 Tristan Bepler1
Tristan Bepler1 Michael D. Barrera2
Michael D. Barrera2 Mary T. Rooney3,4Yawei Xiong3Huihui Kuang1Evan Goodell3Matthew J. Goodwin5Elizabeth Harbron5Riqiang Fu6Mihaela Mihailescu7
Mary T. Rooney3,4Yawei Xiong3Huihui Kuang1Evan Goodell3Matthew J. Goodwin5Elizabeth Harbron5Riqiang Fu6Mihaela Mihailescu7 Aarthi Narayanan8*
Aarthi Narayanan8* Myriam L. Cotten3,9*
Myriam L. Cotten3,9*- 1New York Structural Biology Center, New York, NY, United States
- 2School of Systems Biology, George Mason University, Manassas, VA, United States
- 3Department of Applied Science, William & Mary, Williamsburg, VA, United States
- 4Department of Chemistry, Hofstra University, Hempstead, NY, United States
- 5Department of Chemistry, William & Mary, Williamsburg, VA, United States
- 6National High Magnetic Field Laboratory, Tallahassee, FL, United States
- 7Institute for Bioscience and Biotechnology Research, Rockville, MD, United States
- 8Department of Biology, George Mason University, Manassas, VA, United States
- 9Department of Biochemistry and Biophysics, Oregon State University, Corvallis, OR, United States
Outbreaks of viral diseases are on the rise, fueling the search for antiviral therapeutics that act on a broad range of viruses while remaining safe to human host cells. In this research, we leverage the finding that the plasma membranes of host cells and the lipid bilayers surrounding enveloped viruses differ in lipid composition. We feature Piscidin 1 (P1), a cationic host defense peptide (HDP) that has antimicrobial effects and membrane activity associated with its N-terminal region where a cluster of aromatic residues and copper-binding motif reside. While few HDPs have demonstrated antiviral activity, P1 acts in the micromolar range against several enveloped viruses that vary in envelope lipid composition. Notably, it inhibits HIV-1, a virus that has an envelope enriched in cholesterol, a lipid associated with higher membrane order and stability. Here, we first document through plaque assays that P1 boasts strong activity against SARS-CoV-2, which has an envelope low in cholesterol. Second, we extend previous studies done with homogeneous bilayers and devise cholesterol-containing zwitterionic membranes that contain the liquid disordered (Ld; low in cholesterol) and ordered (Lo, rich in cholesterol) phases. Using dye leakage assays and cryo-electron microscopy on vesicles, we show that P1 has dramatic permeabilizing capability on the Lo/Ld, an effect matched by a strong ability to aggregate, fuse, and thin the membranes. Differential scanning calorimetry and NMR experiments demonstrate that P1 mixes the lipid content of vesicles and alters the stability of the Lo. Structural studies by NMR indicate that P1 interacts with the Lo/Ld by folding into an α-helix that lies parallel to the membrane surface. Altogether, these results show that P1 is more disruptive to phase-separated than homogenous cholesterol-containing bilayers, suggesting an ability to target domain boundaries. Overall, this multi-faceted research highlights how a peptide that interacts strongly with membranes through an aromatic-rich N-terminal motif disrupt viral envelope mimics. This represents an important step towards the development of novel peptides with broad-spectrum antiviral activity.
Introduction
The Coronavirus disease 2019 (COVID-19) pandemic has had a devastating impact worldwide. According to the World Health Organization, the death toll exceeded 6.9 million at the end of 2023. While vaccination against the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) that causes COVID-19 has saved tens of millions of lives globally, concerns remain that even mild infections, including breakthrough infections in vaccinated subjects, may lead to lingering health issues (Amanatidou et al., 2022; Lipsitch et al., 2022; Rahman et al., 2022; Zheng et al., 2022; Davis et al., 2023; Perumal et al., 2023). It is also the case that a large portion of the world population is not vaccinated against SARS-CoV-2 and emerging viruses constitute a growing threat to human health (Sallam, 2021; Rahman et al., 2022; Lazarus et al., 2023). This context is fueling the search for broad-spectrum antiviral therapeutics that could complement existing vaccines in the fight against emerging and reemerging viruses (Harrison, 2008; Porotto et al., 2010; Wolf et al., 2010; Miller et al., 2011; Schoggins and Randall, 2013; Vigant et al., 2013; Hollmann et al., 2014; Vigant et al., 2015; Zhao et al., 2016; Park and Gallagher, 2017; Wang et al., 2018; Momattin et al., 2019; Wiehe et al., 2019; Baglivo et al., 2020; Chowdhury et al., 2020; Li and De Clercq, 2020; Schütz et al., 2020; Zeng et al., 2020; Saud et al., 2022). In this research, we focus on enveloped viruses and explore the ability of membrane-active agents, such as motif-containing host defense peptides (HDPs), to disrupt their envelopes.
Enveloped viruses dangerous to humans include SARS-CoV-2 as well as Human Immunodeficiency Virus Type 1 (HIV-1). These viruses infect cells by fusing their lipid envelope with the plasma or endosomal membrane of the host (Lorizate and Kräusslich, 2011; Plemper, 2011; Schoggins and Randall, 2013). While interactions between host and viral proteins play a major role in viral infection, other key players include the lipid envelope that surrounds virions (Pollock et al., 2010; Wolf et al., 2010; Martín-Acebes et al., 2013; Miguel et al., 2013; Schoggins and Randall, 2013; Vigant et al., 2015; Yang et al., 2016; Yan et al., 2019). Viruses acquire lipids for their envelopes in a way that depends on the assembly and egress pathways involving host components (Brügger et al., 2006; Jolly and Sattentau, 2007; Heaton and Randall, 2011; Lorizate and Kräusslich, 2011; Hogue et al., 2014; Alsaadi and Jones, 2019; Ghosh et al., 2020; Saud et al., 2022). With great strides made recently in lipidomics, a growing number of viral lipodomes have become available, shedding light on the complex chemical composition of viral envelopes (Chan et al., 2010; Heaton and Randall, 2011; Ivanova et al., 2015; Kyle, 2021). In particular, viral envelopes and plasma membranes differ in chemical content, and therefore physicochemical properties (e.g., curvature; fluidity). Notably, the envelope of HIV-1 is enriched in cholesterol (Chol), sphingomyelin (SM), and phosphatidylserine (PS) compared to the plasma membrane from which it buds (Brügger et al., 2006; Jolly and Sattentau, 2007). With a high Chol content, the HIV-1 envelope is expected to contain ordered microdomains, also called lipid rafts. In contrast to HIV-1, SARS-CoV-2 follows a lysosomal egress pathway after leaving the endoplasmic reticulum (ER)/Golgi intermediate complex (Ghosh et al., 2020). A recent study suggests that its envelope features lower Chol and higher phosphatidylinositol (PI) content than the ER and Golgi (Saud et al., 2022). For instance, SARS-CoV-2 derived from Vero and A549 cells contains 18% and 37% PI, respectively, while the plasma membrane content is typically below 10%. In these two respective cell lines, the Chol/phospholipid ratios of the virus are 0.0006 and 0.005 while this ratio is 0.76 in the plasma membrane. Another differentiating factor when considering host and viral membranes is that the biogenic reparative ability associated with metabolically active cells is lacking in viruses (Hanson et al., 2016; Zeng et al., 2020; Saud et al., 2022). Overall, the differentiating factors between the membranes of enveloped viruses and host cell organelles provide fertile ground to design agents that specifically target enveloped viruses over host cells. Recently, several studies have indicated that broad-spectrum antiviral action can be achieved through membrane-active compounds that disrupt viral envelopes (Wolf et al., 2010; Vigant et al., 2013; Saud et al., 2022).
In this study, we explore a peptide-based approach to disrupt enveloped viruses such as SARS-CoV-2 and HIV-1. For this purpose, the focus is on Piscidin 1 (P1, FFHHIFRGIVHVGKTIHRLVTG), an amphipathic HDP derived from fish (Silphaduang and Noga, 2001; Lauth et al., 2002). Most membrane-active HDPs, including P1, are cationic, enabling them to preferentially associate with the anionic membranes of bacteria and spare mammalian cells given that the outer leaflet of their plasma membrane is dominated by zwitterionic phospholipids (McCafferty et al., 1999; Anantharaman et al., 2010; Mansour et al., 2015; Bolosov et al., 2017; Li et al., 2017; Wu et al., 2017; Kampshoff et al., 2019; Ruden et al., 2019; Sheard et al., 2019; Li et al., 2020; Duong et al., 2021; Xia et al., 2021; Greve and Cowan, 2022; Howell et al., 2022; Zhu et al., 2022). The mechanisms of membrane disruption by HDPs are highly debated, but some consensus exists on a few points (Peschel and Sahl, 2006; Rea et al., 2010; Kong et al., 2012; Mansour et al., 2015; Pérez-Cobas et al., 2015; Kumar et al., 2018; Izoré et al., 2019; Kintses et al., 2019; Mookherjee et al., 2020; Silveira et al., 2021; Torres et al., 2021). First, binding by many membrane-active peptides induces positive membrane curvature strain and thinning by intercalating with the lipid headgroups and expanding the local surface area of the bilayer at constant hydrocarbon density. Second, once the peptides reach a critical threshold concentration, they reorient to insert more deeply and permeabilize the membranes, forcing the cellular/vesicular content to leak out. Structures that have been proposed to induce permeabilization include toroidal pores, carpets, and surface defects. In addition to direct membrane permeabilizing effects, HDPs can induce changes in the physicochemical properties of pathogenic cell membranes, which would indirectly affect the function of their membrane proteins (Wenzel et al., 2014; Omardien et al., 2018). In this regard, P1 was recently shown to lower the threshold at which mechanosensitive channels are activated in E. coli model membranes (Comert et al., 2019).
In the fight against enveloped viruses, HDPs present multiple advantages. Their membrane activity spans a broad range of membrane compositions and they are fit to work on protective barriers such as the respiratory tract and skin, as shown from studies with cathelicidins, defensins, histatins, neuropeptide Y, magainins, piscidins, aureins, pleurocidin, and pardaxin (El Karim et al., 2008; Hans and Madaan Hans, 2014; Conlon, 2015; Mahlapuu et al., 2020; Mookherjee et al., 2020). Furthermore, HDPs have dual antimicrobial and immunomodulatory effects, and thus have emerged as a possible answer to the unmet need of a dual-function therapeutic approach to address both infection and inflammation (Beisswenger and Bals, 2005; Gordon et al., 2005; Yount and Yeaman, 2012; Haney and Hancock, 2013; Hilchie et al., 2013; Mansour et al., 2014; Mansour et al., 2015; Boto et al., 2018). Several studies have reported on HDPs that have efficacy in vitro and in vivo in the context of multiple respiratory viral diseases (Zambrowicz et al., 2013; Uhlig et al., 2014; Mansour et al., 2015; Boto et al., 2018). However, only a fraction of HDPs (6%) have documented antiviral action (Wang et al., 2010; Wang, 2013). A possible reason for this low statistic includes that due to their origin, viral envelopes feature a much lower content of anionic lipids compared to bacterial cell membranes, and therefore favorable electrostatic interactions with cationic HDPs are reduced. It is also the case that a number of viral envelopes are enriched in Chol (Brügger et al., 2006; Chan et al., 2010; Heaton and Randall, 2011; Ivanova et al., 2015).
Chol plays a crucial role in determining the phase behavior of membranes, including the formation of lipid rafts in the plasma membranes of mammalian cells (Brown and London, 1998; Pike, 2003; Tulenko et al., 2007; Simons and Sampaio, 2011; Krause and Regen, 2014; Nicolson, 2014; Lorent et al., 2017). Due to its rigid ring structure, it preferentially interacts with the saturated acyl chains of lipids and prevents their trans-gauche isomerization (Krause and Regen, 2014; Yang et al., 2016; Hao et al., 2018). This reduces the dynamics and fluidity of the membrane and increases its order and stability (Mihailescu et al., 2011). In model membrane systems, the equivalent of the lipid rafts is believed to be the liquid ordered (Lo) phase that, in comparison to the liquid disordered (Ld) phase, is enriched in Chol and phospholipids with saturated fatty acid chains (Vereb et al., 2003; Engelman, 2005; van Meer et al., 2008; Krause and Regen, 2014; Nicolson, 2014). The higher order and stability that Chol imparts to the Lo signifies a protective role against membrane-active peptides, including HDPs (Katsu et al., 1990; Matsuzaki et al., 1995; Epand et al., 2006; Wessman et al., 2008; McHenry et al., 2012; Qian and Heller, 2015). However, some membrane-active peptides are more disruptive when phase-separated (“heterogenous”) Lo/Ld membranes versus single-phase (“homogenous”) bilayers are used. They appear to concentrate at domain boundaries in heterogenous membranes (Schön et al., 2008; Blicher et al., 2009; Apellániz et al., 2011; Hao et al., 2018; Nasr et al., 2020; Kumar et al., 2022; Utterström et al., 2022). This interface features curvature strain due to the hydrophobic mismatch between the thicker Lo and thinner Ld, and thus is conducive to defect formation (Brender et al., 2008; Sciacca et al., 2016; Hao et al., 2018). By associating preferentially with this more susceptible region of the membrane, the peptides can achieve enhanced membrane activity. This mechanism has been investigated in the context of several peptide families, including fusogenic peptides, amyloid peptides, and fusion inhibitors (Schön et al., 2008; Blicher et al., 2009; Apellániz et al., 2011; Hao et al., 2018; Nasr et al., 2020; Kumar et al., 2022; Utterström et al., 2022). Still, little is known about its applicability to membrane-active and antiviral HDPs. In-depth mechanistic studies must be performed on these peptides before they can be developed into more potent and selective antiviral agents.
Piscidins are unique templates to accelerate discovery of novel anti-infective therapeutics (Lauth et al., 2002; Noga and Silphaduang, 2003; Lauth et al., 2005; Dezfuli et al., 2012). They are active against many bacteria and viruses, including coronaviruses, methicillin-resistant Staphylococcus aureus (MRSA), and Streptococcus pneumoniae (S. pneumoniae) (Silphaduang and Noga, 2001; Lauth et al., 2002; Noga and Silphaduang, 2003; Menousek et al., 2012; Wang, 2013; Lee et al., 2014; Huang et al., 2015a; Huang et al., 2015b; Lei et al., 2018; Hu et al., 2019; Oludiran et al., 2019). P1, the most widely studied piscidin isoform, has activity against bacterial endotoxin and anti-inflammatory effects. It inactivates multiple classes of enveloped viruses, including the HIV-1 virus, pseudorabies virus, porcine CoV, and channel catfish virus (Chinchar et al., 2004; Wang et al., 2010; Wang, 2013; Lei et al., 2018; Hu et al., 2019). P1 is directly active against pseudorabies and porcine CoV and it is much more effective on the latter than lactoferricin, another important HDP (Lei et al., 2018; Hu et al., 2019). Another advantage of P1 is that it is biocompatible and has favorable pharmacokinetic/pharmacodynamics metrics in vivo, including in the context of CoV infections (Chen et al., 2015; Kumar et al., 2017; Lei et al., 2018; Hu et al., 2019). In particular, P1 was safely used in mice infected with the pseudorabies, leading to lower mortality (Hu et al., 2019).
Mechanistic studies focused on the direct action of P1 on pathogenic cells have revealed that it adopts an amphipathic α-helical structure in the membrane-bound state. Upon inserting into the membrane in an orientation parallel to the membrane surface, it disrupts the packing of the phospholipids. As part of this disordering effect, the lipid acyl chains form “baskets” to wrap around the peptide and fill the gap under it. An important consequence of this conformational rearrangement of the acyl chains is a pronounced thinning of the membrane (Perrin et al., 2014; Hayden et al., 2015; Perrin et al., 2015; Mihailescu et al., 2019; Comert et al., 2021; Liu et al., 2023). Prior work has also revealed that P1 induces and leverages membrane heterogeneity to disrupt homogeneous bilayers containing Chol (Comert et al., 2019). Its mechanism of action involves pushing away Chol, leading to lipid demixing (phase separation) and enhanced peptide accumulation in the more disordered region of the membrane.
Notably, the antimicrobial and membrane activities of P1 are associated with its N-terminal region that has a cluster of aromatic residues (e.g., histidines; phenylalanines) and an amino-terminal-copper-and-nickel (ATCUN) motif (Lee et al., 2007; Libardo et al., 2017; Kim et al., 2018; Paredes et al., 2020; Fu et al., 2021). Aromatic residues have been identified to be important for membrane interaction and membrane-remodeling, and thus could point at important design principles to develop new therapeutics that act at membranes (Epand et al., 2005; Apellániz et al., 2011; Zhou and Xu, 2012; Fantini and Barrantes, 2013; Paterson et al., 2017). For instance, Chol can establish favorable π-stacking interactions with aromatic sidechains. Once bound to Chol-containing membranes, P1 can induce positive curvature strain (Perrin et al., 2015). In the context of viral infections, this would counteract the negative spontaneous curvature that characterizes viral fusion intermediates (Risselada, 2017; Shekunov et al., 2023). The capability of P1 to interact and disrupt Chol-containing membranes sets an interesting stage when it comes to investigating how it directly disrupts enveloped viruses.
Here, we demonstrate that the antiviral activity of P1 against SARS-CoV-2 includes direct action on the virus. To better understand the ability of P1 to act on viruses that differ significantly in membrane compositions, we extend the studies previously done with homogeneous Ld bilayers. Zwitterionic Chol-containing membrane mimics featuring the Lo and Lo/Ld phases are designed. Using complementary biophysical methods, including circular dichroism (CD), solid-state nuclear magnetic resonance (SS-NMR), cryogenic electron microscopy (Cryo-EM), differential scanning calorimetry (DSC), dynamic light scattering (DLS), and machine learning (ML) tools, we investigate how the peptide interacts with and disrupts viral envelope mimics that feature a high Chol content. Through these studies, we reveal novel insights into the mechanism that enables P1, an HDP with antiviral action, to disrupt membranes featuring a broad range of lipid compositions relevant to viral envelopes. This is an important step towards the rational design of broad-spectrum antiviral therapeutics.
Methods
Materials
Carboxyamidated P1 (FFHHIFRGIVHVGKTIHRLVTG, MW 2,571) was obtained from the University of Texas Southwestern Medical Center. Fmoc solid-phase peptide synthesis was performed to produce the peptide in the unlabeled and 15N-labeled states, as described previously (Chekmenev et al., 2006a; Chekmenev et al., 2006b). The labeled amino acids (15N, 98%) were purchased from Cambridge Isotope Laboratories (Tewksbury, MA, United States). Purification of the crude peptide was performed by HPLC as previously reported to yield 98% pure peptide, as characterized by HPLC and mass spectrometry (Chekmenev et al., 2006b; Oludiran et al., 2019). Next, P1 was dissolved in dilute HCl to substitute chloride ions for trifluoroacetate ions. Excess chloride ions were removed by dialysis. The lyophilized peptide was dissolved in nanopure water prior to using it in the studies presented here. The concentrations of the stocks were determined by amino acid analysis performed at the Protein Chemistry Center at Texas A&M (College Station, TX). Metallation of P1 in a stoichiometric ratio with Cu2+ was achieved using copper chloride, as described previously (Libardo et al., 2017; Rai et al., 2019; Paredes et al., 2020; Comert et al., 2021; Fu et al., 2021). Reagents such as sodium hydroxide, buffers, salts, EDTA, chloroform, and copper chloride were purchased from Fisher Scientific (Hampton, NH). Calcein dye was obtained from Sigma-Aldrich (Saint Louis, MO). The Millipore MilliQ system (Sigma Aldrich, Saint Louis, MO) was used to obtain Nanopure water.
Lipids were acquired from Avanti Polar Lipids (Alabaster, AL). Non-deuterated lipids were dissolved in deuterated chloroform and concentrations quantified by solution NMR.
Cell culture
Vero E6 cells (ATCC, CRL-1586, Manassas, VA, United States) were cultured with Eagle’s minimum essential medium (EMEM, Quality Biological, 112–016,101CS, Gaithersburg, MD, United States) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Gibco, Thermo Fisher Scientific, Waltham, MA), 10 U/mL penicillin and 10 μg/mL streptomycin antibiotics (Corning 30–002-CI, Corning, NY, United States), and 2 mM L-glutamine (Corning, 25–005-CI, Corning NY, United States). Vero cells (ATCC, CCL-81, Manassas, VA, United States) were cultured with Dulbecco’s Modified Eagle’s Medium (DMEM, Quality Biological, 112–013,101CS, Gaithersburg, MD, United States) supplemented with 5% heat-inactivated FBS, 10 U/mL penicillin and 10 μg/mL streptomycin antibiotics (Corning 30–002-CI, Corning, NY, United States), and 2 mM L-glutamine (Corning, 25–005-CI, Corning NY, United States). All cell lines were cultivated at 37°C and 5% CO2.
Cell viability assay
Vero E6 cells were seeded at 1.5 × 104 cells per well in 96-well plates and incubated at 37°C and 5% CO2 for 24 h. Cells were then treated with 20 μg/mL or 10 μg/mL of P1 for 24 h and cell viability was determined using CellTiter-glo Luminescent Cell Viability Assay (Promega, G7570, Madison, WI, United States) following the manufacturer’s instructions and compared to vehicle control.
SARS-CoV-2 infections
SARS-CoV-2 (Washington strain 2019-nCoV/USA-WA1/2020) was obtained from BEI Resources (NR-52281) and was used for all infections. Vero E6 cells were seeded at 1.5 × 104 cells per well in 96-well plates 24 h prior to infection. P1 was dissolved in water and diluted in culture media to the indicated concentrations. For direct viral treatment, the virus was diluted to a multiplicity of infection (MOI) of 0.1 in media containing P1 at 10 μg/mL or vehicle control. For pre-treatment and post-treatment infections, cells were pretreated with P1 at 10 μg/mL or H2O vehicle control for 1 h at 37°C and 5% CO2. Pre-treatment was removed and cells were infected at an MOI of 0.1 in culture media for 1 h at 37°C and 5% CO2. After infection, inoculum was removed and post-treatment, P1 at 10 μg/mL or H2O vehicle, was applied. Treated inoculum was incubated at 37°C and 5% CO2 for 1 h and then applied to cells for 1 h. After infection, inoculum was removed, and culture media was applied to the cells. For infections combing pre-treatment, direct viral treatment, and post-treatment with P1, the above steps with P1 were combined. For post-treatment only infections, cells were infected at an MOI of 0.1 for 1 h. After infection the inoculum was removed and media containing P1 at 10 μg/mL or H2O vehicle control was added to the cells and incubated at 37°C and 5% CO2. After 24 h post infection (hpi), viral supernatants were collected and used immediately for assays or stored at −80°C.
Plaque assay
Vero cells were plated in 12-well plates at a density of 2 × 105 cells per well and incubated for 24 h. Infection supernatants were serially diluted to 10−6 in culture media and overlaid on cells for 1 h. Cells were covered with Eagle’s Minimum Essential Medium (without phenol red (Quality Biological 115–073-101, Gaithersburg, MD, United States), supplemented with 5% FBS, 1% nonessential amino acids, 1 mM sodium pyruvate (VWR, 45,000–710, Dixon, CA, United States), 2 mM L-glutamine, 20 U/mL penicillin, and 20 μg/mL streptomycin) with 0.6% agarose (ThermoFisher, 16,500,100, Waltham, MA, United States). At 48 hpi, cells were fixed with 10% formaldehyde (ThermoFisher, F79P-4, Waltham, MA, United States) for 1 h. Medium was removed, wells were washed with diH2O and stained with a 1% crystal violet (ThermoFisher, C581-25, Waltham, MA, United States) and 20% ethanol solution (ThermoFisher, BP2818-4, Waltham, MA, United States).
Circular dichroism
The secondary structure of P1 interacting with the Lo and Lo/Ld viral envelope mimics was accomplished by CD in potassium phosphate buffer (5 mM, pH 7.4). LUVs were made from lipid films, as described previously (Chekmenev et al., 2006b; Paredes et al., 2020). Briefly, the films were made using DPPC, POPC, DPPE, POPE, and Chol (Avanti Polar Lipids, Alabaster, AL) mixed in chloroform in the amounts needed to form the Lo or Lo/Ld. The solvent was evaporated under N2 gas and the films dried under vacuum overnight. The films were then hydrated with phosphate buffer (5 mM, pH 7.4), subjected to freeze-thaw cycles, and extruded using an extruder (Avanti Polar Lipids, Alabaster, AL) and 0.1 µm size membranes (Whatman, Florham Park, NJ). The vesicles were then diluted to 3 mM. Each spectrum was performed at fixed peptide concentration (20 µM). P1 was added to reach various P/L ratios, ranging from 1:20 to 1:200. CD spectra were acquired at 298 K on a Jasco J-1500 spectrometer (Jasco Analytical Instruments, Easton, MD). Experimental parameters included a wavelength range of 190–260 nm, scan speed of 100 nm/min, 1 nm bandwidth, and number of scans equal to four. The samples were run in duplicates. A blank containing buffer and lipids but no peptide was recorded and subtracted from the signal obtained for the peptide-containing sample, as needed to account for the background signal. Peptide helicity was obtained using the molar ellipticity measured at 222 nm and assuming an ellipticity of −32,000 deg·cm2/dmol for an ideal α-helix (Lee et al., 2007).
Permeabilization via dye leakage release from large unilamellar vesicles
The permeabilization assays were performed as previously described (Mihailescu et al., 2019; Liu et al., 2023). Briefly, for each lipid composition, either Lo or Lo/Ld, a film was made in round bottom flask using DPPC, POPC, DPPE, POPE, and Chol (4 µmol total of lipids) mixed at a desired molar ratio in chloroform. After evaporating the organic phase under N2 gas, the film was lyophilized overnight. Next, the film was hydrated with 300 µL of 80 mM calcein dye (pH 7.4) before undergoing three freeze-thaw cycles and extrusion through a mini extruder (Avanti Polar Lipids) using a 0.1 µm polycarbonate membrane. Free dye was removed using a size-exclusion column with a Sephadex G-50 resin stationary phase run with HEPES buffer (50 mM, 100 mM NaCl, 0.3 mM EDTA, pH 7.4). The exact lipid concentration of the LUVs was obtained using a Total Phosphorus Assay (Chen et al., 1956). They were then diluted to a final concentration of ∼35 μM, and distributed in a 96-well plate using 180 µL per well. Next, 20 µL of P1 solution was added to each well in concentrations needed to result in specific peptide-to-lipid ratios (P/L) in the wells to bracket P/L = 1:2 and 1:2048 based on the serial dilution of the P1 stock. Each P/L value was run in triplicate and at least three independent assays were run. Positive and negative controls featured 20 µL of 1% Triton-X detergent or 20 µL nanopure water instead of the peptide solution. Each plate was shaken for 1 hour at 45 °C (or 30 °C) and allowed to cool for 20 min before recording the fluorescence intensity using a BioTek SynergyH1 plate reader (BioTek, Winooski, VT). An excitation wavelength of 490 nm and an emission wavelength of 520 nm was used for calcein. Fractional peptide-induced leakage was obtained using the equation:
where Ix is the fluorescence intensity in a well with a particular peptide concentration, I100% is the average intensity in the positive-control wells (100% leakage) and I0%, is the average intensity in cells with the negative control (0% leakage). The fractional leakage was then plotted against lipid-to-peptide ratio (L/P) and fitted in GraphPad Prism using the Hill equation to identify the EC50 or P/L at which half of the vesicles were lysed (Sani et al., 2014). The 95% confidence interval (CI) was obtained from the program.
Preparation of large unilamellar vesicles for Cryo-EM
Lipid films of samples featuring the Lo or Lo/Ld were made using the needed molar ratio of DPPC, POPC, DPPE, POPE, and Chol in chloroform. After evaporating the solvent and vacuuming overnight, the films were hydrated with potassium phosphate buffer (5 mM, pH 7.4), diluted, and extruded through a mini extruder (Avanti Polar Lipids) using 0.1 µm polycarbonate membrane (Whatman, Florhan Park, NJ). Piscidin was added in varying amounts to reach specific P/L ratios ranging from 1:20 to 1:200. To confirm the size of the LUVs made for Cryo-EM experiments, we used Dynamic Light Scattering (DLS). This involved transferring 540 µL of vesicles into 2 mL Eppendorf tubes. The particle size distribution of the samples was measured at room temperature using a Nicomp N3000 Submicron Particle Sizer (Particle Sizing Systems). Some peptide-containing samples were also tested. In this case, we used 60 µL of peptide solution to reach a specific P/L. For lipid-only samples, 60 µL of nanopure water were used instead of peptide solution.
Cryo-EM experiments
Cryo-EM experiments were performed at the New York Structural Biology Center (NYSBC). Before cryo grids preparation, the samples were warmed up to 50 °C for 5–10 min. Then 4 μL of the solution was immediately added to freshly plasma cleaned (Gatan Solarus plasma cleaner, 75% argon/25% oxygen atmosphere at 15 Watts for 7 s) Au R1.2/1.3 300-mesh (UltrAuFoil®) grids. Grids were blotted for 4 s or 5 s after a 3 s pre-blotting time, then plunge-frozen in liquid ethane using Vitrobot Mark IV with the chamber maintained at 20°C and 100% humidity. Cryopreserved grids were stored in liquid N2 until use. Image collection was accomplished at ∼2 μm under focus on a Thermo Scientific™ Glacios™ Cryo-Transmission Electron Microscope (Cryo-TEM) operated at 200 kV equipped with a Falcon3 operated in linear mode. Data collection was performed in the automatic data collection software Leginon (Suloway et al., 2005) at 2.5 Å or 1.2 Å per pixel. The dose was fractionated over 30 raw frames and collected with the total electron dose around 31.0 e–/Å2. Individual frames were aligned and dose-weighted with MotionCor2 (Zheng et al., 2017).
Cryo-EM data analysis
Membrane segmentation and contour extraction
Membranes were segmented in micrographs collected for each liposome preparation using an implementation of the MemNet algorithm available on GitHub. MemNet uses convolutional neural networks to annotate membranes in micrographs using a U-Net-like neural network architecture. The network was trained and evaluated on a subset of micrographs collected in this study in which membranes were manually annotated using the VGG Image Annotator (Dutta and Zisserman, 2019) and a second set of micrographs containing liposomes provide by Fred Heberle (University of Tennessee, Knoxville).
The neural network outputs pixel masks containing the probability, for each pixel, that it is within a lipid bilayer. Contours were extracted from these by identifying points tiling the positive segmented membrane regions with 40 Å spacing, using the “predict_contours.py” script. Membrane fragments containing fewer than 10 coordinates were removed. Contours were then split into linear fragments and smoothed using a spline fit, the “smooth_contours.py” script. Linear fragments were identified by finding contiguous chains of points. Because these micrographs often contain tightly packed, intersecting, or overlapping membranes for which we could not resolve the full contour of single liposomes, we broke liposomes boundaries into contiguous linear fragments by removing points which could have more than two neighbors (intersecting points) for downstream analysis.
Membrane curvature analysis
In order to estimate the curvature of the membranes at each point in the membrane contours, we first estimated the curve tangent and normal vectors using finite differences. Let the x and y coordinates of the curve be parameterized in terms of an auxiliary variable, t, which denotes the distance along the curve from an arbitrary starting point. The tangent vectors to the curve are
where
Membrane width analysis
Membrane thicknesses were estimated by first linearizing each membrane fragment along its contour to find a centered, linearized membrane profile, similar to Heberle et al. (Heberle et al., 2020) For each point along the membrane contour, these membrane profiles were estimated for 20 nm high and 5 nm wide slices through the membrane. These membrane profiles were then lightly smoothed using a Gaussian filter along the length dimension of the linear profile with a small standard deviation of 1.5 pixels.
To estimate the membrane widths, we first found the bilayer peak signals by fitting an 8th order polynomial to the signal intensities for each linearized membrane slice, using the polyfit algorithm in numpy (Harris et al., 2020). We then found the locations of the peaks by finding the smallest critical points in the fit polynomials. The width of the membrane at each location was then defined as the distance between these peaks in Å.
Statistical analysis of membrane biophysical properties
We compared the biophysical properties of the membranes across liposome preparations spanning a range of P/L ratios and Lo and Lo/Ld phases. For each condition, we extracted membrane contours and estimated membrane curvatures and widths as described above. In total, we analyzed 2,707,744 5 nm membrane fragments across 10 conditions. This added up to more than 1.3 cm of contiguous membrane analyzed.
We examined the differences in the mean curvatures and widths between conditions and assessed their statistical significance using a two-sample Student’s t-test. To understand the effects of ligand concentration and phase on membrane curvature, we also fit ordinary least squares models to predict the observed curvatures from the log of the P/L ratios, phases, and ratio-phase cross terms using the “statsmodels” python package (Seabold and Perktold, 2010).
Preparation of oriented samples for solid-state NMR
Oriented samples were made to characterize the structural and orientational features of P1 that had been exposed to the Lo/Ld viral envelope mimics, and to determine how P1 affects the phase behavior of these model membranes. Aligned phospholipid preparations were achieved using a protocol previously established (Chekmenev et al., 2006a; Perrin et al., 2014). Briefly, the samples were made using the Lo/Ld mixture as viral membrane mimics, i.e. 20:10:12:33:25 DPPC/DPPE/POPC/POPE/Chol. For 2H detection, 5 mg of DPPC-d62 was incorporated. For the sample containing P1, after the lipids were mixed in chloroform, P1 dissolved in trifluoroethanol was added at P/L = 1:100. About 2 mg of 15N-V10G13I16 P1 was used. After co-dissolving all of the components, the solvent was evaporated using nitrogen gas and lyophilized overnight. The next day, the lipids were hydrated with 1 mL of Bis-Tris buffer (3 mM, pH 7.4). The mixture was incubated overnight at 40 °C and then spread on thin glass slides (dimensions 5.7 × 12 × 0.03 mm3 from Matsunami Trading Co., Japan). Each sample was then allowed to equilibrate in humidity chamber (>90% using a saturated potassium sulfate solution). The slides were stacked after hydrating at 42% by weight using 2H-depleted water. The stacked samples were placed in a glass cell (Vitrocom Inc., NJ), sealed with beeswax, and incubated at 45 °C until homogeneously hydrated.
Preparation of unoriented samples for solid-state NMR
Unoriented samples were prepared using a procedure similar to that used for oriented samples. Deuterated DPPC-d62 was weighed out and mixed in a round-bottom flask with appropriate amounts of non-deuterated lipids to form the Lo phase, i.e. 40:18:3:6:33 DPPC/DPPE/POPC/POPE/Chol. The solvent was evaporated, and the film dried on a lyophilizer overnight. The film was then hydrated with Bis-Tris buffer and underwent freeze-thaw cycles to ensure distribution of buffer. The resulting suspension was incubated overnight at 40°C. The suspension was then transferred to a small glass vial and lyophilized. The dry lipid was transferred to a sample tube and hydrated to 42% (v/w) with deuterium-depleted water, so that the final BisTris buffer concentration matched that of the oriented sample. To ensure complete removal of HOD that would give rise to 2H NMR signal, the sample was lyophilized at least 4 h, rehydrated to 42% with the 2H depleted water, and sealed. The sample was then incubated for 24 h before NMR analysis.
2H solid-state NMR
2H spectra were collected at the National High Magnetic Field Laboratory (NHMFL) on a 600 MHz wide-bore magnet equipped with a Bruker Avance I console where the 2H Larmor frequency is 92.12 MHz. Parameters for the quadrupolar echo pulse sequence included a 3.0 µs 90° pulse on 2H, and an echo time of 20 µs. The FIDs were recorded with a dwell time of 2 µs for an acquisition time of 4.096 ms. Samples were first run at 50°C to allow for annealing, then run from 10°C up to 50°C in steps of 5°C. A spectrum was also obtained at 42°C between the 40°C and 45°C spectra. Each sample was allowed to equilibrate at each temperature before 4,096 and 1,536 scans for the oriented and unoriented samples, respectively, were averaged with a recycle delay of 1 s. The data were processed in Topspin (Bruker, Billerica, MA) with 100 Hz of exponential linebroadening and referenced to D2O at 0 kHz. The data from the unoriented Lo sample were de-Paked in NMRPipe (Delaglio et al., 1995) using the dePake macro detailed in Sani et al. (Sani et al., 2013). The FID was left-shifted to the echo maximum before applying a sinebell function, zero-filling, and dePakeing by weighted fast Fourier transform. Basic peak detection within NMRPipe was used to identify the position of resonances and yield the quadrupolar splittings.
1H solid-state NMR
1H spectra were acquired at the NHMFL on a 600 MHz wide-bore magnet equipped with a Bruker Avance I console where the 1H Larmor frequency is 600.13 MHz. A pulse sequence that suppresses the background signal from the probe was implemented (Wang et al., 2021). Samples packed in 3.2 mm rotors with caps containing O-rings (Revolution NMR, Fort Collings, CO) were spun at 10 kHz. Parameters included a 4.0 µs 90° pulse, and a recycle delay of 2 s. Samples were first run at 50°C to allow for annealing, then run from 10°C up to 55°C in steps of 5°C, with a spectrum at 42°C collected between the 40°C and 45°C spectra. The data, corresponding to 32 scans, were processed with no window function and referenced to the methylene signal at 1.30 ppm (Veatch et al., 2004; Polozov et al., 2008) TopSpin (Bruker, Billerica, MA) was used to process the data. Peak picking was used to determine the intensity of the methylene peak at 1.30 ppm. The peak width at half height was determined by measuring from the picked peak to the half-height on the left as the right side was convoluted in spectra.
15N solid-state NMR
2D SAMPI4 (Nevzorov and Opella, 2007) spectra were recorded on a 600 MHz wide bore Bruker instrument with Avance I console at the NHMFL where the 1H and 15N frequencies were 600.13 and 60.82 MHz, respectively. The temperature was set to the indicated temperature ±0.1°C. For each spectrum, a recycle delay of 4 s and 32 t1 increments with 2048 transients each were used. The pulse sequence used a contact time of 810 µs, cross-polarization field of 50.0 kHz, and 1H decoupling of 62.5 kHz (using PWTPPM) (Fu, 2009). Data were processed using Topspin with Gaussian window function (LB = - 50 Hz, GB = 0.1) in the 15N dimension and (LB = - 100 Hz, GB = 0.1) in the 1H-15N dimension. The 15N chemical shifts were referenced to a saturated solution of ammonium sulfate set at 26.8 ppm with respect to ammonia.
Differential scanning calorimetry
Sample preparation
Lipids in chloroform (Avanti Polar Lipids, Alabaster, Alabama) were used for the sample preparations. Individual POPE and DPPC vesicles in water were obtained by separating aliquots of lipid in chloroform in glass vials, removing the organic solvent under a stream of nitrogen gas and placing the samples under vacuum for 2 h. The dry films were reconstituted in water with repeated agitation, followed by 10 min bath sonication at 50°C. The vesicles in water were stored for several hours (less than 24 h) at 4°C before the DSC measurements. Homogenous mixtures of POPE/DPPC at (1:1 M ratio) or POPE/DPPC/Chol (1:1:0.5) were prepared as above by first co-dissolving the lipid and Chol in chloroform in the desired proportions. Sample with P1 peptide were obtained by adding the peptide to lipid vesicles in water just before the measurement.
DSC measurements
Differential scanning calorimetry (DSC) measurements were made on a VP-DSC microcalorimeter (MicroCal Inc., Northampton, MA). The samples were degassed and equilibrated at the starting scan temperature for 5 min, just before measurements. At least three cycles of heating-cooling curves were collected, at a scan rate of 30 °C/hour and in the range of 12°C to 95°C. There was a 5 min equilibrating period prior to starting the experiment and a delay of 5 min between sequential scans to allow for thermal equilibration. Samples with and without peptides in the same category were always prepared from the same stock of vesicle in water, and measured one after the other, following the same measurement routine. DSC curves were analyzed by Origin, version 7.0 (OriginLab Corporation). The scans were corrected by subtracting the background, normalizing to lipid concentration, and applying baseline correction.
Results
Piscidin 1 inhibits SARS-CoV-2 in cell cultures
The potential for piscidins to exert an antiviral activity against SARS-CoV-2 was assessed using Vero E6 cells as an infection model. The Cell Titer Glo assay was used to determine cell viability 24 h post infection (hpi). For the initial assessment, we aimed at determining if the peptide could directly inhibit the ability of the virus to establish a productive infection in cells. For this purpose, the virus (Washington strain 2019-nCoV/USA-WA1/2020) was directly exposed to piscidin or vehicle control before the cells were infected. After 24 hpi, plaque assays were performed to determine if the peptide inhibited viral infection. As shown in Figure 1, P1 at 20.0, 10.0, and 1.0 μg/mL inhibited viral titers by 56.6%, 35.8%, and 18.9% respectively. This indicates that the piscidin peptide exerts a direct inhibitory effect on the ability of the virus to establish a productive infection.
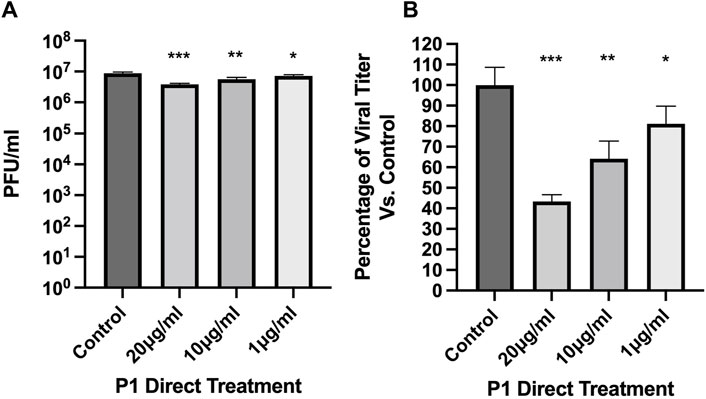
Figure 1. Direct antiviral effects of Piscidin 1 on SARS-CoV-2. The plaque assay was employed on cultured Vero E6 cells to quantify the amount of SARS-CoV-2 infectious titer after direct treatment with Piscidin 1 (P1). The virus was diluted to an MOI of 0.1 in media containing P1 at the indicated concentrations or vehicle control. Cells seeded at a density of 1.5 × 104 per well were then exposed to the virus. (A) Data are reported as the average plaque forming units (Avg PFU/mL) over triplicates; *p > 0.05; **p > 0.01; and ***p > 0.001. (B) The viral titer is plotted as a percentage of the H2O control. P1 demonstrated inhibitory effect on SARS-CoV-2. Under the conditions tested, P1 was more effective than Brilacidin used as a reference antiviral peptide (Supplementary Figure S1).
Next, pre-treatment and post-treatment were used to determine if post-entry mechanisms were also inhibited by P1. As shown in Figure 2, pre- and post-treatment with 10.0 μg/mL of P1 reduced SARS-CoV-2 viral titer by 41.5% compared to the vehicle control. Similarly, post-treatment alone reduced viral titers by 40.0%. Combining pre-treatment, direct viral treatment, and post-treatment showed the highest inhibition at 78.1% compared to the vehicle control. This suggests that continual treatment with P1 is characterized by a concentration to reach 50% viral inhibition (EC50) that is on the order of 5 μg/mL (2 µM). This is stronger than the direct viral treatment described above since the EC50 in that case appeared to be on the order of 20.0 μg/mL (8 µM). Overall, these assays indicate that P1 can inhibit the infection potential of the virus in two ways, by directly disrupting the viral particles and indirectly influencing the post-entry infection mechanisms of the virus in cells.
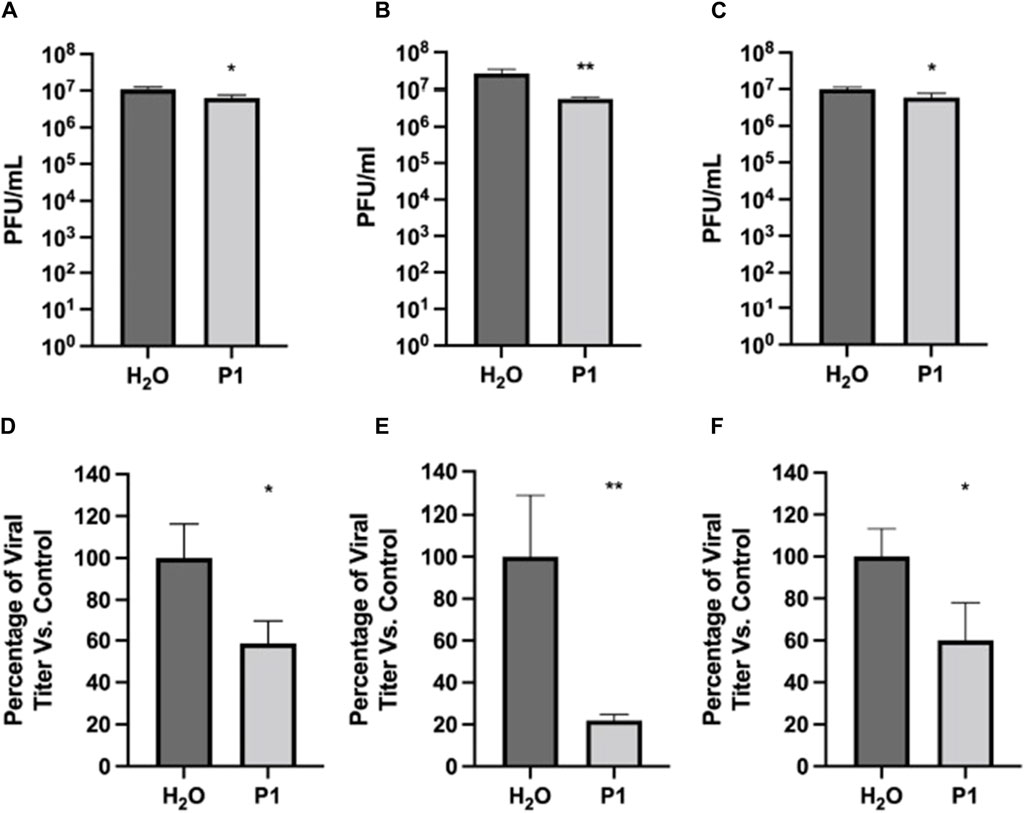
Figure 2. Antiviral effects of Piscidin 1 on SARS-CoV-2 as a function of treatment type. The plaque assay was used on Vero E6 cells to quantify the amount of SARS-CoV-2 infectious titer. Cells were seeded at a density of 1.5 × 104. Infection was done with SARS-CoV-2 at an MOI of 0.1. Three treatment types were tested to complement the direct treatment data in Figure 1. (A–C) Viral titer are reported as the average plaque forming units (Avg PFU/mL) over triplicates; *p > 0.05 and **p > 0.01. (A) Pre- and post-treatment assay: the cell were pretreated for one hour with peptide at 10 μg/mL; the peptide was removed during infection by the virus and added again after infection. (B) Pre-, direct-, and post-treatment assay: the peptide P1 at 10 μg/mL was present at all steps. (C) Post-treatment assay: P1 at 10 μg/mL was added only after infection with SARS-CoV-2 had been completed. (D–F): Same (A–C), respectively, but as a percentage of the H2O control.
The effectiveness of P1 was compared to that of other peptides, such as the copper-bound form of P1 (P1-Cu) and the piscidin isoforms P3 and TP4, to compare its effectiveness, as shown in Supplementary Figure S1. Brilacidin is a peptidomimetic which we have already published as exerting an antiviral activity against SARS-CoV-2 in this infection model and was used as a positive comparison control (Bakovic et al., 2021). In this case, the pre-treatment strategy was used on Vero cells and each peptide was added at a concentration of 10.0 μg/mL. No apparent cell toxicity was noted following peptide treatment as compared to the vehicle-alone control (data not shown). The data in Supplementary Figure S1 demonstrate that P1 and P3 peptide treatment resulted in a statistically significant decrease as compared to the vehicle-alone control, with P1 demonstrating greater inhibition (>90%) than P3. The extent of inhibition observed with P3, HR9 (histidine-rich nona-arginine) and TP4 was comparable. The conjugation of copper decreased the observed inhibitory potential of both P1 and P3 in a comparable manner. Cumulatively, the data support the antiviral potential of P1 and P3 piscidins against SARS-CoV-2 in cell culture. They also show that the peptides have a direct effect on the virus. Previously, P1 was demonstrated to be active in vivo on pseudorabies and porcine CoV (Lei et al., 2018; Hu et al., 2019). It is also inhibitory to HIV-1 in vitro (Wang, 2013). Since these viruses have a lipid envelope and piscidin is membrane active, we performed biophysical experiments on viral envelope mimics to gain further insight into the mechanism of direct inhibition.
Design of viral envelope mimics
As explained in the introduction, enveloped viruses acquire their lipids from host cells, but viral envelopes have lipid compositions that vary from virus to virus and differ from the host plasma membrane. P1 is active on viruses that feature a broad range of lipid compositions, especially with regard to Chol, a lipid that strengthens membranes. It was previously shown to be active on HIV-1 (Wang et al., 2010; Wang, 2013), which has a high Chol content in its envelope (Brügger et al., 2006). The reported EC50 was 2.1 µM (5.4 μg/mL), which is similar to the EC50 observed here against SARS-CoV-2, a virus that has a low Chol content (Saud et al., 2022). Based on their assembly and egress pathway, the low Chol level also applies to the porcine CoV and pseudorabies that P1 directly inhibits (Hogue et al., 2014; Schoeman and Fielding, 2019; Ghosh et al., 2020). Through our previous studies, we showed that P1 permeabilizes both anionic and zwitterionic Ld bilayers. Membranes tested contained lipids such as PC, PE, and Chol, which are relevant to the envelopes of the viruses that P1 inhibits. However, the effect of P1 has not been characterized on the Lo and Lo/Ld bilayers that are relevant to dangerous viruses such as HIV-1 and Influenza A (Brügger et al., 2006; Ivanova et al., 2015). To fill this gap and to extend previous studies beyond the Ld, we designed Chol containing membranes featuring the Lo and Ld.
In preparing bilayers containing the Lo and Ld, we opted for a membrane composition that best represented multiple viruses, including HIV-1 and Influenza A (Brügger et al., 2006; Ivanova et al., 2015). In addition to containing a large amount of Chol, their membranes have an outer leaflet that is enriched in zwitterionic lipids (Brügger et al., 2006). To represent the main lipid headgroups present in these viruses, we used phosphoethanolamine (PE) and phosphocholine (PC), which are also the dominant species in plasma membranes of host cells (Brügger et al., 2006; Ivanova et al., 2015; Lorent et al., 2020). In terms of acyl chains, we used saturated (palmitoyl) and unsaturated (oleoyl) fatty acids, as needed to be able to form both the Lo and Lo/Ld phases in the presence of Chol. Table 1 lists the specific lipids and the molar ratios used to generate Lo and Lo/Ld systems.
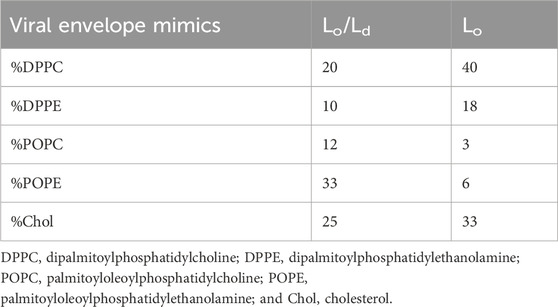
Table 1. Composition of model membranes (mol fraction).
Characterization of Lo/Ld and Lo viral envelope mimics exposed to P1 by solid-state NMR
As a first step with the viral envelope mimics, we leveraged 2H static SS-NMR to characterize their phase behavior and confirm the presence of the Lo. To achieve high resolution, oriented lipid bilayers were made and DPPC-d62 was used as the 2H reporter. Figure 3A and Supplementary Figure S2 show 2H SS-NMR spectra collected at different temperatures to monitor the phase behavior. At the high temperature of 55°C, the methyl region of the 2H spectrum displays a single quadrupolar splitting (distance between the two black arrows at the top of Figure 3). This indicates that DPPC-d62 exists in a bilayer that is well mixed, and thus above its miscibility point. When the temperature is decreased to 45°C, multiple splittings start appearing, indicating the onset of demixing and the co-existence of the Lo and Ld. Essentially, the rate at which the DPPC-d62 exchanges between the Lo and the Ld phases is slow enough on the time scale of the 2H NMR experiments that their splittings are becoming resolved from each other (Veatch et al., 2004). The outer splittings, which are well resolved at 30 °C (green arrows in Figure 3) are 17.2 and 14.1 kHz, clearly corresponding to the sn-1 and sn-2 methyl groups of DPPC-d62 in the Lo phase (Veatch et al., 2004). The same chains in the Ld phase give rise to the inner splitting (8.4 kHz, gray arrows in Figure 3). As previously explained by the Gawrisch lab, the difference between the inner and outer splittings indicate that large Lo domains (>> 160 nm) are present, and thus the samples are experiencing large-scale demixing (Veatch et al., 2004). The onset temperature for this demixing is referred to as Tlow. Using a similar approach, we also investigated the Lo system. As displayed in Supplementary Figure S3, two large quadrupolar splittings (e.g.,; 19.5 and 15.4 kHz at 30 °C) are detected, and therefore the Lo is present throughout the temperature range. Overall, the signals observed by solid-state NMR confirm that the chosen lipid compositions form the expected Lo/Ld or Lo phases.
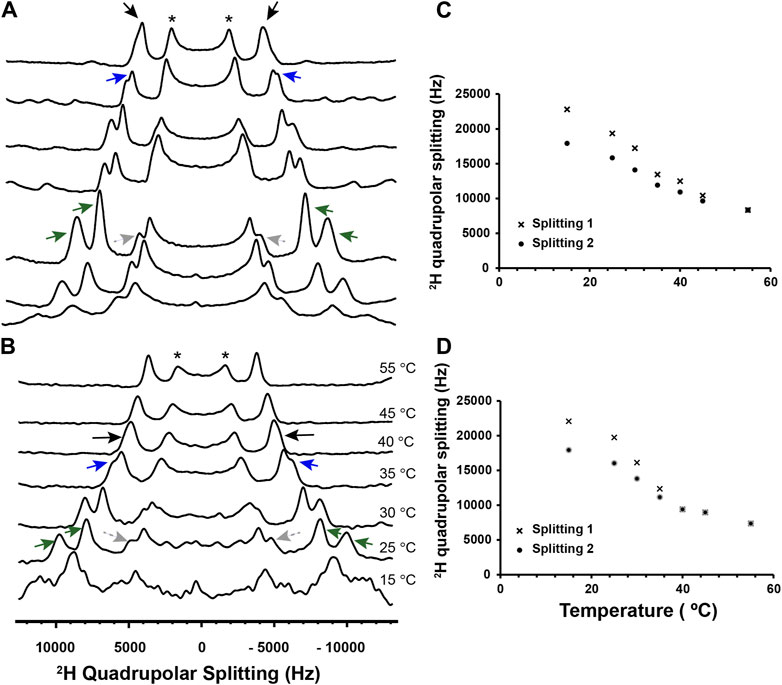
Figure 3. 2H solid-state NMR of Lo/Ld viral envelope mimics in the absence and presence of P1. Multilamellar Lo/Ld vesicles prepared with 20:10:12:33:25 DPPC/DPPE/POPC/POPE/Chol were studied by 2H static solid-state NMR at different temperatures in the absence (A) and presence of P1 at P/L = 1:100 (B). For 2H detection of the phospholipid methyl region, deuterated DPPC (d62) was incorporated in vesicles that were mechanically aligned on glass plates. Quadrupolar splittings arising from deuterated methyl groups give rise to signals that are centered at 0 kHz. Throughout the temperature range, some residual signal from unoriented lipids is present and indicated with an asterisk. At 55°C, the Lo and Ld phases are mixed, with DPPC-d62 giving rise to a single quadrupolar splitting (black arrows). As the temperature decreases, the onset of demixing occurs at Tlow, and DPPC-d62 gives rise to broadened peaks that start separating (blue arrows). Dropping the temperature further enables resolved signals to appear for the Lo and Ld phases. More specifically, the methyl groups of DPPC-d62 in the Lo phase give rise to two distinct outer splittings (green arrows). In the Ld phase, the lipid produces a single splitting, which is smaller and indicated using gray dashed arrows. It is not fully resolved from the signals arising from the unoriented lipids in the same spectral region. The 2H quadrupolar splittings for the Lo phase were extracted from the spectra and plotted as a function of temperature for the lipid-only sample (C) and sample with P1 (D). Splittings 1 and 2 correspond to the larger and smaller of the Lo splittings (green arrows in A and B), respectively. In the presence of P1, the onset of large-scale demixing occurs at Tlow = 35°C ± 5 °C while it is 45°C ± 5 °C in the lipid-only sample. Thus, the peptide extends the range over which the lipid phases are miscible. The ability of P1 to alter the stability of the Lo is confirmed by 1H MAS experiments presented in Supplementary Figures S4, S5.
In the presence of P1 at a peptide-to-lipid ratio (P/L) of 1:100 (Figure 3B), the large-scale demixing of the Lo/Ld occurs at 35°C ± 5°C, which is significantly lower than in the neat bilayer. Thus, P1 exerts a mixing effect on the Lo and Ld, forcing them to mix at a lower temperature than in the neat membrane. In parallel to these 2H NMR experiments, the viral envelope mimics were also subjected to magic angle spinning (MAS), the NMR method commonly used to detect the high-resolution 1H signals of unoriented samples. As shown in Supplementary Figure S4, the intensity of the central band for the methylene signals from the lipid acyl chains keeps increasing throughout the temperature range, indicating that the miscibility point, Tmix, has not been reached by 55 °C. However, in the presence of P1 (Supplementary Figure S5), a plateau is achieved at Tmix = 40°C ± 5°C, showing that the peptide depresses the stability of the Lo, in agreement with the 2H data. We note that the 2H experiments detect large-scale demixing, explaining why its onset occurs at a lower temperature than Tmix (Veatch et al., 2004).
CD-monitored binding of P1 to Lo/Ld and Lo viral envelope mimics
Next, the secondary structure of P1 was investigated in the presence of the viral envelope mimics. P1 is unstructured in aqueous solution while it adopts an α-helical structure bound to membranes (Chekmenev et al., 2006b; Perrin et al., 2014; Hayden et al., 2015; Paredes et al., 2020). CD spectroscopy readily resolves these two states, and thus provides a useful tool to determine the secondary structure of P1 when it is exposed to viral envelope mimics containing the Lo and Lo/Ld. Large unilamellar vesicles were made with a diameter of 0.1 µm to mimic the small size of virions (Lenard and Compans, 1974).
Figure 4A shows the CD data obtained at 45 °C when P1 was added to the Lo/Ld viral mimics at P/L = 1:200 and 1:120. Based on the 2H and 1H data presented in Figure 3 and Supplementary Figure S5, the Lo and Ld are miscible at this temperature. The CD bands detected at 208 and 222 nm demonstrate that P1 is α-helical, and therefore bound to the viral envelope mimics. The binding is stronger at lower P/L values, with 98% of the peptide bound at P/L = 1:200. Similar results are obtained with the Lo sample, with 92% binding detected at P/L = 1:200 (Figure 4B). Below 45°C, the CD data became noisier, preventing a similar characterization. However, NMR data (below) were collected at 30°C, showing that the peptide binds to the Lo/Ld below Tlow.
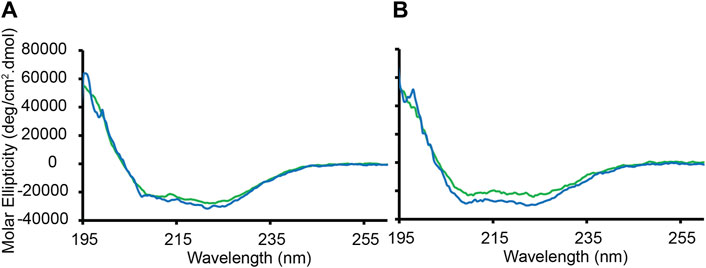
Figure 4. CD-monitored binding of P1 to large unilamellar vesicles containing Lo or Lo/Ld phases. Large unilamellar Lo/Ld and Lo vesiscles were prepared using DPPC/DPPE/POPC/POPE/Chol in ratios of 20:10:12:33:25 and 40:18:3:6:33, respectively. The vesicles prepared in phosphate buffer (5 mM potassium phosphate, pH 7.4) were extruded at a size of 0.1 µm and mixed with the peptide P1 at P/L = 1:120 (green) or 1:200 (blue). The peptide concentration was fixed to 20 μM. The molar ellipticity is displayed as a function of wavelength after subtracting the background signal from a lipid-only sample. (A) Binding to the Lo/Ld mixture. (B) Binding to the Lo only. The data were recorded at 50°C, which is above Tlow and Tmix. Duplicates collected on two different batches of vesicles gave rise to similar data. Based on the molar ellipticity obtained at 222 nm when P/L = 1:200, the amounts of peptide bound to the Lo/Ld and Lo are 98% and 92%, respectively.
Permeabilization effects of P1 on Lo/Ld and Lo viral envelope mimics
Next, we investigated whether P1 permeabilizes viral envelope mimics featuring the Lo/Ld and Lo. We previously used dye leakage assays to characterize the concentration-dependent permeabilization effects of P1 on various Ld membrane mixtures (Perrin et al., 2014; Hayden et al., 2015; Comert et al., 2019). It was found that P1 tends to be more membrane active when PC rather than PE headgroups are combined with anionic phosphatidylglycerol (PG), possibly because PE is more extensively hydrogen bonded and features stronger intrinsic negative curvature compared to PC (Murzyn et al., 2005). We also observed that P1 is active on zwitterionic membranes and Chol does not inhibit its membrane disruptive effects (Comert et al., 2019).
Figure 5 (red) shows the data obtained when we exposed Lo/Ld LUVs containing trapped calcein to increasing peptide concentrations. Based on the fitted dose-response curves, the effective lipid-to-peptide molar ratio (L/P) needed to reach 50% lysis (EC50) is 1769 ± 220 at 45 °C. At 30°C, which is below Tlow, the EC50 became 865 ± 35, and thus remained low. These EC50 values indicate that the peptide is more effective on the Lo/Ld than the previously studied 3:1 PC/PG (EC50 = 22), 1:1 PE/PG (EC50 = 10), PC (EC50 = 166), and 4:1 PC/Chol (EC50 = 130) lipid systems (the experimental error was indicated to be 20%) (Perrin et al., 2014; Hayden et al., 2015; Comert et al., 2019). We also tested P1 on the Lo phase alone. The data in Figure 5 (blue) show that the EC50 decreased compared to the Lo/Ld mixture (EC50 = 463 ± 31 and 206 ± 7 at 45°C and 30°C, respectively) but the peptide remained highly permeabilizing. Remarkably, even at 30°C, P1 is much more effective on the Lo/Ld than the zwitterionic 4:1 PC/Chol Ld. As indicated in the introduction, the interface between domains can be a place of higher susceptibility for disruption by membrane-active agents (Schön et al., 2008; Blicher et al., 2009; Apellániz et al., 2011; Hao et al., 2018; Nasr et al., 2020; Kumar et al., 2022; Utterström et al., 2022).
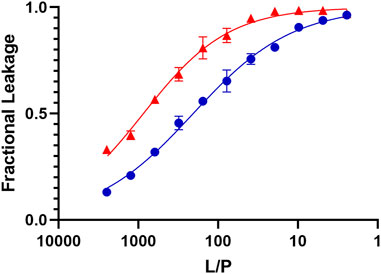
Figure 5. Dye leakage assays for Lo and Lo/Ld phases viral envelope mimics exposed to P1. The fractional leakage is displayed for calcein dye leakage assays performed with P1 and large unilamellar vesicles containing the Lo/Ld (red) and Lo (blue) phases. The normalized fluorescence intensity obtained after incubation at 45 °C is plotted against the lipid-to-peptide (L/P) ratio and fitted using an adaptation of the Hill equation. Each point represents the mean of triplicates and error bars are ± SD. The positive control (100% leakage) is obtained using 0.1% Triton X-100 (see Methods). These curves are used to extract the EC50, which represents the L/P values at which 50% leakage is observed. These values are higher for P1 acting on the Lo/Ld (EC50 = 1769 ± 220) than Lo (EC50 = 463 ± 31). Similar but lower EC50 values were obtained at 30°C, which is below Tlow (Supplementary Figure S6).
Cryo-EM studies of Lo/Ld and Lo viral envelope mimics exposed to P1
Previous studies with membrane-active HDPs, including P1, have showed that their binding to membranes induces thinning (Balla et al., 2004; Ouellet et al., 2006; Mihailescu et al., 2019). Knowing that P1 permeabilizes viral envelope mimics featuring a high Chol content, we employed Cryo-EM to investigate how thickness changes when these model membranes are exposed to the peptide. The Lo and Lo/Ld vesicles used for Cryo-EM were first characterized by DLS (Supplementary Figure S7) to confirm size homogeneity. Figure 6 shows the Cryo-EM images obtained for such LUVs before and after exposure to P1. The data indicate that the membrane is dramatically rearranged when the peptide is present. From a visual inspection of the micrographs, the LUVs become much larger and adopt multilamellar structures, which indicates that aggregation and fusion occur upon peptide addition.

Figure 6. Cryo-EM micrographs of Lo and Lo/Ld phases vesicles exposed to P1. LUVs (Lo/Ld and Lo) extruded at a size of 0.1 µm were exposed to P1 at P/L = 1:40. Lo/Ld (A) and Lo (C) vesicles have normal unilamellar structure and size in the absence of piscidin. Upon adding the peptide at P/L = 1:40 to the Lo/Ld (B) and Lo (D) vesicles, their size increases dramatically and multilamellar structures are observed. A ML process was developed to annotate the vesicles and extract the membrane thickness and curvature (See Methods). Supplementary Figure S8 shows an example of vesicles annotated every 4 nm as part of the ML analysis.
To quantify the changes in membrane thickness and vesicle size that P1 induces to LUVs, we developed a new automated program to annotate the micrographs and characterize their curvature over a distance of 4 nm. This protocol was inspired by previous work done by the Heberle lab where Cryo-EM data collected on LUVs were analyzed to yield bilayer thickness and relative amounts of Lo and Ld (Heberle et al., 2020). In that case, the analysis was done manually. Here, a ML process was leveraged to process large data sets and give direct information on vesicle size since curvature and vesicle size are inversely related. Major benefits of this approach include that it is fast, and since all of the vesicles are analyzed, bias towards prioritizing some vesicles over others is avoided. Supplementary Figure S8 shows examples of vesicles that were annotated by this method.
As shown in Figure 7, a clear relationship exists between the curvature and P/L, with the curvature decreasing (i.e., the membrane becomes more flat as the vesicles become larger) when more peptide is added. This is observed whether the Lo/Ld or Lo system is used. A trend towards reduced membrane thickness is also detected as more peptide is added. For the Lo/Ld samples, the vesicles contain two types of domains, with the Lo phase being thicker due to higher Chol content. Since the analysis did not resolve the Lo from the Ld, the trends represent how the thickness of the overall bilayer, i.e., the mixed Lo and Ld phases, responded to P1. When the Lo is used alone, thinning occurs but less than when the Lo/Ld are mixed, indicating that the bilayer thinning effect of P1 occurs more readily when the peptide has the opportunity to interact with the Ld that it preferentially binds to (Comert et al., 2019). Overall, the statistical analysis indicates that the relationships associated with curvature and thickness are significant across the data sets: P1 decreases the curvature and thickness of the viral envelope mimics containing the ordered phase.
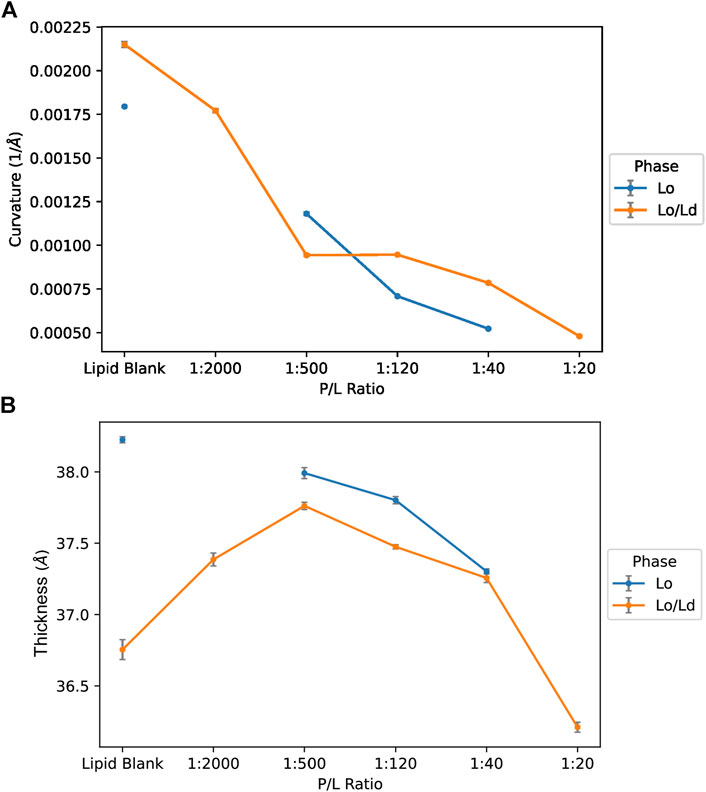
Figure 7. Curvature and thickness trends for vesicles containing Lo or Lo/Ld phases as characterized by Cryo-EM data and ML. The data obtained by Cryo-EM were analyzed using an automated process to characterize the membrane curvature (A) and thickness (B) of the LUVs as a function of the P/L ratio (see Methods). Smaller vesicular curvature corresponds to larger vesicles. Smaller thicknesses indicate thinning of the bilayers. The error bar indicates ±3 SD.
Use of differential scanning calorimetry to investigate the ability of P1 to fuse and mix lipids
Given the strong remodeling effect of piscidin on viral envelope mimics detected by Cryo-EM, we sought to characterize the ability of the peptide to mix lipids from vesicles varying in lipid composition. Investigations by DSC were aimed at observing the effect of P1 on the melting transitions and mixing behavior of lipids commonly found in raft mixtures such as POPE, DPPC and Chol, all of which are used to make our Lo and Lo/Ld viral envelope mimics. Two types of experiments were performed. In the first type, POPE, DPPC and Chol were co-dissolved in chloroform to form a homogeneous lipid mixture that was then used to produce vesicles in water (Figure 8A). Taken separately, POPE and DPPC show the typical (gel-to-fluid) melting transitions at 25°C, and 41°C, respectively. However, when homogeneously mixed at a 1:1 molar ratio, they display a single transition peak at 30.8°C, broader than the two individual transition of the two lipids. The presence of Chol in proportion of 20% (molar fraction) broadened the transition of the mixture while shifting Tm to 29°C. Finally, addition of P1 to an aliquot of the same POPE/DPPC/Chol mixture in water further broadened the transition pointing to a strong lipid mixing and chain disordering effect due to P1. In a second approach, separate aliquots of POPE and DPPC vesicles were produced. We combined them with each other (Figure 8B) and with P1 in water (Figure 8C) just before the DSC measurements. We found that this type of investigation provides further insight into whether P1 has a differentiated effect on the two lipids. A few repeated scans were performed until the peak shapes stabilized (see also Supplementary Figure S9). As shown in Figure 8B, the POPE and DPPC melting peaks, while well separated at the beginning at the scans (Tm at ∼ 22°C and ∼39°C, respectively), eventually merged into a broad central peak (Tm ∼ 27°C), that was very similar to the transition peak for the pre-mixed POPE/DPPC (Figure 8A). However, when aliquots of the same POPE and DPPC vesicle suspensions in water were combined as above and P1 was added to this mixture immediately before the measurement, a much more dramatic mixing effect was observed (Figure 8C). By the third scan, the POPE peak completely disappears with no distinguishable mixed lipid peak. A remnant of the DPPC transition is still observed at 41°C. Overall, the data indicates that the P1 has an immediate effect on the melting and mixing of the two lipids, probably by promoting fusion of vesicles. Also, the POPE component appears to be more readily affected by P1. Not only does P1 cause “fluidization” and lipid mixing in membranes but it also displays affinities for certain components. In some of our previous studies, it was shown, by X-ray diffraction and microscopy in “raft” forming mixtures, that P1 has a preference to associate with fluid domains in membranes and/or causes Chol to segregate away from the P1-associated regions (Comert et al., 2019). Taken together, our DSC results indicate that P1 strongly affects the lipid landscape in mixed membranes such as those found in viruses. These changes in structure and organization are likely to directly affect the viral envelope lipids as well as affect viral protein distribution and function.
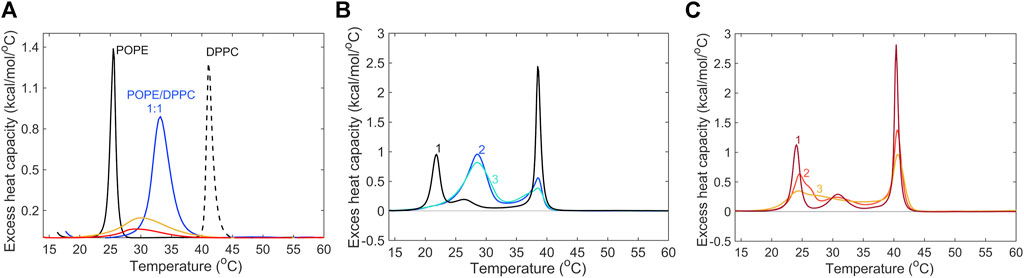
Figure 8. Differential scanning calorimetry data of model membranes exposed to P1. (A) The following conditions were investigated by DSC: gel-to-fluid transition curves for POPE (solid black), DPPC (dashed black), a homogeneous, equimolar mixture of POPE/DPPC (blue), a mixture of POPE/DPPC/Chol (1:1:0.5, molar) (yellow) and the same POPE/DPPC/Chol mixture in the presence of P1 peptide, at P/L = 1:100 (red). The samples were prepared at a target concentration of 3.5 mmol/L. The maxima of the melting transition temperatures (Tm) were found at 25.4°C for POPE, 41.2 °C for DPPC, 32.8°C for POPE/DPPC (1:1), 30.1°C for POPE/DPPC/Chol, and 29.1°C for POPE/DPPC/Chol/P1. (B) Three consecutive heating scans (Lipsitch et al., 2022; Davis et al., 2023; Perumal et al., 2023) were performed on vesicles of POPE and DPPC in water, prepared separately (see Methods) and combined at a 1:1 ratio just before the measurements. The target total lipid concentration was 6.9 mmol/L. (C) DSC was performed on the same mixture of vesicles as in (B), but the P1 peptide in water was added at P/L = 1:100 just before the measurements. See also Supplementary Figure S9 for full, raw heating, and cooling data.
Use of solid-state NMR to characterize the conformational arrangement of P1 bound to Lo/Ld viral envelope mimics
We previously used oriented sample solid-state NMR to solve the structure of P1 bound to various lipid mixtures, including 3:1 PC/PG, 1:1 PE/PG, and 4:1 PC/Chol (Perrin et al., 2014; Comert et al., 2019). Not only does this method provide the structure of the peptide but it also characterizes the tilt and helix rotation in the membrane (Ketchem et al., 1997; Opella et al., 2001; Opella et al., 2002; Nevzorov et al., 2004; Opella and Marassi, 2004). Since P1 interacts with viral envelope mimics as shown by the data presented above, we made oriented samples for NMR structural analysis of P1.
Figure 9 shows the data obtained for 15N-labeled V10G13I16 P1 bound to the viral envelope mimics. Since the labeled positions are spread out along the backbone of the peptide, they report on the overall peptide structure, including G13 where the α-helix is kinked (Perrin et al., 2014). The data were collected at 30°C, which is below the Tlow characterized by solid-state NMR. These spectra provide two orientational restraints per peptide plane: 15N chemical shifts (CSs) and 15N-1H dipolar couplings (DCs). Both of these sets of values (CS near 70–80 ppm and DCs around 8–10 kHz) are consistent with the peptide lying almost parallel to the membrane surface. Based on previous studies of these three 15N-labeled sites (Perrin et al., 2014; Comert et al., 2019), the signals were assigned as follows: V10: 80.5; G13: 70.1; and I16: 66.2 ppm. In comparison to the data obtained with the bacterial cell membrane mimic systems (PC/PG and PE/PG), the chemical shifts and DCs are comparable indicating similar tilt angles in the membrane, i.e., the peptide sits parallel to the membrane surface (Perrin et al., 2014; Comert et al., 2019). Hence, despite the high Chol content, P1 retains a strong ability to partition in these membranes. We also collected data at 45°C, which is above Tmix, and noticed that a new set of resonances appeared in proximity to G13 and I16 while the V10 resonance became broader, and thus weaker (Supplementary Figure S10). This suggests that when the Lo and Ld are miscible based on the 2H data, the peptide adopts a minor conformational state, possibly reflecting that the peptide, which prefers interacting with the Ld, is now able to partition significantly in the Lo. Thus, its conformation or surrounding environment would be slightly different in the Ld than Lo. This could result in different CSs and DCs given the sensitivity of these restraints.
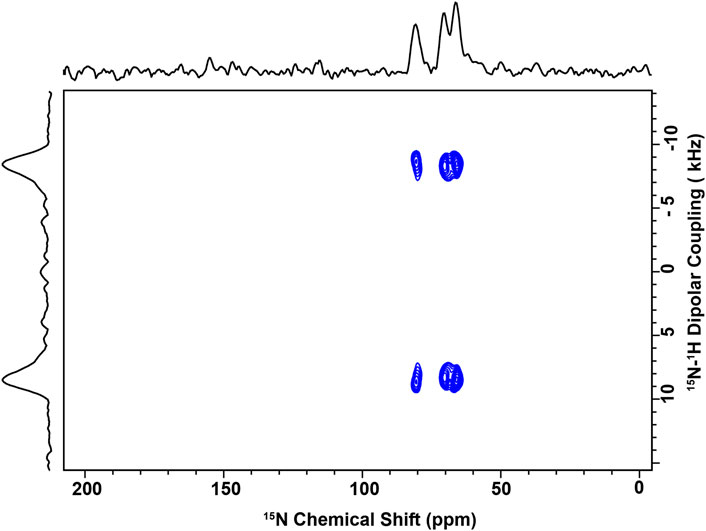
Figure 9. NMR spectrum of P1 bound to Lo/Ld viral envelope mimics. 2D solid-state spectra of 15N-V10G13I16-P1 reconstituted in the Lo/Ld phase. The sample was made at P/L = 1:100 and placed in the NMR probe with the bilayer normal parallel to B0. The data were recorded at 1H and 15N frequencies of 600 MHz and 60.8 MHz, respectively, with 2048 transients per slice of the 2D experiment. Each 15N label gives rise to a splitting in the indirect dimension, allowing for the quantification of the 15N-1H dipolar coupling between each amide 15N label and its attached proton. Based on previous studies of these three sites, the following assignments are made: V10: 80.5; G13: 70.1; and I16: 66.2 ppm. The spectrum was recorded at 30°C, which is below the miscibility point detected by NMR (See Figure 3; Supplementary Figure S5). Above Tmix, a second set of splittings appeared in the indirect dimension suggesting that another peptide conformation was present (see Supplementary Figure S10).
Discussion
The COVID-19 pandemic with its devastating death toll and societal impacts has underscored the crucial need to develop broad-spectrum therapeutics against (re)emerging viruses (Amanatidou et al., 2022; Lipsitch et al., 2022; Rahman et al., 2022; Zheng et al., 2022; Davis et al., 2023; Perumal et al., 2023). In this research, we feature a peptide-based approach to target viral membrane envelopes, given that their lipid composition and reparative abilities differentiate them from host cell membranes. Focusing on the membrane-mediated action of P1, a highly potent HDP with an aromatic-rich motif in its N-terminal region, we tested its antiviral activity on SARS-CoV-2 and investigated on a mechanistic level its disruptive effects on viral envelope mimics containing Chol. P1 demonstrates significant activity on both SARS-CoV-2 and HIV-1 (Wang et al., 2010; Wang, 2013), viruses with low and elevated cholesterol content, respectively (Brügger et al., 2006). We leveraged complimentary biophysical methods to establish structure-activity relationships in P1 acting on Chol-containing Lo and Lo/Ld viral envelope mimics. A major finding from this work is that P1 is more active on phase-separated (Lo/Ld) membranes, indicating that P1 creates and exploits heterogeneity in mixed lipid bilayers by accumulating preferentially in Ld regions (Comert et al., 2019) and possibly, at regions featuring thickness mismatch-driven line tension at the boundaries between Lo/Ld domains (Heberle et al., 2013). Next, we discuss the results and highlight important facets.
A novel feature of our work lies in the incorporation of ML tools to analyze large Cryo-EM data sets collected on lipid vesicles. Previously, Heberle and coauthors manually annotated vesicles as part of a study demonstrating the power of Cryo-EM to characterize the thickness of complex membranes such as Lo/Ld mixtures (Heberle et al., 2020). Building on this, we developed a new methodology that automatically annotates individual vesicles, enabling the rapid tabulation of membrane thickness and curvature. This breakthrough has important ramifications given that vesicles are used in a growing number of fields such as membrane biophysics, medicinal chemistry, and drug delivery (Sheynis et al., 2003; Lo et al., 2015; Pattni et al., 2015; Hou et al., 2021; Liu et al., 2022; Utterström et al., 2022).
In recent years, peptide-based approaches have garnered interest in biomedical research and the pharmaceutical industry (Thundimadathil, 2012; Uhlig et al., 2014; Fosgerau and Hoffmann, 2015; Davenport et al., 2020; Varanko et al., 2020; Muttenthaler et al., 2021). This growth is bolstered by pivotal advances such as lower production cost, novel formulation methods, and the innovative incorporation of non-canonical amino acids that increase plasma half-life. In the context of fighting viruses, antiviral HDPs present several advantages, including the dual ability to decrease viral infectivity rates and exert anti-inflammatory effects on host cells. Among the few HDPs that have demonstrated antiviral action, P1 stands out due to its strong potency (EC50 = 5.4 μg/mL) against HIV-1 (Wang et al., 2010; Wang, 2013), a virus characterized by a Chol-rich envelope (Brügger et al., 2006).
In this study, we show that P1 is similarly active (EC50 ∼ 5 μg/mL) on SARS-CoV-2 and that its activity is partly due to direct action on the virions. Examples of synthetic antimicrobial peptides active on SARS-CoV-2 include MXB-4 and MXB-9 with respective EC50 of 20 and 7 μg/mL (Diamond et al., 2021), and thus less effective than Pl. DP7 is active on the SARS-CoV-2 protein pseudovirus at a potency (EC50 = 73.6 μg/mL) lower than that of P1, which was tested on the whole virus (Zhang et al., 2021). Our assays also show that P1 is more active than Brilacidin, a short HDP-mimetic that has gone through stage 2 clinical trials for its antimicrobial action. Previous reports indicated that P1 is particularly active on porcine epidemic diarrhea CoV (EC50 = 1 μg/mL) and pseudorabies (EC50 = 0.23 μg/mL) (Lei et al., 2018; Hu et al., 2019). Based on their egress pathways, these viruses are bound to have a low Chol content (Hogue et al., 2014; Schoeman and Fielding, 2019; Ghosh et al., 2020). Overall, these studies reveal that P1 is active on a broad range of viruses. Its activity on HIV-1 is particularly significant given that Chol increases the order and stability of membranes (Mihailescu et al., 2011). This led us to investigate on a molecular level how P1 achieves this uncommon effectiveness.
In earlier work that we performed, P1 was demonstrated to be active on Chol-containing zwitterionic membranes (Comert et al., 2019). More specifically, we found that P1 induces phase separation in homogeneous membranes, highlighting a novel mechanism of action where the peptide pushes away cholesterol and concentrates itself in the disordered region. Here, to characterize how P1 exerts its direct antiviral effects, we took an interdisciplinary approach. We employed antiviral and permeabilization activity assays to quantify the activity of P1 on SARS-CoV-2. The results demonstrate that its permeabilization effects have the following ranking: Lo/Ld (20:10:12:33:25 DPPC/DPPE/POPC/POPE/Chol) > Lo (40:18:3:6:33 DPPC/DPPE/POPC/POPE/Chol) > Ld (4:1 POPC/Chol). Furthermore, P1 is active on the Lo alone, demonstrating its ability to interact with Chol-rich regions. Its stronger activity on phase-separated than homogenous bilayers supports a mechanism of disruption where enhanced activity is achieved by targeting the interface between the Lo and Ld phases. This phenomenon has been reported for several peptides involved in viral fusion (Schön et al., 2008; Blicher et al., 2009; Apellániz et al., 2011; Hao et al., 2018; Nasr et al., 2020; Kumar et al., 2022; Utterström et al., 2022). To the best of our knowledge, P1 could be the first HDP shown to interact with the Lo and target the Lo/Ld interfaces.
To investigate the mechanism by which P1 disrupts Lo and Lo/Ld membranes, CD, NMR, Cryo-EM, and DSC were combined. CD and 15N SS-NMR data document that the peptide adopts an α-helical structure that lies parallel to the surface of the viral envelope mimics. Cryo-EM and DSC reveal the strong lipid mixing effect of P1 on these membranes. The disordering effect of P1 is also demonstrated through the 2H SS-NMR experiments that establish the destabilization of the Lo domains in the Lo/Ld mixture. Since P1 is able to interact with the Lo, its inhibitory effect on demixing of the Lo/Ld could be due to its inherent ability to partition in the region between the headgroups and hydrocarbon core, where it can induce disorder in the acyl chains of the lipids (Perrin et al., 2014; Hayden et al., 2015; Perrin et al., 2015; Mihailescu et al., 2019; Comert et al., 2021; Liu et al., 2023). Interestingly, P1 is more active on the Lo than Ld when each of them is used alone. As learned from earlier work with POPC/Chol bilayers, P1 can induce heterogeneity in homogenous membranes by pushing away the cholesterol (Comert et al., 2019), This provides a pathway to enhanced activity via the formation of Chol-depleted domains and defect-prone interfaces between domains. The N-terminal motif of P1, which is rich in aromatic residues, may promote its ability to initiate interactions with the Lo phase via π-stacking interactions (Epand et al., 2005; Apellániz et al., 2011; Zhou and Xu, 2012; Fantini and Barrantes, 2013; Paterson et al., 2017). Other nonpolar residues such as Ile and Val are also expected to play a role in mediating favorable interactions between the peptide and hydrophobic membrane. Another notable feature is that the fast bi-axial diffusion that apo P1 experiences about its α-helical axis and the bilayer normal enables the peptide to travel a distance that is greater than 30 μm per second (Chekmenev et al., 2010). The fast dynamics and compact arrangement of the α-helical peptide could translate into an enhanced capability to migrate to line defects and/or insert and disrupt the bilayer regions where it partitions.
Another aspect that could promote the disruption of viral envelopes by P1 is its preferential interaction with lipids that are important for viral fusion. As shown by the DSC data, the peptide prefers interacting with POPE over DPPC. Due to its inverted conical shape, the PE headgroup has a strong preference for the inverted hexagonal HII phase (Cullis and de Kruijff, 1979). Previous work that we performed showed that P1 inhibits PE from forming the HII phase, an effect that correlates with its ability to induce positive curvature strain (Perrin et al., 2015; Comert et al., 2019). As a result, it could hamper the formation of viral fusion intermediates since these feature negative spontaneous curvature.
Overall, P1 can leverage several effects to enhance its disruptive effects on viral envelopes that contain Chol. It can induce chain disordering, lipid mixing, and Chol-segregation. It exploits phase-separated membranes for enhanced permeabilization. Importantly, these effects can not only directly disintegrate viral envelopes but also indirectly disrupt the functions of key viral proteins embedded in the envelope. In particular, the disruption of lipids surrounding the glycosylated proteins (e.g., Spike proteins) involved in viral fusion could have deleterious effects on infectivity (Wolf et al., 2010; Miguel et al., 2013; Schoggins and Randall, 2013; Vigant et al., 2015; Yang et al., 2016). It would be insightful to investigate the role of these proteins in future mechanistic work on the peptide. Beyond acting on the viral membrane proteins, the peptide may be involved in changing the pH of endosomes, an effect reported for several antiviral peptides (Miller et al., 2011; Zhao et al., 2016). Further studies are needed to better understand the indirect effects of P1. Additional investigations could also be performed to compare P1 to other antiviral peptides, test its antiviral effect on other cell lines, and characterize its selectivity index. Examples of other HDPs with antiviral activity include caerin, indolicidin, LL-37, and maculatin (Wang, 2013; VanCompernolle et al., 2015; Ahmed et al., 2019; Hu et al., 2019; Ahmed et al., 2020).
In conclusion, this investigation unveils that P1, a peptide with an aromatic-rich N-terminal motif, is unique among HDPs by exhibiting promising antiviral properties against viruses such as HIV-1 and SARS-COV-2. It achieves stronger activity on HIV-1 and SARS-CoV-2 than other antimicrobial peptides. Our mechanistic studies on Chol-containing viral envelope mimics underscore several features important to explain its antiviral potency. First, P1 causes a melting of the gel phase of lipids by inserting with high affinity in the hydrocarbon core and inducing lipid mixing and acyl chain disordering. Second, P1 leverages heterogeneity in Chol-containing phase-separated membranes by preferentially interacting with the Chol-depleted region, thus exacerbating the existing line tension between regions and creating defects. Third, it induces positive curvature that counteracts the natural negative curvature of lipids such as PE. Together these local membrane effects could result in the direct disruption of viral envelopes as well as the indirect perturbation of surrounding membrane proteins. ML tools used in this study provided a novel way to rapidly annotate lipid vesicles and characterize their curvature and thickness. Overall, our combination of experimental and ML methods bodes well in the search for novel antiviral compounds that have broad spectrum properties and could help fight existing and (re)emerging viruses.
Data availability statement
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
Ethics statement
Ethical approval was not required for the studies on humans in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used. Ethical approval was not required for the studies on animals in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used.
Author contributions
TB: Formal Analysis, Investigation, Methodology, Visualization, Writing–review and editing, Software. MDB: Formal Analysis, Investigation, Methodology, Visualization, Writing–review and editing. MTR: Formal Analysis, Investigation, Visualization, Writing–review and editing. YX: Formal Analysis, Investigation, Visualization, Writing–review and editing. HK: Formal Analysis, Investigation, Visualization, Writing–review and editing, Methodology. EG: Formal Analysis, Investigation, Visualization, Writing–review and editing. MG: Formal Analysis, Investigation, Visualization, Writing–review and editing. EH: Formal Analysis, Investigation, Resources, Supervision, Validation, Visualization, Writing–review and editing. RF: Formal Analysis, Investigation, Methodology, Validation, Visualization, Writing–review and editing. MM: Conceptualization, Formal Analysis, Investigation, Methodology, Resources, Validation, Visualization, Writing–review and editing. AN: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Resources, Supervision, Validation, Visualization, Writing–review and editing. MLC: Formal Analysis, Investigation, Methodology, Visualization, Writing–review and editing, Conceptualization, Data curation, Funding acquisition, Project administration, Resources, Supervision, Validation, Writing–original draft.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was supported by the National Science Foundation (MCB-1716608 to MLC and MCB-1714164 to MM). The NMR experiments were performed at the National High Magnetic Field Lab supported by the NSF Cooperative agreement (DMR-1644779 and DMR-2128556), and the State of Florida. Some of the work presented here was conducted at the National Resource for Automated Molecular Microscopy located at the New York Structural Biology Center, supported by grants from the NIH National Institute of General Medical Sciences (GM103310) and the Simons Foundation (SF349247).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2024.1379192/full#supplementary-material
References
Ahmed, A., Bakovic, A., Risner, K., Kortchak, S., Der Torossian Torres, M., de la Fuente-Nunez, C., et al. (2020). Synthetic host defense peptides inhibit Venezuelan equine encephalitis virus replication and the associated inflammatory response. Sci. Rep. 10 (1), 21491. doi:10.1038/s41598-020-77990-3
Ahmed, A., Siman-Tov, G., Keck, F., Kortchak, S., Bakovic, A., Risner, K., et al. (2019). Human cathelicidin peptide LL-37 as a therapeutic antiviral targeting Venezuelan equine encephalitis virus infections. Antivir. Res. 164, 61–69. doi:10.1016/j.antiviral.2019.02.002
Alsaadi, E. A. J., and Jones, I. M. (2019). Membrane binding proteins of coronaviruses. Future Virol. 14 (4), 275–286. doi:10.2217/fvl-2018-0144
Amanatidou, E., Gkiouliava, A., Pella, E., Serafidi, M., Tsilingiris, D., Vallianou, N. G., et al. (2022). Breakthrough infections after COVID-19 vaccination: insights, perspectives and challenges. Metab. Open 14, 100180. doi:10.1016/j.metop.2022.100180
Anantharaman, A., Rizvi, M. S., and Sahal, D. (2010). Synergy with rifampin and kanamycin enhances potency, kill kinetics, and selectivity of de novo-designed antimicrobial peptides. Antimicrob. Agents Chemother. 54 (5), 1693–1699. doi:10.1128/aac.01231-09
Apellániz, B., García-Sáez, A., Nir, S., and Nieva, J. L. (2011). Destabilization exerted by peptides derived from the membrane-proximal external region of HIV-1 gp41 in lipid vesicles supporting fluid phase coexistence. Biochim. Biophys. Acta 1808 (7), 1797–1805. doi:10.1016/j.bbamem.2011.02.005
Baglivo, M., Baronio, M., Natalini, G., Beccari, T., Chiurazzi, P., Fulcheri, E., et al. (2020). Natural small molecules as inhibitors of coronavirus lipid-dependent attachment to host cells: a possible strategy for reducing SARS-COV-2 infectivity? Acta Biomed. 91 (1), 161–164. doi:10.23750/abm.v91i1.9402
Bakovic, A., Risner, K., Bhalla, N., Alem, F., Chang, T. L., Weston, W. K., et al. (2021). Brilacidin demonstrates inhibition of SARS-CoV-2 in cell culture. Viruses 13 (2), 271. doi:10.3390/v13020271
Balla, M. S., Bowie, J. H., and Separovic, F. (2004). Solid-state NMR study of antimicrobial peptides from Australian frogs in phospholipid membranes. Eur. Biophys. J. 33, 109–116. doi:10.1007/s00249-003-0342-7
Beisswenger, C. B. R., and Bals, R. (2005). Functions of antimicrobial peptides in host defense and immunity. Curr. Protein Pept. Sci. 6 (3), 255–264. doi:10.2174/1389203054065428
Blicher, A., Wodzinska, K., Fidorra, M., Winterhalter, M., and Heimburg, T. (2009). The temperature dependence of lipid membrane permeability, its quantized nature, and the influence of anesthetics. Biophys. J. 96 (11), 4581–4591. doi:10.1016/j.bpj.2009.01.062
Bolosov, I. A., Kalashnikov, A. A., Panteleev, P. V., and Ovchinnikova, T. V. (2017). Analysis of synergistic effects of antimicrobial peptide arenicin-1 and conventional antibiotics. Bull. Exp. Biol. Med. 162 (6), 765–768. doi:10.1007/s10517-017-3708-z
Boto, A., Pérez de la Lastra, J. M., and González, C. C. (2018). The road from host-defense peptides to a new generation of antimicrobial drugs. Molecules 23 (2), 311. doi:10.3390/molecules23020311
Brender, J. R., Lee, E. L., Cavitt, M. A., Gafni, A., Steel, D. G., and Ramamoorthy, A. (2008). Amyloid fiber formation and membrane disruption are separate processes localized in two distinct regions of IAPP, the type-2-diabetes-related peptide. J. Am. Chem. Soc. 130 (20), 6424–6429. doi:10.1021/ja710484d
Brown, D. A., and London, E. (1998). Functions of lipid rafts in biological membranes. Annu. Rev. Cell Dev. Biol. 14 (1), 111–136. doi:10.1146/annurev.cellbio.14.1.111
Brügger, B., Glass, B., Haberkant, P., Leibrecht, I., Wieland, F. T., and Kräusslich, H. G. (2006). The HIV lipidome: a raft with an unusual composition. Proc. Natl. Acad. Sci. U. S. A. 103 (8), 2641–2646. doi:10.1073/pnas.0511136103
Chan, R. B., Tanner, L., and Wenk, M. R. (2010). Implications for lipids during replication of enveloped viruses. Chem. Phys. Lipids 163 (6), 449–459. doi:10.1016/j.chemphyslip.2010.03.002
Chekmenev, E., Vollmar, B., and Cotten, M. (2010). Can antimicrobial peptides scavenge around a cell in less than a second? Biochim. Biophys. Acta 1798, 228–234. doi:10.1016/j.bbamem.2009.08.018
Chekmenev, E. Y., Jones, S. M., Nikolayeva, Y. N., Vollmar, B. S., Wagner, T. J., Gor’kov, P. L., et al. (2006a). High-field NMR studies of molecular recognition and structure-function relationships in antimicrobial piscidins at the water-lipid bilayer interface. J. Am. Chem. Soc. 128, 5308–5309. doi:10.1021/ja058385e
Chekmenev, E. Y., Vollmar, B. S., Forseth, K. T., Manion, M. N., Jones, S. M., Wagner, T. J., et al. (2006b). Investigating molecular recognition and biological function at interfaces using piscidins, antimicrobial peptides from fish. Biochim. Biophys. Acta 1758, 1359–1372. doi:10.1016/j.bbamem.2006.03.034
Chen, P. S., Toribara, T. Y., and Warner, H. (1956). Microdetermination of Phosphorus. Anal. Chem. 28 (11), 1756–1758. doi:10.1021/ac60119a033
Chen, W. F., Huang, S. Y., Liao, C. Y., Sung, C. S., Chen, J. Y., and Wen, Z. H. (2015). The use of the antimicrobial peptide piscidin (PCD)-1 as a novel anti-nociceptive agent. Biomaterials 53, 1–11. doi:10.1016/j.biomaterials.2015.02.069
Chinchar, V. G., Bryan, L., Silphaduang, U., Noga, E., Wade, D., and Rollins-Smith, L. (2004). Inactivation of viruses infecting ectothermic animals by amphibian and piscine antimicrobial peptides. Virology 323 (2), 268–275. doi:10.1016/j.virol.2004.02.029
Chowdhury, S. M., Talukder, S. A., Khan, A. M., Afrin, N., Ali, M. A., Islam, R., et al. (2020). Antiviral peptides as promising therapeutics against SARS-CoV-2. J. Phys. Chem. B 124 (44), 9785–9792. doi:10.1021/acs.jpcb.0c05621
Comert, F., Greenwood, A., Maramba, J., Acevedo, R., Lucas, L., Kulasinghe, T., et al. (2019). The host-defense peptide piscidin P1 reorganizes lipid domains in membranes and decreases activation energies in mechanosensitive ion channels. J. Biol. Chem. 294 (49), 18557–18570. doi:10.1074/jbc.ra119.010232
Comert, F., Heinrich, F., Chowdhury, A., Schoeneck, M., Darling, C., Anderson, K. W., et al. (2021). Copper-binding anticancer peptides from the piscidin family: an expanded mechanism that encompasses physical and chemical bilayer disruption. Sci. Rep. 11 (1), 12620. doi:10.1038/s41598-021-91670-w
Conlon, J. M. (2015). Host-defense peptides of the skin with therapeutic potential: from hagfish to human. Peptides 67, 29–38. doi:10.1016/j.peptides.2015.03.005
Cullis, P. R., and de Kruijff, B. (1979). Lipid polymorphism and the functional roles of lipids in biological membranes. Biochim. Biophys. Acta 559 (4), 399–420. doi:10.1016/0304-4157(79)90012-1
Davenport, A. P., Scully, C. C. G., de Graaf, C., Brown, A. J. H., and Maguire, J. J. (2020). Advances in therapeutic peptides targeting G protein-coupled receptors. Nat. Rev. Drug Discov. 19 (6), 389–413. doi:10.1038/s41573-020-0062-z
Davis, H. E., McCorkell, L., Vogel, J. M., and Topol, E. J. (2023). Long COVID: major findings, mechanisms and recommendations. Nat. Rev. Microbiol. 21 (3), 133–146. doi:10.1038/s41579-022-00846-2
Delaglio, F., Grzesiek, S., Vuister, G., Zhu, G., Pfeifer, J., and Bax, A. (1995). NMRPipe:A multidimensional spectral processing system based on UNIX pipes. J. Biomol. Nmr. 6, 277–293. doi:10.1007/bf00197809
Dezfuli, B. S., Lui, A., Giari, L., Castaldelli, G., Mulero, V., and Noga, E. J. (2012). Infiltration and activation of acidophilic granulocytes in skin lesions of gilthead seabream, Sparus aurata, naturally infected with lymphocystis disease virus. Dev. Comp. Immunol. 36 (1), 174–182. doi:10.1016/j.dci.2011.06.017
Diamond, G., Molchanova, N., Herlan, C., Fortkort, J. A., Lin, J. S., Figgins, E., et al. (2021). Potent antiviral activity against HSV-1 and SARS-CoV-2 by antimicrobial peptoids. Pharm. (Basel). 14 (4), 304. doi:10.3390/ph14040304
Duong, L., Gross, S. P., and Siryaporn, A. (2021). Developing antimicrobial synergy with AMPs. Front. Med. Technol. 3, 640981. doi:10.3389/fmedt.2021.640981
Dutta, A., and Zisserman, A. (2019). “The VIA annotation software for images, audio and video,” in Proceedings of the 27th ACM international conference on multimedia (Nice, France: Association for Computing Machinery), 2276–2279.
El Karim, I. A., Linden, G. J., Orr, D. F., and Lundy, F. T. (2008). Antimicrobial activity of neuropeptides against a range of micro-organisms from skin, oral, respiratory and gastrointestinal tract sites. J. Neuroimmunol. 200 (1-2), 11–16. doi:10.1016/j.jneuroim.2008.05.014
Engelman, D. M. (2005). Membranes are more mosaic than fluid. Nature 438 (7068), 578–580. doi:10.1038/nature04394
Epand, R. F., Ramamoorthy, A., and Epand, R. M. (2006). Membrane lipid composition and the interaction of pardaxin: the role of cholesterol. Protein Pept. Lett. 13 (1), 1–5. doi:10.2174/0929866510602010001
Epand, R. F., Sayer, B. G., and Epand, R. M. (2005). The tryptophan-rich region of HIV gp41 and the promotion of cholesterol-rich domains. Biochemistry 44 (14), 5525–5531. doi:10.1021/bi0500224
Fantini, J., and Barrantes, F. (2013). How cholesterol interacts with membrane proteins: an exploration of cholesterol-binding sites including CRAC, CARC, and tilted domains. Front. Physiol. 4, 31. doi:10.3389/fphys.2013.00031
Fosgerau, K., and Hoffmann, T. (2015). Peptide therapeutics: current status and future directions. Drug Discov. Today 20 (1), 122–128. doi:10.1016/j.drudis.2014.10.003
Fu, R. (2009). Efficient heteronuclear dipolar decoupling in NMR of static solid samples using phase-wiggled two-pulse phase modulation. Chem. Phys. Lett. 483 (1), 147–153. doi:10.1016/j.cplett.2009.10.039
Fu, R., Rooney, M. T., Zhang, R., and Cotten, M. L. (2021). Coordination of redox ions within a membrane-binding peptide: a tale of aromatic rings. J. Phys. Chem. Lett. 12 (18), 4392–4399. doi:10.1021/acs.jpclett.1c00636
Ghosh, S., Dellibovi-Ragheb, T. A., Kerviel, A., Pak, E., Qiu, Q., Fisher, M., et al. (2020). β-Coronaviruses use lysosomes for egress instead of the biosynthetic secretory pathway. Cell 183 (6), 1520–1535.e14. doi:10.1016/j.cell.2020.10.039
Gordon, Y. J., Romanowski, E. G., and McDermott, A. M. (2005). A review of antimicrobial peptides and their therapeutic potential as anti-infective drugs. Curr. Eye Res. 30, 505–515. doi:10.1080/02713680590968637
Greve, J. M., and Cowan, J. A. (2022). Tackling antimicrobial stewardship through synergy and antimicrobial peptides. RSC Med. Chem. 13 (5), 511–521. doi:10.1039/d2md00048b
Haney, E. F., and Hancock, R. E. W. (2013). Peptide design for antimicrobial and immunomodulatory applications. Biopolymers 6 (6), 572–583. doi:10.1002/bip.22250
Hans, M., and Madaan Hans, V. (2014). Epithelial antimicrobial peptides: guardian of the oral cavity. Int. J. Peptides 2014, 1–13. doi:10.1155/2014/370297
Hanson, J. M., Gettel, D. L., Tabaei, S. R., Jackman, J., Kim, M. C., Sasaki, D. Y., et al. (2016). Cholesterol-enriched domain formation induced by viral-encoded, membrane-active amphipathic peptide. Biophys. J. 110 (1), 176–187. doi:10.1016/j.bpj.2015.11.032
Hao, R., Li, Y., Guan, L., Lu, T., Meng, F., Wang, C., et al. (2018). Cholesterol-sensing role of phenylalanine in the interaction of human islet amyloid polypeptide with lipid bilayers. RSC Adv. 8 (71), 40581–40588. doi:10.1039/c8ra07310d
Harris, C. R., Millman, K. J., van der Walt, S. J., Gommers, R., Virtanen, P., Cournapeau, D., et al. (2020). Array programming with NumPy. Nature 585 (7825), 357–362. doi:10.1038/s41586-020-2649-2
Harrison, S. C. (2008). Viral membrane fusion. Nat. Struct. Mol. Biol. 15 (7), 690–698. doi:10.1038/nsmb.1456
Hayden, R. M., Goldberg, G. K., Ferguson, B. M., Schoeneck, M. W., Libardo, M. D., Mayeux, S. E., et al. (2015). Complementary effects of host defense peptides piscidin 1 and piscidin 3 on DNA and lipid membranes: biophysical insights into contrasting biological activities. J. Phys. Chem. B 119 (49), 15235–15246. doi:10.1021/acs.jpcb.5b09685
Heaton, N. S., and Randall, G. (2011). Multifaceted roles for lipids in viral infection. Trends Microbiol. 19 (7), 368–375. doi:10.1016/j.tim.2011.03.007
Heberle, F. A., Doktorova, M., Scott, H. L., Skinkle, A. D., Waxham, M. N., and Levental, I. (2020). Direct label-free imaging of nanodomains in biomimetic and biological membranes by cryogenic electron microscopy. Proc. Natl. Acad. Sci. U. S. A. 117 (33), 19943–19952. doi:10.1073/pnas.2002200117
Heberle, F. A., Petruzielo, R. S., Pan, J., Drazba, P., Kučerka, N., Standaert, R. F., et al. (2013). Bilayer thickness mismatch controls domain size in model membranes. J. Am. Chem. Soc. 135 (18), 6853–6859. doi:10.1021/ja3113615
Hilchie, A. L., Wuerth, K., and Hancock, R. E. (2013). Immune modulation by multifaceted cationic host defense (antimicrobial) peptides. Nat. Chem. Biol. 9 (12), 761–768. doi:10.1038/nchembio.1393
Hogue, I. B., Bosse, J. B., Hu, J. R., Thiberge, S. Y., and Enquist, L. W. (2014). Cellular mechanisms of alpha herpesvirus egress: live cell fluorescence microscopy of pseudorabies virus exocytosis. PLoS Pathog. 10 (12), e1004535. doi:10.1371/journal.ppat.1004535
Hollmann, A., Castanho, M. A., Lee, B., and Santos, N. C. (2014). Singlet oxygen effects on lipid membranes: implications for the mechanism of action of broad-spectrum viral fusion inhibitors. Biochem. J. 459 (1), 161–170. doi:10.1042/bj20131058
Hou, X., Zaks, T., Langer, R., and Dong, Y. (2021). Lipid nanoparticles for mRNA delivery. Nat. Rev. Mat. 6 (12), 1078–1094. doi:10.1038/s41578-021-00358-0
Howell, M., Wenc, A. K., Donaghy, C. M., Wasche, D. V., Abissi, I., Naing, M. D., et al. (2022). Exploring synergy and its role in antimicrobial peptide biology. Methods Enzymol. 663, 99–130. doi:10.1016/bs.mie.2021.09.017
Hu, H., Guo, N., Chen, S., Guo, X., Liu, X., Ye, S., et al. (2019). Antiviral activity of Piscidin 1 against pseudorabies virus both in vitro and in vivo. Virol. J. 16 (1), 95. doi:10.1186/s12985-019-1199-4
Huang, H. N., Chan, Y. L., Hui, C. F., Wu, J. L., Wu, C. J., and Chen, J. Y. (2015b). Use of tilapia piscidin 3 (TP3) to protect against MRSA infection in mice with skin injuries. Oncotarget 6 (15), 12955–12969. doi:10.18632/oncotarget.4102
Huang, H. N., Chan, Y. L., Wu, C. J., and Chen, J. Y. (2015a). Tilapia piscidin 4 (TP4) stimulates cell proliferation and wound closure in MRSA-infected wounds in mice. Mar. Drugs 13 (5), 2813–2833. doi:10.3390/md13052813
Ivanova, P. T., Myers, D. S., Milne, S. B., McClaren, J. L., Thomas, P. G., and Brown, H. A. (2015). Lipid composition of viral envelope of three strains of influenza virus - not all viruses are created equal. ACS Infect. Dis. 1 (9), 399–452. doi:10.1021/acsinfecdis.5b00040
Izoré, T., Tailhades, J., Hansen, M. H., Kaczmarski, J. A., Jackson, C. J., and Cryle, M. J. (2019). Drosophila melanogaster nonribosomal peptide synthetase Ebony encodes an atypical condensation domain. Proc. Natl. Acad. Sci. U. S. A. 116 (8), 2913–2918. doi:10.1073/pnas.1811194116
Jolly, C., and Sattentau, Q. J. (2007). Human immunodeficiency virus type 1 assembly, budding, and cell-cell spread in T cells take place in tetraspanin-enriched plasma membrane domains. J. Virol. 81 (15), 7873–7884. doi:10.1128/jvi.01845-06
Kampshoff, F., Willcox, M. D. P., and Dutta, D. (2019). A pilot study of the synergy between two antimicrobial peptides and two common antibiotics. Antibiotics 8 (2), 60. doi:10.3390/antibiotics8020060
Katsu, T., Kuroko, M., Morikawa, T., Sanchika, K., Yamanaka, H., Shinoda, S., et al. (1990). Interaction of wasp venom mastoparan with biomembranes. Biochimica Biophysica Acta (BBA) - Biomembr. 1027 (2), 185–190. doi:10.1016/0005-2736(90)90083-z
Ketchem, R. R., Roux, B., and Cross, T. A. (1997). High-resolution polypeptide structure in a lamellar phase lipid environment from solid state NMR derived orientational constraints. Structure 5, 1655–1669. doi:10.1016/s0969-2126(97)00312-2
Kim, S. Y., Zhang, F., Gong, W., Chen, K., Xia, K., Liu, F., et al. (2018). Copper regulates the interactions of antimicrobial piscidin peptides from fish mast cells with formyl peptide receptors and heparin. J. Biol. Chem. 293 (40), 15381–15396. doi:10.1074/jbc.ra118.001904
Kintses, B., Méhi, O., Ari, E., Számel, M., Györkei, Á., Jangir, P. K., et al. (2019). Phylogenetic barriers to horizontal transfer of antimicrobial peptide resistance genes in the human gut microbiota. Nat. Microbiol. 4 (3), 447–458. doi:10.1038/s41564-018-0313-5
Kong, Q., Six, D. A., Liu, Q., Gu, L., Wang, S., Alamuri, P., et al. (2012). Phosphate groups of lipid A are essential for Salmonella enterica serovar Typhimurium virulence and affect innate and adaptive immunity. Infect. Immun. 80 (9), 3215–3224. doi:10.1128/iai.00123-12
Krause, M. R., and Regen, S. L. (2014). The structural role of cholesterol in cell membranes: from condensed bilayers to lipid rafts. Accounts Chem. Res. 47 (12), 3512–3521. doi:10.1021/ar500260t
Kumar, A., Mahajan, M., Awasthi, B., Tandon, A., Harioudh, M. K., Shree, S., et al. (2017). Piscidin-1-analogs with double L- and D-lysine residues exhibited different conformations in lipopolysaccharide but comparable anti-endotoxin activities. Sci. Rep. 7 (1), 39925. doi:10.1038/srep39925
Kumar, K., Sebastiao, M., Arnold, A. A., Bourgault, S., Warschawski, D. E., and Marcotte, I. (2022). In situ solid-state NMR study of antimicrobial peptide interactions with erythrocyte membranes. Biophys. J. 121 (8), 1512–1524. doi:10.1016/j.bpj.2022.03.009
Kumar, P., Kizhakkedathu, J. N., and Straus, S. K. (2018). Antimicrobial peptides: diversity, mechanism of action and strategies to improve the activity and biocompatibility in vivo. Biomolecules 8 (1), 4. doi:10.3390/biom8010004
Kyle, J. E. (2021). How lipidomics can transform our understanding of virus infections. Expert Rev. Proteomics 18 (5), 329–332. doi:10.1080/14789450.2021.1929177
Lauth, X., Babon, J. J., Stannard, J. A., Singh, S., Nizet, V., Carlberg, J. M., et al. (2005). Bass hepcidin synthesis, solution structure, antimicrobial activities and synergism, and in vivo hepatic response to bacterial infections. J. Biol. Chem. 280 (10), 9272–9282. doi:10.1074/jbc.m411154200
Lauth, X., Shike, H., Burns, J. C., Westerman, M. E., Ostland, V. E., Carlberg, J. M., et al. (2002). Discovery and characterization of two isoforms of moronecidin, a novel antimicrobial peptide from hybrid striped bass. J. Biol. Chem. 277 (7), 5030–5039. doi:10.1074/jbc.m109173200
Lazarus, J. V., Wyka, K., White, T. M., Picchio, C. A., Gostin, L. O., Larson, H. J., et al. (2023). A survey of COVID-19 vaccine acceptance across 23 countries in 2022. Nat. Med. 29 (2), 366–375. doi:10.1038/s41591-022-02185-4
Lee, E., Shin, A., Jeong, K. W., Jin, B., Jnawali, H. N., Shin, S., et al. (2014). Role of phenylalanine and valine10 residues in the antimicrobial activity and cytotoxicity of piscidin-1. PLoS One 9 (12), e114453. doi:10.1371/journal.pone.0114453
Lee, S. A., Kim, Y. K., Lim, S. S., Zhu, W. L., Ko, H., Shin, S. Y., et al. (2007). Solution structure and cell selectivity of piscidin 1 and its analogues. Biochemistry 46 (12), 3653–3663. doi:10.1021/bi062233u
Lei, Z., Liu, Q., Zhu, Q., Yang, B., Khaliq, H., Sun, A., et al. (2018). Comparative pharmacokinetics and preliminary pharmacodynamics evaluation of piscidin 1 against PRV and PEDV in rats. Front. Chem. 6, 244. doi:10.3389/fchem.2018.00244
Lenard, J., and Compans, R. W. (1974). The membrane structure of lipid-containing viruses. Biochim. Biophys. Acta 344 (1), 51–94. doi:10.1016/0304-4157(74)90008-2
Li, D., Yang, Y., Tian, Z., Lv, J., Sun, F., Wang, Q., et al. (2017). Synergistic antibiotic effect of looped antimicrobial peptide CLP-19 with bactericidal and bacteriostatic agents. Oncotarget 8 (34), 55958–55966. doi:10.18632/oncotarget.18124
Li, G., and De Clercq, E. (2020). Therapeutic options for the 2019 novel coronavirus (2019-nCoV). Nat. Rev. Drug Discov. 19 (3), 149–150. doi:10.1038/d41573-020-00016-0
Li, J., Fernández-Millán, P., and Boix, E. (2020). Synergism between host defence peptides and antibiotics against bacterial infections. Curr. Top. Med. Chem. 20 (14), 1238–1263. doi:10.2174/1568026620666200303122626
Libardo, M. D. J., Bahar, A. A., Ma, B., Fu, R., McCormick, L. E., Zhao, J., et al. (2017). Nuclease activity gives an edge to host-defense peptide piscidin 3 over piscidin 1, rendering it more effective against persisters and biofilms. FEBS J. 284 (21), 3662–3683. doi:10.1111/febs.14263
Lipsitch, M., Krammer, F., Regev-Yochay, G., Lustig, Y., and Balicer, R. D. (2022). SARS-CoV-2 breakthrough infections in vaccinated individuals: measurement, causes and impact. Nat. Rev. Immunol. 22 (1), 57–65. doi:10.1038/s41577-021-00662-4
Liu, F., Greenwood, A. I., Xiong, Y., Miceli, R. T., Fu, R., Anderson, K. W., et al. (2023). Host defense peptide piscidin and yeast-derived glycolipid exhibit synergistic antimicrobial action through concerted interactions with membranes. JACS Au 3, 3345–3365. doi:10.1021/jacsau.3c00506
Liu, P., Chen, G., and Zhang, J. (2022). A review of liposomes as a drug delivery system: current status of approved products, regulatory environments, and future perspectives. Molecules 27 (4), 1372. doi:10.3390/molecules27041372
Lo, Y.-L., Lee, H.-P., and Tu, W.-C. (2015). The use of a liposomal formulation incorporating an antimicrobial peptide from Tilapia as a new adjuvant to epirubicin in human squamous cell carcinoma and pluripotent testicular embryonic carcinoma cells. Int. J. Mol. Sci. 16 (9), 22711–22734. doi:10.3390/ijms160922711
Lorent, J. H., Diaz-Rohrer, B., Lin, X., Spring, K., Gorfe, A. A., Levental, K. R., et al. (2017). Structural determinants and functional consequences of protein affinity for membrane rafts. Nat. Commun. 8 (1), 1219. doi:10.1038/s41467-017-01328-3
Lorent, J. H., Levental, K. R., Ganesan, L., Rivera-Longsworth, G., Sezgin, E., Doktorova, M., et al. (2020). Plasma membranes are asymmetric in lipid unsaturation, packing and protein shape. Nat. Chem. Biol. 16 (6), 644–652. doi:10.1038/s41589-020-0529-6
Lorizate, M., and Kräusslich, H. G. (2011). Role of lipids in virus replication. Cold Spring Harb. Perspect. Biol. 3 (10), a004820. doi:10.1101/cshperspect.a004820
Mahlapuu, M., Björn, C., and Ekblom, J. (2020). Antimicrobial peptides as therapeutic agents: opportunities and challenges. Crit. Rev. Biotechnol. 40 (7), 978–992. doi:10.1080/07388551.2020.1796576
Mansour, S. C., de la Fuente-Nunez, C., and Hancock, R. E. (2015). Peptide IDR-1018: modulating the immune system and targeting bacterial biofilms to treat antibiotic-resistant bacterial infections. J. Pept. Sci. 21 (5), 323–329. doi:10.1002/psc.2708
Mansour, S. C., Pena, O. M., and Hancock, R. E. (2014). Host defense peptides: front-line immunomodulators. Trends Immunol. 35 (9), 443–450. doi:10.1016/j.it.2014.07.004
Martín-Acebes, M., Vázquez Calvo, Á., Caridi, F., Saiz, J.-C., and Sobrino, F. (2013). Lipid involvement in viral infections: present and future perspectives for the design of antiviral strategies. Lipid Metab., 291–322. doi:10.5772/51068
Matsuzaki, K., Sugishita, K., Fujii, N., and Miyajima, K. (1995). Molecular basis for membrane selectivity of an antimicrobial peptide, magainin 2. Biochemistry 34 (10), 3423–3429. doi:10.1021/bi00010a034
McCafferty, D. G., Cudic, P., Yu, M. K., Behenna, D. C., and Kruger, R. (1999). Synergy and duality in peptide antibiotic mechanisms. Curr. Opin. Chem. Biol. 3 (6), 672–680. doi:10.1016/s1367-5931(99)00025-3
McHenry, A. J., Sciacca, M. F., Brender, J. R., and Ramamoorthy, A. (2012). Does cholesterol suppress the antimicrobial peptide induced disruption of lipid raft containing membranes? Biochim. Biophys. Acta 1818 (12), 3019–3024. doi:10.1016/j.bbamem.2012.07.021
Menousek, J., Mishra, B., Hanke, M. L., Heim, C. E., Kielian, T., and Wang, G. (2012). Database screening and in vivo efficacy of antimicrobial peptides against methicillin-resistant Staphylococcus aureus USA300. Int. J. Antimicrob. Agents 39 (5), 402–406. doi:10.1016/j.ijantimicag.2012.02.003
Miguel, A. M.-A., Ángela, V.-C., Flavia, C., Juan-Carlos, S., and Francisco, S. (2013). “Lipid involvement in viral infections: present and future perspectives for the design of antiviral strategies,” in Lipid metabolism. Editor B. Rodrigo Valenzuela (Rijeka: IntechOpen). Ch. 13.
Mihailescu, M., Sorci, M., Seckute, J., Silin, V. I., Hammer, J., Perrin, B. S., et al. (2019). Structure and function in antimicrobial piscidins: histidine position, directionality of membrane insertion, and pH-dependent permeabilization. J. Am. Chem. Soc. 141 (25), 9837–9853. doi:10.1021/jacs.9b00440
Mihailescu, M., Vaswani, R. G., Jardon-Valadez, E., Castro-Roman, F., Freites, J. A., Worcester, D. L., et al. (2011). Acyl-chain methyl distributions of liquid-ordered and -disordered membranes. Biophys. J. 100 (6), 1455–1462. doi:10.1016/j.bpj.2011.01.035
Miller, E. H., Harrison, J. S., Radoshitzky, S. R., Higgins, C. D., Chi, X., Dong, L., et al. (2011). Inhibition of Ebola virus entry by a C-peptide targeted to endosomes. J. Biol. Chem. 286 (18), 15854–15861. doi:10.1074/jbc.m110.207084
Momattin, H., Al-Ali, A. Y., and Al-Tawfiq, J. A. (2019). A systematic review of therapeutic agents for the treatment of the Middle East respiratory syndrome coronavirus (MERS-CoV). Travel Med. Infect. Dis. 30, 9–18. doi:10.1016/j.tmaid.2019.06.012
Mookherjee, N., Anderson, M. A., Haagsman, H. P., and Davidson, D. J. (2020). Antimicrobial host defence peptides: functions and clinical potential. Nat. Rev. Drug Disc 19 (5), 311–332. doi:10.1038/s41573-019-0058-8
Murzyn, K., Róg, T., and Pasenkiewicz-Gierula, M. (2005). Phosphatidylethanolamine-phosphatidylglycerol bilayer as a model of the inner bacterial membrane. Biophys. J. 88 (2), 1091–1103. doi:10.1529/biophysj.104.048835
Muttenthaler, M., King, G. F., Adams, D. J., and Alewood, P. F. (2021). Trends in peptide drug discovery. Nat. Rev. Drug Discov. 20, 309–325. doi:10.1038/s41573-020-00135-8
Nasr, G., Greige-Gerges, H., Elaissari, A., and Khreich, N. (2020). Liposomal membrane permeability assessment by fluorescence techniques: main permeabilizing agents, applications and challenges. Int. J. Pharm. 580, 119198. doi:10.1016/j.ijpharm.2020.119198
Nevzorov, A. A., Mesleh, M. F., and Opella, S. J. (2004). Structure determination of aligned samples of membrane proteins by NMR spectroscopy. Magnetic Reson. Chem. MRC. 42 (2), 162–171. doi:10.1002/mrc.1320
Nevzorov, A. A., and Opella, S. J. (2007). Selective averaging for high-resolution solid-state NMR spectroscopy of aligned samples. J. Magn. Reson 185 (1), 59–70. doi:10.1016/j.jmr.2006.09.006
Nicolson, G. L. (2014). The Fluid-Mosaic Model of Membrane Structure: still relevant to understanding the structure, function and dynamics of biological membranes after more than 40 years. Biochim. Biophys. Acta 1838 (6), 1451–1466. doi:10.1016/j.bbamem.2013.10.019
Noga, E. J., and Silphaduang, U. (2003). Piscidia: a novel family of peptide antibiotics from fish. Drug News Perspect. 16 (2), 87–92. doi:10.1358/dnp.2003.16.2.829325
Oludiran, A., Courson, D. S., Stuart, M. D., Radwan, A. R., Poutsma, J. C., Cotten, M. L., et al. (2019). How oxygen availability affects the antimicrobial efficacy of host defense peptides: lessons learned from studying the copper-binding peptides piscidins 1 and 3. Int. J. Mol. Sci. 20 (21), 5289. doi:10.3390/ijms20215289
Omardien, S., Drijfhout, J. W., Vaz, F. M., Wenzel, M., Hamoen, L. W., Zaat, S. A. J., et al. (2018). Bactericidal activity of amphipathic cationic antimicrobial peptides involves altering the membrane fluidity when interacting with the phospholipid bilayer. Biochimica Biophysica Acta (BBA) - Biomembr. 1860 (11), 2404–2415. doi:10.1016/j.bbamem.2018.06.004
Opella, S., Ma, C., and Marassi, F. (2001). Nuclear magnetic resonance of membrane-associated peptides and proteins. Methods Enzymol. 339, 285–313. doi:10.1016/s0076-6879(01)39319-9
Opella, S. J., and Marassi, F. M. (2004). Structure determination of membrane proteins by NMR spectroscopy. Chem. Rev. 104, 3587–3606. doi:10.1021/cr0304121
Opella, S. J., Nevzorov, A., Meslesh, M. F., and Marassi, F. M. (2002). Structure determination of membrane proteins by NMR spectroscopy. Biochem. Cell Biol. 80, 597–604. doi:10.1139/o02-154
Ouellet, M., Bernard, G., Voyer, N., and Auger, M. (2006). Insights on the interactions of synthetic amphipathic peptides with model membranes as revealed by 31P and 2H solid-state NMR and infrared spectroscopies. Biophys. J. 90 (11), 4071–4084. doi:10.1529/biophysj.105.077339
Paredes, S. D., Kim, S., Rooney, M. T., Greenwood, A. I., Hristova, K., and Cotten, M. L. (2020). Enhancing the membrane activity of Piscidin 1 through peptide metallation and the presence of oxidized lipid species: implications for the unification of host defense mechanisms at lipid membranes. Biochim. Biophys. Acta Biomembr. 1862 (7), 183236. doi:10.1016/j.bbamem.2020.183236
Park, J. E., and Gallagher, T. (2017). Lipidation increases antiviral activities of coronavirus fusion-inhibiting peptides. Virology 511, 9–18. doi:10.1016/j.virol.2017.07.033
Paterson, D. J., Tassieri, M., Reboud, J., Wilson, R., and Cooper, J. M. (2017). Lipid topology and electrostatic interactions underpin lytic activity of linear cationic antimicrobial peptides in membranes. Proc. Natl. Acad. Sci. U. S. A. 114 (40), E8324–E32. doi:10.1073/pnas.1704489114
Pattni, B. S., Chupin, V. V., and Torchilin, V. P. (2015). New developments in liposomal drug delivery. Chem. Rev. 115 (19), 10938–10966. doi:10.1021/acs.chemrev.5b00046
Pérez-Cobas, A. E., Moya, A., Gosalbes, M. J., and Latorre, A. (2015). Colonization resistance of the gut microbiota against Clostridium difficile. Antibiot. (Basel) 4 (3), 337–357. doi:10.3390/antibiotics4030337
Perrin, B. S., Sodt, A. J., Cotten, M. L., and Pastor, R. W. (2015). The curvature induction of surface-bound antimicrobial peptides piscidin 1 and piscidin 3 varies with lipid chain length. J. Membr. Biol. 248 (3), 455–467. doi:10.1007/s00232-014-9733-1
Perrin, B. S., Tian, Y., Fu, R., Grant, C. V., Chekmenev, E. Y., Wieczorek, W. E., et al. (2014). High-resolution structures and orientations of antimicrobial peptides piscidin 1 and piscidin 3 in fluid bilayers reveal tilting, kinking, and bilayer immersion. J. Am. Chem. Soc. 136 (9), 3491–3504. doi:10.1021/ja411119m
Perumal, R., Shunmugam, L., Naidoo, K., Abdool Karim, S. S., Wilkins, D., Garzino-Demo, A., et al. (2023). Long COVID: a review and proposed visualization of the complexity of long COVID. Front. Immunol. 14, 1117464. doi:10.3389/fimmu.2023.1117464
Peschel, A., and Sahl, H. G. (2006). The co-evolution of host cationic antimicrobial peptides and microbial resistance. Nat. Rev. Microbiol. 4 (7), 529–536. doi:10.1038/nrmicro1441
Pike, L. J. (2003). Lipid rafts: bringing order to chaos. J. Lipid Res. 44 (4), 655–667. doi:10.1194/jlr.r200021-jlr200
Plemper, R. K. (2011). Cell entry of enveloped viruses. Curr. Opin. Virol. 1 (2), 92–100. doi:10.1016/j.coviro.2011.06.002
Pollock, S., Nichita, N. B., Böhmer, A., Radulescu, C., Dwek, R. A., and Zitzmann, N. (2010). Polyunsaturated liposomes are antiviral against hepatitis B and C viruses and HIV by decreasing cholesterol levels in infected cells. Proc. Natl. Acad. Sci. U. S. A. 107 (40), 17176–17181. doi:10.1073/pnas.1009445107
Polozov, I. V., Bezrukov, L., Gawrisch, K., and Zimmerberg, J. (2008). Progressive ordering with decreasing temperature of the phospholipids of influenza virus. Nat. Chem. Biol. 4 (4), 248–255. doi:10.1038/nchembio.77
Porotto, M., Yokoyama, C. C., Palermo, L. M., Mungall, B., Aljofan, M., Cortese, R., et al. (2010). Viral entry inhibitors targeted to the membrane site of action. J. Virol. 84 (13), 6760–6768. doi:10.1128/jvi.00135-10
Qian, S., and Heller, W. T. (2015). Melittin-induced cholesterol reorganization in lipid bilayer membranes. Biochimica Biophysica Acta (BBA) - Biomembr. 1848 (10), 2253–2260. doi:10.1016/j.bbamem.2015.06.012
Rahman, M. M., Masum, M. H. U., Wajed, S., and Talukder, A. (2022). A comprehensive review on COVID-19 vaccines: development, effectiveness, adverse effects, distribution and challenges. Virusdisease 33 (1), 1–22. doi:10.1007/s13337-022-00755-1
Rai, R. K., De Angelis, A., Greenwood, A., Opella, S. J., and Cotten, M. L. (2019). Metal-ion binding to host defense peptide piscidin 3 observed in phospholipid bilayers by magic angle spinning solid-state NMR. ChemPhysChem 20 (2), 295–301. doi:10.1002/cphc.201800855
Rea, M. C., Sit, C. S., Clayton, E., Connor, P. M., Whittal, R. M., Zheng, J., et al. (2010). Thuricin CD, a posttranslationally modified bacteriocin with a narrow spectrum of activity against Clostridium difficile. Proc. Natl. Acad. Sci. U. S. A. 107 (20), 9352–9357. doi:10.1073/pnas.0913554107
Risselada, H. J. (2017). Membrane fusion stalks and lipid rafts: a love-hate relationship. Biophys. J. 112 (12), 2475–2478. doi:10.1016/j.bpj.2017.04.031
Ruden, S., Rieder, A., Chis Ster, I., Schwartz, T., Mikut, R., and Hilpert, K. (2019). Synergy pattern of short cationic antimicrobial peptides against multidrug-resistant Pseudomonas aeruginosa. Front. Microbiol. 10, 2740. doi:10.3389/fmicb.2019.02740
Sallam, M. (2021). COVID-19 vaccine hesitancy worldwide: a concise systematic review of vaccine acceptance rates. Vaccines (Basel) 9 (2), 160. doi:10.3390/vaccines9020160
Sani, M. A., Gagne, E., Gehman, J. D., Whitwell, T. C., and Separovic, F. (2014). Dye-release assay for investigation of antimicrobial peptide activity in a competitive lipid environment. Eur. Biophys. J. 43 (8-9), 445–450. doi:10.1007/s00249-014-0970-0
Sani, M. A., Weber, D. K., Delaglio, F., Separovic, F., and Gehman, J. D. (2013). A practical implementation of de-Pake-ing via weighted Fourier transformation. PeerJ 1, e30. doi:10.7717/peerj.30
Saud, Z., Tyrrell, V. J., Zaragkoulias, A., Protty, M. B., Statkute, E., Rubina, A., et al. (2022). The SARS-CoV2 envelope differs from host cells, exposes procoagulant lipids, and is disrupted in vivo by oral rinses. J. Lipid Res. 63 (6), 100208. doi:10.1016/j.jlr.2022.100208
Schoeman, D., and Fielding, B. C. (2019). Coronavirus envelope protein: current knowledge. Virol. J. 16 (1), 69. doi:10.1186/s12985-019-1182-0
Schoggins, J. W., and Randall, G. (2013). Lipids in innate antiviral defense. Cell Host Microbe 14 (4), 379–385. doi:10.1016/j.chom.2013.09.010
Schön, P., García-Sáez, A. J., Malovrh, P., Bacia, K., Anderluh, G., and Schwille, P. (2008). Equinatoxin II permeabilizing activity depends on the presence of sphingomyelin and lipid phase coexistence. Biophys. J. 95 (2), 691–698. doi:10.1529/biophysj.108.129981
Schütz, D., Ruiz-Blanco, Y. B., Münch, J., Kirchhoff, F., Sanchez-Garcia, E., and Müller, J. A. (2020). Peptide and peptide-based inhibitors of SARS-CoV-2 entry. Adv. Drug Deliv. Rev. 167, 47–65. doi:10.1016/j.addr.2020.11.007
Sciacca, M. F., Lolicato, F., Di Mauro, G., Milardi, D., D'Urso, L., Satriano, C., et al. (2016). The role of cholesterol in driving IAPP-membrane interactions. Biophys. J. 111 (1), 140–151. doi:10.1016/j.bpj.2016.05.050
Seabold, S., and Perktold, J. (2010). “Statsmodels: econometric and statistical modeling with Python,” in Proceedings of the 9th Python in Science Conference, 2010.
Sheard, D. E., O’Brien-Simpson, N. M., Wade, J. D., and Separovic, F. (2019). Combating bacterial resistance by combination of antibiotics with antimicrobial peptides. Pure Appl. Chem. 91 (2), 199–209. doi:10.1515/pac-2018-0707
Shekunov, E. V., Zlodeeva, P. D., Efimova, S. S., Muryleva, A. A., Zarubaev, V. V., Slita, A. V., et al. (2023). Cyclic lipopeptides as membrane fusion inhibitors against SARS-CoV-2: new tricks for old dogs. Antivir. Res. 212, 105575. doi:10.1016/j.antiviral.2023.105575
Sheynis, T., Sykora, J., Benda, A., Kolusheva, S., Hof, M., and Jelinek, R. (2003). Bilayer localization of membrane-active peptides studied in biomimetic vesicles by visible and fluorescence spectroscopies. Eur. J. Biochem. 270, 4478–4487. doi:10.1046/j.1432-1033.2003.03840.x
Silphaduang, U., and Noga, E. J. (2001). Peptide antibiotics in mast cells of fish. Nature 414, 268–269. doi:10.1038/35104690
Silveira, GGOS, Torres, M. D. T., Ribeiro, C. F. A., Meneguetti, B. T., Carvalho, C. M. E., de la Fuente-Nunez, C., et al. (2021). Antibiofilm peptides: relevant preclinical animal infection models and translational potential. ACS Pharmacol. Transl. Sci. 4 (1), 55–73. doi:10.1021/acsptsci.0c00191
Simons, K., and Sampaio, J. L. (2011). Membrane organization and lipid rafts. Cold Spring Harb. Perspect. Biol. 3 (10), a004697. doi:10.1101/cshperspect.a004697
Suloway, C., Pulokas, J., Fellmann, D., Cheng, A., Guerra, F., Quispe, J., et al. (2005). Automated molecular microscopy: the new Leginon system. J. Struct. Biol. 151 (1), 41–60. doi:10.1016/j.jsb.2005.03.010
Thundimadathil, J. (2012). Cancer treatment using peptides: current therapies and future prospects. J. Amino Acids 2012, 1–13. doi:10.1155/2012/967347
Torres, M. D. T., Cao, J., Franco, O. L., Lu, T. K., and de la Fuente-Nunez, C. (2021). Synthetic biology and computer-based frameworks for antimicrobial peptide discovery. ACS Nano 15 (2), 2143–2164. doi:10.1021/acsnano.0c09509
Tulenko, T. N., Lapotofsky, D., Mason, R. P., and Cox, R. H. (2007). “Cholesterol and cell plasma membranes,” in Encyclopedia of gerontology. Editor E. B. James Second Edition (New York: Elsevier), 275–283.
Uhlig, T., Kyprianou, T., Martinelli, F. G., Oppici, C. A., Heiligers, D., Hills, D., et al. (2014). The emergence of peptides in the pharmaceutical business: from exploration to exploitation. EuPA Open Proteomics 4, 58–69. doi:10.1016/j.euprot.2014.05.003
Utterström, J., Barriga, H. M. G., Holme, M. N., Selegård, R., Stevens, M. M., and Aili, D. (2022). Peptide-folding triggered phase separation and lipid membrane destabilization in cholesterol-rich lipid vesicles. Bioconjug Chem. 33 (4), 736–746. doi:10.1021/acs.bioconjchem.2c00115
VanCompernolle, S., Smith, P. B., Bowie, J. H., Tyler, M. J., Unutmaz, D., and Rollins-Smith, L. A. (2015). Inhibition of HIV infection by caerin 1 antimicrobial peptides. Peptides 71, 296–303. doi:10.1016/j.peptides.2015.05.004
van Meer, G., Voelker, D. R., and Feigenson, G. W. (2008). Membrane lipids: where they are and how they behave. Nat. Rev. Mol. Cell Biol. 9 (2), 112–124. doi:10.1038/nrm2330
Varanko, A., Saha, S., and Chilkoti, A. (2020). Recent trends in protein and peptide-based biomaterials for advanced drug delivery. Adv. Drug Deliv. Rev. 156, 133–187. doi:10.1016/j.addr.2020.08.008
Veatch, S. L., Polozov, I. V., Gawrisch, K., and Keller, S. L. (2004). Liquid domains in vesicles investigated by NMR and fluorescence microscopy. Biophys. J. 86 (5), 2910–2922. doi:10.1016/s0006-3495(04)74342-8
Vereb, G., Szöllosi, J., Matkó, J., Nagy, P., Farkas, T., Vigh, L., et al. (2003). Dynamic, yet structured: the cell membrane three decades after the Singer-Nicolson model. Proc. Natl. Acad. Sci. U. S. A. 100 (14), 8053–8058. doi:10.1073/pnas.1332550100
Vigant, F., Lee, J., Hollmann, A., Tanner, L. B., Akyol Ataman, Z., Yun, T., et al. (2013). A mechanistic paradigm for broad-spectrum antivirals that target virus-cell fusion. PLoS Pathog. 9 (4), e1003297. doi:10.1371/journal.ppat.1003297
Vigant, F., Santos, N. C., and Lee, B. (2015). Broad-spectrum antivirals against viral fusion. Nat. Rev. Microbiol. 13 (7), 426–437. doi:10.1038/nrmicro3475
Wang, C., Zhao, L., Xia, S., Zhang, T., Cao, R., Liang, G., et al. (2018). De novo design of α-helical lipopeptides targeting viral fusion proteins: a promising strategy for relatively broad-spectrum antiviral drug discovery. J. Med. Chem. 61 (19), 8734–8745. doi:10.1021/acs.jmedchem.8b00890
Wang, F., Ramakrishna, S. K., Sun, P., and Fu, R. (2021). Triple-pulse excitation: an efficient way for suppressing background signals and eliminating radio-frequency acoustic ringing in direct polarization NMR experiments. J. Magn. Reson 332, 107067. doi:10.1016/j.jmr.2021.107067
Wang, G. (2013). Database-guided discovery of potent peptides to combat HIV-1 or superbugs. Pharmaceuticals 6 (6), 728–758. doi:10.3390/ph6060728
Wang, G., Watson, K. M., Peterkofsky, A., Buckheit, R. W., et al. (2010). Identification of novel human immunodeficiency virus type 1-inhibitory peptides based on the antimicrobial peptide database. Antimicrob. Agents Chemother. 54 (3), 1343–1346. doi:10.1128/aac.01448-09
Wenzel, M., Chiriac, A. I., Otto, A., Zweytick, D., May, C., Schumacher, C., et al. (2014). Small cationic antimicrobial peptides delocalize peripheral membrane proteins. Proc. Natl. Acad. Sci. U. S. A. 111 (14), E1409–E1418. doi:10.1073/pnas.1319900111
Wessman, P., Strömstedt, A. A., Malmsten, M., and Edwards, K. (2008). Melittin-lipid bilayer interactions and the role of cholesterol. Biophys. J. 95 (9), 4324–4336. doi:10.1529/biophysj.108.130559
Wiehe, A., O'Brien, J. M., and Senge, M. O. (2019). Trends and targets in antiviral phototherapy. Photochem Photobiol. Sci. 18 (11), 2565–2612. doi:10.1039/c9pp00211a
Wolf, M. C., Freiberg, A. N., Zhang, T., Akyol-Ataman, Z., Grock, A., Hong, P. W., et al. (2010). A broad-spectrum antiviral targeting entry of enveloped viruses. Proc. Natl. Acad. Sci. U. S. A. 107 (7), 3157–3162. doi:10.1073/pnas.0909587107
Wu, X., Li, Z., Li, X., Tian, Y., Fan, Y., Yu, C., et al. (2017). Synergistic effects of antimicrobial peptide DP7 combined with antibiotics against multidrug-resistant bacteria. Drug Des. Devel Ther. 11, 939–946. doi:10.2147/dddt.s107195
Xia, Y., Cebrián, R., Xu, C., Jong, A., Wu, W., and Kuipers, O. P. (2021). Elucidating the mechanism by which synthetic helper peptides sensitize Pseudomonas aeruginosa to multiple antibiotics. PLoS Pathog. 17 (9), e1009909. doi:10.1371/journal.ppat.1009909
Yan, B., Chu, H., Yang, D., Sze, K. H., Lai, P. M., Yuan, S., et al. (2019). Characterization of the lipidomic profile of human coronavirus-infected cells: implications for lipid metabolism remodeling upon coronavirus replication. Viruses 11 (1), 73. doi:10.3390/v11010073
Yang, S. T., Kreutzberger, A. J. B., Lee, J., Kiessling, V., and Tamm, L. K. (2016). The role of cholesterol in membrane fusion. Chem. Phys. Lipids 199, 136–143. doi:10.1016/j.chemphyslip.2016.05.003
Yount, N. Y., and Yeaman, M. R. (2012). Emerging themes and therapeutic prospects for anti-infective peptides. Annu. Rev. Pharmacol. Toxicol. 52, 337–360. doi:10.1146/annurev-pharmtox-010611-134535
Zambrowicz, A., Timmer, M., Polanowski, A., Lubec, G., and Trziszka, T. (2013). Manufacturing of peptides exhibiting biological activity. Amino Acids 44 (2), 315–320. doi:10.1007/s00726-012-1379-7
Zeng, L., Wang, M. D., Ming, S. L., Li, G. L., Yu, P. W., Qi, Y. L., et al. (2020). An effective inactivant based on singlet oxygen-mediated lipid oxidation implicates a new paradigm for broad-spectrum antivirals. Redox Biol. 36, 101601. doi:10.1016/j.redox.2020.101601
Zhang, R., Jiang, X., Qiao, J., Wang, Z., Tong, A., Yang, J., et al. (2021). Antimicrobial peptide DP7 with potential activity against SARS coronavirus infections. Signal Transduct. Target. Ther. 6 (1), 140. doi:10.1038/s41392-021-00551-1
Zhao, H., Zhou, J., Zhang, K., Chu, H., Liu, D., Poon, V. K., et al. (2016). A novel peptide with potent and broad-spectrum antiviral activities against multiple respiratory viruses. Sci. Rep. 6, 22008. doi:10.1038/srep22008
Zheng, C., Shao, W., Chen, X., Zhang, B., Wang, G., and Zhang, W. (2022). Real-world effectiveness of COVID-19 vaccines: a literature review and meta-analysis. Int. J. Infect. Dis. 114, 252–260. doi:10.1016/j.ijid.2021.11.009
Zheng, S. Q., Palovcak, E., Armache, J. P., Verba, K. A., Cheng, Y., and Agard, D. A. (2017). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14 (4), 331–332. doi:10.1038/nmeth.4193
Zhou, X., and Xu, J. (2012). Free cholesterol induces higher β-sheet content in aβ peptide oligomers by aromatic interaction with Phe19. PLoS One 7 (9), e46245. doi:10.1371/journal.pone.0046245
Zhu, Y., Hao, W., Wang, X., Ouyang, J., Deng, X., Yu, H., et al. (2022). Antimicrobial peptides, conventional antibiotics, and their synergistic utility for the treatment of drug-resistant infections. Med. Res. Rev. 42 (4), 1377–1422. doi:10.1002/med.21879
Glossary

Keywords: antiviral host defense peptide, cholesterol, liquid ordered and disordered phases, lipid mixing, membrane disruption, membrane heterogeneity, membrane thinning, viral envelopes
Citation: Bepler T, Barrera MD, Rooney MT, Xiong Y, Kuang H, Goodell E, Goodwin MJ, Harbron E, Fu R, Mihailescu M, Narayanan A and Cotten ML (2024) Antiviral activity of the host defense peptide piscidin 1: investigating a membrane-mediated mode of action. Front. Chem. 12:1379192. doi: 10.3389/fchem.2024.1379192
Received: 30 January 2024; Accepted: 08 May 2024;
Published: 26 June 2024.
Edited by:
Lai Yue Angeline Chan, The University of Queensland, AustraliaReviewed by:
Karen Garcia Nogueira Oshiro, University of Brasilia, BrazilSunithi Gunasekera, Uppsala University, Sweden
Copyright © 2024 Bepler, Barrera, Rooney, Xiong, Kuang, Goodell, Goodwin, Harbron, Fu, Mihailescu, Narayanan and Cotten. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Myriam L. Cotten, Y290dGVubXlAb3JlZ29uc3RhdGUuZWR1; Aarthi Narayanan, YW5hcmF5YTFAZ211LmVkdQ==