 Vinh Do Cao
Vinh Do Cao Seewon Joung
Seewon Joung- Department of Chemistry, Inha University, Incheon, Republic of Korea
Catalytic hydroboration and hydrosilylation have emerged as promising strategies for the reduction of unsaturated hydrocarbons and carbonyl compounds, as well as for the dearomatization of N-heteroarenes. Various catalysts have been employed in these processes to achieve the formation of reduced products via distinct reaction pathways and intermediates. Among these intermediates, N-silyl enamines and N-boryl enamines, which are derived from hydrosilylation and hydroboration, are commonly underestimated in this reduction process. Because these versatile intermediates have recently been utilized in situ as nucleophilic reagents or dipolarophiles for the synthesis of diverse molecules, an expeditious review of the synthesis and utilization of N-silyl and N-boryl enamines is crucial. In this review, we comprehensively discuss a wide range of hydrosilylation and hydroboration catalysts used for the synthesis of N-silyl and N-boryl enamines. These catalysts include main-group metals (e.g., Mg and Zn), transition metals (e.g., Rh, Ru, and Ir), earth-abundant metals (e.g., Fe, Co, and Ni), and non-metal catalysts (including P, B, and organocatalysts). Furthermore, we highlight recent research efforts that have leveraged these versatile intermediates for the synthesis of intriguing molecules, offering insights into future directions for these invaluable building blocks.
1 Introduction
Enamine is a functional moiety found in a variety of natural products, bioactive molecules, and pharmaceuticals (Burgess et al., 1991; Goldmann and Stoltefuss, 1991; Gordeev et al., 1998; Edafiogho et al., 2007; Sashidhara et al., 2009; Poulsen, 2021). Within the realm of synthetic organic chemistry, enamines serve as versatile intermediates (Fu et al., 2019; Wang et al., 2023) and catalysts (Notz et al., 2004; Mukherjee et al., 2007; Zou et al., 2018) to facilitate the synthesis of diverse chemical structures, including natural products and heterocyclic compounds (Cheng et al., 2004; Hanessian and Chattopadhyay, 2014; Wang, 2015). Traditionally, enamines are prepared through condensation reactions between secondary amines and carbonyl compounds, α,β-elimination of amides, or reductive acylation of ketoximes (Mengya, 2018). However, these conventional methods give low conversion or exhibit narrow functional group tolerance due to their harsh reaction conditions (Dehli et al., 2005). Consequently, numerous alternative methods have been developed, including the amination of alkenes or alkynes (Beller et al., 2002; Ahmed et al., 2003), methylenation of amides (Shen and Porco, 2000), dehydrogenation of tertiary amines (Zhang et al., 2003), cross-coupling of amines and alkenyl bromides (Barluenga et al., 2004), and N-formylation of amines with CO2 and PhSiH3 to synthesize linear enamines (Nakaoka et al., 2023). Furthermore, the cobalt-catalyzed hydrogen transfer of amines (Bolig and Brookhart, 2007) and palladium-catalyzed intramolecular amination of alkenes (Jiang et al., 2017) are utilized to synthesize cyclic enamines. In addition, the hydroboration and hydrosilylation of N-heteroarenes and conjugated imine or nitrile compounds are also used to obtain enamine derivatives, particularly borylated and silylated enamines. In contrast to traditional reductive methods such as metal hydride reduction and hydrogenation, the use of silanes and boranes as reducing agents enables milder reaction conditions, high chemo- and regioselectivity, and compatibility with diverse functional groups (Park and Chang, 2017). Consequently, hydroboration and hydrosilylation have emerged as alternatives to the use of H2, which is a common method in catalytic reduction chemistry (Marciniec, 2005; Arai and Ohkuma, 2008). Moreover, hydroboration and hydrosilylation demonstrate catalytic versatility, as they can be facilitated by various metal-based complexes, metalloids, and non-metal compounds. The initial steps, depending on the activity of the catalyst, may involve the activation of the B–H bond in HBpin and the Si–H bond in silanes, or coordination with heteroatom centers in the reactant substrates. However, the general catalytic mechanisms can be categorized into outer-sphere and inner-sphere pathways (Park and Brookhart, 2010; Iglesias et al., 2014; Corey, 2016; Lipke et al., 2017), which determine the hydride attack pathway through the formation of distinct intermediates. The chemical reactivities, selectivities, and primary mechanistic insights of these hydroboration and hydrosilylation reactions have been extensively discussed in reviews focusing on the hydroelementation of alkene (Du and Huang, 2017), alkyne (Saptal et al., 2020), nitrile (Das et al., 2022), as well as dearomatization of unactivated N-heteroarenes (Park and Chang, 2017; Park, 2020; Escolano et al., 2024). A borane-catalyzed double hydrosilylation for the formation of sp3 C–Si bonds (Park, 2019) and single hydroelementation reaction of N-heteroarenes were also reviewed (Park, 2024) (Figure 1A). However, a review focusing on the synthesis and utility of N-boryl and N-silyl enamines has not yet been reviewed. Thus, in this review, we first outline the formation of N-boryl and N-silyl enamines via the 1,4-reduction of quinolines, conjugated nitriles/aldimines, 1,2-reduction of isoquinoline, and both 1,2- and 1,4-reduction of pyridines. Then, we summarized their subsequent applications in nucleophilic additions and Diels–Alder reactions (Figure 1B).
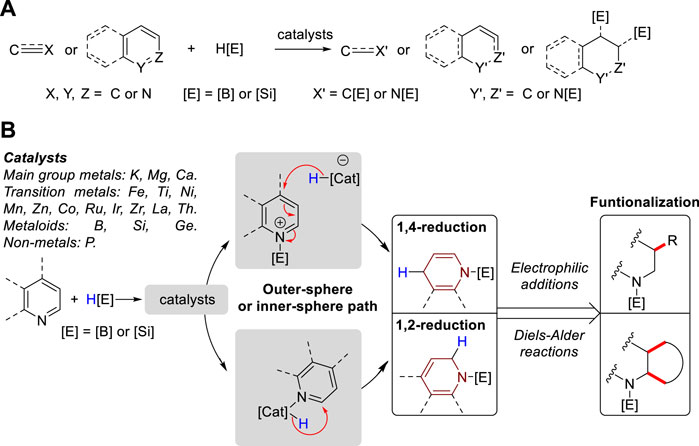
Figure 1. (A) Previous reviews on hydroelementation of pi-bond system. (B) This review: Syntheses of N-boryl and N-silyl enamines using hydroboration and hydrosilylation.
2 Hydroboration in the synthesis of N-boryl enamines
2.1 Alkali- and alkaline-earth-metal-catalyzed hydroboration of N-heteroarenes
The magnesium-catalyzed hydroboration of pyridines and isoquinolines has evolved from mononuclear structures (Mg I) to the dinuclear β-diketiminate magnesium hydrides (Mg II). Recently, a phosphinimino-amido magnesium complex (Mg III) has also been reported (Figure 2A). While the Mg I and Mg II systems exhibited temperature-dependent selectivity between 1,2- and 1,4-hydroboration (Arrowsmith et al., 2011; Intemann et al., 2014), the Mg III catalyst showed highly selective 1,2-hydroboration (Liu et al., 2020). Furthermore, the formation of both regioisomers under Mg I and Mg II conditions indicated that the magnesium hydride complex was not a key step in these mechanisms. Instead, the generation of an intermediate (Int 1, Figure 2) through the reaction of the [Mg]-1,2-reduced intermediate with HBpin directly transfers a hydride from boron to the 2- or 4-position of the pyridine ligand (Arrowsmith et al., 2011; Intemann et al., 2014). Moreover, density functional theory (DFT) calculations using the Mg III catalytic system showed that the dearomatization process resulted in a reasonable energy barrier for the 1,2-reduced intermediate, whereas 1,4-dearomatization presented considerable challenges because of its high energy demand (Liu et al., 2020).
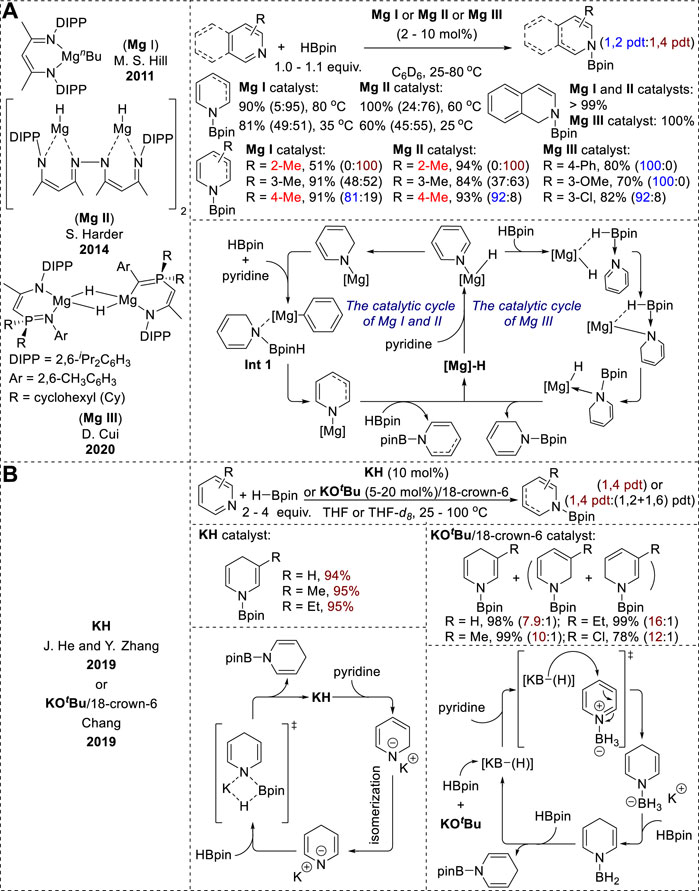
Figure 2. (A) Alkaline-earth-metal-catalyzed hydroboration of N-heteroarenes. (B) Alkali-metal-catalyzed hydroboration of N-heteroarenes.
Recently, Zhang et al. and Chang et al. reported the potassium-catalyzed hydroboration of N-heteroarenes (Figure 2B). Both catalytic conditions resulted in the highly selective 1,4-hydroboration of pyridines (Liu et al., 2019; Jeong et al., 2019). However, the KH catalytic systems reported by Zhang exhibited excellent 1,2-selectivity with quinoline substrates. The use of THF solvents and elevated reaction temperatures enhanced the 1,4-selectivity in the hydroboration of pyridines. This selectivity is due to the formation of a 1,4-reduced intermediate through the thermodynamic isomerization of the 1,2-reduced intermediate (Liu et al., 2019). Conversely, the 1,4-regioselectivity in the KOtBu-promoted hydroboration in Chang’s study originated from an outer-sphere mechanism. KOtBu and HBpin react to form various borohydride species as active hydrides in the presence of BH3. These active hydride species then generate 1,4-reduced intermediates via nucleophilic hydride attack on the pyridine–BH3 adducts. Subsequent hydride transfer generates N-borylated-1,4-dihydropyridines, while liberating BH3 through σ-bond metathesis (Jeong et al., 2019).
2.2 Earth-abundant transition-metal-catalyzed 1,4-hydroboration
In addition to the main group of metals, various earth-abundant transition metal catalysts, such as Ni(acac)2/phosphine ligand, 1-Methylimidazole-based Mn pincer complex [LMn(CO)2], and the heterobinuclear Cu/Fe catalyst (IPr)CuFp exhibited 1,4-selective hydroboration of pyridines and quinolines (Figure 2). In the (IPr)CuFp catalytic system, 3-substituted pyridines with electron-donating groups underwent effective reduction with excellent yields, whereas 2- and 4-substituted pyridines exhibited minimal-to-no conversion (Yu et al., 2020). Conversely, Ni(acac)2/PCyp3 circumvented steric hindrance at the 4-position of pyridine. Furthermore, electron-rich substituents at the 3-position enhanced the reactivity, resulting in high yields, whereas electron-poor pyridines showed no reactivity (Tamang et al., 2018); (LMn(CO)2) exhibited 1,4-hydroboration reactivity on various substituted quinolines, resulting in full conversion with high regioselectivity (Wang et al., 2023).
Kinetic and mechanistic investigations of the Ni(acac)2/PCyp3 catalytic system revealed the formation of bis(heteroarene) complexes as the first step (Tamang et al., 2018). In contrast, the (IPr)CuFp catalytic system formed (IPr)CuH as an active hydride species. Pyridine is then activated to form a pyridyl cation. N-borylated-1,4-dihydropyridines are produced through the interaction of an active hydride species at the 4-position of the pyridyl cation (Int 2, Figure 3) (Yu et al., 2020). Additionally, mechanistic studies of [LMn(CO)2] showed that the 1,2-adduct was kinetically favorable, whereas the 1,4-adduct was more stable and was generated via a thermodynamic process. The unusual 1,4-regioselectivity was derived from the decreased free-energy barrier for the 1,4-hydroboration compared to that for the 1,2-hydroboration. This could be achieved by using a 1-methylimidazole-based pincer Mn catalyst featuring cooperative C–H···N and π···π noncovalent interactions between the 1-methylimidazole moiety and the quinoline substrate (Int 3, Figure 3) (Wang et al., 2023).
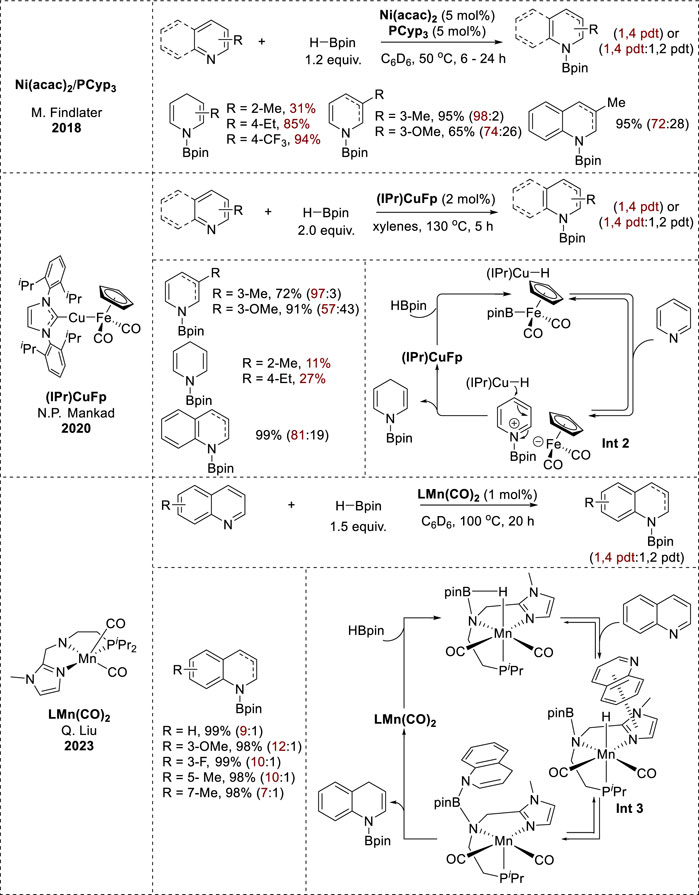
Figure 3. Earth-abundant transition-metal-catalyzed 1,4-hydroboration.
2.3 Earth-abundant transition-metal-catalyzed 1,2-hydroboration
Recent studies have reported the 1,2-selective hydroboration of N-heteroarenes catalyzed by a variety of earth-abundant transition metals (Figure 3). It is generally assumed that the mechanistic pathways involve various hydride intermediate complexes, including metal hydrides and borohydrides, facilitating the hydride shift to the C2-position in N-heteroarenes.
The N2-bridged diiron complex [Cp*-(Ph2PC6H4S)Fe]2 (μ-N2), zinc alkyl complex LZnEt, Cp*Ni(1,2-Ph2PC6H4O), and Mn (hmds)2 demonstrated good reactivity with high 1,2-selectivity towards various substituted pyridines and isoquinolines (Zhang et al., 2017; Liu et al., 2019; Wang et al., 2020; Ghosh and Jacobi von Wangelin, 2021). Next, the zinc hydride NacNacZnH exhibited limited 1,2-hydroboration reactivity with a few pyridine substrates to give cyclic N-boryl enamines (Lortie et al., 2017). Additionally, a cobalt-based complex ((PPh3)3CoH(N2)/pincer) with an N-heterocyclic carbene ligand (MeCNC) showed poor regioselectivity depending on the position of the substituent in the pyridines. Specifically, pyridine derivatives with diverse functional groups at the 4-position afforded excellent yields of the 1,2-hydroborylated products, whereas unsubstituted pyridines and pyridines with C3 substituents showed a notable preference for 1,4-hydroboration products (Meher et al., 2023).
In accordance with the inner-sphere pathway, the hydroboration mechanism of the zinc alkyl complexes LZnEt and Mn (hmds)2 involves the formation of metal hydride species through the interaction of metal catalysts with HBpin in the initial step (Wang et al., 2020; Ghosh and Jacobi von Wangelin, 2021). The insertion of a hydride into C=N in N-heteroarenes (Int 6, Int 7 in Figure 4) generates a 1,2-reduced intermediate complex that undergoes metathesis with HBpin to form N-boryl-dihydropyridines.
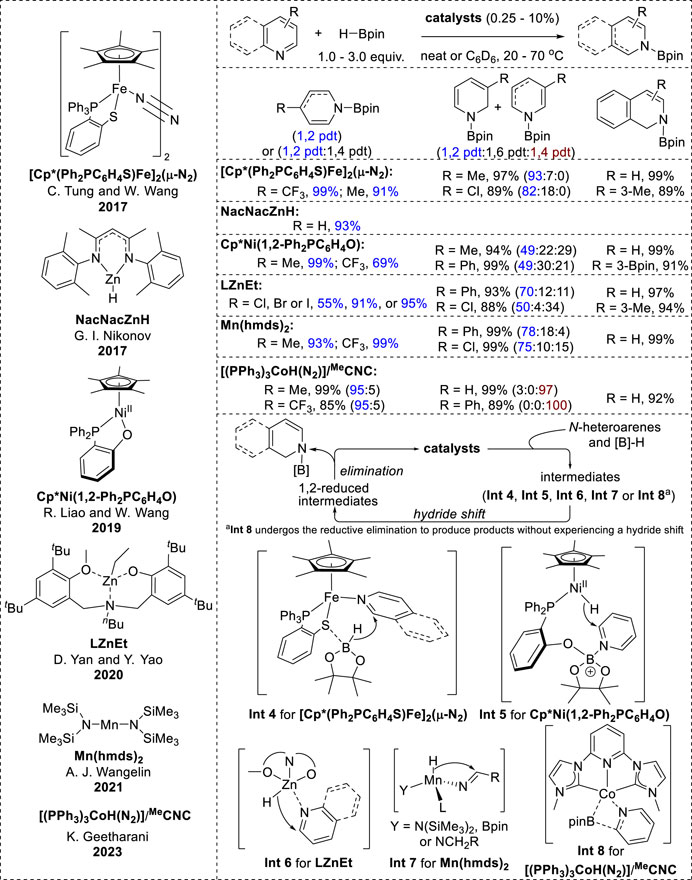
Figure 4. Earth-abundant transition-metal-catalyzed 1,2-hydroboration.
Differently from conventional catalytic mechanisms, (IPr)CuFp activates the B–H bond in HBpin, resulting in the generation of hydride species, whereas the iron–thiolate catalyst coordinates with N-heteroarenes and the thiolate interacts with HBpin. This interaction further facilitates hydride transfer from HBpin to the C2 position of the N-heteroarene ligand (Int 4, Figure 4) (Zhang et al., 2017). Additionally, the catalytic reaction of Cp*Ni(1,2-Ph2PC6H4O) is initiated by the dissociation of the B–H bond via the activation of HBpin and the nickel (II) complex, forming a nickel (II) hydride with an oxygen-stabilized boron moiety. Subsequently, nickel hydride is transferred to the C2-position of pyridine, which coordinates with the boron atom of the ligand (Int 5, Figure 4) (Liu et al., 2019). Eventually, N-borylated-1,2-dihydropyridine is generated through cleavage of the O–B bond. Similarly, the proposed catalytic cycle of (PPh3)3CoH(N2)/(MeCNC) starts with the reaction of the in-situ-formed cobalt hydride [(MeCNC)CoH] with HBpin. This reaction generates [(MeCNC)Co(Bpin)] and simultaneously liberates H2 without a hydride shift to C=N in pyridines. Int 8 (Figure 4) was then formed, as [(MeCNC)Co(Bpin)] was coordinated with pyridine. Subsequent transfer of the Bpin moiety, followed by the reductive elimination of Int 8, resulted in the formation of the N-boryl enamine product (Meher et al., 2023).
2.4 Metalloid and non-metal catalyzed 1,4-hydroboration
Recently, the borane ArF2BMe, NHC-parent silyliumylidene cation complex [(IMe)2SiH]I, and N-heterocyclic germylene LSi(NAr)2GeOTf (L = PhC(NtBu)2 and Ar = 2,6-iPr2C6H3) were utilized for the metalloid-catalyzed hydroboration of pyridines (Figure 4). These catalysts demonstrated highly selective 1,4-hydroboration of various substituted pyridines. Notably, in the borane (ArF2BMe) catalytic system, 3-substituted pyridines with electron-donating groups decreased the reactivity and electron-withdrawing groups accelerated the reactivity with excellent chemoselectivity (Fan et al., 2015). Pyridines bearing C=O and CN groups were tolerated under [(IMe)2SiH]I catalyst conditions (Leong et al., 2019). Moreover, LSi(NAr)2GeOTf exhibited notable efficiency in the 1,4-hydroboration of 3-substituted pyridines bearing both electron-donating and electron-withdrawing groups (Hu et al., 2021). In the ArF2BMe and LSi(NAr)2GeOTf catalytic systems, the mechanism involved an ionic hydroboration. The formation of intermediate Int 9 (Figure 5) and the [ArF2B(H)Me] anion hydride, as well as that of Int 11 (Figure 5) and the germylene hydride, plays a crucial role. 1,4-Reduction occurs via hydride transfer from the [ArF2B(H)Me] anion or germylene hydride to the 4-position of the pyridine in Int 9 or Int 11, resulting in the formation of N-boryl 1,4-dihydropyridine. Notably, the steric effects of the silaamidinate ligand or bulky borohydride may determine the 1,4-selectivity when the hydride attacks the pyridine moiety (Fan et al., 2015; Hu et al., 2021). In contrast, the catalytic mechanism of [(IMe)2SiH]I is initiated by the HBpin-activating silylium catalyst, which facilitates a nucleophilic attack at the C4-position of the pyridines to form the hydroborated intermediate Int 10 (Figure 5). Subsequently, Int 10 underwent hydride transfer from the borane moiety to the C4-position, replacing the C–Si bond and leading to the regeneration of the catalyst and formation of N-boryl-1,4-dihydropyridines (Leong et al., 2019).
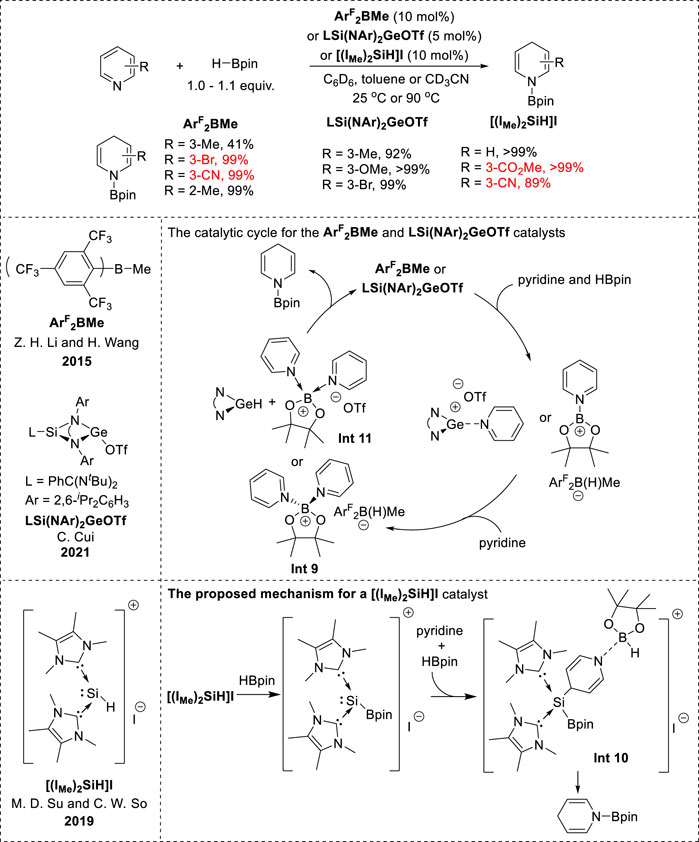
Figure 5. Metalloid-catalyzed 1,4-hydroboration.
In 2018, two distinct non-metal catalytic systems were reported for the 1,4-hydroboration of pyridine. Chong and Kinjo’s group utilized N-heterocyclic phosphenium triflates (NHP-OTf) as catalysts and demonstrated their regioselectivity and tolerance to electron-donating groups (Rao et al., 2018). In contrast, Speed et al. employed a neutral diazaphospholene catalyst that exhibited superior selectivity towards certain electron-deficient pyridines, and the reaction operated well in solvents with low polarity, such as benzene-d6 or diethyl ether, in which Kinjo’s procedure failed (Hynes et al., 2018). The reactions with both cationic and neutral phosphor catalysts resulted in high yields and chemoselectivities. The reaction mechanism for phosphenium triflates (NHP-OTf) was initiated by activation of the B–H bond of HBpin with pyridine and a phosphonium triflate catalyst, leading to the formation of the Py-Bpin-OTf complex. This intermediate complex then coordinates with a second pyridine molecule to afford the boronium [(Py)2∙Bpin]OTf (Int 12, Figure 6). Subsequently, one of the activated pyridine moieties in Int 12 undergoes reduction via hydride transfer from NHP-H, resulting in the formation of either N-boryl 1,2-dihydropyridine or N-boryl 1,4-dihydropyridine, while simultaneously regenerating the phosphonium catalyst (Rao et al., 2018). However, the neutral diazaphospholene catalyst facilitated the interaction of pyridine with H-Bpin, resulting in the formation of a pyridinium borate complex (Int 13, Figure 6), which was not formed during Kinjo’s catalytic cycle, along with diazaphospholene hydride. Subsequently, this Int 13 complex undergoes hydride transfer from the diazaphospholene hydride to the 4-position of the pyridine moiety, yielding a dihydropyridyl borate complex paired with a phosphenium cation. Eventually, the transfer of the hydride from the borate complex to the phosphonium cation regenerates the diazaphospholene hydride and releases N-borylated-1,4-dihydropyridine (Hynes et al., 2018).

Figure 6. Non-metal-catalyzed 1,4-hydroboration.
2.5 Noble d-block transition-metal-catalyzed-hydroboration
AgSbF6 has proven to be an effective catalyst for the hydroboration of various unsaturated functionalities, including isocyanates, pyridines, and quinolines (Pandey et al., 2022). However, only pyridine substrates gave N-boryl enamines via 1,4-hydroboration. Good-to-excellent yields of the 1,4-hydroborated products were obtained from 3-substituted pyridines. In addition, the hydroboration of 3,5-disubstituted pyridines was more efficient for electron-withdrawing than electron-donating substituents, but 2-substituted pyridines failed to undergo hydroboration. In contrast to previous reports, control experiments revealed that silver-salt-catalyzed hydroboration is a radical-mediated process.
The 1,2-selective hydroboration of pyridines using noble d-block transition-metal catalysts involved two distinct catalytic systems (Figure 7). One system utilized the iridium catalyst [Ir (cod)py][SZO], which was synthesized from (cod)IrCl(py) and [Me3Si][SZO300] as part of Conley’s research (Rodriguez and Conley, 2022), whereas the other employed the rhodium catalyst [RhCl(cod)]2 developed by Ohmura and Suginome’s group (Oshima et al., 2012). A comparison of the reactivities of pyridines with the Rh and Ir catalysts revealed notable differences. With the rhodium catalyst, various pyridines exhibited high-to-moderate yields of the corresponding N-borylated 1,2-dihydropyridines with good selectivity (Oshima et al., 2012), while with an iridium catalyst, the pyridines tended to yield mixtures of the 1,2- and 1,4-hydroboration products, or showed a preference for one of the products based on the substituent pattern (Rodriguez and Conley, 2022). In the rhodium-catalyzed hydroboration, the key intermediate is Int 14 (Figure 7), which was formed through the oxidative addition of the B–H bond of HBpin to Rh(I), along with pyridine coordination. Pyridine insertion into the Rh–H bond at the 1,2-positions led to the formation of boryl rhodium amide, and the subsequent reductive elimination yielded the N-boryl enamine and regenerated Rh(I) (Oshima et al., 2012). Further research is needed to understand the mechanistic details of the hydroboration catalyzed by [Ir (cod)py][SZO], which likely proceeds via an inner-sphere pathway involving an iridium boryl hydride intermediate or the interaction of the pyridyl nitrogen with the Ir-BPin species (Rodriguez and Conley, 2022).
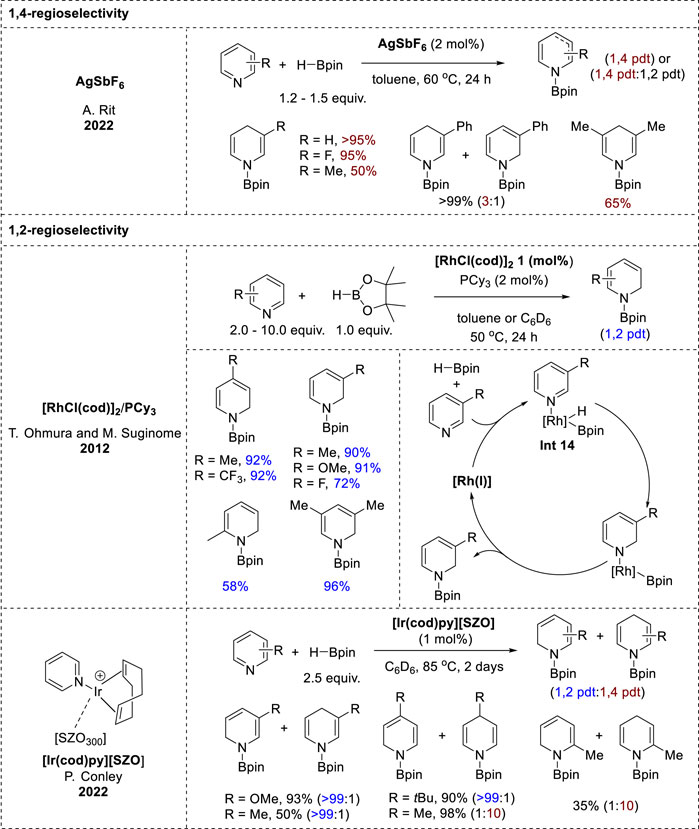
Figure 7. Noble d-block transition-metal-catalyzed-hydroboration.
2.6 f-block transition-metal-catalyzed-hydroboration
Efficient f-block transition-metal catalysts have recently emerged for the 1,2-regioselective hydroboration of pyridines. Notable examples include organolanthanide [Cp*2LaH]2 and thorium complexes such as thorium methyl (C5Me5)2ThMe2 and thorium hydride [(C5Me5)2Th(H) (μ-H)]2 complexes (Figure 8). These catalysts demonstrated similar reactivity profiles and high 1,2-regioselectivity for pyridine substrates. Specifically, ortho-substituted pyridines exhibited negligible activity under both catalytic conditions because of steric hindrance at the 2-position, whereas meta- and para-functionalized pyridines yielded N-boryl dihydropyridines in good yields and excellent selectivities. However, it was observed that the conjugated substituents could not be tolerated under thorium catalysis conditions (Dudnik et al., 2014; Liu et al., 2018). Furthermore, the catalytic mechanism underlying these transformations closely resembles that of other metal-catalyzed hydroboration reactions. It proceeds via an inner-sphere pathway, with the metal hydride species playing a pivotal role in the 1,2-addition of La–H or Th–H to the C=N bond of the coordinated pyridine, leading to the formation of a dihydropyridine complex intermediate. Subsequently, these intermediates undergo σ-bond metathesis with another HBPin molecule to afford N-borylated dihydropyridines (Dudnik et al., 2014; Liu et al., 2018).
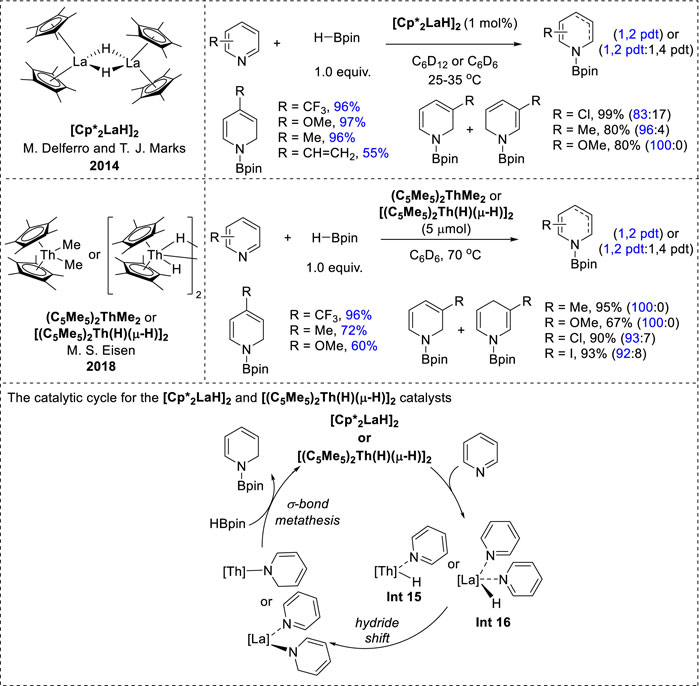
Figure 8. f-Block transition-metal-catalyzed hydroboration.
3 Hydrosilylation in synthesis of N-silyl enamines
3.1 Earth-abundant-metal-catalyzed 1,2-hydrosilylation of N-heteroarenes
Harrod and Samuel’s research on titanocene-catalyzed hydrosilylation began in 1998 and was further explored in a subsequent report in 2001. Their results revealed that Cp2TiMe2 effectively promoted the hydrosilylation of pyridine, yielding high amounts of N-silyl-tetrahydropyridine with MePhSiH2, but was ineffective for hydrosilylation with PhSiH3 or Ph2SiH2 (Hao et al., 1998). In contrast, changing the ligand to Cp*2TiMe2 led to successful hydrosilylation with PhSiH3, whereas using PhMeSiH2 instead of PhSiH3 resulted in a significantly slower reaction rate than that with Cp2TiMe2 (Harrod et al., 2001). The mechanism of titanocene-catalyzed hydrosilylation involves the interaction of Cp2TiMe2 with silane and pyridine to form a hydride complex (Int 17, Figure 9). The formation of Int 17 was not observed directly in the reaction, but was supported by model stoichiometric reactions. Moreover, the hydride complex Int 17 is believed to be the key intermediate in the dearomatization of pyridine, followed by the insertion of the Ti–H bond into the N=C bond of pyridine to form a 1,2-dihydropyridine complex (Hao et al., 1998; Harrod et al., 2001). Eventually, the 1,2-dihydropyridine complex underwent σ-bond metathesis with silane to produce the N-silyl-1,2-dihydropyridine product and regenerate Cp2TiH.
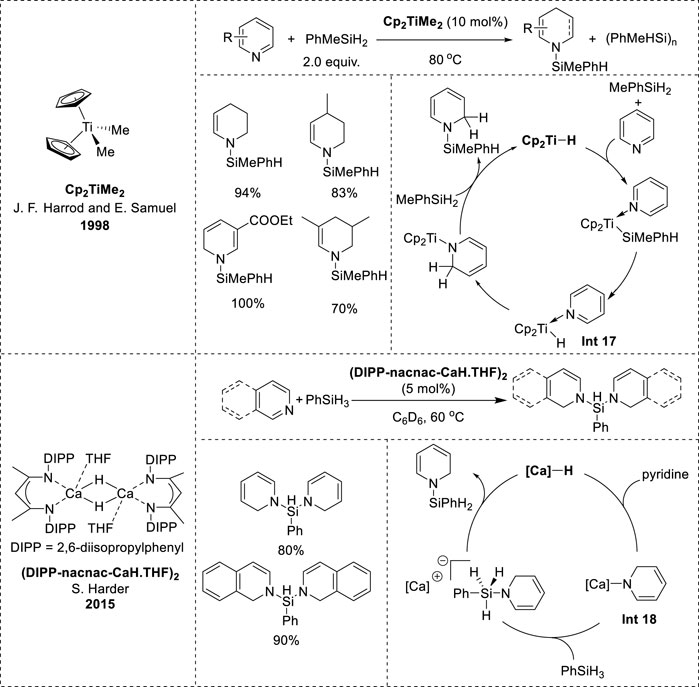
Figure 9. Earth-abundant-metal-catalyzed 1,2-hydrosilylation of N-heteroarenes.
In 2015, Harder et al. embarked on an investigation into the reactivity of the calcium hydride complex (DIPP-nacnac-CaH∙THF)2 ([Ca]–H) based on their prior research on hydroboration catalyzed by magnesium hydride complexes (Intemann et al., 2015). Their initial observations revealed that [Ca]–H exhibited higher reactivity and 1,2-reduction selectivity in the dearomatization of pyridine than the magnesium hydride complex, with 1,2 to 1,4 isomerization upon increasing the temperature (Arrowsmith et al., 2011; Intemann et al., 2014). The formation of the calcium 1,2-DHP complex revealed the catalytic potential of [Ca]–H for hydroboration and hydrosilylation. However, unlike magnesium hydride, [Ca]–H was inactive in the hydroboration of pyridine owing to the predominant formation of B2 (pin)3 rather than the desired hydroboration product, although it proved to be efficient in hydrosilylation. Pyridines and isoquinolines were effectively reduced to afford excellent product yields. Furthermore, the mechanism was elucidated through stoichiometric experiments, initiating the catalytic cycle from the active calcium hydride species. Subsequently, the [Ca]–H species reacts with pyridine to yield the [Ca]-1,2-DHP complex (Int 18, Figure 9), which then reacts with silanes to form an ion pair containing hypervalent silicon species (Intemann et al., 2015). Ultimately, hydride transfer to the cationic calcium species occurs, leading to the release of the N-silyl enamine product and regeneration of the active [Ca]–H complex.
3.2 Borane-catalyzed 1,4-hydrosilylation
Chang et al. recently reported a B(C6F5)3-catalyzed hydrosilylation with a broad substrate scope that encompasses the dearomatization of N-heteroarenes, such as quinoline, isoquinoline, and pyridine, as well as the reduction of conjugated nitriles and imines.
In their catalytic system, the dearomatization of N-heteroarenes proceeded via the formation of N-silyl enamine intermediates prior to yielding 1,3-bis-silylated products (Gandhamsetty et al., 2014; Gandhamsetty et al., 2015a). Moreover, the selectivity of N-silyl enamine formation in the initial step was different for each N-heteroarene. Quinolines and 2- and 3-substituted pyridines underwent 1,4-hydrosilylation selectively. Isoquinolines and 4-substituted pyridines underwent 1,2-hydrosilylation because of the effects of aromaticity and steric hindrance (Gandhamsetty et al., 2014; Gandhamsetty et al., 2015b). Additionally, the formation of linear N-silyl enamine through 1,4-addition was also observed when utilizing α,β-unsaturated nitriles and α,β-unsaturated aldimines (Gandhamsetty et al., 2015a; Kim et al., 2018). Kinetic and control experiments demonstrated the accumulation of N-silyl enamine intermediates until complete consumption of N-heteroarenes occurred. The conversion to the fully reduced products was initiated at the peak N-silyl enamine concentrations (Gandhamsetty et al., 2014; Gandhamsetty et al., 2015a; Gandhamsetty et al., 2015b; Kim et al., 2018). This finding proves that precise control of the amount of silane can achieve exclusive N-silyl enamine formation. N-silyl-1,4-dihydropyridines were synthesized using 1.1 equivalent of Me2PhSiH (1.1 equiv.) and the synthesis of linear N,N-disilyl enamines could be achieved by adjusting the stoichiometry of bulky silanes in Chang’s system (Gandhamsetty et al., 2015b). The utilization of equimolar quantities of HSiMe2Ph resulted in N-silyl-1,4-dihydroquinolines and N-silyl-1,2-isoquinolines (Petrushko and Nikonov, 2020).
Based on experimental studies and density functional theory (DFT) calculations of the reaction mechanism, the formation of N-silyl enamine intermediates in borane-catalyzed hydrosilylation was concluded to occur via an ionic mechanism (Figure 8) (Gandhamsetty et al., 2014; Gandhamsetty et al., 2015a; Gandhamsetty et al., 2015b; Kim et al., 2018). Initially, B(C6F5)3 coordinates to the N center to establish a stable resting species. These species exist in equilibrium with both the free reactants and the active complex (C6F5)3B∙HSiR3. This active complex facilitates the transfer of its silylium cation to the reactants, leading to the formation of iminium salt Int 19 (Figure 10) or Int 20 (Figure 10) along with the borohydride anion [(C6F5)3BH]−. Subsequently, the borohydride anion transfers the hydride to the C4 site of Int 19 or Int 20, forming cyclic N-silyl enamines.
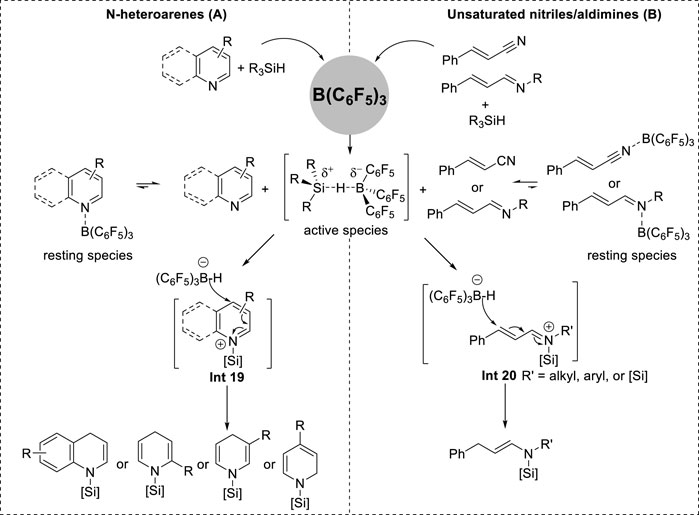
Figure 10. (A) Borane-catalyzed 1,4-hydrosilylation of N-heteroarenes. (B) Borane-catalyzed 1,4-hydrosilylation of unsaturated nitriles/aldimines.
3.3 Transition-metal-catalyzed hydrosilylation
Because of their similar ionic mechanisms, various cationic Ru complexes, including [Cp(iPr3P)Ru(NCCH3)2]+ (Ru I), [Cp(phen)Ru(NCCH3)2]+ (Ru II), and coordinatively unsaturated RuII thiolate (Ru III), exhibit 1,4-selectivity in the hydrosilylation of N-heteroarenes (Figure 11). Ru I and Ru II display good conversions with 3- and 5-substituted pyridines, whereas 2-, 4-, and 6-substituents were ineffective (Gutsulyak et al., 2011; Lee et al., 2013). Conversely, Ru III reduces various N-heteroarenes, including pyridines, isoquinolines, and quinolines, resulting in high regioselectivity and chemoselectivity. Notably, 4-substituted pyridines react effectively to provide high yields of N-silyl-1,4-dihydropyridines (Königs et al., 2013). The proposed mechanism for the formation of Ru I and Ru II complexes begins with the formation of cationic silane complexes from Ru via nitrile dissociation and silane coordination. Cationic silane complexes facilitate the transfer of a silyl cation to the pyridine substrate to form Int 21 (Figure 11) and a reactive Ru hydride species, which then induces hydride transfer to the 4-position to yield the N-silyl enamine product and regenerate the active catalyst complex (Gutsulyak et al., 2011; Lee et al., 2013). In addition, the mechanism for the Ru III complex starts with the formation of a cationic silicon–sulfur intermediate via the activation of silane by the heterolytic cleavage of the Si–H bond. Subsequently, the cationic silicon–sulfur intermediate transferred the cationic silicon to the nitrogen center of pyridine, forming the N-silylpyridinium intermediate (Int 22, Figure 11) and neutral Ru hydride. Eventually, the neutral Ru hydride attacked the C4-position in the N-silyl pyridinium intermediate to generate the N-silyl enamine products (Bähr and Oestreich, 2018).
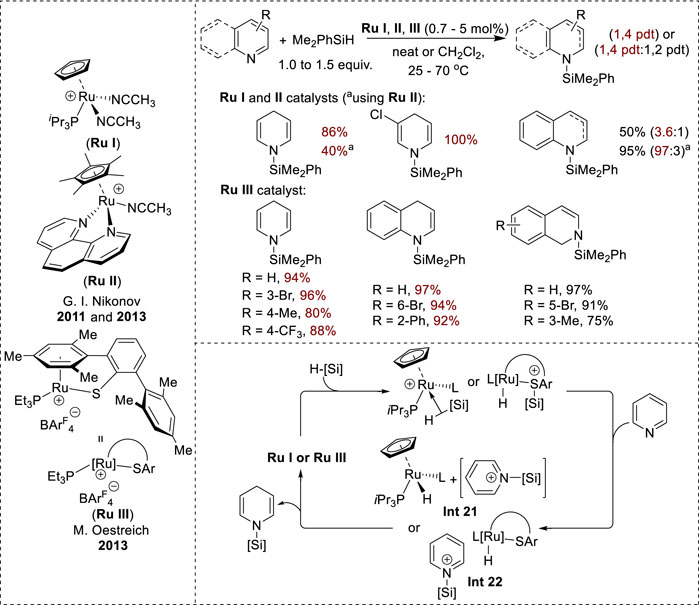
Figure 11. Transition-metal-catalyzed 1,4-hydrosilylation of N-heteroarenes.
In addition to 1,4-hydrosilylation, transition-metal-catalyzed 1,2-hydrosilylation was demonstrated using a metathesis-active ruthenium complex (Ru IV) and an iridium catalyst ([Ir (coe)2Cl]2). These catalysts were versatile, functioned effectively to N-heteroarenes, and displayed excellent tolerance to various functional groups (Jeong et al., 2016; Ma and Nolan, 2023). The mechanisms of both the catalytic systems follow an inner-sphere path. In the iridium complex mechanism, the initial step involves the generation of two isomeric iridium olefin adducts via a reaction between [Ir (coe)2Cl]2 and Et2SiH2. These adducts undergo ligand exchange with N-heteroarenes to form the bimetallic species (Int 23, Figure 12), which undergoes intramolecular insertion of an Ir–H bond into the C=N bond of the N-heteroarene ligand, generating a 1,2-dihydropyridine intermediate. Subsequently, the 1,2-dihydropyridine intermediate produces the N-silyl enamine through reductive elimination (Jeong et al., 2016). In the Ru complex mechanism, the Ru complex transforms into the activated species via PCy3 dissociation and ligand exchange. The N-heteroarene coordinates with the ruthenium species (Int 24, Figure 12), forming a 1,2-reduced intermediate via selective hydride transfer at the 2-position. Eventually, the 1,2-reduced intermediate undergoes σ-bond metathesis with another silane molecule, resulting in the release of N-silyl enamine (Ma and Nolan, 2023).
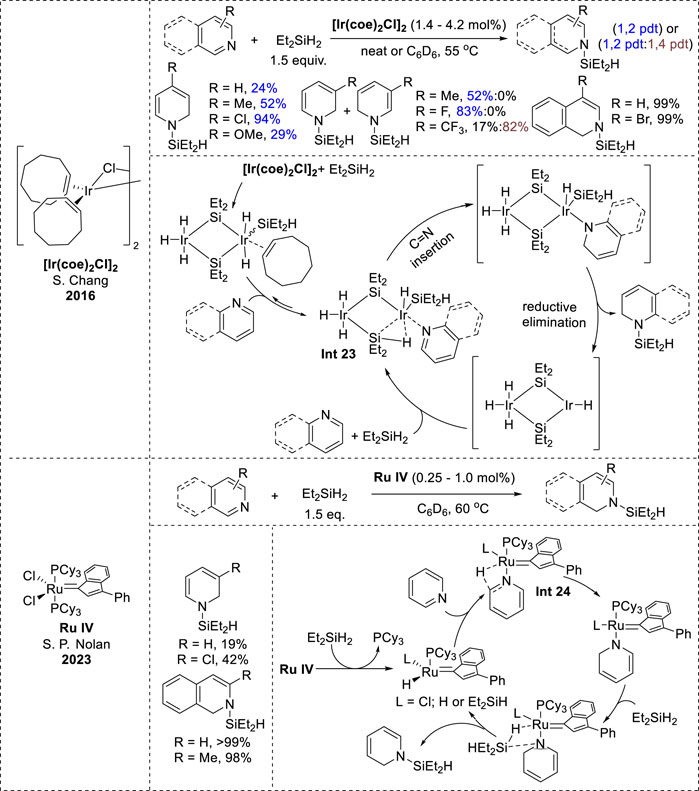
Figure 12. Transition-metal-catalyzed 1,2-hydrosilylation of N-heteroarenes.
4 Transition-metal catalysis of both hydroboration and hydrosilylation
In 2017, Lin et al. reported a zirconium framework, ZrIIIH-BTC, that exhibited high activity and 1,4-selectivity in the dearomatization of pyridines and quinolines using HBpin and triethoxysilane (Figure 13). This selectivity was attributed to the bridging oxo/carboxylate ligands and the site-isolation effect of the MOF, which stabilized the coordinatively unsaturated ZrIIIH centers (Ji et al., 2017). Moreover, 1,2-selective hydroboration and hydrosilylation of N-heteroarenes were achieved under β-diketiminate-supported dimeric zinc hydride [LZnH]2 conditions by Nembena in 2023 (Sahoo et al., 2023). A large range of 3- and 4-substituted pyridines were transformed into the 1,2-hydroborated products in excellent yields. Similar to other 1,2-selective systems, 2-substituted pyridines failed to produce the reductive products. Interestingly, the hydrosilylation of pyridines and isoquinolines in this catalytic system produced bis-hydrosilylated products in quantitative yield. Similar to other metal-hydride-catalyzed hydroboration and hydrosilylation reactions, the reaction mechanism involves a 1,3-hydride transfer to furnish zinc amide intermediates. Further addition of HBpin or a hydrosilane to the amide intermediates produced selective 1,2-hydroborylated and hydrosilylated products.
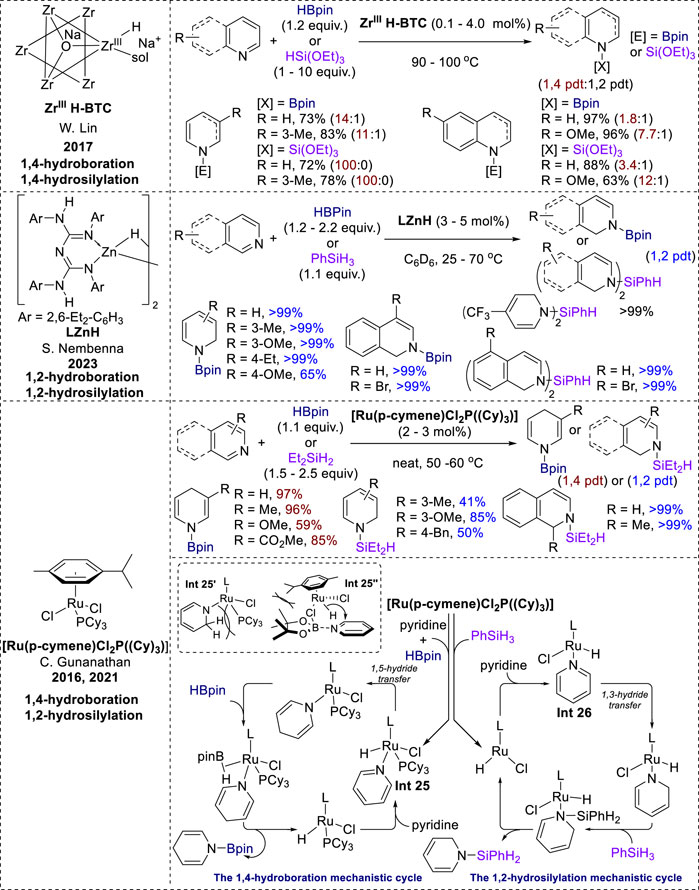
Figure 13. Transition-metal catalysis of both hydroboration and hydrosilylation.
Additionally, the Ru (II) precatalyst [Ru (p-cymene)Cl2(P(Cy)3)] exhibited distinct selectivities for both 1,4-hydroboration and 1,2-hydrosilylation in Gunanathan’s research. The hydroboration of various 3-substituted pyridines in this catalytic system took place with efficient 1,4-selectivity, with good-to-excellent yields. However, 2-substituted and 4-substituted pyridines showed no reactivity towards the ruthenium-catalyzed 1,4-hydroboration (Kaithal et al., 2016). Additionally, this catalyst facilitated the 1,2-hydrosilylation of pyridines, affording moderate product yields for the meta- and para-substituted derivatives. Isoquinoline also underwent efficient conversion, affording N-silyl-1,2-dihydroisoquinoline in quantitative yield (Behera et al., 2021). The formation of the Int 25 (Figure 13) and Int 26 (Figure 13) species determined the distinct selectivity between the two processes. In the hydroboration mechanism, Int 25 is formed by the reaction of a ruthenium precatalyst with HBpin and pyridines in the presence of a phosphine ligand (PCy3) (Kaithal et al., 2016). However, during hydrosilylation, the Ru precatalyst dissociates from the PCy3 ligand before reacting with silane and pyridine to form Int 26. Subsequently, Int 26 undergoes 1,3-hydride transfer to form a dihydropyridine intermediate, whereas Int 25 prefers 1,5-hydride transfer. The different selectivity between 1,2-hydrosilylation and 1,4-hydroboration under the same ruthenium precatalyst is initially attributed to the steric hindrance between the “sp3-CH2” of the 1,2-dihydropyridine ligand and the phosphine ligand (Int 25′, Figure 13) (Kaithal et al., 2016). Additionally, they also performed kinetic studies on this hydroboration to support the intramolecular 1,5-hydride shift mechanism. However, subsequent DFT calculations revealed that the 1,2-hydroboration process was impeded due to steric effects between the methyl groups of HBpin and the p-cymene ligand in a ruthenium catalyst (Int 25″, Figure 13), thereby promoting 1,4-selectivity in hydroboration (Behera et al., 2021). Eventually, the 1,4- or 1,2-dihydropyridine intermediates undergoes subsequent metathesis with HBPin or silane, leading to the formation of the N-boryl or N-silyl enamine products.
5 Applications of N-boryl and N-silyl enamines in organic synthesis
N-Boryl and N-silyl enamines serve as versatile nucleophilic motifs that can react with a broad range of electrophilic reagents. Recent synthetic methodologies have predominantly focused on the functionalization of the C3-position of borylated and silylated enamines through nucleophilic attack on external electrophiles, as well as the facilitation of the construction of complex molecular scaffolds via cycloaddition reactions involving the C=C moiety and various dipoles (Figure 14). Remarkably, the in situ utilization of N-boryl and N-silyl enamines enables efficient one-pot tandem reactions. N-boryl and N-silyl enamines have similar reactivities; the choice of one over the other is dictated by the specific cleavage pathways of their respective silyl and boryl groups in the final product, which may involve rearomatization, carbonylation, or facile N–H bond formation under acidic conditions.
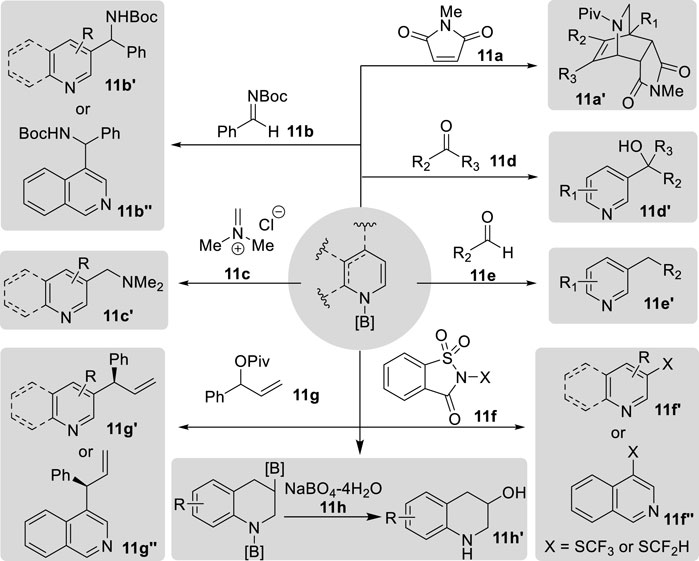
Figure 14. Applications of N-boryl enamines.
The application of borylated enamines in Diels–Alder reactions was described by Suginome et al., who synthesized N-boryl-1,2-dihydropyridines synthesized from pyridines using [RhCl(cod)]2/PCy3 as a catalyst. This N-boryl enamine reacts with N-methyl maleimide 11a via [4 + 2] cycloaddition, to yield isoquinuclidine derivatives 11a′. Subsequent acylation of the B–N bond with pivaloyl chloride afforded the final products in good yields (Oshima et al., 2012). Moreover, Wang et al. reported the C3-selective functionalization of pyridine through a tandem process involving the reaction of N-boryl-1,4-dihydropyridines synthesized via the borane-catalyzed hydroboration of pyridines with various electrophilic reagents, ultimately leading to the desired products via oxidative aromatization of the adducts in air without the need for additional oxidants (Liu et al., 2022; Zhou et al., 2022). When the imine 11b, iminium 11c, and carbonyl compounds 11d–11e were employed as electrophiles, exclusive regioselectivity for C3 carbon–carbon bond formation in 11b′–11e′ or C3 carbon–sulfur bond formation in 11f′ and 11f″ was observed (Liu et al., 2022; Zhou et al., 2022). Specifically, the treatment of 3-substituted pyridine under these conditions yielded moderate-to-high yields of monofunctional derivatives, whereas 4-substituted and 2-substituted pyridines yielded difunctional derivatives at the 3- and 5-positions. Notably, ketones yielded the alcohol products 11e′, whereas aldehydes afforded the dehydroxylated products 11f′ (Liu et al., 2022). Recently, building upon this tandem procedure, the authors successfully synthesized the C3-allylated pyridines 11g′ and 11g″ with allylic ester 11g via enantioselective iridium-catalyzed allylation (Liu et al., 2023). Additionally, the excess HBpin also acted as an electrophilic reagent, which reacts with N-boryl 1,4-dihydroquinolines to produce C3-borylated tetrahydroquinolines in the borane-catalyzed double hydroboration of quinolines. Chang’s group synthesized various 3-hydroxytetrahydroquinolines 11h′ after oxidation of 1,3-bis-borylated tetrahydroquinolines by sodium borate 11h (Kim et al., 2020).
Cyclic N-silyl enamines have been utilized to synthesize polycyclic structures via Diels–Alder reactions (Figure 15). Wanner et al. pioneered the [4 + 2] cycloaddition of N-silyl-1,4-dihydropyridine with cyclopentadiene 12a to synthesize 2-azabicyclo [2.2.2]octane (Schmaunz et al., 2014). The N-silyl-1,4-dihydropyridine precursor was prepared by the reaction of pyridines with triisopropylsilyl triflate, yielding the corresponding pyridinium salts, which were subsequently trapped in diorganomagnesium compounds. After [4 + 2] cycloaddition, 5,5-diarylsubstituted 2-azabicyclo [2.2.2]octane derivatives 12a′ were obtained in good-to-high yields. These derivatives underwent intramolecular electrophilic aromatic substitution reactions to produce 7,8-benzomorphane derivatives upon treatment with acetyl chloride. In contrast, subsequent studies by Joung’s group focused on the [3 + 2] cycloaddition of cyclic N-silyl enamines via the borane-catalyzed hydrosilylation of N-heteroarenes with organic azides 12b, 12c or azomethine imines 12d (Cao et al., 2020a; Cao et al., 2020b; Cao et al., 2022; Jo et al., 2022). Borane-catalyzed hydrosilylations yield a broad range of cyclic N-silyl enamines from various N-heteroarenes. These enamines could be utilized in situ without further purification, thereby facilitating the development of a one-pot procedure. After the [3 + 2] cycloaddition of N-silyl enamines with organic azides, including sulfonyl azide 12b and carbonyl azide 12c, amidines 12b′, 12b″ and 12c′, 12c″ were produced in good-to-high yields. The formation of amidines occurs through the reaction of N-silyl enamines and azides, generating a triazoline intermediate that immediately rearranges to a cyclic amidine via hydride shift and nitrogen gas extrusion (Cao et al., 2020a; Cao et al., 2020b; Jo et al., 2022). In addition, the authors discovered that these N-silyl enamines also underwent [3 + 2] cycloaddition with azomethine imine 12d, resulting in the formation of tetracyclic pyrazolidinone scaffold structures 12d′ in good yield. The [3 + 2] cycloaddition process encompasses both endo and exo pathways (Cao et al., 2022).
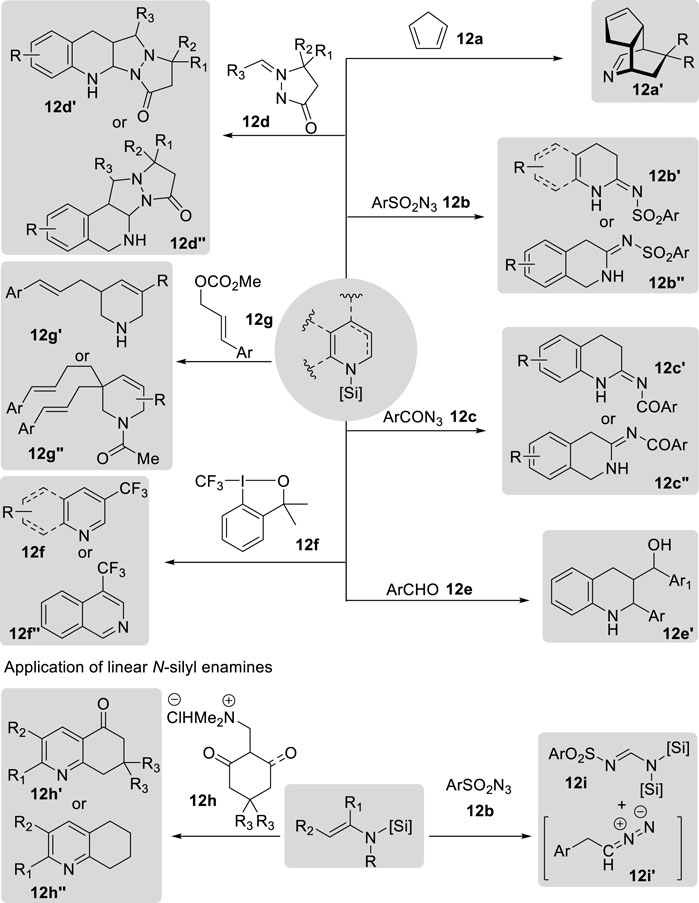
Figure 15. Application of N-silyl enamines.
In addition to their application in cycloaddition reactions, cyclic N-silyl enamines serve as nucleophilic motifs for various chemical transformations. Crudden et al. reported the synthesis of γ-aminoalcohols 12e′ derived from quinoline via dearomatization under borenium-catalyzed hydrosilylation, resulting in the formation of N-silyl-1,4-dihydroquinolines (Clarke et al., 2021). These N-silyl-1,4-dihydroquinolines were then added to aldehyde 12e, followed by reduction using NaBH4 and subsequent deprotection of the silyl group to yield tetrahydroquinoline derivatives in modest-to-good yields. Furthermore, these N-silyl enamines derived from the borane-catalyzed hydrosilylation of N-heteroarenes have also been employed for the production of 3-trifluoromethylated compounds 12f′ and 12f′′ via electrophilic trifluoromethylation with Togni reagent I (12f) (Muta et al., 2022). Remarkably, research conducted by Stoltz et al. utilized N-silyl enamines derived from iridium(I)-catalyzed dearomative 1,2-hydrosilylation as nucleophiles in a subsequent palladium-catalyzed asymmetric allylic alkylation with cinnamyl methyl carbonate 12g, leading to the formation of C3-substituted tetrahydropyridine products 12g′ and 12g′′ (Greßies et al., 2023). The desired products were isolated in moderate yields with excellent enantioselectivity following carbonylation with acetyl chloride. Moreover, bisalkylated tetrahydropyridines 12g′′ were obtained in the presence of benzoic acid and an excess of the allyl carbonate substrate. The formation of the bisalkylated products arose from the tautomerization of the imine intermediate, resulting in an enamine that could participate in additional alkylation.
Similarly, Sakai et al. developed a Yb(OTf)3-catalyzed cyclization process utilizing the nucleophilicity of linear N-silyl enamines to synthesize quinolinone and tetrahydroquinoline derivatives from endione and enone precursors 12h (Sakai et al., 2006). This process yielded the desired cyclic products 12h′ and 12h′′ in good yields; the quinolinone derivatives were further transformed into substituted quinolines upon treatment with NBS, AIBN, and p-toluenesulfonic acid in methanol. Joung et al. reported the use of linear N-silyl enamines in [3 + 2] cycloaddition reactions with sulfonyl azides 12b (Do et al., 2024). The authors synthesized linear N-silyl enamines through borane-catalyzed hydrosilylation of α,β-unsaturated nitriles. However, unlike the rearrangement of a triazoline intermediate from cyclic N-silyl enamines and azides, linear N-silyl enamines reacted with sulfonyl azides to generate a triazoline intermediate, which underwent a distinct retro-(3 + 2) cycloaddition to produce the corresponding formamidine 12i′ and versatile alkyl diazomethane 12i′′.
6 Summary
This review described the formation of N-boryl and N-silyl enamines through the hydroboration and hydrosilylation of a range of conjugated systems employing a variety of catalysts. The hydrides for the reduction process originate from in situ formed catalysts, the dissociation of B–H or Si–H bonds, or coordination of HBPin or silane as ligands of the catalyst. The selectivity between 1,2- and 1,4-addition arises from the formation of distinct intermediates. Specifically, active metal hydrides induce an intramolecular 1,3-hydride shift in the metal-substrate complex, leading to the formation of 1,2-adducts. Bulky active hydrides or sterically hindered ligands promote a hydride shift to the less-hindered C4-position. Isomerization from kinetic 1,2-adducts to thermodynamically stable 1,4-adducts can also afford 1,4-adducts. As an application of these N-boryl and N-silyl enamines, the utilization of the various functional N-heterocyclic structures obtained from the diverse regioselective reductions of N-boryl and N-silyl enamines described above facilitates the development of one-pot or tandem procedures for subsequent chemical transformations of the versatile N-boryl and N-silyl enamines.
Author contributions
VC: Conceptualization, Investigation, Writing–original draft, Writing–review and editing. SJ: Conceptualization, Supervision, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was supported by the National Research Foundation of Korea (NRF-2021R1C1C1012844).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ahmed, M., Seayad, A. M., Jackstell, R., and Beller, M. (2003). Highly selective synthesis of enamines from olefins. Angew. Chem. Int. Ed. 42 (45), 5615–5619. doi:10.1002/anie.200352320
Arai, N., and Ohkuma, T. (2008). Carbonyl hydroboration. Mod. Reduct. Methods, 159–181. doi:10.1002/9783527622115.ch7
Arrowsmith, M., Hill, M. S., Hadlington, T., Kociok-Köhn, G., and Weetman, C. (2011). Magnesium-catalyzed hydroboration of pyridines. Organometallics 30 (21), 5556–5559. doi:10.1021/om2008138
Bähr, S., and Oestreich, M. (2018). A neutral RuII hydride complex for the regio- and chemoselective reduction of N-silylpyridinium ions. Chem. – A Eur. J. 24 (21), 5613–5622. doi:10.1002/chem.201705899
Barluenga, J., Fernández, M. A., Aznar, F., and Valdés, C. (2004). Palladium-catalyzed cross-coupling reactions of amines with alkenyl bromides: a new method for the synthesis of enamines and imines. Chem. – A Eur. J. 10 (2), 494–507. doi:10.1002/chem.200305406
Behera, D., Thiyagarajan, S., Anjalikrishna, P. K., Suresh, C. H., and Gunanathan, C. (2021). Ruthenium(II)-Catalyzed regioselective 1,2-hydrosilylation of N-heteroarenes and tetrel bonding mechanism. ACS Catal. 11 (10), 5885–5893. doi:10.1021/acscatal.1c01148
Beller, M., Breindl, C., Eichberger, M., Hartung, C. G., Seayad, J., Thiel, O. R., et al. (2002). Advances and adventures in amination reactions of olefins and alkynes. Synlett 2002 (10), 1579–1594. doi:10.1055/s-2002-34240
Bolig, A. D., and Brookhart, M. (2007). Activation of sp3 C−H bonds with cobalt(I): catalytic synthesis of enamines. J. Am. Chem. Soc. 129 (47), 14544–14545. doi:10.1021/ja075694r
Burgess, V. A., Davies, S. G., and Skerlj, R. T. (1991). NADH mimics for the stereoselective reduction of benzoylformates to the corresponding mandelates. Tetrahedron Asymmetry 2 (5), 299–328. doi:10.1016/s0957-4166(00)82109-6
Cao, V. D., Jo, D. G., Kim, H., Kim, C., Yun, S., and Joung, S. (2020b). Utilization of borane-catalyzed hydrosilylation as a dearomatizing tool: six-membered cyclic amidine synthesis from isoquinolines and pyridines. Synthesis 53 (04), 754–764. doi:10.1055/s-0040-1707323
Cao, V. D., Kim, H., Kwak, J., and Joung, S. (2022). (3 + 2) cycloaddition reaction of the endocyclic N-silyl enamine and N,N′-Cyclic azomethine imine. Org. Lett. 24 (10), 1974–1978. doi:10.1021/acs.orglett.2c00366
Cao, V. D., Mun, S. H., Kim, S. H., Kim, G. U., Kim, H. G., and Joung, S. (2020a). Synthesis of cyclic amidines from quinolines by a borane-catalyzed dearomatization strategy. Org. Lett. 22 (2), 515–519. doi:10.1021/acs.orglett.9b04275
Cheng, Y., Huang, Z.-T., and Wang, M.-X. (2004). Heterocyclic enamines: the versatile intermediates in the synthesis of heterocyclic compounds and natural products. Curr. Org. Chem. 8 (4), 325–351. doi:10.1002/chin.200429245
Clarke, J. J., Devaraj, K., Bestvater, B. P., Kojima, R., Eisenberger, P., DeJesus, J. F., et al. (2021). Hydrosilylation and Mukaiyama aldol-type reaction of quinolines and hydrosilylation of imines catalyzed by a mesoionic carbene-stabilized borenium ion. Org. Biomol. Chem. 19 (31), 6786–6791. doi:10.1039/d1ob01056e
Corey, J. Y. (2016). Reactions of hydrosilanes with transition metal complexes. Chem. Rev. 116 (19), 11291–11435. doi:10.1021/acs.chemrev.5b00559
Das, S., Maity, J., and Panda, T. K. (2022). Metal/non-metal catalyzed activation of organic nitriles. Chem. Rec. 22 (12), e202200192. doi:10.1002/tcr.202200192
Dehli, J. R., Legros, J., and Bolm, C. (2005). Synthesis of enamines, enol ethers and related compounds by cross-coupling reactions. Chem. Commun. 36 (8), 973–986. doi:10.1039/b415954c
Do, C. V., Lee, S., and Joung, S. (2024). In-situ utilization of non-stabilized diazoalkanes from (3+2) cycloaddition of linear N,N-disilyl enamines and azides. Adv. Synthesis Catal. 366 (1), 114–120. doi:10.1002/adsc.202301213
Du, X., and Huang, Z. (2017). Advances in base-metal-catalyzed alkene hydrosilylation. ACS Catal. 7 (2), 1227–1243. doi:10.1021/acscatal.6b02990
Dudnik, A. S., Weidner, V. L., Motta, A., Delferro, M., and Marks, T. J. (2014). Atom-efficient regioselective 1,2-dearomatization of functionalized pyridines by an earth-abundant organolanthanide catalyst. Nat. Chem. 6 (12), 1100–1107. doi:10.1038/nchem.2087
Edafiogho, I. O., Kombian, S. B., Ananthalakshmi, K. V. V., Salama, N. N., Eddington, N. D., Wilson, T. L., et al. (2007). Enaminones: exploring additional therapeutic activities. J. Pharm. Sci. 96 (10), 2509–2531. doi:10.1002/jps.20967
Escolano, M., Gaviña, D., Alzuet-Piña, G., Díaz-Oltra, S., Sánchez-Roselló, M., and Pozo, C. d. (2024). Recent strategies in the nucleophilic dearomatization of pyridines, quinolines, and isoquinolines. Chem. Rev. 124 (3), 1122–1246. doi:10.1021/acs.chemrev.3c00625
Fan, X., Zheng, J., Li, Z. H., and Wang, H. (2015). Organoborane catalyzed regioselective 1,4-hydroboration of pyridines. J. Am. Chem. Soc. 137 (15), 4916–4919. doi:10.1021/jacs.5b03147
Fu, R.-G., Wang, Y., Xia, F., Zhang, H.-L., Sun, Y., Yang, D.-W., et al. (2019). Synthesis of 2-Amino-5-acylthiazoles by a tertiary amine-promoted one-pot three-component cascade cyclization using elemental sulfur as a sulfur source. J. Org. Chem. 84 (18), 12237–12245. doi:10.1021/acs.joc.9b02032
Gandhamsetty, N., Joung, S., Park, S.-W., Park, S., and Chang, S. (2014). Boron-catalyzed silylative reduction of quinolines: selective sp3 C–Si bond formation. J. Am. Chem. Soc. 136 (48), 16780–16783. doi:10.1021/ja510674u
Gandhamsetty, N., Park, J., Jeong, J., Park, S.-W., Park, S., and Chang, S. (2015b). Chemoselective silylative reduction of conjugated nitriles under metal-free catalytic conditions: β-silyl amines and enamines. Angew. Chem. Int. Ed. 54 (23), 6832–6836. doi:10.1002/anie.201502366
Gandhamsetty, N., Park, S., and Chang, S. (2015a). Selective silylative reduction of pyridines leading to structurally diverse azacyclic compounds with the formation of sp3 C–Si bonds. J. Am. Chem. Soc. 137 (48), 15176–15184. doi:10.1021/jacs.5b09209
Ghosh, P., and Jacobi von Wangelin, A. (2021). Manganese-catalyzed hydroborations with broad scope. Angew. Chem. Int. Ed. 60 (29), 16035–16043. doi:10.1002/anie.202103550
Goldmann, S., and Stoltefuss, J. (1991). 1,4-Dihydropyridines: effects of chirality and conformation on the calcium antagonist and calcium agonist activities. Angewandte Chemie Int. Ed. Engl. 30 (12), 1559–1578. doi:10.1002/anie.199115591
Gordeev, M. F., Patel, D. V., England, B. P., Jonnalagadda, S., Combs, J. D., and Gordon, E. M. (1998). Combinatorial synthesis and screening of a chemical library of 1,4-dihydropyridine calcium channel blockers. Bioorg. Med. Chem. 6 (7), 883–889. doi:10.1016/s0968-0896(98)00048-0
Greßies, S., Süße, L., Casselman, T., and Stoltz, B. M. (2023). Tandem dearomatization/enantioselective allylic alkylation of pyridines. J. Am. Chem. Soc. 145 (22), 11907–11913. doi:10.1021/jacs.3c02470
Gutsulyak, D. V., van der Est, A., and Nikonov, G. I. (2011). Facile catalytic hydrosilylation of pyridines. Angew. Chem. Int. Ed. 50 (6), 1384–1387. doi:10.1002/anie.201006135
Hanessian, S., and Chattopadhyay, A. K. (2014). Iminium ion–enamine cascade cyclizations: facile access to structurally diverse azacyclic compounds and natural products. Org. Lett. 16 (1), 232–235. doi:10.1021/ol403229q
Hao, L., Harrod, J. F., Lebuis, A.-M., Mu, Y., Shu, R., Samuel, E., et al. (1998). Homogeneous catalytic hydrosilylation of pyridines. Angew. Chem. Int. Ed. 37 (22), 3126–3129. doi:10.1002/(sici)1521-3773(19981204)37:22<3126::aid-anie3126>3.0.co;2-q
Harrod, J. F., Shu, R., Woo, H.-G., and Samuel, E. (2001). Titanocene(III) catalyzed homogeneous hydrosilation-hydrogenation of pyridines. Can. J. Chem. 79 (5-6), 1075–1085. doi:10.1139/cjc-79-5-6-1075
Hu, C., Zhang, J., Yang, H., Guo, L., and Cui, C. (2021). Synthesis of cationic silaamidinate germylenes and stannylenes and the catalytic application for hydroboration of pyridines. Inorg. Chem. 60 (18), 14038–14046. doi:10.1021/acs.inorgchem.1c01314
Hynes, T., Welsh, E. N., McDonald, R., Ferguson, M. J., and Speed, A. W. H. (2018). Pyridine hydroboration with a diazaphospholene precatalyst. Organometallics 37 (6), 841–844. doi:10.1021/acs.organomet.8b00028
Iglesias, M., Fernández-Alvarez, F. J., and Oro, L. A. (2014). Outer-sphere ionic hydrosilylation catalysis. ChemCatChem 6 (9), 2486–2489. doi:10.1002/cctc.201402355
Intemann, J., Bauer, H., Pahl, J., Maron, L., and Harder, S. (2015). Calcium hydride catalyzed highly 1,2-selective pyridine hydrosilylation. Chem. – A Eur. J. 21 (32), 11452–11461. doi:10.1002/chem.201501072
Intemann, J., Lutz, M., and Harder, S. (2014). Multinuclear magnesium hydride clusters: selective reduction and catalytic hydroboration of pyridines. Organometallics 33 (20), 5722–5729. doi:10.1021/om500469h
Jeong, E., Heo, J., Park, S., and Chang, S. (2019). Alkoxide-promoted selective hydroboration of N-heteroarenes: pivotal roles of in situ generated BH3 in the dearomatization process. Chem. – A Eur. J. 25 (25), 6320–6325. doi:10.1002/chem.201901214
Jeong, J., Park, S., and Chang, S. (2016). Iridium-catalyzed selective 1,2-hydrosilylation of N-heterocycles. Chem. Sci. 7 (8), 5362–5370. doi:10.1039/c6sc01037g
Ji, P., Feng, X., Veroneau, S. S., Song, Y., and Lin, W. (2017). Trivalent zirconium and hafnium metal–organic frameworks for catalytic 1,4-dearomative additions of pyridines and quinolines. J. Am. Chem. Soc. 139 (44), 15600–15603. doi:10.1021/jacs.7b09093
Jiang, B., Meng, F.-F., Liang, Q.-J., Xu, Y.-H., and Loh, T.-P. (2017). Palladium-catalyzed direct intramolecular C–N bond formation: access to multisubstituted dihydropyrroles. Org. Lett. 19 (4), 914–917. doi:10.1021/acs.orglett.7b00072
Jo, D. G., Kim, C., Lee, S., Yun, S., and Joung, S. (2022). Synthesis of cyclic N-acyl amidines by [3 + 2] cycloaddition of N-silyl enamines and activated acyl azides. Molecules 27 (5), 1696. doi:10.3390/molecules27051696
Kaithal, A., Chatterjee, B., and Gunanathan, C. (2016). Ruthenium-catalyzed regioselective 1,4-hydroboration of pyridines. Org. Lett. 18 (14), 3402–3405. doi:10.1021/acs.orglett.6b01564
Kim, E., Jeon, H. J., Park, S., and Chang, S. (2020). Double hydroboration of quinolines via borane catalysis: diastereoselective one pot synthesis of 3-hydroxytetrahydroquinolines. Adv. Synthesis Catal. 362 (2), 308–313. doi:10.1002/adsc.201901050
Kim, E., Park, S., and Chang, S. (2018). Silylative reductive amination of α,β-unsaturated aldehydes: a convenient synthetic route to β-silylated secondary amines. Chem. – A Eur. J. 24 (22), 5765–5769. doi:10.1002/chem.201800958
Königs, C. D. F., Klare, H. F. T., and Oestreich, M. (2013). Catalytic 1,4-selective hydrosilylation of pyridines and benzannulated congeners. Angew. Chem. Int. Ed. 52 (38), 10076–10079. doi:10.1002/anie.201305028
Lee, S.-H., Gutsulyak, D. V., and Nikonov, G. I. (2013). Chemo- and regioselective catalytic reduction of N-heterocycles by silane. Organometallics 32 (16), 4457–4464. doi:10.1021/om400269q
Leong, B.-X., Lee, J., Li, Y., Yang, M.-C., Siu, C.-K., Su, M.-D., et al. (2019). A versatile NHC-parent silyliumylidene cation for catalytic chemo- and regioselective hydroboration. J. Am. Chem. Soc. 141 (44), 17629–17636. doi:10.1021/jacs.9b06714
Lipke, M. C., Liberman-Martin, A. L., and Tilley, T. D. (2017). Electrophilic activation of silicon–hydrogen bonds in catalytic hydrosilations. Angew. Chem. Int. Ed. 56 (9), 2260–2294. doi:10.1002/anie.201605198
Liu, H., Khononov, M., and Eisen, M. S. (2018). Catalytic 1,2-regioselective dearomatization of N-heteroaromatics via a hydroboration. ACS Catal. 8 (4), 3673–3677. doi:10.1021/acscatal.8b00074
Liu, J., Chen, J.-Y., Jia, M., Ming, B., Jia, J., Liao, R.-Z., et al. (2019b). Ni–O cooperation versus nickel(II) hydride in catalytic hydroboration of N-heteroarenes. ACS Catal. 9 (5), 3849–3857. doi:10.1021/acscatal.8b05136
Liu, T., He, J., and Zhang, Y. (2019a). Regioselective 1,2-hydroboration of N-heteroarenes using a potassium-based catalyst. Org. Chem. Front. 6 (15), 2749–2755. doi:10.1039/c9qo00497a
Liu, X., Li, B., Hua, X., and Cui, D. (2020). 1,2-Hydroboration of pyridines by organomagnesium. Org. Lett. 22 (13), 4960–4965. doi:10.1021/acs.orglett.0c01388
Liu, Z., He, J.-H., Zhang, M., Shi, Z.-J., Tang, H., Zhou, X.-Y., et al. (2022). Borane-catalyzed C3-alkylation of pyridines with imines, aldehydes, or ketones as electrophiles. J. Am. Chem. Soc. 144 (11), 4810–4818. doi:10.1021/jacs.2c00962
Liu, Z., Shi, Z.-J., Liu, L., Zhang, M., Zhang, M.-C., Guo, H.-Y., et al. (2023). Asymmetric C3-allylation of pyridines. J. Am. Chem. Soc. 145 (21), 11789–11797. doi:10.1021/jacs.3c03056
Lortie, J. L., Dudding, T., Gabidullin, B. M., and Nikonov, G. I. (2017). Zinc-catalyzed hydrosilylation and hydroboration of N-heterocycles. ACS Catal. 7 (12), 8454–8459. doi:10.1021/acscatal.7b02811
Ma, X., and Nolan, S. P. (2023). Regioselective 1,2-hydrosilylation of N-heterocycles catalyzed by a ruthenium olefin metathesis catalyst. Organometallics 42 (15), 1978–1984. doi:10.1021/acs.organomet.3c00196
Marciniec, B. (2005). Catalysis by transition metal complexes of alkene silylation–recent progress and mechanistic implications. Coord. Chem. Rev. 249 (21), 2374–2390. doi:10.1016/j.ccr.2005.02.025
Meher, N. K., Verma, P. K., and Geetharani, K. (2023). Cobalt-catalyzed regioselective 1,2-hydroboration of N-heteroarenes. Org. Lett. 25 (1), 87–92. doi:10.1021/acs.orglett.2c03891
Mengya, Y. X. C. S. L. (2018). Synthesis of the functionalized enamine. Prog. Chem. 30 (8), 1082–1096. doi:10.7536/PC171246
Mukherjee, S., Yang, J. W., Hoffmann, S., and List, B. (2007). Asymmetric enamine catalysis. Chem. Rev. 107 (12), 5471–5569. doi:10.1021/cr0684016
Muta, R., Torigoe, T., and Kuninobu, Y. (2022). 3-Position-Selective C–H trifluoromethylation of pyridine rings based on nucleophilic activation. Org. Lett. 24 (44), 8218–8222. doi:10.1021/acs.orglett.2c03327
Nakaoka, K., Guo, C., Saiki, Y., Furukawa, S., and Ema, T. (2023). Synthesis of enamines, aldehydes, and nitriles from CO2: scope of the one-pot strategy via formamides. J. Org. Chem. 88 (21), 15444–15451. doi:10.1021/acs.joc.2c01666
Notz, W., Tanaka, F., and Barbas, C. F. (2004). Enamine-based organocatalysis with proline and diamines: the development of direct catalytic asymmetric aldol, mannich, michael, and Diels−Alder reactions. Accounts Chem. Res. 37 (8), 580–591. doi:10.1021/ar0300468
Oshima, K., Ohmura, T., and Suginome, M. (2012). Regioselective synthesis of 1,2-dihydropyridines by rhodium-catalyzed hydroboration of pyridines. J. Am. Chem. Soc. 134 (8), 3699–3702. doi:10.1021/ja3002953
Pandey, V. K., Sahoo, S., and Rit, A. (2022). Simple silver(i)-salt catalyzed selective hydroboration of isocyanates, pyridines, and quinolines. Chem. Commun. 58 (36), 5514–5517. doi:10.1039/d2cc00491g
Park, S. (2019). B(C6F5)3-Catalyzed sp3 C—Si bond forming consecutive reactions. Chin. J. Chem. 37 (10), 1057–1071. doi:10.1002/cjoc.201900240
Park, S. (2020). Recent advances in catalytic dearomative hydroboration of N-heteroarenes. ChemCatChem 12 (12), 3170–3185. doi:10.1002/cctc.201902303
Park, S. (2024). First-row transition metal-catalyzed single hydroelementation of N-heteroarenes. ChemCatChem 16 (5), e202301422. doi:10.1002/cctc.202301422
Park, S., and Brookhart, M. (2010). Hydrosilylation of carbonyl-containing substrates catalyzed by an electrophilic η1-silane iridium(III) complex. Organometallics 29 (22), 6057–6064. doi:10.1021/om100818y
Park, S., and Chang, S. (2017). Catalytic dearomatization of N-heteroarenes with silicon and boron compounds. Angew. Chem. Int. Ed. 56 (27), 7720–7738. doi:10.1002/anie.201612140
Petrushko, W. D., and Nikonov, G. I. (2020). Mono(hydrosilylation) of N-heterocycles catalyzed by B(C6F5)3 and silylium ion. Organometallics 39 (24), 4717–4722. doi:10.1021/acs.organomet.0c00697
Poulsen, T. B. (2021). Total synthesis of natural products containing enamine or enol ether derivatives. Accounts Chem. Res. 54 (8), 1830–1842.
Rao, B., Chong, C. C., and Kinjo, R. (2018). Metal-free regio- and chemoselective hydroboration of pyridines catalyzed by 1,3,2-diazaphosphenium triflate. J. Am. Chem. Soc. 140 (2), 652–656. doi:10.1021/jacs.7b09754
Rodriguez, J., and Conley, M. P. (2022). A heterogeneous iridium catalyst for the hydroboration of pyridines. Org. Lett. 24 (25), 4680–4683. doi:10.1021/acs.orglett.2c01859
Sahoo, R. K., Sarkar, N., and Nembenna, S. (2023). Intermediates, isolation and mechanistic insights into zinc hydride-catalyzed 1,2-regioselective hydrofunctionalization of N-heteroarenes. Inorg. Chem. 62 (1), 304–317. doi:10.1021/acs.inorgchem.2c03389
Sakai, N., Aoki, D., Hamajima, T., and Konakahara, T. (2006). Yb(OTf)3-catalyzed cyclization of an N-silylenamine with 2-methylene-1,3-cyclohexanedione to afford a 7,8-dihydroquinolin-5(6H)-one derivative and its application to the one-pot conversion to a 2,3,5-trisubstituted quinoline derivative. Tetrahedron Lett. 47 (8), 1261–1265. doi:10.1016/j.tetlet.2005.12.080
Saptal, V. B., Wang, R., and Park, S. (2020). Recent advances in transition metal-free catalytic hydroelementation (E = B, Si, Ge, and Sn) of alkynes. RSC Adv. 10 (71), 43539–43565. doi:10.1039/d0ra07768b
Sashidhara, K. V., Kumar, A., Bhatia, G., Khan, M. M., Khanna, A. K., and Saxena, J. K. (2009). Antidyslipidemic and antioxidative activities of 8-hydroxyquinoline derived novel keto-enamine Schiffs bases. Eur. J. Med. Chem. 44 (4), 1813–1818. doi:10.1016/j.ejmech.2008.08.004
Schmaunz, C. E., Mayer, P., and Wanner, K. T. (2014). Inter- and intramolecular [4+2]-Cycloaddition reactions with 4,4-disubstituted N-Silyl-1,4-dihydropyridines as precursors for N-protonated 2-azabutadiene intermediates. Synthesis 46 (12), 1630–1638. doi:10.1055/s-0033-1341044
Shen, R., and Porco, J. A. (2000). Synthesis of enamides related to the salicylate antitumor macrolides using copper-mediated vinylic substitution. Org. Lett. 2 (9), 1333–1336. doi:10.1021/ol005800t
Tamang, S. R., Singh, A., Unruh, D. K., and Findlater, M. (2018). Nickel-catalyzed regioselective 1,4-hydroboration of N-heteroarenes. ACS Catal. 8 (7), 6186–6191. doi:10.1021/acscatal.8b01166
Wang, M.-X. (2015). Exploring tertiary enamides as versatile synthons in organic synthesis. Chem. Commun. 51 (28), 6039–6049. doi:10.1039/c4cc10327k
Wang, Q., Li, Y., Sun, J., Chen, S., Li, H., Zhou, Y., et al. (2023a). Rh-catalyzed C–H activation/annulation of enaminones and cyclic 1,3-dicarbonyl compounds: an access to isocoumarins. J. Org. Chem. 88 (9), 5348–5358. doi:10.1021/acs.joc.2c02898
Wang, X., Zhang, Y., Yuan, D., and Yao, Y. (2020). Regioselective hydroboration and hydrosilylation of N-heteroarenes catalyzed by a zinc alkyl complex. Org. Lett. 22 (14), 5695–5700. doi:10.1021/acs.orglett.0c02082
Wang, Y., Li, H., Yang, H., Fan, M., and Liu, Q. (2023b). Manganese-catalyzed regioselective hydroboration of quinolines via metal–ligand cooperation. CCS Chem. 0 (0), 1–12. doi:10.31635/ccschem.023.202303289
Yu, H.-C., Islam, S. M., and Mankad, N. P. (2020). Cooperative heterobimetallic substrate activation enhances catalytic activity and amplifies regioselectivity in 1,4-hydroboration of pyridines. ACS Catal. 10 (6), 3670–3675. doi:10.1021/acscatal.0c00515
Zhang, F., Song, H., Zhuang, X., Tung, C.-H., and Wang, W. (2017). Iron-catalyzed 1,2-selective hydroboration of N-heteroarenes. J. Am. Chem. Soc. 139 (49), 17775–17778. doi:10.1021/jacs.7b11416
Zhang, X., Fried, A., Knapp, S., and Goldman, A. S. (2003). Novel synthesis of enamines by iridium-catalyzed dehydrogenation of tertiary amines. Chem. Commun. 16, 2060–2061. doi:10.1002/chin.200345045
Zhou, X.-Y., Zhang, M., Liu, Z., He, J.-H., and Wang, X.-C. (2022). C3-Selective trifluoromethylthiolation and difluoromethylthiolation of pyridines and pyridine drugs via dihydropyridine intermediates. J. Am. Chem. Soc. 144 (32), 14463–14470. doi:10.1021/jacs.2c06776
Keywords: hydroboration, hydrosilylation, N-boryl enamine, N-silyl enamine, N-heteroarene, unsaturated nitriles, aldimines
Citation: Cao VD and Joung S (2024) Synthesis and utility of N-boryl and N-silyl enamines derived from the hydroboration and hydrosilylation of N-heteroarenes and N-conjugated compounds. Front. Chem. 12:1414328. doi: 10.3389/fchem.2024.1414328
Received: 08 April 2024; Accepted: 17 May 2024;
Published: 07 June 2024.
Edited by:
Sehoon Park, Guangdong Technion-Israel Institute of Technology (GTIIT), ChinaReviewed by:
Jung-Woo Park, Seoul National University of Science and Technology, Republic of KoreaEun Jin Cho, Chung-Ang University, Republic of Korea
Copyright © 2024 Cao and Joung. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Seewon Joung, c2Vld29uam91bmdAaW5oYS5hYy5rcg==