 Yury E. Tsvetkov1
Yury E. Tsvetkov1 Timur M. Volkov1
Timur M. Volkov1 Sergei A. Eremin2Oleg D. Sklyarov3Yuri K. Kulakov4
Sergei A. Eremin2Oleg D. Sklyarov3Yuri K. Kulakov4 Vadim B. Krylov1,5
Vadim B. Krylov1,5 Nikolay E. Nifantiev1*
Nikolay E. Nifantiev1*- 1Laboratory of Glycoconjugate Chemistry, N.D. Zelinsky Institute of Organic Chemistry, Russian Academy of Sciences, Moscow, Russia
- 2Faculty of Chemistry, M.V. Lomonosov Moscow State University, Moscow, Russia
- 3Russian State Centre of Quality and Standardization of Veterinary Drugs and Feeds, Moscow, Russia
- 4Laboratory of Brucellosis, N.F.Gamaleya National Research Center of Epidemiology and Microbiology, Moscow, Russia
- 5Laboratory of Synthetic Glycovaccines, N.D. Zelinsky Institute of Organic Chemistry, Russian Academy of Sciences, Moscow, Russia
Brucellosis is a dangerous zoonotic disease caused by bacteria of the genus Brucella. Diagnosis of brucellosis is based on the detection in animal and human sera of antibodies to the O-polysaccharide of Brucella lipopolysaccharide. The currently employed serodiagnosis of brucellosis relies on the use of the Brucella O-polysaccharide as a diagnostic antigen. However, the existence of bacterial species, which also express O-polysaccharides structurally similar to that of Brucella, may decrease the specificity of the brucellosis detection due to false-positive test results. It has been shown that the efficiency of the test can be significantly improved by using synthetic oligosaccharides that correspond to the so-called M epitope of the Brucella O-antigen. This epitope is characterized by an α-(1→3)-linkage between d-perosamine units and is unique to Brucella. Here we report on an efficient approach to the synthesis of oligosaccharides that model the M epitope of the Brucella O-polysaccharide. The approach is based on the use of the α-(1→3)-linked disaccharide thioglycoside as the key donor block. Its application allowed the straightforward assembly of a set of four protected oligosaccharides, which includes a disaccharide, two trisaccharides, and a tetrasaccharide, in five glycosylation steps. The synthesized oligosaccharides are planned to be used in the development of diagnostic tools for identifying brucellosis in humans and domestic animals, as well as a potential vaccine against it.
Introduction
Brucellosis is a bacterial infection of domestic animals, including cattle, sheep, goats, pigs and others. It is caused by Gram-negative bacteria of the genus Brucella. The disease can lead to abortions, infertility, weight loss, and reduced productivity in infected animals. When humans come into contact with infected animals or their products, such as milk, contaminated meat or organs, they can become infected with brucellosis. This can lead to serious acute and chronic illness with non-specific symptoms similar to malaria or influenza, and is therefore a significant public health concern (Franco et al., 2007).
One of the main virulence factors of Brucella is a lipopolysaccharide (LPS), which is located on the surface of the bacterial cell. The lipid part of LPS is embedded in the cell membrane, while the polysaccharide component (O-polysaccharide) extends outwards into the external environment, and thus determines the immunologic properties of the bacterium. O-Polysaccharide is a homopolymer of N-formylated 4-amino-4,6-dideoxy-d-mannose, also known as d-perosamine. N-Formyl-d-perosamine forms an α-(1→2)-linked linear chain terminated with one or more tetrasaccharide units that contain two α-(1→2)- and one central α-(1→3)-glycosidic bonds (Figure 1) (Kubler-Kielb and Vinogradov, 2013). The O-polysaccharide chain has a relatively low molecular weight, with a maximum of 25–30 monosaccharide units (Kubler-Kielb and Vinogradov, 2013). The antigenic determinants, represented by the α-(1→2)-linked chain and the terminal tetrasaccharide with the α-(1→3)-bond, are referred to as the A epitope (prevails in most strains of B. abortus) and the M epitope (is characteristic of B. melitensis), respectively.
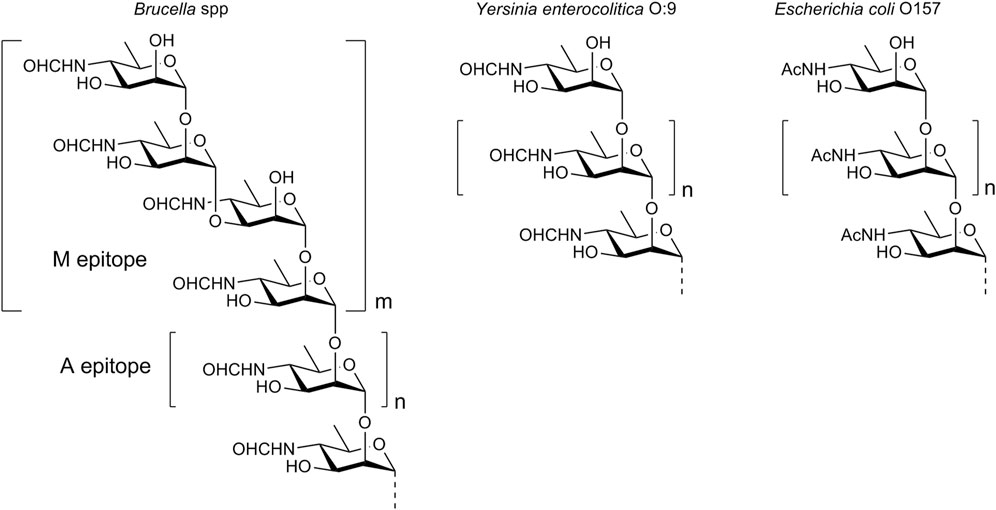
Figure 1. Structure of the O-polysaccharide of Brucella and the O-polysaccharides of other bacteria comprising N-acyl-d-perosamine.
The detection of antibodies specific to the O-polysaccharide in animal and human sera is the basis for the diagnosis of brucellosis (Nielsen, 2002; Godfroid et al., 2010). However, this biopolymer is not readily available through bacterial cultivation. Although the chemical synthesis of polysaccharides is in principal possible (Kochetkov et al., 1987; Tsvetkov et al., 1989), this task is often too complex. It is important that the specificity of the serodiagnosis using Brucella O-polysaccharide as a diagnostic antigen may not be high enough, as there are some bacteria that have O-polysaccharides made up of N-acylated d-perosamine. A prominent example of these bacteria is Yersinia enterocolitica O:9. Its O-polysaccharide consists of α-(1→2)-linked N-formyl-d-perosamine units only (Caroff et al., 1984), and is structurally identical to the A epitope of Brucella. Another example is Escherichia coli O157, whose O-polysaccharide contains α-(1→2)-linked N-acetyl-d-perosamine (Perry and Bundle, 1990). Sera obtained from animals or humans infected with such bacteria may contain antibodies that cross-react with the Brucella O-polysaccharide, leading to false-positive test results.
An increase in the specificity of the serodiagnosis of brucellosis can be achieved through the use of antigens related to the epitope M, which is unique to Brucella and is absent in the O-polysaccharides of other bacteria. It is evident that obtaining such antigens from the natural O-polysaccharide is challenging, if not impossible, and the most promising way to produce them is through chemical synthesis. Nowadays, synthetic oligosaccharides of virtually any structure are accessible and find application in immunological studies, including the development of diagnostic tools and vaccines, as a good alternative for antigenic polysaccharides (for recent examples see Argunov et al., 2015; Gening et al., 2015; Kazakova et al., 2020; Komarova et al., 2018; Krylov et al., 2019; Laverde et al., 2020; Solovev et al., 2023; Wong et al., 2020).
The use of synthetic oligosaccharides that mimic the M epitope of the Brucella O-polysaccharide for serodiagnosis has recently been investigated by Bundle et al. A large group of oligosaccharides with the characteristic for the M epitope (1→3)-glycoside bond between N-formyl-d-perosamine residues, was synthesized (Guiard et al., 2013; Ganesh et al., 2014; Bundle and McGiven, 2017). BSA conjugates of these synthetic oligosaccharides, particularly the (1→3)-linked disaccharide and the tetrasaccharide with the central (1→3)-glycoside bond, have shown high diagnostic sensitivity and specificity, as well as the ability to reliably differentiate between the sera of infected animals and those that give false-positive results when tested using conventional methods (McGiven et al., 2015; Bundle and McGiven, 2017).
However, the use of BSA as a carrier protein may carry risks of obtaining overestimated results. This is due to the presence in human sera of antibodies capable of recognizing BSA (Mogues et al., 2005; Sjöwall et al., 2011) and false-positive signals caused by multimeric presentation of glycoligands in the BSA conjugates (Grant et al., 2014; Temme et al., 2019). In addition, there may be a risk of non-specific interactions with hydrophobic spacers, for example, the 5-carboxypentyl spacer used in the works by Bundle et al. (Guiard et al., 2013; Ganesh et al., 2014; Mandal et al., 2017a; Mandal et al., 2017b; Bundle and McGiven, 2017).
α-(1→2)-Linked oligomers of N-(3-deoxy-L-glycero-tetronoyl)-D-perosamine related to the O-polysaccharide of Vibrio cholerae O1 have been synthesized by the Kováć’s group (Hou and Kovač, 2010; Mukherjee et al., 2019). Recently, we reported the synthesis of the (1→3)-linked disaccharide 4 (Figure 2) (Tsvetkov and Nifantiev, 2023), which corresponds to the unique structural element of the M epitope of the Brucella O-polysaccharide. Here, we report on the synthesis of larger oligosaccharides 1-3 that comprise the (1→3)-linked disaccharide as a fragment. They were prepared with the aim of developing fluorescence polarization–based and other types of diagnostic assays to identify brucellosis in animals and humans and as potential ligands for the design of corresponding vaccines. Additionally, pentasaccharide 6 has been synthesized, representing tetrasaccharide 1 devoid of terminal unsubstituted d-perosamine through its capping with a d-rhamnose residue. We have also prepared monosaccharide 5, as it has been reported that sera from mice immunized with conjugate immunogens based on (1→2)-linked Brucella oligosaccharides and sera from naturally Brucella-infected cattle contained high titers of antibodies specific to the terminal d-perosamine unit (Mandal et al., 2017a; Duncombe et al., 2022). The synthesized compounds were equipped with a short 3-aminipropyl spacer group, which was thought to be less hydrophobic than the 5-carboxypentyl group used by Bundle et al. We hypothesized that the use of the 3-aminopropyl spacer would minimize hydrophobic interactions, which could interfere with antigen-antibody recognition processes.
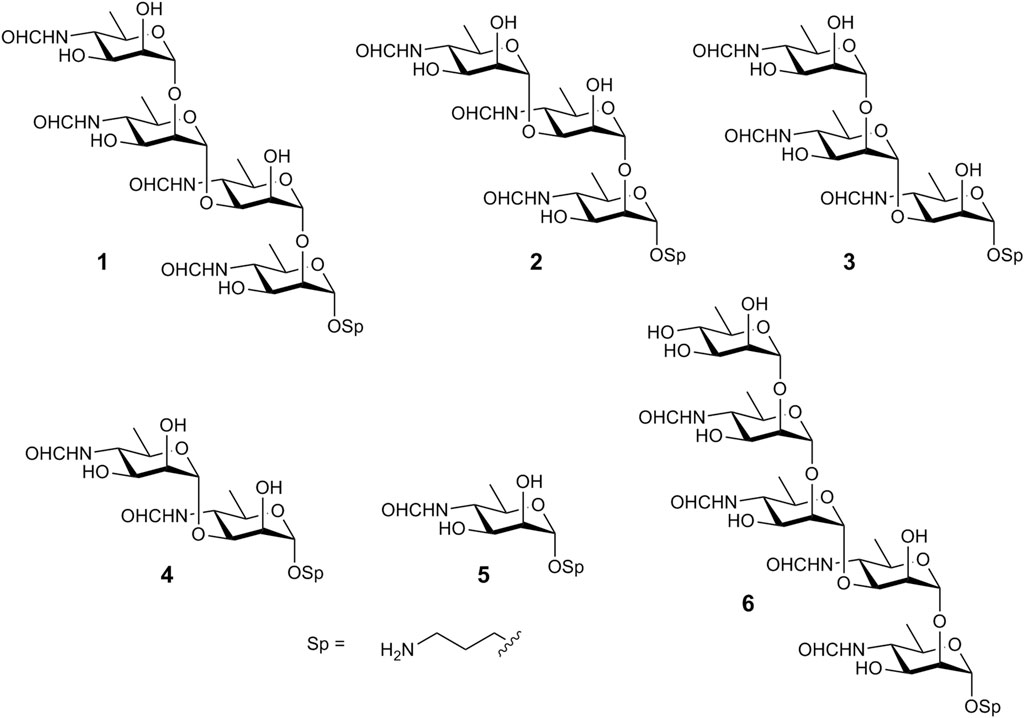
Figure 2. Oligosaccharides synthesized in this work.
Results and discussion
Oligosaccharides 1-4 were synthesized using a disaccharide (1→3)-linked glycosyl donor 15 as the key synthetic block. This choice of the donor allowed the assembly of all four oligosaccharides in a straightforward manner. The synthesis of monosaccharide derivatives, which were used as glycosyl acceptors in the preparation of the target structures, is shown in Scheme 1.
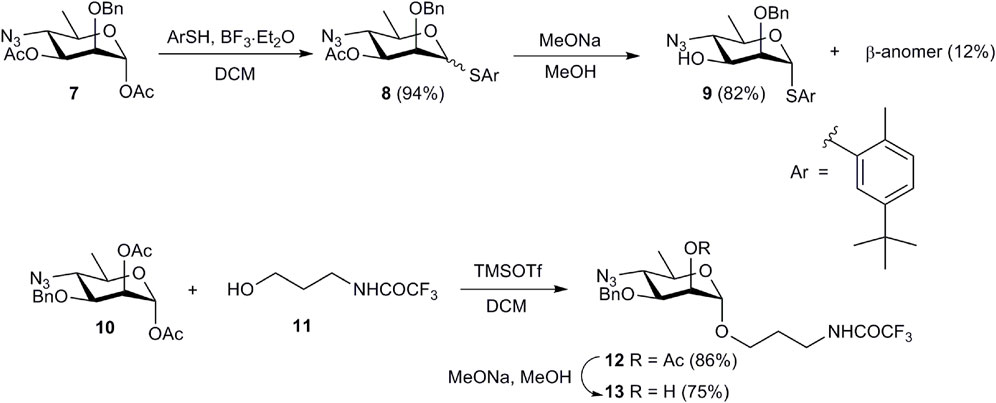
Scheme 1. Preparation of monosaccharide acceptor blocks 9 and 13.
Treatment of known 1,3-diacetate 7 (Mandal et al., 2017b) with 5-(tert-butyl)-2-methylthiophenol in the presence of BF3·Et2O produced an inseparable mixture of α- and β-thioglycosides 8. After conventional deacetylation of 8, the anomers were separated and pure α-anomer 9 was isolated in 82% yield. Similarly, TMSOTf-promoted glycosylation of alcohol 11 with 1,2-diacetate 10 (Bundle et al., 1988) and subsequent deacetylation of glycoside 12 afforded glycosyl acceptor 13.
Using acceptors 9 and 13, and the known trichloroacetimidate 14 (Ariosa-Alvarez et al., 1998) as a glycosyl donor, all oligosaccharides were assembled in five glycosylation steps (Scheme 2).
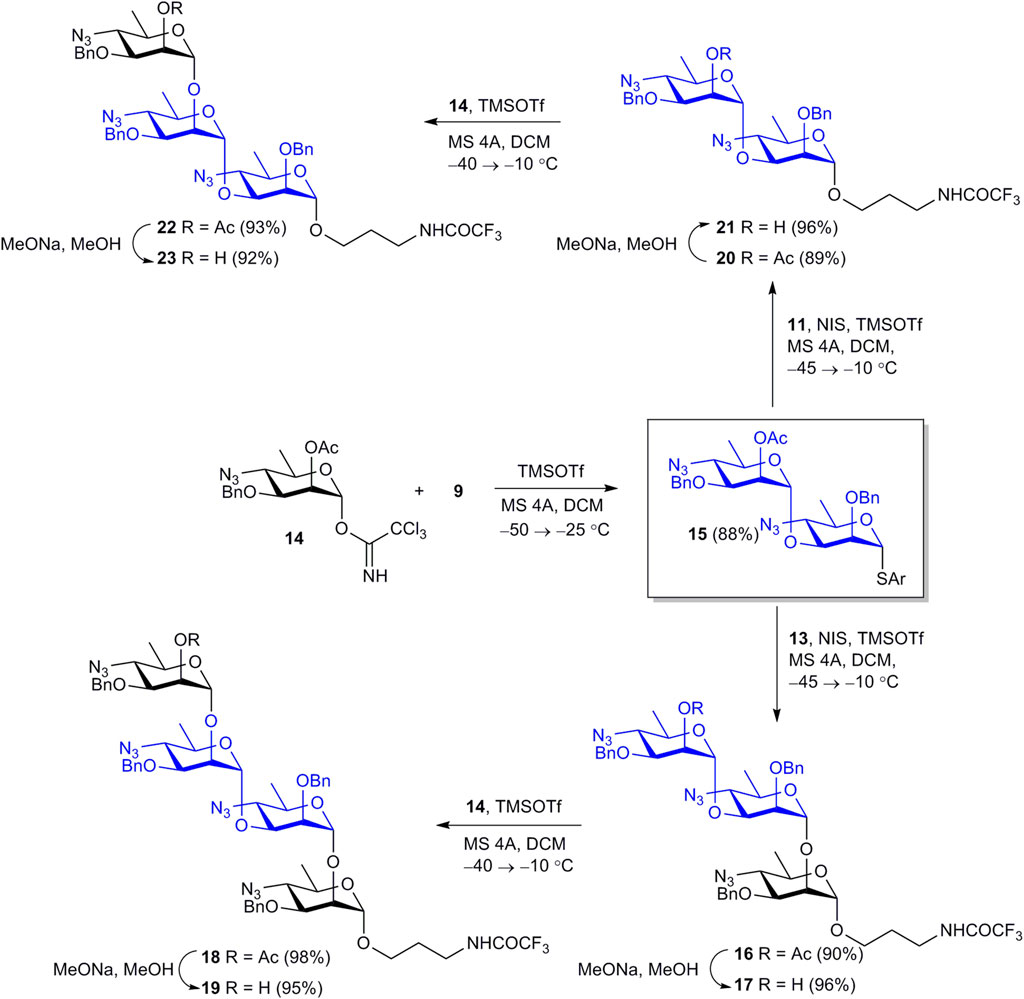
Scheme 2. Assembly of protected oligosaccharides 16, 18, 20, 22.
Orthogonal glycosylation of thioglycoside acceptor 9 with imidate 14 smoothly gave key intermediate 15 in 88% yield. Further NIS–TMSOTf-promoted glycosylation of acceptors 11 and 13 with thioglycoside 15 efficiently produced disaccharide 20 and trisaccharide 16. The 1JC-1,H-1 coupling constant values (170–172 Hz) for the 3-substituted monosaccharide residues in products 16 and 20 confirmed the α-configuration of the newly formed glycoside bonds (Bock and Pedersen, 1974). Despite the presence of a non-participating benzyl group at O-2, donor 15 ensured high α-stereoselectivity of glycosylation, in agreement with published data (Hou and Kovač, 2010). Removal of sole acetyl groups from 16 and 20 provided glycosyl acceptors 17 and 21 almost quantitatively. Final glycosylation of these acceptors with imidate 14 afforded tetrasaccharide 18 and trisaccharide 22 in excellent yields. Deacetylation of 18 and 22 gave alcohols 19 and 23. The structure of protected oligosaccharides 15-23 was confirmed by data of fully assigned 1H and 13C NMR spectra (see Supplementary Material) and high resolution MS.
Then oligosaccharides 17, 19, 21, 23, and monosaccharide 13 were transformed to final products 1-5. The reaction sequence employed for this transformation included reduction of azide groups, N-formylation, hydrogenolytic debenzylation and removal of the N-trifluoroacetyl group. This sequence is shown in Scheme 3 in relation to disaccharide 21. Intermediate products 24-26 on the way from 21 to unprotected 4 were characterized by high resolution mass-spectrometry. Each of the four protected compounds 13, 17, 19, and 23 was processed similarly (for details see Supplementary Material).
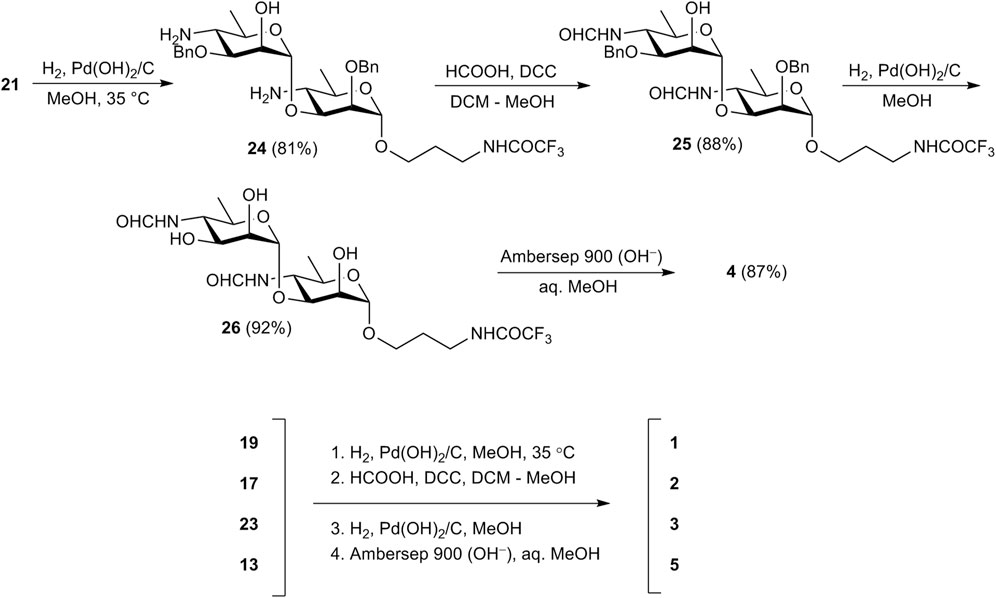
Scheme 3. Synthesis of free mono-(5) and oligosaccharides (1–4).
As hydrogen sulfide applied by Bundle et al. for the reduction of azide moieties (Guiard et al., 2013; Ganesh et al., 2014; Mandal et al., 2017b; Bundle and McGiven, 2017) is toxic and has an unpleasant odor, we used catalytic hydrogenation over Pd(OH)2/C at this step instead. Thus, catalytic reduction of diazide 21 proceeded with only minor affecting benzyl protections (Ariosa-Alvarez et al., 1998) and provided, after chromatographic purification, diamine 24 in 81% yield. N-Formylation of the latter with formic anhydride generated in situ from formic acid and DCC produced bis(formamide) 25 (88%), which was subjected to catalytic debenzylation with the formation of triol 26 (92%). Final basic removal of the N-trifluoroacetyl group and purification by size-exclusion chromatography gave target 3-aminopropyl glycoside 4 in 87% yield.
E/Z-Isomerism of the formamido groups results in the existence of compounds 1-5 as mixtures of 2n isomers, where n is the number of monosaccharides in the molecule (Peters et al., 1990). This strongly complicated the full assignment of NMR spectra, making it virtually impossible for oligosaccharides larger than the disaccharide. Nevertheless, the NMR data of 1-5 provided confirmation of the presence of the main functionalities such as formyl groups, anomeric centers, the spacer moiety, etc. (see Supplementary Material). Particularly, signals for Z- (δH 8.24–8.18 ppm; δC 166.1, 165.8 ppm) and E-isomers (δH 8.06–7.99 ppm; δC 169.0, 168.9 ppm) of the formyl groups, four anomeric centers of the prevailing isomer (δH 5.09, 5.02, 4.98, 4.92 ppm; δC 103.4, 102.9, 101.8; 99.6 ppm), and C-4 of E- (δC 58.2, 58.0, 57.9, 56.7 ppm) and Z-isomers (53.3, 53.1, 52.9, 52.1 ppm) were observed, inter alia, in the 1H and 13C NMR spectra of tetrasaccharide 1.
Pentasaccharide 6, in which the d-perosamine chain is terminated by a d-rhamnose unit, was prepared from tetrasaccharide acceptor 19 (Scheme 4). This pentasaccharide was thought to lose the ability to be recognized by antibodies specific to the terminal unsubstituted d-perosamine residue (Mandal et al., 2017a). Thioglycoside 27, which was used as the glycosyl donor, was synthesized by the reaction of known 1,2-diacetate 26 (Zhang et al., 2015) with 5-(tert-butyl)-2-methylthiophenol in the presence of BF3·Et2O. Following NIS–TMSOTf-promoted glycosylation of 19 with 27 produced pentasaccharide 28 in excellent yield. After deacetylation of 28, product 29 was subjected to deprotection with the use of the common reaction sequence (Scheme 4) resulting in the formation of target pentasaccharide 6.
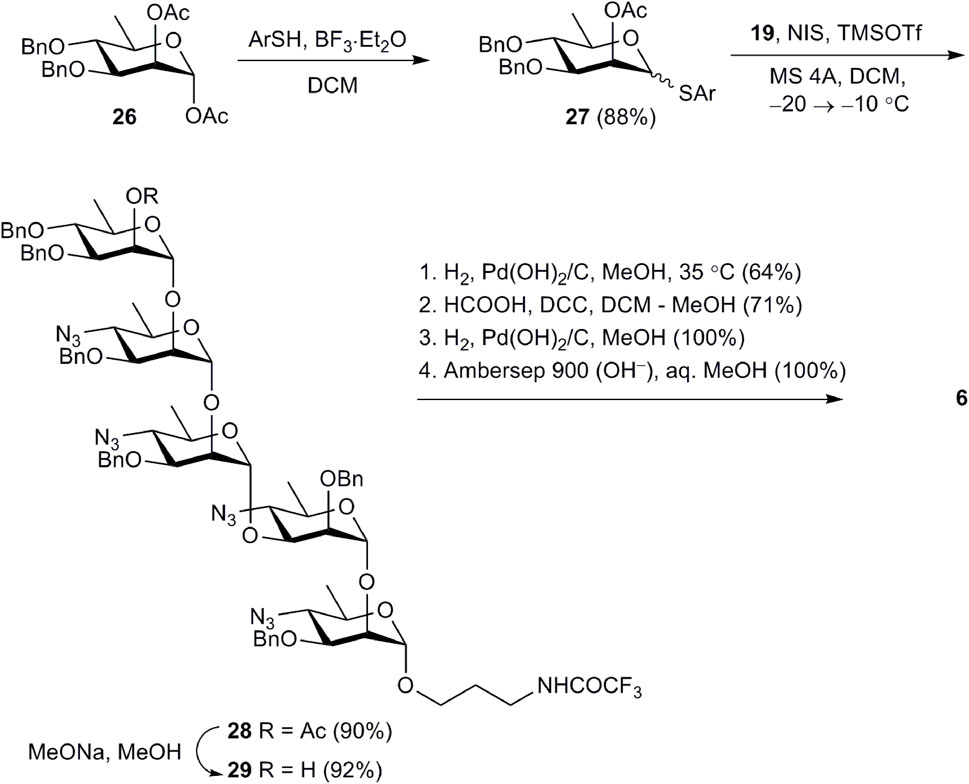
Scheme 4. Synthesis of pentasaccharide 6.
Conclusion
In conclusion, an efficient approach to the synthesis of oligosaccharides related to the M epitope of the Brucella O-polysaccharide has been developed. The approach is based on the use of the (1→3)-linked disaccharide thioglycoside as the key donor block. This disaccharide donor mimics the unique structural fragment of the M epitope with the (1→3)-linkage between two N-formyl-d-perosamine residues. Its application allowed the straightforward assembly of a set of four protected oligosaccharides, which comprised the disaccharide, two trisaccharides, and the tetrasaccharide, in five glycosylation steps. Following deprotection included reduction of azido groups, N-formylation, O-debenzylation, and de-N-trifluoroacetylation and produced free oligosaccharides as 3-aminopropyl glycosides. Using the partially protected tetrasaccharide as the glycosyl acceptor, the pentasaccharide devoid of terminal d-perosamine through its capping with a d-rhamnose residue has been also prepared. The synthesized oligosaccharides will be used for development of a diagnostic assay to identify brucellosis in animals and humans as well as the potential ligands to design vaccine candidates.
Experiment
General
NMR spectra were recorded on a Bruker Avance 600 NMR spectrometer. Protected oligosaccharides were measured in chloroform-d (CDCl3), and 1H NMR chemical shifts were referenced to the solvent residual signal (δH 7.27). 13C chemical shifts were referenced to the central resonance of CDCl3 (δC 77.0). Free oligosaccharides were measured in deuterium oxide (D2O) with suppression of the HOD signal. Acetone (δH 2.225, δC 31.45) was used as an internal standard. Signal assignment was made using COSY, TOCSY, HSQC, and ROESY experiments. In the presentation of NMR data, monosaccharide residues in oligosaccharides are denoted by the capital letters (A, B, C, etc.) starting from the reducing end. NMR spectra of synthesized compounds are presented in the Supplementary Material. High-resolution mass spectrometry (HRMS) with electrospray ionization (ESI) was performed on a MicrOTOF II (Bruker Daltonics) instrument. Optical rotations were measured using a JASCO P-2000 polarimeter at 20°C–25°C in chloroform (protected oligosaccharides) or water (free oligosaccharides). TLC was performed on Silica Gel 60 F254 plates (Merck) and visualization was accomplished using UV light or by charring at ∼150°C with orcinol–phosphoric acid (180 mg of orcinol in a mixture of 85% H3PO4 (10 mL), ethanol (5 mL), and water (85 mL)). Column chromatography was carried out using Silica Gel 60 (40–63μm; Merck Millipore). Gel-permeation chromatography of free oligosaccharides was performed on a Toyopearl TSK HW-40(S) column (2.8 × 80 cm) in 0.1 M acetic acid. A K-2401 refractive index detector (Knauer) was used to monitor gel-permeation chromatography. All moisture-sensitive reactions were carried out using dry solvents under dry argon. Powdered molecular sieve 4 Å was activated at 300°C under vacuum (∼1 mbar) for 30 min directly prior to the reaction. Solutions were concentrated under reduced pressure using a rotatory evaporator at 40°C (bath temperature).
5-(tert-Butyl)-2-methylphenyl 4-azido-2-O-benzyl-4,6-dideoxy-1-thio-α-d-mannopyranoside (9). BF3·Et2O (84 μL, 0.67 mmol) was added to a solution of 1,3-diacetate 7 (203 mg, 0.56 mmol) and 5-(tert-butyl)-2-methylthiophenol (153 μL, 0.84 mmol) in DCM (4 mL) upon chilling in an ice bath. The mixture was stirred for 10 min. with chilling and then at room temperature for 1.5 h. The resulting mixture was diluted with chloroform (50 mL), washed with water (30 mL) and saturated aqueous NaHCO3 (30 mL). The solvent was evaporated and the residue was purified by column chromatography (petroleum ether–EtOAc, 0→5%) to give thioglycoside 8 (255 mg, 94%) as a colorless syrup; α,β ratio ∼6.3:1. 1H NMR (600 MHz,CDCl3): δ 7.62–7.13 (m, 9.3 H, Ar), 5.44 (s, 1 H, H-1α), 5.15 (dd, 1 H, J2,3 = 3.2 Hz, J3,4 = 10.4 Hz, H-3α), 4.91 (d, 0.16 H, J = 11.8 Hz, PhCHaHbβ) 4.84 (dd, 0.16 H, J2,3 = 3.2 Hz, J3,4 = 10.2 Hz, H-3β), 4.78 (d, 0.16 H, J = 11.8 Hz, PhCHaHbβ), 4.73 (s, 1 H, H-1β), 4.70 (d, 1 H, J = 12.2 Hz, PhCHaHbα), 4.52 (d, 1 H, J = 12.2 Hz, PhCHaHbα), 4.25 (d, 0.16 H, J2,3 = 3.2 Hz, H-2β), 4.16–4.11 (m, 2 H, H-2α, H-5α), 3.76–3.71 (m, 1.16 H, H-4α, H-4β), 3.30 (dq, 0.16 H, J5,6 = 6.3 Hz, J4,5 = 9.8 Hz, H-5β), 2.38 (s, 0.5 H, Ar-CH3β), 2.36 (s, 3 H, Ar-CH3α), 2.10 (s, 3 H, CH3COα), 2.05 (s, 0.5 H, CH3COβ), 1.46 (d, 0.5 H, J6,5 = 6.3 Hz, H-6β), 1.40 (d, 3 H, J6,5 = 6.1 Hz, H-6α), 1.32 (s, 10.4 H, Ar-C(CH3)αβ). 13C NMR (150 MHz, CDCl3): δ 169.9 (CH3CO), 149.8, 137.2, 136.4, 132.7, 130.0, 129.8, 129.4, 129.0, 128.5, 128.1, 128.0, 124.8 (Ar), 87.1 (C-1α), 84.8 (C-1β), 77.6 (C-2β), 76.4 (C-2α), 76.0 (PhCH2β), 75.9 (C-3β), 75.3 (C-5β), 72.7 (C-3α), 72.5 (PhCH2α), 68.3 (C-5α), 62.9 (C-4α), 62.3 (C-4β), 31.2 (C(CH3)3), 20.9 (CH3CO), 20.2 (CH3-Ar), 18.7 (C-6β), 18.4 (C-6α).
1 M Sodium methoxide in MeOH (50 μL) was added to a solution of 3-acetate 8 (255 mg, 0.53 mmol) in MeOH (5 mL), and the mixture was stirred for 2 h at room temperature. The mixture was neutralized with Amberlite IR-120 (H+), the resin was filtered off and washed with MeOH (4 × 4 mL), and the filtrate was concentrated. The residue was subjected to column chromatography (petroleum ether–EtOAc, 2→8%) to produce α-anomer 9 (192 mg, 82%) and β-anomer (29 mg, 12%).
α-Anomer 9, colorless syrup, [α]D +95 (c 1, СHCl3). 1H NMR (600 MHz, CDCl3): δ 7.56–7.16 (m, 8 H, Ar), 5.53 (s, 1 H, H-1), 4.77 (d, 1 H, J = 11.8 Hz, PhCHaHb), 4.55 (d, 1 H, J = 11.8 Hz, PhCHaHb), 4.07 (dq, 1 H, J4,5 = 9.9 Hz, J5,6 = 6.3 Hz, H-5), 4.02 (d, 1 H, J2,3 = 3.7 Hz, H-2), 3.94 (dd, 1 H, J2,3 = 3.7 Hz, J3,4 = 9.8 Hz, H-3), 3.40 (t, 1 H, J = 9.8 Hz, H-4), 2.37 (s, 3 H, Ar-CH3), 1.37 (d, 3 H, J6,5 = 6.3 Hz, H-6), 1.33 (s, 9 H, Ar-C(CH3)). 13C NMR (150 MHz, CDCl3): δ 149.8, 136.9, 136.4, 132.6, 130.0, 129.6, 128.7, 128.3, 128.1, 125.0 (Ar), 84.3 (C-1), 78.9 (C-2), 72.3 (PhCH2), 70.9 (C-3), 67.9 (C-5), 66.8 (C-4), 31.3 (C(CH3)3), 20.1 (CH3-Ar), 18.3 (C-6). HRMS (ESI): calcd. for C24H31N3O3S [M + Na]+ m/z 464.1978; found m/z 464.1968.
β-Anomer, colorless syrup, [α]D –96 (c 0.5, СHCl3). 1H NMR (600 MHz, CDCl3): δ 7.63–7.14 (m, 8 H, Ar), 5.16 (d, 1 H, J = 11.5 Hz, PhCHaHb), 4.80 (d, 1 H, J = 11.5 Hz, PhCHaHb), 4.72 (s, 1 H, H-1), 4.10 (d, 1 H, J2,3 = 3.5 Hz, H-2), 3.61 (dd, 1 H, J2,3 = 3.6 Hz, J3,4 = 9.7 Hz, H-3), 3.35 (t, 1 H, J = 9.8 Hz, H-4), 3.23 (dq, 1 H, J4,5 = 9.8 Hz, J5,6 = 6.3 Hz, H-5), 2.40 (s, 3 H, Ar-CH3), 1.44 (d, 3 H, J6,5 = 6.3 Hz, H-6), 1.33 (s, 9 H, Ar-C(CH3)). 13C NMR (150 MHz, CDCl3): δ 149.6, 137.5, 135.7, 133.6, 129.8, 128.7, 128.5, 128.3, 128.1, 124.6 (Ar), 87.1 (C-1), 80.1 (C-2), 76.5 (PhCH2), 75.2 (C-5), 74.3 (C-3), 66.2 (C-4), 31.3 (C(CH3)3), 20.2 (CH3-Ar), 18.7 (C-6). HRMS (ESI): calcd. for C24H31N3O3S [M + Na]+ m/z 464.1978; found m/z 464.1981.
3-Trifluoroacetamidopropyl 2-O-acetyl-4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranoside (12). TMSOTf (0.59 mL, 3.24 mmol) was added to a chilled (ice bath) solution of 1,2-diacetate 10 (906 mg, 2.49 mmol) and alcohol 11 (554 mg, 3.24 mmol) in DCM (15 mL). The mixture was stirred for 20 min. upon chilling and then 2.5 h at room temperature. After dilution with DCM (100 mL), the mixture was washed with saturated aqueous NaHCO3 (50 mL) and water (2 × 50 mL), and concentrated. The residue was purified by column chromatography (petroleum ether–EtOAc, 15→30%) to yield glycoside 12 (1.02 g, 86%) as a colorless syrup, , [α]D +88 (c 1, СHCl3). 1H NMR (600 MHz, CDCl3): δ 7.39–7.29 (m, 5 H, Ar), 6.77 (br. s, 1 H, CF3CONH), 5.32 (br. s, 1 H, H-2), 4.76 (s, 1 H, H-1), 4.68 (d, 1 H, J = 11.0 Hz, PhCHaHb), 4.54 (d, 1 H, J = 11.0 Hz, PhCHaHb), 3.82–3.78 (m, 1 H, OCHaHbCH2CH2N), 3.77 (dd, 1 H, J2,3 = 3.3 Hz, J3,4 = 9.8 Hz, H-3), 3.54–3.45 (m, 4 H, OCHaHbCH2CH2N, H-5), 3.44 (t, 1 H, J = 9.8 Hz, H-4), 2.14 (s, 3 H, CH3CO), 1.96–1.84 (m, OCH2CH2CH2N), 1.34 (d, 3 H, J6,5 = 6.1 Hz, H-6). 13C NMR (150 MHz, CDCl3): δ 170.2 (CH3CO), 137.1, 128.4, 128.2, 128.0 (Ar), 97.9 (C-1), 76.2 (C-3), 71.7 (PhCH2), 67.3 (C-2), 76.2 (C-5), 66.1 (OCH2CH2CH2N), 63.8 (C-4), 38.1 (OCH2CH2CH2N), 28.3 (OCH2CH2CH2N), 20.9 (CH3CO), 18.4 (C-6). HRMS (ESI): calcd. for C20H25F3N4O6 [M + Na]+ m/z 497.1618; found m/z 497.1609.
3-Trifluoroacetamidopropyl 4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranoside (13). 1 M Sodium methoxide in MeOH (100 μL) was added to a solution of 2-acetate 12 (1.02 g, 2.15 mmol) in MeOH (15 mL) and the mixture was stirred for 5 h at room temperature. Amberlite IR-120 (H+) was added until neutrality, the resin was filtered off and washed with MeOH (5 × 4 mL), and the filtrate was concentrated. Column chromatography of the residue (toluene–EtOAc, 15→30%) provided compound 13 (700 mg, 75%) as a colorless syrup, [α]D +104 (c 1, СHCl3). 1H NMR (600 MHz, CDCl3): δ 7.43–7.33 (m, 5 H, Ar), 6.82 (br. s, 1 H, CF3CONH), 4.83 (s, 1 H, H-1), 4.71 (d, 1 H, J = 11.0 Hz, PhCHaHb), 4.67 (d, 1 H, J = 11.0 Hz, PhCHaHb), 3.99 (br. s, 1 H, H-2), 3.85–3.80 (m, 1 H, OCHaHbCH2CH2N), 3.68 (dd, 1 H, J2,3 = 3.3 Hz, J3,4 = 9.3 Hz, H-3), 3.56–3.42 (m, 5 H, OCHaHbCH2CH2N, H-4, H-5), 2.58 (br. s, 1 H, OH), 1.94–1.83 (m, OCH2CH2CH2N), 1.34 (d, 3 H, J6,5 = 5.9 Hz, H-6). 13C NMR (150 Hz, CDCl3): δ 137.1, 128.6, 128.2, 128.0 (Ar), 99.2 (C-1), 78.5 (C-3), 72.1 (PhCH2), 67.1 (C-2), 67.0 (C-5), 66.3 (OCH2CH2CH2N), 63.8 (C-4), 38.4 (OCH2CH2CH2N), 28.2 (OCH2CH2CH2N), 18.4 (C-6). HRMS (ESI): calcd. for C18H23F3N4O5 [M + Na]+ m/z 455.1513; found m/z 455.1506.
4-(tert-Butyl)-2-methylphenyl 2-O-acetyl-4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→3)-4-azido-2-O-benzyl-4,6-dideoxy-1-thio-α-d-mannopyranoside (15). A mixture of acceptor 9 (168 mg, 0.381 mmol), donor 14 (204 mg, 0.438 mmol) and mol. sieve 4 Å (400 mg) in DCM (6 mL) was stirred for 1.5 h at room temperature and cooled to −55°C. TMSOTf (16 μL, 0.088 mmol) was added and the mixture was stirred for 30 min. at −55 ÷ −50°C. Then, over the next 30 min., the temperature was gradually increased to −25°C and the mixture was stirred at this temperature for another 30 min. The reaction was quenched by adding Et3N (50 μL), the mixture was diluted with chloroform (50 mL), the solids were filtered off through a Celite layer and washed with chloroform. The filtrate was washed with water (50 mL), and the solvent was evaporated. The residue was subjected to gel-permeation chromatography on a Bio-Beads S-X3 column (3.5 × 70 cm) in toluene; fractions containing disaccharide 15 were pooled and concentrated. The product obtained was additionally purified by silica gel column chromatography (petroleum ether–EtOAc, 5→8%) to produce disaccharide 15 (251 mg, 88%) as a colorless syrup, [α]D +129 (c 1, СHCl3). 1H NMR (600 MHz, CDCl3): δ 7.52–7.14 (m, 13 H, Ar), 5.53 (br. s, 1 H, H-2B), 5.48 (s, 1 H, H-1A), 5.12 (s, 1 H, H-1B), 4.72 (d, 1 H, J = 11.0 Hz, PhCHaHb), 4.68 (d, 1 H, J = 12.0 Hz, PhCHaHb′), 4.54 (d, 1 H, J = 11.0 Hz, PhCHaHb), 4.53 (d, 1 H, J = 12.0 Hz, PhCHaHb′), 4.04 (dq, 1 H, J4,5 = 9.8 Hz, J5,6 = 6.1 Hz, H-5A), 4.01 (br. s, 1 H, H-2A), 3,96 (dd, 1 H, J2,3 = 3.0 Hz, J3,4 = 10.0 Hz, H-3A), 3.88 (dd, 1 H, J2,3 = 3.2 Hz, J3,4 = 9.8 Hz, H-3B), 3.72 (t, 1 H, J = 9.8 Hz, H-4A), 3.67 (dq, 1 H, J4,5 = 10.2 Hz, J5,6 = 6.3 Hz, H-5B), 3.46 (t, 1 H, J = 10.0 Hz, H-4B), 2.36 (s, 3 H, Ar-CH3), 2.17 (s, 3 H, CH3CO), 1.39 (d, 3 H, J6,5 = 6.1 Hz, H-6A), 1.32 (s, 9 H, Ar-C(CH3)), 1.28 (d, 3 H, J6,5 = 6.3 Hz, H-6B). 13C NMR (150 MHz, CDCl3): δ 170.0 (CH3CO), 149.9, 137.2, 136.4, 132.6, 130.0, 129.4, 128.6, 128.4, 128.3, 128.0, 127.9, 127.5, 125.0 (Ar), 99.8 (C-1B), 84.3 (C-1A), 79.6 (C-3A), 78.5 (C-2A), 75.8 (C-3B), 71.7, 71.5 (2 PhCH2), 68.5 (C-5A), 67.9 (C-5B), 67.3 (C-2B), 64.4 (C-4A), 63.8 (C-4B), 31.3 (C(CH3)3), 20.9 (CH3CO), 20.2 (CH3-Ar), 18.6 (C-6B), 18.4 (C-6A). HRMS (ESI): calcd. for C39H48N6O7S [M + NH4]+ m/z 762.3643; found m/z 762.3636.
3-Trifluoroacetamidopropyl 2-O-acetyl-4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→3)-4-azido-2-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranoside (16). A mixture of donor 15 (251 mg, 0.337 mmol), acceptor 13 (132 mg, 0.306 mmol) and mol. sieve 4 Å (500 mg) in DCM (6 mL) was stirred for 1 h at room temperature, and then was cooled to −45°C. NIS (152 mg, 0.674 mmol) and TMSOTf (12 μL) were added and the mixture was stirred for 1 h, gradually increasing the temperature to −10°C. The reaction was quenched by adding pyridine (50 μL), the mixture was diluted with chloroform (10 mL), the mol. sieve was filtered off and washed with chloroform (4 × 5 mL). The filtrate was washed with 0.5 M Na2S2O3 solution (30 mL) and water (50 mL), and concentrated. Two consecutive column chromatographies (first in toluene–EtOAc (10→15%) and then in petroleum ether–EtOAc (15→25%)) resulted in trisaccharide 16 (273 mg, 90%) as a colorless foam, [α]D +75 (c 1, СHCl3). 1H NMR (600 MHz, CDCl3): δ 7.39–7.12 (m, 15 H, Ar), 6.68 (poorly resolved t, 1 H, NHCOCF3), 5.49 (br. s, H-2C), 5.08 (s, 1 H, H-1B), 5.06 (br. s, 1 H, H-1C), 4.72–4.68 (m, 3 H, H-1A, PhCHaHb, PhCHaHb′), 4.57 (d, 1 H, J = 10.8 Hz, PhCHaHb), 4.50 (d, 1 H, J = 11.0 Hz, PhCHaHb′), 4.24 (d, 1 H, J = 11.8 Hz, PhCHaHb′′), 4.15 (d, 1 H, J = 11.8 Hz, PhCHaHb′′), 3.99 (br. s, 1 H, H-2A), 3.90 (dd, 1 H, J2,3 = 3.0 Hz, J3,4 = 9.8 Hz, H-3B), 3.82–3.78 (m, 2 H, H-3C, OCHaHbCH2CH2N), 3.72–3.68 (m, 2 H, C-2B, C-3A), 3.61 (t, 1 H, J = 10.0 Hz, H-4B), 3.58–3.43 (m, 6 H, H-5A, H-5B, H-5C, OCHaHbCH2CH2N), 3,41 (t, 1 H, J = 9.8 Hz, H-4C), 3.38 (t, 1 H, J = 10.0, H-4A), 2.14 (s, 3 H, CH3CO), 1.93–1.85 (m, 2 H, OCH2CH2CH2N), 1.35 (d, 3 H, J6,5 = 6.2 Hz, H-6B), 1.34 (d, 3 H, J6,5 = 6.3 Hz, H-6A), 1.29 (d, 3 H, J6,5 = 6.1 Hz, H-6C). 13C NMR (150 MHz, CDCl3): δ 170.0 (CH3CO), 137.3, 137.2, 137.1, 128.5, 128.4, 128.3, 128.2, 128.0, 127.9, 127.8, 127.4, 99.6 (C-1C), 99.1 (C-1A), 98.2 (C-1B), 78.4 (C-3A), 78.3 (C-3B), 76.2 (C-2B), 75.8 (C-3C), 72.9 (C-2A), 72.5, 71.7, 71.5 (3 PhCH2), 68.0 (C-5B), 67.7 (C-5C), 67.5 (C-5A), 67.3 (C-2C), 65.9 (OCH2CH2CH2N), 64.4 (C-4A), 64.1 (C-4B), 63.7 (C-4C), 38.1 (OCH2CH2CH2N), 28.3 (OCH2CH2CH2N), 20.9 (CH3CO), 18.6, 18.5 (×2) (C-6A, C-6B, C-6C). HRMS (ESI): calcd. for C46H55F3N10O12 [M + NH4]+ m/z 1014.4291; found m/z 1014.4291.
3-Trifluoroacetamidopropyl 4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→3)-4-azido-2-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranoside (17). 1 M sodium methoxide in MeOH (60 μL) was added to a solution of monoacetate 16 (273 mg, 0.337 mmol) in MeOH (6 mL), and the mixture was stirred for 4 h at room temperature. The solution was made neutral by adding Amberlite IR-120 (H+), the resin was filtered off and washed with MeOH (4 × 4 mL). The filtrate was concentrated, and the residue was purified by column chromatography (toluene–EtOAc, 15→25%) to produce deacetylated trisaccharide 17 (250 mg, 96%) as a colorless foam, [α]D +57 (c 1, СHCl3). 1H NMR (600 MHz, CDCl3): δ 7.42–7.13 (m, 15 H, Ar), 6.70 (poorly resolved t, 1 H, NHCOCF3), 5.11 (s, 1 H, H-1C), 5.10 (s, 1 H, H-1B), 4.74–4.65 (m, 4 H, H-1A, PhCH2, PhCHaHb′), 4.58 (d, 1 H, J = 10.8 Hz, PhCHaHb′), 4.23 (d, 1 H, J = 11.8 Hz, PhCHaHb′′), 4.17 (br. s, 1 H, H-2C), 4.13 (d, 1 H, J = 11.8 Hz, PhCHaHb′′), 4.00 (br. s, 1 H, H-2A), 3.92 (dd, 1 H, J2,3 = 2.8 Hz, J3,4 = 9.6 Hz, H-3B), 3.82–3.78 (m, 1 H, OCHaHbCH2CH2N), 3.72-3.69 (m, 3 H, H-2B, H-3A, H-3C), 3.60 (t, 1 H, J = 9.8 Hz, H-4B), 3.58–3.44 (m, 6 H, H-5A, H-5B, H-5C, OCHaHbCH2CH2N), 3.43 (t, 1 H, J = 10.0 Hz, H-4C), 3.39 (t, 1 H, J = 9.8 Hz, H-4A), 2.50 (s, 1 H, OH), 1.94-1.85 (m, 2 H, OCH2CH2CH2N), 1.36–1.33 (m, 6 H, H-6A, H-6B), 1.20 (d, 3 H, J6,5 = 6.1 Hz, H-6C). 13C NMR (150 MHz, CDCl3): δ 137.4, 137.2, 128.6, 128.5, 128.3, 128.2, 128.1, 128.0, 127.8, 127.4 (Ar), 101.2 (C-1C), 99.1 (C-1A), 98.2 (C-1B), 78.5, 78.1 (×2) (C-3A, C-3B, C-3C), 76.4 (C-2A), 72.6, 71.9, 71.7 (3 PhCH2), 68.0 (C-5B), 67.5 (C-5A), 67.4 (C-5C), 67.2 (C-2C), 65.9 (OCH2CH2CH2N), 64.5 (C-4A), 64.2 (C-4B), 63.6 (C-4C), 38.1 (OCH2CH2CH2N), 28.3 (OCH2CH2CH2N), 18.6, 18.5 (C-6A, C-6B), 18.4 (C-6C). HRMS (ESI): calcd. for C44H53F3N10O11 [M + NH4]+ m/z 972.4186; found m/z 972.4195.
3-Trifluoroacetamidopropyl 2-O-acetyl-4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→3)-4-azido-2-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranoside (18). A mixture of imidate 14 (53 mg, 0.113 mmol), acceptor 17 (90 mg, 0.094 mmol) and mol. sieve 4 Å (200 mg) in DCM (2 mL) was stirred for 1 h at room temperature and cooled to −40°C. TMSOTf (4.2 μL, 23 μmol) was added, and the mixture was stirred for 1.5 h, while the temperature was gradually increased to −10°C. After adding Et3N (20 μL), the mixture was diluted with chloroform (10 mL), the solids were filtered off and washed with chloroform (4 × 4 mL). The filtrate was washed with 1 M HCl (10 mL), water (30 mL), and the solvent was evaporated. The residue was purified by column chromatography (petroleum ether–EtOAc, 20→25%) to give tetrasaccharide 18 (116 mg, 98%) as a colorless foam, [α]D +59 (c 1, СHCl3). 1H NMR (600 MHz, CDCl3): δ 7.40–7.25 (m, 20 H, Ar), 6.64 (poorly resolved t, 1 H, NHCOCF3), 5.43 (br. s, 1 H, H-2D), 5.09 (s, 1 H, H-1B), 5.00 (s, 1 H, H-1C), 4.90 (s, 1 H, H-1D), 4.75-4.69 (m, 3 H, H-1A, PhCHaHb, PhCHaHb′), 4.65 (d, 1 H, J = 11.6 Hz, PhCHaHb′′), 4.62 (d, 1 H, J = 11.6 Hz, PhCHaHb′′), 4.57 (d, 1 H, J = 11.2 Hz, PhCHaHb), 4.55 (d, 1 H, J = 11.8 Hz, PhCHaHb′), 4.25 (d, 1 H, J = 12.0 Hz, PhCHaHb′′′), 4.15(d, 1 H, J = 12.0 Hz, PhCHaHb′′′), 4.05 (br. s, 1 H, H-2C), 4.00 (br. s, 1 H, H-2A), 3.85 (dd, 1 H, J2,3 = 3.0 Hz, J3,4 = 9.4 Hz, H-3B), 3.82–3.78 (m, 2 H, H-3D, OCHaHbCH2CH2N), 3.71 (dd, 1 H, J2,3 = 3.0 Hz, J3,4 = 9.6 Hz, H-3A), 3.69 (dd, 1 H, J2,3 = 3.0 Hz, J3,4 = 9.6 Hz, H-3C), 3.64–3.59 (m, 2 H, H-2B, H-5D), 3.59–3.52 (m, 2 H, H-4B), H-5B), 3.50-3.38 (m, 6 H, H-4D, H-5A, H-5C, OCHaHbCH2CH2N), 3.36 (t, 1 H, J = 10.0 Hz, H-4A), 3.35 (t, 1 H, J = 10.0 Hz, H-4C), 2.09 (s, 3 H, CH3CO), 1.93-1.85 (m, 2 H, OCH2CH2CH2N), 1.35–1.32 (m, 9 H, H-6A, H-6B, H-6D), 1.18 (d, 3 H, J6,5 = 6.1 Hz, H-6C). 13C NMR (150 MHz, CDCl3): δ 169.7 (CH3CO), 137.4, 137.3, 137.1, 128.6, 128.5, 128.4, 128.2, 128.0, 127.8, 127.5 (Ar), 100.7 (C-1C), 99.4 (C-1D), 99.1 (C-1A), 98.3 (C-1B), 78.4 (C-3A), 77.5 (C-3C), 77.3 (C-3B), 76.3 (C-2B), 75.5 (C-3D), 73.3 (C-2C), 72.8 (C-2A), 72.5, 71.9, 71.8, 71.6 (4 PhCH2), 68.0, (C-5B), 67.8 (C-5C), 67.7 (C-5D), 67.5 (C-5A), 67.1 (C-2D), 65.9 (OCH2CH2CH2N), 64.5 (C-4B), 64.4 (C-4A), 63.8 (C-4D), 63.7 (C-4C), 38.1 (OCH2CH2CH2N), 28.4 (OCH2CH2CH2N), 20.9 (CH3CO), 18.5 (×2), 18.4 (×2) (C-6A, C-6B, C-6C, C-6D). HRMS (ESI): calcd. for C59H70F3N13O15 [M + NH4]+ m/z 1275.5405; found m/z 1275.5399.
3-Trifluoroacetamidopropyl 4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→3)-4-azido-2-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranoside (19). 1 M sodium methoxide (50 μL) was added to a solution of compound 18 (298 mg, 0.237 mmol) in MeOH (5 mL), and the mixture was stirred for 4.5 h at room temperature. The solution was made neutral by adding Amberlite IR-120 (H+), the resin was filtered off and washed with MeOH (4 × 4 mL). The filtrate was evaporated, and the residue was subjected to column chromatography (toluene–EtOAc, 10→25%) to produce deacetylated tetrasaccharide 19 (274 mg, 95%) as a colorless foam, [α]D +73 (c 1, СHCl3). 1H NMR (600 MHz, CDCl3): δ 7.43–7.26 (m, 20 H, Ar), 6.65 (br. s, 1 H, NHCOCF3), 5.09 (s, 1 H, H-1B), 5.03 (s, 1 H, H-1C), 5.00 (s, 1 H, H-1D), 4.74 (d, 1 H, J = 11.2 Hz, PhCHaHb), 4.72 (s, 1 H, H-1A), 4.71 (d, 1 H, J = 10.8 Hz, PhCHaHb′), 4.69 (d, 1 H, J = 11.2 Hz, PhCHaHb), 4.63 (s, 2 H, PhCH2″), 4.68 (d, 1 H, J = 10.8 Hz, PhCHaHb′), 4.25 (d, 1 H, J = 12.0 Hz, PhCHaHb‴), 4.16 (d, 1 H, J = 12.0 Hz, PhCHaHb‴), 4.09 (br. s, 1 H, H-2C), 4.00 (br. s, 2 H, H-2A, H-2D), 3.86 (dd, 1 H, J2,3 = 3.0 Hz, J3,4 = 9.5 Hz, H-3B), 3.83–3.78 (m, 1 H, OCHaHbCH2CH2N), 3.75–3.69 (m, 3 H, H-3A, H-3C, H-3D), 3.66–3.60 (m, 2 H, H-2B, H-5D), 3.60–3.52 (m, 3 H, H-4B, H-5B, OCHaHbCH2CH2N), 3.51-3.39 (m, 5 H, H-4D, H-5A, H-5С, OCH2CH2CH2N), 3.37 (t, 1 H, J = 10.0 Hz, H-4A), 3.32 (t, 1 H, J = 9.8 Hz, H-4C), 2.32 (br. s, 1 H, OH), 1.94-1.86 (m, 2 H, OCH2CH2CH2N), 1.36–1.33 (m, 9 H, H-6A, H-6B, H-6D), 1.19 (d, 3 H, J6,5 = 6.1 Hz, H-6C). 13C NMR (150 MHz, CDCl3): δ 137.4, 137.1, 128.6, 128.5, 128.4, 128.3, 128.2, 128.0, 127.8, 127.5 (Ar), 100.8 (C-1C), 100.7 (C-1D), 99.1 (C-1A), 98.4 (C-1B), 78.4 (C-3A), 77.7, 77.6 (C-3C, C-3D), 77.4 (C-3B), 76.3 (C-2B), 73.3 (C-2C), 72.8 (C-2A), 72.5, 72.1, 72.0, 71.8 (PhCH2), 68.1 (C-5B), 67.9 (C-5C), 67.5 (C-5A), 67.4 (C-5D), 67.1 (C-2D), 65.9 (OCH2CH2CH2N), 64.5 (×2) (C-4A, C-4B), 63.9 (C-4С), 63.8 (C-4D), 38.1 (OCH2CH2CH2N), 28.4 (OCH2CH2CH2N), 18.5 (×3), 18.4 (C-6A, C-6B, C-6C, C-6D). HRMS (ESI): calcd. for C57H68F3N13O14 [M + Na]+ m/z 1238.4853; found m/z 1238.4854.
3-Trifluoroacetamidopropyl 3-O-acetyl-4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→3)-4-azido-2-O-benzyl-4,6-dideoxy-α-d-mannopyranoside (20). A mixture of thiglycoside 15 (220 mg, 0.295 mmol), alcohol 11 (76 mg, 0.444 mmol) and mol. sieve 4 Å in DCM (5 mL) was stirred for 1 h at room temperature and then cooled to −45°C. NIS (132 mg, 0.587 mmol) and TMSOTf (22 μL, 0.121 mmol) were added, and the mixture was stirred for 1 h with gradual increase of the temperature to −10°C. Pyridine (50 μL) was added, the mixture was diluted with chloroform (10 mL), the solids were filtered off and washed with chloroform (4 × 4 mL). The filtrate was washed with 0.5 M Na2S2O3 solution (30 mL) and water (50 mL), and concentrated. Column chromatography of the residue (petroleum ether–EtOAc, 20→30%) produced glycoside 20 (194 mg, 89%) as a colorless syrup, [α]D +83 (c 1, СHCl3). 1H NMR (600 MHz, CDCl3): δ 7.39–7.27 (m, 10 H, Ar), 6.57 (br. s, 1 H, NHCOCF3), 5.48 (br, s, 1 H, H-2B), 5.04 (s, 1 H, H-1B), 4.78 (s, 1 H, H-1A), 4.70 (d, 1 H, J = 11.2 Hz, PhCHaHb), 4.65 (d, 1 H, J = 11.8 Hz, PhCHaHb′), 4.60 (d, 1 H, J = 11.8 Hz, PhCHaHb′), 4.53 (d, 1 H, J = 11.2 Hz, PhCHaHb), 3.86 (dd, 1 H, J2,3 = 3.2 Hz, J3,4 = 9.8 Hz, H-3B), 3.83 (dd, 1 H, J2,3 = 3.2 Hz, J3,4 = 10.2 Hz, H-3A), 3.78-3.74 (m, 1 H, OCHaHbCH2CH2N), 3.71 (br. s, 1 H, H-2A), 3.69 (dq, 1 H, J4,5 = 10.0 Hz, J5,6 = 6.1 Hz, H-5B), 3.62 (t, 1 H, J = 10.0 Hz, H-4A), 3.53–3.38 (m, 5 H, H-4B, H-5A, OCHaHbCH2CH2N), 2.14 (s, 3 H, CH3CO), 1.88–1.83 (m, 2 H, OCH2CH2CH2N), 1.35 (d, 3 H, J6,5 = 6.1 Hz, H-6A), 1.27 (d, 3 H, J6,5 = 6.1 Hz, H-6B). 13C NMR (150 MHz, CDCl3): δ 170.1 (CH3CO), 137.5, 137.2, 128.5, 128.4, 128.3, 128.0, 127.9, 127.5 (Ar), 99.7 (C-1B), 97.2 (C-1A), 79.2 (C-3A), 76.5 (C-2A), 75.8 (C-3B), 72.6, 71.5 (2 PhCH2), 67.9 (C-5B), 67.6 (C-5A), 67.4 (C-2B), 65.7 (OCH2CH2CH2N), 64.2 (C-4A), 63.8 (C-4B), 37.9 (OCH2CH2CH2N), 28.4 (OCH2CH2CH2N), 20.9 (CH3CO), 18.6 (C-6B), 18.5 (C-6A). HRMS (ESI): calcd. for C33H40F3N7O9 [M + Na]+ m/z 758.2732; found m/z 758.2732.
3-Trifluoroacetamidopropyl 4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→3)-4-azido-2-O-benzyl-4,6-dideoxy-α-d-mannopyranoside (21). 1 M sodium methoxide (25 μL) was added to a solution of compound 20 (77 mg, 0.105 mmol) in MeOH (2.5 mL), and the mixture was stirred for 3 h at room temperature. The solution was made neutral by adding Amberlite IR-120 (H+), the resin was filtered off and washed with MeOH (4 × 4 mL). The filtrate was evaporated, and the residue was subjected to column chromatography (toluene–EtOAc, 15→25%) to give deacetylated product 21 (70 mg, 96%) as a colorless syrup, [α]D +89 (c 1, СHCl3). 1H NMR (600 MHz, CDCl3): δ 7.43–7.32 (m, 10 H, Ar), 6.60 (br. s, 1 H, NHCOCF3), 5.09 (s, 1 H, H-1B), 4.80 (s, 1 H, H-1A), 4.70 (d, 1 H, J = 11.6 Hz, PhCHaHb), 4.68 (d, 1 H, J = 11.6 Hz, PhCHaHb), 4.67 (d, 1 H, J = 11.6 Hz, PhCHaHb′), 4.60 (d, 1 H, J = 11.6 Hz, PhCHaHb′), 4.19 (br. s, 1 H, H-2B), 3.84 (dd, 1 H, J2,3 = 3.2 Hz, J3,4 = 10.0 Hz, H-3A), 3.79–3.74 (m, 2 H, H-3B, OCHaHbCH2CH2N), 3.72 (br. s, 1 H, H-2A), 3.68 (dq, 1 H, J4,5 = 10.0 Hz, J5,6 = 6.3 Hz, H-5B), 3.60 (t, 1 H, J = 10.2 Hz, H-4A), 3.52–3.40 (m, 5 H, H-4B, H-5A, OCHaHbCH2CH2N), 2.49 (s, 1 H, OH), 1.89–1.84 (m, 2 H, OCH2CH2CH2N), 1.35 (d, 3 H, J6,5 = 6.3 Hz, H-6A), 1.28 (d, 3 H, J6,5 = 6.3 Hz, H-6B). 13C NMR (150 MHz, CDCl3): δ 137.5, 137.2, 128.6, 128.5, 128.2, 128.1, 128.0, 127.5 (Ar), 101.4 (C-1B), 97.2 (C-1A), 78.9 (C-3A), 78.0 (C-3B), 76.8 (C-2A), 72.6, 71.9 (2 PhCH2), 67.5 (×2) (C-5A, C-5B), 67.2 (C-2B), 65.7 (OCH2CH2CH2N), 64.3 (C-4A), 63.7 (C-4B), 37.9 (OCH2CH2CH2N), 28.4 (OCH2CH2CH2N), 18.5 (×2) (C-6A, C-6B). HRMS (ESI): calcd. for C31H38F3N7O8 [M + Na]+ m/z 716.2626; found m/z 716.2628.
3-Trifluoroacetamidopropyl 2-O-acetyl-4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→3)-4-azido-2-O-benzyl-4,6-dideoxy-α-d-mannopyranoside (22). Mol. sieve 4 Å (500 mg) was added to a solution of imidate 14 (150 mg, 0.32 mmol) and acceptor 21 (195 mg, 0.28 mmol) in DCM (5 mL), the resulting mixture was stirred for 1 h at room temperature, and then cooled to −40°C. TMSOTf (11.6 μL, 0.064 mmol) was added, and the mixture was stirred for 1 h with gradual increasing the temperature to −10°C, after which the reaction was quenched with Et3N (20 μL). The mixture was diluted with chloroform (10 mL), and the solids were filtered off and washed with chloroform (4 × 4 mL). The filtrate was washed with 1 M HCl (20 mL) and water (30 mL), and the solvent was evaporated. The residue was purified by column chromatography (petroleum ether–EtOAc, 20→30%) to yield trisaccharide 22 (260 mg, 93%) as a colorless foam, [α]D +70 (c 1, СHCl3). 1H NMR (600 MHz, CDCl3): δ 7.40–7.27 (m, 15 H, Ar), 6.55 (poorly resolved t, 1 H, NHCOCF3), 5.44 (br. s, 1 H, H-2C), 5.00 (s, 1 H, H-1B), 4.92 (s, 1 H, H-1C), 4.79 (s, 1 H, H-1A), 4.74 (d, 1 H, J = 11.0 Hz, PhCHaHb), 4.68 (d, 1 H, J = 11.6 Hz, PhCHaHb′), 4.65 (d, 1 H, J = 11.8 Hz, PhCHaHb′′), 4.64 (d, 1 H, J = 11.6 Hz, PhCHaHb′), 4.60 (d, 1 H, J = 11.8 Hz, PhCHaHb′′), 4.55 (d, 1 H, J = 11.0 Hz, PhCHaHb), 4.07 (br. s, 1 H, H-2B), 3.81–3.74 (m, 4 H, H-3A, H-3B, H-3C, OCHaHbCH2CH2N), 3.65 (br. s, 1 H, H-2A), 3.62–3.54 (m, 3 H, H-4A, H-5B, H-5C), 3.53–3.40 (m, 5 H, H-4C, H-5A, OCHaHbCH2CH2N), 3.38 (t, 1 H, J = 9.8 Hz, H-4B), 2.10 (s, 3 H, CH3CO), 1.89–1.84 (m, 2 H, OCH2CH2CH2N), 1.35 (d, 3 H, J6,5 = 6.3 Hz, H-6A), 1.33 (d, 3 H, J6,5 = 6.1 Hz, H-6C), 1.26 (d, 3 H, J6,5 = 6.1 Hz, H-6B). 13C NMR (150 MHz, CDCl3): δ 169.8 (CH3CO), 137.4, 137.1, 128.6, 128.5, 128.4, 128.0, 127.9, 127.6 (Ar), 101.0 (C-1B), 99.3 (C-1C), 97.4 (C-1A), 78.2 (C-3A), 77.4 (C-3B), 76.7 (C-2A), 75.4 (C-3C), 73.2 (C-2B), 72.7, 71.9, 71.6 (3 PhCH2), 68.0 (C-5B), 67.7 (C-5C), 67.6 (C-5A), 67.1 (C-2C), 65.7 (OCH2CH2CH2N), 64.7 (C-4A), 63.8 (×2) (C-4B, C-4C), 37.8 (OCH2CH2CH2N), 28.4 (OCH2CH2CH2N), 18.6, 18.5, 18.4 (C-6A, C-6B, C-6C). HRMS (ESI): calcd. for C46H55F3N10O12 [M + Na]+ m/z 1019.3845; found m/z 1019.3846.
3-Trifluoroacetamidopropyl 4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→3)-4-azido-2-O-benzyl-4,6-dideoxy-α-d-mannopyranoside (23). 1 M sodium methoxide (70 μL) was added to a solution of compound 22 (347 mg, 0.348 mmol) in MeOH (7 mL). After being stirred for 4 h at room temperature, the mixture was made neutral with Amberlite IR-120 (H+) and the resin was filtered off and washed with MeOH (4 × 4 mL). The filtrate was concentrated, and the residue was purified by column chromatography (toluene–EtOAc, 10→20%) to afford deacetylated trisaccharide 23 (306 mg, 92%) as a colorless foam, [α]D +86 (c 1, СHCl3). 1H NMR (600 MHz, CDCl3): δ 7.43-7.30 (m, 15 H, Ar), 6.58 (poorly resolved t, 1 H, NHCOCF3), 5.03 (s, 1 H, H-1B), 5.01 (s, 1 H, H-1C), 4.79 (s, 1 H, H-1A), 4.74 (d, 1 H, J = 11.2 Hz, PhCHaHb), 4.70 (d, 1 H, J = 11.2 Hz, PhCHaHb), 4.65 (s, 2 H, PhCH2′), 4.65 (d, 1 H, J = 11.8 Hz, PhCHaHb′′), 4.61 (d, 1 H, J = 11.8 Hz, PhCHaHb′′), 4.12 (br. s, 1 H, H-2B), 4.01 (s, 1 H, H-2C), 3.81–3.74 (m, 3 H, H-3A, H-3B, OCHaHbCH2CH2N), 3.73 (dd, 1 H, J2,3 = 3.2 Hz, J3,4 = 9.8 Hz, H-3C), 3.66 (br. s, 1 H, H-2A), 3.64–3.55 (m, 3 H, H-4A, H-5B, H-5C), 3.54–3.40 (m, 5 H, H-4C, H-5A, OCHaHbCH2CH2N), 3.36 (t, 1 H, J = 9.8 Hz, H-4B), 2.35 (br. s, 1 H, OH), 1.90–1.84 (m, 2 H, OCH2CH2CH2N), 1.36 (d, 3 H, J6,5 = 6.1 Hz, H-6A), 1.34 (d, 3 H, J6,5 = 6.1 Hz, H-6C), 1.27 (d, 3 H, J6,5 = 6.3 Hz, H-6B). 13C NMR (150 MHz, CDCl3): δ 137.4, 137.3, 137.1, 128.6, 128.5, 128.3, 128.2, 128.0, 127.6 (Ar), 101.1 (C-1B), 100.7 (C-1C), 97.4 (C-1A), 78.2 (C-3A), 77.6 (C-3C), 77.5 (C-3B), 76.7 (C-2A), 73.2 (C-2B), 72.7, 72.0 (×2) (3 PhCH2), 67.9 (C-5B), 67.6 (C-5A), 67.4 (C-5C), 67.1 (C-2C), 65.7 (OCH2CH2CH2N), 64.7 (C-4A), 64.0 (C-4B), 63.8 (C-4C), 37.9 (OCH2CH2CH2N), 28.4 (OCH2CH2CH2N), 18.6 (C-6B), 18.5 (C-6A), 18.3 (C-6C). HRMS (ESI): calcd. for C44H53F3N10O11 [M + NH4]+ m/z 972.4186; found m/z 972.4170.
5-(tert-Butyl)-2-methylphenyl 2-O-acetyl-3,4-di-O-benzyl-1-thio-α,β-d-rhamnopyranoside (27). BF3·Et2O (68 μL, 0.55 mmol) was added to a chilled (ice bath) solution of 1,2-diacetate 27 (195 mg, 0.455 mmol) and 5-(tert-butyl)-2-methylthiophenol in DCM (4 mL). The mixture was stirred for 15 min., then the cooling bath was removed and stirring was continued at room temperature for next 2 h. The mixture was diluted with chloroform (30 mL), washed with water (30 mL) and saturated NaHCO3 solution, and concentrated. Column chromatography of the residue (petroleum ether–EtOAc, 0→10%) produced an anomeric mixture (α:β ratio ∼1.3:1) of thiglycosides 27 (219 mg, 88%) as a colorless syrup. 1H NMR (600 MHz, CDCl3): δ 7.67–7.13 (m, 30 H, Ar), 5.84 (d, 1H, J2,3 = 3.2 Hz, H-2β), 5.68 (br. s, 1.3 H, H-2α), 5.37 (s, 1.3 H, H-1α), 4.97 (d, 1.3 H, J = 11.0 Hz, PhCHaHbα), 4.95 (d, 1 H, J = 11.0 Hz, PhCHaHbβ), 4.84 (d, 1 H, J = 11.0 Hz, PhCHaHbα′), 4.81 (s, 1 H, H-1β), 4.77 (d, 1.3 H, J = 11.2 Hz, PhCHaHbα′), 4.67 (d, 1.3 H, J = 11.0 Hz, PhCHaHbα), 4.65 (d, 1 H, J = 11.0 Hz, PhCHaHbβ), 4.61 (d, 1.3 H, J = 11.2 Hz, PhCHaHbα′), 4.53 (d, 1 H, J = 11.0 Hz, PhCHaHbβ′), 4.32 (dq, 1.3 H, J4,5 = 9.4 Hz, H-5α), 4.00 (dd, 1.3 H, J2,3 = 3.2 Hz, J3,4 = 9.4 Hz, H-3α), 3.68 (dd, 1 H, J3,4 = 8.7 Hz, H-3β), 3.55 (t, 1.3 H, J = 9.4 Hz, H-4α), 3.51 (t, 1 H, J = 9.2 Hz, H-4β), 3.49 (dq, 1 H, J4,5 = 9.2 Hz, H-5β), 2.42 (s, 4 H, CH3-Arα), 2.39 (s, 3 H, CH3-Arβ), 2.28 (s, 3 H, CH3COβ), 2.18 (s, 4 H, CH3COα), 1.49 (d, 3 H, J5,6 = 5.7 Hz, H-6β), 1.38 (d, 3 H, J5,6 = 6.1 Hz, H-6α), 1.33 (s, 21 H, C(CH3)3). ). 13C NMR (150 MHz, CDCl3): δ 170.4, 170.3 (CH3CO), 149.7, 149.6, 138.4, 138.2, 137.7, 137.6, 136.7, 135.8, 133.2, 132.6, 130.0, 129.8, 128.4, 128.3, 128.1, 128.0, 127.8, 127.7, 125.0, 124.5 (Ar), 85.8 (C-1α), 84.8 (C-1β), 81.3 (C-3β), 80.2 (C-4α), 79.3 (C-4β), 78.5 (C-3α), 76.1 (C-5β), 75.5, 71.9, 71.8 (PhCH2), 70.9 (C-2α), 70.7 (C-2β), 69.1 (C-5α), 31.3 (C(CH3)3), 21.1 (CH3COα), 20.9 (CH3COβ), 20.2 (CH3-Ar), 18.3 (C-6β), 17.9 (C-6α). HRMS (ESI): calcd. for C33H40O5S [M + Na]+ m/z 571.2489; found m/z 571.2482.
3-Trifluoroacetamidopropyl 2-O-acetyl-3,4-di-O-benzyl-α-d-rhamnopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→3)-4-azido-2-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranoside (28). A mixture of thiglycoside 27 (58 mg, 0.106 mmol), acceptor 19 (103 mg, 0.085 mmol) and vol. sieve 4 Å (150 mg) in DCM (3 mL) was for 1 h at room temperature and then cooled to −20°C. NIS (48 mg, 0.212 mmol) and TMSOTf (3.8 μL, 0.021 mmol) were added, and stirring was continued for 1 h at −20°C. The reaction was quenched with pyridine (20 μL), the mixture was diluted with chloroform (10 mL), and the solids were filtered off and washed with chloroform (4 × 4 mL). The filtrate was washed with 0.5 M Na2S2O3 solution (20 mL) and water (30 mL), and concentrated. Column chromatography of the residue (toluene–EtOAc, 8→13%) produced pentasaccharide 28 (121 mg, 90%) as a colorless foam, [α]D +49 (c 1, СHCl3). 1H NMR (600 MHz, CDCl3): δ 7.42–7.22 (m, 30 H, Ar), 6.66 (poorly resolved t, 1 H, NHCOCF3), 5.46 (br. s, 1 H, H-2E), 5.08 (s, 1 H, H-1B), 5.04 (s, 1 H, H-1D), 4.99 (s, 1 H, H-1C), 4.90 (d, 1 H, J = 10.8 Hz, benzylic H), 4.85 (s, 1 H, H-1E), 4.76-4.51 (m, 10 H, 9 benzylic H, H-1A), 4.23 (d, 1 H, J = 11.8 Hz, PhCHaHb), 4.15 (d, 1 H, J = 11.8 Hz, PhCHaHb), 4.03 (br. s, 1 H, H-2C), 4.00 (br. s, 1 H, H-2A), 3.94–3.90 (m, 2 H, H-2D, H-3E), 3.85 (dd, 1 H, J3,2 = 2.8 Hz, J3,4 = 9.4 Hz, H-3B), 3.83–3.78 (m, 1 H, OCHaHbCH2CH2N), 3.78–3.69 (m, 3 H, H-3A, H-3D, H-5E), 3.66 (dd, 1 H, J3,2 = 2.8 Hz, J3,4 = 10.0 Hz, H-3C), 3.61 (br. s, 1 H, H-2B), 3.59–3.52 (m, 4 H, H-4B, H-5B, H-5D, OCH2CH2CHaHbN), 3.51–3.32 (m, 7 H, H-4A, H-4D, H-4E, H-5A, H-5C, OCHaHbCH2CH2N, OCH2CH2CHaHbN), 3.25 (t, 1 H, J = 9.8 Hz, H-4C), 2.14 (s, 3 H, CH3CO), 1.93–1.88 (m, 2 H, OCH2CH2CH2N), 1.36–1.33 (m, 6 H, H-6A, H-6D), 1.31 (d, 3 H, J6,5 = 6.1 Hz, H-6B), 1.22 (d, 3 H, J6,5 = 6.3 Hz, H-6E), 1.17 (d, 3 H, J6,5 = 6.3 Hz, H-6C). 13C NMR (150 MHz, CDCl3): δ 169.9 (CH3CO), 138.4, 137.0, 137.5, 137.3, 137.1, 128.6, 128.5, 128.4, 128.3, 128.2, 128.1, 128.0, 127.9, 127.8, 127.7, 127.6, 127.5 (Ar), 100.8 (C-1C), 100.5 (C-1D), 99.3 (С-1E), 99.1 (C-1A), 98.4 (C-1B), 79.9 (C-4E), 78.4 (C-3A), 77.5 (C-3E), 77.2 (C-3B), 77.1 (C-3C), 76.9 (C-3D), 76.2 (C-2B), 75.4 (PhCH2), 72.8 (C-2A), 72.5 (PhCH2), 71.9, 71.8 (×2), 71.7 (4 PhCH2), 68.8 (C-2E), 68.1, 67.9, 67.8 (C-5B, C-5C, C-5D), 67.5 (C-5A), 65.9 (OCH2CH2CH2N), 64.6 (C-4B), 64.4 (C-4A), 64.0, 63.9 (C-4C, C-4D), 38.1 (OCH2CH2CH2N), 28.4 (OCH2CH2CH2N), 21.1 (CH3CO), 18.0 (×4) (C-6A, C-6B, C-6C, C-6D), 17.8 (C-6E). HRMS (ESI): calcd. for C79H92F3N13O19 [M + NH4]+ m/z 1601.6923; found m/z 1601.6906.
3-Trifluoroacetamidopropyl 3,4-di-O-benzyl-α-d-rhamnopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→3)-4-azido-2-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranoside (29). 1 M sodium methoxide (20 μL) was added to a solution of compound 28 (121 mg, 0.076 mmol) in MeOH (2 mL). After being stirred for 4 h at room temperature, the mixture was made neutral with Amberlite IR-120 (H+) and the resin was filtered off and washed with MeOH (4 × 4 mL). The filtrate was concentrated, and the residue was purified by column chromatography (toluene–EtOAc, 8→15%) to yield deacetylated pentasaccharide 29 (109 mg, 92%) as a colorless foam, [α]D +59 (c 1, СHCl3). 1H NMR (600 MHz, CDCl3): δ 7.42–7.23 (m, 30 H, Ar), 6.63 (br. s, 1 H, NHCOCF3), 5.08 (s, 1 H, H-1B), 5.05 (s, 1 H, H-1D), 5.00 (s, 1 H, H-1C), 4.97 (s, 1 H, H-1E), 4.88 (d, 1 H, J = 11.0 Hz, benzylic H), 4.75–4.62 (m, 8 H, H-1A, 7 benzylic H), 4.57 (d, 1 H, J = 11.0 Hz, benzylic H), 4.54 (d, 1 H, J = 11.6 Hz, benzylic H), 4.24 (d, 1 H, J = 11.8 Hz, PhCHaHb), 4.17 (d, 1 H, J = 11.8 Hz, PhCHaHb), 4.06 (br. s, 1 H, H-2E), 4.03 (br. s, 1 H, H-2C), 4.00–3.97 (m, 2 H, H-2A, H-2D), 3.87–3.82 (m, 2 H, H-3B, H-3E), 3.82–3.77 (m, 1 H, OCHaHbCH2CH2N), 3.77–3.69 (m, 3 H, H-3A, H-3D, H-5E), 3.67 (dd, 1 H, , J3,2 = 2.6 Hz, J3,4 = 9.8 Hz, H-3C), 3.61 (br. s, 1 H, H-2B), 3.58–3.51 (m, 4 H, H-4B, H-5B, H-5D, OCH2CH2CHaHbN), 3.51–3.43 (m, 4 H, H-4E, H-5A, OCHaHbCH2CH2N, OCH2CH2CHaHbN), 3.40–3.34 (m, 3 H, H-4A, H-4D, H-5C), 3.27 (t, 1 H, J = 9.8 Hz, H-4C), 2.40 (br. s, 1 H, OH), 1.94–1.85 (m, 2 H, OCH2CH2CH2N), 1.36-1.32 (m, 6 H, H-6A, H-6D), 1.31 (d, 3 H, J6,5 = 6.1 Hz, H-6B), 1.20 (d, 3 H, J6,5 = 6.1 Hz, H-6E), 1.17 (d, 3 H, J6,5 = 6.1 Hz, H-6C). 13C NMR (150 MHz, CDCl3): δ 138.4, 137.9, 137.4, 137.3, 137.2, 128.6, 128.5, 128.4, 128.3, 128.2, 128.1, 128.0, 127.8, 127.7, 127.5 (Ar), 100.8 (C-1C), 100.7 (×2), (C-1D, C-1E), 99.1 (C-1A), 98.5 (C-1B), 79.9 (C-4E), 79.5 (C-3E), 78.4 (C-3A), 77.2 (C-3B), 77.1 (×2) (C-3C, C-3D), 76.4 (С-2B), 75.4 (PhCH2), 73.4 (C-2C), 73.2 (C-2D), 72.9 (C-2A), 72.5, 72.2, 72.1, 71.9, 71.8 (5 PhCH2), 68.6 (C-2E), 68.1 (×2), 67.9 (×2) (C-5B, C-5C, C-5D, C-5E), 67.5 (C-5A), 65.9 (OCH2CH2CH2N), 64.6 (C-4B), 64.5 (C-4A), 64.2 (C-4D), 64.0 (C-4C), 38.1 (OCH2CH2CH2N), 28.4 (OCH2CH2CH2N), 18.5 (×4) (C-6A, C-6B, C-6C, C-6D), 17.7 (C-6E). HRMS (ESI): calcd. for C77H90F3N13O18 [M + NH4]+ m/z 1559.6817; found m/z 1559.6804.
3-Aminopropy 4,6-dideoxy-4-formamido-α-d-mannopyranosyl-(1→3)-4,6-dideoxy-4-formamido-α-d-mannopyranoside (4). A mixture of diazide 21 (95 mg, 0.137 mmol) and Pd(OH)2/C (30 mg) in MeOH was stirred in a hydrogen atmosphere at 35°C for 1.5 h. The catalyst was filtered off and washed with MeOH (4 × 4 mL), and the filtrate was concentrated. Column chromatography (DCM–MeOH, 0→8%) of the residue produced diamine 24 (71 mg, 81%) as a colorless syrup. HRMS (ESI): calcd. for C31H42F3N3O8 [M + H]+ m/z 642.2997; found m/z 642.2991.
Formic acid (25.5 μL, 0.663 mmol) and DCC (69 mg, 0.334 mmol) were added to a solution of diamine 24 (71 mg, 0.111 mmol) in a mixture of DCM–MeOH (9:1, 4 mL). The mixture was stirred for 1 h, the solvents were evaporated, and the residue was suspended in DCM (4 mL). Dicyclohexylurea was filtered off and washed with DCM (3 × 3 mL), the filtrate was concentrated, and the residue was purified by column chromatography (DCM–MeOH, 0→7%) to give bis(formamide) 25 (68 mg, 88%) as a white amorphous solid. HRMS (ESI): calcd. for C33H42F3N3O10 [M + NH4]+ m/z 715.3161; found m/z 715.3167.
A mixture of compound 25 (68 mg, 0.097 mmol) and Pd(OH)2/C (20 mg) in MeOH (3 mL) was stirred under hydrogen for 2 h at room temperature. The catalyst was remove by filtration and washed with MeOH (4 × 4 mL), and the filtrate was concentrated. Column chromatography of the residue afforded triol 26 (46 mg, 92%) as a white amorphous solid. HRMS (ESI): calcd. for C19H30F3N3O10 [M + Na]+ m/z 540.1775; found m/z 540.1780.
Ambersep 900 (OH−) (3 mL) was added to a solution of compound 26 (46 mg, 0.089 mmol) in 50% aqueous MeOH (2 mL). The mixture was kept for 1 h with periodic shaking, and then the resin was filtered off and washed with 50% aqueous MeOH (6 × 3 mL). The filtrate was concentrated and the residue was subjected to gel-permeation chromatography to give title compound 4 (36 mg, 92%) as a white amorphous solid. The data of NMR and mass-spectra were identical to those described previously (Tsvetkov and Nifantiev, 2023).
3-Aminopropy 4,6-dideoxy-4-formamido-α-d-mannopyranosyl-(1→2)-4,6-dideoxy-4-formamido-α-d-mannopyranosyl-(1→3)-4,6-dideoxy-4-formamido-α-d-mannopyranosyl-(1→2)-4,6-dideoxy-4-formamido-α-d-mannopyranoside (1). The title compound was synthesized in four steps from protected precursor 19 in total yield of 55% (for details see Supplementary Material). A white fluffy solid; contained a non-stoichiometric amount (0.45 equiv.) of AcOH; [α]D +55 (c 1, water). 1H NMR (600 MHz, D2O): δ 8.24–8.18 (m, 2.8 H, HZCON), 8.06-7.99 (m, 1.2 H, HECON), 5.11–4.92 (m, 4 H, 4 H-1), 4.20–3.77 (m, 15.8 H, 4 H-2, 4 H-3, 4 H-4Z, 4 H-5, CHaHbCH2CH2N), 3.61–3.56 (m, 1 H, OCHaHbCH2CH2N), 3.51–3.35 (m, 1.2 H, 4 H-4E), 3.16–3.07 (m, 2 H, OCH2CH2CH2N), 2.02–1.95 (m, 2 H, OCH2CH2CH2N), 1.90 (s, ∼1.4 H, CH3COOH), 1.30-1.21 (m, 12 H, 4 H-6). 13C NMR (150 MHz, D2O): δ 169.0, 168.9 (HECON), 166.1, 165.8 (HZCON), 103.5, 103.4, 103.1, 102.9, 101.9, 99.6 (4 C-1), 79.4, 79.2, 79.0, 78,7, 78.6, 78,4, 77.5 (C-2A, C-2C, C-3B), 70.1,70.0, 69.9, 69.6, 69.5, 69.2, 69.0, 68.9, 68.7, 68.6, 68.5, 68.3 (C-3A, C-5A, C-2B, C-5B, C-3C, C-5C, C-2D, C-3D, C-5D), 66.4 (OCH2CH2CH2N), 58.2, 58.0, 57.9, 56.7 (4 C-4E), 53.3, 53.1, 52.9, 52.2, 52.1 (4 C-4Z), 38.6 (OCH2CH2CH2N), 27.9 (OCH2CH2CH2N), 18.4, 18.2, 18.1, 18.0, 17.9 (4 C-6). HRMS (ESI): calcd. for C31H53N5O17 [M + H]+ m/z 768.3509; found m/z 768.3510.
3-Aminopropyl 4,6-dideoxy-4-formamido-α-d-mannopyranosyl-(1→3)-4,6-dideoxy-4-formamido-α-d-mannopyranosyl-(1→2)-4,6-dideoxy-4-formamido-α-d-mannopyranoside (2). The title compound was obtained in four steps from protected progenitor 17 in total yield of 54% (for details see Supplementary Material). A white fluffy solid; contained a non-stoichiometric amount (0.40 equiv.) of AcOH; [α]D +60 (c 1, water). 1H NMR (600 MHz, D2O): δ 8.23–8.18 (m, 2.1 H, 3 HZCON), 8.06–8.01 (m, 0.9 H, 3 HECON), 5.04-4.92 (m, 3 H, 3 H-1), 4.21–3.79 (m, 12.1 H, 3 H-2, 3 H-3, 3 H-4Z, 3 H-5, OCHaHbCH2CH2N), 3.61–3.56 (m, 1 H, OCHaHbCH2CH2N), 3.48 (t, 0.3 H, J = 10.1 Hz, H-4E), 3.43 (t, 0.3 H, J = 10.3 Hz, H-4E), 3.36 (t, 0.3 H, J = 10.5 Hz, H-4E), 3.17–3.07 (m, 2 H, OCH2CH2CH2N), 2.02–1.95 (m, 2 H, OCH2CH2CH2N), 1.91 (s, 1.2 H, 0.4 CH3COOH), 1.30–1.21 (m, 9 H, 3 H-6). 13C NMR (150 MHz, D2O): δ 169.0, 168.9 (3 HECON), 165.9, 165.8 (3 HZCON), 103.4, 103.2, 103.1, 102.9 (C-1B, C-1C), 99.4 (C-1A), 78.8, 77.8, 77.5, 77.4 (C-2A, C-3B), 70.3, 70.0, 69.8, 69.7, 69.2, 69.1 69.0, 68.8, 68.7, 68.6, 68.2, (C-2B, C-2C, C-3A, C-3C, C-5A, C-5B, C-5C), 66.2 (OCH2CH2CH2N), 57.9, 57.7, 56.5 (C-4EA, C-4EB, C-4EC), 53.1, 52.7, 51.6 (C-4ZA, C-4ZB, C-4ZC), 38.4 (OCH2CH2CH2N), 27.7 (OCH2CH2CH2N), 24.3 (CH3COOH), 17.9, 17.7 (C-6A, C-6B, C-6C). HRMS (ESI): calcd. for C24H42N4O13 [M + H]+ m/z 595.2821; found m/z 595.2819.
3-Aminopropyl 4,6-dideoxy-4-formamido-α-d-mannopyranosyl-(1→2)-4,6-dideoxy-4-formamido-α-d-mannopyranosyl-(1→3)-4,6-dideoxy-4-formamido-α-d-mannopyranoside (3). The title trisaccharide was synthesized in four steps from protected precursor 23 in overall yield of 52% (for details see Supplementary Material). A white fluffy solid; contained a non-stoichiometric amount (0.35 equiv.) of AcOH; [α]D +65 (c 1, water). 1H NMR (600 MHz, D2O): δ 8.22–8.19 (m, 2.2 H, 3 HZCON), 8.05-8.01 (m, 0.8 H, 3 HECON), 5.09–4.98 (m, 2 H, 2 H-1), 4.83 (d, 1 H, J1,2 = 2.0 Hz, H-1), 4.15–3.91 (m, 12.2 H, 3 H-2, 3 H-3, 3 H-4Z, 3 H-5, OCHaHbCH2CH2N), 3.62–3.57 (m, 1 H, OCHaHbCH2CH2N), 3.46 (t, 0.25 H, J = 10.3 Hz, H-4E), 3.43-3.35 (m, 0.5 H, 2 H-4E), 3.17–3.07 (m, 2 H, OCH2CH2CH2N), 2.03-1.95 (m, 2 H, OCH2CH2CH2N), 1.91 (s, ∼1 H, 0.35 CH3COOH), 1.29–1.20 (m, 9 H, 3 H-6). 13C NMR (150 MHz, D2O): δ 169.1, 169.0 (3 HECON), 166.2, 166.1 165.9 (3 HZCON), 103.6, 103.5, 103.1, 102.1, 102.0, 100.9, 100.8 (C-1A, C-1B, C-1C), 79.4, 79.3, 78.7, 78.3, 77.9 (C-3A, C-2B), 70.2, 70.0, 69.7, 69.5, 69.3, 69.2, 69.1, 69.0, 68.9, 68.7, 68.6, 68.5, 68.2, 68.1 (C-2A, C-5A, C-3B, C-5B, C-2C, C-3C, C-5C), 66.2 (OCH2CH2CH2N), 58.1, 57.9, 56.7 (C-4EA, C-4EB, C-4EC), 53.1, 53.0, 52.4 (C-4ZA, C-4ZB, C-4ZC), 38.6 (OCH2CH2CH2N), 27.9 (OCH2CH2CH2N), 24.4 (CH3COOH), 18.4, 18.3, 18.2, 18.0 (C-6A, C-6B, C-6C). HRMS (ESI): calcd. for C24H42N4O13 [M + H]+ m/z 595.2821; found m/z 595.2815.
3-Aminopropyl 4,6-dideoxy-4-formamido-α-d-mannopyranoside (5). The title glycoside was synthesized in four steps from protected compound 13 in total yield of 57% (for details see Supplementary Material). A colorless glassy solid; contained a non-stoichiometric amount (0.4 equiv.) of AcOH; [α]D +42 (c 1, water). 1H NMR (600 MHz, D2O): δ 8.19 (s, 0.7 H, HZCON), 8.02 (s, 0.4 H, HECON), 4.85–4.84 (m, 1 H, H-1), 3.98 (dd, 0.3 H, J2,1 = 2.0 Hz, J2,3 = 3.2 Hz, H-2E), 3.96 (t, 0.7 H, J = 2.4 Hz, H-2Z), 3.91-3.77 (m, 3.7 H, H-3, H-4Z, H-5, OCHaHbCH2CH2N), 3.62–3.57 (m, 1 H, OCHaHbCH2CH2N), 3.36 (t, 0.3 H, J = 10.1 Hz, H-4E), 3.17–3.08 (m, 2 H, OCH2CH2CH2N), 2.03–1.95 (m, 2 H, OCH2CH2CH2N), 1.90 (s, 1.2 H, CH3COO–), 1.26 (d, 0.9 H, J6,5 = 6.4 Hz, H-6E), 1.22 (d, 2.1 H, J6,5 = 6.3 Hz, H-6Z). 13C NMR (150 MHz, D2O): δ 169.0 (HECON), 166.1 (HZCON), 100.9 (C-1), 70.3 (C-2), 69.2 (C-3), 68.5 (C-5Z), 68.2 (C-5E), 66.2 (OCH2CH2CH2N), 57.9 (C-4E), 53.2 (C-4Z), 38.7 (OCH2CH2CH2N), 27.8 (OCH2CH2CH2N), 18.0 (C-6Z), 17.9 (C-6E). HRMS (ESI): calcd. for C10H20N2O5 [M + H]+ m/z 249.1445; found m/z 249.1449.
3-Aminopropy α-d-rhamnopyranosyl-(1→2)-4,6-dideoxy-4-formamido-α-d-mannopyranosyl-(1→2)-4,6-dideoxy-4-formamido-α-d-mannopyranosyl-(1→3)-4,6-dideoxy-4-formamido-α-d-mannopyranosyl-(1→2)-4,6-dideoxy-4-formamido-α-d-mannopyranoside (6). The title pentasaccharide was obtained in four steps from protected precursor 29 in total yield of 45% (for details see Supplementary Material). A white fluffy solid; contained a non-stoichiometric amount (0.35 equiv.) of AcOH; [α]D +55 (c 1, water). 1H NMR (600 MHz, D2O): δ 8.25–8.19 (m, 2.9 H, 3 HZCON), 8.07–8.00 (m, 1.1 H, 3 HECON), 5.19–4.93 (m, 5 H, 5 H-1), 4.20-3.73 (m, 20 H, H, 5 H-2, 5 H-3, 4 H-4Z, H-4E, 5 H-5, OCHaHbCH2CH2N), 3.62–3.57 (m, 1 H, OCHaHbCH2CH2N), 3.52–3.39 (m, 1.1 H, 4 H-4E), 3.15–3.06 (m, 2 H, OCH2CH2CH2N), 2.01–1.95 (m, 2 H, OCH2CH2CH2N), 1.91 (s, 1.4 H, CH3COOH), 1.31–1.22 (m, 15 H, 5 H-6). 13C NMR (150 MHz, D2O): δ 169.0, 168.9 (3 HECON), 166.1, 165.7 (3 HZCON), 103.3, 103.1, 103.0, 102.1, 102.0, 101.9, 101.7, 99.6 (5 C-1), 79.0, 78.8, 78.6, 78.2, 77.9, 77.6, 77.4 (C-2A, C-3B, C-2C, C-2D), 73.2, 71.4, 71.3, 70.4, 70.0, 69.9, 69.7, 69.5, 69.4, 68.9, 68.7, 68.4 (C-3A, C-5A, C-2B, C-5B, C-3C, C-5C, C-3D, C-5D, C-2E, C-3E, C-4E, C-5E), 66.4 (OCH2CH2CH2N), 58.2, 58.1, 58.0, 56.6 (4 C-4E), 53.3, 53.2, 53.1, 52.2, 52.1 (4 C-4Z), 38.6 (OCH2CH2CH2N), 28.1 (OCH2CH2CH2N), 18.5, 18.3, 18.1, 18.0, 17.9, 17.8 (5 C-6). HRMS (ESI): calcd. for C37H63N5O21 [M + H]+ m/z 914.4088; found m/z 914.4089.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author contributions
YT: Investigation, Writing–original draft. TV: Investigation, Writing–original draft. SE: Investigation, Writing–original draft. OS: Writing–original draft, Investigation. YK: Investigation, Writing–original draft. VK: Investigation, Writing–original draft. NN: Conceptualization, Funding acquisition, Writing–original draft.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Russian Science Foundation (Grant 19-73-30017-P).
Acknowledgments
The authors are grateful to Dr. A. S. Dmitrenok and Dr. A. O. Chizhov for recording NMR and mass spectra of synthesized compounds, respectively.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2024.1424157/full#supplementary-material
References
Argunov, D. A., Krylov, V. B., and Nifantiev, N. E. (2015). Convergent synthesis of isomeric heterosaccharides related to the fragments of galactomannan from Aspergillus fumigatus. Org. Biomol. Chem. 13, 3255–3267. doi:10.1039/c4ob02634a
Ariosa-Alvarez, A., Arencibia-Mohar, A., Madrazo-Alonso, O., Garcia-Imia, L., Sierra-Gonzalez, G., and Verez-Bencomo, V. (1998). Synthesis of the Vibrio cholerae O1 ogawa and inaba terminal disaccharides with dioxolane-type spacers and their coupling to proteins. J. Carbohydr. Chem. 17, 1307–1320. doi:10.1080/07328309808002355
Bock, K., and Pedersen, C. (1974). A study of 13CH coupling constants in hexopyranoses. J. Chem. Soc. Perkin Trans. 2, 293–297. doi:10.1039/P29740000293
Bundle, D. R., Gerken, M., and Peters, T. (1988). Synthesis of antigenic determinants of the Brucella A antigen, utilizing methyl 4-Azido-4,6-Dideoxy-α-D-Mannopyranoside efficiently derived from D-mannose. Carbohydr. Res. 174, 239–251. doi:10.1016/0008-6215(88)85094-8
Bundle, D. R., and McGiven, J. (2017). Brucellosis: improved diagnostics and vaccine insights from synthetic glycans. Acc. Chem. Res. 50, 2958–2967. doi:10.1021/acs.accounts.7b00445
Caroff, M., Bundle, D. R., and Perry, M. B. (1984). Structure of the O-chain of the phenol-phase soluble cellular lipopolysaccharide of Yersinia enterocolitica serotype O:9. Eur. J. Biochem. 139, 195–200. doi:10.1111/j.1432-1033.1984.tb07994.x
Duncombe, L., Howells, L., Haughey, A., Taylor, A. V., Kaveh, D., Gűrbilek, S. E., et al. (2022). The tip of Brucella O-polysaccharide is a potent epitope in response to brucellosis infection and enables short synthetic antigens to Be superior diagnostic reagents. Microorganisms 10, 708. doi:10.3390/microorganisms10040708
Franco, M. P., Mulder, M., Gilman, R. H., and Smits, H. L. (2007). Human brucellosis. Lancet Infect. Dis. 7, 775–786. doi:10.1016/S1473-3099(07)70286-4
Ganesh, V. N., Sadowska, J. M., Sarkar, S., Howells, L., McGiven, J., and Bundle, D. R. (2014). Molecular recognition of Brucella A and M antigens dissected by synthetic oligosaccharide glycoconjugates leads to a disaccharide diagnostic for brucellosis. J. Am. Chem. Soc. 136, 16260–16269. doi:10.1021/ja5081184
Gening, M. L., Kurbatova, E. A., Tsvetkov, Y. E., and Nifantiev, N. E. (2015). Development of approaches to a third-generation carbohydrate-conjugate vaccine againstStreptococcus pneumoniae: the search for optimal oligosaccharide ligands. Russ. Chem. Rev. 84, 1100–1113. doi:10.1070/RCR4574
Godfroid, J., Nielsen, K., and Saegerman, C. (2010). Diagnosis of brucellosis in livestock and wildlife. Croat. Med. J. 51, 296–305. doi:10.3325/cmj.2010.51.296
Grant, O. C., Smith, H. M. K., Firsova, D., Fadda, E., and Woods, R. J. (2014). Presentation, presentation, presentation! Molecular-level insight into linker effects on glycan array screening data. Glycobiology 24, 17–25. doi:10.1093/glycob/cwt083
Guiard, J., Paszkiewicz, E., Sadowska, J. M., and Bundle, D. R. (2013). Design and synthesis of a universal antigen to detect brucellosis. Angew. Chem. Int. Ed. 52, 7181–7185. doi:10.1002/anie.201302303
Hou, S.-j., and Kovač, P. (2010). Enhanced stereoselectivity of α-mannosylation under thermodynamic control using trichloroacetimidates. Carbohydr. Res. 345, 999–1007. doi:10.1016/j.carres.2010.03.025
Kazakova, E. D., Yashunsky, D. V., Krylov, V. B., Bouchara, J.-P., Cornet, M., Valsecchi, I., et al. (2020). Biotinylated oligo-α-(1→4)-D-galactosamines and their N-acetylated derivatives: α-stereoselective synthesis and immunology application. J. Am. Chem. Soc. 142, 1175–1179. doi:10.1021/jacs.9b11703
Kochetkov, N. K., Nifant’ev, N. E., and Backinowsky, L. V. (1987). Synthesis of the capsular polysaccharide of type 14. Tetrahedron 43, 3109–3121. doi:10.1016/S0040-4020(01)86852-6
Komarova, B. K., Wong, S. S., Orekhova, M. V., Tsvetkov, Y. E., Krylov, V. B., Beauvais, A., et al. (2018). Chemical synthesis and application of biotinylated oligo-α-(1→3)-D-glucosides to study the antibody and cytokine response against the cell wall α-(1→3)-D-glucan of Aspergillus fumigatus. J. Org. Chem. 83, 12965–12976. doi:10.1021/acs.joc.8b01142
Krylov, V. B., Solovev, A. S., Argunov, D. A., Latgé, J.-P., and Nifantiev, N. E. (2019). Reinvestigation of carbohydrate specificity of EB-A2 monoclonal antibody used in the immune detection of Aspergillus fumigatus galactomannan. Heliyon 5, e01173. doi:10.1016/j.heliyon.2019.e01173
Kubler-Kielb, J., and Vinogradov, E. (2013). Reinvestigation of the structure of Brucella O-antigens. Carbohydr. Res. 378, 144–147. doi:10.1016/j.carres.2013.03.021
Laverde, D., Romero-Saavedra, F., Argunov, D. A., Enotarpi, J., Krylov, V. B., Kalfopoulou, E., et al. (2020). Synthetic oligomers mimicking capsular polysaccharide diheteroglycan are potential vaccine candidates against encapsulated enterococcal infections. ACS Infect. Dis. 6, 1816–1826. doi:10.1021/acsinfecdis.0c00063
Mandal, S. S., Duncombe, L., Ganesh, N. V., Sarkar, S., Howells, L., Hogarth, P. J., et al. (2017a). Novel solutions for vaccines and diagnostics to combat brucellosis. ACS Cent. Sci. 3, 224–231. doi:10.1021/acscentsci.7b00019
Mandal, S. S., Ganesh, N. V., Sadowska, J. M., and Bundle, D. R. (2017b). Synthetic glycoconjugates characterize the fine specificity of Brucella A and M monoclonal antibodies. Org. Biomol. Chem. 15, 3874–3883. doi:10.1039/c7ob00445a
McGiven, J., Howells, L., Duncombe, L., Stack, J., Ganesh, N. V., Guiard, J., et al. (2015). Improved serodiagnosis of bovine brucellosis by novel synthetic oligosaccharide antigens representing the capping M epitope elements of Brucella O-polysaccharide. J. Clin. Microbiol. 53, 1204–1210. doi:10.1128/JCM.03185-14
Mogues, T., Li, J., Coburn, J., and Kuter, D. J. (2005). IgG antibodies against bovine serum albumin in humans—their prevalence and response to exposure to bovine serum albumin. J. Immunol. Methods 300, 1–11. doi:10.1016/j.jim.2005.01.022
Mukherjee, M. M., Xu, P., Stevens, E. D., and Kováć, P. (2019). Towards the complete synthetic O-antigen of Vibrio cholerae O1, serotype inaba: improved synthesis of the conjugation-ready upstream terminal hexasaccharide determinant. RSC Adv. 9, 36440–36454. doi:10.1039/c9ra08232h
Nielsen, K. (2002). Diagnosis of brucellosis by serology. Vet. Microbiol. 90, 447–459. doi:10.1016/S0378-1135(02)00229-8
Perry, M. B., and Bundle, D. R. (1990). Antigenic relationships of the lipopolysaccharides of Escherichia hermannii strains with those of Escherichia coli O157:H7, Brucella melitensis, and Brucella abortus. Infect. Immun. 58, 1391–1395. doi:10.1128/iai.58.5.1391-1395.1990
Peters, V., Brisson, J.-R., and Bundle, D. R. (1990). Conformational analysis of key disaccharide components of Brucella A and M antigens. Can. J. Chem. 68, 979–988. doi:10.1139/v90-154
Sjöwall, C., Kastbom, A., Almroth, G., Wetterö, J., and Skogh, T. (2011). Beware of antibodies to dietary proteins in “antigen-specific” immunoassays! Falsely positive anticytokine antibody tests due to reactivity with bovine serum albumin in rheumatoid arthritis (the Swedish TIRA project). J. Rheumatol. 38, 215–220. doi:10.3899/jrheum.100690
Solovev, A. S., Denisova, E. M., Kurbatova, E. A., Kutsevalova, O. Y., Boronina, L. G., Ageevets, V. A., et al. (2023). Synthesis of methylphosphorylated oligomannosides structurally related to lipopolysaccharide O-antigens of Klebsiella pneumoniae serotype O3 and their application for detection of specific antibodies in rabbit and human sera. Org. Biomol. Chem. 21, 8306–8319. doi:10.1039/d3ob01203d
Temme, J. S., Campbell, C. T., and Gildersleeve, J. C. (2019). Factors contributing to variability of glycan microarray binding profiles. Faraday Discuss. 219, 90–111. doi:10.1039/c9fd00021f
Tsvetkov, Y. E., Backinowsky, L. V., and Kochetkov, N. K. (1989). Synthesis of a common polysaccharide antigen of Pseudomonas aeruginosa as the 6-aminohexyl glycoside. Carbohydr. Res. 193, 75–90. doi:10.1016/0008-6215(89)85108-0
Tsvetkov, Y. E., and Nifantiev, N. E. (2023). Synthesis of a spacer-armed disaccharide structurally related to the M antigenic fragment of Brucella O-polysaccharides. Russ. Chem. Bull. 72, 2731–2737. doi:10.1007/s11172-023-4079-4
Wong, S. S. W., Krylov, V. B., Argunov, D. A., Karelin, A. A., Bouchara, J.-P., Fontaine, T., et al. (2020). Potential of chemically synthesized oligosaccharides to define the carbohydrate moieties of the fungal cell wall responsible for the human immune response, using Aspergillus fumigatus galactomannan as a model. Ex. Aspergillus fumigatus Cell wall galactomannan. mSphere 5 (e5), e00688. doi:10.1128/mSphere.00688-19
Keywords: Brucella, lipopolysaccharide, O-polysaccharide, M epitope, 4-formamido, 4,6-dideoxy-D-mannose, oligosaccharides
Citation: Tsvetkov YE, Volkov TM, Eremin SA, Sklyarov OD, Kulakov YK, Krylov VB and Nifantiev NE (2024) New synthesis of oligosaccharides modelling the M epitope of the Brucella O-polysaccharide. Front. Chem. 12:1424157. doi: 10.3389/fchem.2024.1424157
Received: 27 April 2024; Accepted: 30 May 2024;
Published: 21 June 2024.
Edited by:
Steve Suib, University of Connecticut, United StatesCopyright © 2024 Tsvetkov, Volkov, Eremin, Sklyarov, Kulakov, Krylov and Nifantiev. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nikolay E. Nifantiev, bmVuQGlvYy5hYy5ydQ==