 Hidetoshi Noda
Hidetoshi Noda Yasuko Asada
Yasuko Asada
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem., 03 November 2023
Sec. Organic Chemistry
Volume 11 - 2023 | https://doi.org/10.3389/fchem.2023.1271896
This article is part of the Research TopicRecent Advances in Synthesizing and Utilizing Nitrogen-containing HeterocyclesView all 12 articles
In this study, the reactivity of the alkyl nitrenes, generated from the substituted hydroxylamine precursors, was determined using the same rhodium catalyst. The results revealed that in competitive C–H insertion experiments, the regioselectivity between benzylic and tertiary C–H bonds could be modulated by adding Brønsted acids or changing the substituents on oxygen. This study enhances our understanding of the metallonitrene structures and provides valuable insights for further development of selective N-heterocycle syntheses.
Nitrogen-containing compounds prevail over biologically active compounds (Lovering et al., 2009; Vitaku et al., 2014). Therefore, the synthetic chemists have made considerable efforts to introduce nitrogen atoms at desired positions in the molecular skeleton (Park et al., 2017; Trowbridge et al., 2020). Among various approaches, utilizing nitrenes is preferred, as they can functionalize the otherwise inert C–H bonds (Müller and Fruit, 2003; Díaz-Requejo and Pérez, 2008; Darses et al., 2017). Given the high reactivity of free nitrenes, metallonitrenes are primarily used for nitrogen insertion as their reactivity can be regulated by the structure of the metal complexes (Ju and Schomaker, 2021). Metalated nitrenes are typically generated from oxidized precursors, either prepared in situ or from those containing a labile bond (Breslow and Gellman, 1982; Nägeli et al., 1997; Lebel et al., 2005).
The substituents on the nitrogen can be used to classify nitrene structures such as carbamoyl (Cui and He, 2004), sulfamoyl (Espino et al., 2001), aryl (Stokes et al., 2007), acyl (Hong et al., 2018), and alkyl (Hennessy and Betley, 2013) nitrenes. The class of nitrenes determines the structure of the resulting product. For instance, intramolecular C–H insertion of sulfamoyl nitrenes provides a 1,3-aminoalcohol unit, whereas that of alkyl nitrenes delivers a saturated N-heterocycle. Therefore, the advancement in nitrene chemistry is directed toward expanding the product structures and its evolution has resulted in the development of efficient catalysts and new precursors (Roizen et al., 2012; Alderson et al., 2017; Hong et al., 2021). The chemoselective amination reactions have garnered considerable interest in this area that has triggered the identification of various catalyst-controlled aminations (Noda et al., 2021). However, there are limited studies in the literature investigating the reactivity difference between various nitrene classes using identical catalysts. The comparison of the same nitrene class obtained from different precursors is also lacking. This could be attributed to the lack of a suitable system for studying the reactivities.
We previously reported that substituted isoxazolidin-5-ones (Annibaletto et al., 2017; Noda, 2021) acted as alkyl nitrene precursors in the presence of rhodium (Yu et al., 2019) or copper catalysts (Tak et al., 2021a). The generated metallonitrene reacted intramolecularly with an aromatic ring (Tak et al., 2021b) or C(sp3)–H bond (Espinosa et al., 2019) to afford the corresponding unprotected cyclic β-amino acids. In our study to synthesize remotely decorated trisubstituted pyrrolidines via C(sp3)–H insertion (Tang et al., 2022) using Rh2(esp)2, a Du Bois catalyst (Espino et al., 2004), it was observed that the alkyl nitrene derived from the heterocycle selectively aminated the C(sp3)–H bond at the allylic position without touching the double bond (Scheme 1A), which was in contrast to the sulfamoyl nitrene favoring aziridination over C(sp3)–H insertion, using the same rhodium catalyst (Scheme 1B) (Fiori et al., 2009). These results highlighted the unique nature of the nitrene reactivities associated with their structural classes.
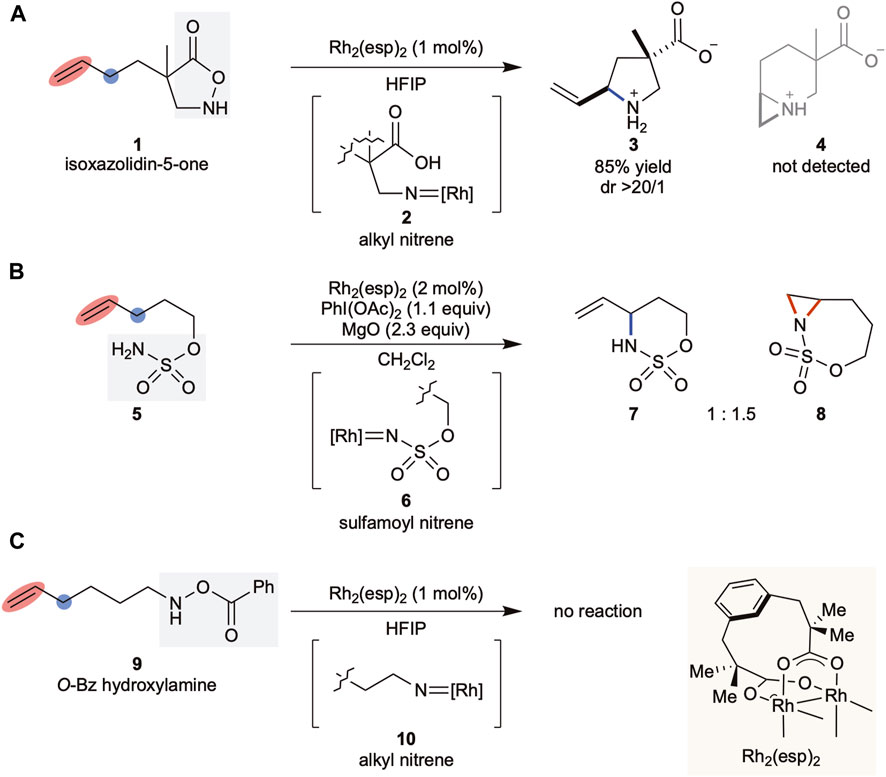
SCHEME 1. (A–C) Background of this work. esp: α, α, α′, α′-tetramethyl-1,3-benzenedipropionic acid, HFIP: 1,1,1,3,3,3-hexafluoroisopropanol.
Driven by the significance of saturated N-heterocycles in drug discovery programs, we further investigated alkyl nitrenes and identified O-benzoyl hydroxylamines as efficient alkyl nitrene precursors for the transformation of a linear primary amine into the corresponding five-membered cyclic amine (Noda et al., 2020). When we subjected substrate 9 to the catalytic conditions to explore the scope of the method, no reaction was observed, resulting in full recovery of the substrate (Scheme 1C). As both substrates 1 and 9 were expected to generate similar alkyl rhodium nitrene species 2 and 10, respectively, as shown in Scheme 2, this difference in the outcomes could be attributed to the precursor structure. However, the lack of insight into the structure-reactivity relationship between nitrene precursors and the reaction conditions required a further detailed examination of these factors. Herein, we report our study on the reactivity of alkyl nitrenes derived from substituted hydroxylamines.
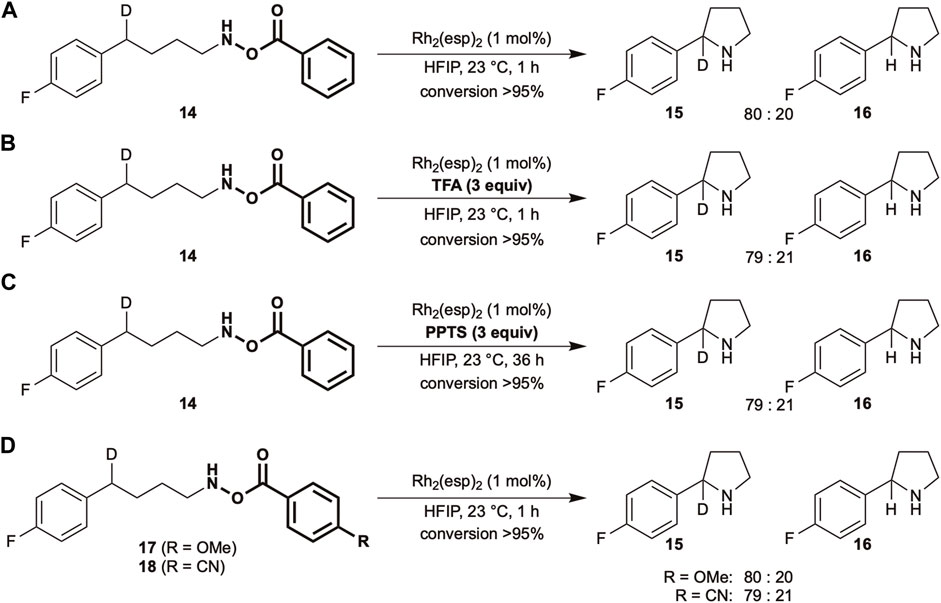
SCHEME 2. KIE experiments using Rh2(esp)2 as a catalyst under various conditions.
From the outset, we focused on the reactivities of alkyl nitrenes, as the products obtained from the intramolecular amination are medicinally important saturated N-heterocycles. In addition to isoxazolidin-5-ones and O-Bz hydroxylamines, alkyl azides (Thacker et al., 2016; Bagh et al., 2017; Shing et al., 2018; Qin et al., 2019), and O-Ts hydroxylamines (Munnuri et al., 2017) act as nitrene precursors. Owing to their stability and facile structural modification, O-Bz hydroxylamines were used as model substrates in this study, and Rh2(esp)2 was used as the catalyst. Examination of the presumed nitrene structures 2 and 10 implied that a suitably located acidic proton in 2 played an important role in determining the reactivity, which was the driving force to investigate the additive effect using Brønsted acids and bases.
Table 1 summarizes the influence of additives on the regioselectivity of reactions, where the selectivity between benzylic and tertiary C–H bonds was used as a reactivity probe. In the absence of additives, the site selectivity of 11aA is close to 1:4, favoring the tertiary C–H bond (entry 1). The ratio marginally decreases in the presence of trifluoroacetic acid (TFA) (entry 2), suggesting that an acidic proton source plays a vital role in the selectivity-determining transition state. Higher acid loadings do not decrease the selectivity further (entry 3). It was observed that all the acids do not lower the selectivity, and the addition of a stronger acid, trifluoromethanesulfonic acid (TfOH), creates a strong bias in the reaction site for the tertiary C–H bond, although with a considerably slower kinetics (entry 4). Similar trend is observed for pyridinium p-toluenesulfonate (PPTS), which is a milder Brønsted acid compared to the TFA (entry 5). In contrast to acidic additives, the addition of a Brønsted base does not lead to a noticeable shift in the regioselectivity (entries 6–8).
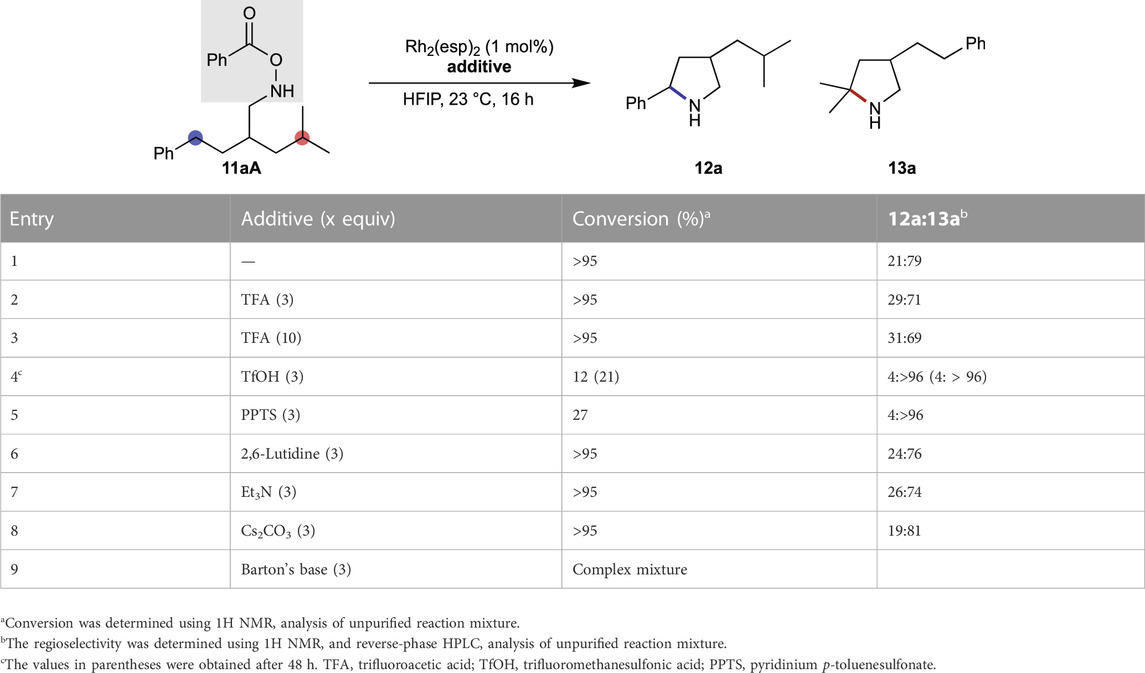
TABLE 1. Evaluation of additives on the regioselectivity of Rh-alkyl nitrene.
A change in the selectivity is often accompanied by a change in the reaction mechanism. Therefore, to understand the nature of reactive intermediates under Brønsted acidic conditions, kinetic isotope effect (KIE) experiments were conducted (Scheme 2). Fluorine-substituted compounds were used for this purpose as the high sensitivity of 19F nuclei in nuclear magnetic resonance (NMR) is beneficial for determining the selectivity. Under standard conditions, using 1 mol% Rh2(esp)2 as a catalyst in 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) at ambient temperature, a KIE value of 4.0 is obtained (Scheme 2A). The value remains the same in the presence of TFA (Scheme 2B), suggesting similar properties of the reactive intermediate in both cases. Despite the distinct selectivity trend observed in Table 1, the addition of PPTS does not change the KIE value (Scheme 2C). The KIE value of a similar N-Boc-O-Ts substrate was reported to be 5.3 (Munnuri et al., 2017), therefore, electronically tuned benzoate substrates were subjected to identical conditions. The obtained values are approximately same to those of the unmodified substrate (Scheme 2D).
We questioned whether the difference in the KIE values between O-Bz and O-Ts hydroxylamines would be translated into a difference in regioselectivities. Although the reactivity trend of O-Ts hydroxylamines has been previously studied using a different rhodium catalyst, a comparison with three classes of substrates was carried out for ranking the reactivities of various C–H bonds (benzylic, tertiary, secondary, and primary). In addition to regioselectivity, diastereoselectivity was utilized as the other reactivity probe.
The instability of the N-H form of O-Ts hydroxylamines requires in situ removal of a protective group from the nitrogen. Following the previous reports, the N-Boc group was used to mask the nitrogen atom and TFA was used as the proton source in TFE. Thus, TFA was included in the reactions with O-Bz substrates for a fair comparison (Table 2). All the products were isolated after conversion to their N-Ts forms. Examining the regioselectivity between benzylic and tertiary C–H bonds, a marginally lower selectivity is obtained using the O-Ts substrate compared to that of O-Bz substrate, although the diastereoselectivity is similar for both (entries 1 and 2). Owing to their diminished reactivity toward metallonitrene, cyclization products are not obtained at the secondary or primary C–H bonds for either of the substrates. However, better diastereoselectivities are recorded with O-Ts substrates compared to those with O-Bz substrates (entries 3 vs. 4 and 5 vs. 6).
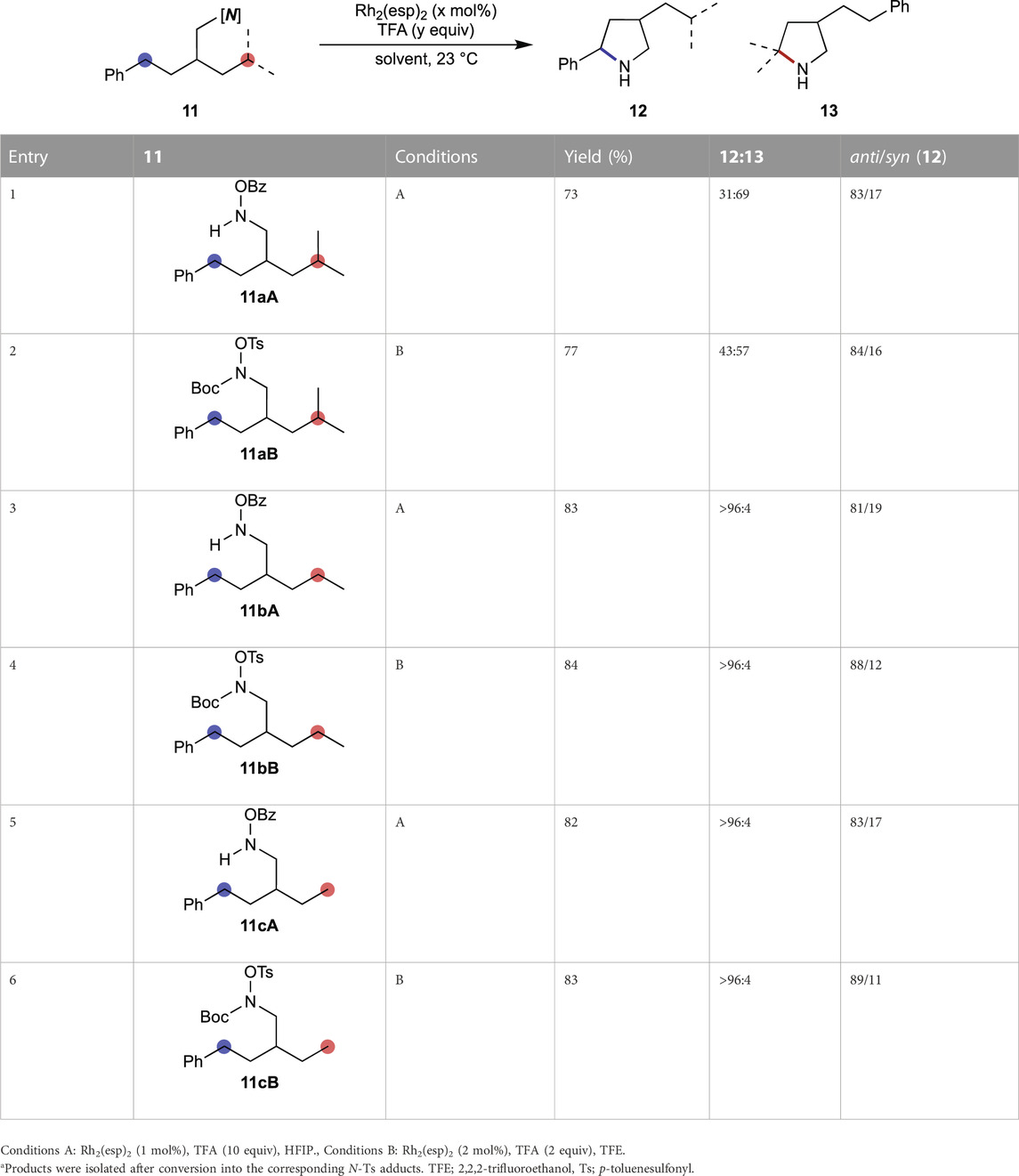
TABLE 2. Comparison between O-Bz and O-Ts hydroxylamines using rhodium catalyst.a
Falck and coworkers reported aziridination of olefins with O-(2,4-dinitrophenyl)hydroxylamine under the influence of a Rh2(esp)2 catalyst (Jat et al., 2014). The group revealed in a subsequent report that the combination of the same rhodium catalyst with O-Ts hydroxylamine aminated an aromatic C–H bond keeping the olefin moiety intact (Scheme 3A) (Paudyal et al., 2016). The authors proposed that the rhodium nitrene generated upon N–O bond cleavage was in equilibrium with the protonated nitrenium ion and the two species exhibited distinct reactivity, which justified the remarkable chemoselectivity switch (Scheme 3B). Moreover, acidity of the liberated Brønsted acids determined the equilibrium positions; the nitrophenol was not strong enough to protonate metallonitrenes, whereas a stronger sulfonic acid propelled the equilibrium forward to yield nitrenium ions.
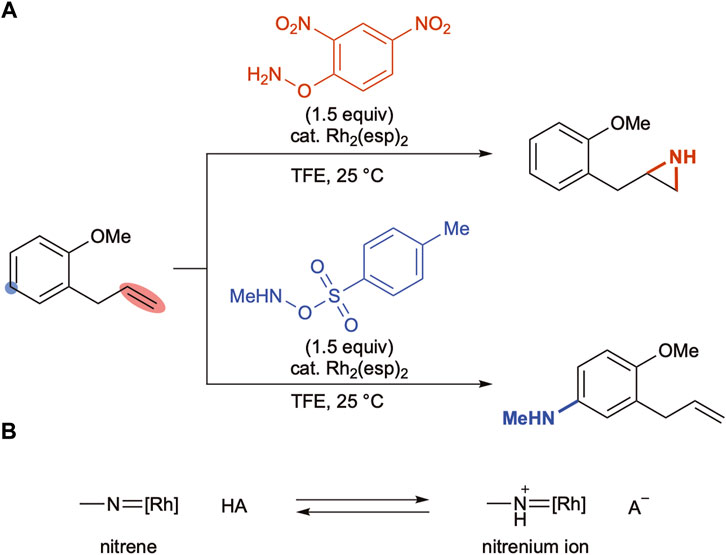
SCHEME 3. (A) Reagent-controlled chemoselectivity switch under rhodium-catalyzed conditions. (B) Possible interconversion between nitrenes and nitrenium ions.
Given that the acidity of the reaction medium is responsible for the equilibrium position, it was hypothesized that the addition of external Brønsted acids should play an identical role. The results in Table 1 reveal that the inclusion of sulfonic acids drastically changes the regioselectivity trend (entries 4,5, Table 1), which can be explained by a possible shift in the equilibrium. The observed lower reactivity could be ascribed to either the lower C–H insertion activity of the nitrenium ion intermediate or the slower N–O bond cleavage to form a reactive intermediate. However, the addition of external Brønsted acids does not alter the KIE values (Scheme 2A–C), suggesting that a similar reactive intermediate is involved in the C–H amination step in the absence or presence of acids.
Over the years, KIE experiments have been used to assess and elucidate the nature of reactive intermediates in various C–H functionalization reactions. It is well-known in nitrene chemistry that KIE values lower than three indicate the presence of a concerted mechanism involving a singlet nitrene. For instance, the KIE values of sulfamoyl nitrenes using a similar rhodium catalyst were in the range of 1.9–2.9 (Fiori et al., 2009; Varela-Álvarez et al., 2016). In contrast, a stepwise mechanism displays higher KIE values (Badiei et al., 2008; Harvey et al., 2011). The difference in KIE values between O-Bz (4.0) and O-Ts (5.3) hydroxylamines using the same catalyst inferred that precursor structures play a significant role in determining the nature of reactive intermediate but the acidity of the reaction medium. As a slight modification of the benzoic acid derivatives (pKa in H2O: p-CN-C6H4 3.55, C6H5 4.21, p-MeO-C6H4 4.47) did not affect the KIE values (Scheme 2D), a considerably drastic change in the acidity of the leaving groups might be required to induce a change.
The precursor structure-dependent KIE value implies that the reactive intermediate generated from O-Ts substrates possesses a triplet, radical-like nature. Radical-like intermediates typically follow the bond dissociation energy (BDE) order, which is the enthalpy change associated with the homolytic scission of the bond (Figure 1). The results summarized in Table 2 agree with this trend. Therefore, the O-Ts substrate undergoes the benzylic amination preferably compared to that for O-Bz (entry 1 vs. 2, Table 2). Comparable results are obtained in HFIP using 11aB (76% yield, 12a:13a 41:59, anti/syn 83/17), excluding the possibility that the choice of solvent governs the selectivity. The observed diastereoselectivities support the different natures of the reactive intermediates generated from O-Bz and O-Ts hydroxylamines.
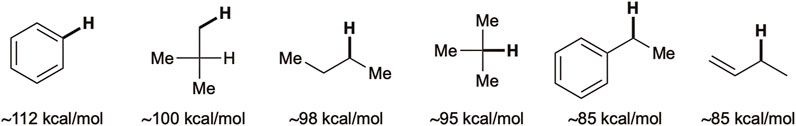
FIGURE 1. BDEs for various representative C–H bonds.
We have investigated the reactivity of rhodium alkyl nitrenes generated from substituted hydroxylamines. The addition of Brønsted acids modulated the regioselectivity of the intramolecular C–H insertion between the benzylic and tertiary positions. Despite the distinct regioselectivities, approximately identical KIE values were observed for various Brønsted acids. In contrast to external acids, the KIE values fluctuated as a function of the precursor structures. The conditions that produced the more radical-like reactive intermediate followed the expected reaction tendency. Although further efforts are required to completely understand the nature of reactive intermediates, particularly with the external addition of Brønsted acids, our results comprehensively confirmed that the reactivities of seemingly similar reactive intermediates could be regulated by incorporating additives or changing the precursor structures. This work significantly enhances our understanding of the rhodium nitrene structures, which are typically devoid of precursor residues, and opens up new avenues for a substrate-controlled approach to fine-tune the reactivity, as with other hydroxylamine-involving reactions (Noda et al., 2014; Niu and Buchwald, 2015).
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
HN: Writing–original draft, Writing–review and editing. YA: Writing–original draft. MS: Writing–review and editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the JSPS KAKENHI grant numbers JP20K06957 and JP22H05383 (Digi-TOS). HN is grateful to the Naito Foundation and the Takahashi Industrial and Economic Research Foundation for their financial support.
We are grateful to Dr. Ryuichi Sawa, Yumiko Kubota, Dr. Kiyoko Iijima, and Dr. Shinya Adachi at the Institute of Microbial Chemistry for their technical assistance with the NMR and MS analyses.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2023.1271896/full#supplementary-material
Alderson, J. M., Corbin, J. R., and Schomaker, J. M. (2017). Tunable, chemo- and site-selective nitrene transfer reactions through the rational design of silver(I) catalysts. Acc. Chem. Res. 50, 2147–2158. doi:10.1021/acs.accounts.7b00178
Annibaletto, J., Oudeyer, S., Levacher, V., and Brière, J.-F. (2017). Catalytic enantioselective syntheses of isoxazolidin-5-ones. Synthesis 49, 2117–2128. doi:10.1055/s-0036-1588765
Badiei, Y. M., Dinescu, A., Dai, X., Palomino, R. M., Heinemann, F. W., Cundari, T. R., et al. (2008). Copper–nitrene complexes in catalytic C–H amination. Angew. Chem. Int. Ed. 47, 9961–9964. doi:10.1002/anie.200804304
Bagh, B., Broere, D. L. J., Sinha, V., Kuijpers, P. F., van Leest, N. P., de Bruin, B., et al. (2017). Catalytic synthesis of N-Heterocycles via direct C(sp3)–H amination using an Air-stable Iron(III) species with a redox-active ligand. J. Am. Chem. Soc. 139, 5117–5124. doi:10.1021/jacs.7b00270
Breslow, R., and Gellman, S. H. (1982). Tosylamidation of cyclohexane by a cytochrome P-450 model. J. Chem. Soc. Chem. Commun. 1982, 1400–1401. doi:10.1039/c39820001400
Cui, Y., and He, C. (2004). A silver-catalyzed intramolecular amidation of saturated C–H bonds. Angew. Chem. Int. Ed. 43, 4210–4212. doi:10.1002/anie.200454243
Darses, B., Rodrigues, R., Neuville, L., Mazurais, M., and Dauban, P. (2017). Transition metal-catalyzed iodine(iii)-mediated nitrene transfer reactions: efficient tools for challenging syntheses. Chem. Commun. 53, 493–508. doi:10.1039/c6cc07925c
Díaz-Requejo, M. M., and Pérez, P. J. (2008). Coinage metal catalyzed C-H bond functionalization of hydrocarbons. Chem. Rev. 108, 3379–3394. doi:10.1021/cr078364y
Espino, C. G., Fiori, K. W., Kim, M., and Du Bois, J. (2004). Expanding the scope of C−H amination through catalyst design. J. Am. Chem. Soc. 126, 15378–15379. doi:10.1021/ja0446294
Espino, C. G., Wehn, P. M., Chow, J., and Du Bois, J. (2001). Synthesis of 1,3-difunctionalized amine derivatives through Selective C−H bond oxidation. J. Am. Chem. Soc. 123, 6935–6936. doi:10.1021/ja011033x
Espinosa, M., Noda, H., and Shibasaki, M. (2019). Synthesis of unprotected spirocyclic β-Prolines and β-Homoprolines by Rh-catalyzed C-H insertion. Org. Lett. 21, 9296–9299. doi:10.1021/acs.orglett.9b03198
Fiori, K. W., Espino, C. G., Brodsky, B. H., and Du Bois, J. (2009). A mechanistic analysis of the Rh-catalyzed intramolecular C–H amination reaction. Tetrahedron 65, 3042–3051. doi:10.1016/j.tet.2008.11.073
Harvey, M. E., Musaev, D. G., and Du Bois, J. (2011). A diruthenium catalyst for selective, intramolecular allylic C–H amination: reaction development and mechanistic insight gained through experiment and theory. J. Am. Chem. Soc. 133, 17207–17216. doi:10.1021/ja203576p
Hennessy, E. T., and Betley, T. A. (2013). Complex N-heterocycle synthesis via iron-catalyzed, direct C-H bond amination. Science 340, 591–595. doi:10.1126/science.1233701
Hong, S. Y., Hwang, Y., Lee, M., and Chang, S. (2021). Mechanism-guided development of transition-metal-catalyzed C-N bond-forming reactions using dioxazolones as the versatile amidating source. Acc. Chem. Res. 54, 2683–2700. doi:10.1021/acs.accounts.1c00198
Hong, S. Y., Park, Y., Hwang, Y., Kim, Y. B., Baik, M.-H., and Chang, S. (2018). Selective formation of γ-lactams via C–H amidation enabled by tailored iridium catalysts. Science 359, 1016–1021. doi:10.1126/science.aap7503
Jat, J. L., Paudyal, M. P., Gao, H., Xu, Q. L., Yousufuddin, M., Devarajan, D., et al. (2014). Direct stereospecific synthesis of unprotected N-H and N-Me aziridines from olefins. Science 343, 61–65. doi:10.1126/science.1245727
Ju, M., and Schomaker, J. M. (2021). Nitrene transfer catalysts for enantioselective C–N bond formation. Nat. Rev. Chem. 5, 580–594. doi:10.1038/s41570-021-00291-4
Lebel, H., Huard, K., and Lectard, S. (2005). N-Tosyloxycarbamates as a source of metal nitrenes: rhodium-catalyzed C-H insertion and aziridination reactions. J. Am. Chem. Soc. 127, 14198–14199. doi:10.1021/ja0552850
Lovering, F., Bikker, J., and Humblet, C. (2009). Escape from flatland: increasing saturation as an approach to improving clinical success. J. Med. Chem. 52, 6752–6756. doi:10.1021/jm901241e
Müller, P., and Fruit, C. (2003). Enantioselective catalytic aziridinations and asymmetric nitrene insertions into CH bonds. Chem. Rev. 103, 2905–2920. doi:10.1021/cr020043t
Munnuri, S., Adebesin, A. M., Paudyal, M. P., Yousufuddin, M., Dalipe, A., and Falck, J. R. (2017). Catalyst-controlled diastereoselective synthesis of cyclic amines via C-H functionalization. J. Am. Chem. Soc. 139, 18288–18294. doi:10.1021/jacs.7b09901
Nägeli, I., Baud, C., Bernardinelli, G., Jacquier, Y., Moraon, M., and Müllet, P. (1997). Rhodium(II)-catalyzed CH insertions with [(4-Nitrophenyl)sulfonyl]iminophenyl-λ3-iodane. Helv. Chim. Acta 80, 1087–1105. doi:10.1002/hlca.19970800407
Niu, D., and Buchwald, S. L. (2015). Design of modified amine transfer reagents allows the synthesis of α-chiral secondary amines via CuH-catalyzed hydroamination. J. Am. Chem. Soc. 137, 9716–9721. doi:10.1021/jacs.5b05446
Noda, H. (2021). Imbuing an old heterocycle with the power of modern catalysis: an isoxazolidin-5-one Story. Chem. Pharm. Bull. 69, 1160–1169. doi:10.1248/cpb.c21-00750
Noda, H., Asada, Y., and Shibasaki, M. (2020). O-Benzoylhydroxylamines as alkyl nitrene precursors: synthesis of saturated N-heterocycles from primary amines. Org. Lett. 22, 8769–8773. doi:10.1021/acs.orglett.0c02842
Noda, H., Erős, G., and Bode, J. W. (2014). Rapid ligations with equimolar reactants in water with the potassium acyltrifluoroborate (KAT) amide formation. J. Am. Chem. Soc. 136, 5611–5614. doi:10.1021/ja5018442
Noda, H., Tang, X., and Shibasaki, M. (2021). Catalyst-controlled chemoselective nitrene transfers. Helv. Chim. Acta 104, e2100140. doi:10.1002/hlca.202100140
Park, Y., Kim, Y., and Chang, S. (2017). Transition metal-catalyzed C–H amination: scope, mechanism, and applications. Chem. Rev. 117, 9247–9301. doi:10.1021/acs.chemrev.6b00644
Paudyal, M. P., Adebesin, A. M., Burt, S. R., Ess, D. H., Ma, Z., Kürti, L., et al. (2016). Dirhodium-catalyzed C-H arene amination using hydroxylamines. Science 353, 1144–1147. doi:10.1126/science.aaf8713
Qin, J., Zhou, Z., Cui, T., Hemming, M., and Meggers, E. (2019). Enantioselective intramolecular C–H amination of aliphatic azides by dual ruthenium and phosphine catalysis. Chem. Sci. 10, 3202–3207. doi:10.1039/c9sc00054b
Roizen, J. L., Harvey, M. E., and Du Bois, J. (2012). Metal-catalyzed nitrogen-atom transfer methods for the oxidation of aliphatic C-H bonds. Acc. Chem. Res. 45, 911–922. doi:10.1021/ar200318q
Shing, K.-P., Liu, Y., Cao, B., Chang, X.-Y., You, T., and Che, C.-M. (2018). N-heterocyclic carbene Iron(III) porphyrin-catalyzed intramolecular C(sp3)-H amination of alkyl azides. Angew. Chem. Int. Ed. 57, 12123–12127. doi:10.1002/ange.201806059
Stokes, B. J., Dong, H., Leslie, B. E., Pumphrey, A. L., and Driver, T. G. (2007). Intramolecular C−H amination reactions: exploitation of the Rh2(II)-catalyzed decomposition of azidoacrylates. J. Am. Chem. Soc. 129, 7500–7501. doi:10.1021/ja072219k
Tak, R. K., Amemiya, F., Noda, H., and Shibasaki, M. (2021a). Generation and application of Cu-bound alkyl nitrenes for the catalyst-controlled synthesis of cyclic β-amino acids. Chem. Sci. 12, 7809–7817. doi:10.1039/d1sc01419f
Tak, R. K., Noda, H., and Shibasaki, M. (2021b). Ligand-Enabled, copper-catalyzed electrophilic amination for the asymmetric synthesis of β-amino acids. Org. Lett. 23, 8617–8621. doi:10.1021/acs.orglett.1c03328
Tang, X., Tak, R. K., Noda, H., and Shibasaki, M. (2022). A missing link in multisubstituted pyrrolidines: remote stereocontrol forged by Rhodium-alkyl nitrene. Angew. Chem. Int. Ed. 61, e202212421. doi:10.1002/anie.202212421
Thacker, N. C., Lin, Z., Zhang, T., Gilhula, J. C., Abney, C. W., and Lin, W. (2016). Robust and porous β-diketiminate-functionalized metal–organic frameworks for earth-abundant-metal-catalyzed C–H amination and hydrogenation. J. Am. Chem. Soc. 138, 3501–3509. doi:10.1021/jacs.5b13394
Trowbridge, A., Walton, S. M., and Gaunt, M. J. (2020). New strategies for the transition-metal catalyzed synthesis of aliphatic amines. Chem. Rev. 120, 2613–2692. doi:10.1021/acs.chemrev.9b00462
Varela-Álvarez, A., Yang, T., Jennings, H., Kornecki, K. P., MacMillan, S. N., Lancaster, K. M., et al. (2016). Rh2(II,III) catalysts with chelating carboxylate and carboxamidate supports: electronic structure and nitrene transfer reactivity. J. Am. Chem. Soc. 138, 2327–2341. doi:10.1021/jacs.5b12790
Vitaku, E., Smith, D. T., and Njardarson, J. T. (2014). Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J. Med. Chem. 57, 10257–10274. doi:10.1021/jm501100b
Keywords: nitrene, N-heterocycle, rhodium, C-H insertion, kinetic isotope effect
Citation: Noda H, Asada Y and Shibasaki M (2023) Examining the effects of additives and precursors on the reactivity of rhodium alkyl nitrenes generated from substituted hydroxylamines. Front. Chem. 11:1271896. doi: 10.3389/fchem.2023.1271896
Received: 03 August 2023; Accepted: 23 October 2023;
Published: 03 November 2023.
Edited by:
Hsyueh-Liang Wu, National Taiwan Normal University, TaiwanReviewed by:
Rajendra Rohokale, University of Florida, United StatesCopyright © 2023 Noda, Asada and Shibasaki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hidetoshi Noda, aG5vZGFAYmlrYWtlbi5vci5qcA==; Masakatsu Shibasaki, bXNoaWJhc2FAYmlrYWtlbi5vci5qcA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.