 Patrick Floß
Patrick Floß Christopher S. Reilly
Christopher S. Reilly Daniel J. Auerbach1,3
Daniel J. Auerbach1,3 Rainer D. Beck
Rainer D. Beck- 1Institute of Chemical Sciences and Engineering (ISIC), École Polytechnique Fédérale de Lausanne (EPFL), Lausanne, Switzerland
- 2Max Planck-EPFL Center for Molecular Nanoscience and Technology, Lausanne, Switzerland
- 3Max Planck Institute for Multidisciplinary Sciences, Göttingen, Germany
Recent state-to-state experiments of methane scattering from Ni(111) and graphene-covered Ni(111) combined with quantum mechanical simulations suggest an intriguing correlation between the surface-induced vibrational energy redistribution (SIVR) during the molecule/surface scattering event and the catalytic activity for methane dissociation of the target surface (Werdecker, Phys. Rev. Res., 2020, 2, 043251). Herein, we report new quantum state and angle-resolved measurements for methane scattering from Ni(111) and Au(111) probing the extent of
1 Introduction
State-to-state scattering experiments provide a powerful and well-established technique to elucidate the atomic-level details of what happens when molecules collide with other gas-phase molecules or with surfaces (Auerbach et al., 2021). Often, the connection between the measured final state distributions and the property under study is direct and obvious. For example, the degree of rotational excitation in a collision is directly related to the angular anisotropy of the potential energy surface (PES) of the system under study and can be used to test theoretical methods for calculating the PES. In other cases, the connection of the scattering data to the underlying question of what is going on is more subtle. For example, highly vibrationally excited NO or CO molecules scatter almost vibrationally elastically from insulator surfaces but undergo facile multi-quantum vibrational relaxation if they strike a metal surface. Qualitatively, these observations show that the presence of a continuum of low-lying electronically excited states in the metal is being excited by molecular vibrations-a strong violation of the Born–Oppenheimer approximation. More detailed theoretical work established that charge transfer in the collisions is the key driving factor and that the propensity for charge transfer therefore correlates with the degree of vibrational relaxation (Wodtke et al., 2008; Shenvi et al., 2009; Wagner et al., 2019).
An intriguing example of this more subtle connection of observations to dynamical propensities emerges from the first state-to-state measurements of methane scattering from surfaces. When CH4 (
Surprisingly, SIVR was not observed for CH4 (
In this paper, we present new measurements to test this hypothesis. We extend the range of surfaces studied to include the closely packed Au(111) surface which, based on density function calculations1 of the barrier to dissociative adsorption, is expected to have a catalytic activity intermediate to that of Ni(111) (Jackson and Nave, 2013) and Gr/Ni(111) (Li et al., 2018).
In addition to providing data on a new system with catalytic activity intermediate between Ni(111) and Gr/Ni(111), we have also improved the method of determining the “branching ratio”
Our measurements show that the value of
2 Materials and methods
Figure 1 shows a schematic illustration of the experimental setup. Full details can be found in Reilly et al. (2023). Briefly, a continuous supersonic expansion from a temperature-controlled nozzle into a vacuum is used to create a molecular beam of methane with the mean kinetic energy of 100 meV and a FWHM spread of 30%. Approximately 10% of the methane molecules in the molecular beam are prepared in the
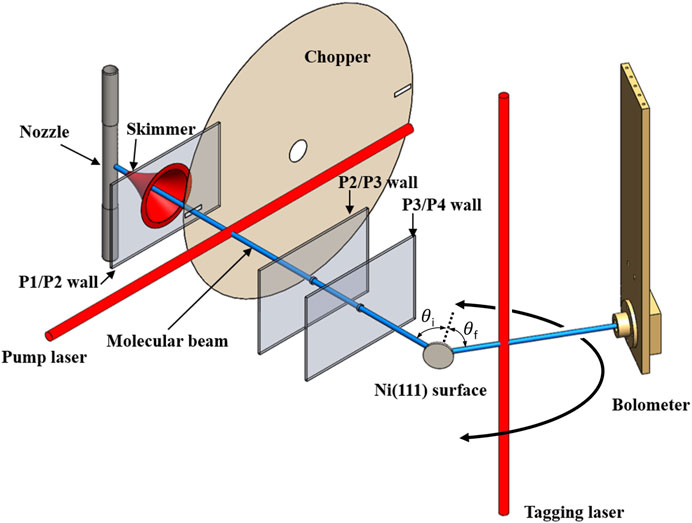
FIGURE 1. Schematic of the experimental setup used for state-to-state surface scattering of methane. A molecular beam is generated in vacuum chamber P1 and traverses the differential pumping stages P2 and P3. Quantum state-specific preparation of methane in the molecular beam occurs in P2 which also contains a chopper disk for beam modulation. The state-prepared molecular beam collides in P4 with the single crystal target surface at an incident angle
The state-prepared molecular beam collides with a single crystal surface sample in the ultrahigh vacuum surface science chamber with a base pressure of 1 × 10−10 mbar. A 4-axis manipulator permits displacement the surface in three dimensions and rotation of the incident angle
Scattered molecules are detected and resolved by the quantum state and scattering angle using the bolometer infrared laser tagging (BILT) technique (Reilly et al., 2023). Here, a second, tunable, single-mode, CW IR OPO (TOPO TOPTICA Photonics, “tagging laser” in Figure 1) laser beam is chopped at 237 Hz with 50% duty cycle using an optical chopper wheel (not shown) and crosses the scattering plane perpendicularly 16 mm from the target surface. By tuning the tagging laser into resonance with different rovibrational transitions, we selectively excite the methane molecules that have scattered from the initially prepared state into the lower state of the resonant transition, transferring to those molecules one quantum of
Using a lock-in amplifier, we record the response of the detector to the chopping of the tagging laser. To the raw lock-in output, we apply a series of corrections to obtain a “scattering intensity”
Synchronized, computer-controlled rotation of the bolometer and the tagging laser beam enables variation of the measured final angle
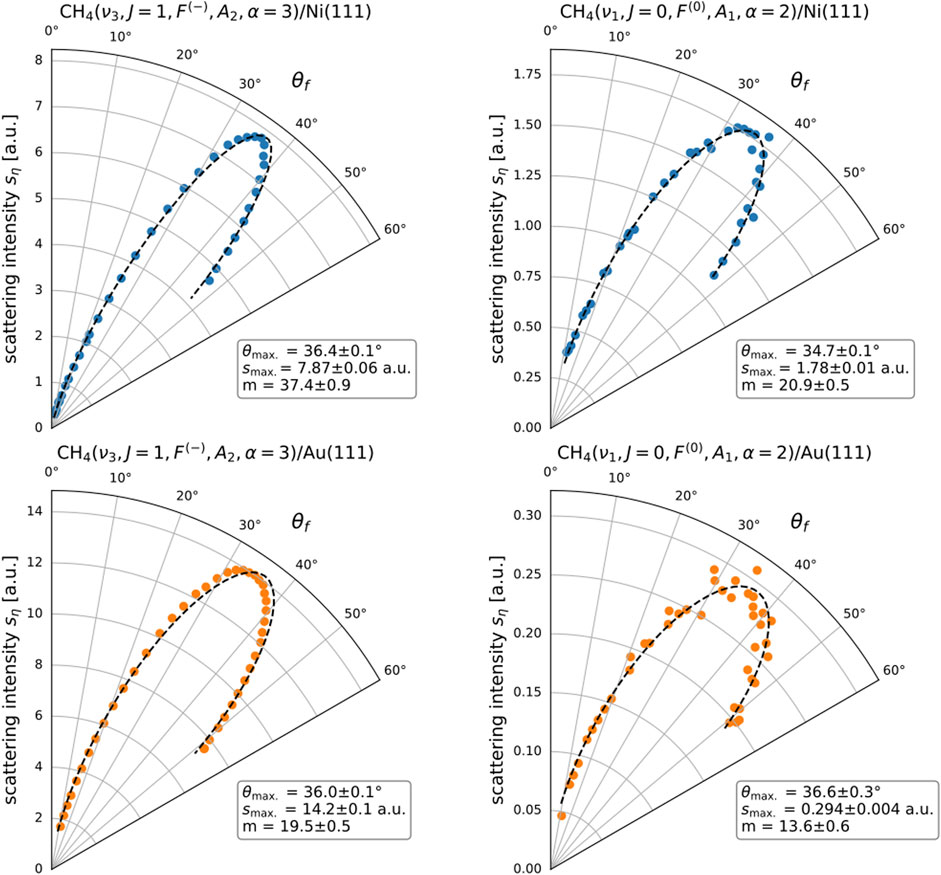
FIGURE 2. State-resolved in-plane angular distributions
For preparation of atomically flat Ni(111) and Au(111) surfaces, we use Ar+ sputtering followed by annealing to 973 K and 773 K, respectively. Crystallographic order of prepared surfaces is verified using low-energy electron diffraction (LEED), and the absence of surface contaminants is verified using Auger electron spectroscopy. After conducting all experiments, the contamination levels were verified to be below the detection threshold of approximately 1% ML.
3 Results and discussion
For both the Ni(111) and Au(111) surfaces, we recorded 14 state-resolved angular distributions. To identify the rovibrational level being tagged, we use the quantum numbers (
Figure 2 illustrates a selected subset of the measured angular distributions. The complete set of measured distributions is available in Supplementary Section S1. The rotational states tagged include all meta-CH4 states in the
In the following sections, we present an analysis and discussion of the measured distributions. First, in Section 3.1, we treat the central topic of SIVR before moving on to a detailed look at aspects of scattering dynamics relating to the scattering angle (Section 3.2) and rotational state (Section 3.3).
3.1 Surface-induced vibrational energy redistribution
The principle objective of the current work is to test the hypothesis advanced in Werdecker et al. and discussed in the Introduction of the current work. We remind the reader that this hypothesis asserts a positive correlation between the efficacy of
The branching ratios

TABLE 1. Scattering angle-integrated branching ratios
To assess the repeatability of the branching ratio measurements the full set of measurements for the Au(111) surface was performed twice. The data from the second run showed a somewhat better signal-to-noise ratio and so we present the branching ratio of 6.6% obtained from this run in Table 1. The first run yielded a branching ratio of 7.9%. The percentage deviation of
3.2 Trends in state-resolved angular distributions
In computing the branching ratios, we integrate over the scattering angle and sum over rotational states and thus discard any dynamical information contained in the angular and rotational state dependence of the scattering intensities. To explore in finer detail the dynamics of the molecule–surface collision, we return in this section to the state-resolved angular distributions. In the following section (Section 3.3), we narrow our attention to the gross rotational dynamics via the scattering angle-integrated state fluxes
We have found it useful to summarize individual state-resolved angular distributions by three parameters, namely,
That the chosen fit function gives a good description of our observations can be verified by inspection of Figure 2 which shows measured data overlaid with their associated best-fit curves (see also the Supplementary Materials for full set of angular distributions and fits).
Figure 3 shows the best-fit parameter values

FIGURE 3. Fitting parameters obtained from fitting Eq. 1 to the measured state-resolved in-plane angular distributions
In addition to our classification of the scattering as direct/impulsive, we also identify three distinct trends from the analysis of the angular distributions. First, the exponents
Third, the peak angles
Going beyond the trends just discussed, we can also make a few additional qualitative observations. We first point out the anomalously large exponent
In another sense, that the vibrationally and rotationally elastic scattering peak is so much narrower than the others and should imply something about the molecule–surface interaction potential and associated scattering dynamics. We offer the following simple microscopic explanation for a strong and anomalously narrow elastic scattering distribution peaking near specular (
To qualitatively summarize the explanation just presented, our observations can be interpreted as implying a certain order in which the different aspects of the molecule–surface interaction “turn on” as the molecule approaches the surface, with a two-body repulsive force preceding the onset of dispersive forces, which in turn precedes the onset of forces acting to distort or rotate the molecule.
We close the section with a discussion regarding the differences in peak widths
Compare for example the best-fit
Concerning the Au(111) surface data, while the form of the distributions among
3.3 Rotational dynamics
Figure 4 shows the scattering angle-integrated rovibrational state populations
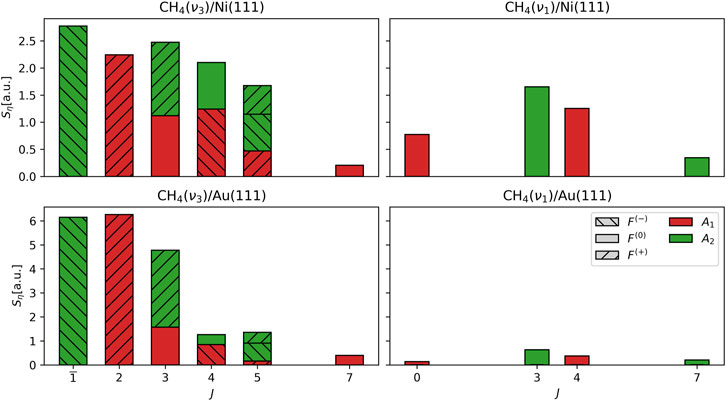
FIGURE 4. Distribution of scattering angle-integrated rovibrational state fluxes
One feature that is directly observable is the differences in the forms of the distributions of the vibrationally elastic (
The extracted mean rotational energies
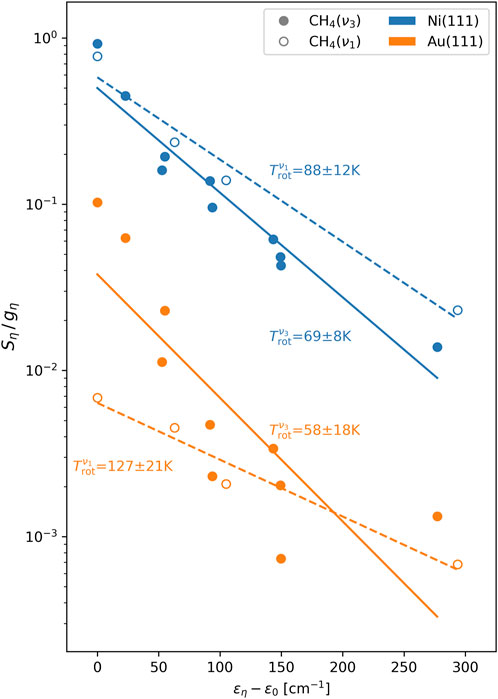
FIGURE 5. Boltzmann fits to the rotational-level population. Plotted are the scattering angle-integrated state-resolved fluxes
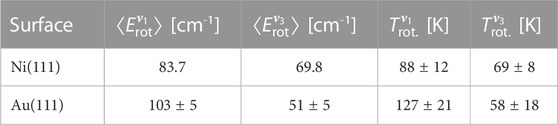
TABLE 2. Extracted mean rotational energies
Perhaps the most interesting result emerging from this analysis is that the ratio of the degree of rotational excitation for molecules scattered into the
Why do the ratios
In the reduced-dimensional CH4/Ni(111) SIVR calculations presented in Werdecker et al., the surface was held frozen and the molecule was forced to assume at all times the minimum energy orientation so that energy released in the
The last observation we wish to make about the rotational distributions concerns the
At the present time, the authors have no explanation for the form and extent of this internal alignment of molecular rotation observed in the scattering. We plan to present more extensive results on these interesting alignment phenomena in CH4 surface scattering in a future publication.
4 Summary and future directions
A number of promising avenues exist for further testing the connection between SIVR and catalytic activity. One extension of the work presented here would be a study of SIVR efficiency for a range of different incident kinetic energies
Another direction, one which would offer a more direct validation of the theoretical results reported in Werdecker et al., would be to measure the variation in SIVR across the different microscopic impact sites of the same macroscopic surface. Although it is of course not feasible to experimentally control the impact site of a molecule, one can vary the relative concentration of different sites. In the calculations of Werdecker et al., the authors find an increase in SIVR at sites where a surface Ni atom is displaced from equilibrium. Such displacements are also expected to increase a site’s catalytic activity (Nave et al., 2010). By raising/lowering the surface temperature, one can increase/decrease the density of thermally generated lattice distortions and test for the expected increase/decrease in SIVR.
Another more controlled means of varying the density of different surface sites is to vary the exposed crystal plane, characterized by Miller indices (ijk) (Kolasinski, 2012). For fcc metals (e.g., Ni, Au, and Pt), the atoms of the (111) (or “close-packed”) surface are highly coordinated and therefore present sites of relatively low catalytic activity. The stepped fcc(211) surface, however, exposes under-coordinated atoms at step sites, offering lower barrier sites for methane dissociative chemisorption, as observed in experiments and calculations for CH4 sticking experiments on platinum (Chadwick et al., 2018). A measurement of the branching ratios for different surface planes of the same crystal could thus serve as a compelling experimental verification of the theoretically observed connection between a site’s SIVR efficiency and its catalytic activity.
We conclude with a brief summary. For the Ni(111) and Au(111) surfaces, we measured rovibrational state-resolved angular distributions for 10 (4) rotational levels of the
Analysis of the shape of angular distributions reveals for both surfaces a broadening of the scattering distributions with increasing total angular momentum quantum number
From the distribution of scattering angle-integrated state fluxes, the Ni(111) surface produces more highly rotationally excited molecules in the
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Author contributions
RB conceptualized the project, secured funding, and supervised the project. DA designed the experimental setup with support from RB. PF and CR performed the experiments, and PF analyzed the data. CR wrote the manuscript with contributions from PF, DA, and RB. All authors contributed to the article and approved the submitted version.
Acknowledgments
The authors acknowledge financial support by the Swiss National Science Foundation through Grant No. 200497 and by the Max Planck-EPFL Center for Molecular Nanoscience and Technology. The authors thank the EPFL ISIC machine shop and Bo-Jung Chen for their invaluable contributions to the construction of the apparatus used to perform the experiments presented in this manuscript. The authors would also like to thank Bret Jackson for his calculations of the CH4/Au(111) dissociation barrier and valuable discussions. DA gratefully acknowledges support from the International Center for Advanced Energy Conversion in Göttingen, Germany.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2023.1238711/full#supplementary-material
Footnotes
1Jackson, B. (2023) Private communication.
References
Al Taleb, A., and Farías, D. (2017). Coherent quantum scattering of CH 4 from Ni(111). Phys. Chem. Chem. Phys. 19, 21267–21271. doi:10.1039/c7cp04559j
Auerbach, D. J., Tully, J. C., and Wodtke, A. M. (2021). Chemical dynamics from the gas-phase to surfaces. Nat. Sci. 1, e10005. doi:10.1002/ntls.10005
Bhati, M., Dhumal, J., and Joshi, K. (2022). Lowering the C–H bond activation barrier of methane by means of SAC@Cu(111): Periodic DFT investigations. New J. Chem. 46, 70–74. doi:10.1039/d1nj04525c
Boudon, V., Rey, M., and Loëte, M. (2006). The vibrational levels of methane obtained from analyses of high-resolution spectra. J. Quant. Spectrosc. Radiat. Transf. 98, 394–404. doi:10.1016/j.jqsrt.2005.06.003
Brown, L. R., Margolis, J., Champion, J., Hilico, J., Jouvard, J., Loete, M., et al. (1992). Methane and its isotopes: Current status and prospects for improvement. J. Quant. Spectrosc. Radiat. Transf. 48, 617–628. doi:10.1016/0022-4073(92)90126-o
Chadwick, H., Guo, H., Gutiérrez-González, A., Menzel, J. P., Jackson, B., and Beck, R. D. (2018). Methane dissociation on the steps and terraces of Pt(211) resolved by quantum state and impact site. J. Chem. Phys. 148, 014701. doi:10.1063/1.5008567
Chadwick, H., Hundt, P. M., van Reijzen, M. E., Yoder, B. L., and Beck, R. D. (2014). Quantum state specific reactant preparation in a molecular beam by rapid adiabatic passage. J. Chem. Phys. 140, 034321. doi:10.1063/1.4861054
di Lauro, C. (2020). “Spectra of spherical top molecules,” in Rotational structure in molecular infrared spectra (China: Elsevier), 223–242.
Gerrits, N., Chadwick, H., and Kroes, G.-J. (2019). Dynamical study of the dissociative chemisorption of CHD3 on Pd(111). J. Phys. Chem. C 123, 24013–24023. doi:10.1021/acs.jpcc.9b05757
Gordon, I. E., Rothman, L., Hargreaves, R., Hashemi, R., Karlovets, E., Skinner, F., et al. (2022). The HITRAN2020 molecular spectroscopic database. J. Quant. Spectrosc. Radiat. Transf. 277, 107949. doi:10.1016/j.jqsrt.2021.107949
Hecht, K. T. (1961b). The vibration-rotation energies of tetrahedral XY4 molecules: Part I. Theory of spherical top molecules. J. Mol. Spectrosc. 5, 355–389. doi:10.1016/0022-2852(61)90102-3
Hecht, K. T. (1961a). Vibration-rotation energies of tetrahedral XY4 molecules: Part II. The fundamental ν3 of CH4. J. Mol. Spectrosc. 5, 390–404. doi:10.1016/0022-2852(61)90103-5
Jackson, B., and Nave, S. (2013). The dissociative chemisorption of methane on Ni(111): The effects of molecular vibration and lattice motion. J. Chem. Phys. 138, 174705. doi:10.1063/1.4802008
Jackson, B. (2022). Quantum studies of methane-metal inelastic diffraction and trapping: The variation with molecular orientation and phonon coupling. Chem. Phys. 559, 111516. doi:10.1016/j.chemphys.2022.111516
Kolasinski, K. (2012). “Surface and adsorbate structure,” in Surface science (China: John Wiley and Sons, Ltd), 9–49. doi:10.1002/9781119941798.ch1
Kondo, T., Al Taleb, A., Anemone, G., and Farías, D. (2018). Low-energy methane scattering from Pt(111). J. Chem. Phys. 149, 084703. doi:10.1063/1.5044744
Li, K., Li, H., Yan, N., Wang, T., and Zhao, Z. (2018). Adsorption and dissociation of CH4 on graphene: A density functional theory study. Appl. Surf. Sci. 459, 693–699. doi:10.1016/j.apsusc.2018.08.084
Milot, R., Kleyn, A. W., and Jansen, A. P. J. (2001). Energy dissipation and scattering angle distribution analysis of the classical trajectory calculations of methane scattering from a Ni(111) surface. J. Chem. Phys. 115, 3888–3894. doi:10.1063/1.1388224
Nave, S., Tiwari, A. K., and Jackson, B. (2010). Methane dissociation and adsorption on Ni(111), Pt(111), Ni(100), Pt(100), and Pt(110)-(1 x 2): Energetic study. J. Chem. Phys. 132, 054705. doi:10.1063/1.3297885
Reilly, C. S., Floß, P., Chen, B.-J., Auerbach, D. J., and Beck, R. D. (2023). Quantum state-resolved methane scattering from Ni(111) and NiO(111) by bolometer infrared laser tagging: The effect of surface oxidation. J. Chem. Phys. 158, 214202. doi:10.1063/5.0150009
Rettner, C. T., and Auerbach, D. J. (1994). Distinguishing the direct and indirect products of A gas-surface reaction. Science 263, 365–367. doi:10.1126/science.263.5145.365
Shenvi, N., Roy, S., and Tully, J. C. (2009). Nonadiabatic dynamics at metal surfaces: Independent-electron surface hopping. J. Chem. Phys. 130, 174107. doi:10.1063/1.3125436
Virtanen, P., Gommers, R., Oliphant, T. E., Haberland, M., Reddy, T., Cournapeau, D., et al. (2020). SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 17, 261–272. doi:10.1038/s41592-019-0686-2
Wagner, V., Park, G. B., Wallrabe, M., Wodtke, A. M., and Schäfer, T. (2019). Electron transfer mediates vibrational relaxation of CO in collisions with Ag(111). Phys. Chem. Chem. Phys. 21, 1650–1655. doi:10.1039/c8cp06041j
Werdecker, J., Chen, B. J., Van Reijzen, M. E., Farjamnia, A., Jackson, B., and Beck, R. D. (2020). State-to-state methane-surface scattering as a probe of catalytic activity. Phys. Rev. Res. 2, 043251. doi:10.1103/physrevresearch.2.043251
Werdecker, J., van Reijzen, M. E., Chen, B.-J., and Beck, R. D. (2018). Vibrational energy redistribution in a gas-surface encounter: State-to-State scattering of CH 4 from Ni(111). Phys. Rev. Lett. 120, 053402. doi:10.1103/physrevlett.120.053402
Wodtke, A. M., Matsiev, D., and Auerbach, D. J. (2008). Energy transfer and chemical dynamics at solid surfaces: The special role of charge transfer. Prog. Surf. Sci. 83, 167–214. doi:10.1016/j.progsurf.2008.02.001
Zhang, L., and Jiang, B. (2019). Efficient vibrational energy redistribution between stretching modes: State-to-State quantum scattering of H 2 O from Cu(111). Phys. Rev. Lett. 123, 106001. doi:10.1103/physrevlett.123.106001
Keywords: methane dissociation, state-to-state scattering, angular distributions, surface-induced vibrational energy redistribution, optothermal spectroscopy, bolometer infrared laser tagging, heterogeneous catalysis
Citation: Floß P, Reilly CS, Auerbach DJ and Beck RD (2023) Surface-induced vibrational energy redistribution in methane/surface scattering depends on catalytic activity. Front. Chem. 11:1238711. doi: 10.3389/fchem.2023.1238711
Received: 12 June 2023; Accepted: 05 July 2023;
Published: 25 July 2023.
Edited by:
Marco Sacchi, University of Surrey, United KingdomReviewed by:
Bret Jackson, University of Massachusetts Amherst, United StatesHirokazu Ueta, Japan Atomic Energy Agency, Japan
Copyright © 2023 Floß, Reilly, Auerbach and Beck. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rainer D. Beck, cmFpbmVyLmJlY2tAZXBmbC5jaA==
†These authors have contributed equally to this work and share first authorship