
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem., 25 October 2023
Sec. Theoretical and Computational Chemistry
Volume 11 - 2023 | https://doi.org/10.3389/fchem.2023.1301690
This article is part of the Research TopicTheoretical Study of Two-Dimensional Materials for Photocatalysis and PhotovoltaicsView all 8 articles
 Xihao Chen1,2,3
Xihao Chen1,2,3 Zonghang Liu4
Zonghang Liu4 Jiang Cheng2Jiwen Li5Donglin Guo2Liang Zhang6Xianghong Niu7Ning Wang1,3Guangzhao Wang8Peng Gao9,10*
Jiang Cheng2Jiwen Li5Donglin Guo2Liang Zhang6Xianghong Niu7Ning Wang1,3Guangzhao Wang8Peng Gao9,10*Under the background of energy crisis, hydrogen owns the advantage of high combustion and shows considerable environment friendliness; however, to fully utilize this novel resource, the major hurdle lies in its delivery and storage. The development of the in-depth yet systematical methodology for two-dimensional (2D) storage media evaluation still remains to be challenging for computational scientists. In this study, we tried our proposed evaluation protocol on a 2D material, g-C3N5, and its hydrogen storage performance was characterized; and with addition of Li atoms, the changes of its electronical and structural properties were detected. First-principles simulations were conducted to verify its thermodynamics stability; and, its hydrogen adsorption capacity was investigated qualitatively. We found that the charges of the added Li atoms were transferred to the adjacent nitrogen atoms from g-C3N5, with the formation of chemical interactions. Thus, the isolated metallic sites tend to show considerable electropositivity, and can easily polarize the adsorbed hydrogen molecules, and the electrostatic interactions can be enhanced correspondingly. The maximum storage capacity of each primitive cell can be as high as 20 hydrogen molecules with a gravimetric capacity of 8.65 wt%, which surpasses the 5.5 wt% target set by the U.S. Department of Energy. The average adsorption energy is ranged from −0.22 to −0.13 eV. We conclude that the complex 2D material, Li-decorated g-C3N5 (Li@C3N5), can serve as a promising media for hydrogen storage. This methodology provided in this study is fundamental yet instructive for future 2D hydrogen storage materials development.
Under the context of global energy crisis, development of clean yet renewable resource is highly needed. Hydrogen, which is regarded as the fuel of future, owns the advantage of high renew-ability, and its combustion has zero-emission of CO2 (Allendorf et al., 2018; Züttel, 2004; U.S Department of Energy, 2020); thus it can be widely applied in many fields. However, in real practice, the main limitation lies in the fact that we are lack of high-efficiency storage media (U.S Department of Energy, 2020; Huang and Autrey, 2012; Züttel, 2003). Solid state materials based media are preferred due to their superior properties. In the current stage, some researchers tend to use metal-organic frameworks (MOFs) or metal hydrides based materials for this task (Bogdanovi and Schwickardi, 1997; Züttel et al., 2003; Sakintuna et al., 2007; Graetz, 2009; Gao et al., 2022); and at the same time, hydrocarbons or BN compounds had also been reported to own high content of hydrogen (Campbell et al., 2010; Luo et al., 2011a; Luo et al., 2011b; Teichmann et al., 2012; Gao et al., 2020; Gao and Zhang, 2020; Gao and Zhang, 2021). To successfully develop a high-performance hydrogen storage media, not only the storage capacity should be highlighted, manufacturing convenience is also crucial.
Within the past few decades, 2D carbon materials based storage media are promisingly emerging due to their high adsorption ability (Jürgens et al., 2003; Holst and Gillan, 2008; Thomas et al., 2008; Wei and Jacob, 2013; Algara-Siller et al., 2014; Dong et al., 2014; Fina et al., 2015; Hussain et al., 2016; Liu et al., 2019; Wang et al., 2019; Gao et al., 2021a; Wang et al., 2023; Zhang et al., 2023). These kinds of materials own superior aperture structures that enable themselves to be easily doped metal atoms or superalkali clusters for properties optimization and performance enhancement (Zhang et al., 2009; Wu et al., 2013; Zhu et al., 2014; Mahmood et al., 2015; Nair et al., 2015; Ruan et al., 2015; Wei et al., 2016; Gao et al., 2019; Panigrahi et al., 2020; Gao et al., 2021b; Gao et al., 2021c; Chen et al., 2021). Moreover, their original structure features can be qualitatively correlated with their optimizability and functions; thus computational investigation and evaluation of the novel derivatization units can be highly instructive for experimental practice (Wang et al., 2018; Bafekry et al., 2021). The graphene-like 2D unit, g-C3N5, is reported to be one of the most promising structures for electronic and optical devices development; and, it is proposed to own higher adsorption carrier, making itself suitable for storage media design.
Moreover, a systematic computation protocol for 2D materials’ hydrogen storage performance evaluation is of great importance for future studies; in this study, we proposed a density functional theory (DFT) calculations based methodology for the solution of structural and electronic properties of the pristine and metal-doped 2D materials, and the rules of the correlation between the properties and functions were summarized as reference. We tried our methodology upon Li@C3N5; and its hydrogen adsorption mechanism was successfully summarized. We anticipate that the fundamental insights provided in this study will be highly instructive for future development of 2D energy storage media.
First-principles calculations were carried out within the Vienna Ab-initio Simulation Package (VASP) under periodic boundary conditions (Kresse and Furthmüller, 1996; Grimme, 2006). Considering the balance between cost and efficiency, g-C3N5 primitive cell was selected as a prototype, and the applied lattice parameters were a = b = 15 Å. The electronic states were explored with the plane wave basis projector augmented wave (PAW) method (Blöchl, 1994), the cutoff of energy is set to 520 eV. The exchange-correlation energies were obtained with the generalized gradient approximation, by the PBE method (Perdew–Burke–Ernzerhof) (Perdew and Wang, 1992; Perdew et al., 1996). The convergence of the Hellmann-Feynman forces is 0.01 eV/Å. The conjugate-gradient algorithm was employed for structural relaxations with an energy convergence criteria of 1 × 10−5 eV. The post-processing of figures were conducted by VESTA. In our calculations, Van der Waals (VDW) corrections are included to describe long-range interactions with the DFT-D2 method (Grimme, 2006). To avoid the interactions between the periodic slabs along the z-axis, the vacuum layer with the magnitude of 20 Å was inserted. Bader analysis was employed to estimate the amount of charge transferred among the pristine g-C3N5 surface and Li atoms (Bader, 1990). And, a 3 × 3 × 1 Gamma-centered k-point grid was used for sampling within the Brillouin zone (Monkhorst and Pack, 1976; Chadi, 1977).
The thermodynamics stability of Li@C3N5 was investigated by first-principles molecular dynamics (MD) simulations under the canonical (NVT) ensemble (Martyna et al., 1992). The adsorption energy of Li atom upon g-C3N5 were obtained by the equation below:
where k represents the number of Li atoms, and E is the energy term. The averaged adsorption energy per hydrogen molecule was calculated by the following equation:
where n indicates the number of adsorbed hydrogen molecules upon the surface of Li@C3N5. The hydrogen storage capacity can be obtained by the following equation:
where m(X) is the number of X (X = C, N, Li, and H) atom, and M(X) is the corresponding molar mass. The desorption temperature (TD) between hydrogen molecules and the substrate can be defined as:
Here, R and KB are the universal gas constant and Boltzman constant, respectively; ΔS is the change of entropy for hydrogen molecules from the gas to liquid state (75.44 J mol−1 K−1); while P is the pressure of equilibrium (1 atm).
The optimized configurations of pristine and Li@C3N5 were presented in Figure 1. To systematically estimate its thermodynamics stability, ab initio molecular dynamics (AIMD) simulations were conducted, and the results were shown in Figure 2 for reference. It is notable that under the temperature of 400 K, the fluctuation of the total energy mainly oscillates at the equilibrium point; and from the snapshot that reflects the complex structure of the last frame, we noticed that the Li atom can stably bind with the N atom of the g-C3N5. All the observations indicate the high stability of this complex 2D material. At the same time, the mechanical properties of the pristine g-C3N5 (see in Figure 1A) were also investigated, and the simulated independent elastic constants are: C11 = 17.05 N/m, C12 = 15.24 N/m, and C66 = 0.91 N/m, respectively. These obtained values satisfy the Born-Huang criterion, revealing that the such a CN bonding network displays considerable mechanical hardness.
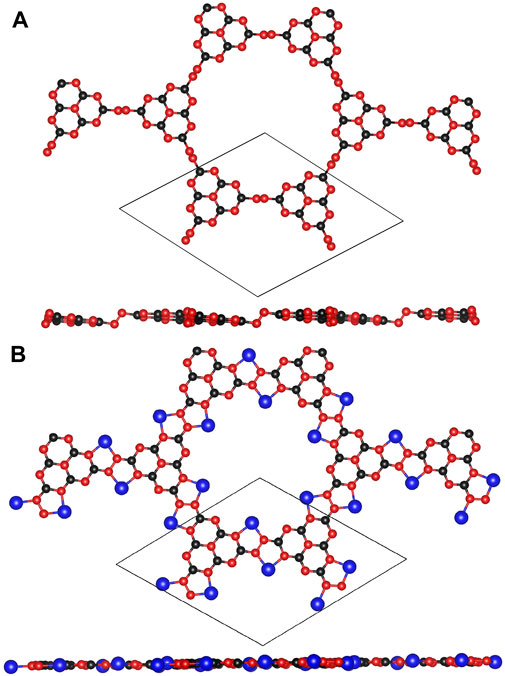
FIGURE 1. The optimized structures of (A) g-C3N5 primitive cell and (B) Li@C3N5 primitive cell. The black, red and blue balls represent C, N and Li atoms, respectively.
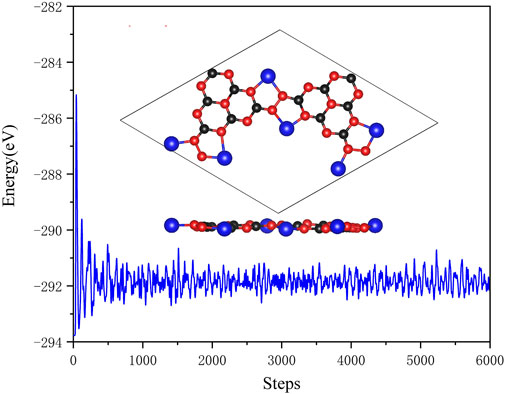
FIGURE 2. The AIMD simulations of Li@C3N5. The black, red and blue balls represent C, N and Li atoms, respectively.
To in-depth solve the binding mechanism of g-C3N5 monolayer, the electronic localization function (ELF) and charge density difference calculations were carried out, the results are presented in Figures 3A, B. The crystal face was obtained by cutting the g-C3N5 primitive cell based on the Mille index (−2.528, 1, 379.194). One can clearly see that the calculated ELF value of N-C binding is around 0.8 that is, within the reasonable range of covalent bonds. To further investigate the strength of this kind of binding, the Crystal Orbital Hamilton Population (COHP) calculations were employed, and the calculated results of N-N and N-C bonds are −0.46 and −2.61 eV, respectively (more details can be found in Figures 4A, B). With addition of Li atoms, we noticed that both the N-N (changed to −0.28 eV) and N-C (changed to −2.43 eV) bonds become weaker (more details can be found in Figures 5A–C), further indicating the fact that the charges of Li atoms are effectively transferred to the antibonding orbitals of C-N and N-N bonds. And for pristine g-C3N5, we notice the anitbonding states move down to the Fermi level, further indicating its capability of adsorbing metallic atoms that own ability of electron donating. The partial density of states (PDOSs) were also calculated for reference, and the results are shown in Figure 6. We can notice that the state density component of conduction band bottom and valence band top were both dominated by the 2p orbitals of N atoms, while less contributions were from 2p orbitals of C atoms.
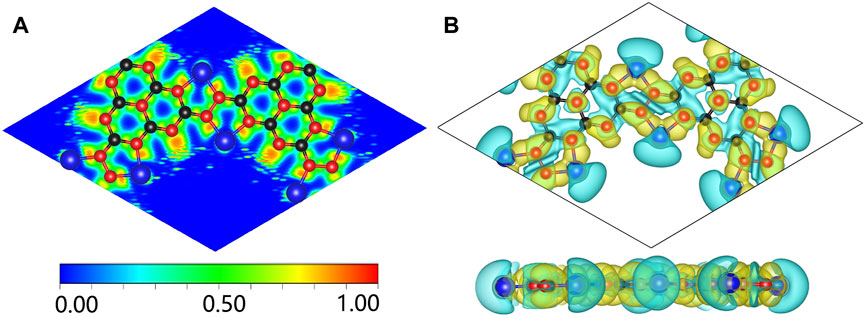
FIGURE 3. (A) The simulated ELF of Li@C3N5; the isosurface value is set to be 0.0013. (B) The calculated charge density difference of Li@C3N5. The black, red and blue balls represent C, N and Li atoms, respectively.
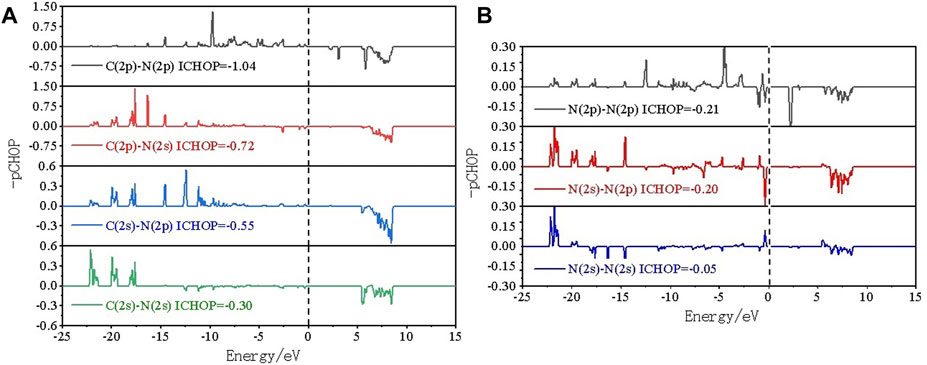
FIGURE 4. Crystal orbital Hamilton population COHP analysis of (A) C-N and (B) N-N bonds within pristine g-C3N5.
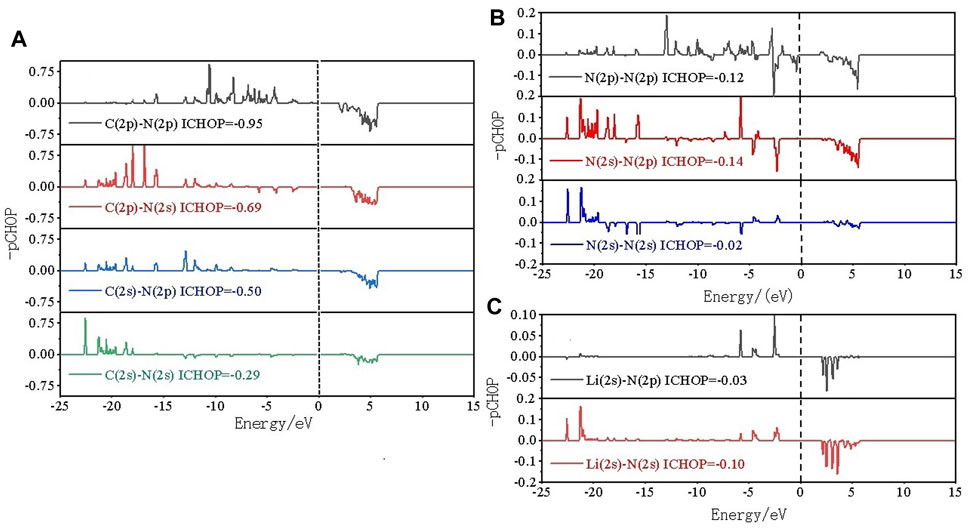
FIGURE 5. COHP analysis of (A) C-N, (B) N-N and (C) Li-N bonds within Li@C3N5.
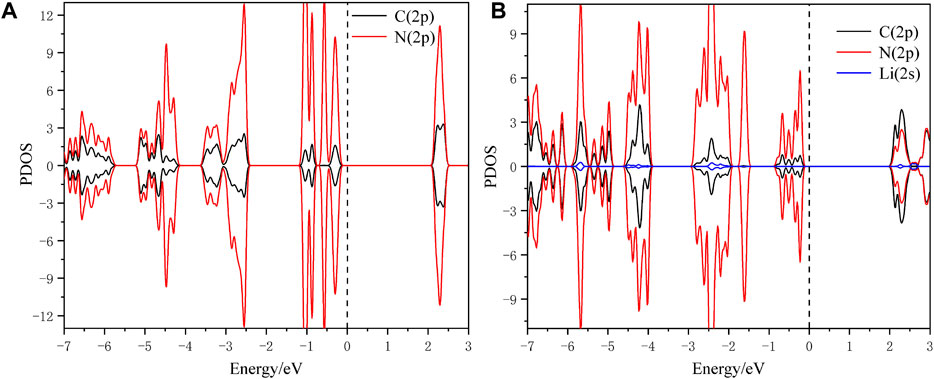
FIGURE 6. The simulated PDOS of (A) pristine g-C3N5 and (B) Li@C3N5.
The N-2p and C-2p orbitals overlap at the energy interval, indicating chemical interactions among these two atoms; such an observation is consistent with the conclusion obtained by ELF calculations. We hope these theoretical insights could be instructive for future experiment work upon this novel 2D material.
With the addition of Li atoms (the configuration is shown in Figure 1B), we first estimated the absorption energy per metal atom upon this 2D material; the obtained value is around −4.88 eV, within the reasonable range to overcome the risk of cohesive effects. Secondly, we notice that its 2s orbital can effectively overlap with the 2p orbitals of N atoms from g-C3N5 within the ranges of −2.5 ∼ −2 eV and −4.5 ∼ −4 eV, respectively. (details can be found from the simulated PDOSs shown in Figure 6), indicating the binding availability between these atoms. And also, from the calculated results of ELF, we can confirm that there exist chemical interactions between the added Li atoms and g-C3N5 monolayer. With the assistance of Barder analysis, we figure out each Li atom transfers 0.89 e− to g-C3N5, and displays electropositivty. Such an observation further indicates the possible potency of these metallic sites for gas molecules adsorption due to enhanced electrostatic interactions.
Then we optimized the geometric configurations of Li@C3N5 with adsorbed hydrogen molecules by first-principles calculations. We can clearly see that the adsorbed hydrogen molecules can be easily polarized by the metallic sites that display considerable electropositivity, consistent with our previous conclusion. We can also see from the calculated reduced density gradient (RDG) shown in Figure 7, the electrostatic interactions between these polarized hydrogen molecules and Li atoms can be correspondingly enhanced. And from the results shown in Figure 7, the main interaction between the adsorbed hydrogen molecules and Li atoms can be attributed to van der Waals interaction.
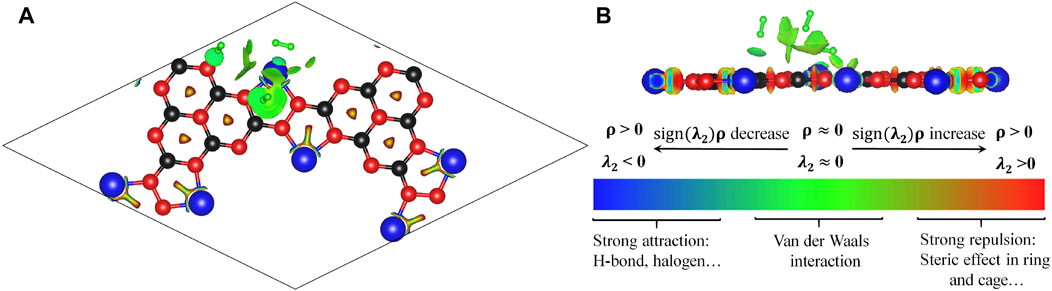
FIGURE 7. (A,B). The simulated RDG of Li@C3N5 with adsorbed hydrogen molecules.
The optimized configurations of Li@C3N5 with multiple adsorbed hydrogen molecules are presented in Figure 8 for reference. The corresponding results are listed in Table 1. The averaged adsorption energy per H2 molecule is ranged from −0.22 to −0.13 eV, with the storage capacity from 0.43 to 8.65 wt%. And for the adsorbed hydrogen molecules, the binding length of H-H is predicted to be slightly stretched between 0.753 Å and 0.755 Å. As stated before, the behind reason can be attributed to the induced polarization by addition of metal atoms. We also anticipated that the storage capacity can be further increased, with the decoration of extra metal atoms. We conclude that the overall electronic structures of 2D monolayer materials can be favorably improved with respect to specific needs of adsorption tasks in real practice, further indicating a promising methodology by computational evaluation for functional materials design.
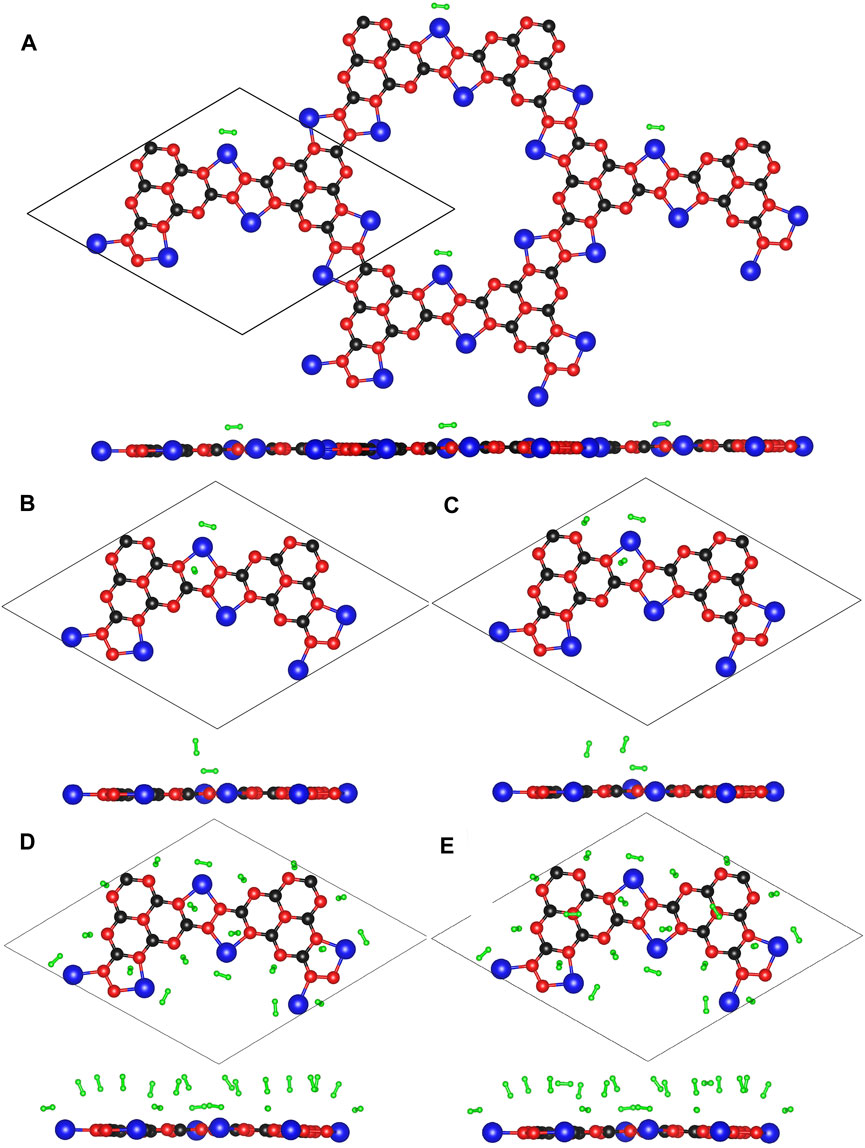
FIGURE 8. (A–E) The optimized configurations of Li@C3N5 with multiple adsorbed hydrogen molecules.
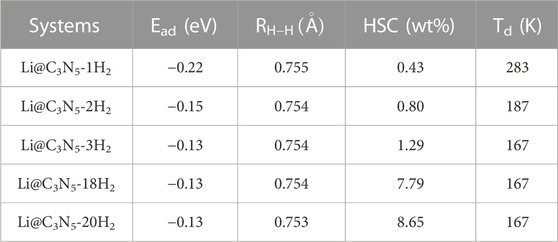
TABLE 1. The average adsorption energy Ead, the average bond length RH-H, the desorption temperature Td and the hydrogen storage density (HSC) for Li@C3N5 monolayer.
In summary, we proposed a systematic methodology for 2D materials’ hydrogen storage performance evaluation; and we decently investigated the adsorption mechanism of Li@C3N5 monolayer. DFT computational studies reveal that the pristine g-C3N5 cannot form strong interactions with hydrogen molecules; by decoration of Li atoms, its structure and electronic properties can be modified. Binding interaction between the added Li atoms and the adjacent N atoms from g-C3N5 is proved to be stable. Bader charge analysis reveals that each Li atom can transfer 0.89 e− to the pristine g-C3N5 and display considerable electropositivity. These metallic sites can essentially polarize the adsorbed hydrogen molecules and generate stronger electrostatic interactions, leading to the enhanced hydrogen adsorption performance. The average adsorption energy per hydrogen molecule on the Li@C3N5 complex 2D material is up to −0.22 eV that is, within a reasonable range; and each primitive cell can adsorb up to 20 hydrogen molecules, and the overall storage capacity can reach to 8.65 wt%. The insights obtained by this study indicated that the Li@C3N5 can serve as a promising storage media; and moreover, the proposed methodology is highly instructive for future studies.
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
XC: Conceptualization, Investigation, Software, Writing–original draft, Writing–review and editing. ZL: Formal Analysis, Writing–original draft. JC: Formal Analysis, Writing–original draft. JL: Formal Analysis, Writing–original draft. DG: Formal Analysis, Writing–original draft. Liang Zhang: Formal Analysis, Writing–original draft. XN: Formal Analysis, Writing–original draft. NW: Data curation, Writing–original draft. GW: Data curation, Writing–original draft. PG: Writing–original draft.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Algara-Siller, G., Severin, N., Chong, S. Y., Björkman, T., Palgrave, R. G., Laybourn, A., et al. (2014). Triazine-based graphitic carbon nitride: a two-dimensional semiconductor. Angew. Chem. Int. Ed. Engl. 53, 7450–7455. doi:10.1002/anie.201402191
Allendorf, M. D., Hulvey, Z., Gennett, T., Ahmed, A., Autrey, T., Camp, J., et al. (2018). An assessment of strategies for the development of solid-state adsorbents for vehicular hydrogen storage. Energy Environ. Sci. 11, 2784–2812. doi:10.1039/c8ee01085d
Bafekry, A., Faraji, M., Fadlallah, M. M., Abdolhosseini Sarsari, I., Jappor, H. R., Fazeli, S., et al. (2021). Two-dimensional porous graphitic carbon nitride C6N7 monolayer: first-principles calculations. Appl. Phys. Lett. 119, 142102. doi:10.1063/5.0060496
Blöchl, P. E. (1994). Projector augmented-wave method. Phys. Rev. B 50, 17953–17979. doi:10.1103/physrevb.50.17953
Bogdanovi, B., and Schwickardi, M. (1997). Ti-doped alkali metal aluminium hydrides as potential novel reversible hydrogen storage materials1: invited paper presented at the international symposium on metal−hydrogen systems. J. Alloy Comp. 253-254, 1–9. doi:10.1016/s0925-8388(96)03049-6
Campbell, P. G., Zakharov, L. N., Grant, D. J., Dixon, D. A., and Liu, S.-Y. (2010). Hydrogen storage by boron-nitrogen heterocycles: a simple route for spent fuel regeneration. J. Am. Chem. Soc. 132, 3289–3291. doi:10.1021/ja9106622
Chadi, D. J. (1977). Special points for brillouin-zone integrations. Phys. Rev. B 16, 1746–1747. doi:10.1103/physrevb.16.1746
Chen, X., wen Li, J., Dou, X., and Gao, P. (2021). Computational evaluation of mg-decorated g-cn as clean energy gas storage media. Int. J. Hydrogen Energy 46, 35130–35136. doi:10.1016/j.ijhydene.2021.08.071
Dong, G., Zhang, Y., Pan, Q., and Qiu, J. (2014). A fantastic graphitic carbon nitride (g-C3N4) material: electronic structure, photocatalytic and photoelectronic properties. J. Photochem Photobiol. C. Photochem Rev. 20, 33–50. doi:10.1016/j.jphotochemrev.2014.04.002
Fina, F., Callear, S. K., Carins, G. M., and Irvine, J. T. S. (2015). Structural investigation of graphitic carbon nitride via XRD and neutron diffraction. Chem. Mater 27, 2612–2618. doi:10.1021/acs.chemmater.5b00411
Gao, P., Huang, Z., and Yu, H. (2020). Exploration of the dehydrogenation pathways of ammonia diborane and diammoniate of diborane by molecular dynamics simulations using reactive force fields. J. Phys. Chem. A 124, 1698–1704. doi:10.1021/acs.jpca.9b10441
Gao, P., Li, J., and Wang, G. (2021a). Computational evaluation of superalkali-decorated graphene nanoribbon as advanced hydrogen storage materials. Int. J. Hydrogen Energy 46, 24510–24516. doi:10.1016/j.ijhydene.2021.05.023
Gao, P., Li, J., Zhang, J., and Wang, G. (2021c). Computational exploration of magnesium-decorated carbon nitride (g-C3N4) monolayer as advanced energy storage materials. Int. J. Hydrogen Energy 46, 21739–21747. doi:10.1016/j.ijhydene.2021.04.049
Gao, P., Liu, Z., and Zhang, F. (2021b). Computational evaluation of li-doped g-c2n monolayer as advanced hydrogen storage media. Int. J. Hydrogen Energy 47, 3625–3632. doi:10.1016/j.ijhydene.2021.11.003
Gao, P., Zhang, J., Liu, Z., and Hu, X. (2022). Computational insights into the energy storage of ultraporous mofs nu-1501-m (m = al or fe): protonization revealing and performance improving by decoration of superalkali clusters. Int. J. Hydrogen Energy 47, 41034–41045. Future Energy Materials. doi:10.1016/j.ijhydene.2022.09.166
Gao, P., and Zhang, J. (2020). Understanding the dehydrogenation pathways of ammonium octahydrotriborate (NH4B3H8) by molecular dynamics simulations with the reactive force field (ReaxFF). Adv. Theory Simul. 3, 2000139. doi:10.1002/adts.202000139
Gao, P., and Zhang, J. (2021). Understanding the intra-molecular proton transfer of octahydrotriborate and exploring the dehydrogenation pathways of NH4B3H8 by dft calculations. Adv. Theory Simul. 4, 2000287. doi:10.1002/adts.202000287
Gao, Z., Wang, L., Wang, L., Huang, J., She, H., and Wang, Q. (2019). Construction of heterostructured g-C3N4@TiATA/Pt composites for efficacious photocatalytic hydrogen evolution. Int. J. Hydrogen Energy 44, 24407–24417. doi:10.1016/j.ijhydene.2019.07.211
Graetz, J. (2009). New approaches to hydrogen storage. Chem. Soc. Rev. 38, 73–82. doi:10.1039/b718842k
Grimme, S. (2006). Semiempirical gga-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 27, 1787–1799. doi:10.1002/jcc.20495
Holst, J. R., and Gillan, E. G. (2008). From triazines to heptazines: deciphering the local structure of amorphous nitrogen-rich carbon nitride materials. J. Am. Chem. Soc. 130, 7373–7379. doi:10.1021/ja709992s
Huang, Z., and Autrey, T. (2012). Boron–nitrogen–hydrogen (BNH) compounds: recent developments in hydrogen storage, applications in hydrogenation and catalysis, and new syntheses. Energy Environ. Sci. 5, 9257–9268. doi:10.1039/c2ee23039a
Hussain, T., Hankel, M., and Searles, D. J. (2016). Computational evaluation of lithium-functionalized carbon nitride (g-C6N5) monolayer as an efficient hydrogen storage material. J. Phys. Chem. C 120, 25180–25188. doi:10.1021/acs.jpcc.6b06182
Jürgens, B., Irran, E., Senker, J., Kroll, P., Müller, H., and Schnick, W. (2003). Melem (2,5,8-Triamino-tri-s-triazine), an important intermediate during condensation of melamine rings to graphitic carbon nitride: synthesis, structure determination by X-ray powder diffractometry, solid-state NMR, and theoretical studies. J. Am. Chem. Soc. 125, 10288–10300. doi:10.1021/ja0357689
Kresse, G., and Furthmüller, J. (1996). Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186. doi:10.1103/physrevb.54.11169
Liu, G., Xue, M., Liu, Q., Yang, H., and Zhou, Y. (2019). Facile synthesis of C-doped hollow spherical g-C3N4 from supramolecular self-assembly for enhanced photoredox water splitting. Int. J. Hydrogen Energy 44, 25671–25679. doi:10.1016/j.ijhydene.2019.08.056
Luo, W., Campbell, P. G., Zakharov, L. N., and Liu, S.-Y. (2011b). A single-component liquid-phase hydrogen storage material. J. Am. Chem. Soc. 133, 19326–19329. doi:10.1021/ja208834v
Luo, W., Zakharov, L. N., and Liu, S.-Y. (2011a). 1,2-BN cyclohexane: synthesis, structure, dynamics, and reactivity. J. Am. Chem. Soc. 133, 13006–13009. doi:10.1021/ja206497x
Mahmood, J., Lee, E. K., Jung, M., Shin, D., Jeon, I.-Y., Jung, S.-M., et al. (2015). Nitrogenated holey two-dimensional structures. Nat. Commun. 6, 6486. doi:10.1038/ncomms7486
Martyna, G. J., Klein, M. L., and Tuckerman, M. (1992). Nosé–hoover chains: the canonical ensemble via continuous dynamics. J. Chem. Phys. 97, 2635–2643. doi:10.1063/1.463940
Monkhorst, H. J., and Pack, J. D. (1976). Special points for brillouin-zone integrations. Phys. Rev. B 13, 5188–5192. doi:10.1103/physrevb.13.5188
Nair, A. A., Sundara, R., and Anitha, N. (2015). Hydrogen storage performance of palladium nanoparticles decorated graphitic carbon nitride. Int. J. Hydrogen Energy 40, 3259–3267. doi:10.1016/j.ijhydene.2014.12.065
Panigrahi, P., Kumar, A., Karton, A., Ahuja, R., and Hussain, T. (2020). Remarkable improvement in hydrogen storage capacities of two-dimensional carbon nitride (g-C3N4) nanosheets under selected transition metal doping. Int. J. Hydrogen Energy 45, 3035–3045. doi:10.1016/j.ijhydene.2019.11.184
Perdew, J. P., Burke, K., and Ernzerhof, M. (1996). Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868. doi:10.1103/physrevlett.77.3865
Perdew, J. P., and Wang, Y. (1992). Pair-distribution function and its coupling-constant average for the spin-polarized electron gas. Phys. Rev. B 46, 12947–12954. doi:10.1103/physrevb.46.12947
Ruan, L., Xu, G., Gu, L., Li, C., Zhu, Y., and Lu, Y. (2015). The physical properties of Li-doped g-C3N4 monolayer sheet investigated by the first-principles. Mater. Res. Bull. 66, 156–162. doi:10.1016/j.materresbull.2015.02.044
Sakintuna, B., Lamari-Darkrim, F., and Hirscher, M. (2007). Metal hydride materials for solid hydrogen storage: a review. Int. J. Hydrogen Energy 32, 1121–1140. doi:10.1016/j.ijhydene.2006.11.022
Teichmann, D., Stark, K., Müller, K., Zöttl, G., Wasserscheid, P., and Arlt, W. (2012). Energy storage in residential and commercial buildings via liquid organic hydrogen carriers (LOHC). Energy Environ. Sci. 5, 9044–9054. doi:10.1039/c2ee22070a
Thomas, A., Fischer, A., Goettmann, F., Antonietti, M., Müller, J.-O., Schlögl, R., et al. (2008). Graphitic carbon nitride materials: variation of structure and morphology and their use as metal-free catalysts. J. Mater. Chem. 18, 4893–4908. doi:10.1039/b800274f
U.S Department of Energy (2020). Fuel cell technologies office multi-year research, development and demonstration plan. Available at: http://energy.gov/eere/fuelcells.
Wang, G., Zhou, F., Yuan, B., Xiao, S., Kuang, A., Zhong, M., et al. (2019). Strain-tunable visible-light-responsive photocatalytic properties of two-dimensional CdS/g-C3N4: a hybrid density functional study. Nanomater. (Basel) 9, 244. doi:10.3390/nano9020244
Wang, K., Ren, K., Hou, Y., Cheng, Y., and Zhang, G. (2023). Magnon–phonon coupling: from fundamental physics to applications. Phys. Chem. Chem. Phys. 25, 21802–21815. doi:10.1039/d3cp02683c
Wang, Y., Wu, C., Tian, Y., Yan, L., Tan, H., and Su, Z. (2018). The electronic and optical properties of carbon nitride derivatives: a first principles study. Appl. Surf. Sci. 453, 442–448. doi:10.1016/j.apsusc.2018.05.051
Wei, J., Huang, C., Wu, H., and Kan, E. (2016). High-capacity hydrogen storage in li-adsorbed g-C3N4. Mater Chem. Phys. 180, 440–444. doi:10.1016/j.matchemphys.2016.06.028
Wei, W., and Jacob, T. (2013). Strong excitonic effects in the optical properties of graphitic carbon nitride g-C3N4 from first principles. Phys. Rev. B 87, 085202. doi:10.1103/physrevb.87.085202
Wu, M., Wang, Q., Sun, Q., and Jena, P. (2013). Functionalized graphitic carbon nitride for efficient energy storage. J. Phys. Chem. C 117, 6055–6059. doi:10.1021/jp311972f
Zhang, C., Ren, K., Wang, S., Luo, Y., Tang, W., and Sun, M. (2023). Recent progress on two-dimensional van der waals heterostructures for photocatalytic water splitting: a selective review. J. Phys. D Appl. Phys. 56, 483001. doi:10.1088/1361-6463/acf506
Zhang, Y., Sun, H., and Chen, C. (2009). New template for metal decoration and hydrogen adsorption on graphene-like C3N4. Phys. Lett. A 373, 2778–2781. doi:10.1016/j.physleta.2009.05.046
Zhu, G., Lü, K., Sun, Q., Kawazoe, Y., and Jena, P. (2014). Lithium-doped triazine-based graphitic C3N4 sheet for hydrogen storage at ambient temperature. Comput. Mater Sci. 81, 275–279. doi:10.1016/j.commatsci.2013.08.015
Züttel, A. (2004). Hydrogen storage methods. Naturwissenschaften 91, 157–172. doi:10.1007/s00114-004-0516-x
Züttel, A. (2003). Materials for hydrogen storage. Mater. Today 6, 24–33. doi:10.1016/s1369-7021(03)00922-2
Keywords: hydrogen storage, reversible, g-C3N5, Li-decorated, DFT
Citation: Chen X, Liu Z, Cheng J, Li J, Guo D, Zhang L, Niu X, Wang N, Wang G and Gao P (2023) First-principles study of Li-doped planar g-C3N5 as reversible H2 storage material. Front. Chem. 11:1301690. doi: 10.3389/fchem.2023.1301690
Received: 25 September 2023; Accepted: 11 October 2023;
Published: 25 October 2023.
Edited by:
Kai Ren, Nanjing Forestry University, ChinaReviewed by:
Lili Liu, Chongqing Three Gorges University, ChinaCopyright © 2023 Chen, Liu, Cheng, Li, Guo, Zhang, Niu, Wang, Wang and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peng Gao, cGdhbzE3N0BnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.