many metal oxides that undergo proton-coupled electron transfer (PCET) reactions, redox reactions can involve more than one proton per electron (Burke and Lyons, 1986; Dincă et al., 2010; Gambardella et al., 2011; Fleischmann et al., 2020; Mayer, 2023). Thus, we argue that the equal stoichiometry of LCET is also not guaranteed and cannot be assumed.
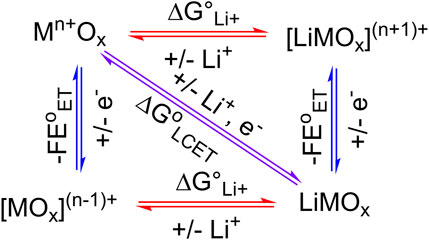
Figure 1. Square scheme of LCET. Here, 1:1 Li-to-electron stoichiometry is assumed.
The Li+-to-electron stoichiometry is fundamental in deriving the thermodynamics of the LCET reaction, which defines the minimal energy required to charge and discharge electrical energy in LIBs. This is conceptually analogous to the thermodynamic potential of an electrocatalytic reaction (Bard and Faulkner, 2001). Optimal electrocatalysts exhibit high catalytic activity at the thermodynamic potential (E
To this day, reports on LCET reaction thermodynamics remain rare in the literature, particularly when compared to those related to PCET reactions. Free energies of PCET reactions have long been examined both experimentally and computationally on molecular species, metals, and even binary and ternary material surfaces (Nørskov et al., 2005; Strmcnik et al., 2008; Seh et al., 2017; Wise et al., 2020; Agarwal et al., 2021; Fertig et al., 2021; Noh and Mayer, 2022; Fortunato et al., 2023; Nedzbala et al., 2024). A few reports related to LCET of cobalt, nickel, vanadium, tungsten, and many other oxides suggest that the reaction free energy (ΔG
Herein, we report the successful incorporation of tungsten oxides (WOx) that undergo LCET reactions within the pores of the Zr-based metal−organic framework, MOF-808 (the MOF-WOx composite here onwards will be referred to as WOx@MOF-808; see Figure 2 for the parent MOF structure) (Furukawa et al., 2014). MOF-808 and many other MOFs have been widely applied in the field of energy and environmental applications (Zhang et al., 2017; Zhao et al., 2020; Shi et al., 2024). Hydrated tungsten oxides are of particular interest as they have been demonstrated to undergo Faradaic reactions in the presence of protons, lithium, and other cations (He et al., 2016; Mitchell et al., 2017; Zheng et al., 2018; Mitchell et al., 2019). The chemical stability of Zr-based MOFs ensures that the porous MOF backbone remains intact throughout all measurements (Bai et al., 2016; Howarth et al., 2016; Shi et al., 2023). The electrochemical and thermochemical analysis of the MOF pore-confined WOx should be pivotal to understanding the fundamental role of the physical properties of electrodes on LCET thermodynamics, alluding to the design principles of energy storage materials.
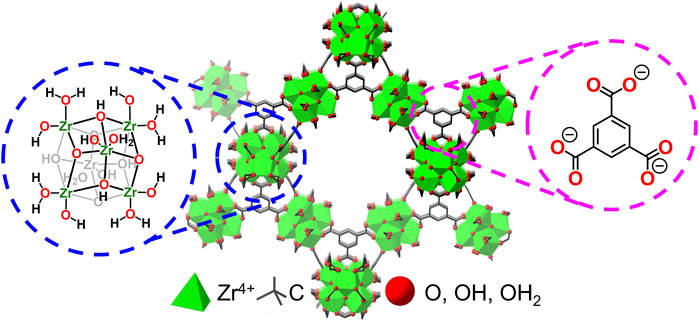
Figure 2. Crystal structure of Zr-based MOF, MOF-808, and its inorganic node and the organic linker.
Zr-based MOF-808 was synthesized and thermally activated according to the reported procedure (Liu et al., 2021). Subsequent incorporation of WOx within the MOF pores through acid precipitation described below is modified from that reported by Freedman (Freedman, 1959). Briefly, freshly synthesized Zr-MOF-808 (12 mg) was submerged into 1 mL of a pH 7-adjusted aqueous buffer consisting of 100 mM of 3-(N-morpholino)propanesulfonic acid (MOPS). Under vigorous stirring, 16.4 mg of Na2WO4
All electrochemical measurements of WOx@MOF-808 were measured in a MeCN-based electrolyte that contains LiClO4 with concentrations ranging between 25 and 250 mM. The total ionic strength of the electrolyte was kept consistent at 1 M using TBAClO4. Pt wire and Ag/Ag+ were used as the counter and pseudoreference electrodes, respectively. A small amount of ferrocene (Fc) was added at the end of each experiment to calibrate the measured electrochemical potential. Unless otherwise noted, all cyclic voltammograms (CVs) were measured at a scan rate (υ) of 100 mV/s.
N2-adsorption-desorption isotherm of WOx@MOF-808 exhibited a significant decrease in the N2 uptake, resulting in a decrease in the Brauner-Emmett-Teller (BET) area from 2,000 to 1,200 m2/g (Figure 3A). This decrease can be attributed to 1) the incorporation of WOx within otherwise vacant MOF pores or 2) an increase in molar mass. Pristine MOF-808 has a pore size of 18.8 Å; this pore decreased to 18.4 Å upon WOx incorporation. Post-synthetic WOx deposition also led to the formation of pores with diameters of ∼14 and 17 Å (Figure 3B). Scanning electron microscopy (SEM) images of MOF-808 and WOx@MOF-808 (Supplementary Figure S2) show that after WOx incorporation, the surfaces of the MOF crystallites are roughened, which may be due to surface WOx or due to the acidic tungstic acid present during the synthesis of WOx@MOF-808 (see Section 4.1 for more details). Energy dispersive X-ray spectroscopy (EDS) revealed that essentially all [WO4]2− were incorporated into the MOF network per Zr6 node, nearly doubling the molecular mass; the molar masses of MOF-808 and WOx@MOF-808 are 1,303 and at least 2,400 g/mol, respectively (Furukawa et al., 2014). The apparent decrease in N2 uptake can be ascribed to both factors. The powder X-ray diffraction pattern (PXRD) confirmed that the bulk crystallinity of the MOF backbone was retained during the WOx incorporation (Supplementary Figure S3).
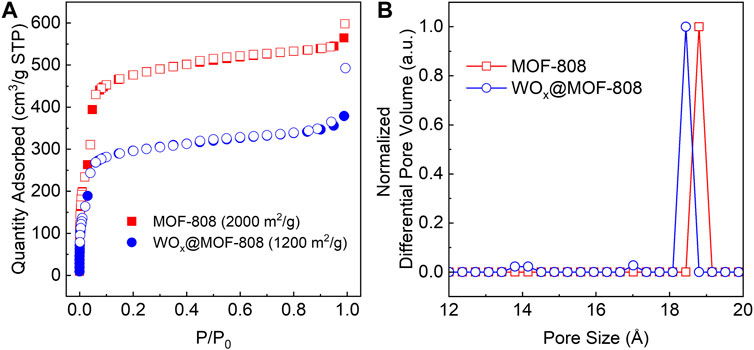
Figure 3. (A) N2-adsorption desorption isotherm and (B) DFT-calculated pore size distributions of pristine MOF-808 and WOx@MOF-808.
CVs of WOx@MOF-808 in LiClO4/TBAClO4-containing electrolyte showed two cathodic and one anodic features (Figure 4A). Amongst the three Faradaic features, the second cathodic feature (labeled B in Figure 4A) was ascribed to the LCET reaction of pore-confined WOx. The peak cathodic potential (Ep,c) scaled in a roughly Nernstian fashion with respect to the concentration of Li+ in the electrolyte {i.e., ca. −59 mV/log [(Li+)]; Figure 4B}. This feature was absent when CVs in 1 M TBAClO4 with no Li+ were measured. CVs measured with electrolytes containing Li+ beyond the range described above were quite distinct from others and hence were not considered (see Supplementary Figure S5 and the Discussion section for details). Nevertheless, within the reported concentration range, the Nernstian shift of the cathodic peak potentials indicates that this feature is associated with the LCET reaction. According to the Nernst equation, the ca. −59 mV/log [(Li+)] slope indicates a 1:1 stoichiometry of Li+ and e−; this stoichiometry is explicitly shown in Eqs 3, 4. We note these are essentially identical to Eqs 1, 2, but with the explicit notation of Li-to-electron stoichiometry and the redox-active metal oxide, WOx.
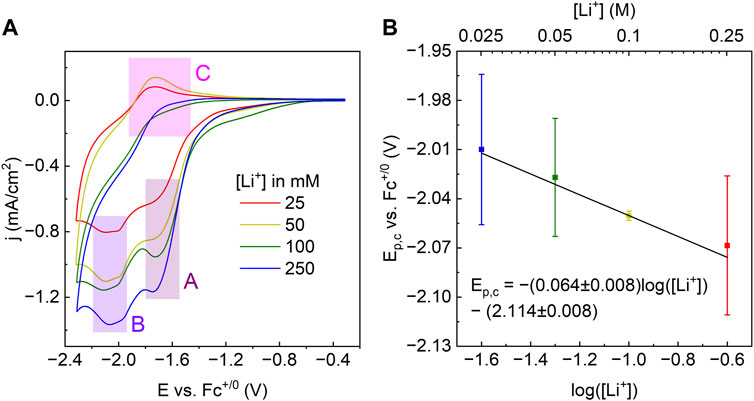
Figure 4. (A) CVs of WOx@MOF-808 in various concentrations of Li-ions. (B) Plot of Ep,c of feature labeled B in (A) against the log of [Li+].
The peak potential of the cathodic Faradaic feature prior to that of the LCET reaction (labeled A in Figure 4A) did not scale with the Li-ion concentration (Supplementary Figure S6). As shown in Supplementary Figure S8, CVs of pristine MOF-808 also exhibited a single cathodic feature with a peak potential closely resembling that of feature A. Because Zr-nodes within the MOF are redox-innocent, we have ascribed this feature to be due to the reduction of the BTC linker.
The anodic Faradaic feature observed in all CVs of WOx@MOF-808 resembles the anodic Faradaic feature observed when bulk WO3
Electrochemical treatments did not result in any changes to the PXRD pattern of the WOx@MOF-808 film, suggesting that the MOF backbone structure was retained. In contrast, the PXRD patterns of bulk WO3
Here onwards, we will focus on the Faradaic feature with peak potentials scaling in a roughly Nernstian fashion with respect to the Li-ion concentration.
CVs of WOx@MOF-808 in the electrolyte containing 100 mM LiClO4 and 900 mM TBAClO4 were measured at scan rates between 10 and 100 mV/s (Supplementary Figure S7). The dependence of the peak currents vs. the measured scan rates is indicative of the LCET mechanism. When the logarithms of the two values linearly scale with a slope of 0.5, the LCET reaction rate is diffusion-controlled, where the “diffusion” of electrons (and in this case equimolar amounts of Li+) through the MOF lattice limits the overall reaction rate. When the slope is instead one, the reaction is kinetically controlled (Bard and Faulkner, 2001). As shown in Supplementary Figure S7, the logarithms of the peak cathodic current density (jp,c) values scaled against that of the log of the measured scan rate with a slope of ∼0.44, suggesting that the LCET reaction rate of WOx@MOF-808 is likely controlled by the diffusion of Li+ and e− within the pore-confined WOx.
Compared to peak potentials, current densities had significantly larger sample-to-sample variation (see Supplementary Figure S4). These observations have been previously reported for MOF-based electrodes yielded from a simple drop-casting method (Chen et al., 2021). Error analysis due to these inconsistencies can be found in the Discussion section. Subtraction of background capacitive current led to somewhat more consistent jp,c, as described above in the scan rate dependence studies. The LCET Faradaic feature after the background subtraction was used to estimate the total amount of electroactive WOx to be 11 ± 6 nmol/cm2. With the separate 1H NMR measurement determining the total amount of deposited WOx@MOF-808, ca. 0.2% of total WOx on the electrode was found to be electroactive (see the SI for the details).
An ideal Faradaic feature exhibits a full-width-at-half-maximum (FWHM) value of 90 mV (Laviron, 1974; Laviron, 1979). At all concentrations measured in this work, the LCET Faradaic feature exhibited FWHM values of 120–150 mV (Supplementary Table S1). This contrasts with the wide FWHM values observed in all CVs of WO3
MOF-808 and many other Zr-based MOFs have high chemical stability over a wide range of pH (Bai et al., 2016; Howarth et al., 2016; Shi et al., 2023). Yet, our initial attempts in addition of more than the reported equivalence of Na2WO4
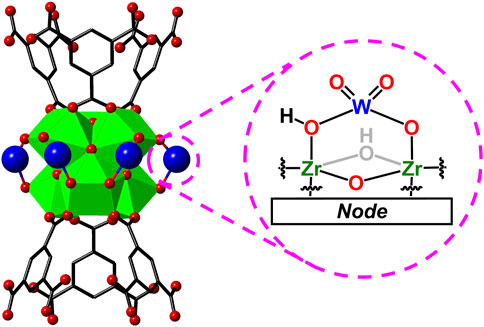
Figure 5. Proposed structure of tungstate on MOF-808 node, prior to acid condensation.
WOx@MOF-808 exhibited multiple Faradaic features; the peak potentials of each feature had a unique dependence with respect to the concentration of Li-ions within the electrolyte. In this report, we focused on the cathodic feature that scaled in roughly Nernstian fashion with an order of magnitude change in Li-ion concentration {i.e., ∼59 mV/log [(Li+)]}. This established the Li-to-electron stoichiometry involved in this reduction reaction to be 1:1 (Bard and Faulkner, 2001).
FWHM of a Faradaic feature is indicative of the chemical nature of the adsorbates formed and released during the heterogeneous redox reaction. An ideal FWHM value of 90 mV suggests that the adsorbates are chemically identical and do not laterally interact with each other (Gileadi, 1967; Laviron, 1974; Laviron, 1979). CVs of WOx@MOF-808 measured in 25–100 mM Li-ion concentrations revealed that the FWHM values of LCET Faradaic features were around 120 mV, which is quite similar to the ideal value. Scan rate dependence studies suggested that the observed LCET reaction is diffusion-controlled. Perhaps, the Li+/e− pairs that are ‘adsorbed’ within the MOF pore-confined WOx can diffuse to a site until the lateral interactions between the two sites are negligible. An increase in FWHM to 150 mV upon an increase in Li-ion concentration further supports this mechanism. Slight deviations from 90 mV may also arise from structural heterogeneity.
In general, CVs of tungsten and many other metal oxides that undergo LCET reactions exhibit multiple Faradaic features; some of them are reversible, while others are irreversible (Ding et al., 2014; Wu and Yao, 2017; Zhang et al., 2020; Tao et al., 2023; Wang et al., 2024; Zheng et al., 2024). This can be, at least in part, attributed to the bulk structural changes of metal oxides upon insertion of Li+ and other cations. For example, bulk WO3 in a monoclinic phase undergoes a dynamic structural transformation where the symmetry increases to orthorhombic, tetrahedral, and finally to cubic phase with increasing amounts of proton/lithium cations inserted within its lattice structure. Excess intercalation leads to an irreversible amorphization (He et al., 2016; Wang et al., 2016). We have also confirmed the irreversible structural change induced by Li-ion insertion by examining the PXRD pattern of WO3
The establishment of the stoichiometry of the reactants and products is fundamental to calculations of the electronic structure of different lithiated phases, and as demonstrated in the next section, thermochemical analysis (Mayer, 2023). LCET stoichiometry can be electrochemically established by measuring CVs in various concentrations of Li cations. This is a direct application of the approach in the PCET literature (Noh and Mayer, 2022). The CV-derived redox potentials at various proton activity yields the so-called Pourbaix diagram which can be used to determine the free energy of H-atom transfer (Pourbaix, 1974; Pourbaix, 1990). While the exact redox potentials of the MOF-confined WOx cannot be determined due to its irreversibility, Figure 4B is a step toward a Pourbaix diagram for LCET reactions. For estimations of the free energy of LCET reactions and the associated errors, see Sections 4.4, 4.5.
CVs of metal oxides in various Li-ion concentrations required for the Pourbaix diagram are not reported often. This may be due to the complex phase transformation of many metal oxides as noted above. The lack of “well-defined” Faradaic features that can be ascribed to LCET further precludes this analysis; indeed, our attempts to measure LCET with bulk WO3
Nikitina et al. (2017) reported the well-defined, reversible LCET Faradaic features of LiMn2O4 to shift by 75 and 163 mV per unit change in log [(Li+)]. Using the Nernst equation (Eq. 2), this >59 mV shift suggests that more than one Li ions are involved per electron transfer. In the PCET literature, this is often referred to as the “super-Nernstian” dependence (Fleischmann et al., 2020). The additional positive charge has been speculated to be compensated by coupling the charge transfer reactions with anions within the electrolyte (Birss et al., 1991; Mayer, 2023). This super-Nernstian dependence is particularly prevalent for layered double hydroxides (LDHs) and other forms of layered metal oxides, which can intercalate ions (Burke and Lyons, 1986; Dincă et al., 2010; Lyons et al., 2012). The overall process, however, is quite complex involving partial or complete de-solvation of ions, intra-lattice diffusion, and others (Augustyn, 2017; Xu et al., 2022; Hu et al., 2023). In general, there lacks a fundamental theory that can correlate this super-Nernstian behavior to charge transfer thermochemistry. To the best of our knowledge, there are only a handful of reports explicitly determining the 1:1 cation-to-electron stoichiometry other than those in the PCET reactions; see the following references (Lyon and Hupp, 1995; Valdez et al., 2018; Saouma et al., 2019).
Determination of thermochemistry, by definition, requires the chemical process to be at standard state and thermodynamically reversible. LCET reaction of WOx@MOF-808 reported here does not strictly follow these requirements.
Measurements beyond 250 mM in Li-ion concentrations proved unsuccessful due to the significant change in the Faradaic features. The apparent diffusion coefficient (Dapp) of Li-ions within the layered metal oxides significantly decreases at high concentrations, leading to a change in Faradaic features. These are typically observed at concentrations ≥1 M for bulk metal oxides (Nikitina et al., 2017; Finney et al., 2021). In the reported system, the Li-ions would have to de-solvate (at least partially) to diffuse into the MOF pores, even before reaching the redox-active sites (Sogawa et al., 2019). This may be the reason why 250 mM was the limit for WOx@MOF-808.
While the common reference electrode for Li-ion batteries and other LCET-related systems is Li metal, its potentials are unstable, varying as high as 0.5 V over the course of the reaction (Cengiz et al., 2021). Instead, we relied on the Fc+/0 redox couple as our reference in non-aqueous solvents (such as MeCN) due to their superior stability (Gagne et al., 1980; Gritzner and Kuta, 1984). Nevertheless, the standard potential of Li+/0 can be used to determine the free energy of lithiation, i.e., the addition of a “Li-atom.” In essence, this is treating the [WO−
All values shown in Figure 6 used for the estimation of ΔGapp,Li are at standard state [i.e., (Li+) = 1 M and 298 K in temperature]. The estimation of ΔGapp,Li requires the difference in solvation free energies of Li+ in H2O vs. MeCN (ΔG
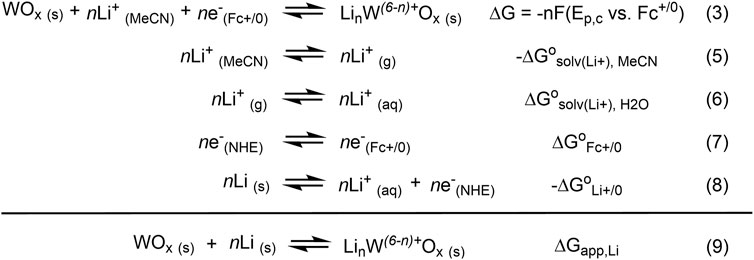
Figure 6. Scheme illustrating the derivation of ΔGapp,Li.
To benchmark this value against other metal oxides that can undergo a net Li-atom transfer, we estimated the standard free energy of lithiation (ΔG
The above comparison highlights the power of using ΔGapp,Li/ΔG
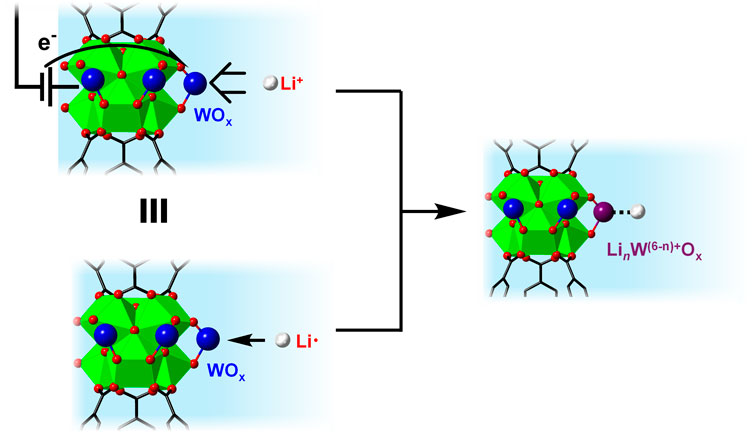
Figure 7. Schematic illustration showing the thermochemical analogy between LCET reaction vs. Li-atom addition on WOx within MOF-808.
We conclude this section by discussing the associated errors in estimating the free energy of LCET.
Thermodynamic reversibility is strictly required for the determination of ΔG
Between different samples, Ep,c values were consistent with standard errors <1 kcal mol−1. jp,c values upon background subtraction were also somewhat consistent. However, the overall current had a large sample-to-sample variation. This large variation in currents has been previously observed for MOF-based electrodes yielded from a simple drop-casting method (Chen et al., 2021). While additions of conductive materials like carbon black and polymeric binders usually improve the consistency, these can convolute the thermochemical analysis. Carbon black is essentially the anode of LIBs and polymeric binders can slow the diffusion (Zhao et al., 2011). The observed inconsistency may also arise from the inhomogeneous distribution of hydrated WO3 within MOF-808 yielded after the acid condensation. Thus, for this report, we primarily focus on the thermodynamics of LCET and not its kinetics. Structural determination of the MOF-confined WO3 is beyond the scope of this work.
Redox-active WOx was successfully incorporated into the Zr-based MOF, MOF-808. In Li-containing electrolytes, CVs of the composite, WOx@MOF-808 exhibited multiple cathodic and anodic features, common for bulk metal oxides under similar electrochemical conditions. One of the reductive features scaled in a close-to-Nernstian fashion with respect to log [(Li+)], suggesting that this reductive process involves one Li cation per every electron transferred. Using this established stoichiometry, we estimated the free energy of lithiation to be roughly −36 kcal mol−1, which was comparable to the estimated values of other reported metal oxides, though the range was large (90 kcal mol−1).
Deposition of WOx within MOF-808 resulted in an ancillary benefit of yielding a more “well-behaved” electrochemical system for thermochemical analysis. Faradaic features of bulk tungsten oxides and many other metal oxides do not exhibit Nernstian dependence with respect to Li-ion concentrations altogether precluding derivation of LCET thermochemistry. The reported success in estimating ΔG
We emphasize the robustness of the parameter, ΔG
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
HG: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Writing–original draft, Writing–review and editing. HN: Funding acquisition, Methodology, Project administration, Supervision, Validation, Writing–original draft, Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was supported by the University of Oklahoma Startup Fund.
HG acknowledges the support of the Fulbright Scholarship. Financial support for publication was provided by the University of Oklahoma Libraries Open Access Fund. PXRD and microscopy data collections were performed at the Samuel Roberts Noble Microscopy Laboratory, an OU core facility supported by the Vice President for Research and Partnerships. HG and HN thank Prof. Daniel T. Glatzhofer for his help in providing many chemicals for this project.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2024.1427536/full#supplementary-material
Adams, D. M., Brus, L., Chidsey, C. E. D., Creager, S., Creutz, C., Kagan, C. R., et al. (2003). Charge transfer on the nanoscale: current status. J. Phys. Chem. B 107, 6668–6697. doi:10.1021/jp0268462
Agarwal, R. G., Coste, S. C., Groff, B. D., Heuer, A. M., Noh, H., Parada, G. A., et al. (2021). Free energies of proton-coupled electron transfer reagents and their applications. Chem. Rev. 122, 1–49. doi:10.1021/acs.chemrev.1c00521
Augustyn, V. (2017). Tuning the interlayer of transition metal oxides for electrochemical energy storage. J. Mater. Res. 32, 2–15. doi:10.1557/jmr.2016.337
Bai, Y., Dou, Y., Xie, L.-H., Rutledge, W., Li, J.-R., and Zhou, H.-C. (2016). Zr-based metal–organic frameworks: design, synthesis, structure, and applications. Chem. Soc. Rev. 45, 2327–2367. doi:10.1039/c5cs00837a
Bard, A. J., and Faulkner, L. R. (2001) Electrochemical methods: fundamentals and applications. New York, NY: John Wiley and Sons Inc.
Birss, V. I., Elzanowska, H., and Gottesfeld, S. (1991). Quartz crystal microbalance measurements during oxidation/reduction of hydrous Ir oxide electrodes. J. Electroanal. Chem. Interfacial Electrochem. 318, 327–333. doi:10.1016/0022-0728(91)85314-f
Burke, L. D., and Lyons, M. E. G. (1986). “Electrochemistry of hydrous metal oxide films,” in Modern aspects of Electrochemistry. Editors C. G. Vayenas, R. E. White, and M. E. Gamboa-Aldeco (New York, NY: Springer).
Carvalho, N. F., and Pliego, J. R. (2015). Cluster-continuum quasichemical theory calculation of the lithium ion solvation in water, acetonitrile and dimethyl sulfoxide: an absolute single-ion solvation free energy scale. Phys. Chem. Chem. Phys. 17, 26745–26755. doi:10.1039/c5cp03798k
Cengiz, E. C., Rizell, J., Sadd, M., Matic, A., and Mozhzhukhina, N. (2021). Review—reference electrodes in Li-ion and next generation batteries: correct potential assessment, applications and practices. J. Electrochem. Soc. 168, 120539. doi:10.1149/1945-7111/ac429b
Chen, K., Downes, C. A., Schneider, E., Goodpaster, J. D., and Marinescu, S. C. (2021). Improving and understanding the hydrogen evolving activity of a cobalt Dithiolene metal–organic framework. ACS Appl. Mater. Interfaces 13, 16384–16395. doi:10.1021/acsami.1c01727
Chen, Y., Ahn, S., Mian, M. R., Wang, X., Ma, Q., Son, F. A., et al. (2022). Modulating chemical environments of metal–organic framework-supported molybdenum(VI) catalysts for insights into the structure–activity relationship in cyclohexene epoxidation. J. Am. Chem. Soc. 144, 3554–3563. doi:10.1021/jacs.1c12421
Choi, H. C., Jung, Y. M., Noda, I., and Kim, S. B. (2003). A study of the mechanism of the electrochemical reaction of lithium with CoO by two-dimensional soft X-ray absorption spectroscopy (2D XAS), 2D Raman, and 2D heterospectral XAS−Raman correlation analysis. J. Phys. Chem. B 107, 5806–5811. doi:10.1021/jp030438w
Dahéron, L., Dedryvère, R., Martinez, H., Ménétrier, M., Denage, C., Delmas, C., et al. (2008). Electron transfer mechanisms upon lithium deintercalation from LiCoO2 to CoO2 investigated by XPS. Chem. Mater. 20, 583–590. doi:10.1021/cm702546s
Dincă, M., Surendranath, Y., and Nocera, D. G. (2010). Nickel-borate oxygen-evolving catalyst that functions under benign conditions. Proc. Natl. Acad. Sci. 107, 10337–10341. doi:10.1073/pnas.1001859107
Ding, L., He, S., Miao, S., Jorgensen, M. R., Leubner, S., Yan, C., et al. (2014). Ultrasmall SnO2 nanocrystals: hot-bubbling synthesis, encapsulation in carbon layers and applications in high capacity Li-ion storage. Sci. Rep. 4, 4647. doi:10.1038/srep04647
Feng, R., Vasiliu, M., Peterson, K. A., and Dixon, D. A. (2017). Acidity of M(VI)O2(OH)2 for M = group 6, 16, and U as central atoms. J. Phys. Chem. A 121, 1041–1050. doi:10.1021/acs.jpca.6b11889
Fertig, A. A., Brennessel, W. W., McKone, J. R., and Matson, E. M. (2021). Concerted multiproton–multielectron transfer for the reduction of O2 to H2O with a polyoxovanadate cluster. J. Am. Chem. Soc. 143, 15756–15768. doi:10.1021/jacs.1c07076
Finney, A. R., McPherson, I. J., Unwin, P. R., and Salvalaglio, M. (2021). Electrochemistry, ion adsorption and dynamics in the double layer: a study of NaCl(aq) on graphite. Chem. Sci. 12, 11166–11180. doi:10.1039/d1sc02289j
Fleischmann, S., Mitchell, J. B., Wang, R., Zhan, C., Jiang, D.-e., Presser, V., et al. (2020). Pseudocapacitance: from fundamental understanding to high power energy storage materials. Chem. Rev. 120, 6738–6782. doi:10.1021/acs.chemrev.0c00170
Fortunato, J., Shin, Y. K., Spencer, M. A., van Duin, A. C. T., and Augustyn, V. (2023). Choice of electrolyte impacts the selectivity of proton-coupled electrochemical reactions on hydrogen titanate. J. Phys. Chem. C 127, 11810–11821. doi:10.1021/acs.jpcc.3c01057
Freedman, M. L. (1959). The tungstic acids. J. Am. Chem. Soc. 81, 3834–3839. doi:10.1021/ja01524a009
Furukawa, H., Gándara, F., Zhang, Y.-B., Jiang, J., Queen, W. L., Hudson, M. R., et al. (2014). Water adsorption in porous metal–organic frameworks and related materials. J. Am. Chem. Soc. 136, 4369–4381. doi:10.1021/ja500330a
Gagne, R. R., Koval, C. A., and Lisensky, G. C. (1980). Ferrocene as an internal standard for electrochemical measurements. Inorg. Chem. 19, 2854–2855. doi:10.1021/ic50211a080
Gambardella, A. A., Bjorge, N. S., Alspaugh, V. K., and Murray, R. W. (2011). Voltammetry of diffusing 2 nm iridium oxide nanoparticles. J. Phys. Chem. C 115, 21659–21665. doi:10.1021/jp206987z
Gileadi, E. (1967). “Adsorption in Electrochemistry,” in Electrosorption. Editor E. Gileadi (Boston, MA: Springer).
Gritzner, G., and Kuta, J. (1984). Recommendations on reporting electrode potentials in nonaqueous solvents (Recommendations 1983). Pure Appl. Chem. 56, 461–466. doi:10.1351/pac198456040461
He, Y., Gu, M., Xiao, H., Luo, L., Shao, Y., Gao, F., et al. (2016). Atomistic conversion reaction mechanism of WO3 in secondary ion batteries of Li, Na, and Ca. Angew. Chem. Int. Ed. 55, 6244–6247. doi:10.1002/anie.201601542
Howarth, A. J., Liu, Y., Li, P., Li, Z., Wang, T. C., Hupp, J. T., et al. (2016). Chemical, thermal and mechanical stabilities of metal–organic frameworks. Nat. Rev. Mater. 1, 15018. doi:10.1038/natrevmats.2015.18
Hu, H., Li, J., Zhang, Q., Ding, G., Liu, J., Dong, Y., et al. (2023). Non-concentrated electrolyte with weak anion coordination enables low Li-ion desolvation energy for low-temperature lithium batteries. Chem. Eng. J. 457, 141273. doi:10.1016/j.cej.2023.141273
Ingram, Z. J., Lander, C. W., Oliver, M. C., Altınçekiç, N. G., Huang, L., Shao, Y., et al. (2024). Hydrogen-atom binding energy of structurally well-defined Cerium oxide nodes at the Metal−Organic framework-liquid interfaces. Journal of Physical Chemistry C, doi:10.1021/acs.jpcc.4c02409
Itkis, D., Cavallo, L., Yashina, L. V., and Minenkov, Y. (2021). Ambiguities in solvation free energies from cluster-continuum quasichemical theory: lithium cation in protic and aprotic solvents. Phys. Chem. Chem. Phys. 23, 16077–16088. doi:10.1039/d1cp01454d
Kerisit, S., Rosso, K. M., Yang, Z., and Liu, J. (2009). Dynamics of coupled lithium/electron diffusion in TiO2 polymorphs. J. Phys. Chem. C 113, 20998–21007. doi:10.1021/jp9064517
Kim, T., Song, W., Son, D.-Y., Ono, L. K., and Qi, Y. (2019). Lithium-ion batteries: outlook on present, future, and hybridized technologies. J. Mater. Chem. A 7, 2942–2964. doi:10.1039/c8ta10513h
Kuppan, S., Xu, Y., Liu, Y., and Chen, G. (2017). Phase transformation mechanism in lithium manganese nickel oxide revealed by single-crystal hard X-ray microscopy. Nat. Commun. 8, 14309. doi:10.1038/ncomms14309
Laviron, E. (1974). Surface linear potential sweep voltammetry: equation of the peaks for a reversible reaction when interactions between the adsorbed molecules are taken into account. J. Electroanal. Chem. Interfacial Electrochem. 52, 395–402. doi:10.1016/0368-1874(74)85054-9
Laviron, E. (1979). The use of linear potential sweep voltammetry and of a.c. voltammetry for the study of the surface electrochemical reaction of strongly adsorbed systems and of redox modified electrodes. J. Electroanal. Chem. Interfacial Electrochem. 100, 263–270. doi:10.1016/s0022-0728(79)80167-9
Lin, F., Nordlund, D., Weng, T.-C., Zhu, Y., Ban, C., Richards, R. M., et al. (2014). Phase evolution for conversion reaction electrodes in lithium-ion batteries. Nat. Commun. 5, 3358. doi:10.1038/ncomms4358
Liu, H., Zhu, Z., Huang, J., He, X., Chen, Y., Zhang, R., et al. (2019). Elucidating the limit of Li insertion into the spinel Li4Ti5O12. ACS Mater. Lett. 1, 96–102. doi:10.1021/acsmaterialslett.9b00099
Liu, X., Kirlikovali, K. O., Chen, Z., Ma, K., Idrees, K. B., Cao, R., et al. (2021). Small molecules, Big effects: tuning adsorption and catalytic properties of metal–organic frameworks. Chem. Mater. 33, 1444–1454. doi:10.1021/acs.chemmater.0c04675
Lyon, L. A., and Hupp, J. T. (1995). Energetics of semiconductor electrode/solution interfaces: EQCM evidence for charge-compensating cation adsorption and intercalation during accumulation layer formation in the titanium dioxide/acetonitrile system. J. Phys. Chem. 99, 15718–15720. doi:10.1021/j100043a005
Lyons, M. E. G., Doyle, R. L., Godwin, I., O’Brien, M., and Russell, L. (2012). Hydrous nickel oxide: redox switching and the oxygen evolution reaction in aqueous alkaline solution. J. Electrochem. Soc. 159, H932–H944. doi:10.1149/2.078212jes
Manthiram, A. (2020). A reflection on lithium-ion battery cathode chemistry. Nat. Commun. 11, 1550. doi:10.1038/s41467-020-15355-0
Mayer, J. M. (2004). Proton-coupled electron transfer: a reaction chemist's view. Annu. Rev. Phys. Chem. 55, 363–390. doi:10.1146/annurev.physchem.55.091602.094446
Mayer, J. M. (2023). Bonds over electrons: proton coupled electron transfer at solid–solution interfaces. J. Am. Chem. Soc. 145, 7050–7064. doi:10.1021/jacs.2c10212
Mayur, M., Yagci, M. C., Carelli, S., Margulies, P., Velten, D., and Bessler, W. G. (2019). Identification of stoichiometric and microstructural parameters of a lithium-ion cell with blend electrode. Phys. Chem. Chem. Phys. 21, 23672–23684. doi:10.1039/c9cp04262h
Medford, A. J., Vojvodic, A., Hummelshøj, J. S., Voss, J., Abild-Pedersen, F., Studt, F., et al. (2015). From the Sabatier principle to a predictive theory of transition-metal heterogeneous catalysis. J. Catal. 328, 36–42. doi:10.1016/j.jcat.2014.12.033
Mitchell, J. B., Geise, N. R., Paterson, A. R., Osti, N. C., Sun, Y., Fleischmann, S., et al. (2019). Confined interlayer water promotes structural stability for high-rate electrochemical proton intercalation in tungsten oxide hydrates. ACS Energy Lett. 4, 2805–2812. doi:10.1021/acsenergylett.9b02040
Mitchell, J. B., Lo, W. C., Genc, A., LeBeau, J., and Augustyn, V. (2017). Transition from battery to pseudocapacitor behavior via structural water in tungsten oxide. Chem. Mater. 29, 3928–3937. doi:10.1021/acs.chemmater.6b05485
Myeong, S., Cho, W., Jin, W., Hwang, J., Yoon, M., Yoo, Y., et al. (2018). Understanding voltage decay in lithium-excess layered cathode materials through oxygen-centred structural arrangement. Nat. Commun. 9, 3285. doi:10.1038/s41467-018-05802-4
Nedzbala, H. S., Westbroek, D., Margavio, H. R. M., Yang, H., Noh, H., Magpantay, S. V., et al. (2024). Photoelectrochemical proton-coupled electron transfer of TiO2 thin films on silicon. J. Am. Chem. Soc. 146, 10559–10572. doi:10.1021/jacs.4c00014
Ng, B., Peng, X., Faegh, E., and Mustain, W. E. (2020). Using nanoconfinement to inhibit the degradation pathways of conversion-metal oxide anodes for highly stable fast-charging Li-ion batteries. J. Mater. Chem. A 8, 2712–2727. doi:10.1039/c9ta11708c
Nikitina, V. A., Zakharkin, M. V., Vassiliev, S. Y., Yashina, L. V., Antipov, E. V., and Stevenson, K. J. (2017). Lithium ion coupled electron-transfer rates in superconcentrated electrolytes: exploring the bottlenecks for fast charge-transfer rates with LiMn2O4 cathode materials. Langmuir 33, 9378–9389. doi:10.1021/acs.langmuir.7b01016
Nocera, D. G. (2022). Proton-coupled electron transfer: the engine of energy conversion and storage. J. Am. Chem. Soc. 144, 1069–1081. doi:10.1021/jacs.1c10444
Noh, H., Cui, Y., Peters, A. W., Pahls, D. R., Ortuño, M. A., Vermeulen, N. A., et al. (2016). An exceptionally stable metal–organic framework supported molybdenum(VI) oxide catalyst for cyclohexene epoxidation. J. Am. Chem. Soc. 138, 14720–14726. doi:10.1021/jacs.6b08898
Noh, H., and Mayer, J. M. (2022). Medium-independent hydrogen atom binding isotherms of nickel oxide electrodes. Chem 8, 3324–3345. doi:10.1016/j.chempr.2022.08.018
Nørskov, J. K., Bligaard, T., Logadottir, A., Kitchin, J. R., Chen, J. G., Pandelov, S., et al. (2005). Trends in the exchange current for hydrogen evolution. J. Electrochem. Soc. 152, J23–J26. doi:10.1149/1.1856988
Okubo, M., Hosono, E., Kim, J., Enomoto, M., Kojima, N., Kudo, T., et al. (2007). Nanosize effect on high-rate Li-ion intercalation in LiCoO2 electrode. J. Am. Chem. Soc. 129, 7444–7452. doi:10.1021/ja0681927
Pan, A., Zhang, J.-G., Nie, Z., Cao, G., Arey, B. W., Li, G., et al. (2010). Facile synthesized nanorod structured vanadium pentoxide for high-rate lithium batteries. J. Mater. Chem. 20, 9193–9199. doi:10.1039/c0jm01306d
Pegis, M. L., Roberts, J. A. S., Wasylenko, D. J., Mader, E. A., Appel, A. M., and Mayer, J. M. (2015). Standard reduction potentials for oxygen and carbon Dioxide couples in acetonitrile and N,N-dimethylformamide. Inorg. Chem. 54, 11883–11888. doi:10.1021/acs.inorgchem.5b02136
Peper, J. L., and Mayer, J. M. (2019). Manifesto on the thermochemistry of nanoscale redox reactions for energy conversion. ACS Energy Lett. 4, 866–872. doi:10.1021/acsenergylett.9b00019
Poizot, P., Laruelle, S., Grugeon, S., and Tarascon, J. M. (2002). Rationalization of the low-potential reactivity of 3d-metal-based inorganic compounds toward Li. J. Electrochem. Soc. 149, A1212. doi:10.1149/1.1497981
Pourbaix, M. (1974) Atlas of electrochemical equilibria in aqueous solution. Houston, TX: National Association of Corrosion Engineers.
Pourbaix, M. (1990). Thermodynamics and corrosion. Corros. Sci. 30, 963–988. doi:10.1016/0010-938x(90)90205-j
Prather, K. V., Stoffel, J. T., and Tsui, E. Y. (2023). Redox reactions at colloidal semiconductor nanocrystal surfaces. Chem. Mater. 35, 3386–3403. doi:10.1021/acs.chemmater.3c00481
Qian, D., Hinuma, Y., Chen, H., Du, L.-S., Carroll, K. J., Ceder, G., et al. (2012). Electronic spin transition in nanosize stoichiometric lithium cobalt oxide. J. Am. Chem. Soc. 134, 6096–6099. doi:10.1021/ja300868e
Saouma, C. T., Tsou, C.-C., Richard, S., Ameloot, R., Vermoortele, F., Smolders, S., et al. (2019). Sodium-coupled electron transfer reactivity of metal–organic frameworks containing titanium clusters: the importance of cations in redox chemistry. Chem. Sci. 10, 1322–1331. doi:10.1039/c8sc04138e
Seh, Z. W., Kibsgaard, J., Dickens, C. F., Chorkendorff, I., Nørskov, J. K., and Jaramillo, T. F. (2017). Combining theory and experiment in electrocatalysis: insights into materials design. Science 355, eaad4998. doi:10.1126/science.aad4998
Shi, L., Kirlikovali, K. O., Chen, Z., and Farha, O. K. (2024). Metal-organic frameworks for water vapor adsorption. Chem 10, 484–503. doi:10.1016/j.chempr.2023.09.005
Shi, L., Yang, Z., Sha, F., and Chen, Z. (2023). Design, synthesis and applications of functional zirconium-based metal-organic frameworks. Sci. China Chem. 66, 3383–3397. doi:10.1007/s11426-023-1809-8
Sogawa, M., Sawayama, S., Han, J., Satou, C., Ohara, K., Matsugami, M., et al. (2019). Role of solvent size in ordered ionic structure formation in concentrated electrolytes for lithium-ion batteries. J. Phys. Chem. C 123, 8699–8708. doi:10.1021/acs.jpcc.9b01038
Strmcnik, D., Tripkovic, D., van der Vliet, D., Stamenkovic, V., and Marković, N. M. (2008). Adsorption of hydrogen on Pt(111) and Pt(100) surfaces and its role in the HOR. Electrochem. Commun. 10, 1602–1605. doi:10.1016/j.elecom.2008.08.019
Tao, B., McPherson, I. J., Daviddi, E., Bentley, C. L., and Unwin, P. R. (2023). Multiscale Electrochemistry of lithium manganese oxide (LiMn2O4): from single particles to ensembles and degrees of electrolyte wetting. ACS Sustain. Chem. Eng. 11, 1459–1471. doi:10.1021/acssuschemeng.2c06075
Valdez, C. N., Delley, M. F., and Mayer, J. M. (2018). Cation effects on the reduction of colloidal ZnO nanocrystals. J. Am. Chem. Soc. 140, 8924–8933. doi:10.1021/jacs.8b05144
Van Noorden, R. (2014). The rechargeable revolution: a better battery. Nature 507, 26–28. doi:10.1038/507026a
Wang, L., Mukherjee, A., Kuo, C.-Y., Chakrabarty, S., Yemini, R., Dameron, A. A., et al. (2024). High-energy all-solid-state lithium batteries enabled by Co-free LiNiO2 cathodes with robust outside-in structures. Nat. Nanotechnol. 19, 208–218. doi:10.1038/s41565-023-01519-8
Wang, X., Hou, Y., Zhu, Y., Wu, Y., and Holze, R. (2013). An aqueous rechargeable lithium battery using coated Li metal as anode. Sci. Rep. 3, 1401. doi:10.1038/srep01401
Wang, Z., He, Y., Gu, M., Du, Y., Mao, S. X., and Wang, C. (2016). Electron transfer governed crystal transformation of tungsten trioxide upon Li ions intercalation. ACS Appl. Mater. Interfaces 8, 24567–24572. doi:10.1021/acsami.6b06581
Wise, C. F., Agarwal, R. G., and Mayer, J. M. (2020). Determining proton-coupled standard potentials and X–H bond dissociation free energies in nonaqueous solvents using open-circuit potential measurements. J. Am. Chem. Soc. 142, 10681–10691. doi:10.1021/jacs.0c01032
Wu, X., and Yao, S. (2017). Flexible electrode materials based on WO3 nanotube bundles for high performance energy storage devices. Nano Energy 42, 143–150. doi:10.1016/j.nanoen.2017.10.058
Xu, J., Liu, K., Khan, M. A., Wang, H., He, T., Zhao, H., et al. (2022). Sub-zero temperature electrolytes for lithium-sulfur batteries: functional mechanisms, challenges and perspectives. Chem. Eng. J. 443, 136637. doi:10.1016/j.cej.2022.136637
Zhang, H., Nai, J., Yu, L., and Lou, X. W. (2017). Metal-organic-framework-based materials as platforms for renewable energy and environmental applications. Joule 1, 77–107. doi:10.1016/j.joule.2017.08.008
Zhang, J., Hughes, T. F., Steigerwald, M., Brus, L., and Friesner, R. A. (2012). Realistic cluster modeling of electron transport and trapping in solvated TiO2 nanoparticles. J. Am. Chem. Soc. 134, 12028–12042. doi:10.1021/ja3013787
Zhang, S., Wang, G., Wang, B., Wang, J., Bai, J., and Wang, H. (2020). 3D carbon nanotube network bridged hetero-structured Ni-Fe-S nanocubes toward high-performance lithium, sodium, and potassium storage. Adv. Funct. Mater. 30, 2001592. doi:10.1002/adfm.202001592
Zhao, Q., Majsztrik, P., and Benziger, J. (2011). Diffusion and interfacial transport of water in nafion. J. Phys. Chem. B 115, 2717–2727. doi:10.1021/jp1112125
Zhao, R., Wu, Y., Liang, Z., Gao, L., Xia, W., Zhao, Y., et al. (2020). Metal–organic frameworks for solid-state electrolytes. Energy and Environ. Sci. 13, 2386–2403. doi:10.1039/d0ee00153h
Zheng, J., Xia, R., Yaqoob, N., Kaghazchi, P., ten Elshof, J. E., and Huijben, M. (2024). Simultaneous enhancement of lithium transfer kinetics and structural stability in dual-phase TiO2 electrodes by ruthenium doping. ACS Appl. Mater. Interfaces 16, 8616–8626. doi:10.1021/acsami.3c15122
Keywords: metal-organic framework, Li-coupled electron transfer, Li-ion battery, electrochemistry, thermochemistry
Citation: Ghuffar HA and Noh H (2024) Lithium-coupled electron transfer reactions of nano-confined WOx within Zr-based metal–organic framework. Front. Chem. 12:1427536. doi: 10.3389/fchem.2024.1427536
Received: 03 May 2024; Accepted: 23 May 2024;
Published: 14 June 2024.
Edited by:
Jian Liu, Rochester Institute of Technology (RIT), United StatesReviewed by:
Zhijie Chen, Zhejiang University, ChinaCopyright © 2024 Ghuffar and Noh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hyunho Noh, aHl1bmhvLm5vaC0xQG91LmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.