 João Augusto Pereira da Rocha1,2,3,4*
João Augusto Pereira da Rocha1,2,3,4* Renato Araújo da Costa3,5
Renato Araújo da Costa3,5 Andreia do Socorro Silva da Costa3Elaine Cristina Medeiros da Rocha1,2,3,4Anderson José Bahia Gomes5Alencar Kolinski Machado6
Andreia do Socorro Silva da Costa3Elaine Cristina Medeiros da Rocha1,2,3,4Anderson José Bahia Gomes5Alencar Kolinski Machado6 Solange Binotto Fagan6Davi do Socorro Barros Brasil3,4
Solange Binotto Fagan6Davi do Socorro Barros Brasil3,4 Anderson Henrique Lima e Lima2,4
Anderson Henrique Lima e Lima2,4- 1Laboratory of Modeling and Computational Chemistry, Federal Institute of Education, Science and Technology of Paraná (IFPA) Campus Bragança, Bragança, Brazil
- 2Laboratório de Planejamento e Desenvolvimento de Fármacos, Instituto de Ciências Exatas e Naturais, Universidade Federal do Pará, Belém, Brazil
- 3Laboratory of Biosolutions and Bioplastics of the Amazon, Graduate Program in Science and Environment, Institute of Exact and Natural Sciences, Federal University of Pará (UFPA), Belém, Brazil
- 4Graduate Program in Chemistry, Institute of Exact and Natural Sciences, Federal University of Pará, Belém, Brazil
- 5Laboratory of Molecular Biology, Evolution and Microbiology, Federal Institute of Education Science and Technology of Paraná (IFPA) Campus Abaetetuba, Abaetetuba, Brazil
- 6Graduate Program in Nanosciences, Franciscana University, Santa Maria, Brazil
SARS-CoV-2 (Severe Acute Respiratory Syndrome Coronavirus 2) is the etiological agent responsible for the global outbreak of COVID-19 (Coronavirus Disease 2019). The main protease of SARS-CoV-2, Mpro, is a key enzyme that plays a vital role in mediating viral replication and transcription. In this study, a comprehensive computational approach was employed to investigate the binding affinity, selectivity, and stability of natural product candidates as potential new antivirals acting on the viral polyprotein processing mediated by SARS-CoV-2 Mpro. A library of 288 flavonoids extracted from Brazilian biodiversity was screened to select potential Mpro inhibitors. An initial filter based on Lipinski’s rule of five was applied, and 204 compounds that did not violate any of the Lipinski rules were selected. The compounds were then docked into the active site of Mpro using the GOLD program, and the poses were subsequently re-scored using MM-GBSA (Molecular Mechanics Generalized Born Surface Area) binding free energy calculations performed by AmberTools23. The top five flavonoids with the best MM-GBSA binding free energy values were selected for analysis of their interactions with the active site residues of the protein. Next, we conducted a toxicity and drug-likeness analysis, and non-toxic compounds were subjected to molecular dynamics simulation and free energy calculation using the MM-PBSA (Molecular Mechanics Poisson-Boltzmann Surface Area) method. It was observed that the five selected flavonoids had lower MM-GBSA binding free energy with Mpro than the co-crystal ligand. Furthermore, these compounds also formed hydrogen bonds with two important residues, Cys145 and Glu166, in the active site of Mpro. Two compounds that passed the drug-likeness filter showed stable conformations during the molecular dynamics simulations. Among these, NuBBE_867 exhibited the best MM-PBSA binding free energy value compared to the crystallographic inhibitor. Therefore, this study suggests that NuBBE_867 could be a potential inhibitor against the main protease of SARS-CoV-2 and may be further examined to confirm our results.
Introduction
In late 2019, a previously unknown zoonotic disease called Coronavirus Disease 2019 (COVID-19) surfaced as a viral respiratory infection in Wuhan, China. Coronavirus 2 (SARS-CoV-2), rapidly spreading worldwide. In March 2020, the World Health Organization (WHO) declared this infectious disease a pandemic (Chauhan, 2020). As of 25 October 2023, there have been 771,549,718 confirmed cases of COVID-19, resulting in 6,974,473 deaths worldwide, according to the WHO (https://covid19.who.int, accessed on 27 October 2023). Although COVID-19 vaccines have been developed remarkably quickly, it remains uncertain how well they will perform in the long term and whether they will be effective against potential future variants of SARS-CoV-2 and other coronaviruses (Heaton, 2020; Jackson et al., 2020; Yu et al., 2020; Panda et al., 2023). With commercially available antiviral treatments proving to be extremely ineffective (Liu et al., 2022; Purohit et al., 2023), the need for the search for potentially more effective antivirals arises.
The primary goal of antiviral drug research for SARS-CoV-2 is to target the viral entry mechanism or viral genome replication (Asselah et al., 2021). SARS-CoV-2 possesses an RNA genome encoding two polyproteins, pp1a and pp1ab, four structural proteins, and various accessory proteins (Qiao et al., 2021). The main protease of SARS-CoV-2 (Mpro, also known as 3CL protease) cleaves the polyproteins at 11 different sites to release mature non-structural proteins (nsps), which play crucial roles in viral RNA replication and immune evasion (Jin et al., 2020; Owen et al., 2021; Wu et al., 2022). Inhibiting Mpro would essentially disrupt viral proliferation throughout the human body (Zhang et al., 2020; Singh et al., 2022). Considering the essential role of Mpro in maturing nsps and SARS-CoV-2 replication, combined with the absence of known human proteases with similar cleavage specificity, makes it an attractive target for the development of antivirals without adverse effects (Qiao et al., 2021; Hu et al., 2022; Liu et al., 2022; Huang et al., 2023). Several SARS-CoV-2 Mpro inhibitors have undergone clinical trials, and nirmatrelvir and ensitrelvir have received regulatory approval (Owen et al., 2021; Mukae et al., 2022; Mukae et al., 2023). However, mutations conferring resistance to nirmatrelvir and ensitrelvir have already been identified in SARS-CoV-2 Mpro (Heilmann et al., 2022), highlighting the importance of seeking new Mpro inhibitors.
Due to the urgent need to find an effective therapy to combat SARS-CoV-2, several studies have emphasized the importance of Natural Products (NPs) as potential antiviral agents (Antonio et al., 2020; Islam et al., 2020; Mani et al., 2020; Wang and Yang, 2020; Rubio-Martínez et al., 2021). Furthermore, some research has focused on the use of NPs as inhibitors of the main protease of SARS-CoV-2 (Mpro) (Aanouz et al., 2021; Ray et al., 2022; Ahamed and Arif, 2023; Purohit et al., 2023).
Among readily accessible Natural Products (NPs), flavonoids stand out, which are naturally occurring polyphenolic compounds found in fruits and vegetables, and have demonstrated significant antiviral activities (Zakaryan et al., 2017; Cataneo et al., 2019; Wang and Yang, 2020). These compounds play a relevant role in enhancing the host’s defense system against viral infections, reducing infection, and inhibiting cytokine generation (Jo et al., 2020; Solnier and Fladerer, 2021; Alzaabi et al., 2022). The potential of flavonoids as antiviral agents against SARS-CoV-2 is reported in the literature (Cherrak et al., 2020; Wang and Yang, 2020; Gogoi et al., 2021; Liskova et al., 2021; Owis et al., 2021; Alzaabi et al., 2022; Liu et al., 2022; Chatterjee et al., 2023). Jo and co-workers (Jo et al., 2020) identified a series of flavonoids as effective inhibitors of SARS-CoV-2 replication in in vitro experiments. Furthermore, Mohapatra and colleagues discussed the specific importance of the Cys145 residue in likely covalent interactions with natural products (Mohapatra et al., 2023). Recently, in a SARS-CoV-2 replication assay and molecular dynamics studies, flavonoids were identified as inhibitors of Mpro (Lin et al., 2023). Recent reviews highlight flavonoids with confirmed in vitro anti-SARS-CoV-2 Mpro activity (Solnier and Fladerer, 2021; Antonopoulou et al., 2022). Therefore, exploring flavonoids as potential inhibitors of SARS-CoV-2 Mpro emerges as a promising strategy in the search for alternative therapies based on natural products.
The present study aimed to employ computational approaches to screen flavonoids derived from Brazilian biodiversity with the purpose of identifying potential inhibitors of the main protease (Mpro) of SARS-CoV-2. The results of this study will guide future efforts in identifying and developing Mpro inhibitors based on natural flavonoids. Additional experimental studies will be necessary to validate computational predictions and assess the activity of these compounds in cellular models.
Materials and methods
Flavonoid-based virtual screening for SARS-CoV-2 protease inhibitors
Initially, a library containing 288 flavonoids obtained from the Natural Products database extracted from Brazilian biodiversity (NuBBEDB) was used in the virtual screening (Valli et al., 2013; Pilon et al., 2017). Subsequently, all compound structures were filtered based on the Lipinski “Rule of 5” (RO5) physicochemical filter (Lipinski et al., 2001) to obtain drug-like molecules. Compounds that did not violate Lipinski’s rule were considered for the virtual screening study.
The 3D crystal structure of SARS-CoV-2 Mpro with PDB ID 6M2N complexed with the crystallographic inhibitor Baicalein (3WL) was used for virtual screening (Su et al., 2020). The Chimera software was used for protein preparation, removing the crystallographic ligand 3WL and water molecules. Hydrogen atoms were added, and protonation states at pH 7.0 were assigned to the protein using the H++ server (Gordon et al., 2005).
The GOLD program (Verdonk et al., 2003) was used to perform molecular docking simulations. First, the crystallographic ligand 3WL was re-docked, and the RMSD between the crystallographic ligand and the generated pose was calculated to validate the molecular docking protocol. The docking spatial search docking sphere was defined using the coordinates of the 3WL ligand in the protein’s crystallographic structure with a radius of 10 Å using the GoldScore scoring function (Verdonk et al., 2003). After the validation step, natural products from the flavonoid classes were subjected to molecular docking following the same protocol as validation. Subsequently, the poses were rescored according to the workflow proposed by Harutyun Sahakyan, 2021; Sahakyan, 2021), which uses the iPBSA code to reclassify the poses generated in molecular docking through the calculation of binding free energy using the MM-PB (GB) SA methods.
For the calculation of MM-GBSA binding free energy, protein and ligand parameterization was performed first, then the complexes were minimized, and finally, MM-GBSA calculations were performed using AmberTools23 (Case et al., 2023). Ligand charges were calculated using the AM1-BCC method ((Jakalian et al., 2002). The ff14SB force field was used to describe protein parameters, and the General Amber Force Field (GAFF) for ligands (Wang et al., 2004; Maier et al., 2015). Required input files (coordinates and topologies) with mbondi3 radii were prepared using tLEaP. Minimization was carried out in generalized implicit solvent models for each protein-ligand complex using the sander mechanism (Salomon-Ferrer et al., 2013). For each complex, the maximum number of minimization cycles was set at 1,000, with 500 steps using steepest descent and another 500 steps using the conjugate gradient algorithm. The final snapshot of the minimized protein-ligand complex was used to rescore the poses through MM-GBSA binding free energy calculation.
Molecular dynamics simulation
The compounds that passed the toxicity analysis were then subjected to a molecular dynamics (MD) simulation study to analyze the stability and potential conformational changes of the target protein complexed with flavonoids using Amber22 (Costa et al., 2023).
The structures of the protein and ligands were treated using the Amber ff14SB and the general Amber force field (GAFF), respectively. For MD, the systems were solvated in the tLeap module in a truncated octahedral box with a 12Å edge and filled with TIP3P water (Price and Brooks, 2004). Counter ions were added as needed to neutralize the systems. Subsequently, the systems were minimized in four steps, gradually removing position restraints on atom groups. The minimization process starts with the steepest descent algorithm and switches to the conjugate gradient. Next, slow heating for 500 ps to 300 K at constant volume with position restraints on the solute and unrestricted equilibration for 500 ps at constant pressure were performed. Using a collision frequency of 2 cm−1 and linked to a Langevin thermostat, the temperature was maintained at 300 K. The SHAKE algorithm (Ryckaert et al., 1977) and Particle Mesh Ewald (PME) (Darden et al., 1993) were used to limit bond lengths involving hydrogen atoms, and a 10 Å cutoff was applied for non-bonded interactions. Finally, a 100 ns production run was conducted without positional restraints at a constant temperature of 300 K.
Binding free energy calculations
To describe the binding affinity of the natural products and the crystallographic inhibitor with the Mpro protein, we performed binding free energy calculations using the MM/PBSA method (Genheden and Ryde, 2015) available in the AmberTools23 package (Case et al., 2023), while the mathematical description was published in previous studies (Costa et al., 2022). The last 10 ns of the MD simulation trajectories were used to calculate the binding free energy and the decomposition of the binding free energy.
Toxicity analysis
The five flavonoids were analyzed for different types of toxicities, including tumorigenic, mutagenic, reproductive effectiveness, irritant, and drug-likeness using ORISIS Data Warrior 5.2.1 (Sander et al., 2015).
Results and discussion
Virtual screening and molecular docking
Although some natural products (NPs) have bioactivity or drug-like activity, many NPs have low solubility or chemical instability, making their transformation into parenteral drugs challenging. Additionally, the complex structures of many NPs result in high molecular weights, which are likely to have negative effects on intestinal absorption (Li et al., 2019).
In this study, the bioavailability of the 288 flavonoids was evaluated based on their physicochemical properties using the Lipinski’s rule. This step was carried out within the NuBBEDB. It was found that 204 flavonoids did not violate any of the Lipinski’s rules and were therefore selected for the virtual screening phase.
Before the virtual screening of the flavonoids, the molecular docking protocol was validated by redocking the co-crystallized ligand 3WL into the active site of Mpro. It was found that the redocked conformation of the ligand perfectly overlapped with the co-crystallized ligand, with an RMSD value of 0.636 Å (Supplementary Figure S2). This result suggests that the GOLD program exhibits satisfactory accuracy in repositioning the 3WL ligand within the active site of Mpro. The docking protocol is considered satisfactory when the RMSD value between the docking pose and the crystallographic ligand pose is less than 2.0 Å (Wang et al., 2016; Costa et al., 2023). Figure 1 illustrates the overlapping alignment of the redocked pose and the crystallographic ligand.
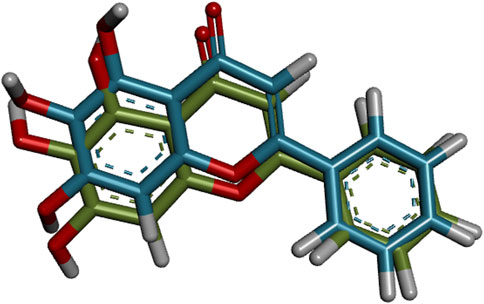
FIGURE 1. Validation of molecular docking protocols using the GOLD program for the crystal structure of the COVID-19 main protease (Mpro) in complex with 3WL inhibitor. Blue is the co-crystal ligand and green is the docking pose.
The validated docking protocol was subsequently used for virtual screening based on molecular docking of 204 flavonoids extracted from the Brazilian biodiversity available in NuBBEDB (Pilon et al., 2017). After being scored by the GoldScore scoring function in the GOLD program, the poses were rescored using the method of binding free energy (∆Gbind) calculation MM/GBSA using AmberTools23.
Virtual screening has become a widely applied method in early-stage enzymatic inhibitor discovery projects. The success in predicting hit candidates is closely related to the scoring function applied. However, docking scores have limitations as they often show a weak correlation with experimental results, with only a small fraction of hits exhibiting activity in in vitro validation assays (Rastelli and Pinzi, 2019). Because of this, in our study, we applied the rescoring method using MM/GBSA binding free energy calculations as it is a more accurate method for the selection of the best molecules in virtual screening studies (Rastelli and Pinzi, 2019; Poli et al., 2020; Sahakyan, 2021).
After molecular docking and rescoring of the poses, the top five ranked flavonoids based on the MM/GBSA values were selected, and the interactions of these structures with the amino acid residues in the active site of the Mpro protein were analyzed and compared with those of the crystallographic ligand 3WL, used as a reference compound in this study.
Table 1 shows the ∆Gbind MM/GBSA values of the top five rescored compounds. A- Nubbe_867 (de Sousa et al., 2014); B- Nubbe_1884 (Braz Filho and Gottlieb, 1971); C- Nubbe_1310 (dos Santos et al., 2009); D- Nubbe_1890 (Braz Filho and Gottlieb, 1971); E- Nubbe_2328 (Ferracin et al., 1998), as well as the reference inhibitor 3WL. Supplementary Table S1 shows the ∆Gbind values obtained by the MM/GBSA method for all flavonoids used in this study.
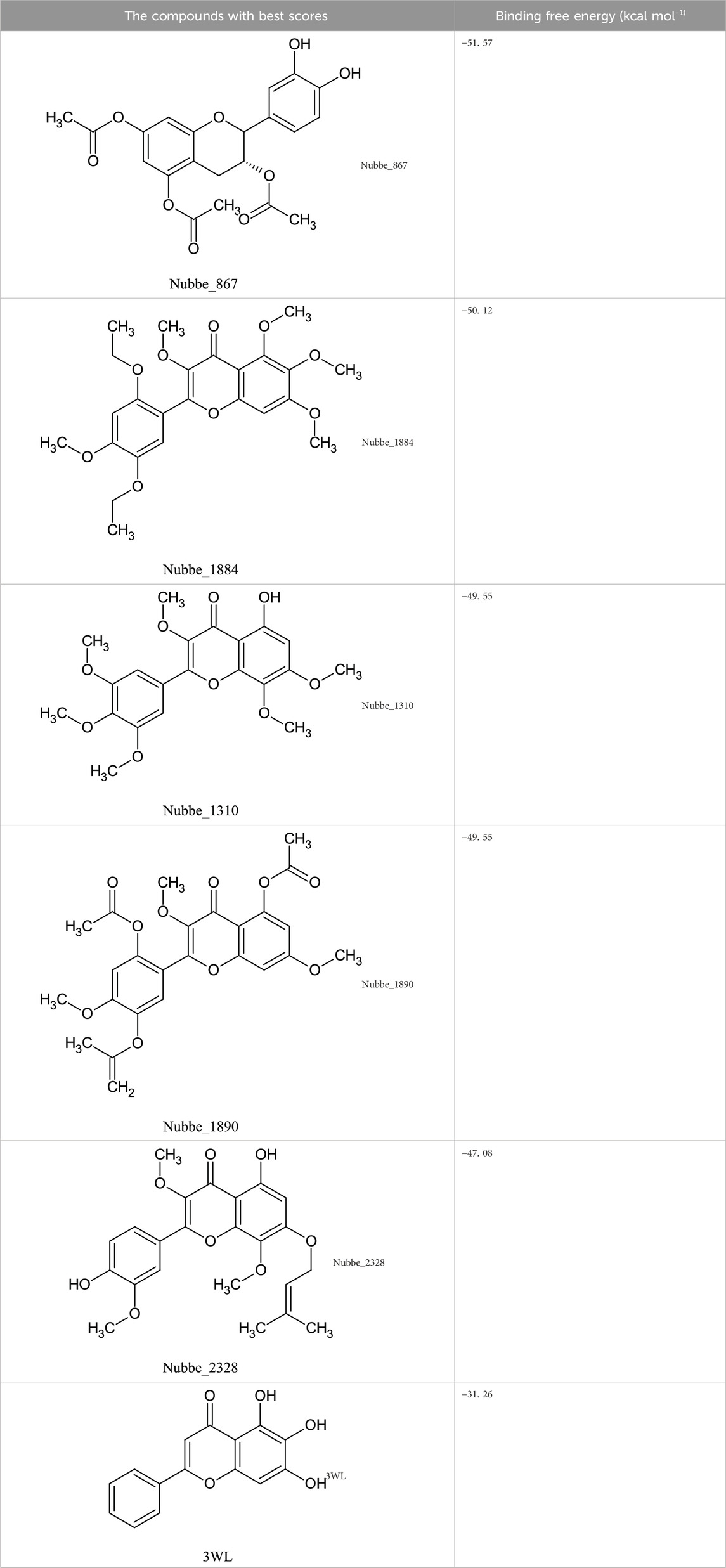
TABLE 1. Binding free energies of the top 5 rescored hits along with the co-crystallized ligand 3WL obtained using the MM/GBSA method.
The analysis of the new MMGBSA scores, used to avoid false predictions, revealed that the five selected compounds have better binding affinities with the Mpro enzyme, ranging from −51.57 to −47.08 kcal mol−1, when compared to the reference inhibitor (3WL: −31.26 kcal mol−1), as shown in Table 1.
We analyzed the binding modes of these compounds to the active site of the Mpro enzyme. The interactions made by the compounds and the inhibitor 3WL are shown in Figure 2. Our results demonstrated that the selected compounds formed an equal or greater number of hydrogen bonds with the target protein compared to the crystallographic inhibitor 3WL. This explains the higher binding affinity of these flavonoids to the Mpro protein than 3WL (Gogoi et al., 2021).
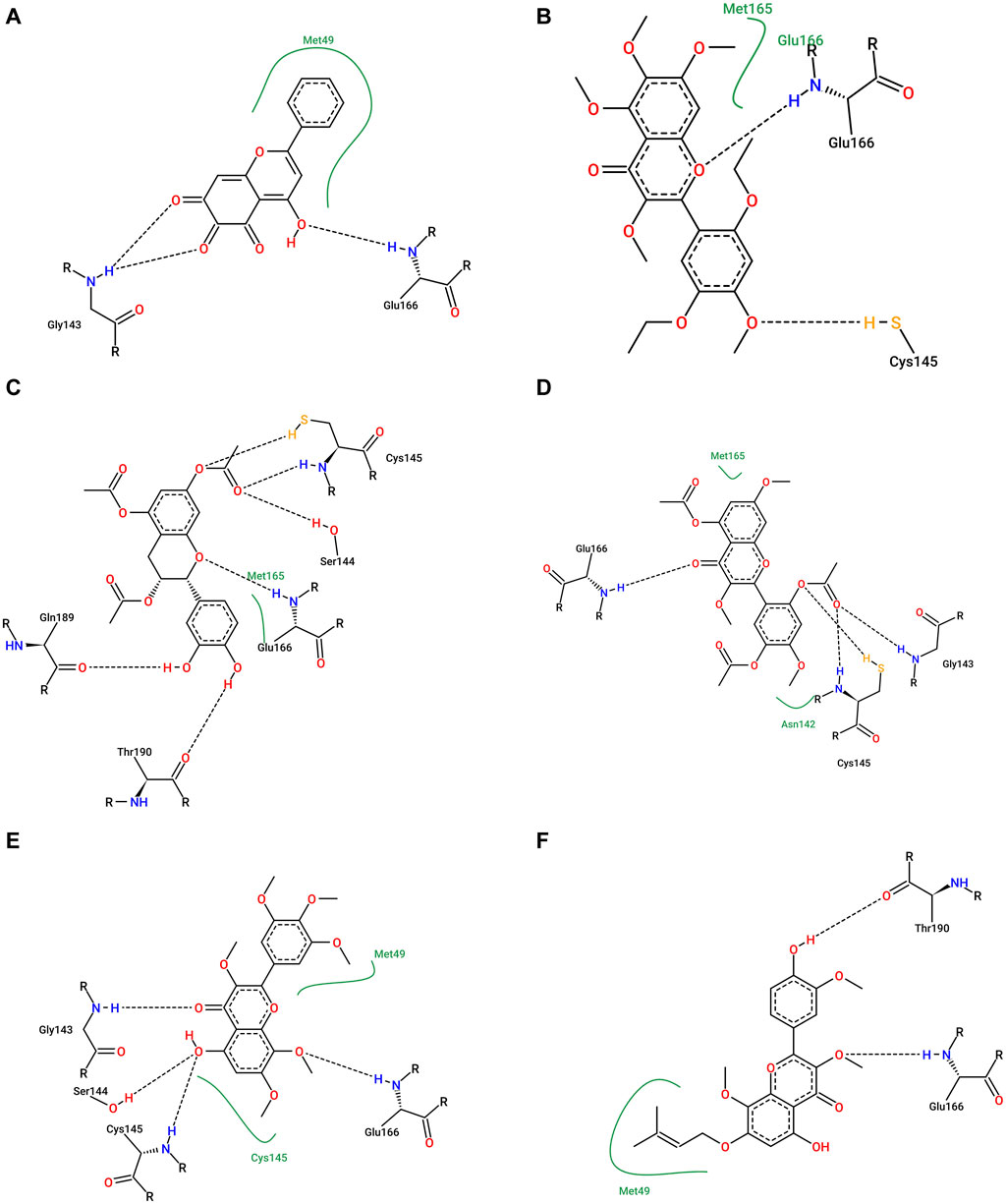
FIGURE 2. Figure 1: (A) 3WL, (B) NuBBE_1884, (C) NuBBE_867, (D) NuBBE_1890, (E) NuBBE_1310, and (F) NuBBE_2328 binding interactions with Mpro. Dashed lines indicate hydrogen bonds between the ligands and interacting Mpro residues.
The analysis of the residues involved in the interactions between the compounds and Mpro showed that NuBBE_867 interacted with the residues Ser144, Cys145, Glu166, Gln192, Gln149, and Thr190. NuBBE_1310 formed interactions with the residues Cys145 and Glu166. NuBBE_1884 interacted with Gly143, Ser144, Cys145, and Glu166. NuBBE_1890 interacted with the residues Gly143, Cys145, and Glu166, and NuBBE_2328 interacted with Glu166 and Thr190. All of these residues are part of the protein’s active site and are responsible for stabilizing these ligands (Su et al., 2020).
The flavonoids NuBBE_867, NuBBE_1310, NuBBE_1884, and NuBBE_1890 form hydrogen bonds with the important catalytic residue Cys145, which is part of the S1 binding pocket present in the catalytic domains I and II of the SARS-CoV-2 Mpro. Along with residue His41, they form the catalytic dyad within the substrate-binding site that actively participates in the catalytic function of the protein. Therefore, binding of these flavonoids to this residue may reduce the catalytic activities of Mpro, ultimately leading to a reduction in viral replication (Gogoi et al., 2021).
Another important residue that interacts with all selected flavonoids is Glu166, a key amino acid involved in the dimerization of the Mpro protein and assists in creating the S1 substrate binding pocket (Goyal and Goyal, 2020; Silvestrini et al., 2021). The formation of a hydrogen bond with the Glu166 residue is reported in the study by Duong and Nguyen (Duong and Nguyen, 2023) as one of the key interactions of non-covalent Mpro inhibitors. It is interesting to note that the crystallographic inhibitor 3WL, used as a reference in this study, also formed a hydrogen bond with the Glu166 residue, which corroborates our results (Su et al., 2020).
The residues Cys145 and Glu166 have been recently reported in molecular docking studies of inhibitors involving natural flavonoids as key residues in the binding process at the Mpro site (Cherrak et al., 2020; Su et al., 2020; Lin et al., 2023), and also with different compounds, corroborating with our results (Gahlawat et al., 2020; Kumar et al., 2020; Gogoi et al., 2021; Ngo et al., 2021; Patel et al., 2021; Antonopoulou et al., 2022; Salarizadeh et al., 2022; Ahamed and Arif, 2023; Rampogu et al., 2023). It is important to mention that all ligands fit perfectly into the protein’s binding pocket, revealing that all ligands form a stable complex with the target receptor.
Toxicity analysis
The five flavonoids were subjected to drug-likeness evaluation and assessment of mutagenic, tumorigenic, reproductive effective, and irritant parameters using ORISIS Data Warrior. It was observed that two flavonoids, NuBBE_867 and NuBBE_1890, showed no toxicity based on the toxicity parameters used in the study. On the other hand, NuBBE_1310 exhibited mutagenic and tumorigenic properties, while NuBBE_2328 displayed mutagenic, tumorigenic, and irritant properties. NuBBE_1884 showed a negative drug-likeness probability. The results of toxicity prediction and drug-likeness property analysis are presented in Table 2. Based on the data obtained from ORISIS Data Warrior (Sander et al., 2015), compounds with higher or positive drug-likeness probability values are considered good drug candidates. Since compounds NuBBE_1310 and NuBBE_2328 exhibited toxic effects, and NuBBE_1884 had a negative drug-likeness probability, they were not considered for further analysis. The non-toxic compounds, NuBBE_867 and NuBBE_1890, underwent molecular dynamics simulation studies.
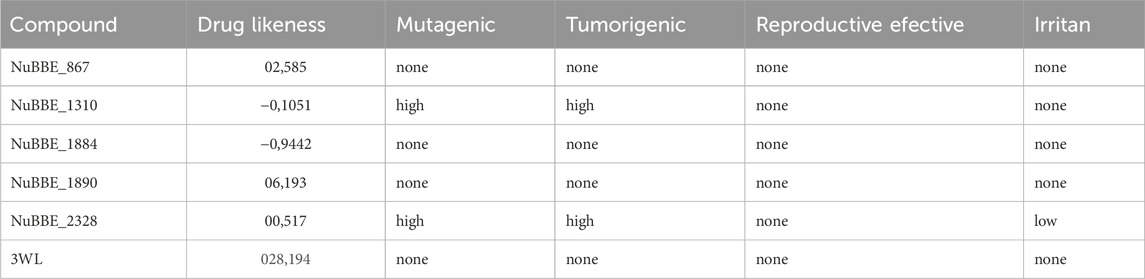
TABLE 2. Toxicity and drug-likeness analysis.
Molecular dynamics simulation
Molecular Dynamics (MD) simulation was performed to assess the flexibility, and stability of the protein-ligand complex, and the interactions of ligands with the SARS-CoV-2 Mpro protein. In this study, MD simulations were conducted for the Mpro-NuBBE_867 and Mpro-NuBBE_1890 complexes, selected based on toxicity and drug-likeness analyses, as well as the Mpro-3WL (reference inhibitor) complex and the unbound form of the protein (Apo). To predict the dynamic behavior and structural changes in the complexes, MD simulations were carried out over a 100 ns and the resulting trajectories were used to investigate complex RMSD, RMSF, and the number of hydrogen bonds.
RMSD analysis was performed to monitor the conformational stability of the ligand-protein complexes (Mpro). The RMSD plots for the ligand-protein complexes (Mpro-NuBBE_867, Mpro_NuBBE_1890, and Mpro-3WL) and the Apo form are shown in Figure 3. The stability of the complexes is directly related to the RMSD values, with lower deviation indicating greater structural stability. The range that justifies complex stability is approximately ∼3 Å (Rafique et al., 2023). Our results demonstrate that the average RMSD values for all complexes fall within the range of 1.47–2.14 Å, indicating that all complexes remain stable during the 100 ns simulation trajectory (Figure 3).
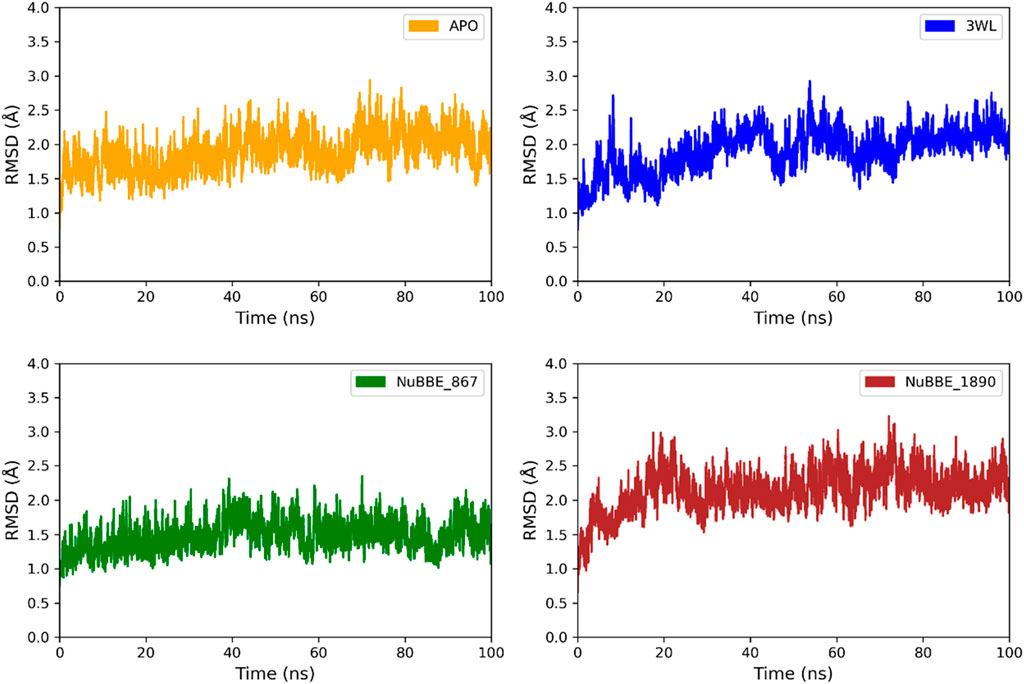
FIGURE 3. RMSD of the Apo form of Mpro and the Mpro complexes with 3WL, NuBBE_867, and NuBBE_1890 over a 100 ns MD simulation time.
The average RMSD values for the Apo protein were 1.89 Å, 2.04 Å for the reference complex (Mpro-3WL), 1.47 Å for Mpro-NuBBE_867, and 2.14 Å for Mpro-NuBBE_1890. The RMSD values revealed that the natural products (NPs) NuBBE_867 enhanced the structural stability of the Mpro protein (Figure 3), inhibiting its active site when compared to the crystallographic inhibitor used as a reference. On the other hand, NuBBE_1890 showed an average RMSD value higher than the Apo protein and the reference inhibitor, indicating that the presence of the ligand influenced the stability of the Mpro enzyme and altered its dynamic behavior. The results of RMSD values showed that the inhibitors formed stable bonds with the active site residues of the protein.
One essential component in determining the stability of a protein-ligand complex is the Root Mean Square Fluctuation (RMSF), which measures the flexibility of Cα atoms within a protein. Residues play a crucial role in forming a stable and strong binding complex between a protein and a ligand (Surti et al., 2020; Singh et al., 2022). The RMSF for the Apo protein and all simulated complexes are displayed in Figure 4.
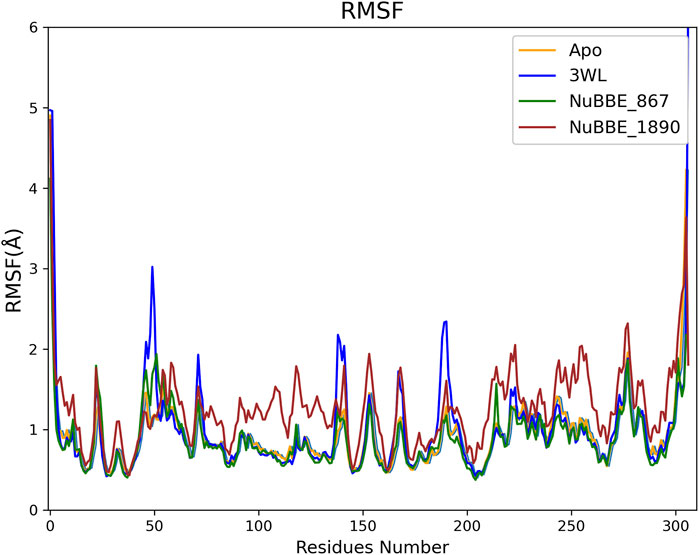
FIGURE 4. Root Mean Square Fluctuations of the Apo protein and Mpro-ligand complexes’ residues.
The RMSF of 3WL showed slightly higher fluctuations when compared to the other simulated systems, suggesting that this system is less stable than the others, a hypothesis that is later supported by the free energy calculations. In general, regions with lower fluctuations included the active site regions of Mpro, including Glu166, Cys145, Asn142, Phe140, Met165, Met49, His41, and Leu141, which comprise more stable regions. In regions where the simulated ligand interacts with amino acid residues of the protein, lower fluctuation values indicate greater stability in those interactions. Particularly noteworthy are the regions of the Ser46, Met49, Cys145, and Gln189 residues.
The fluctuations of the binding site residues Glu166, Cys145, Asn142, Phe140, Met165, Met49, His41, and Leu141 were lower in all Mpro complexes. Small fluctuations in these binding site residues indicated high stability of the Mpro complexes. Furthermore, the data suggested that the residues involved in the binding were responsible for the stable RMSD of the Mpro complexes.
Hydrogen bonding
The analysis of hydrogen bonds is an essential parameter that provides information about the binding affinity of drug candidates to a protein. Therefore, the presence of a significant number of H-bonds indicates a strong interaction between a ligand-protein complex. It is also responsible for drug metabolism and specificity (Rafique et al., 2023).
We investigated the hydrogen interactions of the flavonoids NuBBE_867 and NuBBE_1890 with the Mpro protein’s site residues and compared them to the crystallographic inhibitor 3WL during a 100 ns MD simulation (Figure 5).
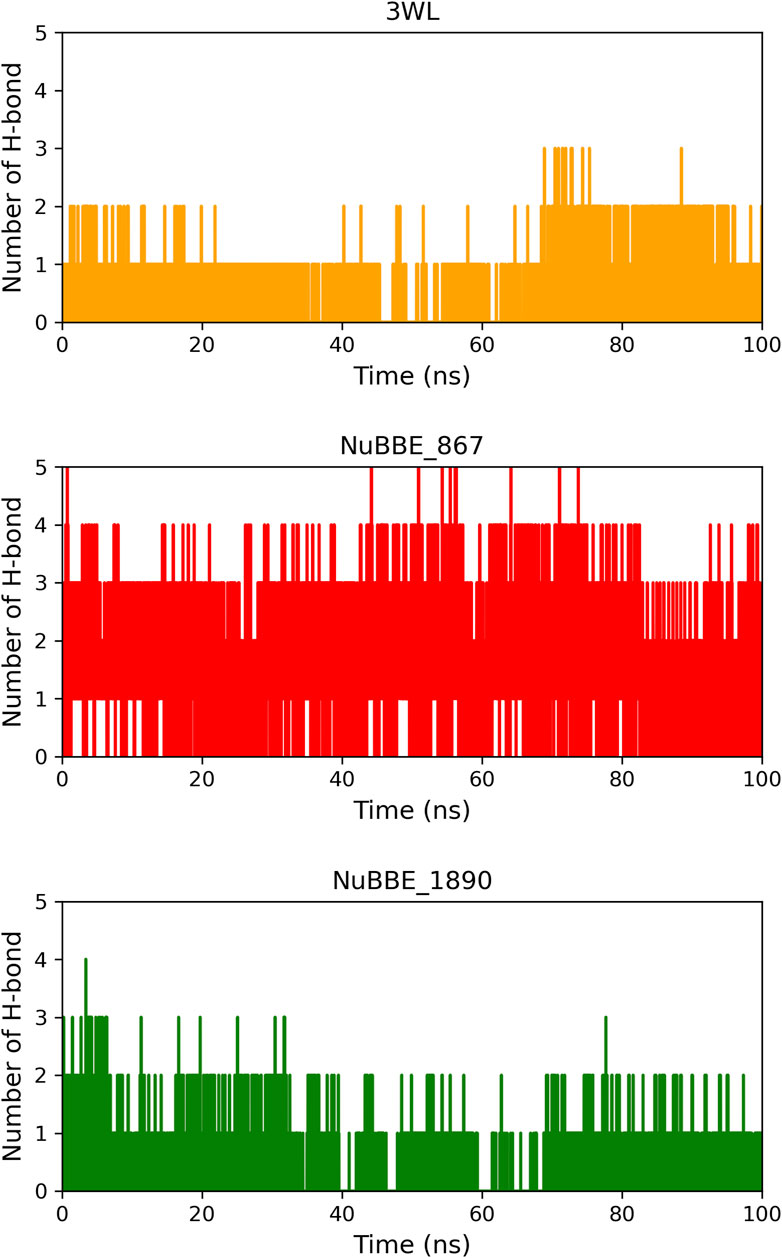
FIGURE 5. Number of hydrogen bonds between the ligands and Mpro protein during the 100 ns MD simulation time.
Our results show that NuBBE_867 formed five hydrogen bonds with the active site of Mpro throughout the simulation, a higher number of interactions compared to NuBBE_1890 and the crystallographic inhibitor 3WL, which had three and two interactions throughout the simulation, respectively.
During the simulation period, NuBBE_867 interacted with the site residues Asp187, Glu166, Ser144, Gly143, and Cys145 with occupancies of 54.19%, 43.06%, 12.28%, 12.24%, and 11.48%, respectively. NuBBE_1890 interacted with the residues Asn142, Thr24, and Glu166 with occupancies of 17.82%, 7.89%, and 6.29%, respectively. In contrast, the crystallographic inhibitor interacted with the residues Arg188 and Glu166 with occupancies of 21.17% and 11.10%, respectively. The formation of multiple hydrogen bonds (H-bonds) between the NuBBE_867 inhibitor and the Mpro protein contributed to the greater stabilization of the structural complex, as shown in the RMSD analysis. The residues Gly143, Ser144, and Cys145 that interacted with NuBBE_867 constitute the “oxyanion hole” of this cysteine protease and play a fundamental role in catalyzing the hydrolysis of protein substrates, in line with studies on Mpro inhibitors by flavonoids (Lin et al., 2023). It is important to note that the interactions with the residues Cys145 and Glu166 are consistent with molecular docking. The Cys25 residue is essential for the catalytic function of Mpro, while the Glu166 residue is crucial for Mpro dimerization and the formation of the substrate binding pocket (Cherrak et al., 2020).
In a previous study, Yoshino and colleagues ((Yoshino et al., 2020) conducted molecular dynamics simulations involving different inhibitors complexed with Mpro to verify the necessary interactions with the active site residues of Mpro for considering a compound as an inhibitor. They revealed that residues Glu166, Gly143, Ser144, and Cys145 were among the main interacting residues, which corroborates our results. Similar interactions have also been observed in previous studies involving natural flavonoids, further reinforcing our findings (Cherrak et al., 2020; Su et al., 2020; Lin et al., 2023).
Our results demonstrate that NuBBE_867 effectively interacted with the binding pocket of the Mpro protein, is highly stable, and is expected to be a potential candidate as an inhibitor of the main protease of SARS-CoV-2.
MM/PBSA binding free energy
To confirm the inhibitory capacity of the ligands for the Mpro protein in the ligand-protein complexes, binding free energy calculations were performed. The calculated results of MM-PBSA binding free energy are presented in Table 3.
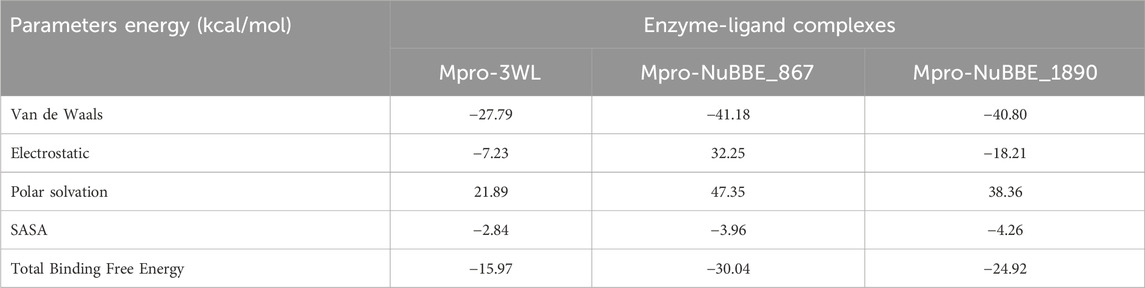
TABLE 3. MM-PBSA binding free energies (∆Gbind) and their components for the complexes under study. All values are reported in kcal.mol−1.
The MM-PBSA calculations results for NuBBE_867, NuBBE_1890, and the inhibitor 3WL are shown in Table 3. From the results, the ∆Gbind of the Mpro-3WL complex was −15.97 kcal/mol. The ∆Gbind of Mpro-NuBBE_867 and Mpro-NuBBE_1890 was −30.04 kcal/mol and −24.92 kcal/mol, respectively. It was observed from the MM-PBSA analysis that the complexes formed between the flavonoids and Mpro had a lower ∆Gbind than the reference complex, Mpro-3WL. This indicates the formation of stable complexes with higher binding affinity for these flavonoids in the active site of Mpro. It's worth noting that NuBBE_867 exhibited the highest binding affinity to Mpro, suggesting a more potent inhibitor of Mpro when compared to the 3WL inhibitor.
Notably, these results are in line with the findings in the analysis of hydrogen bonds, which show that NuBBE_867 formed interactions with key residues in the active site of Mpro, explaining the higher affinity with Mpro. The MM-PBSA method has been effectively used in previous computational studies to estimate and select more potent inhibitors of the Mpro protein from natural sources, further supporting our results (Ahmad et al., 2021; Gogoi et al., 2021; Sharma et al., 2022; Lin et al., 2023; Rafique et al., 2023).
Additionally, energy terms such as van der Waals energy, electrostatic energy, SASA energy, and polar solvation energy are crucial contributors to the total binding free energy of the complexes. We can see in Table 3 that van der Waals, electrostatic, and SASA energies contributed significantly to the total binding energies of the complexes. On the other hand, polar solvation energy had an unfavorable impact on the binding energy.
To delineate the specific contribution of each amino acid residue to ΔGbind, we conducted an energy decomposition analysis for each residue within the protein. Residues contributing to binding with values of −1.00 kcal/mol or lower were deemed significant for the binding process. The energetic contributions of each residue in the Mpro-NuBBE_867 and Mpro-NuBBE_1890 complexes were compared to those in the complexes formed by the reference compounds Mpro-3WL, as illustrated in Figure 6.
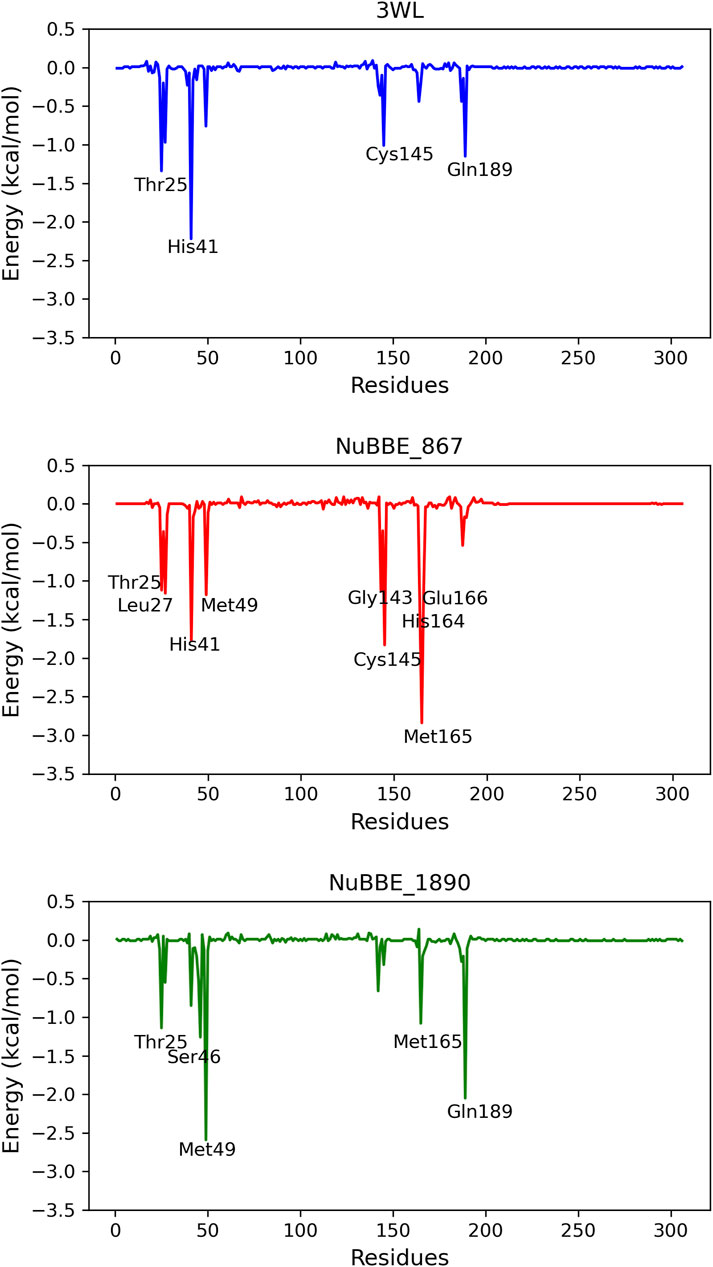
FIGURE 6. Graphical representation of the interaction energy per residue.
The energy decomposition analysis by residues unveiled that NuBBE_867 engages with His41 (ΔGtotal = −1.77) and Cys145 (ΔGtotal = −1.83), both of which constitute the catalytic dyad within this cysteine protease. Additionally, it forms interactions with Gly143 (ΔGtotal = −1.83), a residue that, alongside Cys145, plays a pivotal role in the oxyanion hole responsible for ligand stabilization. Moreover, it interacts with Glu166 (ΔGtotal = −1.14), which contributes to substrate recognition (Antonopoulou et al., 2022; Lin et al., 2023). Furthermore, it establishes connections with residues Thr25 (ΔGtotal = −1.12), Leu27 (ΔGtotal = −1.16), Met49 (ΔGtotal = −1.18), His164 (ΔGtotal = −1.43), and Met165 (ΔGtotal = −2.8). All these residues belong to the catalytic site and are classified as key residues of Mpro (Rampogu et al., 2023).
The residues contributing to the stability of NuBBE_1890 at the Mpro site are Thr25 (ΔGtotal = −1.10), Ser46 (ΔGtotal = −1.26), Met49 (ΔGtotal = −2.59), Met165 (ΔGtotal = −1.07), and Gln189 (ΔGtotal = −2.05), all situated in the active site of Mpro (Rampogu et al., 2023). In the case of the reference compound 3WL, the principal contributions to ΔGbind originated from residues Thr25 (ΔGtotal = −1.34), His41 (ΔGtotal = −2.21), Cys145 (ΔGtotal = −1.10), and Gln189 (ΔGtotal = −1.15).
Figure 6 depicts a greater number of residues with substantial contributions to the flavonoid NuBBE_867 in comparison to the reference ligands 3WL and NuBBE_1890. This observation elucidates the heightened binding affinity of this ligand with the Mpro enzyme, as indicated in Table 3, thus solidifying the validity of our findings.
Our in silico results propose that the molecule NuBBE_867 holds significant promise as an inhibitor of the Mpro enzyme.
Conclusion
We have shown that Flavonoids may be effective to inhibit the SARS-CoV-2 Mpro protein. Molecular docking results indicate that all the compounds exhibit reasonably strong binding energies, ranging from −5.9 to −7.3 kcal/mol, and lower predicted inhibition constants compared to two FDA-approved antiviral drugs, remdesivir and nirmatrelvir, currently used in SARS-CoV-2 treatment. Subsequent MD simulations confirm the stability of the ligand-protein complexes. Thus, RMSD, RMSF, radius of gyration, and hydrogen bond interactions demonstrate the maintenance of structural stability and productive interactions. In conclusion, based on the collective findings of this study, it is evident that these compounds hold promise as candidates for the development of effective medications to inhibit the SARS-CoV-2 Mpro protein. Further in vitro and in vivo experiments are warranted to validate and advance these results, potentially contributing to the development of novel antiviral agents.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
JR: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Software, Visualization, Writing–original draft, Writing–review and editing. RC: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Software, Visualization, Writing–original draft, Writing–review and editing. AC: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Software, Visualization, Writing–original draft, Writing–review and editing. ER: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Software, Visualization, Writing–original draft, Writing–review and editing. AG: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Software, Visualization, Writing–original draft, Writing–review and editing. AM: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing–original draft, Writing–review and editing. SF: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing–original draft, Writing–review and editing. DB: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing–original draft, Writing–review and editing. AL: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. PROPESP-UFPA. Chamada No. 002/2021-Programa de Infraestrutura para Jovens Pesquisadores-Programa Primeiros Projetos-PPPFAPESPA/CNPq.
Acknowledgments
We would like to thank Fundação Amazônia de Amparo a Estudos (FAPESPA) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) for funding our research under Chamada No. 002/2021-Programa de Infraestrutura para Jovens Pesquisadores-Programa Primeiros Projetos-PPPFAPESPA/CNPq and Postgraduate Pro-research (PROPESP/UFPA).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2024.1336001/full#supplementary-material
References
Aanouz, I., Belhassan, A., El-Khatabi, K., Lakhlifi, T., El-Ldrissi, M., and Bouachrine, M. (2021). Moroccan Medicinal plants as inhibitors against SARS-CoV-2 main protease: computational investigations. J. Biomol. Struct. Dyn. 39, 2971–2979. doi:10.1080/07391102.2020.1758790
Ahamed, A., and Arif, I. A. (2023). Finding potential inhibitors for Main protease (Mpro) of SARS-CoV-2 through Virtual screening and MD simulation studies. Saudi J. Biol. Sci. 30, 103845. doi:10.1016/j.sjbs.2023.103845
Ahmad, S., Abbasi, H. W., Shahid, S., Gul, S., and Abbasi, S. W. (2021). Molecular docking, simulation and MM-PBSA studies of nigella sativa compounds: a computational quest to identify potential natural antiviral for COVID-19 treatment. J. Biomol. Struct. Dyn. 39, 4225–4233. doi:10.1080/07391102.2020.1775129
Alzaabi, M. M., Hamdy, R., Ashmawy, N. S., Hamoda, A. M., Alkhayat, F., Khademi, N. N., et al. (2022). Flavonoids are promising safe therapy against COVID-19. Phytochem. Rev. 21, 291–312. doi:10.1007/s11101-021-09759-z
Antonio, A. da S., Wiedemann, L. S. M., and Veiga-Junior, V. F. (2020). Natural products’ role against COVID-19. RSC Adv. 10, 23379–23393. doi:10.1039/D0RA03774E
Antonopoulou, I., Sapountzaki, E., Rova, U., and Christakopoulos, P. (2022). Inhibition of the main protease of SARS-CoV-2 (Mpro) by repurposing/designing drug-like substances and utilizing nature’s toolbox of bioactive compounds. Comput. Struct. Biotechnol. J. 20, 1306–1344. doi:10.1016/j.csbj.2022.03.009
Asselah, T., Durantel, D., Pasmant, E., Lau, G., and Schinazi, R. F. (2021). COVID-19: discovery, diagnostics and drug development. J. Hepatology 74, 168–184. doi:10.1016/j.jhep.2020.09.031
Braz Filho, R., and Gottlieb, O. R. (1971). The flavones of Apuleia leiocarpa. Phytochemistry 10, 2433–2450. doi:10.1016/S0031-9422(00)89891-X
Case, D. A., Aktulga, H. M., Belfon, K., Cerutti, D. S., Cisneros, G. A., Cruzeiro, V. W. D., et al. (2023). AmberTools. J. Chem. Inf. Model 63, 6183–6191. doi:10.1021/acs.jcim.3c01153
Cataneo, A. H. D., Kuczera, D., Koishi, A. C., Zanluca, C., Silveira, G. F., Arruda, T. B. de, et al. (2019). The citrus flavonoid naringenin impairs the in vitro infection of human cells by Zika virus. Sci. Rep. 9, 16348. doi:10.1038/s41598-019-52626-3
Chatterjee, S., Pyne, N., and Paul, S. (2023). In silico screening of flavonoids unearthed Apigenin and Epigallocatechin Gallate, possessing antiviral potentiality against Delta and Omicron variants of SARS-CoV-2. Nucleus. doi:10.1007/s13237-023-00431-9
Chauhan, S. (2020). Comprehensive review of coronavirus disease 2019 (COVID-19). Biomed. J. 43, 334–340. doi:10.1016/j.bj.2020.05.023
Cherrak, S. A., Merzouk, H., and Mokhtari-Soulimane, N. (2020). Potential bioactive glycosylated flavonoids as SARS-CoV-2 main protease inhibitors: a molecular docking and simulation studies. PLoS One 15, e0240653. doi:10.1371/journal.pone.0240653
Costa, R. A. D., Costa, A. D. S. S. D., Rocha, J. A. P. D., Lima, M. R. D. C., Rocha, E. C. M. D., Nascimento, F. C. A., et al. (2023). Exploring natural alkaloids from Brazilian biodiversity as potential inhibitors of the Aedes aegypti juvenile hormone enzyme: a computational approach for vector mosquito control. Molecules 28, 6871. doi:10.3390/molecules28196871
Costa, R. A. da, Rocha, J. A. P. da, Pinheiro, A. S., Costa, A. do S. S. D., Rocha, E. C. M. da, Josino, L. P. C., et al. (2022). In silico identification of novel allosteric inhibitors of Dengue virus NS2B/NS3 serine protease: scientific paper. J. Serbian Chem. Soc. 87, 693–706. doi:10.2298/JSC210929011D
Darden, T., York, D. M., and Pedersen, N. L. (1993). Particle mesh Ewald: an N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092. doi:10.1063/1.464397
de Sousa, L. R. F., Ramalho, S. D., Burger, M. C. de M., Nebo, L., Fernandes, J. B., da Silva, M. F., et al. (2014). Isolation of arginase inhibitors from the bioactivity-guided fractionation of byrsonima coccolobifolia leaves and stems. J. Nat. Prod. 77, 392–396. doi:10.1021/np400717m
dos Santos, D. A. P., Braga, P. A. de C., da Silva, M. F., das, G. F., Fernandes, J. B., Vieira, P. C., et al. (2009). Anti-African trypanocidal and antimalarial activity of natural flavonoids, dibenzoylmethanes and synthetic analogues. J. Pharm. Pharmacol. 61, 257–266. doi:10.1211/jpp.61.02.0017
Duong, C. Q., and Nguyen, P. T. V. (2023). Exploration of SARS-CoV-2 Mpro noncovalent natural inhibitors using structure-based approaches. ACS Omega 8, 6679–6688. doi:10.1021/acsomega.2c07259
Ferracin, R. J., das, G. F., da Silva, M. F., Fernandes, J. B., and Vieira, P. C. (1998). Flavonoids from the fruits of Murraya paniculata. Phytochemistry 47, 393–396. doi:10.1016/S0031-9422(97)00598-0
Gahlawat, A., Kumar, N., Kumar, R., Sandhu, H., Singh, I. P., Singh, S., et al. (2020). Structure-based virtual screening to discover potential lead molecules for the SARS-CoV-2 main protease. J. Chem. Inf. Model. 60, 5781–5793. doi:10.1021/acs.jcim.0c00546
Genheden, S., and Ryde, U. (2015). The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 10, 449–461. doi:10.1517/17460441.2015.1032936
Gogoi, N., Chowdhury, P., Goswami, A. K., Das, A., Chetia, D., and Gogoi, B. (2021). Computational guided identification of a citrus flavonoid as potential inhibitor of SARS-CoV-2 main protease. Mol. Divers 25, 1745–1759. doi:10.1007/s11030-020-10150-x
Gordon, J. C., Myers, J. B., Folta, T., Shoja, V., Heath, L. S., and Onufriev, A. (2005). H++: a server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res. 33, W368–W371. doi:10.1093/nar/gki464
Goyal, B., and Goyal, D. (2020). Targeting the dimerization of the main protease of coronaviruses: a potential broad-spectrum therapeutic strategy. ACS Comb. Sci. 22, 297–305. doi:10.1021/acscombsci.0c00058
Heaton, P. M. (2020). The covid-19 vaccine-development multiverse. N. Engl. J. Med. 383, 1986–1988. doi:10.1056/NEJMe2025111
Hu, Q., Xiong, Y., Zhu, G.-H., Zhang, Y.-N., Zhang, Y.-W., Huang, P., et al. (2022). The SARS-CoV-2 main protease (Mpro): structure, function, and emerging therapies for COVID-19. MedComm 3, e151. doi:10.1002/mco2.151
Huang, C., Shuai, H., Qiao, J., Hou, Y., Zeng, R., Xia, A., et al. (2023). A new generation Mpro inhibitor with potent activity against SARS-CoV-2 Omicron variants. Sig Transduct. Target Ther. 8, 128–213. doi:10.1038/s41392-023-01392-w
Islam, M. T., Sarkar, C., El-Kersh, D. M., Jamaddar, S., Uddin, S. J., Shilpi, J. A., et al. (2020). Natural products and their derivatives against coronavirus: a review of the non-clinical and pre-clinical data. Phytotherapy Res. 34, 2471–2492. doi:10.1002/ptr.6700
Jackson, L. A., Anderson, E. J., Rouphael, N. G., Roberts, P. C., Makhene, M., Coler, R. N., et al. (2020). An mRNA vaccine against SARS-CoV-2 - preliminary report. N. Engl. J. Med. 383, 1920–1931. doi:10.1056/NEJMoa2022483
Jakalian, A., Jack, D. B., and Bayly, C. I. (2002). Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 23, 1623–1641. doi:10.1002/jcc.10128
Jin, Z., Du, X., Xu, Y., Deng, Y., Liu, M., Zhao, Y., et al. (2020). Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 582, 289–293. doi:10.1038/s41586-020-2223-y
Jo, S., Kim, S., Shin, D. H., and Kim, M.-S. (2020). Inhibition of SARS-CoV 3CL protease by flavonoids. J. Enzyme Inhibition Med. Chem. 35, 145–151. doi:10.1080/14756366.2019.1690480
Kumar, S., Sharma, P. P., Shankar, U., Kumar, D., Joshi, S. K., Pena, L., et al. (2020). Discovery of new hydroxyethylamine analogs against 3CLpro protein target of SARS-CoV-2: molecular docking, molecular dynamics simulation, and structure–activity relationship studies. J. Chem. Inf. Model. 60, 5754–5770. doi:10.1021/acs.jcim.0c00326
Li, F., Wang, Y., Li, D., Chen, Y., and Dou, Q. P. (2019). Are we seeing a resurgence in the use of natural products for new drug discovery? Expert Opin. Drug Discov. 14, 417–420. doi:10.1080/17460441.2019.1582639
Lin, L., Chen, D.-Y., Scartelli, C., Xie, H., Merrill-Skoloff, G., Yang, M., et al. (2023). Plant flavonoid inhibition of SARS-CoV-2 main protease and viral replication. iScience 26, 107602. doi:10.1016/j.isci.2023.107602
Lipinski, C. A., Lombardo, F., Dominy, B. W., and Feeney, P. J. (2001). Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings 1PII of original article: S0169-409X(96)00423-1. The article was originally published in Advanced Drug Delivery Reviews 23 (1997) 3–25. 1. Adv. Drug Deliv. Rev. 46, 3–26. doi:10.1016/s0169-409x(00)00129-0
Liskova, A., Samec, M., Koklesova, L., Samuel, S. M., Zhai, K., Al-Ishaq, R. K., et al. (2021). Flavonoids against the SARS-CoV-2 induced inflammatory storm. Biomed. Pharmacother. 138, 111430. doi:10.1016/j.biopha.2021.111430
Liu, H., Iketani, S., Zask, A., Khanizeman, N., Bednarova, E., Forouhar, F., et al. (2022). Development of optimized drug-like small molecule inhibitors of the SARS-CoV-2 3CL protease for treatment of COVID-19. Nat. Commun. 13, 1891. doi:10.1038/s41467-022-29413-2
Maier, J. A., Martinez, C., Kasavajhala, K., Wickstrom, L., Hauser, K. E., and Simmerling, C. (2015). ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 11, 3696–3713. doi:10.1021/acs.jctc.5b00255
Mani, J. S., Johnson, J. B., Steel, J. C., Broszczak, D. A., Neilsen, P. M., Walsh, K. B., et al. (2020). Natural product-derived phytochemicals as potential agents against coronaviruses: a review. Virus Res. 284, 197989. doi:10.1016/j.virusres.2020.197989
Mohapatra, P. K., Chopdar, K. S., Dash, G. C., Mohanty, A. K., and Raval, M. K. (2023). In silico screening and covalent binding of phytochemicals of Ocimum sanctum against SARS-CoV-2 (COVID 19) main protease. J. Biomol. Struct. Dyn. 41, 435–444. doi:10.1080/07391102.2021.2007170
Mukae, H., Yotsuyanagi, H., Ohmagari, N., Doi, Y., Imamura, T., Sonoyama, T., et al. (2022). A randomized phase 2/3 study of ensitrelvir, a novel oral SARS-CoV-2 3C-like protease inhibitor, in Japanese patients with mild-to-moderate COVID-19 or asymptomatic SARS-CoV-2 infection: results of the phase 2a part. Antimicrob. Agents Chemother. 66, e0069722. doi:10.1128/aac.00697-22
Mukae, H., Yotsuyanagi, H., Ohmagari, N., Doi, Y., Sakaguchi, H., Sonoyama, T., et al. (2023). Efficacy and safety of ensitrelvir in patients with mild-to-moderate coronavirus disease 2019: the phase 2b part of a randomized, placebo-controlled, phase 2/3 study. Clin. Infect. Dis. 76, 1403–1411. doi:10.1093/cid/ciac933
Ngo, S. T., Tam, N. M., Pham, M. Q., and Nguyen, T. H. (2021). Benchmark of popular free energy approaches revealing the inhibitors binding to SARS-CoV-2 Mpro. J. Chem. Inf. Model. 61, 2302–2312. doi:10.1021/acs.jcim.1c00159
Owen, D. R., Allerton, C. M. N., Anderson, A. S., Aschenbrenner, L., Avery, M., Berritt, S., et al. (2021). An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science 374, 1586–1593. doi:10.1126/science.abl4784
Owis, A. I., El-Hawary, M. S., Amir, D. E., Refaat, H., Alaaeldin, E., Aly, O. M., et al. (2021). Flavonoids of Salvadora persica L. (meswak) and its liposomal formulation as a potential inhibitor of SARS-CoV-2. RSC Adv. 11, 13537–13544. doi:10.1039/D1RA00142F
Panda, S. K., Gupta, P. S. S., and Rana, M. K. (2023). Potential targets of severe acute respiratory syndrome coronavirus 2 of clinical drug fluvoxamine: docking and molecular dynamics studies to elucidate viral action. Cell Biochem. Funct. 41, 98–111. doi:10.1002/cbf.3766
Patel, C. N., Jani, S. P., Jaiswal, D. G., Kumar, S. P., Mangukia, N., Parmar, R. M., et al. (2021). Identification of antiviral phytochemicals as a potential SARS-CoV-2 main protease (Mpro) inhibitor using docking and molecular dynamics simulations. Sci. Rep. 11, 20295. doi:10.1038/s41598-021-99165-4
Pilon, A. C., Valli, M., Dametto, A. C., Pinto, M. E. F., Freire, R. T., Castro-Gamboa, I., et al. (2017). NuBBEDB: an updated database to uncover chemical and biological information from Brazilian biodiversity. Sci. Rep. 7, 7215. doi:10.1038/s41598-017-07451-x
Poli, G., Granchi, C., Rizzolio, F., and Tuccinardi, T. (2020). Application of MM-PBSA methods in virtual screening. Molecules 25, 1971. doi:10.3390/molecules25081971
Price, D. J., and Brooks, C. L. (2004). A modified TIP3P water potential for simulation with Ewald summation. J. Chem. Phys. 121, 10096–10103. doi:10.1063/1.1808117
Purohit, P., Sahoo, P. S., Panda, M., Kabasi, K., Senapati, S. K., and Meher, B. R. (2023). Evaluating the antiviral potential of phytocompounds from mesua ferrea against SARS-CoV-2 main protease: structure-based virtual screening and molecular dynamics simulation investigations. ChemistrySelect 8, e202302295. doi:10.1002/slct.202302295
Qiao, J., Li, Y.-S., Zeng, R., Liu, F.-L., Luo, R.-H., Huang, C., et al. (2021). SARS-CoV-2 Mpro inhibitors with antiviral activity in a transgenic mouse model. Science 371, 1374–1378. doi:10.1126/science.abf1611
Rafique, A., Muhammad, S., Iqbal, J., Al-Sehemi, A. G., Alshahrani, M. Y., Ayub, K., et al. (2023). Exploring the inhibitory potential of novel piperidine-derivatives against main protease (Mpro) of SARS-CoV-2: a hybrid approach consisting of molecular docking, MD simulations and MMPBSA analysis. J. Mol. Liq. 382, 121904. doi:10.1016/j.molliq.2023.121904
Rampogu, S., Jung, T. S., Ha, M. W., and Lee, K. W. (2023). Repurposing and computational design of PARP inhibitors as SARS-CoV-2 inhibitors. Sci. Rep. 13, 10583. doi:10.1038/s41598-023-36342-7
Rastelli, G., and Pinzi, L. (2019). Refinement and rescoring of virtual screening results. Front. Chem. 7, 498. doi:10.3389/fchem.2019.00498
Ray, A. K., Sen Gupta, P. S., Panda, S. K., Biswal, S., Bhattacharya, U., and Rana, M. K. (2022). Repurposing of FDA-approved drugs as potential inhibitors of the SARS-CoV-2 main protease: molecular insights into improved therapeutic discovery. Comput. Biol. Med. 142, 105183. doi:10.1016/j.compbiomed.2021.105183
Rubio-Martínez, J., Jiménez-Alesanco, A., Ceballos-Laita, L., Ortega-Alarcón, D., Vega, S., Calvo, C., et al. (2021). Discovery of diverse natural products as inhibitors of SARS-CoV-2 Mpro protease through virtual screening. J. Chem. Inf. Model. 61, 6094–6106. doi:10.1021/acs.jcim.1c00951
Ryckaert, J.-P., Ciccotti, G., and Berendsen, H. J. C. (1977). Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341. doi:10.1016/0021-9991(77)90098-5
Sahakyan, H. (2021). Improving virtual screening results with MM/GBSA and MM/PBSA rescoring. J. Comput. Aided Mol. Des. 35, 731–736. doi:10.1007/s10822-021-00389-3
Salarizadeh, N., Aallaei, M. R., Zarei, A., Malekshah, R. E., Molaakbari, E., and Farajnezhadi, A. (2022). Docking and molecular dynamics simulations of flavonoids as inhibitors of infectious agents: rutin as a coronavirus protease inhibitor. ChemistrySelect 7, e202202043. doi:10.1002/slct.202202043
Salomon-Ferrer, R., Case, D. A., and Walker, R. C. (2013). An overview of the Amber biomolecular simulation package. WIREs Comput. Mol. Sci. 3, 198–210. doi:10.1002/wcms.1121
Sander, T., Freyss, J., von Korff, M., and Rufener, C. (2015). DataWarrior: an open-source program for Chemistry aware data visualization and analysis. J. Chem. Inf. Model. 55, 460–473. doi:10.1021/ci500588j
Sharma, P., Joshi, T., Mathpal, S., Joshi, T., Pundir, H., Chandra, S., et al. (2022). Identification of natural inhibitors against Mpro of SARS-CoV-2 by molecular docking, molecular dynamics simulation, and MM/PBSA methods. J. Biomol. Struct. Dyn. 40, 2757–2768. doi:10.1080/07391102.2020.1842806
Silvestrini, L., Belhaj, N., Comez, L., Gerelli, Y., Lauria, A., Libera, V., et al. (2021). The dimer-monomer equilibrium of SARS-CoV-2 main protease is affected by small molecule inhibitors. Sci. Rep. 11, 9283. doi:10.1038/s41598-021-88630-9
Singh, R., Bhardwaj, V. K., Das, P., Bhattacherjee, D., Zyryanov, G. V., and Purohit, R. (2022). Benchmarking the ability of novel compounds to inhibit SARS-CoV-2 main protease using steered molecular dynamics simulations. Comput. Biol. Med. 146, 105572. doi:10.1016/j.compbiomed.2022.105572
Solnier, J., and Fladerer, J.-P. (2021). Flavonoids: a complementary approach to conventional therapy of COVID-19? Phytochem. Rev. 20, 773–795. doi:10.1007/s11101-020-09720-6
Su, H., Yao, S., Zhao, W., Li, M., Liu, J., Shang, W., et al. (2020). Anti-SARS-CoV-2 activities in vitro of Shuanghuanglian preparations and bioactive ingredients. Acta Pharmacol. Sin. 41, 1167–1177. doi:10.1038/s41401-020-0483-6
Surti, M., Patel, M., Adnan, M., Moin, A., Ashraf, S. A., Siddiqui, A. J., et al. (2020). Ilimaquinone (marine sponge metabolite) as a novel inhibitor of SARS-CoV-2 key target proteins in comparison with suggested COVID-19 drugs: designing, docking and molecular dynamics simulation study. RSC Adv. 10, 37707–37720. doi:10.1039/D0RA06379G
Valli, M., dos Santos, R. N., Figueira, L. D., Nakajima, C. H., Castro-Gamboa, I., Andricopulo, A. D., et al. (2013). Development of a natural products database from the biodiversity of Brazil. J. Nat. Prod. 76, 439–444. doi:10.1021/np3006875
Verdonk, M. L., Cole, J. C., Hartshorn, M. J., Murray, C. W., and Taylor, R. D. (2003). Improved protein-ligand docking using GOLD. Proteins 52, 609–623. doi:10.1002/prot.10465
Wang, J., Wolf, R. M., Caldwell, J. W., Kollman, P. A., and Case, D. A. (2004). Development and testing of a general amber force field. J. Comput. Chem. 25, 1157–1174. doi:10.1002/jcc.20035
Wang, Z., Sun, H., Yao, X., Li, D., Xu, L., Li, Y., et al. (2016). Comprehensive evaluation of ten docking programs on a diverse set of protein–ligand complexes: the prediction accuracy of sampling power and scoring power. Phys. Chem. Chem. Phys. 18, 12964–12975. doi:10.1039/C6CP01555G
Wang, Z., and Yang, L. (2020). Turning the tide: natural products and natural-product-inspired chemicals as potential counters to SARS-CoV-2 infection. Front. Pharmacol. 11, 1013. doi:10.3389/fphar.2020.01013
Wu, C., Yin, W., Jiang, Y., and Xu, H. E. (2022). Structure genomics of SARS-CoV-2 and its Omicron variant: drug design templates for COVID-19. Acta Pharmacol. Sin. 43, 3021–3033. doi:10.1038/s41401-021-00851-w
Yoshino, R., Yasuo, N., and Sekijima, M. (2020). Identification of key interactions between SARS-CoV-2 main protease and inhibitor drug candidates. Sci. Rep. 10, 12493. doi:10.1038/s41598-020-69337-9
Yu, J., Tostanoski, L. H., Peter, L., Mercado, N. B., McMahan, K., Mahrokhian, S. H., et al. (2020). DNA vaccine protection against SARS-CoV-2 in rhesus macaques. Science 369, 806–811. doi:10.1126/science.abc6284
Zakaryan, H., Arabyan, E., Oo, A., and Zandi, K. (2017). Flavonoids: promising natural compounds against viral infections. Arch. Virol. 162, 2539–2551. doi:10.1007/s00705-017-3417-y
Keywords: SARS-CoV-2, main protease, flavonoids, drug-likeness, natural products, molecular docking, molecular dynamics, MMPBSA
Citation: da Rocha JAP, da Costa RA, da Costa AdSS, da Rocha ECM, Gomes AJB, Machado AK, Fagan SB, Brasil DdSB and Lima e Lima AH (2024) Harnessing Brazilian biodiversity database: identification of flavonoids as potential inhibitors of SARS-CoV-2 main protease using computational approaches and all-atom molecular dynamics simulation. Front. Chem. 12:1336001. doi: 10.3389/fchem.2024.1336001
Received: 09 November 2023; Accepted: 31 January 2024;
Published: 22 February 2024.
Edited by:
Cleydson Breno Rodrigues dos Santos, Universidade Federal do Amapá, BrazilReviewed by:
Pranab Kishor Mohapatra, C. V. Raman Global University, IndiaMalay Kumar Rana, Indian Institute of Science Education and Research Berhampur (IISER), India
Copyright © 2024 da Rocha, da Costa, da Costa, da Rocha, Gomes, Machado, Fagan, Brasil and Lima e Lima. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: João Augusto Pereira da Rocha, am9hby5yb2NoYUBpZnBhLmVkdS5icg==