 Nikhil Srivastava
Nikhil Srivastava Hyun-Joon Ha
Hyun-Joon Ha
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem., 19 October 2023
Sec. Organic Chemistry
Volume 11 - 2023 | https://doi.org/10.3389/fchem.2023.1280633
This article is part of the Research TopicRecent Advances in Synthesizing and Utilizing Nitrogen-containing HeterocyclesView all 12 articles
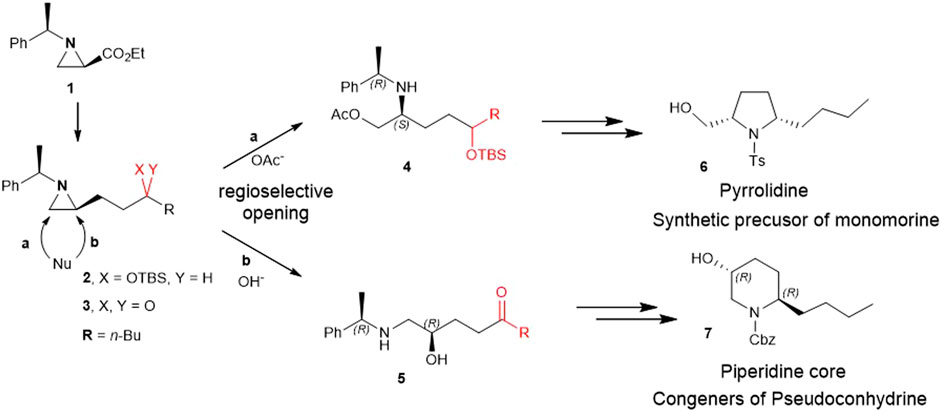
Aziridine had different regioselective ring openings depending on the functional group of its alkyl substituent. In the case of the alkyl group bearing γ-ketone at the C2 substituent of aziridine, the ring opening by the hydroxy nucleophile from H2O occurred by attacking the aziridine carbon at the C2 position. This reaction proceeded efficiently in the presence of CF3CO2H. Interestingly, the same starting aziridine ring bearing the alkyl substituent at the C2 position with the γ-silylated hydroxy group instead of γ-ketone led to the ring-opening reaction by the same oxygen nucleophile at the unsubstituted C3 position, with the breakage of the bond between aziridine N1 nitrogen and carbon at C3. These reaction products were cyclized to afford substituted pyrrolidine and piperidine rings with representative examples of congeners of pseudoconhydrine and monomorine.
GRAPHICAL ABSTRACT | Regioselective ring opening of aziridine for synthesizing azaheterocycle.
Aziridine, a nitrogen-containing three-membered ring, has been used for the synthesis of various azaheterocycles based on its chemical reactivity and unique regio- and stereoselectivity. In our lab, we have synthesized various azaheterocycles utilizing chiral aziridine in its optically pure forms (Ha et al., 2014; D’hooghe and Ha, 2016). Biologically active compounds including alkaloids with azaheterocycles have been prepared from enantiopure aziridines via aziridine ring formation from its acyclic compounds or its ring transformation (Srivastava et al., 2020). Transformation is mostly based on the formation of aziridinium ion with proper electrophiles and the subsequent ring-opening by nucleophiles either at C2 (pathway b in Scheme 1) or C3 (pathway a in Scheme 1) (Dolfen et al., 2016; Choi et al., 2017; Ranjith and Ha, 2021; Ranjith and Ha, 2022). The nucleophilic ring opening at C2 or C3 is controlled by substituents at the aziridine ring, electrophiles, and nucleophiles to provide acyclic amine A or B. The substituent cyclization gives rise to Cyc-A or Cyc-B (Scheme 1) (Eum et al., 2015; Yadav et al., 2016; Srivastava et al., 2020).
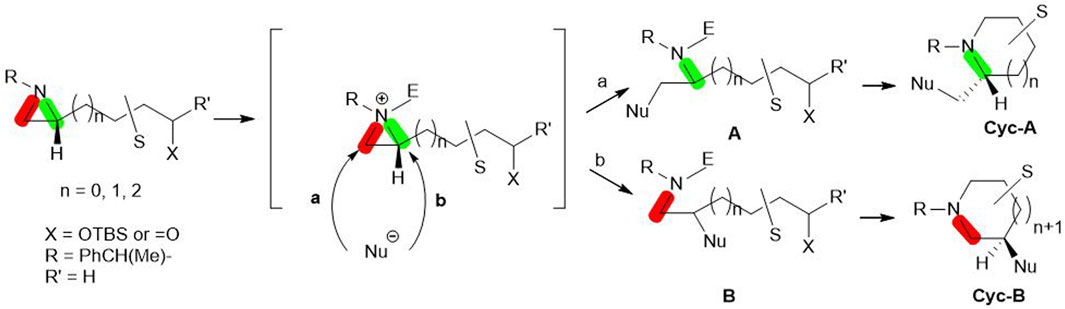
SCHEME 1. Aziridine ring-opening reactions by nucleophiles (either a or b) with breakage of bond C3-N (red) or C2-N (Green) to yield A or B. Subsequent cyclization of A or B decorated with the various functional groups(S) gives rise to either Cyc-A or Cyc-B.
Herein, for the first time, we report the involvement of functionalization of 2-substituted aziridine-2-carboxylate to give piperidine alkaloids in an efficient regiochemical pathway of ring-opening reaction. More specifically, 2-(3 silylated-hydroxy and 3-keto alkyl) aziridine (2 and 3) prepared from the same chiral aziridine-2-carboxylate (1) reacted with nucleophiles for the ring-opening. These reactions proceeded in a regioselective manner through either pathway a or b to yield 4 or 5 which was then cyclized to give pyrrolidine (6) or piperidine (7) (Scheme 2).
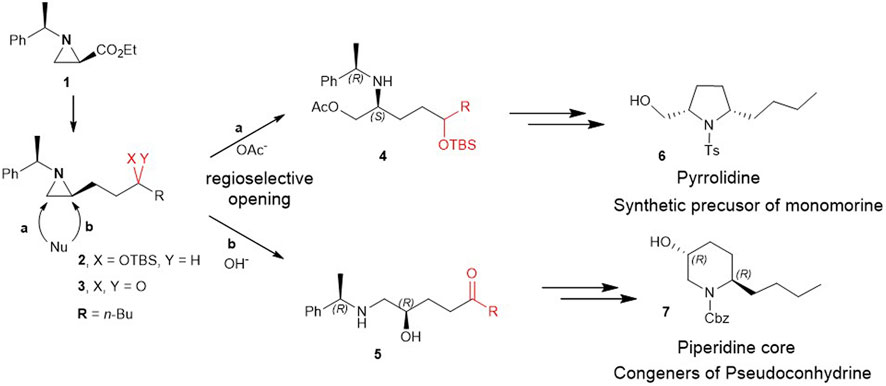
SCHEME 2. Ring-opening of 2-(3-hydroxy and 3-keto alkyl)aziridine (2 and 3) prepared from the same chiral aziridine-2-carboxylate (1) proceeds in a regioselective manner through either pathway a or b to yield 4 or 5 which is then cyclized to give pyrrolidine (6) or piperidine (7).
At first, the ring opening of aziridine was performed to give rise to either terminal amine or internal amines with the breakage of bonds N1-C2 or N1-C3, respectively. For the preparation of piperidine ring, screening of ring-opening reactions were carried out with the compound 1-((S)-1-((R)-1-phenylethyl)aziridin-2-yl)oct-7-en-3-one (3a) as a model substrate in Table 1, which was derived from (2R)-aziridine-2-carboxylates (1) (Its synthetic procedure is described in experimental section).
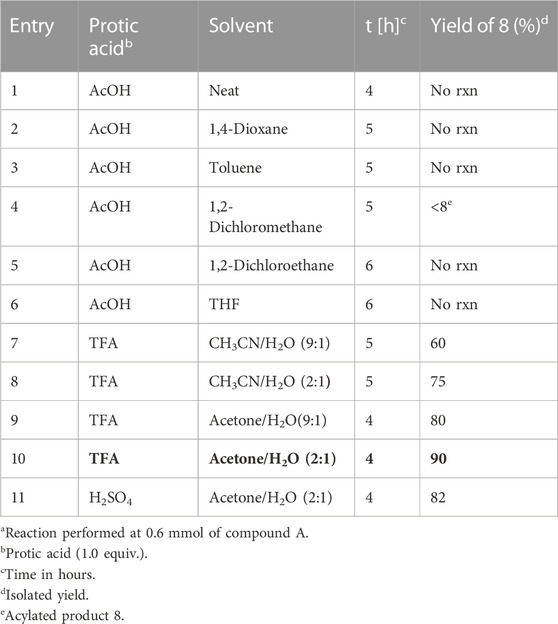
TABLE 1. Screening of regioselective opening of ((R)-1-phenylethyl)aziridin-2-yl)oct-7-en-3-one (3a).
Starting with a typical protocol for the ring openings of aziridines with neat acetic acid for 4 h at room temperature over the compound ((R)-1-phenylethyl)aziridin-2-yl)oct-7-en-3-one 3a, did not afford any ring-opening product (Table 1, entry 1). Up to date, almost all regiochemical pathway takes to yield internal amines regardless of substituents at the side chain functionality including simple halogen, amine, and hydroxyl groups (Stankovic et al., 2012; Ha et al., 2014). Therefore, we expected internal amine as a ring-opening product with breakage of the bond between N1 and C3 without any substituent. After varying solvents such as 1,4-dioxane and toluene and performing the reaction with acetic acid (1 equiv) at room temperature for 5 h, no ring-opened product was obtained (Table 1, entries 2 and 3) with all starting material stayed as they were. We then switched to another solvent, dichloromethane. A regioselective product with OAc group (R)-5-oxo-1-(((R)-1-phenylethyl)amino)dec-9-en-2-yl acetate 8 was obtained in less than 10% yield (Table 1, entry 4). To increase the yield, the experiment was further carried out with the same acetic acid (1 equiv) in 1,2-dichloroethane and THF for 6 h. However, reactions ended up with the recovery of starting material only (Table 1, entries 5 and 6). Then we switched over to trifluoracetic acid (TFA) as another protic acid. When the representative substrate ((R)-1-phenylmethyl)aziridine-2-yl)oct-7-en-3-one 3a was treated with TFA (1 equiv) in CH3CN:H2O (9:1) for 5 h at the same room temperature, unexpectedly, only regioselective product (R)-2-hydroxy-1-(((R)-1-phenylmethyl)amino)dec-9-en-5-one 8 was obtained in 60% yield (Table 1, entry 7). Changing the mixed solvent ratio of CH3CN:H2O from 9:1 to 2:1, the yield for compound 8 was improved to 75% under the same condition with TFA (1 equiv) (Table 1, entry 8). Inspired by this outcome, the starting compound 3a was treated with the TFA (1 equiv) at room temperature under acetone instead of CH3CN. The yield was further improved to 80%. All these observations can be explained by the solubility and the way of the association between TFA and aziridine. This was further justified by changing the ratio of the acetone and H2O solvent system with the ratio from 9:1 to 2:1 to give its ring-opened product (R)-2-hydroxy-1-(((R)-1-phenylmethyl)amino)dec-9-en-5-one (8) as a single isomer in a 90% yield (Table 1, entry 10). In continuation, switching to strong bronsted acid i,e. sulfuric acid (H2SO4) (1N) (1 equiv) also gave efficient regioselective ring opened product 8 in 82% yield under same acetone and H2O (2:1) solvent system (Table 1, entry 11).
On the basis of the above experiments described in Table 1, a plausible mechanism is proposed as shown in Figure 1. Possibly the reaction is mediated by the protonation of aziridine-nitrogen to form in situ aziridinium ion via a transition state (Ta). In this transition state, nitrogen attached proton, simultaneously interacted with oxygen attached to carbonyl carbon-oxygen via hydrogen bonding. Due to this transition state (Ta), the hydroxy nucleophile from H2O selectively approaches to attack at C2 carbon of the aziridinium ring exclusively in a regioselective manner (Figure 1) (Lopez and Salazar, 2013; Dalabehera et al., 2020). Under the same reaction conditions, other aziridinyl ketones such as 2-β- or 2-δ-ketoalkyl substituted aziridine yielded the mixture of the regioisomers of the ring-opened products which means that the conformation with fluorine is a driving force to determine the position of the nucleophilic attacks.
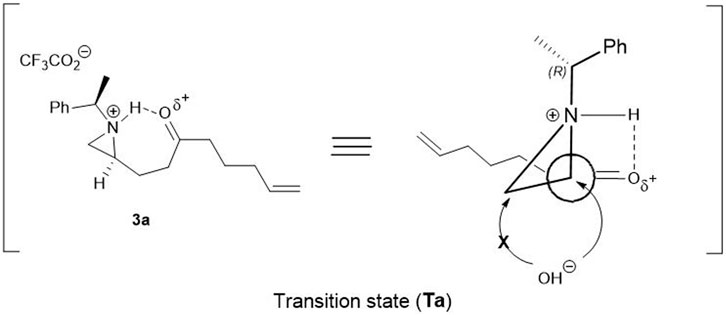
FIGURE 1. A schematic representation of plausible transition state (Ta) with keto compound 3a for the regioselective opening of aziridine moiety.
In order to support the plausible mechanistic pathway proposed above, we investigated 1H-NMR spectral change studies for the regioselective ring opening reaction over compound 3a with TFA under deuterated acetone (d6)/D2O (2:1) solvent system (See in Supplementary Material S2; Figure 1). It was interesting to observe firstly an increase in δ ppm values in both N- benzylic (quartet at 4.00 ppm) and methyl protons (doublet at 1.85 ppm) after 20 min. It happened due to the formation of in situ N-aziridinium ion via transition state (Ta). Then after 1 h, the formation of a new quartet (at 4.63 ppm) of N-benzylic and a doublet of newly methyl protons at 1.81 ppm were observed, which confirmed the regioselective ring opening reaction process happened in a concerted manner. Finally, after 4 h formation of product 8 in a protonated form, via the regioselective ring opened process was fully confirmed during 1H-NMR change studies (See in Supplementary Material S2, Figure 1).
After establishing standard reaction conditions for regioselective aziridine ring opening, we then utilized this method for the synthesis of piperidine core 7 bearing substituents at C2. Those structures are cores of congeners analog to pseudoconhydrine (Scheme 3). Accordingly, we started our synthesis from chiral (2R)-aziridine-2-carboxylates (1a) as a starting material. The preparation of compound (E)-methyl 3-((S)-1-((R)-1-phenylethyl)aziridin-2-yl)acrylate (1c) was easily achieved from Swern oxidation of ((R)-1-((R)-1-phenylethyl)aziridin-2-yl)methanol (1b), followed by a Wittig reaction for the two carbon extension, resulting compound 1c in a yield of 82% of isomers with the cis:trans ratio 88:12 using protocols for previously known sequential reactions (Scheme 3) (Lee and Ha, 2003; Yoon et al., 2010). After treatment with 2-nitrobenzenesulfonylhydrazide (NBSH), this olefinic product 1c was then saturated to afford compound 1d in 95% yield (Lee et al., 2009). The saturated methyl ester 1d was then converted to Weinreb amide 1e, followed by a reaction with n-C4H9MgCl, which afforded γ-aziridinyl ketone 2 in 80% yield (Macha and Ha, 2019). Based on the outcomes shown in Table 1, we then treated compound 2 following our established protocol for the regioselective aziridine ring-opening reaction with TFA under the mixed solvent system, acetone and H2O (2:1), at room temperature for 4 h. This yielded a hydroxy opened product 5 in 90% yield. Compound (R)-2-hydroxy-1-(((R)-1-phenylethyl)amino)nonan-5-one (5) was then treated with atmospheric H2 at room temperature under the catalytic amount of Pd(OH)2, yielding cyclic compound, followed by the reaction with CbzCl. These sequential reactions afforded N-Cbz-protected (2S,5R)-benzyl 2-butyl-5-hydroxypiperidine-1-carboxylate (7) (dr > 98:2) in 80% yield of two steps. The formation of compound 7 as a piperidine core skeleton (Macha et al., 2019) was obtained as a congener of pseudoconhydrine piperidine alkaloid (Bates et al., 2011). The formation of piperidine core 7 was executed with one-pot sequential debenzylation under hydrogenolysis followed by in situ cyclized via intramolecular reductive amination. Diastereoselectivity and assigned stereochemistry at the newly created center were then accessed. It was found that hydrogenation of a more stable intermediate imine could derive from a less hindered β-face of the molecule, resulting in a 2,5-trans-piperidine (Lee et al., 2003; Rao and Kumar, 2006) (Figure 2).
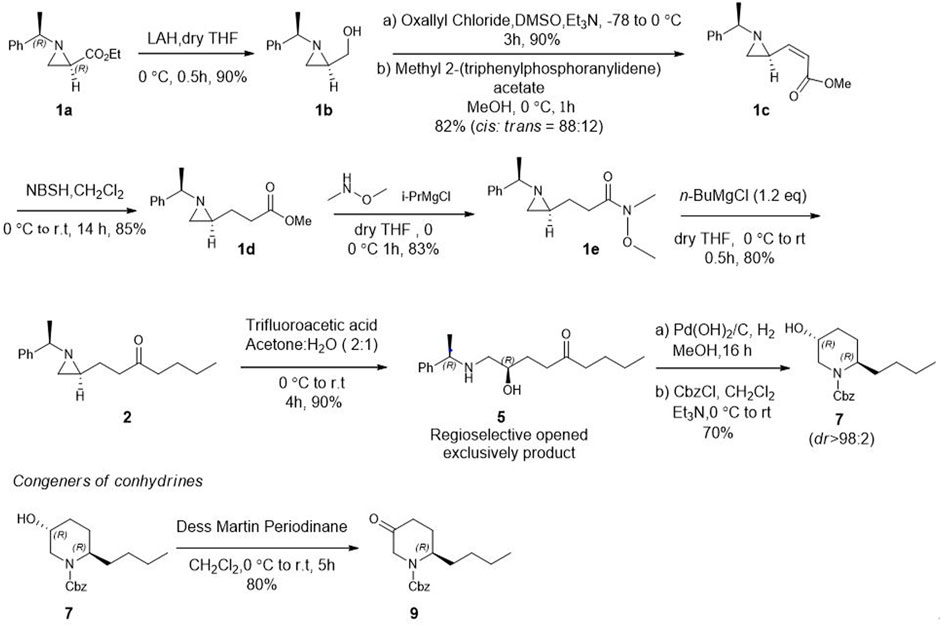
SCHEME 3. Asymmetric synthesis of congeners of pseudoconhydrine from (2R)-aziridine-2-carboxylate (1) via regioselective aziridine ring opening reaction as a key step.
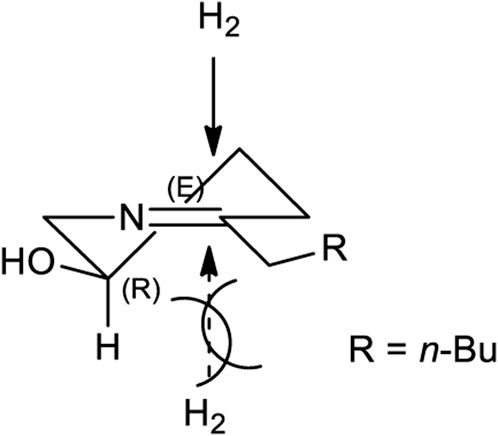
FIGURE 2. A Stereochemical aspect for one-pot hydrogenation of reductive amination for the synthesis of 2,5-trans piperidine core (7).
In addition, when compound 7 was further treated for oxidation under Dess-Martin periodinane, it produced 2-butyl-5-oxopiperidine (9) in 80% yield. Synthesizing this structural type of piperidone 9 (Ying et al., 2018; Lin et al., 2022) is an important intermediate to preparing various other biologically important 3-substituted piperidines (Vataku et al., 2014). The synthesis of 2-alkyl 5-hydroxy piperidine (7) and 2- alkyl 5-piperidone (9) with diverse substituents at C2 can be achieved efficiently using our regioselective opening protocol for γ-aziridinyl ketone which is prepared from commercially available (2R) aziridine −2 carboxylate at ease.
Encouraged by the synthesis of piperidine core, we decided to synthesize pyrrolidine core moiety 11 in which two alkyl groups at C2 and C5 of pyrrolidine are in a cis-stereo relationship (Scheme 4). Accordingly, a regioselective aziridine ring opening driven by the breakage of the bond between N1 and C3 of 2-alkyl aziridine is needed. This could be again prepared from the same starting substrate 1d, which was achieved from chiral aziridine-2-carboxylate (1a) at ease. Ester 1d was reduced to aldehyde by DIBAL and then alkylated with n-butyl magnesium chloride in dry THF to give alcohol (10) in 82% yield for both diastereomers. The stereoselectivity of this alcohol was almost 1:1, which was not important because it would be oxidized for the formation of a pyrrolidine ring (Scheme 4). Both diastereomeric products 10 were treated with TBSOTf under 2,6-lutidine to afford silyl-protected 11 with a yield of 85%. Compound 11 was then subject to a regioselective aziridine ring-opening reaction under acetic acid conditions to give acetate-opened product 12 in a 90% yield (Singh et al., 2011). The drastic difference in the regioselectivity of 11 from the case of γ-ketoalkyl substituent (2) could be explained by the difference in the transition state (Figures 1, 3). A plausible transition state for the regioselective opening of 11 is only protonation of aziridine ring to form aziridinium ion in transition state 11, followed by opening of aziridinium ion by oxygen nucleophiles from less-hindered C3 site (Figure 3).
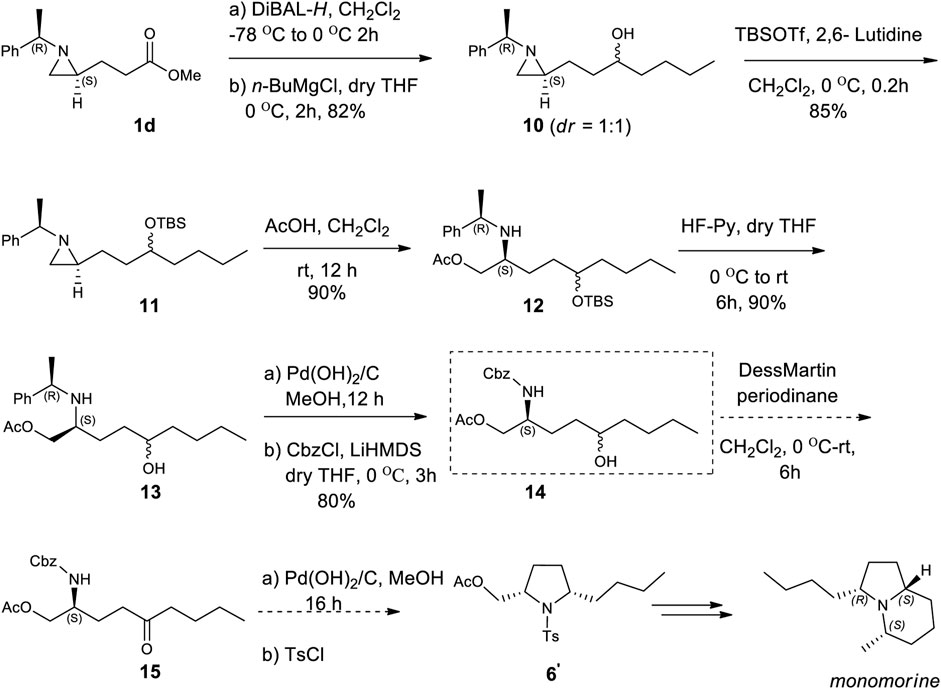
SCHEME 4. Asymmetric synthesis of monomorine from (2R)-aziridine-2-carboxylate (1) via regioselective aziridine ring opening reaction as a key step.
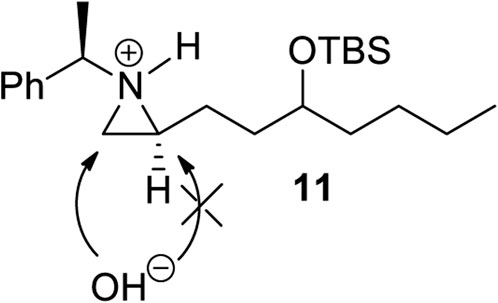
FIGURE 3. A plausible transition state for a regioselective opening of 11.
As expected from the previous studies, we observed a ring-opened product with breakage of N1 and C3 bearing substituent which was different from a ring-opened product of γ-keto aziridine. To determine the regiochemical difference by acid, we treated compound 11 with CF3CO2H as shown in entry 10 of Table 1. It failed to give a ring-opened product with a decent yield. After achieving acetate compound 12, it was treated under HF. Pyridine condition in dry THF to give hydroxy compound 13 in a yield of 80%. Surprisingly, the triers to oxidize 13 under various oxidation conditions were unable to yield an oxidized product in a decent yield. Changing the 2-phenylethyl to N-Cbz as protecting group from sequential reactions consisting of debenzylation followed by CbzCl in LiHMDS base at 0 °C afforded compound 14 in 80% yield. With the known established protocol of oxidation (Srivastava and Ha, 2022) followed by deprotection of Cbz and cyclization under hydrogen in catalytic Pd(OH)2 gave 2,5-cis pyrrolidine (6′), the essential core skeleton of many pyrrolidines and indolizidine alkaloids including monomorine (Wang et al., 2009; Michael, 2016).
In conclusion, we have successfully developed a regioselective ring opening reaction of aziridine moiety. The key feature includes the involvement of the functional group (γ-ketone or γ-silylated hydroxy group) present at the alkyl substituent of aziridine, which played a crucial role in the regioselective ring opening reaction of aziridine. In the case of γ-ketone group, the ring opening of aziridine occurred at carbon C2 position by hydroxy nucleophile from H2O under TFA condition in an efficient manner. Interestingly, in the case of γ-silylated hydroxy group present at the alkyl substituent of aziridine, the regioselective ring-opening reaction occurred at unsubstituted carbon C3 position of aziridine by the acetate oxygen nucleophile under acetic acid condition. These reaction products were cyclized to afford substituted pyrrolidine and piperidine rings with representative examples of congeners of pseudoconhydrine and monomorine.
General Remarks. Chiral aziridine-2-carboxylates were obtained as their methyl ester from Sigma-Aldrich as reagents and from Imagene Co., Ltd. (http://www.imagene.co.kr/) in bulk quantities. Their corresponding ethyl esters were also obtained either from the transesterification of methyl ester or from Imagene (http://www.imagene.co.kr/) in bulk quantities. All commercially available compounds were used as received unless stated otherwise. All reactions were carried out under an atmosphere of nitrogen in oven-dried glassware with a magnetic stirrer. Reactions were monitored by thin layer chromatography (TLC) with 0.25 mm·E. Merck pre-coated silica gel plates (60 F254). Visualization was accomplished with either UV light or by immersion in a solution of ninhydrin, p-anisaldehyde, or phosphomolybdic acid (PMA) followed by heating on a hot plate for about 10 s. Purification of the reaction product was carried out by flash chromatography using Kieselgel 60 Art 9385 (230–400 mesh). 1H NMR and 13C NMR spectra were obtained using Varian unity INOVA 400WB (400 MHz) or Bruker AVANCE III HD (400 MHz) spectrometer. Chemical shifts are reported relative to chloroform (δ = 7.26) for 1H NMR and chloroform (δ = 77.0) for 13C{1H} proton-decoupled carbon NMR. Data are reported as (br = broad, s = singlet, d = doublet, t = triplet, q = quartet, p = quintet, m = multiplet). Coupling constants are given in Hz. Ambiguous assignments were resolved based on standard one-dimensional proton decoupling experiments. Optical rotations were obtained using a Rudolph Autopol III digital polarimeter and a JASCO P-2000. Optical rotation data are reported as follows [α]20 (concentration c = g/100 mL, solvent). High-resolution mass spectra were recorded on a 4.7 T Ion Spec ESI-TOFMS, JEOL (JMS-700). An AB Sciex 4800 Plus MALDI TOFTM (2,5-dihydroxybenzoic acid (DHB) matrix was used to prepare samples for MS. Data were obtained in the reflector positive mode with internal standards for calibration.
To a stirred solution of chiral N-methoxy-N-methyl-3-((S)-1-((R)-1-phenylethyl)aziridin-2-yl)propanamide (1e) (500 mg, 1.9 mmol, 1.0 equiv.) in dry THF (25 mL) at 0°C, 4-pentenylmagnesium bromide solution C5H9MgBr (4.0 mL, 0.5 M in THF, 3.81 mmol) was added. The reaction mixture was slowly warmed to room temperature and stirred for 0.5 h. After completion as per TLC indication, the reaction was quenched with saturated NH4Cl (3 mL). The crude mixture was extracted with EtOAc (3 × 15 mL), dried over anhydrous Na2SO4, and concentrated under vacuum to afford crude product 3a, which was then purified by column chromatography to give compound 1-((S)-1-((R)-1-phenylethyl)aziridin-2-yl)oct-7-en-3-one (3a) as a viscous liquid (415 mg, 80% yield), TLC Rf (30% EtOAc/hexane = 0.30).
[α]20D = + 25.8 (c = 0.7, MeOH); 1H NMR (400 MHz, CDCl3): δ 7.40–7.25 (m, 5 H), 5.87–5.73 (m, 1 H), 5.08–4.97 (m, 2 H), 2.68–2.57 (m, 2 H), 2.48 (t, J = 7.4 Hz, 2 H), 2.41 (q, J = 6.2 Hz, 1 H), 2.13–2.04 (m, 1 H), 2.02–1.89 (m, 1 H), 1.80–1.66 (m, 2 H), 1.60–1.53 (m, 2 H), 1.51–1.48 (m, 1 H), 1.43 (d, J = 6.6 Hz, 3 H), 1.32–1.27 (m, 1 H); 13C NMR (101 MHz, CDCl3): 210.43, 144.44, 137.95, 128.24, 126.90, 126.73, 115.20, 69.60, 41.83, 40.48, 39.32, 33.47, 33.06, 27.05, 23.31, 22.70; HRMS-ESI (m/z) [M+H]+ calcd. for C18H26NO+ 272.2014; found 272.2006.
To a stirred solution of 1-((S)-1-((R)-1-phenylethyl)aziridin-2-yl)oct-7-en-3-one (3a) (165 mg, 0.60 mmol) in acetone (2.0 mL) and water (1.0 mL) solvent system, trifluoro acetic acid (TFA) (0.05 mL, 0.87 mmol, 1.0 equiv.) was added at 0°C. The mixture was then stirred at room temperature for 4 h. After completion of the reaction per TLC indication, the reaction was quenched with a saturated solution of NaHCO3 (1 mL) and extracted with CH2Cl2 (4 × 10 mL). After concentrating under vacuum, the crude product was purified by flash column chromatography on silica gel to afford a pure product 8 (157 mg, 90%) TLC Rf (90% EtOAc/hexane = 0.1).
[α]20D = + 18.2 (c = 0.14, MeOH); 1H NMR (400 MHz, CDCl3): δ 7.40–7.20 (m, 5 H), 5.86–5.68 (m, 1 H), 5.06–4.91 (m, 2 H), 3.81–3.70 (m, 1 H), 3.53–3.37 (m, 1 H), 2.63–2.45 (m, 2 H), 2.45–2.36 (m, 2 H), 2.11–1.96 (m, 2 H), 1.74–1.51 (m, 3 H), 1.42–1.33 (m, 3 H); 13C NMR (101 MHz, CDCl3): δ 211.26, 144.82, 137.94, 128.55, 127.15, 126.45, 115.18, 69.36, 58.61, 53.38, 41.97, 38.91, 33.04, 28.55, 23.83, 22.77; HRMS-ESI (m/z) [M+H]+calcd. for C18H28NO2+ 290.2120; found 290.2115.
To a stirred solution of oxalyl chloride (3.62 mL, 42.31 mmol, 1.5 equiv) in CH2Cl2 (80 mL) at −78°C, dimethyl sulfoxide (6.01 mL, 84.63 mmol, 3.0 equiv.) was added over 15 min. The resulting mixture was stirred for another 40 min. Then a solution of ((R)-1-((R)-1-phenylethyl)aziridin-2-yl)methanol 1b (5.0 g, 28.21 mmol) in CH2Cl2 (30 mL) was added dropwise at the same temperature. The reaction mixture was stirred for 1.5 h at the same temperature. Triethylamine (11.81 mL, 42.31 mmol, 3.0 equiv.) was then added at −78°C and allowed to stir at the same temperature for 30 min. It was then warmed to 0°C and stirred for 30 min. After completion of the reaction per TLC indication, the reaction mixture was quenched with water (80 mL) and extracted with CH2Cl2 (2 × 100 mL). Combined organic layers were dried over anhydrous Na2SO4 and concentrated under reduced pressure to obtain a crude aldehyde which was used for the Wittig reaction without further purification.
To a solution of [(1R)-phenylethylaziridine]-(2R)-carboxaldehyde (3.0 g, 17.12 mmol, 1.0 equiv) in MeOH (34 mL), methyl triphenylphosphoranylideneacetate (6.86 g, 20.54 mmol, 1.2 equiv.) was added at 0°C. The resulting mixture was allowed to stir for 1 h at 0°C. After completion of the reaction, methanol was removed under vacuum and H2O (2 × 50 mL) was added to the reaction mixture. The organic layer was then extracted with CH2Cl2 (3 × 100 mL), dried over anhydrous Na2SO4, and concentrated under a vacuum. The crude product was purified by flash column chromatography on silica gel to afford a pure product (Z)-methyl 3-((S)-1-((R)-1-phenylethyl) aziridin-2-yl)acrylate 1c (3.2 g, 82%) TLC Rf (30% EtOAc/hexane = 0.4).
[α]D = +58.3 (c = 16.20, in CHCl3); 1H NMR (400 MHz, CDCl3): δ 7.43–7.22 (m, 6 H), 6.78 (dt, J = 13.1, 6.6 Hz, 1 H), 6.20–6.09 (m, 1 H), 5.98–5.82 (m, 2 H), 3.78 (s, 3 H), 2.66–2.59 (m, 1 H), 2.21–2.10 (m, 1 H), 1.86–1.72 (m, 2 H), 1.70–1.63 (m, 1 H), 1.47 (t, J = 6.8 Hz, 1 H), 1.43 (dd, J = 6.5, 1.6 Hz, 1 H); 13C NMR (101 MHz, CDCl3): δ 166.57, 150.51, 148.54, 143.78, 128.25, 127.01, 126.64, 126.61, 121.22, 120.17, 69.62, 51.09, 39.45, 37.32, 22.89; HRMS-ESI (m/z) [M+1]+calcd. for C14H18NO2+ 231.1337; found C14H18NO2+- H+ 231.1331.
To a stirred solution of olefin, 1c (2.0 g, 8.64 mmol) in CH2Cl2 (24 mL) at 0°C, 2-nitrobenzenesulfonylhydrazide (NBSH) (7.5 g, 34.58 mmol, 4.0 equiv) was added. Then triethylamine (10.0 mL, 69.17 mmol, 8.0 equiv) was added in a dropwise manner at the same 0°C. The reaction mixture was then allowed to stir at room temperature for an additional 12 h. After completion of starting material as confirmed by TLC, the reaction mixture was quenched with saturated NaHCO3 solution, extracted with CH2Cl2 (3 × 50 mL), and concentrated under vacuum to give a crude product 1d, which was then purified by column chromatography to give compound methyl 3-((S)-1-((R)-1-phenylethyl)aziridin-2-yl)propanoate (1d) as a viscous liquid (1.72 g, 85% yield), TLC Rf (40% EtOAc/hexane = 0.30).
[α]20D = + 35.4 (c = 0.32, MeOH); 1H NMR (400 MHz, CDCl3): δ 7.40–7.23 (m, 5 H), 3.72 (s, 3 H), 2.58–2.51 (m, 2 H), 2.42 (q, J = 6.5 Hz, 1 H), 2.02–1.93 (m, 1 H), 1.65–1.58 (m, 2 H), 1.52 (d, J = 3.2 Hz, 1H), 1.44 (d, J = 6.6 Hz, 3 H), 1.36–1.27 (m, 1 H); 13C NMR (101 MHz, CDCl3) δ 173.71, 144.49, 128.22, 126.87, 126.71, 69.60, 51.52, 39.10, 33.41, 31.91, 28.26, 23.27; HRMS-ESI (m/z) [M+H]+calcd. for C14H20NO2+ 234.1494; found 234.1486.
To a stirred solution of saturated ester 1d (1.5 g, 6.42 mmol, 1.0 equiv.) and N,O-dimethylhydroxylamine hydrochloride (1.15 g, 9.64 mmol, 1.5 equiv.) in dry THF (22 mL) at 0°C, i-PrMgCl (9.6 mL, 2.0 M in THF, 19.28 mmol, 3.0 equiv.) was slowly added. The reaction mixture was stirred for 1 h at the same 0°C. After completion of the reaction as per TLC, the reaction mixture was quenched with saturated NH4Cl solution (2 mL) and extracted with EtOAc (4 × 50 mL). Combined organic layers were dried over anhydrous Na2SO4. Under vacuum, solvents were removed to obtain a crude amide product, which was purified by column chromatography to give a pure Weinreb amide product (1e) (1.40 g, 83% yield), TLC Rf (90% EtOAc/hexane = 0.1).
[α]20D = + 28.3 (c = 0.70, MeOH); 1H NMR (400 MHz, CDCl3): δ 7.37–7.15 (m, 5 H), 3.63 (d, J = 14.7 Hz, 3 H), 3.17–3.09 (m, 3 H), 2.60 (dd, J = 14.3, 8.6 Hz, 2 H), 2.37 (q, J = 6.5 Hz, 1 H), 1.98–1.86 (m, 1 H), 1.65–1.51 (m, 2 H), 1.46 (d, J = 3.3 Hz, 1 H), 1.39 (d, J = 6.6 Hz, 3 H), 1.25 (d, J = 6.3 Hz, 1 H);13C NMR (101 MHz, CDCl3): δ 173.76, 144.20, 127.90, 126.54, 126.43, 69.23, 60.81, 39.24, 33.25, 31.83, 29.36, 27.46, 23.01; HRMS-ESI (m/z) [M+H]+calcd. for C15H23N2O2+ 263.1759; found 263.1751.
To a stirred solution of Weinreb amide 1e (1.0 g, 3.8 mmol, 1.0 equiv.) in dry THF (50 mL) at 0°C, n-Butylmagnesium chloride solution C4H9MgCl (1.9 mL, 2.0 M in Ether, 1.90 mmol) was added. The reaction mixture was slowly warmed to room temperature and stirred for 0.5 h. After completion of the reaction as per TLC, the reaction mixture was quenched with NH4Cl solution and extracted with EtOAc (3 × 80 mL). Combined organic layers were dried over Na2SO4 and concentrated in vacuo, which was then purified by silica gel column chromatography to afford ketone product 2 (830 mg, 80% yield) TLC Rf (60% EtOAc/hexane = 0.5).
[α]20D = + 51.0 (c = 0.06, MeOH); 1H NMR (400 MHz, CDCl3): δ 7.42–7.32 (m, 3 H), 7.31–7.21 (m, 2 H), 2.70–2.58 (m, 1 H), 2.47 (dd, J = 9.3, 5.6 Hz, 2 H), 2.41 (q, J = 6.5 Hz, 1 H), 2.01–1.89 (m, 3 H), 1.66–1.53 (m, 1 H), 1.52–1.48 (m, 1 H), 1.43 (d, J = 6.6 Hz, 3 H), 1.40–1.26 (m, 1 H), 0.98–0.90 (m, 3 H); 13C NMR (101 MHz, CDCl3): δ 210.77, 144.51, 128.26, 126.91, 126.75, 69.64, 42.52, 40.40, 39.36, 33.49, 27.10, 25.94, 23.32, 22.35, 13.85; HRMS-ESI (m/z) [M+H]+calcd. for C17H26NO+ 260.2014; found 260.2003.
The procedure was analogous to that used for preparing compound 8 using 1-((S)-1-((R)-1-phenylethyl)aziridin-2-yl)heptan-3-one (2) (800 mg, 3.084 mmol, 1.0 equiv.) in an acetone (10.0 mL) and water (5.0 mL) solvent system with trifluoro acetic acid (TFA) (0.23 mL, 3.08 mmol, 1.0 equiv) to afford compound (R)-2-hydroxy-1-(((R)-1-phenylethyl)amino)nonan-5-one (5) (760 mg, 90% yield), TLC Rf (90% EtOAc/hexane = 0.1).
[α]20D = + 87.2 (c = 0.14, MeOH); 1H NMR (400 MHz, CDCl3): 7.49–7.11 (m, 5H), 3.87–3.66 (m, 1H), 3.54–3.32 (m, 1H), 2.63–2.43 (m, 3H), 2.40–2.31 (m, 3H), 1.77–1.63 (m, 1H), 1.61–1.46 (m, 3H), 1.44–1.36 (m, 3H), 1.32–1.20 (m, 2H), 0.96–0.82 (m, 3H); 13C NMR (101 MHz, CDCl3): δ 211.63, 144.80, 128.57, 127.19, 126.48, 69.35, 58.61, 53.37, 42.64, 38.82, 28.56, 25.96, 23.79, 22.32, 13.83; HRMS-ESI (m/z) [M+H]+calcd. for C17H28NO2+ 278.2120; found 278.2114.
Compound 5 (600 mg, 2.16 mmol) was taken in MeOH (10 mL) and degassed with N2 for 2 h. Then 20% Pd(OH)2/C (242 mg, 1.7 mmol, 0.8 equiv) was added and the mixture was hydrogenated under an atmospheric pressure of hydrogen for 16 h. After completion of the reaction as per TLC, the reaction mixture was diluted with MeOH (20 mL) and filtered on a pad of Celite using MeOH as solvent. The filtrate was concentrated under vacuum and crude product was used for N-Cbz protection reaction without purification.
To a solution of crude product (325 mg) in dry CH2Cl2 (5 mL), triethyl amine (0.5 mL, 3.25 mmol, 1.6 equiv.) and benzyl chloroformate (CbzCl) (0.43 mL, 3.05 mmol, 1.5 equiv.) were added at 0°C. Then resulting mixture was allowed to stir at room temperature for 3 h. After completion of the reaction, the reaction was quenched with H2O (2 mL). The organic layer was then extracted with CH2Cl2 (2 × 20 mL), dried over anhydrous Na2SO4, and concentrated under vacuum to give an N-Cbz protected piperidine crude product, which was then purified by flash column chromatography on silica gel to afford a pure product (2R,5R)-benzyl 2-butyl-5-hydroxypiperidine-1-carboxylate (7) (412 mg, 70% yield for 2 steps) TLC Rf (40% EtOAc/hexane = 0.3).
[α]20D = −9.3 (c = 0.30, MeOH); 1H NMR (400 MHz, CDCl3): δ 7.44–7.29 (m, 5H), 5.16 (q, J = 12.3 Hz, 2H), 4.33 (s, 1H), 4.12 (s, 1H), 3.90 (d, J = 38.5 Hz, 1H), 3.05 (d, J = 14.0 Hz, 1H), 2.21–1.93 (m, 2H), 1.84–1.58 (m, 3H), 1.46–1.11 (m, 6H), 0.88 (t, J = 6.4 Hz, 3H); 13C NMR (101 MHz, CDCl3): δ 156.52, 136.76, 128.42, 127.89, 127.77, 67.08, 64.47, 50.59, 44.76, 28.73, 28.46, 25.40, 22.54, 14.05; HRMS-ESI (m/z) [M+H]+calcd. for C17H26NO3+ 292.1912; found 292.1905.
To a stirred solution of 7 (200 mg, 0.68 mmol, 1.0 equiv) in CH2Cl2 (15 mL), Dess-Martin periodinane (DMP) (581 mg, 1.37 mmol, 2.0 equiv) was added at 0°C. After 10 min, the reaction mixture was allowed to stir for an additional 8 h at room temperature. After completion of the reaction as per TLC, the reaction mixture was filtered on a pad of Celite using CH2Cl2 (2 × 5 mL) as solvent. The saturated NaHCO3 (5 mL) was used to treat the filtrate. Combined organic extracts were washed with CH2Cl2 (2 × 15 mL), dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel to provide keto compound 9 (158 mg, 80%) as a liquid, TLC Rf (30% EtOAc/hexane = 0.5).
[α]20D = +48.0 (c = 0.55, MeOH); 1H NMR (400 MHz, CDCl3): δ 7.50–7.32 (m, 5H), 5.28–5.08 (m, 2H), 4.68–4.44 (m, 1H), 4.40–4.18 (m, 1H), 3.62 (d, J = 17.7 Hz, 1H), 2.44 (t, J = 6.7 Hz, 2H), 2.24 (m, 1H), 1.68 (m, 2H), 1.54 (m, 1H), 1.32 (m, 4H), 0.98–0.84 (m, 3H); 13C NMR (101 MHz, CDCl3): δ 207.37, 166.95, 136.29, 128.52, 128.16, 127.99, 67.50, 50.81, 50.40, 35.69, 27.90, 22.51, 13.97; HRMS-ESI (m/z) [M+H]+calcd. for C17H24NO3+ 290.1756; found 290.1748.
To a stirred solution of methyl 3-((S)-1-((R)-1-phenylethyl) aziridine-2-yl)propanoate (1d) (2 g, 8.57 mmol, 1.0 equiv) in dry CH2Cl2 (40 mL), a solution of diisobutylaluminum hydride solution (DIBAL-H) (9.4 mL, 1.0 M in Toluene, 9.43 mmol) was added dropwise at—78°C. The reaction mixture was allowed to stir for 2 h at the same temperature. After completion of the reaction as per TLC indication, the reaction was quenched with a saturated solution of Na-k tartarate (10 mL) at 0°C. The reaction mixture was then stirred for an additional 1 h. The crude mixture was extracted with CH2Cl2 (3 × 20 mL), dried over anhydrous Na2SO4, and concentrated under vacuum to afford crude aldehyde, which was used for the next step without further purification.
To a stirred solution of crude aldehyde in dry THF (40 mL) at 0°C, n-butylmagnesium chloride solution C4H9MgCl (4.1 mL, 2.0 M in ether, 4.0 mmol) was added. The reaction mixture was allowed to stir for 2 h at the same temperature. After completion of the reaction per TLC indication, the reaction mixture was quenched with saturated NH4Cl solution and extracted with EtOAc (2 × 100 mL). Combined organic layers were dried over Na2SO4 and concentrated in vacuo to afford a crude product which was then purified by silica gel column chromatography to afford diastereomeric (1:1) hydroxy compound 10 (1.82 g, 80% yield for two steps) at the same Rf, TLC Rf (40% EtOAc/hexane = 0.5).
[α]20D = + 55.2 (c = 0.11, MeOH);1H NMR (400 MHz, CDCl3): δ 7.43–7.24 (m, 10H), 3.67–3.52 (m, 1H), 2.56–2.50 (m, 1H), 2.49–2.43 (m, 1H), 2.06–1.88 (m, 1H), 1.79–1.69 (m, 2H), 1.69–1.64 (m, 2H), 1.60 (qd, J = 4.0, 1.9 Hz, 3H), 1.54–1.42 (m, 11H), 1.41–1.31 (m, 4H), 1.30–1.26 (m, 1H), 1.04–0.85 (m, 6H); 13C NMR (101 MHz, CDCl3): δ 144.22, 144.04, 128.31, 127.00, 126.75, 71.56, 69.81, 39.96, 39.68, 37.34, 33.88, 33.21, 28.82, 28.67, 27.97, 23.37, 22.84, 14.08; HRMS-ESI (m/z) [M+H]+calcd. for C17H27NO+ 262.2171; found 262.2160.
Tert-butyldimethylsilyltrifluoromethane sulfonate (TBSOTf) (1.5 mL, 6.73 mmol, 1.1 equiv) was slowly added to a cooled solution (0°C) of alcohol 10 (1.6 g, 6.12 mmol, 1.0 equiv) and 2,6-lutidine (1.4 mL, 12.24 mmol, 2.0 equiv) in dry CH2Cl2 (30 mL). After 20 min, the reaction mixture was diluted with CH2Cl2 (20 mL), quenched with water, and extracted with CH2Cl2 (2 × 15 mL). Combined organic layers were dried over anhydrous Na2SO4 and concentrated in vacuo to obtain a crude product which was purified by flash column chromatography to afford TBS protected compound 11 (1.92 g, 85% yield) TLC Rf (20% EtOAc/hexane = 0.6).
[α]20D = + 26.2 (c = 0.5, MeOH); 1H NMR (400 MHz, CDCl3): δ 7.46–7.26 (m, 10H), 3.90–3.53 (m, 2H), 2.46–2.34 (m, 2H), 1.85–1.51 (m, 3H), 1.51–1.43 (m, 10H), 1.39–1.23 (m, 6H), 0.96–0.86 (m, 18H), 0.12–0.06 (m, 6H), 0.04 (s, 6H); 13C NMR (101 MHz, CDCl3): δ 144.65, 128.24, 126.84, 72.27, 72.21, 71.92, 71.84, 69.89, 40.76, 37.12, 36.57, 35.14, 34.98, 33.36, 29.42, 28.64, 27.50, 25.95, 25.91, 25.70, 25.66, 23.29, 22.87, 18.14, 18.11, 14.11, −2.96, −4.37; HRMS-ESI (m/z) [M+H]+calcd. for C23H42NOSi+ 376.3035; found 376.3023.
To (2S)-2-(3-((tert-butyldimethylsilyl)oxy)heptyl)-1-((R)-1-phenylethyl)aziridine (11) (800 mg, 2.12 mmol) in CH2Cl2 (10 mL), acetic acid (0.3 mL, 4.25 mmol, 2.5 equiv) was added at 0°C. The reaction mixture was allowed to stir at room temperature for 12 h. After completion of the reaction per TLC indication, the reaction mixture was quenched with a saturated solution of NaHCO3 (5 mL), extracted with CH2Cl2 (3 × 15 mL), and concentrated under vacuum to obtain a crude product which was then purified by flash column chromatography on silica gel to afford a pure product 12 (820 mg, 89%) TLC Rf (20% EtOAc/hexane = 0.5).
[α]20D = + 38.2 (c = 0.70, MeOH);1H NMR (400 MHz, CDCl3): δ 7.40–7.16 (m, 10H), 4.05–3.94 (m, 1H), 3.92–3.81 (m, 3H), 3.71–3.53 (m, 1H), 2.72–2.45 (m, 1H), 2.07–2.01 (m, 6H), 1.62–1.53 (m, 3H), 1.50–1.38 (m, 3H), 1.36–1.31 (m, 4H), 1.30–1.21 (m, 3H), 0.92–0.85 (m, 18H), 0.05–0.00 (m, 12H); 13C NMR (101 MHz, CDCl3): δ 171.01, 145.76, 145.72, 128.40, 126.93, 126.52, 72.06, 72.02, 66.86, 66.83, 55.23, 55.17, 53.61, 36.62, 32.59, 27.49, 26.50, 25.91, 24.79, 22.86, 20.92, 18.10, 14.11, −4.47; HRMS-ESI (m/z) [M+H]+calcd. for C25H46NO3Si+ 436.3247; found 436.3234.
To a stirred solution of 12 (500 mg, 1.14 mmol) in dry THF (12 mL) in a polypropylene vial HF-Py complex (70%, 0.4 mL) at 0°C was added. The reaction mixture was slowly raised to room temperature and stirred for 12 h. After completion of the reaction per TLC indication, the reaction mixture was cautiously quenched with saturated aqueous NaHCO3 and stirred for 20 min. Then both layers were separated. The aqueous layer was further extracted with EtOAc (2 × 20 mL). Combined organic layers were washed with saturated aqueous CuSO4 (5 mL), water (5 mL), and brine (5 mL), dried over Na2SO4, and concentrated in vacuo to obtain a crude product, which was then purified by flash column chromatography on silica gel to afford a pure product 13 (295 mg, 80%) TLC Rf (70% EtOAc/hexane = 0.2).
[α]20D =—51.2 (c = 0.22, MeOH); 1H NMR (400 MHz, CDCl3): δ 7.51–7.12 (m, 10H), 4.17–4.02 (m, 2H), 4.02–3.94 (m, 2H), 3.93–3.81 (m, 2H), 3.63–3.43 (m, 1H), 3.06–2.81 (m, 1H), 2.81–2.60 (m, 1H), 2.13–1.94 (m, 6H), 1.84–1.45 (m, 4H), 1.46–1.16 (m, 9H), 1.03–0.71 (m, 6H); 13C NMR (101 MHz, CDCl3): δ 170.77, 170.67, 144.95, 144.65, 128.51, 128.47, 127.18, 127.12, 126.38, 126.26, 71.66, 71.25, 66.17, 65.82, 55.22, 54.92, 54.20, 52.68, 37.22, 37.15, 34.45, 32.82, 29.59, 27.95, 27.92, 27.59, 23.78, 23.00, 22.71, 22.69, 20.71, 13.99; HRMS-ESI (m/z) [M+H]+calcd. for C19H32NO3+ 322.2382; found 322.2372.
Compound 13 (250 mg, 0.77 mmol) was taken in MeOH (10 mL) and degassed with N2 for 1 h. Then 20% Pd(OH)2/C (218 mg, 1.55 mmol, 2.0 equiv) was added and the mixture was hydrogenated under an atmospheric pressure of hydrogen for 16 h. The reaction mixture was diluted with MeOH (15 mL) and filtered on a pad of Celite using MeOH as a solvent. The filtrate was concentrated under a vacuum to obtain a crude product which was used for the N-Cbz protection reaction without purification.
To a solution of crude product in dry THF (8 mL), LiHMDS (0.4 mL, 0.44 mmol, 1.2 equiv), benzyl chloroformate (CbzCl) (0.2 mL, 0.58 mmol, 1.6 equiv) were added at 0°C. The resulting mixture was allowed to stir at the same temperature for 3 h. After completion of the reaction, the mixture was quenched with H2O (3 mL). The organic layer was extracted with EtOAc (2 × 15 mL), dried over anhydrous Na2SO4, and concentrated under vacuum to obtain an N-Cbz protected crude product, which was purified by flash column chromatography on silica gel to afford a pure product 14 (120 mg, 80% yield for 2 steps) TLC Rf (80% EtOAc/hexane = 0.2).
[α]20D = + 28.5 (c = 0.07, MeOH); 1H NMR (400 MHz, CDCl3): δ 7.51–7.31 (m, 10H), 5.24–5.08 (m, 4H), 4.28–4.08 (m, 5H), 3.67–3.52 (m, 2H), 3.38–3.15 (m, 2H), 1.99–1.90 (m, 6H), 1.77–1.49 (m, 3H), 1.47–1.19 (m, 10H), 1.18–1.08 (m, 3H), 1.01–0.73 (m, 6H); 13C NMR (101 MHz, CDCl3) δ 170.18, 170.15, 155.08, 155.07, 134.91, 128.66, 128.61, 128.44, 71.40, 69.93, 69.89, 69.45, 69.18, 48.36, 48.32, 37.30, 34.43, 33.11, 33.04, 27.81, 27.75, 27.46, 23.20, 22.64, 14.76, 14.00; HRMS-ESI (m/z) [M+H]+calcd. for C19H30NO5+ 352.2124; found 352.2114.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
NS: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing–original draft, Writing–review and editing. H-JH: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Validation, Visualization, Writing–original draft, Writing–review and editing.
The authors declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by a grant (NRF-2012M3A7B4049645 with Centre for New Directions in Organic Synthesis) of the National Research Foundation of Korea and HUFS fund 2023.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2023.1280633/full#supplementary-material
Bates, R. W., Sivarajan, K., and Straub, B. F. (2011). A Synthesis of pseudoconhydrine and its epimer via hydroformylation and dihydroxylation. J. Org. Chem. 76 (16), 6844–6848. doi:10.1021/jo2008912
Choi, J., Yadav, N. N., and Ha, H.-J. (2017). Preparation of a stable bicyclic aziridinium ion and its ring expansion toward piperidines and azepanes. Asian J. Org. Chem. 6, 1292–1307. doi:10.1002/ajoc.201700080
Dalabehera, N. R., Meher, S., Palai, B. B., and Sharma, N. K. (2020). Instability of amide bond with trifluoroacetic acid (20%): synthesis, conformational analysis, and mechanistic insights into cleavable amide bond comprising β-troponylhydrazino acid. ACS Omega 5, 26141–26152. doi:10.1021/acsomega.0c03729
D’hooghe, M., and Ha, H.-J. (2016). Synthesis of 4- to 7-Membered heterocycles by ring expansion: aza-, oxo-and thiaheterocyclic Small-Ring Systems. Heidelberg. Springer, 1–367.
Dolfen, J., Yadav, N. N., De Kimpe, N., D’hooghe, M., and Ha, H.-J. (2016). Front cover picture: bicyclic aziridinium ions in azaheterocyclic chemistry – preparation and synthetic application of 1-Azoniabicyclo[n.1.0]alkanes (adv. Synth. Catal. 22/2016). Adv. Synth. Catal. 358, 3483–3511. doi:10.1002/adsc.201601081
Eum, H., Choi, J., Cho, C.-G., and Ha, H.-J. (2015). Cover picture: regiochemistry-directed syntheses of polyhydroxylated alkaloids from chiral aziridines (asian J. Org. Chem. 12/2015). Asian J. Org. Chem. 4, 1325–1340. doi:10.1002/ajoc.201500380
Ha, H.-J., Jung, J.-H., and Lee, W. K. (2014). Application of regio- and stereoselective functional group transformations of chiral aziridine-2-carboxylates. Asian J. Org. Chem. 3, 1020–1035. doi:10.1002/ajoc.201402098
Lee, B. K., Lee, W. K., Yoon, D.-H., and Ha, H.-J. (2009). Selective reduction of C-C double bonds of 2-vinylaziridnes: preparation of enationmerically pure 2-alkylaziridines. Bull. Kor. Chem. Soc. 30, 3123–3126. doi:10.5012/bkcs.2009.30.12.3123
Lee, W.-K., and Ha, H.-J. (2003). High light of the chemistry of enantiomerically pure aziridine-2-carboxylates. Aldrichimica Acta 36 (2), 57–63. doi:10.1002/chin.200428252
Lee, Y.-S., Shin, Y.-H., Kim, Y.-H., Lee, K.-Y., Oh, C.-Y., Pyun, S.-J., et al. (2003). Stereoselective synthesis of (+)-spectaline. Tetrahedron Asymm 14, 87–93. doi:10.1016/S0957-4166(02)00794-2
Lin, S., Liu, C., Zhao, X., Han, X., Li, X., Ye, Y., et al. (2022). Recent advances of pyridinone in medicinal chemistry. Front. Chem. 23, 869860. doi:10.3389/fchem.2022.869860
Lopez, S. E., and Salazar, J. (2013). Trifluoroacetic acid: uses and recent applications in organic synthesis. J. Fluor. Chem. 156, 73–100. doi:10.1016/j.jfluchem.2013.09.004
Macha, L., D'hooghe, M., and Ha, H.-J. (2019). Deployment of aziridines for the synthesis of alkaloids and their derivatives. Synthesis 51, 1491–1515. doi:10.1055/s-0037-1611715
Macha, L., and Ha, H.-J. (2019). Total synthesis and absolute stereochemical assignment of microgrewiapine A and its stereoisomers. J. Org. Chem. 84, 94–103. doi:10.1021/acs.joc.8b02342
Michael, J. P. (2016). Simple indolizidine and quinolizidine alkaloids. Alkaloids Chem. Biol. 75, 1–498. doi:10.1016/bs.alkal.2014.12.001
Ranjith, J., and Ha, H.-J. (2022). Strained aziridinium ion, more synthetic approaches to nonaromatic nitrogen heterocycles, 1. John Wiley and Sons, Ltd., 499–528. doi:10.1002/9781119757153.ch15
Ranjith, J., and Ha, H.-J. (2021). Synthetic Applications of aziridinium ions. Molecules 26 (6), 1774. doi:10.3390/molecules26061774
Rao, V. K. S., and Kumar, P. (2006). An asymmetric aminohydroxylation route to cis-2,6-disubstituted piperidine-3-ol: application to the synthesis of (−)-deoxocassine. Tetrahedron 62, 9942–9948. doi:10.1016/j.tet.2006.08.014
Singh, A., Kim, B., Lee, W. K., and Ha, H.-J. (2011). Asymmetric synthesis of 1-deoxyazasugars from chiral aziridines. Org. Biomol. Chem. 9, 1372–1380. doi:10.1039/C0OB00730G
Srivastava, N., and Ha, H.-J. (2022). Highly efficient and stereoselective Mukaiyama aldol reaction with chiral aziridine-2-carboxaldehyde and its synthetic applications. Asian J. Org. Chem. 11 (1), 72–81. doi:10.1002/ajoc.202100567
Srivastava, N., Macha, L., and Ha, H.-J. (2020). Stereoselective synthesis of 2,6-disubstituted piperidine alkaloids. Org. Biomol. Chem. 18, 5493–5512. doi:10.1039/D0OB00918K
Stankovic, S., D’hooghe, M., Catak, S., Eum, H., Waroquier, M., Van Speybroeck, V., et al. (2012). Regioselectivity in the ring opening of non-activated aziridines. Chem. Soc. Rev. 41, 643–665. doi:10.1039/C1CS15140A
Vataku, E., Smith, D. T., and Njardarson, J. T. (2014). Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J. Med. Chem. 57, 10257–10274. doi:10.1021/jm501100b
Wang, Y.-G., Kumano, T., Kano, T., and Maruoka, K. (2009). Organocatalytic approach to enantioselective one-pot synthesis of pyrrolidine, hexahydropyrrolizine, and octahydroindolizine Core Structures. Org. Lett. 11 (9), 2027–2029. doi:10.1021/ol900477x
Yadav, N. N., Choi, J., and Ha, H.-J. (2016). One-pot multiple reactions: asymmetric synthesis of 2,6-cis-disubstituted piperidine alkaloids from chiral aziridine. Org. Biomol. Chem. 14, 6426–6434. doi:10.1039/C6OB00806B
Ying, X., Zhang, J., Wang, C., Huang, M., Ji, Y., Cheng, F., et al. (2018). Characterization of a carbonyl reductase from rhodococcus erythropolis WZ010 and its variant Y54F for the asymmetric synthesis of (S)-N- Boc-3-hydroxypiperidine. Molecules 23 (12), 3117. doi:10.3390/molecules23123117
Keywords: aziridine, regioselectivity, ring opening, pyrrolidine, piperidine
Citation: Srivastava N and Ha H-J (2023) Regioselective ring opening of aziridine for synthesizing azaheterocycle. Front. Chem. 11:1280633. doi: 10.3389/fchem.2023.1280633
Received: 21 August 2023; Accepted: 02 October 2023;
Published: 19 October 2023.
Edited by:
Hasan Küçükbay, İnönü University, TürkiyeReviewed by:
Turan Ozturk, Istanbul Technical University, TürkiyeCopyright © 2023 Srivastava and Ha. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hyun-Joon Ha, aGpoYUBodWZzLmFjLmty
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.