 Naga Venkateswara Rao Nulakani
Naga Venkateswara Rao Nulakani Mohamad Akbar Ali
Mohamad Akbar Ali- 1Department of Chemistry, Khalifa University of Science and Technology, Abu Dhabi, United Arab Emirates
- 2Center for the Catalyst and Separations, Khalifa University of Science and Technology, Abu Dhabi, United Arab Emirates
Aminomethanol is released into the atmosphere through various sources, including biomass burning. In this study, we have expounded the chemical kinetics of aminomethanol in the reaction pathways initiated by the hydroxyl radical (
1 Introduction
Amines, especially those bearing the –NH2 functional group (RNH2), find extensive applications in the chemical industry, including uses in solvents, catalysis, surfactants, pharmaceuticals, adhesives, dyes and pigments, etc. (Ge et al., 2011a). For instance, carbon capture and storage (CCS) technology utilizes amine solvents to separate the CO2 emission from the acid gas treatment of natural gas and fossil fuel power plants (Dai et al., 2012; Yamada, 2021). The rapid development of CCS technology consequently leads to a significant increase in alkanolamines emissions into the atmosphere due to their high vapor pressure (Kapteina et al., 2005). Additionally, the amines are also released into the atmosphere by various other sources such as thorough industrial emission, wastewater treatment, animal husbandry and automotive activities, emissions from ocean organisms, biomass combustion and the degradation of proteins, etc. (Ge et al., 2011b). These amines undergo degradation and participate in conversion reactions both in gas and aqueous phase (clouds, raindrops, fog) in the atmosphere. As a result, they have detrimental effects on air quality, leading to issues like acid rain, urban smog and tropospheric ozone. Also, the amines and alkanolamines have the potential to contribute to greenhouse gas formation, notably nitrous oxide (N2O) and the production of highly carcinogenic compounds and nitrosamines (Schade and Crutzen, 1995; Ge et al., 2011a; Ge et al., 2011b). Hence, elucidating the transformation, mechanisms and kinetics of amines is of potential importance.
The Strecker synthesis, involving a series of chemical reactions, has received great scholarly attention as a potential technique to synthesize chiral α-amino acids using ammonia (NH3), hydrogen cyanide (HCN) and aldehyde (RCHO) (Strecker, 1850; Gröger, 2003; Nájera and Sansano, 2007; Wang et al., 2011). However, aminomethanol (NH2CH2OH), a pivotal intermediate for the synthesis of the simplest amino acid namely, glycine, has not been observed in the laboratories (Nielsen et al., 1979; Schutte et al., 1993). This is may be due to the decomposition of aminomethanol to methanimine (CH2NH) and water (H2O). Previous theoretical investigations have indicated that aminomethanol is kinetically stable in the gas phase with a substantial barrier of 230 kJ/mol toward dehydration to methanimine (Feldmann et al., 2005). Our recent study also supports that the formation of methanimine and water from aminomethanol is negligibly small and forbidden by an energy barrier of 234 kJ/mol (Ali, 2019). Bossa et al., have observed aminomethanol at low temperatures through the thermal reaction between ammonia and formaldehyde (Bossa et al., 2009). They have also pointed out that aminomethanol may exist in hot corinos in gas phase. The recent experimental study on aminomethanol in astrophysical-like conditions also validates that aminomethanol could be generated from amines on ice grains in proto-stellar cores or protoplanetary disks (Singh et al., 2022). These studies strongly corroborate the existence of aminomethanol in the atmosphere.
In our recent study (Ali, 2019), the calculated rate constant of CH2O + NH3 has suggested that aminomethanol could potentially form at higher temperatures rather than under atmospheric conditions, as illustrated in Figure 1. Additionally, the significant lifetime (∼4 days) of aminomethanol could facilitate the initiation of various chemical reactions with other atmospheric species (Ali, 2019). Nevertheless, the atmospheric chemistry of aminomethanol remains largely unexplored to date, primarily due to the considerable challenges associated with experimental synthesis. Therefore, it is crucial to delve into the reaction mechanisms and kinetics of aminomethanol to evaluate the potential formation of various compounds, including toxicants or carcinogenic byproducts. However, there has been a lack of experimental studies specifically examining the reaction kinetics of the photo-oxidation of aminomethanol. In light of this gap, theoretical approaches, coupled with state-of-the-art computational methodologies and advanced statistical rate theories, are essential for exploring the intricate atmospheric chemistry of aminomethanol, particularly in extreme environmental conditions.
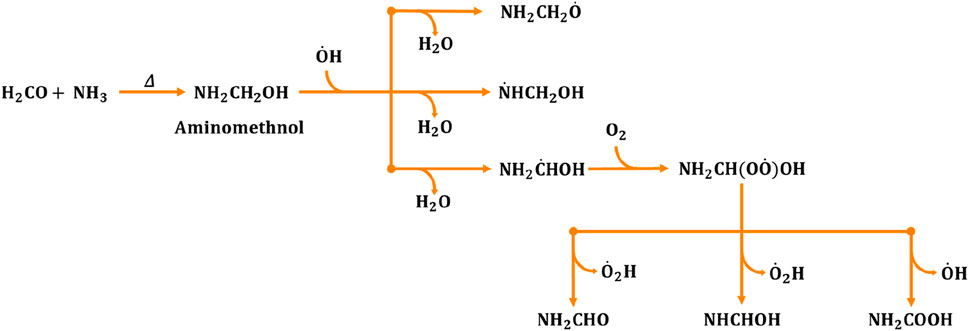
Figure 1. Generalized reaction mechanism for the formation of formamide (NH2CHO), formimidic acid (NHCHOH) and amino formic acid (NH2COOH) during the aminomethanol +
The fate of aminomethanol, in general, is greatly influenced by its gas-phase oxidation by various oxidizing agents. Hydroxyl radicals (
Among these product species, formamide is simplest and naturally occurring amide molecule. It is a potential precursor for the synthesis of a broad range of organic molecules (Saladino et al., 2012a; Saitta and Saija, 2014), which are vital for life as it poses all the important elements including carbon C), hydrogen H), oxygen O) and nitrogen N) except the heavy elements like sulphur S) and phosphorus P) (Saladino et al., 2005; 2007; 2012b). Additionally, formamide has an amide functional (-N-C (=O)-) group, which is essential for the formation of the chains of amino acids in order to build up proteins. It promotes the synthesis of four nucleobases namely, adenine, guanine, cytosine and uracil of ribonucleic acid (RNA) (Saladino et al., 2005; Ferus et al., 2015). It also serves as the pre-cursor for carboxylic acids, amino acids and sugars (Botta et al., 2018). These points clearly demonstrating the potential role of formamide in biochemical processes, that are essential for life’s sustenance and evolution.
Also, formamide is a highly ubiquitous molecule in the Universe. It was first detected in space dates back to 1971 when Rubin et al., utilized the 140-foot telescope at the National Radio Astronomy Observatory (NRAO) to scrutinize the Sagittarius B2 region (Sgr B2), situated proximate to the Galactic center (Rubin et al., 1971). Their observations distinctly revealed the three hyperfine components (∆F = 0) of the 21,1–21,2 rotational transition of NH2CHO, resonating at 4.62 GHz (6.5 cm). Sgr B2, renowned as the most prodigious star formation region within our Milky Way and marked the maiden identification of an interstellar compound harboring carbon C), hydrogen H), oxygen O), and nitrogen N). Subsequent detections of formamide have been made in diverse astronomical locales, including the vicinity of the young stellar entity W33A (Schutte et al., 1999), within cometary bodies such as C/1995 O1 (Bockelée-Morvan et al., 2000) and C/1996 B2 (Lis et al., 1997) and within the broader expanse of the interstellar medium (Solomon, 1973).
A plethora of experimental and theoretical inquiries have been undertaken to elucidate the formation of formamide along with other organic compounds in the interstellar medium (ISM) by different methods (Woon, 2002; Navarro-González and Raulin, 2004; Pastorek et al., 2019; Ferus et al., 2023). These investigations spanning from the proton irradiation (PI) of gas mixtures containing methane and nitrogen (Koike et al., 2003), ultraviolet irradiation (UV) of ice mixtures containing hydrogen cyanide (HCN), water (H2O) and ammonia (NH3) (Gerakines et al., 2004), pyrolysis of mixtures comprising carbon monoxide (CO), NH3, and H2O (Takano et al., 2004) and the photolysis of ices (Bernstein et al., 1997). Similarly, under terrestrial conditions, formamide (NH2CHO) can be synthesized from combinations of low molecular weight compounds such as NH3, formic acid (HCOOH), formic ester derivatives (HCOOR), CO and alcohols, under both catalyzed and uncatalyzed experimental setups (Deschamps, 1931).
In the current investigation, our focus lies in exploring the intricate mechanistic aspects and branching ratios corresponding to the pivotal stages of the
2 Theoretical methodology
2.1 Electronic structure calculations
All gas-phase ab initio/density functional theory (DFT) calculations were performed using the Gaussian 16 suite of programs (Frisch et al., 2019). The stationary points of all molecular systems, including reactants, products, intermediates and transition states on the potential energy surface (PES) were optimized using the Minnesota 2006 exchange correlation functional such as M06-2X, in conjunction with the Pople’s split-valence 6-311++G (3df, 3pd) basis set (Frisch et al., 1984; Zhao and Truhlar, 2008). The long-range van der Waals interactions between the reactive species were accounted using Grimme’s empirical dispersion (GD3) corrections (Grimme et al., 2010). Previous studies have demonstrated that the current level of theory exhibits a reliable performance in addressing noncovalent interactions between gaseous molecules and in locating the transition states of atmospheric and combustion reactions (Ali et al., 2016; 2018; 2022; Ali, 2019; 2020; Dash and Ali, 2022; 2023; Ali and Balaganesh, 2023). Tight convergence criteria were applied during the wave function optimization of the reactive species, complexes, products and transition states. Unscaled vibrational frequencies at the same level of theory (M06-2X/6-311++G (3df, 3pd)) were utilized to compute zero-point energy (ZPE) corrections to the total energies of all molecular systems, to characterize the stationary points on the PES and for rate-constant calculations. Vibrational frequency analysis confirmed all positive frequencies for the reactants, complexes, intermediates and products, while a single imaginary frequency was observed for the transition states.
Additionally, single-point energy calculations were conducted at a higher-level of theory on the molecular structures optimized at a lower-level of theory to ensure an accurate description of the energetic parameters. Specifically, the CCSD(T)/6-311++G (3df, 3pd) level of theory was utilized to estimate the single-point energies of the gas-phase molecular geometries, which were initially optimized at the M06-2X/6-311++G (3df, 3pd) level of theory (Raghavachari et al., 1989). The basis set superposition error (BSSE) calculations were also performed using the counterpoise (CP) corrected method (Boys and Bernardi, 1970; Simon et al., 1996). The <Ŝ2> eigenvalues were monitored to evaluate the spin contamination for the wavefunction of the open-shell radicals. The T1-diagnostic values obtained at the CCSD(T)/6-311++G (3df, 3pd) level of theory were analyzed to validate the single-reference method and were found to be within the acceptable range (i.e., ≤0.02) for all important species (Lee and Taylor, 1989). Overall, the combination of CCSD(T)//M06-2X functionals has been employed in numerous research studies, providing a reasonably accurate description of the thermochemistry and chemical kinetics of many atmospheric reactions (Ali et al., 2016; Ali et al., 2018; Ali, 2019; Ali, 2020; Ali et al., 2022; Dash and Ali, 2022; Ali and Balaganesh, 2023; Dash and Ali, 2023).
2.2 Chemical kinetic analysis
Comprehensive chemical kinetic calculations for the
where, k1 and k-1 are the forward and reverse rate constant for the first bimolecular reaction and the k2 is the rate constant for the second unimolecular reaction. The kinetic rate constants for these bimolecular (k, in cm3 molecule−1 s-1) and unimolecular (kuni, s-1) reactions in the high-pressure limit defined by transition state theory are represented as follows,
Assuming that the pre-reactive complex was in equilibrium with the reactants and was at a steady state, then the overall rate constants is expressed as;
If k-1 >> k2, the rate constant is rewritten as
This kinetic model is reasonably correct at the high-pressure limit, where the pre-reactive complex can be stabilized by collisions with other atmospherics species. This approach was widely used in literature for the water-assisted reaction and the predicted rate coefficients are reasonably good agreement with the experimental values (Ali, 2019; Ali et al., 2022, Ali and Balaganesh 2023).
The different parameters of Eqs 3, 6 were breakdown and elaborately discussed the specifics of each component in Supplementary Material to prevent redundancy from earlier research. The k and k2 for the other plausible oxidation reactions of
The temperature- and pressure-dependent microscopic rate constants k(E) have also been computed for the O2 addition reaction to the aminomethanol radicals generated in Eq. 2. This was accomplished using the Rice−Ramsperger−Kassel−Marcus (RRKM)/master equation (ME) theory, implemented in the MultiWell suite of programs. The MultiWell code facilitates the computation of non-steady-state effects including unimolecular decomposition processes, isomerization, collision energy transfer and chemical activation for the complex rate-constant calculations. To perform these calculations, molecular and energetic parameters such as vibrational frequencies, moments of inertia and reaction barriers are required as input data. Using this data, the MultiWell suite computes sum and density-of-states, followed by the evaluation of microscopic rate-constant k(E). The RRKM/ME microscopic rate-constant k(E) is defined as follows,
The details of each term can be found in the Supplementary Material. Temperature and pressure-dependent rate constants and branching ratios of the products were evaluated by incorporating N2 gas as the bath gas. The collisional energy transfer process was addressed using the conventional temperature-dependent exponential-down model with a <ΔE > down parameter (which represents the average energy loss per the collision of the active compound with the bath gas molecule), with an approximate value of ∼200*(T/300)0.85 cm−1 (Goldsmith et al., 2012). Lennard-Jones (L-J) parameters were employed to account for the frequency of collisions between the active compound and the bath gas (N2) collider. The L-J parameters for N2 gas, specifically σ (N2) = 3.74 Å and ε/kB (N2) = 82 K, were sourced from the literature, while the same parameters for all wells were adopted from our previous study (Dash and Ali, 2022).
For the barrierless reactions i.e.,
The pressure-dependent total rate constants kbimol (T, M) for aminomethanol radical (
where, Γ represents the quantum mechanical tunneling corrections,
Finally, the calculated rate constants were fitted at the high-pressure limit (
where A is the pre-exponential factor, T is the temperature, n is the temperature exponent and Ea is the activation energy. The coordinates of equilibrium geometries, vibrational harmonic frequencies and rational constants of all important species involved in the
3 Results and discussion
3.1 Rotational conformers of aminomethanol
The oxidation reaction between the aminomethanol (AM) and
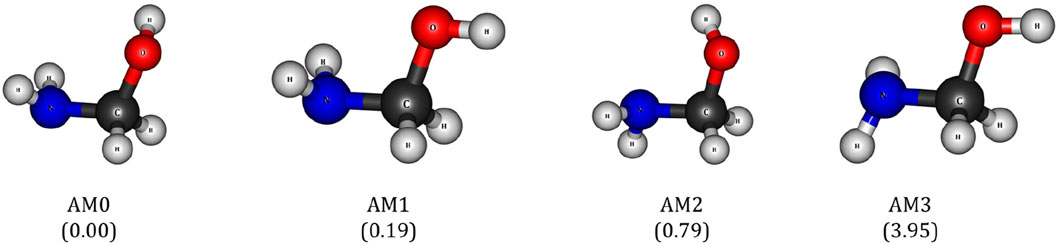
Figure 2. Different rotational conformations of aminomethanol (AM). The values in the parenthesis indicate the relative energies in kcal/mol.
3.2 Potential energy surface of aminomethanol +
The zero-point energy (ZPE) corrected potential energy surface (PES) for the H-abstraction reaction of the most stable conformation of aminomethanol (AM0) by the
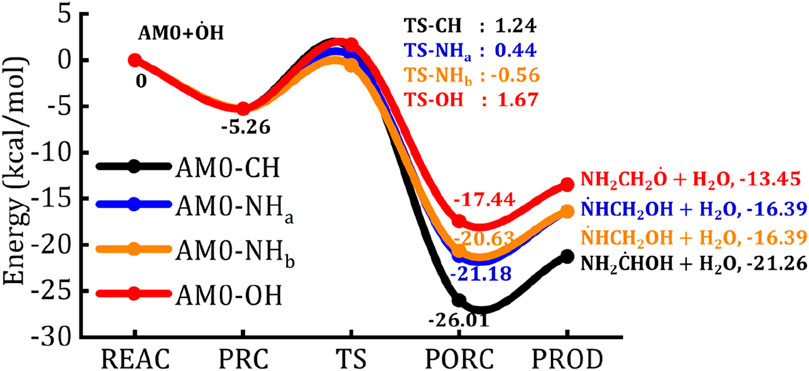
Figure 3. The zero-point energy corrected potential energy surface of AM0 +
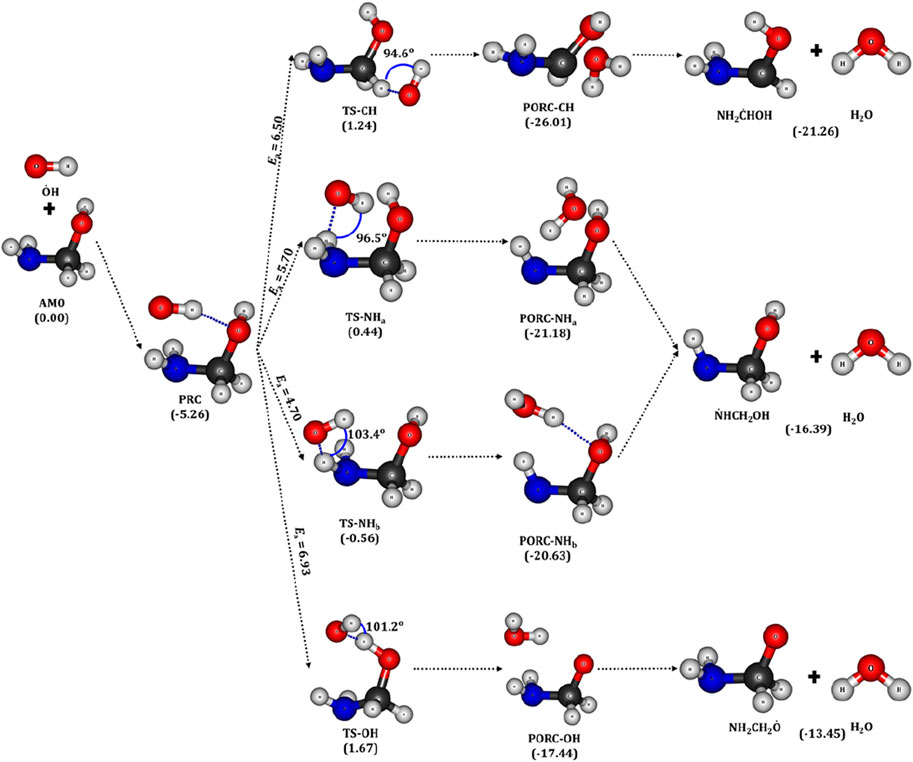
Figure 4. Optimized geometries of aminomethanol (AM0), PRC, TSs, PORCs and the radicals of
For example, the bond angle between the H atom of the –CH2 group and
To account for the effect of other rotational conformers on the oxidation reaction, the energies of PRCs, TSs and PORCs have been computed for the abstraction of H atom from –CH2, –NH2 and –OH groups of other rotational conformations of AM (i.e., AM1 and AM2). The complete reaction profile for the H-abstraction reaction of AM1 along with the energies is presented Supplementary Figure S1. The H abstraction from the –CH2 group of AM1 proceeds via three TSs such as TS-CHa TS-CHb and TS-CHc. Among these TSs, TS-CHa is linked to the PRC1 while TS-CHb and TS-CHc are linked to the PRC2 (see Supplementary Figure S2). However, all these–CH TSs are linked to the same product complex and lead to the formation of a single NH2
Finally, the energetics and the barrier height values of H abstraction reactions from AM0, AM1 and AM2 by
It is worth to mention here that, the TS energies for the major H-abstraction pathways i.e., from the –CH2 and –NH2 channels of NH2CH2OH +
The rate constant (
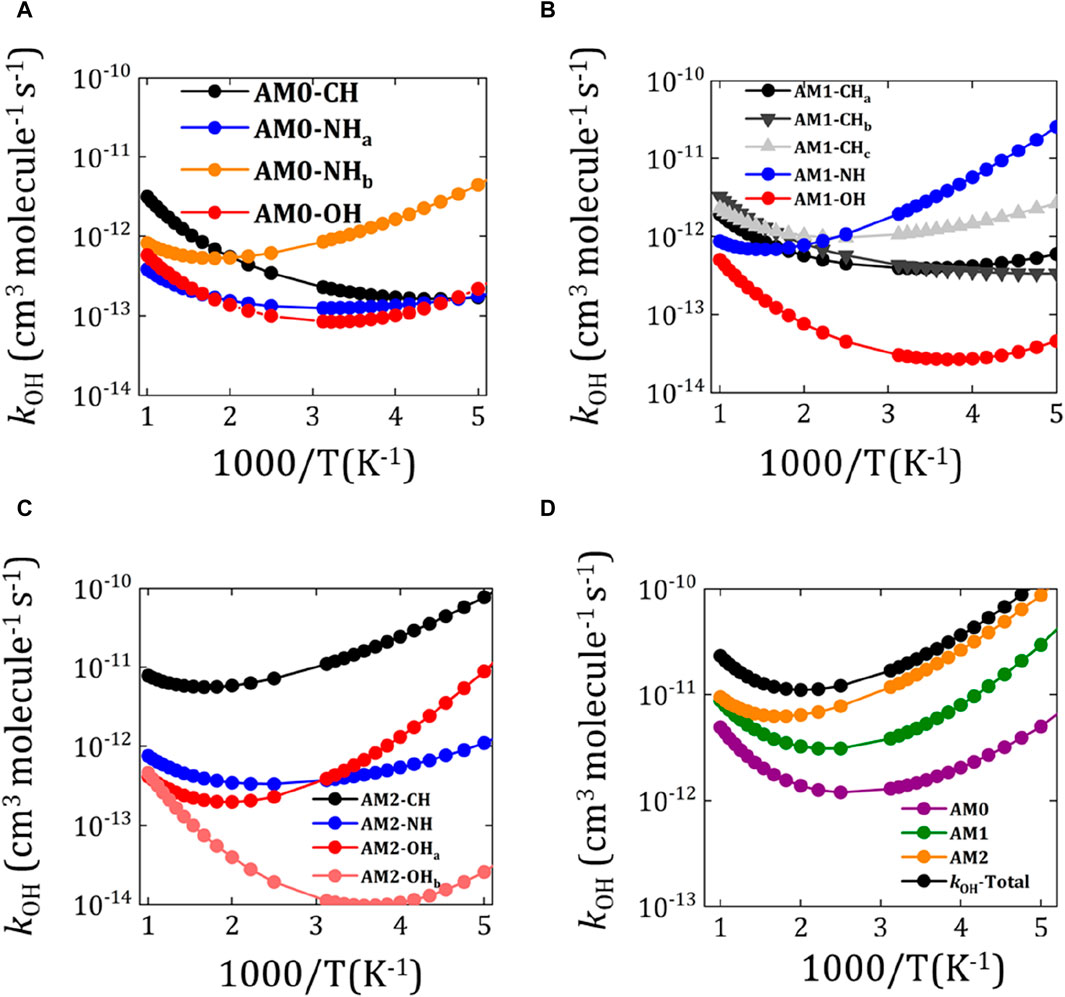
Figure 5. Calculated rate constants (
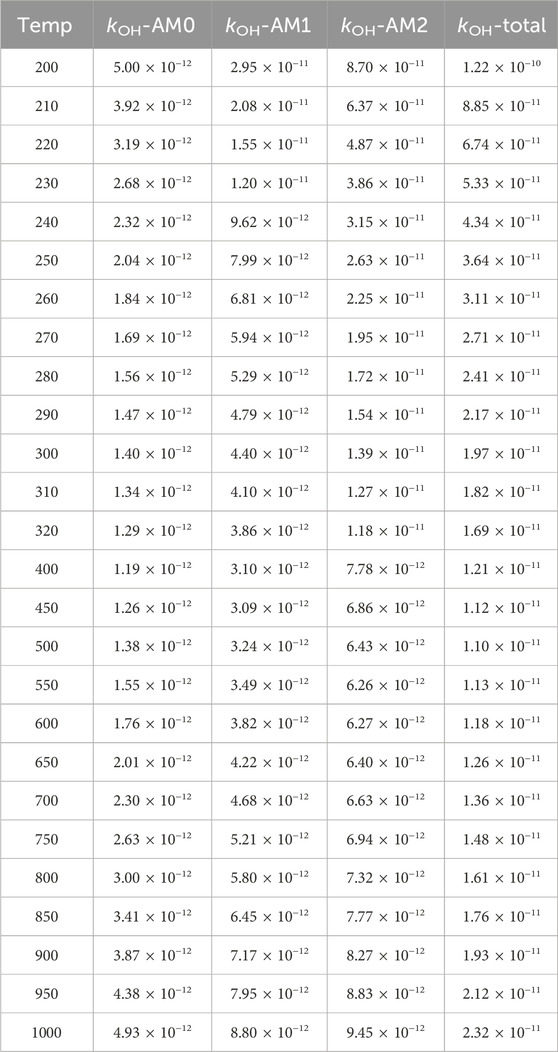
Table 1. Calculated temperature dependent rate constants (kOH, cm3 molecule−1 s−1) for the
The bimolecular reactions between the aminomethanol (NH2CH2OH) +
In recent decades, significant advances in atmospheric chemistry have spurred the development of new theoretical approaches for exploring intricate details of ground-state chemical reactions and their underlying mechanisms. Nevertheless, an equivalent synergy between theory and experimentation remains absent in the realm of atmospheric photochemistry involving electronically excited states. The modeling of molecular photochemistry necessitates a meticulous consideration of non-adiabatic effects, specifically, the coupling between electronic states and molecular motion. This presents formidable challenges, as it contradicts several conventional approximations in theoretical chemistry. Notably, non-adiabatic effects challenge the venerable Born-Oppenheimer approximation, while classical treatments of nuclear dynamics may prove inadequate and non-equilibrium phenomena can challenge established reaction rate theories.
A plethora of methodologies have emerged to address these challenges, including MCTDH (Multi Configuration Time Dependent Hartree) (Manthe et al., 1992; Beck, 2000; Meyer, 2012), trajectory surface hopping (TSH) (Tully and Preston, 1971; Hammes-Schiffer and Tully, 1994; Fabiano et al., 2008) and ab initio multiple spawning (AIMS) (Ben-Nun et al., 2000; Ben-Nun and Martínez, 2002; Yang and Martínez, 2011). However, the application of these techniques to investigate atmospheric photochemistry encounters various challenges. These encompass complexities in simulating spectroscopically relevant choices and atmospheric modeling interests, the intricate electronic architecture of multichromophoric volatile organic compounds (VOCs), the diverse excited-state dynamics triggered by solar irradiation, the protracted dynamics of excited VOCs, the prominence of intersystem crossings or collisional processes and the modulating influence of aqueous environments such as those found in atmospheric aerosols and clouds. Given that non-adiabatic dynamic approaches demand considerable computational resources beyond our current capacity and each method carries its own limitations, we envision these studies as prospects for future exploration.
3.3 Potential energy surface of NH2
The aminomethanol radicals produced in the initial reactions of aminomethanol +
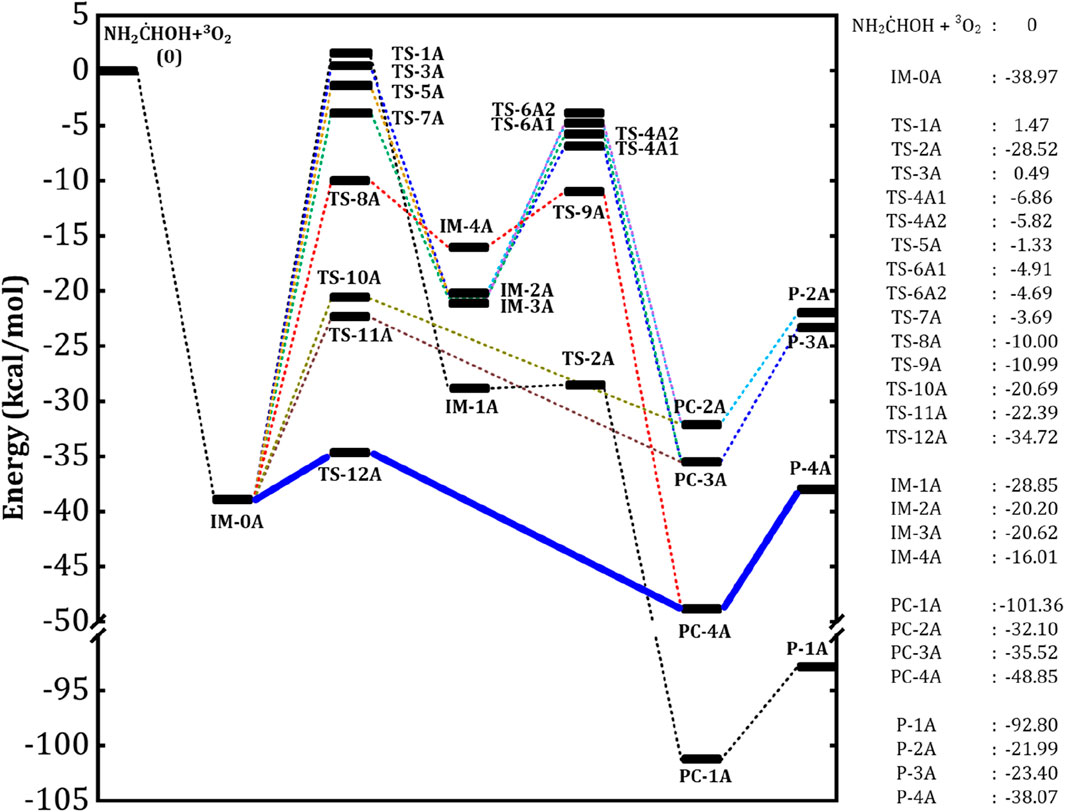
Figure 6. The zero-point energy (ZPE) corrected potential energy surface of AM radical (NH2
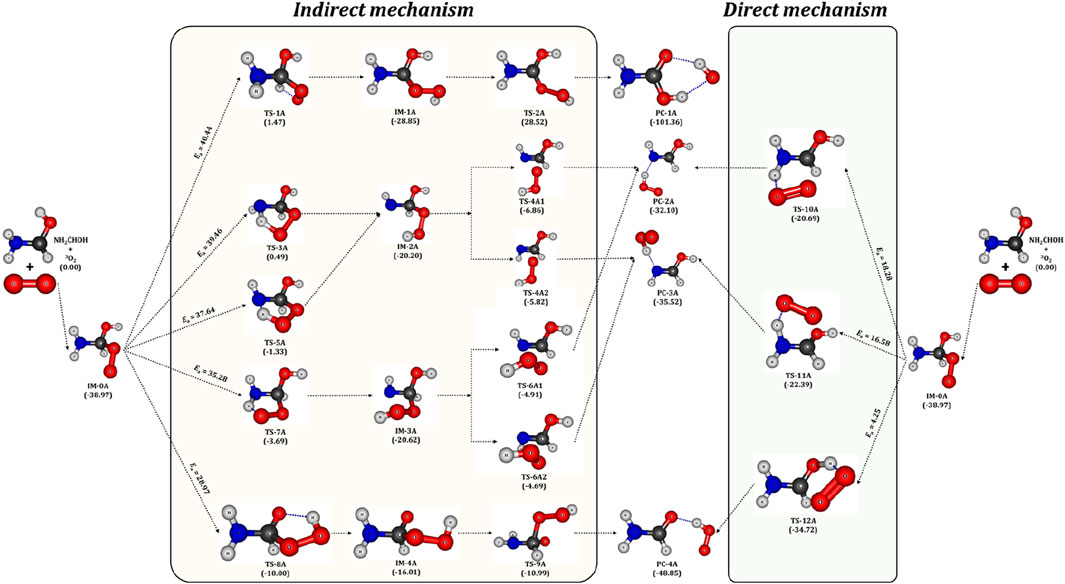
Figure 7. Optimized geometries of intermediates (IMs), transition states (TSs) and product complexes (PCs) of 3O2 + NH2
In detail, the transfer of H atom from –CH and –OH groups to the O-site of peroxy radical (IM-0A) is favored through the transition states TS-1A and TS-8A. On the other hand, the presence of two different H atoms in –NH2 group and different attacking directions of 3O2 lead to three different reaction pathways, which proceed through the TSs, TS-3A, TS-5A and TS-7A for the H abstraction from –NH2 group. Among all these TSs, the TS-1A and TS-3A are situated above the reactants with an energy of 1.5 and 0.5 kcal/mol followed by TS-5A, TS-7A and TS-8A (−1.3, −3.7 and −10.0 kcal/mol). It indicates that the H-transfer from –CH group to O-site of IM-0A requires a high energy of ∼40 kcal/mol to form the intermediate IM-1A. The transfer of two different H atoms from –NH2 group respectively requires 40 (via TS-3A) and 35 (via TS-7A) kcal/mol to form the intermediates, IM-2A and IM-3A. Further, different spatial arrangement of O2 also facilitates a distinct transition state, TS-5A for the H transfer from –NH2 group and it leads to the previous intermediate, IM-2A. The reaction proceed through TS-5A shows a barrier height more than 35 kcal/mol. The H transfer process from that of –OH group requires a relatively low energy of 29 kcal/mol via the TS-8A to form IM-4A. The barrier heights for these H-transfer reactions strongly comply with that of H transfer from the similar functional groups. The intermediates IM-1A to IM-4A are stabilized by ∼ -29, −20.2, −20.6 and −16 kcal/mol with respect to the reactants, respectively. The breaking of O-O and C-O bond in IM-1A and IM-4A through the transition states TS-2A and TS-9A form the product complexes, PC-1A and PC-4A, respectively. While, PC-2A and PC-3A can be formed by the breaking of C-O bond of IM-2A via the TSs, TS-4A1 and TS-4A2. The IM-3A also leads to the same product complexes, PC-2A and PC-4A via the TSs, TS-6A1 and TS-6A2. Figure 6 shows that, the IM-1A is almost barrierlessly (0.3 kcal/mol) dissociated into the product complex PC-1A, which is composed of hydrogen bonded amino formic acid (H2NCOOH) and
On the other hand, the reactions proceed via the transition states, TS-10A, TS-11A and TS-12A directly form the product complexes. In detail, these TSs initiates the simultaneous transfer of H atom from –NH2 and –OH groups to the O-site of peroxy radical and the breaking of C-O bond to form the post-reactive product complexes, PC-2A, PC-3A and PC-4A, respectively. Similar to TS-3A and TS-7A, the TS-10A and TS-11A involve the transfer of inequivalent H atoms of –NH2 to the O-site. The energy barriers have been found to be 18, 17 and 4.3 kcal/mol for the transfer of H atom from –NH2 (via TS-10A, TS-11A) and –OH (via TS-12A) groups to the O-site of peroxy radical. Overall, it is evident from Figure 6 that, the reaction pathway proceeding through the transition state, TS-12A to form P-4A (NH2CHO) is energetically more favorable followed by that proceeding through TS-10A and TS-11A to form P-2A and P-3A (NHCHOH) by the simultaneous H transfer and CO bond breaking process rather than reaction pathways that proceeding through TS-1A, TS-3A, TS-5A, TS-7A and TS-8A.
We have compared the energetics of important reactive species of NH2
Overall, the computed results suggest that the reaction pathways, which proceed through the TS-1A, TS-3A, TS-5A, TS-7A and TS-8A from the intermediate IM0-A exhibit high energy barriers. These energy barriers are in the range of (30-40 kcal/mol) as shown in Figure 7. Hence, these reaction pathways are excluded from the further studies of reaction kinetics and branching ratio analysis due to the high energy barriers. Relatively low energy barrier pathways which occurred via the transition states TS-10A, TS-11A and TS-12A were adopted for further kinetics and branching ratio calculations.
The rate constants and the branching ratio values of 3O2 + NH2
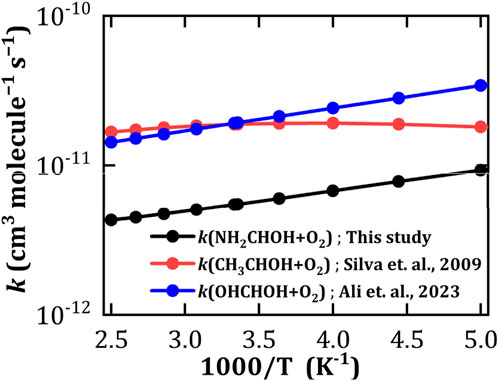
Figure 8. Computed rate constants for the NH2
We have also computed the ZPE corrected PES of nitrogen centered radical, namely,
3.4 Molecular dynamics simulations
We have conducted Born-Oppenheimer molecular dynamics (BOMD) simulations to investigate the formation of formamide at 300 K through the reactions involving the reactive species (
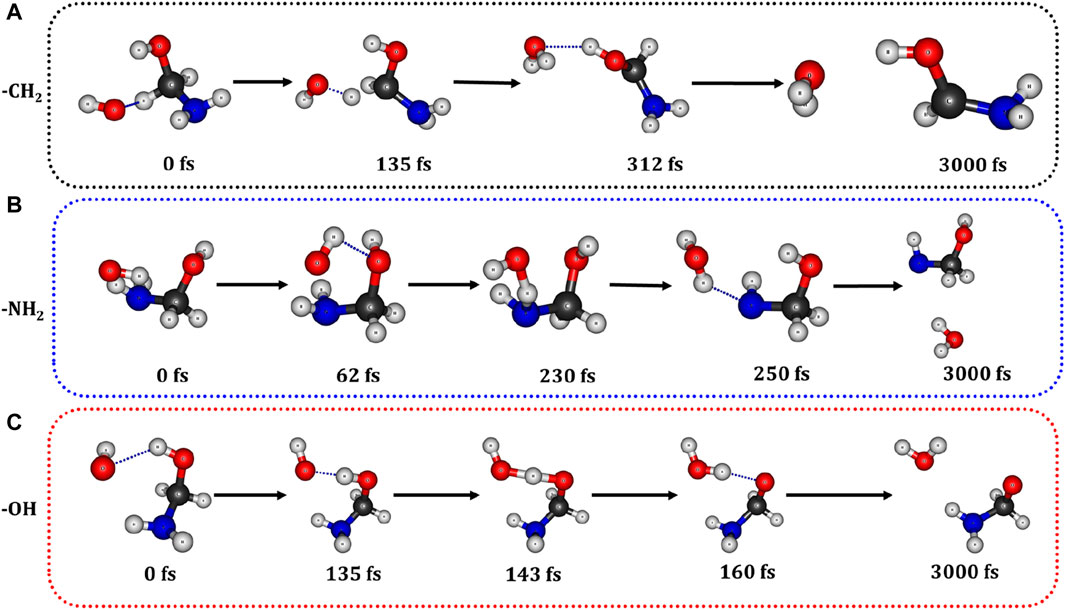
Figure 9. Snapshots taken at different time intervals during the BOMD simulations between
3.5 Atmospheric implications
Our previous investigation (Ali, 2019) has demonstrated the occurrence of a high-temperature reaction between ammonia (NH₃) and formaldehyde (H2CO) during biomass burning, resulting in the formation of aminomethanol (NH₂CH₂OH). This prompts a keen interest in elucidating the oxidation reaction mechanism and kinetics of aminomethanol, leading to the formation of various compounds, including potentially hazardous or carcinogenic byproducts. The possible atmospheric decomposition pathway resulting from
where, k denotes the rate constant of NH₂CH₂OH +
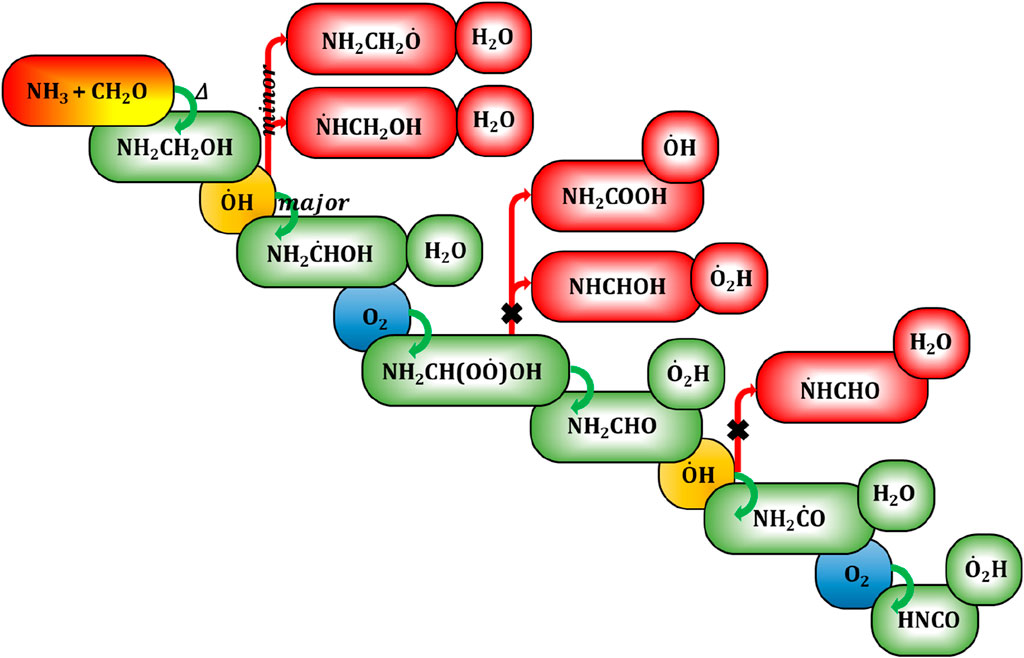
Figure 10. Generalized reaction mechanism for the formation of aminomethanol from ammonia and formaldehyde and its subsequent decomposition reaction pathways to potentially hazardous isocyanic acid.
Barnes et al., have reviewed the mechanistic details and atmospheric chemistry of amides (Barnes et al., 2010). They showed that the formamide could further react with
On the other hand, the branching fraction (20%) of N-centered radicals indicates the formation of
Overall, investigating the branching fraction and ensuing rate constants associated with the hydrogen abstraction from –CH2, –NH2, and –OH groups of aminomethanol by hydroxyl radicals offers initial insights into the predominant formation of formamide. Additionally, this study provides additional insights into the formation of N-centered radicals in significant quantities. Nevertheless, conducting further investigations on the reactions of O2, NO and NO2 with N-centered radicals would undoubtedly contribute to significantly understand their impact on the atmosphere.
4 Conclusion
In this study, we have comprehensively explored the mechanistic details of aminomethanol oxidation reaction initiated by atmospheric
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author contributions
NN: Conceptualization, Data curation, Formal Analysis, Methodology, Writing–original draft, Writing–review and editing. MA: Conceptualization, Formal Analysis, Funding acquisition, Investigation, Software, Supervision, Validation, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. The work is supported by the faculty startup grant #8474000461 at Khalifa University of Science and Technology, Abu Dhabi, UAE.
Acknowledgments
NN and MA thank the supercomputer facility and Department of Chemistry at the College of Engineering and Physical Sciences at Khalifa University of Science and Technology, at Abu Dhabi UAE for their support. MA thanks Khalifa University of Science and Technology, at Abu Dhabi UAE for Faculty Start-up grant #8474000461.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2024.1407355/full#supplementary-material
References
Abdel-Rahman, M. A., Shibl, M. F., El-Nahas, A. M., Abdel-Azeim, S., El-demerdash, S. H., and Al-Hashimi, N. (2021). Mechanistic insights of the degradation of an O-anisidine carcinogenic pollutant initiated by OH radical attack: theoretical investigations. New J. Chem. 45, 5907–5924. doi:10.1039/D0NJ06248K
Ali, M. A. (2019). Theoretical study on the gas phase reaction of CH2O + NH3: the formation of CH2 O⋯NH3, NH2CH2OH, or CH2NH + H2O. Phys. Chem. Chem. Phys. 21, 19242–19251. doi:10.1039/C9CP02777G
Ali, M. A. (2020). Computational studies on the gas phase reaction of methylenimine (CH2NH) with water molecules. Sci. Rep. 10, 10995. doi:10.1038/s41598-020-67515-3
Ali, M. A., and Balaganesh, M. (2023). Effect of formic acid on O2 + OH˙CHOH → HCOOH + HO2 reaction under tropospheric condition: kinetics of cis and trans isomers. Phys. Chem. Chem. Phys. 25, 9965–9978. doi:10.1039/D2CP05874J
Ali, M. A., Balaganesh, M., Al-Odail, F. A., and Lin, K. C. (2021). Effect of ammonia and water molecule on OH + CH3OH reaction under tropospheric condition. Sci. Rep. 11, 12185. doi:10.1038/s41598-021-90640-6
Ali, M. A., Balaganesh, M., and Lin, K. C. (2018). Catalytic effect of a single water molecule on the OH + CH2NH reaction. Phys. Chem. Chem. Phys. 20, 4297–4307. doi:10.1039/C7CP07091H
Ali, M. A., Dash, M. R., and Al Maieli, L. M. (2022). Catalytic effect of CO2 and H2O molecules on •CH3 + 3O2 reaction. Catalysts 12, 699. doi:10.3390/catal12070699
Ali, M. A., Sonk, J. A., and Barker, J. R. (2016). Predicted chemical activation rate constants for HO2 + CH2NH: the dominant role of a hydrogen-bonded pre-reactive complex. J. Phys. Chem. A 120, 7060–7070. doi:10.1021/acs.jpca.6b06531
Baidya, B., Lily, M., and Chandra, A. K. (2018). Theoretical insight into the kinetics of H-abstraction reaction of CHF2CH2OH with OH radical, atmospheric lifetime and global warming potential. ChemistrySelect 3, 6136–6144. doi:10.1002/slct.201800491
Barker, J. R. (2001). Multiple-Well, multiple-path unimolecular reaction systems. I. MultiWell computer program suite. Int. J. Chem. Kinet. 33, 232–245. doi:10.1002/kin.1017
Barker, J. R. (2009). Energy transfer in master equation simulations: a new approach. Int. J. Chem. Kinet. 41, 748–763. doi:10.1002/kin.20447
Barker, J. R., Nguyen, T. L., Stanton, J. F., Aieta, C., Ceotto, M., Gabas, F., et al. (2023) MultiWell-2023 software SuiteBarker. Ann Arbor, Michigan, USA: J. R. University of Michigan. Available at: https://multiwell.engin.umich.edu.
Barnes, I., Solignac, G., Mellouki, A., and Becker, K. H. (2010). Aspects of the atmospheric chemistry of amides. ChemPhysChem 11, 3844–3857. doi:10.1002/cphc.201000374
Beck, M. (2000). The multiconfiguration time-dependent Hartree (MCTDH) method: a highly efficient algorithm for propagating wavepackets. Phys. Rep. 324, 1–105. doi:10.1016/S0370-1573(99)00047-2
Ben-Nun, M., and Martínez, T. J. (2002). Ab initio quantum molecular dynamics. Adv. Chem. Phys. 121, 439–512. doi:10.1002/0471264318.ch7
Ben-Nun, M., Quenneville, J., and Martínez, T. J. (2000). Ab initio multiple spawning: photochemistry from first principles quantum molecular dynamics. J. Phys. Chem. A 104, 5161–5175. doi:10.1021/jp994174i
Bernstein, M. P., Allamandola, L. J., and Sandford, S. A. (1997). Complex organics in laboratory simulations of interstellar/cometary ices. Adv. Space Res. 19, 991–998. doi:10.1016/S0273-1177(97)00340-2
Bockelée-Morvan, D., Lis, D. C., Wink, J. E., Despois, D., Crovisier, J., Bachiller, R., et al. (2000). New molecules found in comet C/1995 O1 (Hale-Bopp). Investigating the link between cometary and interstellar material. Astron Astrophys. 353, 1101–1114.
Borduas, N., Abbatt, J. P. D., and Murphy, J. G. (2013). Gas phase oxidation of monoethanolamine (MEA) with OH radical and ozone: kinetics, products, and particles. Environ. Sci. Technol. 47, 6377–6383. doi:10.1021/es401282j
Bossa, J. B., Theule, P., Duvernay, F., and Chiavassa, T. (2009). NH 2 CH2 OH thermal formation in interstellar ices contribution to the 5-8 μm region toward embedded protostars. Astrophys. J. 707, 1524–1532. doi:10.1088/0004-637X/707/2/1524
Botta, L., Saladino, R., Bizzarri, B. M., Cobucci-Ponzano, B., Iacono, R., Avino, R., et al. (2018). Formamide-based prebiotic chemistry in the phlegrean fields. Adv. Space Res. 62, 2372–2379. doi:10.1016/j.asr.2017.07.017
Boys, S. F., and Bernardi, F. (1970). The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 19, 553–566. doi:10.1080/00268977000101561
Bunkan, A. J. C., Mikoviny, T., Nielsen, C. J., Wisthaler, A., and Zhu, L. (2016). Experimental and theoretical study of the OH-initiated photo-oxidation of formamide. J. Phys. Chem. A 120, 1222–1230. doi:10.1021/acs.jpca.6b00032
Dai, N., Shah, A. D., Hu, L., Plewa, M. J., McKague, B., and Mitch, W. A. (2012). Measurement of nitrosamine and nitramine formation from NO x reactions with amines during amine-based carbon dioxide capture for postcombustion carbon sequestration. Environ. Sci. Technol. 46, 9793–9801. doi:10.1021/es301867b
Dash, M. R., and Ali, M. A. (2022). Effect of a single water molecule on ˙CH 2 OH + 3 O 2 reaction under atmospheric and combustion conditions. Phys. Chem. Chem. Phys. 24, 1510–1519. doi:10.1039/D1CP03911C
Dash, M. R., and Ali, M. A. (2023). Can a single ammonia and water molecule enhance the formation of methanimine under tropospheric conditions? kinetics of •CH2NH2 + O2 (+NH3/H2O). Front. Chem. 11, 1243235. doi:10.3389/fchem.2023.1243235
Deschamps, G. (1931). Preparation of formamide and its conversion into hydrocyanic acid. Chimie Industrie, 589–597.
Evans, M. G., and Polanyi, M. (1938). Inertia and driving force of chemical reactions. Trans. Faraday Soc. 34, 11. doi:10.1039/tf9383400011
Fabiano, E., Keal, T. W., and Thiel, W. (2008). Implementation of surface hopping molecular dynamics using semiempirical methods. Chem. Phys. 349, 334–347. doi:10.1016/j.chemphys.2008.01.044
Feldmann, M. T., Widicus, S. L., Blake, G. A., Kent, D. R., and Goddard, W. A. (2005). Aminomethanol water elimination: theoretical examination. J. Chem. Phys. 123, 034304. doi:10.1063/1.1935510
Ferus, M., Knížek, A., Cassone, G., Rimmer, P. B., Changela, H., Chatzitheodoridis, E., et al. (2023). Simulating asteroid impacts and meteor events by high-power lasers: from the laboratory to spaceborne missions. Front. Astron Space Sci. 10. doi:10.3389/fspas.2023.1186172
Ferus, M., Nesvorný, D., Šponer, J., Kubelík, P., Michalčíková, R., Shestivská, V., et al. (2015). High-energy chemistry of formamide: a unified mechanism of nucleobase formation. Proc. Natl. Acad. Sci. 112, 657–662. doi:10.1073/pnas.1412072111
Firaha, D. S., Döntgen, M., Berkels, B., and Leonhard, K. (2018). Pressure-dependent rate constant predictions utilizing the inverse Laplace transform: a victim of deficient input data. ACS Omega 3, 8212–8219. doi:10.1021/acsomega.8b00311
Franco, B., Blumenstock, T., Cho, C., Clarisse, L., Clerbaux, C., Coheur, P.-F., et al. (2021). Ubiquitous atmospheric production of organic acids mediated by cloud droplets. Nature 593, 233–237. doi:10.1038/s41586-021-03462-x
Frisch, M. J., Pople, J. A., and Binkley, J. S. (1984). Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys. 80, 3265–3269. doi:10.1063/1.447079
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2019) Gaussian 16, revision C.01. Wallingford CT: Gaussian, Inc.
Ge, X., Wexler, A. S., and Clegg, S. L. (2011a). Atmospheric amines – Part I. A review. Atmos. Environ. 45, 524–546. doi:10.1016/j.atmosenv.2010.10.012
Ge, X., Wexler, A. S., and Clegg, S. L. (2011b). Atmospheric amines – Part II. Thermodynamic properties and gas/particle partitioning. Atmos. Environ. 45, 561–577. doi:10.1016/j.atmosenv.2010.10.013
Gerakines, P. A., Moore, M. H., and Hudson, R. L. (2004). Ultraviolet photolysis and proton irradiation of astrophysical ice analogs containing hydrogen cyanide. Icarus 170, 202–213. doi:10.1016/j.icarus.2004.02.005
Goldsmith, C. F., Green, W. H., and Klippenstein, S. J. (2012). Role of O 2 + QOOH in low-temperature ignition of propane. 1. Temperature and pressure dependent rate coefficients. J. Phys. Chem. A 116, 3325–3346. doi:10.1021/jp210722w
González, D., Lema-Saavedra, A., Espinosa, S., Martínez-Núñez, E., Fernández-Ramos, A., Canosa, A., et al. (2022). Reaction of OH radicals with CH 3 NH 2 in the gas phase: experimental (11.7–177.5 K) and computed rate coefficients (10–1000 K). Phys. Chem. Chem. Phys. 24, 23593–23601. doi:10.1039/D2CP03414J
Grimme, S., Antony, J., Ehrlich, S., and Krieg, H. (2010). A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104. doi:10.1063/1.3382344
Gröger, H. (2003). Catalytic enantioselective strecker reactions and analogous syntheses. Chem. Rev. 103, 2795–2828. doi:10.1021/cr020038p
Hammes-Schiffer, S., and Tully, J. C. (1994). Proton transfer in solution: molecular dynamics with quantum transitions. J. Chem. Phys. 101, 4657–4667. doi:10.1063/1.467455
Hays, B. M., Weaver, W., and Susanna, L. (2013). Theoretical examination of O(1 D) insertion reactions to form methanediol, methoxymethanol, and aminomethanol. J. Phys. Chem. A 117, 7142–7148. doi:10.1021/jp400753r
Kapteina, S., Slowik, K., Verevkin, S. P., and Heintz, A. (2005). Vapor pressures and vaporization enthalpies of a series of ethanolamines. J. Chem. Eng. Data 50, 398–402. doi:10.1021/je049761y
Karl, M., Dye, C., Schmidbauer, N., Wisthaler, A., Mikoviny, T., D’Anna, B., et al. (2012). Study of OH-initiated degradation of 2-aminoethanol. Atmos. Chem. Phys. 12, 1881–1901. doi:10.5194/acp-12-1881-2012
Koike, T., Kaneko, T., Kobayashi, K., Miyakawa, S., and Takano, Y. (2003). Formation of organic compounds from simulated Titan atmosphere: perspectives of the Cassini mission. Biol. Sci. Space 17, 188–189.
Lee, T. J., and Taylor, P. R. (1989). A diagnostic for determining the quality of single-reference electron correlation methods. Int. J. Quantum Chem. 36, 199–207. doi:10.1002/qua.560360824
Lis, D. C., Mehringer, D. M., Benford, D., Gardner, M., Phillips, T. G., Bockelée-Morvan, D., et al. (1997). New molecular species in comet C/1995 O1 (Hale-Bopp) observed with the caltech ssubmillimeter observatory. Earth Moon Planets 78, 13–20. doi:10.1023/A:1006281802554
Manthe, U., Meyer, H.-D., and Cederbaum, L. S. (1992). Wave-packet dynamics within the multiconfiguration Hartree framework: general aspects and application to NOCl. J. Chem. Phys. 97, 3199–3213. doi:10.1063/1.463007
Mazarei, E., and Barker, J. R. (2022). CH2 + O2: reaction mechanism, biradical and zwitterionic character, and formation of CH2OO, the simplest Criegee intermediate. Phys. Chem. Chem. Phys. 24, 914–927. doi:10.1039/D1CP04372B
Meyer, H. (2012). Studying molecular quantum dynamics with the multiconfiguration time-dependent Hartree method. WIREs Comp. Mol. Sci. 2, 351–374. doi:10.1002/wcms.87
Nájera, C., and Sansano, J. M. (2007). Catalytic asymmetric synthesis of α-amino acids. Chem. Rev. 107, 4584–4671. doi:10.1021/cr050580o
Navarro-González, R., and Raulin, F. (2004). Steps toward the origin(s) of life: endogenous sources and chemistry. Adv. Space Res. 33, 79–80. doi:10.1016/j.asr.2003.11.002
Nguyen, T. L., Ruscic, B., and Stanton, J. F. (2019). A master equation simulation for the •OH + CH3OH reaction. J. Chem. Phys. 150, 084105. doi:10.1063/1.5081827
Nielsen, A. T., Moore, D. W., Ogan, M. D., and Atkins, R. L. (1979). Structure and chemistry of the aldehyde ammonias. 3. Formaldehyde-ammonia reaction. 1,3,5-Hexahydrotriazine. J. Org. Chem. 44, 1678–1684. doi:10.1021/jo01324a021
Onel, L., Blitz, M. A., and Seakins, P. W. (2012). Direct determination of the rate coefficient for the reaction of OH radicals with monoethanol amine (MEA) from 296 to 510 K. J. Phys. Chem. Lett. 3, 853–856. doi:10.1021/jz300200c
Onel, L., Thonger, L., Blitz, M. A., Seakins, P. W., Bunkan, A. J. C., Solimannejad, M., et al. (2013). Gas-phase reactions of OH with methyl amines in the presence or absence of molecular oxygen. An experimental and theoretical study. J. Phys. Chem. A 117, 10736–10745. doi:10.1021/jp406522z
Pastorek, A., Hrnčířová, J., Jankovič, L., Nejdl, L., Civiš, S., Ivanek, O., et al. (2019). Prebiotic synthesis at impact craters: the role of Fe-clays and iron meteorites. Chem. Comm. 55, 10563–10566. doi:10.1039/C9CC04627E
Priya, A. M., and Lakshmipathi, S. (2017). DFT study on abstraction reaction mechanism of oh radical with 2-methoxyphenol. J. Phys. Org. Chem. 30, e3713. doi:10.1002/poc.3713
Raghavachari, K., Trucks, G. W., Pople, J. A., and Head-Gordon, M. (1989). A fifth-order perturbation comparison of electron correlation theories. Chem. Phys. Lett. 157, 479–483. doi:10.1016/S0009-2614(89)87395-6
Rahbar, A., Zahedi, E., Aghaie, H., Giahi, M., and Zare, K. (2021). DFT insight into the kinetics and mechanism of the OH . -initiated atmospheric oxidation of catechol: OH . Addition and hydrogen abstraction pathways. Chem. Sel. 6, 3875–3883. doi:10.1002/slct.202100524
Rissanen, M. P., Eskola, A. J., Nguyen, T. L., Barker, J. R., Liu, J., Liu, J., et al. (2014). CH 2 NH 2 + O 2 and CH 3 CHNH 2 + O 2 reaction kinetics: photoionization mass spectrometry experiments and master equation calculations. J. Phys. Chem. A 118, 2176–2186. doi:10.1021/jp411238e
Robertson, S. H., Pilling, M. J., Baulch, D. L., and Green, N. J. B. (1995). Fitting of pressure-dependent kinetic rate data by master equation/inverse Laplace transform analysis. J. Phys. Chem. 99, 13452–13460. doi:10.1021/j100036a020
Rubin, R. H., Swenson Jr, G. W., Benson, R. C., Tigelaar, H. L., and Flygare, W. H. (1971). Microwave detection of interstellar formamide. Astrophysical J. 169, L39. doi:10.1086/180810
Saitta, A. M., and Saija, F. (2014). Miller experiments in atomistic computer simulations. Proc. Natl. Acad. Sci. 111, 13768–13773. doi:10.1073/pnas.1402894111
Saladino, R., Botta, G., Pino, S., Costanzo, G., and Di Mauro, E. (2012a). Genetics first or metabolism first? The formamide clue. Chem. Soc. Rev. 41, 5526. doi:10.1039/c2cs35066a
Saladino, R., Crestini, C., Ciciriello, F., Costanzo, G., and Di Mauro, E. (2007). Formamide chemistry and the origin of informational polymers. Chem. Biodivers. 4, 694–720. doi:10.1002/cbdv.200790059
Saladino, R., Crestini, C., Costanzo, G., and DiMauro, E. (2005). “On the prebiotic synthesis of nucleobases, nucleotides, oligonucleotides, pre-RNA and pre-DNA molecules,” in Prebiotic chemistry. Editor P. Walde (Berlin/Heidelberg: Springer-Verlag), 29–68. doi:10.1007/b136152
Saladino, R., Crestini, C., Pino, S., Costanzo, G., and Di Mauro, E. (2012b). Formamide and the origin of life. Phys. Life Rev. 9, 84–104. doi:10.1016/j.plrev.2011.12.002
Schade, G. W., and Crutzen, P. J. (1995). Emission of aliphatic amines from animal husbandry and their reactions: potential source of N2O and HCN. J. Atmos. Chem. 22, 319–346. doi:10.1007/BF00696641
Schutte, W. A., Allamandola, L. J., and Sandford, S. A. (1993). An experimental study of the organic molecules produced in cometary and interstellar ice analogs by thermal formaldehyde reactions. Icarus 104, 118–137. doi:10.1006/icar.1993.1087
Schutte, W. A., Boogert, A. C. A., Tielens, A., Whittet, D. C. B., Gerakines, P. A., Chiar, J. E., et al. (1999). Weak ice absorption features at 7.24 and 7.41 MU M in the spectrum of the obscured young stellar object W 33A. Astron Astrophys. 343, 966–976.
Silva, G. D. (2012). Atmospheric chemistry of 2-aminoethanol (MEA): reaction of the NH2 •CHCH2 OH radical with O2. J. Phys. Chem. A 116, 10980–10986. doi:10.1021/jp307726w
Silva, G. da, Bozzelli, J. W., Liang, L., and Farrell, J. T. (2009). Ethanol oxidation: kinetics of the α-hydroxyethyl radical + O2 reaction. J. Phys. Chem. A 113, 8923–8933. doi:10.1021/jp903210a
Simon, S., Duran, M., and Dannenberg, J. J. (1996). How does basis set superposition error change the potential surfaces for hydrogen-bonded dimers? J. Chem. Phys. 105, 11024–11031. doi:10.1063/1.472902
Singh, S. K., Zhu, C., La Jeunesse, J., Fortenberry, R. C., and Kaiser, R. I. (2022). Experimental identification of aminomethanol (NH2CH2OH)—the key intermediate in the Strecker Synthesis. Nat. Commun. 13, 375. doi:10.1038/s41467-022-27963-z
Strecker, A. (1850). Ueber die künstliche Bildung der Milchsäure und einen neuen, dem Glycocoll homologen Körper. Justus Liebigs Ann. Chem. 75, 27–45. doi:10.1002/jlac.18500750103
Takano, Y., Tsuboi, T., Kaneko, T., Kobayashi, K., and Marumo, K. (2004). Pyrolysis of high-molecular-weight complex organics synthesized from a simulated interstellar gas mixture irradiated with 3 MeV proton beam. Bull. Chem. Soc. Jpn. 77, 779–783. doi:10.1246/bcsj.77.779
Tian, W., Wang, W., Zhang, Y., and Wang, W. (2009). Direct dynamics study on the mechanism and the kinetics of the reaction of CH3NH2 with OH. Int. J. Quantum Chem. 109, 1566–1575. doi:10.1002/qua.22000
Tully, J. C., and Preston, R. K. (1971). Trajectory surface hopping approach to nonadiabatic molecular collisions: the reaction of H+ with D2. J. Chem. Phys. 55, 562–572. doi:10.1063/1.1675788
VandeVondele, J., Krack, M., Mohamed, F., Parrinello, M., Chassaing, T., and Hutter, J. (2005). Quickstep: fast and accurate density functional calculations using a mixed Gaussian and plane waves approach. Comput. Phys. Commun. 167, 103–128. doi:10.1016/j.cpc.2004.12.014
Wang, J., Liu, X., and Feng, X. (2011). Asymmetric strecker reactions. Chem. Rev. 111, 6947–6983. doi:10.1021/cr200057t
Woon, D. E. (2002). Ab initio quantum chemical studies of reactions in astrophysical ices. 4. Reactions in ices involving HCOOH, CH2NH, HCN, HNC, NH3, and H2O. Int. J. Quantum Chem. 88, 226–235. doi:10.1002/qua.10082
Xie, H.-B., Li, C., He, N., Wang, C., Zhang, S., and Chen, J. (2014). Atmospheric chemical reactions of monoethanolamine initiated by OH radical: mechanistic and kinetic study. Environ. Sci. Technol. 48, 1700–1706. doi:10.1021/es405110t
Xu, L., Tsona, N. T., Tang, S., Li, J., and Du, L. (2019). Role of (H 2 O) n (n = 1–2) in the gas-phase reaction of ethanol with hydroxyl radical: mechanism, kinetics, and products. ACS Omega 4, 5805–5817. doi:10.1021/acsomega.9b00145
Yamada, H. (2021). Amine-based capture of CO2 for utilization and storage. Polym. J. 53, 93–102. doi:10.1038/s41428-020-00400-y
Yang, S., and Martínez, T. J. (2011). “Ab initio multiple spawning: first principles dynamics around conical intersections,” in Conical intersections (Singapore: World Scientific), 347–374. doi:10.1142/9789814313452_0009
Zhao, Y., and Truhlar, D. G. (2008). The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 120, 215–241. doi:10.1007/s00214-007-0310-x
Keywords: aminomethanol, photooxidalion, DFT, transition state theory, formamide, rate constant
Citation: Nulakani NVR and Ali MA (2024) Unveiling the chemical kinetics of aminomethanol (NH2CH2OH): insights into
Received: 26 March 2024; Accepted: 08 May 2024;
Published: 30 May 2024.
Edited by:
Steve Suib, University of Connecticut, United StatesReviewed by:
Federico Palazzetti, University of Perugia, ItalyRyan C. Fortenberry, University of Mississippi, United States
Giuseppe Cassone, National Research Council (CNR), Italy
Copyright © 2024 Nulakani and Ali. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mohamad Akbar Ali, YWtiYXIubW9oYW1hZEBrdS5hYy5hZQ==