
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. , 06 December 2023
Sec. Catalytic Reactions and Chemistry
Volume 11 - 2023 | https://doi.org/10.3389/fchem.2023.1260726
 Marco Sirignano1
Marco Sirignano1 Assunta D’Amato1
Assunta D’Amato1 Chiara Costabile1*
Chiara Costabile1* Annaluisa Mariconda2*Alessandra Crispini3
Annaluisa Mariconda2*Alessandra Crispini3 Francesca Scarpelli3
Francesca Scarpelli3 Pasquale Longo1
Pasquale Longo1Imines are valuable key compounds for synthesizing several nitrogen-containing molecules used in biological and industrial fields. They have been obtained, as highly regioselective Markovnikov products, by reacting several alkynes with arylamines in the presence of three new N-Heterocyclic carbene gold(I) complexes (3b, 4b, and 6b) together with the known 1-2b and 7b gold complexes as well as silver complexes 1-2a. Gold(I) complexes were investigated by means of NMR, mass spectroscopy, elemental analysis, and X-ray crystallographic studies. Accurate screening of co-catalysts and solvents led to identifying the best reaction conditions and the most active catalyst (2b) in the model hydroamination of phenylacetylene with aniline. Complex 2b was then tested in the hydroamination of alkynes with a wide variety of arylamines yielding a lower percentage of product when arylamines with both electron-withdrawing and electron-donating substituents were involved. Computational studies on the rate-determining step of hydroamination were conducted to shed light on the significantly different yields observed when reacting arylamines with different substituents.
Nowadays, considerable chemical research is aimed at developing greener strategies for synthesizing value-added compounds from low-cost reagents and using environmentally sustainable processes. The hydroamination reaction is an excellent example of this focus (Müller et al., 2008). This reaction, able to build C-N bonds by adding amines to multiple carbon bonds, can be eulogized as a model of a modern sustainable catalytically promoted chemical process with a 100% atom economy. The production of N-containing compounds (amines, imines, enamines, etc.) represents an important branch of the pharmaceutical and chemical industries, due to the importance of these molecules as scaffolds in the synthesis of biologically active compounds, drugs, N-heterocycles, polymers, bulk, and fine chemicals (Pohlki and Doye, 2003; Severin and Doye, 2007; Hartwig, 2008; Müller et al., 2008; Huang et al., 2015; Patel et al., 2017; Huo et al., 2019).
The amine addition to double and triple carbon bonds requires very high activation barriers for the repulsion between electron-rich species (Müller et al., 2008). Nevertheless, metal complexes can decrease these barriers by coordinating one or more nucleophilic species (Scheme 1).
SCHEME 1. General intermolecular hydroamination of terminal alkyne.
Many systems based on metal compounds are able to catalyze the reactions of alkaline and alkaline-earth metals (Jaspers and Doye, 2011; Mukherjee et al., 2011; Wixey and Ward, 2011), rare earth metals and actinides (Reznichenko et al., 2010b; 2010a), and early (groups 4 and 5) (Shi et al., 2001; Odom, 2005; Manna et al., 2011) and late transition (groups 8–12) (Hesp et al., 2010; Hesp and Stradiotto, 2010; Liu et al., 2018) metals. The latter, being less oxophilic and therefore more tolerant to air and moisture, are generally chosen for the hydroamination of alkynes to give enamines or imines, although high temperatures, long reaction times, and significant loads of catalysts are often required to achieve good conversions (Sevov et al., 2014; Gurak et al., 2016; Yang et al., 2017; Baron et al., 2018; Yahata et al., 2020).
The metal plays the important role of activating the double (or triple) C-C bond or the amine. As for alkaline and alkaline-earth metals, rare earth metals, and early transition metals, activation of the amine has been hypothesized. The catalytic cycle would involve the insertion of multiple C-C bonds into an M-N bond followed by fast protonolysis by other amino substrates (Michael et al., 2003; Reznichenko et al., 2010b; 2010a; Wixey and Ward, 2011).
On the other hand, two possible mechanisms have been proposed in the presence of late transition metals (E. Müller et al., 1999; Shanbhag and Halligudi, 2004; Katari et al., 2012): 1) the activation of the olefin (alkene or alkyne) would occur through its coordination to the metal, followed by the nucleophilic attack by the amine (Zhang et al., 2006); and 2) activation of the amine through oxidative addition to the metal center would be followed by the insertion of an unsaturated C–C bond into the M–N bond, where reductive elimination of the obtained intermediate would generate the hydroamination product and restore the catalyst (Zhao et al., 2005; Tsipis and Kefalidis, 2006).
Due to their carbophilicity and Lewis acidity, gold(I) and gold (III) complexes have recently received increased attention and have been used in homogeneous phase catalytic reactions to activate multiple C-C bonds that could undergo nucleophilic attack (Campeau et al., 2021). The most studied reactions are those that lead to the synthesis of nitrogen-containing heterocycles, the hydration (Biasiolo et al., 2015; Gatto et al., 2016; 2018b), alkoxylation (Gatto et al., 2018a), or hydroarylation (Tubaro et al., 2013; Baron and Biffis, 2019; Leung et al., 2020; Biffis et al., 2021) reactions of alkynes and the A3 (alkyne, aldehyde, and amine) coupling reactions (Visbal et al., 2018). Gold complexes can also catalyze the addition of an amine to an alkyne (Duan et al., 2009; Liu and Che, 2009; Alvarado et al., 2012; Sarcher et al., 2012; Gonell et al., 2013). Pioneering studies by Tanaka and coworkers reported the catalytic activity of (PPh3)AuCH3 in adding aniline to internal and terminal alkynes (Mizushima et al., 2003).
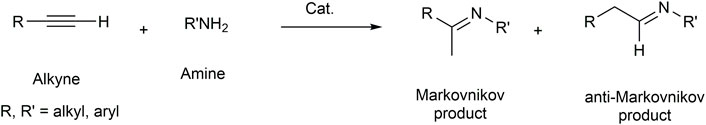
In the last 30 years, since the first isolation of free carbene by Arduengo (Arduengo et al., 1991), N-heterocyclic carbenes (NHCs) have frequently replaced phosphines for their ability to coordinate and stabilize transition metals as strong σ and π-donors and π-acceptors and their easily tunable electronic and steric properties (Jacobsen et al., 2009; Hopkinson et al., 2014; Nolan, 2014). NHC gold complexes have been extensively used to promote the hydroamination of alkynes (Mariconda et al., 2022). Recently, Bertrand et al. compared gold complexes bearing a variety of phosphine and carbene ligands, highlighting the superior performances of the latter (Yazdani et al., 2020). Herein we report a comparison among the activities of complexes 1a-b, 2a-b, and 3-8b (Figure 1) with the addition of aniline to alkynes. Complexes 3b, 4b, and 6b are described here for the first time, while all other complexes have already been reported in the literature (Napoli et al., 2013; Saturnino et al., 2016; Mariconda et al., 2020; Sirignano et al., 2021). Complexes 1, 2, 3b, and 4b differ in the substituents on the backbone of the NHC, while for complexes 5b–6b and 7b–8b, the authors have decided to study two effects: substituents on the backbone and modification of a substituent on a nitrogen atom. The choice of different substituents influences the electronic properties of the NHC, as reflected in the catalytic activity. In addition, the presence of the pendant hydroxyl group could be used as an advantageous linker functionality for immobilizing catalysts onto solid supports. Complex 2b, which was found to be the best performing, was tested in the hydroamination of phenylacetylene, diphenylacetylene, and 4-octyne with a large variety of arylamines. Density Functional Theory (DFT) studies were conducted to understand the dramatical yield differences observed when reacting variously substituted arylamines.
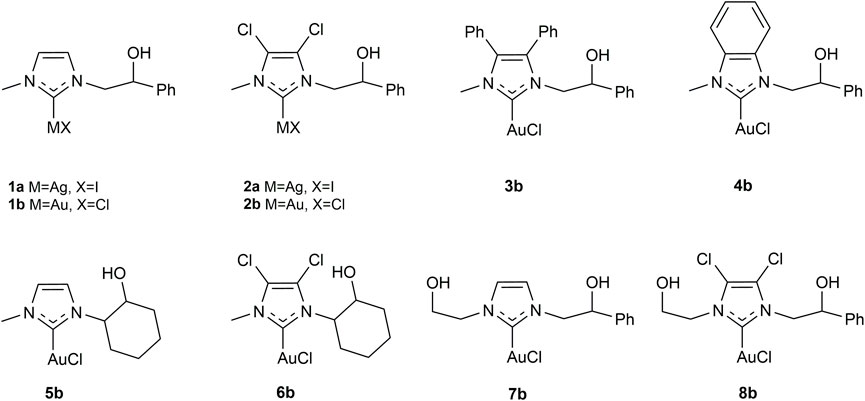
FIGURE 1. N-Heterocyclic carbene silver(I) and gold(I) complexes tested in the hydroamination reaction of phenylacetylene.
The synthesis of NHC metal complexes were conducted following the procedure reported in the literature: 1a (Napoli et al., 2013), 1b and 5b (Saturnino et al., 2016), 2a and 2b (Mariconda et al., 2020), and 7b and 8b (Sirignano et al., 2021). The synthetic routes used for the preparation of imidazolium salts and their relative silver(I) and gold(I) complexes are illustrated in Scheme 2. The NHC proligands were obtained following the procedure reported by Tacke (Patil et al., 2010) and modified for our purposes (Napoli et al., 2013; Saturnino et al., 2016; Mariconda et al., 2020; Sirignano et al., 2021). The sp3 hybridized nitrogen atom of imidazole-derivative was deprotonated by potassium carbonate to produce the nucleophilic species. Next, it was reacted with styrene oxide or cyclohexene oxide to give the monoalkylated product, to which an excess of iodomethane was added, to lead to the alkylation of the sp2-hybridized nitrogen atom and the formation of imidazolium salts (PL-3, PL-4, and PL-6).
SCHEME 2. Synthetic routes for the synthesis of NHC-silver(I) (3a, 4a, 6a) and -gold(I) (3b, 4b, 6b) complexes.
All the new-synthesized proligands and their relative silver(I) and gold(I) complexes were characterized by nuclear magnetic resonance (1H- and13C-NMR), mass spectrometry (ESI or MALDI), and elemental analysis (see Materials and Methods). Moreover, crystals of 4b suitable for X-ray diffractometry were also grown. In 1H-NMR spectra, the acid protons of imidazolium salts PL-3, PL-4, and PL-6 show signals at 9.50, 9.77, and 9.72 ppm, respectively, while in 13C-NMR spectra, signals at 138.88, 144.00, and 135.67 ppm are due to the carbocationic carbons (NCHN). The attribution of signals in the 13C-NMR spectra was supported by DEPT 135 experiments (see Supplementary Material).
MALDI-MS analyses present the signal attributable to the cationic portion of imidazolium salts.
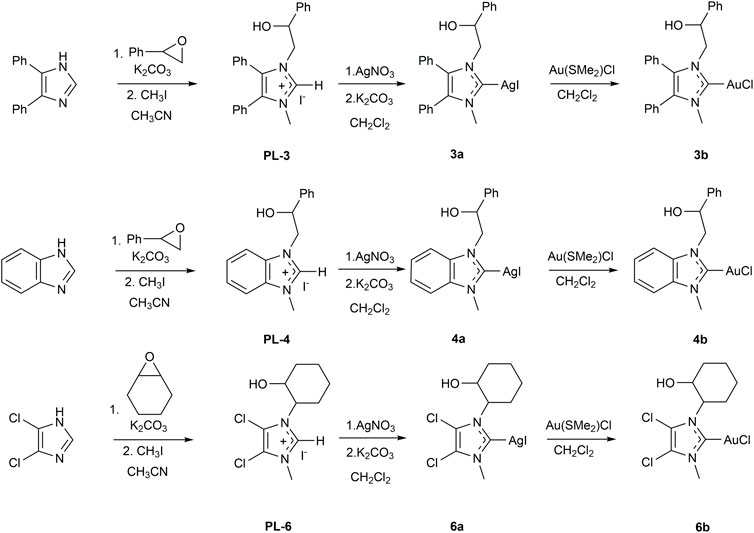
The NHC silver(I) complexes were obtained by reacting the corresponding proligand with silver nitrate, in the presence of K2CO3, following the procedure published by Nolan and Gimeno (Collado et al., 2013; Visbal et al., 2013). The absence of the singlet signal of the acid proton in 1H-NMR spectra demonstrates the deprotonation and the consequent formation of carbene species, while the formation of the silver complexes is confirmed by 13C-NMR spectroscopy. In fact, the signals at 181.38, 190.91, and 182.90 ppm of carbene carbons of 3a, 4a, and 6a, respectively, were observed. MS analyses of three silver complexes indicate several signals of a bis-carbene structure [Ag(NHC)2]+. The presence of double signals for 3a and 4a is due to the silver isotopes 107Ag and 109Ag, almost equally abundant. Due to isotopes of chlorine (35Cl 75%, 37Cl 25%), the spectrum of 6a is more intricate.
The presence bis-carbenic structures of Ag-complexes is well established in the literature, as also observed by solid state analysis (Mariconda et al., 2014). On the other hand, it is also well accepted that bis-carbenic species are in fast equilibrium with NHC-Ag(I)X complexes (Scheme 3).
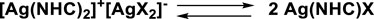
SCHEME 3. Possible dynamic equilibrium between bis and mono NHC-Ag(I) complex.
This equilibrium is mainly influenced by the nature of the counterion (Lin and Vasam, 2007; Hintermair et al., 2011) as well as by the steric and donating properties of the N-heterocyclic carbene (Garrison and Youngs, 2005).
NHC-Au(I) complexes were synthesized by reaction among NHC-Ag(I) (3a, 4a, and 6a) complexes and chloro-gold(I)dimethyl-sulfide [Au(SMe2)Cl]. The gold complexes were obtained as yellow powder in good yield: 70% for 3b, 75% for 4b, and 78% for 6b, respectively. 1H-NMR spectra of gold(I) complexes exhibited all the expected signals, corresponding to NHC-silver complexes. However, some differences were observed in the 13C-NMR spectra (see Supporting Information). As for 13C-NMR, carbene carbons of 3b, 4b, and 6b were related to signals at 169.46, 177.41, and 171.20 ppm, respectively. The up-field shift of the carbene carbon signals is due to the different nature of the metal center as well as the different electronic properties of counterion bound to the metal center (iodide for silver vs. chloride for gold) (Frenking et al., 1997; 2005; Baker et al., 2006). The mass spectra of gold complexes show peaks at 905.31858 for 3b, 701.22194 for 4b, and 693.06319 for 6b m/z, confirming the presence of bis-carbene structures. All the complexes are stable in moisture and light. In fact, the spectra of all complexes in DMSO/D2O (90/10) stay unchanged after 24 h also when exposed to light.
The X-ray single crystal molecular structure of complex 4b, with the atomic numbering scheme, is shown in Figure 2. Selected bond distances and angles are shown in Table 1.
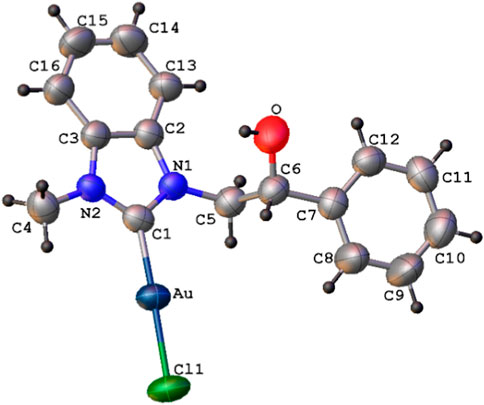
FIGURE 2. ORTEP diagram (thermal ellipsoids drawn at 50% probability) of complex 4b with atomic numbering scheme (labels of hydrogen atoms omitted for clarity).
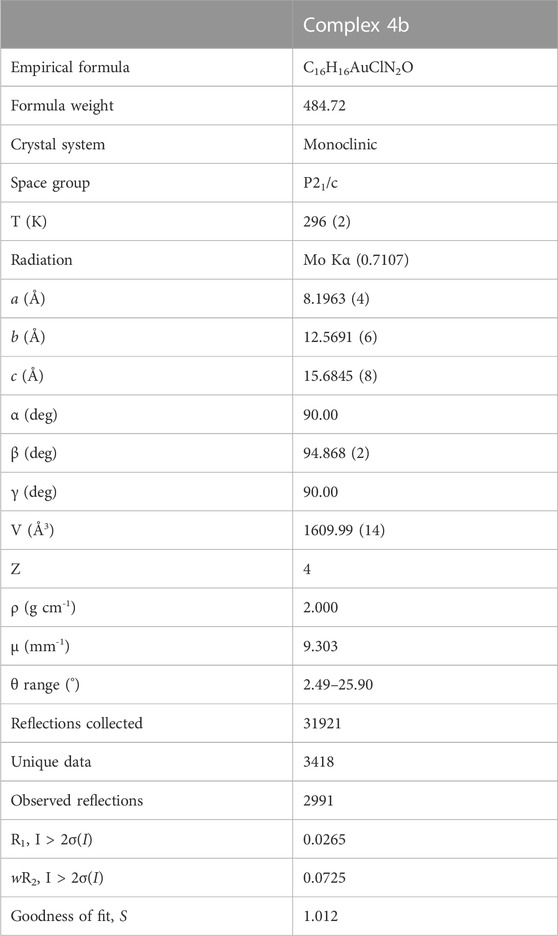
TABLE 1. Details of data collection and structure refinements for complex 4b.
Complex 4b, found in the solid crystalline state in the neutral mono-carbenic form, presents a two-coordinate Au(I) atom in a nearly linear geometry, with Cl (1)-Au-C (1) bond angle of 178.3 (1)°. Both Au-C (1) and Au-Cl (1) bond lengths of 1.983 (5) and 2.289 (2) Å, respectively, are comparable with those already reported for similar Au(I)-carbene complexes (de Frémont et al., 2005; Hashmi et al., 2012; Zargaran et al., 2018; Costabile et al., 2021; Savchuk et al., 2022). Complex 4b adopts a trans conformation around the C (5)-C (6) bond, as shown by the N (1)-C (5)-C (6)-C (7) torsion angle of 176.4 (4)°.
The 3D packing of 4b is characterized by the presence of dimers of discrete molecules, built up by aurophilic interactions with Au … Au contacts of 3.449 (5) Å (Figure 3).
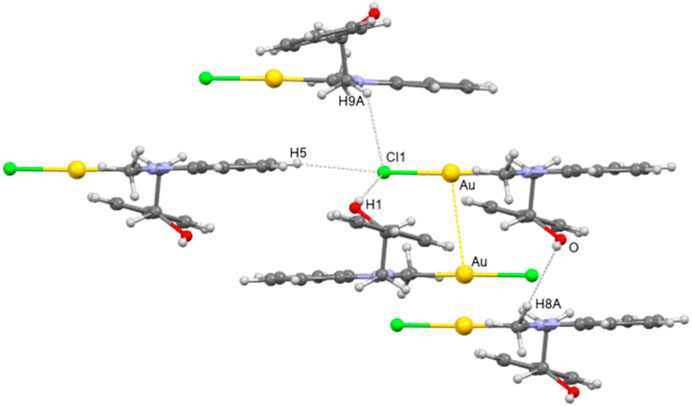
FIGURE 3. Crystal packing view of 4b showing the main intermolecular interactions.
Intermolecular hydrogen bonds between the–OH group and the chlorine atom of each molecule cooperate with the aurophilic interaction to the assembling of monomers (O … Cl (1)i distance of 3.276 (1) Å and O-H … Cl (1) angle of 166.33°, i = -x+1, -y+1, -z). Each dimer is connected to the others mainly through C-H … Cl weak hydrogen bond interactions, involving both aromatic and a methylene hydrogen atom. Finally, C-H … O weak hydrogen bonds involving a methyl hydrogen atom are a further structural feature in the 3D crystal packing of 4b.
NHC-Au(I) complexes, as shown in Figure 1, have been tested for the intermolecular hydroamination of phenylacetylene with aniline (Scheme 4), giving regioselective Markovinkov imine products (see Scheme 1).
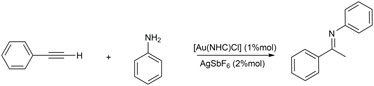
SCHEME 4. Hydroamination reaction of phenylacetylene with aniline.
The reaction is co-catalyzed by two equivalents of silver salt in order to precipitate the chloride anion coordinated to the metal center and generate in situ the catalytic species a (Scheme 5). To select the best cocatalyst, different silver salts (hexafluoroantimonate, hexafluorophosphate, nitrate, and acetate) were tested using gold complex 7b as catalytic precursor in the reaction between phenylacetylene and aniline. Screening of silver salts is reported in Table 2. The performances of the gold(I) complex were strongly influenced by the silver co-catalyst, and the best catalytic activities were found with non-coordinating anions such as hexafluoroantimonate (best performing) and hexafluorophosphate (Entries 1 and 2, Table 2). Low yields were obtained for reactions carried out in the presence of oxyanions (acetate and nitrate). Baron et al. (Baron et al., 2018) have reported similar results with different oxyanions (TsO−, TfO−). They asserted that the differences in catalytic activities could be the output of different reaction mechanisms or diverse rate-determining steps.
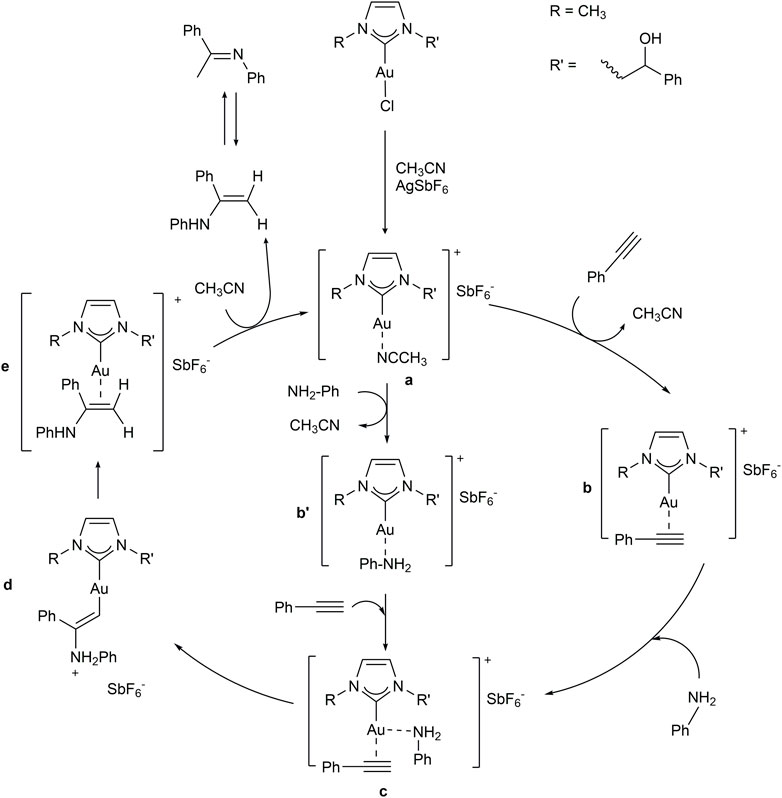
SCHEME 5. Proposed mechanism for the hydroamination reaction of phenylacetylene with aniline.
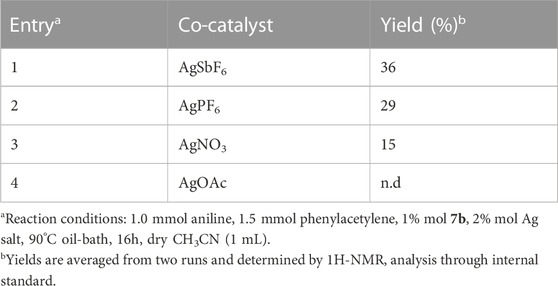
TABLE 2. Screening, promoted by 7b, of the silver salt co-catalyst for the hydroamination reaction of phenylacetylene with aniline.
An additional study conducted on the solvent identified acetonitrile as the most effective solvent in the hydroamination reaction using complex 2b. Results are listed in Table 3 and agree with those reported in the literature (Dash et al., 2010; Nuevo et al., 2018; Kumar et al., 2020). The best catalytic performance in CH3CN (Entry 1, Table 3) is possibly caused by the coordination and stabilization of the catalytic species a (Scheme 5), after the abstraction of chloride anion. Indeed, Nolan and co-workers suggested that the use of a coordinating solvent can avoid the decomposition of gold(I) complexes to give colloidal gold (0) (de Frémont et al., 2009).
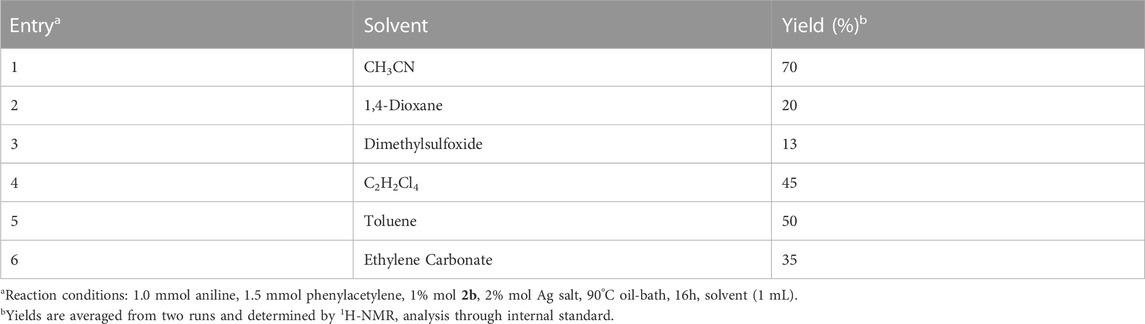
TABLE 3. Screening of the solvent in the reaction of hydroamination promoted by 2b.
Consequently, AgSbF6 co-catalyst and acetonitrile were chosen to compare the activity of gold complexes (1-8b), chloro-gold(I)dimethyl-sulfide, and silver complexes 1a and 2a in the hydroamination reaction of phenylacetylene with aniline.
As shown in Table 4, all complexes were able to promote hydroamination. Gold complexes (1b and 2b) showed a higher catalytic activity than the silver analogues (compare entries 1 and 2 with entries 10 and 11, Table 4), highlighting the role of gold(I) complexes in this kind of reaction. The low catalytic activity of the chloro-gold(I)dimethyl-sulfide (Entry 11, Table 4) suggests the relevance of the NHC ligand on the stabilization and activation of the catalytic species. NHC-gold(I) complexes with chlorine atoms on the backbone of the ligand (2b, 6b and 8b) were more active than other gold(I) complexes. The better catalytic activity of these complexes could be caused by the ability of the chlorine atoms to reduce the σ-donor ability of the carbene, making the metal center more electrophilic. This would possibly allow faster coordination of the olefin to the metal at the early stages of the catalytic cycle (Mariconda et al., 2020; Sirignano et al., 2021) (see Scheme 5). An additional comment on the role of chlorine atoms can be found in the “Molecular modeling studies” section.

TABLE 4. Catalytic activity of NHC complexes in the hydroamination reaction of phenylacetylene with aniline.
Once 2b was identified as the most efficient complex, the catalyst/co-catalyst ratio was screened, identifying a 1:1 ratio as the optimal proportion. Lowering the co-catalyst to 1% mol caused an increase of the yield from 70% to 99% (compare Entry 13 with Entry 2, Table 4). Once the optimal conditions were identified, the hydroamination reaction scope was extended to a large variety of primary arylamines, keeping AgSbF6 as co-catalyst and acetonitrile as solvent.
As depicted in Table 5, all substrates generated the expected imines. Quantitative yields are obtained with aniline and methyl substituted anilines (entries 1–4, Table 5). Yields decrease when isopropyl substituted anilines and 2-naphthylamine are used (Entries 5–8, Table 5), as well as with 4-methoxyaniline (51%). Deactivating amines with electron-withdrawing groups on the aryl ring (entries 10, 11, 12 in Table 5) showed yields of 51 and 64% when substituted with halogens and only 20% yield when bearing the nitro group. Finally, the reaction between an internal alkyne (diphenylacetylene or 4-octyne) and aniline leads to a drastic reduction of the yield of the corresponding imine (15% and 5%, respectively).
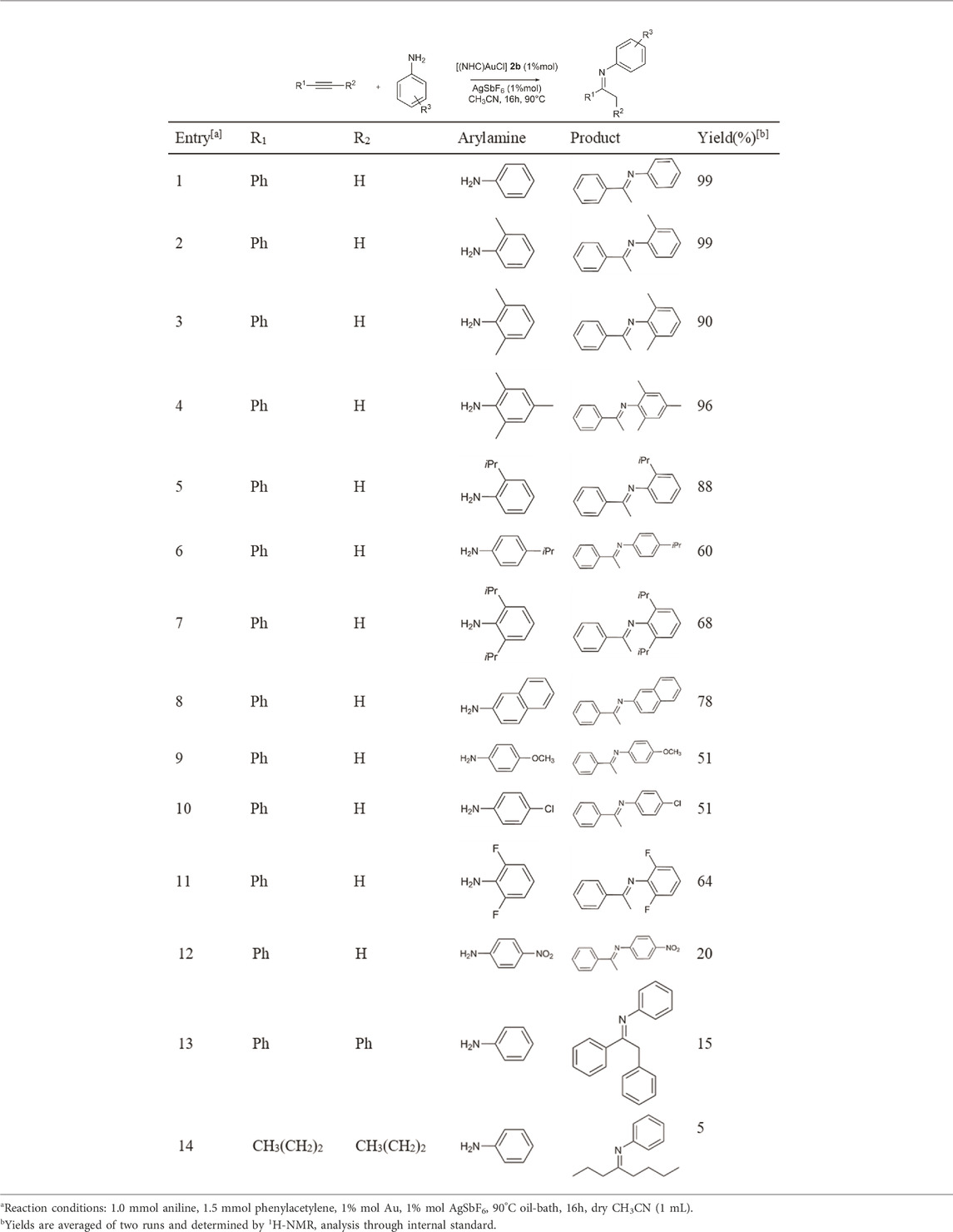
TABLE 5. Substrate Screening in the hydroamination reaction with 2b.
According to experimental studies, when phenylacetylene reacts in the presence of catalyst 2b, quantitative yield percentage was observed only with aniline and orto-methyl aniline (Table 5). All other substituted anilines gave lower percentage yield independently from the nature and position of the substitution. This means that electron-donating activating aryl substituents on aniline influence the kinetics of the reaction in the same direction as the electron-withdrawing activating substituents and the electron-withdrawing deactivating substituents. To investigate this intriguing outcome, DFT (Density Functional Theory) studies at the PBE0/6–311-G (d,p) level were conducted.
The hydroamination reaction mechanism in the presence of NHC-Au complexes was extensively studied by Ghosh et al. (Katari et al., 2012). In detail, Ghosh and co-workers investigated the hydroamination reaction between MeC≡CH and PhNH2, as representative substrates, in the presence of [1,3-dimethylimidazol-2-ylidene] gold chloride. In this study, the hydrogen transfer (reaction d→e of Scheme 5) from nitrogen to carbon enabling the formation of the enamine [(NHC)Au(PhNHMeC = CH2)]+ (e) from the intermediate [(NHC)AuCH = CMeNH2Ph]+ (d) was revealed to be the rate-determining step.
In detail, this proton transfer can be assisted by a water molecule or a PhNH2 substrate (Katari et al., 2012). As shown by the authors, the proton transfer assisted by a water molecule presents a slightly lower energy with respect to that assisted by PhNH2 substrate, occurs in only one step, and would possibly occur even in presence of only traces of water.
In the hydroamination reactions conducted in this work, we can assume the presence of traces of water since substrates employed were not dried before the reaction.
As a consequence, we investigated the proton transfer assisted by water from [(NHC)AuCH = CPhNH2Ph]+(d) to [(NHC)Au(PhNHPhC = CH2)]+(e) for selected substrates in the presence of 2b. Substrates were chosen taking into account results of Table 5, in order to compare entries 1, 5, 10 and 12, involving phenylamines with different electron-withdrawing and donating groups and steric hindrance. Minimum energy intermediates (d in Scheme 6) and transition states (TS) of proton transfer ([d-e]≠ of Scheme 6) were calculated and compared with the energy of the solvent coordinated initiating species [(NHC)AuCH3CN]+ (a in Scheme 5).
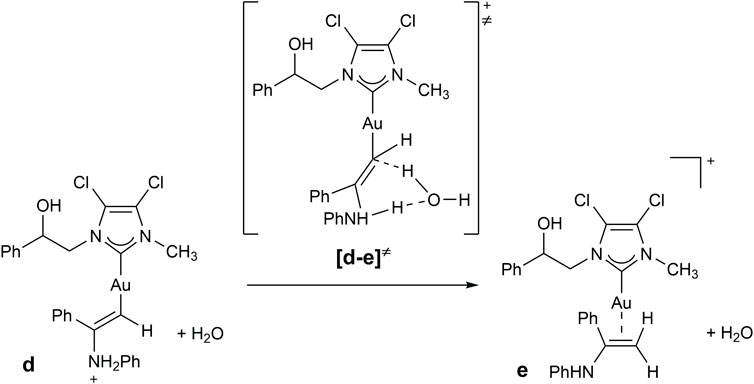
SCHEME 6. Proposed mechanism for the hydroamination reaction of phenylacetylene with aniline.
Geometries and free energies calculated in acetonitrile have been reported in Figure 4.
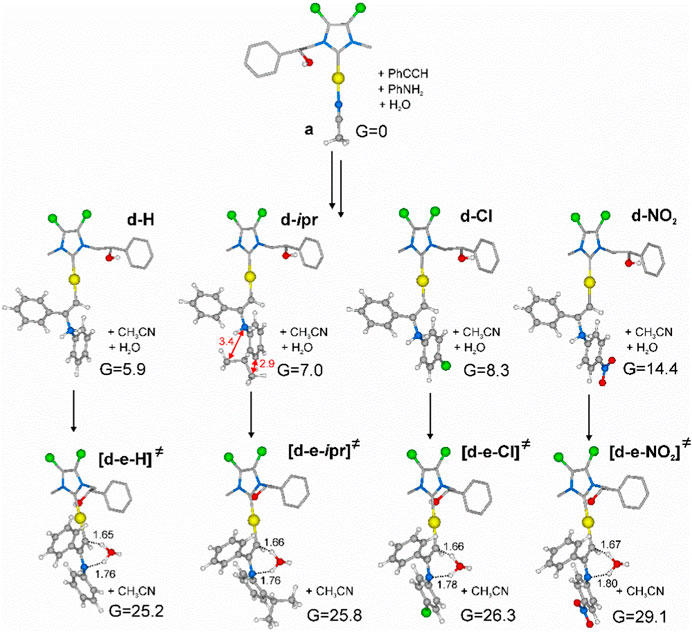
FIGURE 4. Minimum energy intermediates (d-H, d-ipr, d-Cl, d-NO2) and transition states relative to proton transfer ([d-e-H]≠, [d-e-ipr]≠, [d-e-Cl]≠, [d-e-NO2]≠) located according to Scheme 6, starting from minimum energy active species a of Scheme 5. Free energies calculated at PBE0/6–311-G (d,p) in CH3CN are in kcal/mol. Distances are in Å. Some hydrogens of the NHC ligand skeleton were omitted for clarity.
According to molecular modeling results, intermediate d-H and TS [d-e-H]≠, involving aniline, present higher energies with respect to those calculated by Ghosh and co-workers (Katari et al., 2012), possibly due to the higher steric hindrance of phenylacetylene with respect to propyne and to the higher electron-withdrawing NHC bearing chlorines on the backbone.
Shifting to a comparison among intermediates d, we can observe an increase of free energy for substituted phenylamines. As for d-ipr, the electro-donating ability of the isopropyl group does not compensate its steric hindrance, which introduces internal repulsions as reported in Figure 4. On the other hand, electron-withdrawing groups in para position, such as chlorine or -NO2, lead to intermediates with higher energy, possibly due to electron depletion of the metal.
The same energy trend was seen for hydrogen transfer TS ([d-e]≠), although energy differences were reduced. This is possibly due to an increase of acidity of the hydrogen bound to the nitrogen, especially in the presence of electron-withdrawing groups.
To gain details on the electronic effects of the various substrates on the percentage yields, we calculated the Au charge for all intermediates and TS by carrying out a natural bond orbital (NBO) analysis. In Table 6, experimental percentage yield, intermediate and free energy barriers in gas phase and acetonitrile, and Au charges were collected. Electron depletion of the Au can be observed for amine with electron-withdrawing groups and could be responsible for the increase of energy barriers for these substrates.
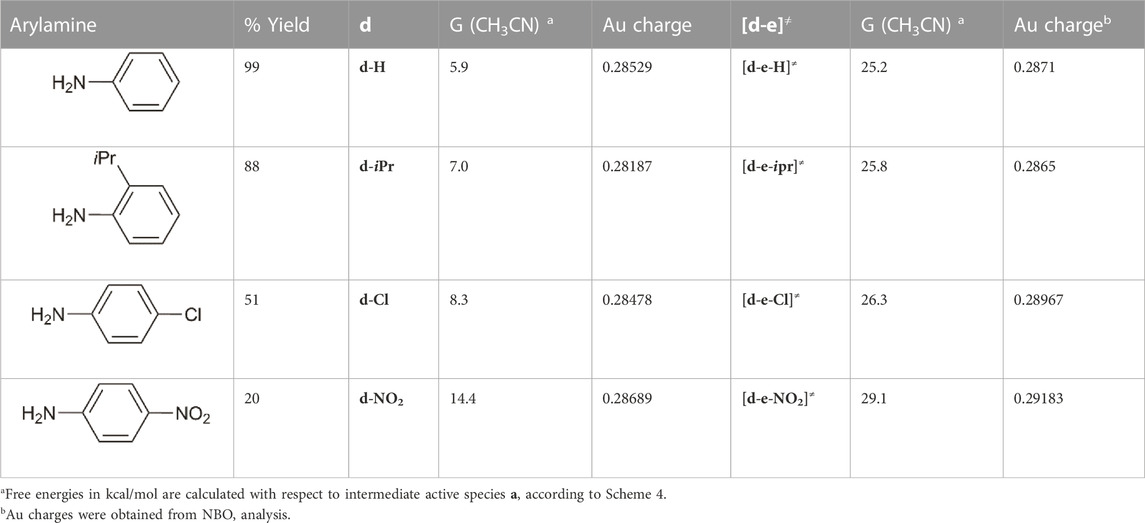
TABLE 6. Comparison among percentage yield of hydroamination and calculated free energies and Au charges for intermediate d (d-H, d-iPr, d-Cl, d-NO2) and TS [d-e]≠ ([d-e-H]≠, [d-e-ipr]≠, [d-e-Cl]≠, [d-e-NO2]≠) relative to water-assisted proton transfer according to Scheme 6.
On the other hand we can speculate that, besides the steric effects, the presence of electron-donating substituents increases the energy barrier due to a decrease of the amine acidity since electron-withdrawing substituents actually give lower energy barriers.
According to computational studies, the non-monotonic trend observed for the reactivity of substituted aniline, where a drop in the percentage yield has been observed with both electron-donating or withdrawing substituents, would be the balance of two contrasting effects: the stabilization of the intermediate preceding the rate-determining step barrier (favored for electron-donating substituted anilines) and the decrease of the barrier itself (favored for electron-withdrawing substituted anilines due to their acidity). The global effect leads to the highest percentage yield for non-substituted aniline where these two factors find the best balance.
In this framework, the higher performances of catalysts with chlorine on the backbone could be rationalized supposing that electron-withdrawing substituents on NHC would increase the acidity of the ammonium intermediate d decreasing the overall [d-e]≠ barrier and favoring the hydrogen transfer.
This study confirms, as previously reported by Ghosh (Katari et al., 2012), that the hydrogen transfer represents a key step in the alkyne hydroamination, and variables able to decrease the energy of this step can be responsible for the increase of percentage yields. It is important to underline that the hypothesis that the proton transfer from the amine to the carbon bonded to the metal is mediated by water is also supported by the fact that when conducting the reaction between aniline and phenylacetylene as in Entry 1, but in the presence of molecular sieves to eliminate traces of H2O, no product is obtained.
NHC-Au(I) complexes (1-8b) have been tested as catalysts in the hydroamination reaction of alkynes to give imines. New complexes 3b, 4b, and 6b were characterized by NMR, mass spectroscopy, elemental analysis, and, as for 4b, by X-ray diffraction analysis. All complexes were shown to be active in the hydroamination reaction of phenylacetylene. During the optimization of the reaction conditions, AgSbF6 was selected as the best co-catalyst, and acetonitrile as the best solvent. Complex 2b was revealed as the most active catalyst and was chosen to study the different catalytic behaviors on varying the involved arylamine. The presence of the two chlorine atoms (electron-withdrawing) positively influences the catalytic activity. Quantitative yields were obtained only by reacting aniline and o-methyl aniline. All other substituted arylamines gave lower yields independently from the electron-donating or -withdrawing ability of the substituents. This non-monotonic reactivity was investigated by DFT studies. According to theoretical results, the low reaction yields in the presence of electron-withdrawing groups is caused by the destabilization of the intermediate [(NHC)AuCH = CPhNH2Ph]+ preceding the water-assisted hydrogen transfer from nitrogen to carbon as consequence of electron depletion of the metal induced by electron-withdrawing groups on N-phenyl group of intermediate. On the other hand, the low reaction yield observed with electron-donating arylamine substituents would be the consequence of a decrease of acidity of the ammonium group of intermediate [(NHC)AuCH = CPhNH2Ph]+ that could increase the TS barrier of the hydrogen transfer. In the framework, aniline and o-methyl aniline perfectly balance these two opposite effects, giving the best yields.
All the reactions were carried out using Schlenk and glove-box techniques, under a dry nitrogen atmosphere. Solvents were dehydrated by heating at reflux temperature over suitable drying agents. Reagents were purchased from Merck KGaA (Darmstadt, Germany) and TCI Chemicals (Tokyo, Japan), and they were used as received. NMR spectra were recorded on Brucker AM 300 spectrometers (300 MHz for 1H; 75 MHz for 13C) and Brucker AVANCE 400 spectrometer (400 MHz for 1H; 100 MHz for 13C) using DMSO-d6 and CDCl3 as solvents. The chemical shifts are referenced to tetramethylsilane (SiMe4, δ = 0) by using residual protons impurities of deuterated solvents as internal standards. The multiplicities of spectra are abbreviated in the following manner: singlet (s), doublet (d), triplet (t), multiplet (m), broad (br), and overlapped (o). Elemental analyses for C, H, and N were recorded with Thermo-Finnigan Flash EA 1112 following microanalytical procedures. Chloride and iodide were determined by the reaction of AgNO3 with halogen, precipitation of AgX (X = Cl, I), which was dissolved in Na2S2O3. The content of silver was determined by flame atomic absorption spectroscopy (FAAS), and halogen content was calculated by using the content of silver.
ESI-MS measurements of organic compounds were acquired on a Waters Quattro Micro triple quadrupole mass spectrometer equipped with an electrospray ion source. MALDI-MS was performed using a Brucker SolariX XR Fourier transform ion cyclotron resonance mass spectrometer (Brucker DaltonikGmBH, Bremen, Germany) equipped with a 7T refrigerated actively shielded superconducting magnet (Brucker Biospin, Wissembourg, France). The mass range is set to m/z 200–3000. To improve the mass accuracy, the sample spectra were calibrated internally by matrix ionization (2,5-dihydroxybenzoic acid). Single crystal X ray diffraction data of complex 4b were collected at room temperature with a Bruker-Nonius X8APEXII CCD area detector system equipped with a graphite monochromator with radiation Mo Kα (λ = 0.71073 Å). Data were processed through the SAINT (SAINT, 2023) reduction and SADABS (Sheldrick, 2003) absorption software. The structure was solved by direct methods and refined by full matrix least-squares based on F2 through the SHELX and SHELXTL-2018 structure determination package (Sheldrick, 2008; Sheldrick, 2015). All non-hydrogen atoms were refined anisotropically, and hydrogen atoms were included as idealized riding atoms. All graphical representations were obtained by using Olex2 (Dolomanov et al., 2009) and CCDC Mercury 4.0 (Macrae et al., 2020). Details of data and structure refinements are reported in Table 1. The supplementary crystallographic data were deposited as CCDC 2260506.
Proligands PL-3, PL-4, and P-L6 were synthesized following the strategy developed by Tacke (Patil et al., 2010) and employing the procedures reported in the literature (Napoli et al., 2013; Mariconda et al., 2014; 2020; Saturnino et al., 2016). Imidazole ring (1 eq) was dissolved in CH3CN and deprotonated by K2CO3 (2 eq), then it was reacted with (1.2 eq) epoxy-alkylating agent (styrene oxide or cyclohexene oxide) for 12 h at refluxing temperature. Afterwards, the reaction mixture was filtered, and iodomethane (5 eq) was added. The reaction mixture was stirred for 8 h. The imidazolium salts were recovered by removing the solvent, and precipitation in acetone. Finally, the product was washed with hexane (3 × 30 mL) and diethyl ether (2 × 30 mL).
Iodo [4,5-diphenyl N-methyl, N’-(2-hydroxy-2-phenyl) ethyl-imidazole-2-ylidine] PL-3.
Yield: 1.92 g; 88%
1H-NMR (400 MHz, DMSO-d6): δ 9.50 (s, 1H, NCHN); 7.46–7.11 (m, 15H, Ph rings); 6.12 (s, 1H, OH); 4.72 (m, 1H, OCH, Janti 7.37 Hz, Jgauche 5.80 Hz); 4.26–4.11 (m, 2H, NCH2, Jgem14.5 Hz, Janti 7.37 Hz, Jgauche5.80 Hz); 3.84 (s, 3H, NCH3).
13C-NMR (100 MHz, DMSO-d6): δ 140.7 (ipso aromatic carbon, Ph ring); 136.8 (NCN); 131.3–125.5 (aromatic carbons, Ph rings), 125.5–125.0 (backbone carbons, NCPh = CPhN), 70.0 (OCH); 54.0 (NCH2); 34.4 (NCH3).
MALDI-ToF (m/z): 355.18080 Da attributable to the cationic portion of the imidazolium salt [C24H23N2O]+.
Elemental Analysis: calculated for C24H23I N2O, C 59.76, H 4.81, I 26.31, N 5.81, O 3.32; Found: C 59.60, H 4.70, I 26.58, N 5.60, O. 3.43.
Iodo [N-methyl, N’-(2-hydroxy-2-phenyl) ethyl-benzoimidazol-2-ylidine] PL-4.
Yield: 2.85 g; 85%
1H-NMR (400 MHz, DMSO-d6): δ 9.77 (s, 1H, NCHN); 8.06–7.31 (m, 9H, Ph rings); 5.99 (d, 1H, OH); 5.10–5.07 (dd, 1H, OCH, Janti 7.58 Hz, Jgauche 5.50 Hz); 4.61–4.52 (m, 2H, NCH2, Jgem14.7 Hz, Janti 7.58 Hz, Jgauche5.50 Hz); 4.14 (s, 3H, NCH3).
13C-NMR (100 MHz, DMSO-d6): δ 144.0 (NCN); 141.9 (ipso aromatic carbon, Ph ring); 131.6–131.3 (backbone carbons, NC = CN); 128.3, 127.9, 126.3, 126.1 (aromatic carbons, Ph rings); 114.0, 113.4 (aromatic carbons, Ph rings); 70.0 (OCH); 54.0 (NCH2), 34.4 (NCH3).
MALDI-ToF (m/z): 253.13474 Da attributable to the cationic portion of the imidazolium salt [C16H17N2O]+.
Elemental Analysis: calculated for C16H17I N2O, C 50.44, H 4.10, I 33.38, N 7.37, O 4.41; Found: C 50.50, H 4.50, I 33.20, N 7.40, O. 4.40.
Iodo [4,5-dichloro l N-methyl, N’-(cyclohexane-2-ol) imidazole-2-ylidine] PL-6.
Yield: 1.88 g; 70%
1H-NMR (400MHz, DMSO-d6): δ 9.72 (s, 1H, NCHN); 5.25 (m, 1H, OH); 4.09 (m, 1H, OCH, Jax-eq 4.81 Hz, Jax-eq 4.50 Hz, Jeq-eq 2.8Hz); 3.84 (s, 3H, NCH3); 3.70 (m, 1H, NCH, Jax-ax 11.5 Hz, Jax-eq 4.81 Hz, Jeq-eq 2.7 Hz); 2.08–1.36 (m, 8H, Cyclohexyl protons).
13C-NMR (100 MHz, DMSO-d6): δ 135.6 (NCN); 118.8 (backbone carbons, NCCl = CClN); 71.5 (OCH); 65.2 (NCH); 35.2 (NCH3); 34.0, 30.68, 24.2, 23.5 (Cyclohexyl carbons).
MALDI-ToF (m/z): 249.05614 Da attributable to the cationic portion of the imidazolium salt [C10H15Cl2N2O]+.
Elemental Analysis: calculated for C10H15Cl2IN2O, C 31.86, H 4.01, Cl 18.80, I 33.60, N 7.43, O 4.24; Found: C 31.70, H 4.00, Cl 18.96, I 33.50, N 7.48, O. 4.29.
NHC-Ag(I) complexes were synthesized using the procedure published in literature by Nolan and Gimeno (Collado et al., 2013; Visbal et al., 2013) and slightly modified by us. The imidazolium salt (1 eq) and AgNO3 (1 eq) were suspended in dichloromethane (25 mL) and stirred for 2 h. Then, K2CO3 (10 eq) was added, and the resulting mixture was stirred for 6 h with the exclusion of light. Then, the reaction mixture was filtered through Celite, and the complex was obtained removing the solvent in vacuo.
Iodo [4,5-diphenyl N-methyl, N’-(2-hydroxy-2-phenyl) ethyl-imidazole-2-yden]silver(I) 3a.
Yield: 0.412 g, 70%
1H-NMR (400 MHz, DMSO-d6): δ 7.46–7.04 (m, 15H, Ph rings); 4.76 (m, 1H, OCH, Janti 7.73 Hz, Jgauche 5.00 Hz); 4.19 (m, 2H, NCH2, Jgem14.0 Hz, Janti 7.73 Hz, Jgauche5.00 Hz); 3.82 (s, 3H, NCH3).
13C-NMR (100 MHz, DMSO-d6): δ 181.4 (NCN); 142.2 (ipso aromatic carbon, Ph ring); 131.9–125.6 (aromatic carbons, Ph rings); 72.2 (OCH); 56.0 (NCH2); 37.4 (NCH3).
ESI-MS (m/z): 647.45829 Da attributable to a bis-carbene structure [C35H30AgN4O2]+.
Elemental Analysis: calculated for C24H22AgIN2O, C, 48.92; H, 3.76; Ag, 18.31; I, 21.54; N, 4.75; O, 2.72; Found: C 48.60, H 3.70, Ag 18.30, I 21.84, N 5.66, O 2.72.
Iodo [N-methyl, N’-(2-hydroxy-2-phenyl) ethyl-benzoimidazol-2-yden]silver(I) 4a.
Yield: 0.267 g, 55%
1H-NMR (400 MHz, DMSO-d6): δ 7.82–7.24 (m, 9H, Ph rings); 5.86 (s, 1H, OH); 5.09–5.07 (dd, 1H, OCH, Janti 7.85 Hz, Jgauche 5.40 Hz); 4.65 (m, 2H, NCH2, Jgem 13.83 Hz, Janti 7.85 Hz, Jgauche 5.40 Hz); 4.04 (s, 3H, NCH3).
13C-NMR (100 MHz, DMSO-d6): δ 190.9 (NCN); 142.3 (ipso aromatic carbon, Ph-ring); 134.4, 133.9 (backbone carbons, NC = CN); 128.2, 127.5, 126.2, 123.6 (aromatic carbons, Ph-rings); 112.6, 111.7 (aromatic carbons, Ph rings); 72.0 (OCH); 55.8 (NCH2); 35.5 (NCH3).
MALDI-ToF (m/z): 517.26179 Da attributable to bis-carbene structure [C26H25AgN4O]+.
Elemental Analysis: calculated for C16H16IN2O, C 39.45, H 3.31, Ag 22.15, I 26.05, N 5.75, O 3.28; Found: C 39.00, H 3.30, Ag 22.00, I 26.50, N 5.60, O 3.43.
Iodo [4,5-dichloro N-methyl, N’-(cyclohexane-2-ol) imidazole-2-yden] silver (I) 6a.
Yield: 0.310 g, 64%
1H-NMR (400 MHz, DMSO-d6): δ 4.93 (b, 1H, OH); 4.00 (m, 1H, HOCH Jax-eq 4.81 Hz, Jax-eq 4.50 Hz, Jeq-eq 2.8Hz); 3.86 (o, 4H, NCH, NCH3); 1.96–1.31 (m, 8H, Cyclohexyl protons).
13C-NMR (100 MHz, DMSO-d6): δ 182.9 (NCN); 118.6, 117.3 (backbone carbons, NCCl = CClN); 76.1 (CHOH); 70.4 (NCH); 38.8 (NCH3); 35.0, 30.7, 24.2, 23.9 (Cyclohexyl carbons).
MALDI-ToF (m/z): 606.99642 Da attributable to [C20H28AgCl4N4O2]+.
Elemental Analysis: calculated for C10H14AgCl2IN2O, C 24.82, H 2.92, Ag 22.29, Cl 14.65 I 26.22, N 5.79, O 3.31; Found: C 24.53, H 2.92, Ag 22.00, Cl 14.35, I 26.30, N 5.81, O 3.51.
NHC-gold(I) complexes were prepared by the trans-metalation route, following the procedure published in the literature (Lin and Vasam, 2007). The suitable Ag-NHC complex (1.00 eq) was reacted with chloro (dimethylsulfide) gold(I) (1 eq) in dry CH2Cl2 (25 mL) for 4 h at room temperature. Afterward, the mixture was filtered, through Celite, to remove the AgI byproduct. NHCAu complex was obtained after removal of the solvent in vacuo.
Chloro [4,5-diphenyl N-methyl, N’-(2-hydroxy-2-phenyl) ethyl-imidazole-2-ylidine] gold(I) 3b
Yield: 0.16 g, 70%
1H-NMR (400 MHz, DMSO-d6): δ 7.36–7.14 (m, 15H, Ph rings); 5.13 (m, 1H, OCH, Janti 7.00 Hz, Jgauche 5.30 Hz); 4.11 (m, 2H, NCH2, Jgem 13.89 Hz, Janti 7.00 Hz, Jgauche 5.30Hz); 3.72 (s, 3H, NCH3).
13C-NMR (100 MHz, DMSO-d6): δ 169.5 (NCN); 141.8 (ipso aromatic carbon, Ph ring); 131.7–125.5 (aromatic carbons, Ph rings); 72.3 (OCH); 55.6 (NCH2); 36.9 (NCH3).
MALDI-ToF (m/z): 905.31858 Da attributable to bis-carbene structure [C48H44AuN4O2]+.
Elemental Analysis: calculated for C24H22AuClN2O, C 49.12, H 3.78, Au 33.56, Cl 6.04, N 4.77, O 2.73; Found: C 49.00, H 3.70, Cl 6.10, N 4.70.
Chloro [N-methyl, N’-(2-hydroxy-2-phenyl) ethyl-benzimidazol-2-ylidine] gold(I) 4b
Yield: 0.150 g, 60%
1H-NMR (400 MHz, DMSO-d6): δ 7.80–7.28 (m, 9H, Ph rings); 5.76 (s, 1H, OH); 5.22 (m, 1H, OCH, Janti 7.68 Hz, Jgauche 5.89 Hz); 4.60 (m, 2H, NCH2, Jgem 14.65 Hz, Janti 7.68 Hz, Jgauche 5.89Hz); 4.00 (s, 3H, NCH3).
13C-NMR (100 MHz, DMSO-d6): δ 177.4 (NCN); 141.9 (ipso aromatic carbon, Ph ring); 133.5, 133.1 (backbone carbons, NC = CN); 128.3, 127.7, 126.0, 124.1 (aromatic carbons, Ph rings); 112.8, 111.8 (aromatic carbons, Ph rings); 72.5 (OCH); 55.4 (NCH2); 35.0 (NCH3).
MALDI-ToF (m/z): 701.22194 Da attributable to bis-carbene structure [C32H32AuN4O2]+.
Elemental Analysis: calculated for C16H16AuClN2O, C 39.65, H 3.33, Au 40.63, Cl 7.31, N 5.78, O 3.30; Found: C 40.00, H 3.00, Cl 7.41, N 5.80.
Chloro [4,5-dichloro N-methyl, N’-(2-hydroxy-2-phenyl) ethyl-benzimidazol-2-ylidine] gold(I) 6b
Yield: 0.220 g, 90%.
1H-NMR (400 MHz, DMSO-d6): δ 4.17–4.12 (m, 1H, HOCH); 3.89–3.86 (m, 1H, NCH); 3.73 (s, 3H, NCH3); 1.97–1.29 (Cyclohexyl protons).
13C-NMR (100 MHz, DMSO-d6): δ 171.2 (NCN); 119.6, 117.2 (backbone carbons, NCCl = CClN); 77.1 (CHOH); 71.4 (NCH); 39.1 (NCH3); 35.1, 30.6, 24.3, 23.8 (Cyclohexyl carbons).
MALDI- ToF: 695.05472 Da attributable to bis-carbene structure [C20H28AuCl4N4O2]+
Elemental Analysis: calculated for C10H14AuCl3N2O, C 24.94, H 2.93, Au 40.90, Cl 22.08, N 5.82, O 3.32; Found: C 24.90, H 3.00, Cl 22.30, N 5.22.
In a Schlenk tube under an inert atmosphere, arylamine (1.0 mmol), alkyne (1.5 mmol), Au-NHC precatalyst (1% mol), silver salt (2% mol), and 1.0 mL of the solvent (listed in Table 2) were loaded. The flask was placed in preheated oil bath at 90°C and stirred for 16 h. After this time, the solvent was removed under reduced pressure, and at crude oil was added the internal standard (CH2Br2 1.0 mmol in 0.7 mL of CDCl3). The yield of imines (listed in Table 4) was determined by 1H-NMR spectroscopy. The degree conversion was determined by integrating the 1H NMR signals of the two protons of the internal standard (CH2Br2) and the methyl group of the imine products. 1H NMR characterization of the isolated imines has been reported in the literature (Anderson et al., 2004; Wang et al., 2006; Chen et al., 2010; Peeters et al., 2013; Kumar et al., 2020).
The structural data is deposited in the CCDC database repository: https://www.ccdc.cam.ac.uk/structures/ (CCDC 2260506).
MS: Investigation, Methodology, Writing–original draft. AD: Investigation, Methodology, Writing–original draft. CC: Conceptualization, Writing–review and editing. AM: Conceptualization, Writing–review and editing. AC: Investigation, Writing–original draft. FS: Investigation, Writing–original draft. PL: Conceptualization, Writing–review and editing.
The author(s) declare that no financial support was received for the research, authorship, and of this article.
FS is grateful to the project PON “Ricerca e Innovazione” 2014–2020, Asse IV “Istruzione e ricerca per il recupero,” and Azione IV.6 “Contratti di ricerca su tematiche Green” (CUP: H25F21001230004; identification code: 1062_R8_GREEN). The authors are grateful to Patrizia Oliva and Patrizia Iannece for technical assistance.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2023.1260726/full#supplementary-material
Alvarado, E., Badaj, A. C., Larocque, T. G., and Lavoie, G. G. (2012). N-heterocyclic carbenes and imidazole-2-thiones as ligands for the gold(I)-Catalysed hydroamination of phenylacetylene. Chem. – A Eur. J. 18, 12112–12121. doi:10.1002/chem.201201448
Anderson, L. L., Arnold, J., and Bergman, R. G. (2004). Catalytic hydroamination of alkynes and norbornene with neutral and cationic tantalum imido complexes. Org. Lett. 6, 2519–2522. doi:10.1021/ol0492851
Arduengo, A. J., Harlow, R. L., and Kline, M. (1991). A stable crystalline carbene. J. Am. Chem. Soc. 113, 361–363. doi:10.1021/ja00001a054
Baker, M. V., Barnard, P. J., Berners-Price, S. J., Brayshaw, S. K., Hickey, J. L., Skelton, B. W., et al. (2006). Cationic, linear Au(i) N-heterocyclic carbene complexes: synthesis, structure and anti-mitochondrial activity. Dalton Trans. 3708, 3708–3715. doi:10.1039/b602560a
Baron, M., Battistel, E., Tubaro, C., Biffis, A., Armelao, L., Rancan, M., et al. (2018). Single-step synthesis of dinuclear neutral gold(I) complexes with bridging di(N-heterocyclic carbene) ligands and their catalytic performance in cross coupling reactions and alkyne hydroamination. Organometallics 37, 4213–4223. doi:10.1021/acs.organomet.8b00531
Baron, M., and Biffis, A. (2019). Gold(I) complexes in ionic liquids: an efficient catalytic system for the C-H functionalization of arenes and heteroarenes under mild conditions. Eur. J. Org. Chem. 2019, 3687–3693. doi:10.1002/ejoc.201900529
Biasiolo, L., Del Zotto, A., and Zuccaccia, D. (2015). Toward optimizing the performance of homogeneous L-Au-X catalysts through appropriate matching of the ligand (L) and counterion (X –). Organometallics 34, 1759–1765. doi:10.1021/acs.organomet.5b00308
Biffis, A., Baron, M., Tubaro, C., Rancan, M., Armelao, L., Marchenko, A., et al. (2021). Gold(I) complexes with multifunctional phosphane ligands: synthesis and catalysis. Inorganica Chim. Acta 517, 120218. doi:10.1016/j.ica.2020.120218
Campeau, D., León Rayo, D. F., Mansour, A., Muratov, K., and Gagosz, F. (2021). Gold-catalyzed reactions of specially activated alkynes, allenes, and alkenes. Chem. Rev. 121, 8756–8867. doi:10.1021/acs.chemrev.0c00788
Chen, D., Wang, Y., and Klankermayer, J. (2010). Enantioselective hydrogenation with chiral frustrated Lewis pairs. Angew. Chem. 122, 9665–9668. doi:10.1002/ange.201004525
Collado, A., Gómez-Suárez, A., Martin, A. R., Slawin, A. M. Z., and Nolan, S. P. (2013). Straightforward synthesis of [Au(NHC)X] (NHC = N-heterocyclic carbene, X = Cl, Br, I) complexes. Chem. Commun. 49, 5541. doi:10.1039/c3cc43076f
Costabile, C., Mariconda, A., Sirignano, M., Crispini, A., Scarpelli, F., and Longo, P. (2021). A green approach for A 3 -coupling reactions: an experimental and theoretical study on NHC silver and gold catalysts. New J. Chem. 45, 18509–18517. doi:10.1039/D1NJ03444H
Dash, C., Shaikh, M. M., Butcher, R. J., and Ghosh, P. (2010). Highly convenient regioselective intermolecular hydroamination of alkynes yielding ketimines catalyzed by gold(I) complexes of 1,2,4-triazole based N-heterocyclic carbenes. Inorg. Chem. 49, 4972–4983. doi:10.1021/ic100087d
de Frémont, P., Marion, N., and Nolan, S. P. (2009). Cationic NHC–gold(I) complexes: synthesis, isolation, and catalytic activity. J. Organomet. Chem. 694, 551–560. doi:10.1016/j.jorganchem.2008.10.047
de Frémont, P., Scott, N. M., Stevens, E. D., and Nolan, S. P. (2005). Synthesis and structural characterization of N -heterocyclic carbene gold(I) complexes. Organometallics 24, 2411–2418. doi:10.1021/om050111c
Dolomanov, O. V., Bourhis, L. J., Gildea, R. J., Howard, J. A. K., and Puschmann, H. (2009). OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 42, 339–341. doi:10.1107/S0021889808042726
Duan, H., Sengupta, S., Petersen, J. L., Akhmedov, N. G., and Shi, X. (2009). Triazole−Au(I) complexes: a new class of catalysts with improved thermal stability and reactivity for intermolecular alkyne hydroamination. J. Am. Chem. Soc. 131, 12100–12102. doi:10.1021/ja9041093
Frenking, G., Fau, S., Marchand, C. M., and Grützmacher, H. (1997). The π-donor ability of the halogens in cations and neutral molecules. A theoretical study of ax 3+, ah 2 X +, yx 3, and YH 2 X (A = C, Si, Ge, Sn, Pb; Y = B, Al, Ga, in, Tl; X = F, Cl, Br, I). J. Am. Chem. Soc. 119, 6648–6655. doi:10.1021/ja970335p
Frenking, G., Solà, M., and Vyboishchikov, S. F. (2005). Chemical bonding in transition metal carbene complexes. J. Organomet. Chem. 690, 6178–6204. doi:10.1016/j.jorganchem.2005.08.054
Garrison, J. C., and Youngs, W. J. (2005). Ag(I) N-heterocyclic carbene complexes: synthesis, structure, and application. Chem. Rev. 105, 3978–4008. doi:10.1021/cr050004s
Gatto, M., Baratta, W., Belanzoni, P., Belpassi, L., Del Zotto, A., Tarantelli, F., et al. (2018a). Hydration and alkoxylation of alkynes catalyzed by NHC–Au–OTf. Green Chem. 20, 2125–2134. doi:10.1039/C8GC00508G
Gatto, M., Belanzoni, P., Belpassi, L., Biasiolo, L., Del Zotto, A., Tarantelli, F., et al. (2016). Solvent-Silver-and acid-free NHC-Au-X catalyzed hydration of alkynes. The pivotal role of the counterion. ACS Catal. 6, 7363–7376. doi:10.1021/acscatal.6b01626
Gatto, M., Del Zotto, A., Segato, J., and Zuccaccia, D. (2018b). Hydration of alkynes catalyzed by L–Au–X under solvent- and acid-free conditions: new insights into an efficient, general, and green methodology. Organometallics 37, 4685–4691. doi:10.1021/acs.organomet.8b00689
Gonell, S., Poyatos, M., and Peris, E. (2013). Triphenylene-based tris(N-heterocyclic carbene) ligand: unexpected catalytic benefits. Angew. Chem. 125, 7147–7151. doi:10.1002/ange.201302686
Gurak, J. A., Yang, K. S., Liu, Z., and Engle, K. M. (2016). Directed, regiocontrolled hydroamination of unactivated alkenes via protodepalladation. J. Am. Chem. Soc. 138, 5805–5808. doi:10.1021/jacs.6b02718
Hartwig, J. F. (2008). Carbon–heteroatom bond formation catalysed by organometallic complexes. Nature 455, 314–322. doi:10.1038/nature07369
Hashmi, A. S. K., Yu, Y., and Rominger, F. (2012). Efficient one-pot synthesis of unsymmetrical gold(I) N-heterocyclic carbene complexes and their use as catalysts. Organometallics 31, 895–904. doi:10.1021/om2008919
Hesp, K. D., and Stradiotto, M. (2010). Stereo- and regioselective gold-catalyzed hydroamination of internal alkynes with dialkylamines. J. Am. Chem. Soc. 132, 18026–18029. doi:10.1021/ja109192w
Hesp, K. D., Tobisch, S., and Stradiotto, M. (2010). [Ir(COD)Cl] 2 as a catalyst precursor for the intramolecular hydroamination of unactivated alkenes with primary amines and secondary alkyl- or arylamines: a combined catalytic, mechanistic, and computational investigation. J. Am. Chem. Soc. 132, 413–426. doi:10.1021/ja908316n
Hintermair, U., Englert, U., and Leitner, W. (2011). Distinct reactivity of mono- and bis-NHC silver complexes: carbene donors versus carbene–halide exchange reagents. Organometallics 30, 3726–3731. doi:10.1021/om101056y
Hopkinson, M. N., Richter, C., Schedler, M., and Glorius, F. (2014). An overview of N-heterocyclic carbenes. Nature 510, 485–496. doi:10.1038/nature13384
Huang, L., Arndt, M., Gooßen, K., Heydt, H., and Gooßen, L. J. (2015). Late transition metal-catalyzed hydroamination and hydroamidation. Chem. Rev. 115, 2596–2697. doi:10.1021/cr300389u
Huo, J., He, G., Chen, W., Hu, X., Deng, Q., and Chen, D. (2019). A minireview of hydroamination catalysis: alkene and alkyne substrate selective, metal complex design. BMC Chem. 13, 89. doi:10.1186/s13065-019-0606-7
Jacobsen, H., Correa, A., Poater, A., Costabile, C., and Cavallo, L. (2009). Understanding the M(NHC) (NHC=N-heterocyclic carbene) bond. Coord. Chem. Rev. 253, 687–703. doi:10.1016/j.ccr.2008.06.006
Jaspers, D., and Doye, S. (2011). Potassium hydroxide catalyzed addition of arylamines to styrenes. Synlett 2011, 1444–1448. doi:10.1055/s-0030-1260555
Katari, M., Rao, M. N., Rajaraman, G., and Ghosh, P. (2012). Computational insight into a gold(I) N-heterocyclic carbene mediated alkyne hydroamination reaction. Inorg. Chem. 51, 5593–5604. doi:10.1021/ic2024605
Kumar, A., Singh, C., Tinnermann, H., and Huynh, H. V. (2020). Gold(I) and gold(III) complexes of expanded-ring N-heterocyclic carbenes: structure, reactivity, and catalytic applications. Organometallics 39, 172–181. doi:10.1021/acs.organomet.9b00718
Leung, C. H., Baron, M., and Biffis, A. (2020). Gold-catalyzed intermolecular alkyne hydrofunctionalizations—mechanistic insights. Catalysts 10, 1210. doi:10.3390/catal10101210
Lin, I. J. B., and Vasam, C. S. (2007). Preparation and application of N-heterocyclic carbene complexes of Ag(I). Coord. Chem. Rev. 251, 642–670. doi:10.1016/j.ccr.2006.09.004
Liu, D., Nie, Q., Zhang, R., and Cai, M. (2018). Regiospecific hydroamination of unsymmetrical electron-rich and electron-poor alkynes with anilines catalyzed by gold(I) immobilized in MCM-41. Adv. Synth. Catal. 360, 3940–3948. doi:10.1002/adsc.201800621
Liu, X.-Y., and Che, C.-M. (2009). Highly enantioselective synthesis of chiral secondary amines by gold(I)/Chiral brønsted acid catalyzed tandem intermolecular hydroamination and transfer hydrogenation reactions. Org. Lett. 11, 4204–4207. doi:10.1021/ol901443b
Macrae, C. F., Sovago, I., Cottrell, S. J., Galek, P. T. A., McCabe, P., Pidcock, E., et al. (2020). Mercury 4.0: from visualization to analysis, design and prediction. J. Appl. Crystallogr. 53, 226–235. doi:10.1107/S1600576719014092
Manna, K., Xu, S., and Sadow, A. D. (2011). A highly enantioselective zirconium catalyst for intramolecular alkene hydroamination: significant isotope effects on rate and stereoselectivity. Angew. Chem. Int. Ed. 50, 1865–1868. doi:10.1002/anie.201006163
Mariconda, A., Grisi, F., Costabile, C., Falcone, S., Bertolasi, V., and Longo, P. (2014). Synthesis, characterization and catalytic behaviour of a palladium complex bearing a hydroxy-functionalized N-heterocyclic carbene ligand. New J. Chem. 38, 762–769. doi:10.1039/C3NJ01281F
Mariconda, A., Sirignano, M., Costabile, C., and Longo, P. (2020). New NHC- silver and gold complexes active in A3-coupling (aldehyde-alkyne-amine) reaction. Mol. Catal. 480, 110570. doi:10.1016/j.mcat.2019.110570
Mariconda, A., Sirignano, M., Troiano, R., Russo, S., and Longo, P. (2022). N-heterocyclic carbene gold complexes active in hydroamination and hydration of alkynes. Catalysts 12, 836. doi:10.3390/catal12080836
Michael, F. E., Duncan, A. P., Sweeney, Z. K., and Bergman, R. G. (2003). Mechanisms of allene stereoinversion by imidozirconium complexes. J. Am. Chem. Soc. 125, 7184–7185. doi:10.1021/ja0348389
Mizushima, E., Hayashi, T., and Tanaka, M. (2003). Au(I)-Catalyzed highly efficient intermolecular hydroamination of alkynes. Org. Lett. 5, 3349–3352. doi:10.1021/ol0353159
Mukherjee, A., Nembenna, S., Sen, T. K., Sarish, S. P., Ghorai, P.Kr., Ott, H., et al. (2011). Assembling zirconium and calcium moieties through an oxygen center for an intramolecular hydroamination reaction: a single system for double activation. Angew. Chem. Int. Ed. 50, 3968–3972. doi:10.1002/anie.201100022
Müller, T. E., Hultzsch, K. C., Yus, M., Foubelo, F., and Tada, M. (2008). Hydroamination: direct addition of amines to alkenes and alkynes. Chem. Rev. 108, 3795–3892. doi:10.1021/cr0306788
Müller, T. E., and Pleier, A.-K. (1999). Intramolecular hydroamination of alkynes catalysed by late transition metals. J. Chem. Soc. Dalton Trans. 583, 583. doi:10.1039/a808938h
Napoli, M., Saturnino, C., Cianciulli, E. I., Varcamonti, M., Zanfardino, A., Tommonaro, G., et al. (2013). Silver(I) N-heterocyclic carbene complexes: synthesis, characterization and antibacterial activity. J. Organomet. Chem. 725, 46–53. doi:10.1016/j.jorganchem.2012.10.040
S. P. Nolan (Editor) (2014). N-heterocyclic carbenes: effective tools for organometallic synthesis. 1 (Weinheim: Wiley VCH).
Nuevo, D., Poyatos, M., and Peris, E. (2018). A dinuclear Au(I) complex with a pyrene-di-N-heterocyclic carbene linker: supramolecular and catalytic studies. Organometallics 37, 3407–3411. doi:10.1021/acs.organomet.8b00087
Odom, A. L. (2005). New C–N and C–C bond forming reactions catalyzed by titanium complexes. Dalton Trans., 225–233. doi:10.1039/B415701J
Patel, M., Saunthwal, R. K., and Verma, A. K. (2017). Base-mediated hydroamination of alkynes. Acc. Chem. Res. 50, 240–254. doi:10.1021/acs.accounts.6b00449
Patil, S., Claffey, J., Deally, A., Hogan, M., Gleeson, B., Menéndez Méndez, L. M., et al. (2010). Synthesis, cytotoxicity and antibacterial studies of p -Methoxybenzyl-Substituted and benzyl-substituted N-heterocyclic carbene–silver complexes. Eur. J. Inorg. Chem. 2010, 1020–1031. doi:10.1002/ejic.200900889
Peeters, A., Valvekens, P., Ameloot, R., Sankar, G., Kirschhock, C. E. A., and De Vos, D. E. (2013). Zn–Co double metal cyanides as heterogeneous catalysts for hydroamination: a structure–activity relationship. ACS Catal. 3, 597–607. doi:10.1021/cs300805z
Pohlki, F., and Doye, S. (2003). The catalytic hydroamination of alkynes. Chem. Soc. Rev. 32, 104–114. doi:10.1039/b200386b
Reznichenko, A. L., Nguyen, H. N., and Hultzsch, K. C. (2010a). Asymmetric intermolecular hydroamination of unactivated alkenes with simple amines. Angew. Chem. Int. Ed. 49, 8984–8987. doi:10.1002/anie.201004570
Reznichenko, A. L., Nguyen, H. N., and Hultzsch, K. C. (2010b). Asymmetrische intermolekulare Hydroaminierung nichtaktivierter Alkene mit einfachen Aminen. Angew. Chem. 122, 9168–9171. doi:10.1002/ange.201004570
Sarcher, C., Lühl, A., Falk, F. C., Lebedkin, S., Kühn, M., Wang, C., et al. (2012). [2.2]Paracyclophanediyldiphosphane complexes of gold. Eur. J. Inorg. Chem. 2012, 5033–5042. doi:10.1002/ejic.201200751
Saturnino, C., Barone, I., Iacopetta, D., Mariconda, A., Sinicropi, M. S., Rosano, C., et al. (2016). N -heterocyclic carbene complexes of silver and gold as novel tools against breast cancer progression. Future Med. Chem. 8, 2213–2229. doi:10.4155/fmc-2016-0160
Savchuk, M., Bocquin, L., Albalat, M., Jean, M., Vanthuyne, N., Nava, P., et al. (2022). Transition metal complexes bearing atropisomeric saturated NHC ligands. Chirality 34, 13–26. doi:10.1002/chir.23378
Severin, R., and Doye, S. (2007). The catalytic hydroamination of alkynes. Chem. Soc. Rev. 36, 1407. doi:10.1039/b600981f
Sevov, C. S., Zhou, J., Steve), , and Hartwig, J. F. (2014). Iridium-catalyzed, intermolecular hydroamination of unactivated alkenes with indoles. J. Am. Chem. Soc. 136, 3200–3207. doi:10.1021/ja412116d
Shanbhag, G. V., and Halligudi, S. B. (2004). Intermolecular hydroamination of alkynes catalyzed by zinc-exchanged montmorillonite clay. J. Mol. Catal. A Chem. 222, 223–228. doi:10.1016/j.molcata.2004.08.010
Sheldrick, G. M. (2008). A short history of SHELX. Acta Crystallogr. A Found. Crystallogr. 64, 112–122. doi:10.1107/S0108767307043930
Sheldrick, G. M. (2015). Crystal structure refinement with SHELXL. Acta Crystallogr. C Struct. Chem. 71, 3–8. doi:10.1107/S2053229614024218
Shi, Y., Ciszewski, J. T., and Odom, A. L. (2001). Ti(NMe 2) 4 as a precatalyst for hydroamination of alkynes with primary amines. Organometallics 20, 3967–3969. doi:10.1021/om010566b
Sirignano, M., Mariconda, A., Vigliotta, G., Ceramella, J., Iacopetta, D., Sinicropi, M. S., et al. (2021). Catalytic and biological activity of silver and gold complexes stabilized by NHC with hydroxy derivatives on nitrogen atoms. Catalysts 12, 18. doi:10.3390/catal12010018
Tsipis, C. A., and Kefalidis, C. E. (2006). How efficient are the hydrido-bridged diplatinum catalysts in the hydrosilylation, hydrocyanation, and hydroamination of alkynes: a theoretical analysis of the catalytic cycles employing electronic structure calculation methods. Organometallics 25, 1696–1706. doi:10.1021/om0509342
Tubaro, C., Baron, M., Biffis, A., and Basato, M. (2013). Alkyne hydroarylation with Au N-heterocyclic carbene catalysts. Beilstein J. Org. Chem. 9, 246–253. doi:10.3762/bjoc.9.29
Visbal, R., Graus, S., Herrera, R. P., and Gimeno, M. C. (2018). Gold catalyzed multicomponent reactions beyond A3 coupling. Molecules 23, 2255. doi:10.3390/molecules23092255
Visbal, R., Laguna, A., and Gimeno, M. C. (2013). Simple and efficient synthesis of [MCI(NHC)] (M = Au, Ag) complexes. Chem. Commun. 49, 5642. doi:10.1039/c3cc42919a
Wang, Z., Ye, X., Wei, S., Wu, P., Zhang, A., and Sun, J. (2006). A highly enantioselective Lewis basic organocatalyst for reduction of N-aryl imines with unprecedented substrate spectrum. Org. Lett. 8, 999–1001. doi:10.1021/ol060112g
Wixey, J. S., and Ward, B. D. (2011). Chiral calciumcatalysts for asymmetric hydroamination/cyclisation. Chem. Commun. 47, 5449–5451. doi:10.1039/C1CC11229E
Yahata, K., Kaneko, Y., and Akai, S. (2020). Cobalt-catalyzed intermolecular Markovnikov hydroamination of nonactivated olefins: N2 -selective alkylation of benzotriazole. Org. Lett. 22, 598–603. doi:10.1021/acs.orglett.9b04375
Yang, X.-H., Lu, A., and Dong, V. M. (2017). Intermolecular hydroamination of 1,3-dienes to generate homoallylic amines. J. Am. Chem. Soc. 139, 14049–14052. doi:10.1021/jacs.7b09188
Yazdani, S., Junor, G. P., Peltier, J. L., Gembicky, M., Jazzar, R., Grotjahn, D. B., et al. (2020). Influence of carbene and phosphine ligands on the catalytic activity of gold complexes in the hydroamination and hydrohydrazination of alkynes. ACS Catal. 10, 5190–5201. doi:10.1021/acscatal.0c01352
Zargaran, P., Wurm, T., Zahner, D., Schießl, J., Rudolph, M., Rominger, F., et al. (2018). A structure-based activity study of highly active unsymmetrically substituted NHC gold(I) catalysts. Adv. Synth. Catal. 360, 106–111. doi:10.1002/adsc.201701080
Zhang, J., Yang, C.-G., and He, C. (2006). Gold(I)-Catalyzed intra- and intermolecular hydroamination of unactivated olefins. J. Am. Chem. Soc. 128, 1798–1799. doi:10.1021/ja053864z
Keywords: N-heterocyclic carbene, gold, hydroamination, alkynes, DFT modelling
Citation: Sirignano M, D’Amato A, Costabile C, Mariconda A, Crispini A, Scarpelli F and Longo P (2023) Hydroamination of alkynes catalyzed by NHC-Gold(I) complexes: the non-monotonic effect of substituted arylamines on the catalyst activity. Front. Chem. 11:1260726. doi: 10.3389/fchem.2023.1260726
Received: 18 July 2023; Accepted: 24 October 2023;
Published: 06 December 2023.
Edited by:
David Morales-Morales, National Autonomous University of Mexico, MexicoReviewed by:
Sushobhan Mukhopadhyay, University of Florida, United StatesCopyright © 2023 Sirignano, D’Amato, Costabile, Mariconda, Crispini, Scarpelli and Longo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chiara Costabile, Y2Nvc3RhYmlsZUB1bmlzYS5pdA==; Annaluisa Mariconda, YW5uYWx1aXNhLm1hcmljb25kYUB1bmliYXMuaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.