 Matthew E. Thornton
Matthew E. Thornton Jake Hemsworth1
Jake Hemsworth1 Sam Hay
Sam Hay Patrick Parkinson
Patrick Parkinson Louise S. Natrajan
Louise S. Natrajan- 1Department of Chemistry, Faculty of Science and Engineering, The University of Manchester, Manchester, United Kingdom
- 2Department of Chemistry, Faculty of Science and Engineering, Manchester Institute of Biotechnology, The University of Manchester, Manchester, United Kingdom
- 3Department of Physics and Astronomy and the Photon Science Institute, Faculty of Science and Engineering, The University of Manchester, Manchester, United Kingdom
- 4Department of Chemistry, Chemistry Research Laboratory, The University of Oxford, Oxford, United Kingdom
Macrocyclic lanthanide complexes have become widely developed due to their distinctive luminescence characteristics and wide range of applications in biological imaging. However, systems with sufficient brightness and metal selectivity can be difficult to produce on a molecular scale. Presented herein is the stepwise introduction of differing lanthanide ions in a bis-DO3A/DTPA scaffold to afford three trinuclear bimetallic [Ln2Ln’] lanthanide complexes with site-specific, controlled binding [(Yb2Tb), (Eu2Tb), (Yb2Eu)]. The complexes display simultaneous emission from all LnIII centers across the visible (TbIII, EuIII) and near infra-red (YbIII) spectrum when excited via phenyl ligand sensitization at a wide range of temperatures and are consequently of interest for exploiting imaging in the near infra-red II biological window. Analysis of lifetime data over a range of excitation regimes reveals intermetallic communication between TbIII and EuIII centers and further develops the understanding of multimetallic lanthanide complexes.
1 Introduction
The photophysical properties of lanthanide ions in the common +III oxidation state (LnIII) has become a wide and complex field of research across multiple disciplines and applications, particularly LnIII luminescence for bioimaging (Bünzli and Piguet, 2005; Bünzli, 2010; Monteiro, 2020). Previous works have exploited “windows” of attenuation in the absorption profiles of biological tissues at near-infrared (NIR) wavelengths, allowing deeper penetration of such wavelengths and therefore enhanced imaging (Hemmer et al., 2016; Fan and Zhang, 2019). The LnIII ions are of particular interest due to their characteristic line-like emission across the visible and NIR range, low autofluorescence and photobleaching and high signal-to-noise ratios in comparison to classic organic fluorophores. A caveat to their use is intrinsically poor extinction coefficient as a result of partially-forbidden f-f transitions, which is mitigated by sensitization strategies via organic chromophores (the antenna effect) or d- and f-block metal complexes (Faulkner and Pope, 2003; Natrajan et al., 2009). Furthermore, X-H (X = O, N, C) oscillators are well documented in their ability to vibrationally quench LnIII excited states due to significant overlap between vibrational overtones of these bonds and emissive LnIII energy levels (Doffek et al., 2012).
This work further develops a previously designed ligand based on a multi-macrocyclic architecture of 1,4,7,10-tetraazacyclododecane-1,4,7-triacetic acid (DO3A) and diethylenetriaminepentaacetic acid (DTPA) (Faulkner and Pope, 2003). Polyamino carboxylate ligands are well known to securely bind lanthanides in a range of environments and have displayed the ability to act as trinuclear heterometallic binding ligands in a site-selective [TbYbTb] arrangement. Herein, we develop the scaffold to include two [LnTbLn] complexes (LnIII = YbIII, EuIII) and a complementary [YbEuYb] species (Figure 1) and further investigate the photophysical capabilities of the ligand across various temperature windows and excitation regimes. The metal selection results in clear LnIII–centred emission across the visible (TbIII, EuIII) and NIR (YbIII) spectrum, which overlaps with both NIR-I (650–950 nm) and NIR-II (1000–1350 nm) imaging windows and therefore invites application in imaging technologies (Foucault-Collet et al., 2013; Jin et al., 2022). The simultaneous response from both LnIII centres has further application as a dual-modal device; the unique magnetic and photophysical behaviour of these metals facilitates use as both MRI contrast agents and optical probes (Rivas et al., 2013; Xu et al., 2013).
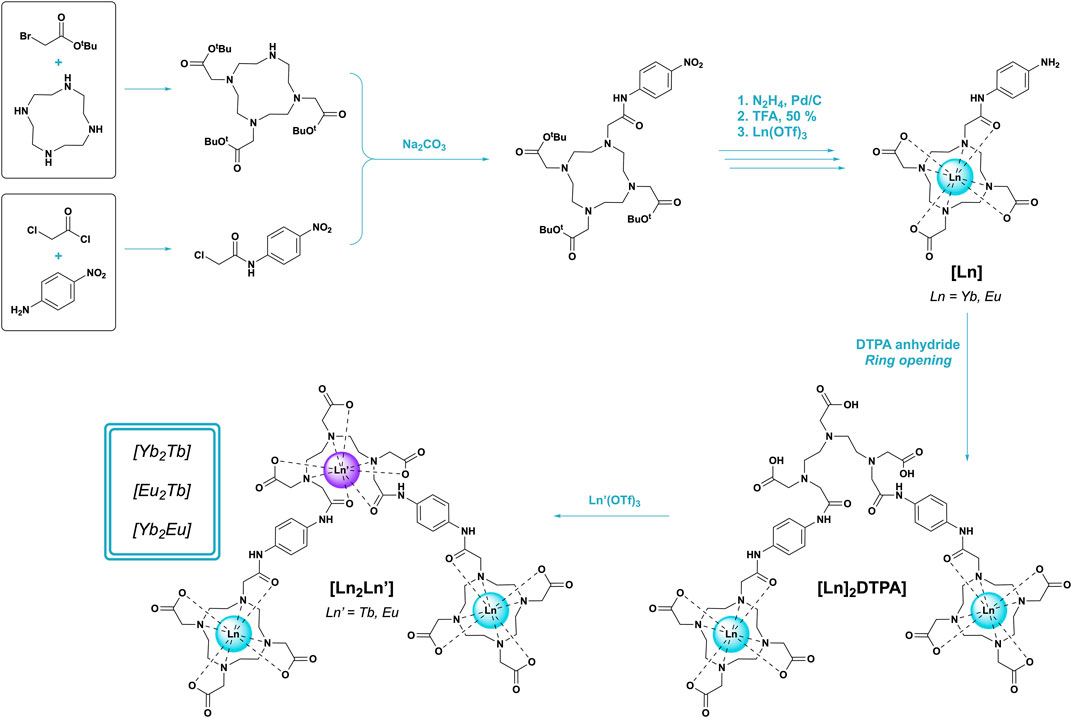
FIGURE 1. Synthetic scheme of the stepwise approach used in synthesis of [Ln2Ln’] trinuclear bimetallics.
Low temperature, solid-state and deuterated media measurements are prioritized to mitigate non-emissive quenching mechanisms. These experiments were also designed to facilitate observation of energy transfer (ET) between metal centers; the relative excited state energies of the chosen metal combinations have been exploited for ET processes in numerous multimetallic LnIII systems (Bispo-Jr et al., 2018; Abad Galán et al., 2021). This work contributes toward the growing library of bimetallic trinuclear LnIII systems that have been studied to elucidate the characteristics of intermetallic communication in discrete molecular complexes (Aboshyan-Sorgho et al., 2012; Zaïm et al., 2014; Tropiano et al., 2015; Maniaki et al., 2023). Finally, the presence of ET in molecular LnIII systems is also useful for imaging applications such as those employing two-photon processes like upconversion (Aboshyan-Sorgho et al., 2011; Nonat et al., 2019).
2 Materials and methods
2.1 Synthesis and characterization
The overall synthesis of target complexes [Yb2Tb], [Eu2Tb], and [Yb2Eu] was adapted and modified from a previously reported literature procedure (Natrajan et al., 2009). Experimental procedures and characterization are detailed on pages S2-S41.
1,4,7,10-tetraazacyclododecane (cyclen) was purchased from CheMatech and used without further purification. All other reagents and solvents were purchased from Sigma-Aldrich, Fluorochem Ltd. or Apollo Scientific Ltd. and used without further purification. Electrospray +/− (ES-MS) spectra were recorded on a Thermo Orbitrap Exactive Plus mass spectrometer. MALDI-TOF spectra were recorded on a Shimadzu Biotech Axima Confidence mass spectrometer. FT-IR spectra were recorded on a Bruker ALPHA I FT-IR spectrometer. Elemental analysis data were recorder using a Thermo Scientific FlashSmart Elemental Analyzer.
NMR spectra were recorded on a Bruker AVIII HD 500 MHz spectrometer (BBFO inverse probe) in deuterated chloroform, deuterium oxide or deuterated methanol and analyzed using MestReNova 14.1.0. Chemical shifts in parts per million (ppm–δ) are reported relative to residual proton resonances and an internal tetramethylsilane reference. Splitting abbreviations: s: singlet, br. s: broad singlet, d: doublet, dd: doublet of doublets, t: triplet, dt: doublet of triplets, m: multiplet. Blank sections of spectra or those containing solvent resonances are omitted in certain spectra for clarity. Due to complex isomerism between the square and twisted square antiprismatic (SAP
Energy minimization of crystal structures was carried out using Avogadro 1.2.0. Compound structures were downloaded as mol2 files from the Cambridge Crystallographic Data Centre (CCDC) and Ln…C distances measured directly from crystallographic data without any further modification to the structure. The superimposed YbIII—GdIII structure was achieved via manual manipulation of each structure to minimize steric clash and bonding of the two structures. Bond angles and lengths were maintained during any new bond formation. The auto optimization tool was then used to minimize the energy of the new structure (UFF force field, steepest descent algorithm). Intermetallic measurements were then conducted on this new, minimzed structure.
2.2 Luminescence spectroscopy
Luminescence spectra were recorded on an Edinburgh Instruments FLS1000 Photoluminescence Spectrometer. Solid-state and low temperature spectra were recorded using a cryostat attachment with the sample deposited on a fused silica slide and were the default method of measurement unless stated otherwise. Solution measurements were recorded using a Hellma quartz glass 3.5 mL cuvette with a 1 cm path length and a sample absorbance of 0.1. UV-VIS spectra were recorded using a Mettler-Toledo UV5Bio spectrometer. Samples were excited using a 450 W Xe lamp with a long-pass filter on the detection arm and emission captured by PMT-900 (visible) and PMT-1700 (NIR) detectors. Any direct comparison of spectra used identical settings; excitation/emission monochromator slit widths and post-collection processing were identical for all. Lifetime measurements were collected using a microsecond flash lamp operating at 40 Hz (TbIII, EuIII data) or 100 Hz (YbIII data). Plotting, fitting and analysis of data was carried out using Origin 2019b. All data were fitted with exponential decay models starting with the fewest terms (mono-, bi-exponential) until sufficiently good fit residuals were achieved. In particular, 2-component fits were always compared against tri-exponential alternatives and found to better fit the data via residual, visual, Chi2 and R2 analysis.
Shorter–lived lifetimes from YbIII emission require consideration of the instrument response function (IRF) of the excitation source, both of which are on a microsecond timescale. An IRF trace was recorded at 100 Hz in line with the procedure detailed by the instrument manufacturer (Edinburgh Instruments) by matching excitation and detection wavelengths (λ = 280 nm) and recording the decay. This was repeated across different temperatures, both with the sample present and separately using milk powder to provide scatter (in both solid and solution-state). The variation in IRF trace between different variables is minimal. Detector response of the (NIR) PMT-1700 was also considered by recording a pseudo-IRF detecting scattered 2λ light from visible excitation (λex = 600 nm, λem = 1200 nm), to provide a similar result. The convolution tool in Origin was used to generate a decay trace with the appropriate IRF taken into consideration, which was then fitted to an exponential decay profile in the same manner as all other lifetime fits. Fitting of TbIII and EuIII signals used the IRF as a benchmark to ensure fitting parameters were only applied after the instrument response had decayed to background and therefore could not contribute to the resulting lifetime.
3 Results
3.1 Photophysical properties of the near infra-red-visible emitting complex [Yb2Tb]
Initial excitation via the phenyl linker (λex = 280 nm) affords sensitized visible emission from the TbIII metal center 5D4
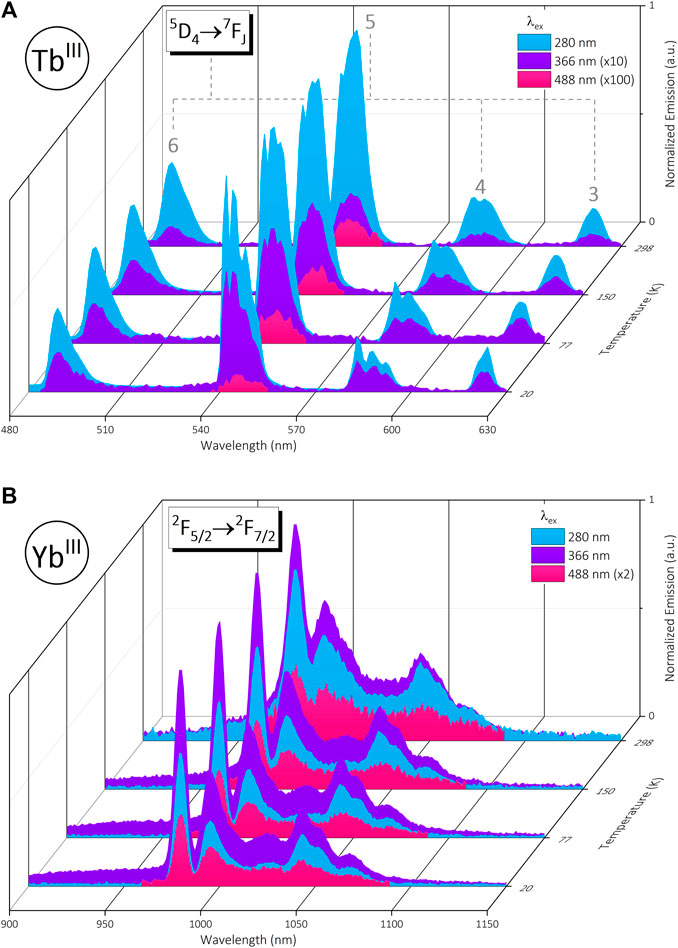
FIGURE 2. (A): Normalized visible emission of trinuclear bimetallic [Yb2Tb] under 280 nm (blue), 366 nm (purple) and 488 nm (pink) excitation in the solid state from 20—298 K. (B): Simultaneous normalized NIR emission from [Yb2Tb] under the same conditions. Data are normalized at each temperature relative to the emission maximum (∼545 nm @ λex = 280 nm for 2A, ∼980 nm @ λex = 366 nm for 2B).
Simultaneous NIR YbIII emission from the 2F5/2
Solid-state lifetime measurements of the primary TbIII emission at 545 nm fit a bi-exponential decay profile across all temperatures and λex, determined via analysis of R2 values and fit residuals (Supplementary Figures S2.8–S2.10). For simplicity, we report the global fluorescence lifetime, τn, of each decay component which is an average across variable λex (280, 366, 488 nm) and temperature (20, 77, 150, and 298 K). This value for the two exponents of TbIII decay are 0.14 ± 0.01 ms and 0.70 ± 0.03 ms for τ1 and τ2 respectively (fitting after the IRF). The relative contribution of each component varies marginally across the variable excitation and temperature series, but the long-lived τ2 component remains the most significant (Supplementary Table S2). Calculation of the average lifetime, τavg, considers the variation in percentage contribution from each τn component toward an overall average value (Supplemetary equations S1, S2). Factoring in all data across the excitation and temperature range results in τavg = 0.55 ± 0.03 m for the TbIII center (Table 1).
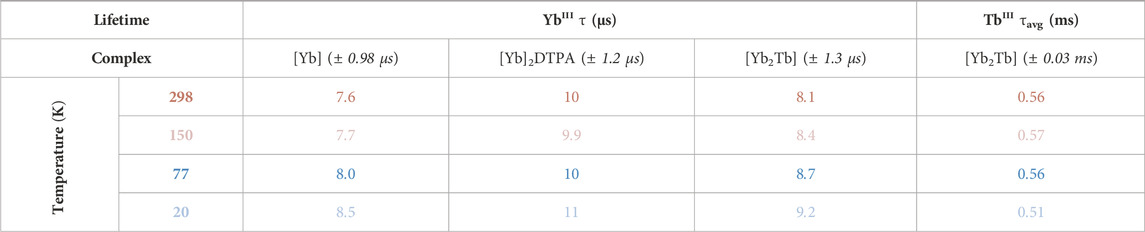
TABLE 1. Solid-state lifetimes of LnIII centers at multiple temperatures from the [Yb] complex series including trinuclear bimetallic [Yb2Tb], averaged across variable excitation wavelengths (298 K: dark red, 150 K: light red, 77 K: dark blue, 20 K: light blue). TbIII values are calculated average lifetimes from a bi-exponential fit.
The 980 nm–centered YbIII emission fits a mono-exponential decay following convolution with the IRF signal (Supplemetary Figures S2.11—S2.14) and has a global fluorescence lifetime τ = 8.6 ± 0.4 µs across variable λex and temperature. This is comparable to lifetime values for YbIII precursors (±0.4 µs); τ [Yb] = 7.9 µs, τ [Yb]2DTPA = 10 µs (Table 1).
3.2 Emission behavior of the dual-visible emitting complex [Eu2Tb]
Insertion of EuIII into the DO3A binding pocket results in strong visible emission (480—700 nm) from the same 7FJ TbIII transitions present in [Yb2Tb], in addition to the 5D0
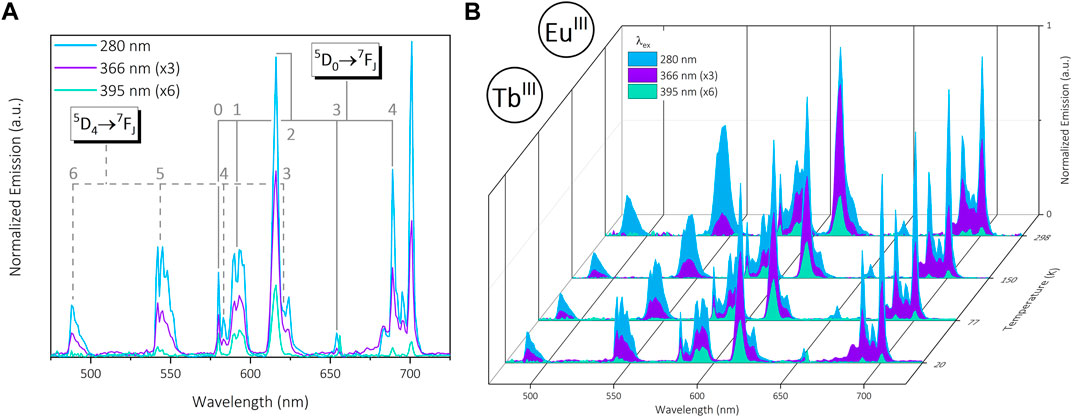
FIGURE 3. (A): Normalized visible emission of trinuclear bimetallic [Eu2Tb] under 280 nm (blue), 366 nm (purple) and 395 nm (green) excitation in the solid state at 20 K. (B): Normalized visible emission from [Eu2Tb] at 20—298 K across the same set of excitations. Data are normalized at each temperature relative to the emission maximum (∼701 nm @ λex = 280 nm).
Fitting of the 545 nm TbIII decay results in a bi-exponential fit analogous to the [Yb2Tb] species (τ1 = 0.11 ± 0.01 ms, τ2 = 0.56 ± 0.07 ms). The percentage contribution of each lifetime component is more evenly distributed than in the corresponding [Yb2Tb] complex, resulting in a shorter τavg value of 0.33 ± 0.02 ms (Table 2). However, comparison of the longer-lived τ2 component in both TbIII-containing complexes highlights similarity between the two (Supplementary Table S3).
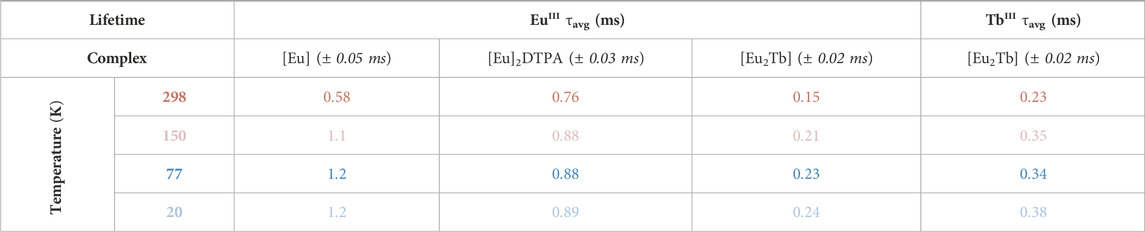
TABLE 2. Solid-state lifetimes of LnIII centers at multiple temperatures from the [Eu] complex series, averaged across variable excitation wavelengths (298 K: dark red, 150 K: light red, 77 K: dark blue, 20 K: light blue). Both EuIII and TbIII values are calculated average lifetimes from a bi-exponential fit.
The EuIII lifetimes were measured by fitting the decay of the primary 615 nm emission (5D0
Comparison of EuIII precursors [Eu] and [Eu]2DTPA with the trinuclear target species [Eu2Tb] highlights significant differences in solid-state excitation profiles arising from 615 nm emission (Supplementary Figure S2.20). The DO3A complex [Eu] displays strong absorption bands corresponding to 5D4
3.3 Photophysical properties of the near infra-red-visible emitting complex [Yb2Eu]
Synthesis of the {Yb(DO3A)}2-{Eu (DTPA)} complex [Yb2Eu] results in simultaneous NIR and visible emission from the two metals (λex = 280, 366 and 395 nm) analogous to [Yb2Tb] and [Eu2Tb], with an expected decrease in emission intensity at higher temperatures (Supplementary Figures S2.23, S2.24). Excitation at 366 nm was investigated despite the absence of TbIII to generate results comparable with other data sets. The EuIII 7F2 transition displays a minor spectral shift, and the 7F4 presents a change in splitting pattern compared to [Eu2Tb], both of which are sensitive to coordination environment and suggest EuIII is selectively bound in the DTPA site (Figure 4A). The EuIII center exhibits a greater response at λex = 395 nm compared to [Eu2Tb], which is consistent across a range of temperatures (Figure 4B). Solid-state excitation spectra (λem = 615 nm) show a large ligand-centered signal in addition to distinct EuIII bands analogous to [Eu] and rationalize this behavior (Supplementary Figure S2.25). Solution-state excitation spectra of [Yb2Eu] present 5D4
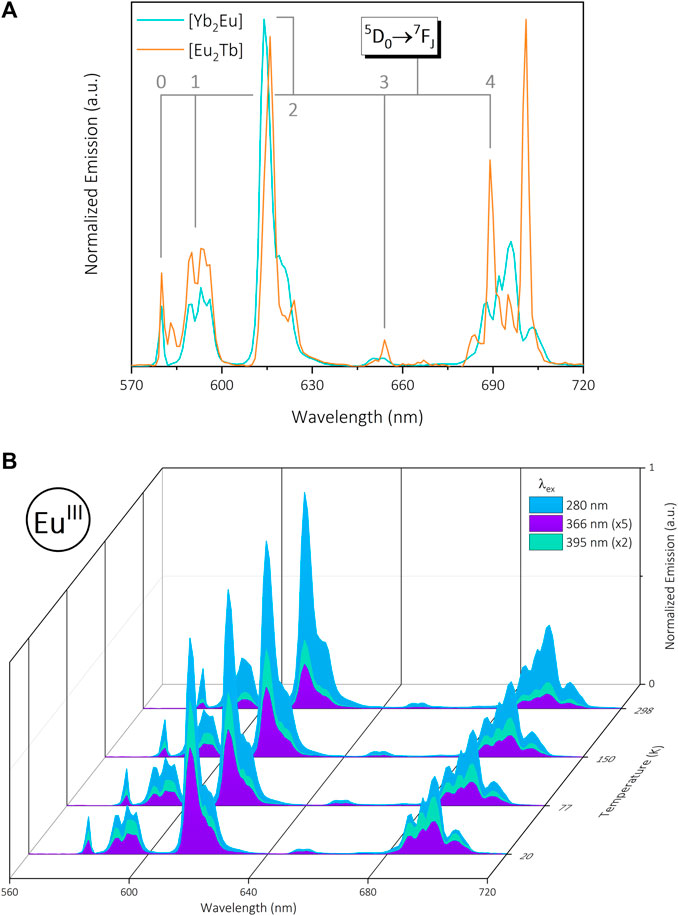
FIGURE 4. (A): Normalized visible emission of trinuclear bimetallic [Yb2Eu] (green) and [Eu2Tb] (orange) under 280 nm excitation in the solid state at 20 K. Data are normalized for each complex relative to the emission maximum. (B): Normalized visible emission from [Yb2Eu] at 20—298 K under 280 nm (blue), 366 nm (purple) and 395 nm (green) excitation in the solid state (normalized to ∼615 nm @ λex = 280 nm).
The YbIII emission intensity from the 2F5/2
Lifetime data for EuIII emission in [Yb2Eu] is similar to previous samples with τ1 = 0.17 ± 0.02 ms, τ2 = 0.74 ± 0.03 ms across the temperature and excitation range. The individual component contribution reflects the previous DTPA-bound metal (TbIII [Yb2Tb]), with a clear dominance of the long-lived τ2 decay (Supplementary Table S5) which gives rise to a longer τavg value of 0.63 ± 0.09 ms. YbIII lifetimes remain on the same order of magnitude as previous measurements; τ = 9.8 ± 0.6 µs across variable λex and temperature. A summary of temperature-dependent lifetime data for each LnIII across the trinuclear bimetallic series is presented in Table 3 and highlights the general observation of longer lifetimes at lower temperatures.
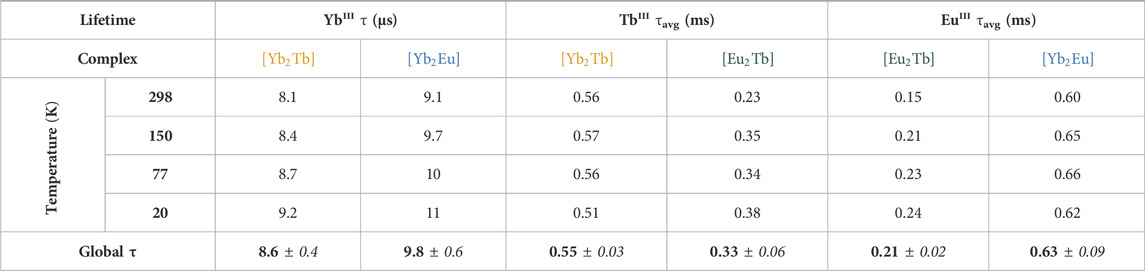
TABLE 3. Solid-state lifetimes of LnIII centers at multiple temperatures from the [Ln2Ln’] complex series, averaged across variable excitation wavelengths ([Yb2Tb] = orange, [Eu2Tb] = green, [Yb2Eu] = blue). Both EuIII and TbIII values are calculated average lifetimes from a bi-exponential fit.
3.4 Solution-state measurements
Solution-state measurements in water and D2O result in lifetimes with mono-exponential decay profiles for all metals; the shorter-lived τ1 component in the bi-exponential TbIII and EuIII solids are not present. Solution measurements are likely probing an average lifetime arising from minor changes in hydration state and exchange between the square and twisted square antiprismatic (SAP
Comparison of the luminescence decay in water and D2O (Table 4) shows q values change depending on both metal choice and binding environment, with the lowest arising from DO3A-bound YbIII in [Yb2Tb] (q = 0.21) and [Yb2Eu] (q = 0.29). The small size of YbIII due to the lanthanide contraction and availability of hard Lewis basic N- and O- donors in the octadentate DO3A precludes access to nearby solvent molecules (Faulkner and Pope, 2003). In complex [Yb2Tb], the neighboring DTPA-bound TbIII is more readily accessible due to a larger ionic radius and reduction in kinetic stability of the DTPA binding pocket, resulting in ∼1 bound water molecule (Idée et al., 2009; Sørensen and Faulkner, 2018). Similarly, in the complex [Eu2Tb], q = 1.0 for the DO3A EuIII center as a result of the slightly larger 9 coordinate ionic radius (Sastri et al., 2003) which competes with the steric bulk of the DO3A macrocycle to allow inclusion of 1 solvent donor. In the case of the TbIII(DTPA) centre in [Eu2Tb], an apparent q value of 2.4 is determined. However, in the case of substantial energy transfer from the assumedly 5D4 excited state of TbIII to the 5D0 state of EuIII occuring, phonon assisted energy transfer processes through the O-H vibrational manifold are in direct competition with those that act to quench the 5D4 → 7FJ transitions. Given that both of these quenching pathways will possess different rate constants, it follows that Horrocks equation is no longer appropriate (Beeby et al., 1999). Analysis of lifetime data for the analogous [Yb2Eu] complex indicates that any competitive intermolecular energy transfer from the 5D0 EuIII excited state to the 7F5/2 excited state of YbIII is inconsequential, and q of EuIII bound in the DTPA coordination pocket is calculated as 1.1 as expected.
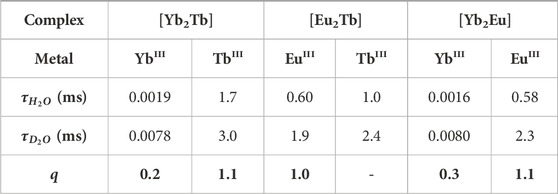
TABLE 4. Solution-state lifetimes of LnIII centers in [Ln2Ln’] complexes and associated q values for each, at 298 K (λex = 280 nm, λem = 980 nm (YbIII), 545 nm (TbIII), 615 nm (EuIII).
4 Discussion
The potential of this molecular scaffold to facilitate energy transfer between two bound LnIII ions was a key factor in both the ligand and experimental design. Measurement of the Gd-(DO3A)-aminophenyl acetamide complex [Gd] (analogous to [Yb] and [Eu]) facilitates characterization of the triplet state T1 of the phenyl linker. The primary Gd 6P7/2 state is too high in energy to be sensitized, therefore emission of [Gd] is entirely ligand-centered. Solid-state measurements at 298 K display broad fluorescence (λem = 431 nm), while phosphorescence from the triplet state is observed at 20 K (λem = 476 nm, Supplementary Figure S2.32). Crucially, this is high enough in energy to sensitize TbIII, EuIII and YbIII emissive states (T1 = 21,008 cm-1, TbIII 5D4 = 20,453 cm-1, EuIII 5D0 = 17,227 cm-1, YbIII 2F7/2 = 2924 cm-1) (Carnall et al., 1968b; 1968a; Sastri et al., 2003) (Figure 5). Additionally, the relative energies and stoichiometry of potential donor and acceptor states (2:1 acceptor:donor ratio) of the metal centers are conducive toward intermetallic ET pathways in [Yb2Tb] (TbIII 5D4
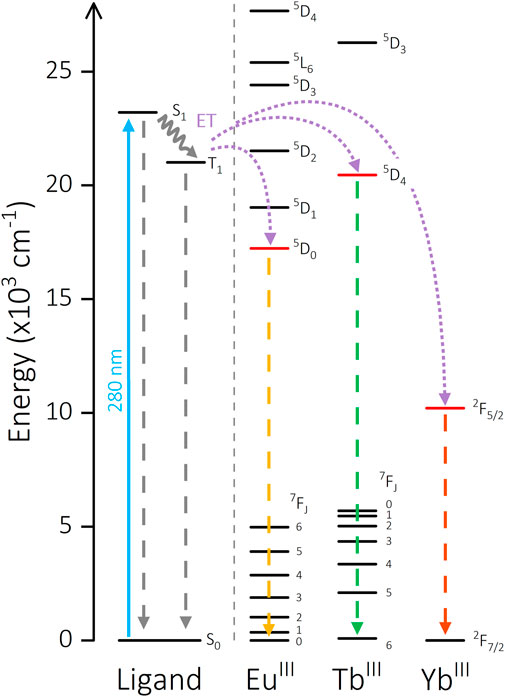
FIGURE 5. Energy level diagram showing the sensitization pathways of [Ln2Ln’] complexes. Solid arrow: excitation, waved arrow: non-radiative decay, dashed arrow: emission, dotted arrow: energy transfer pathway. Ligand energy levels are calculated from [Gd] measurements. EuIII, TbIII, and YbIII energies are taken from literature for comparison (Carnall et al., 1968a; Carnall et al., 1968b; Sastri et al., 2003).
Existence of energy transfer between bound metals is often strongly correlated with changes in the luminescence lifetime of excited states, where an energetic pathway from the donor excited state to an acceptor results in an overall reduction in lifetime in the former. An observed reduction in lifetime is apparent as temperature increases for each metal site due to increased X-H (X = C, N, O) vibrations and therefore quenching of excited states. Recorded lifetimes for the potential TbIII 5D4 donor state are largely consistent across a range of excitation bands. There is however a marked decrease in TbIII τavg values between target complexes [Yb2Tb] and [Eu2Tb] (0.55 ms vs. 0.33 ms). There is also a significant reduction of EuIII τavg in [Eu2Tb] compared to the EuIII complex series (0.64 ms, ∼75%) which is not present in the analogous YbIII(DO3A) acceptor series, suggesting the existence of an relaxation pathway present in [Eu2Tb] that is absent in [Yb2Tb]. Furthermore, Table 3 indicates a 48% decrease in EuIII τavg from [Yb2Eu] to [Eu2Tb] and supports evidence toward a EuIII-TbIII interaction. This is illustrated more clearly when considering the observed rate constants kobs (1/τavg), for EuIII emission in various systems. There is an increase in kobs upon addition of TbIII compared to the EuIII–only complex series ([Eu] kobs = 1000 µs-1 [Eu]2DTPA kobs = 1180 µs-1 [Eu2Tb] kobs = 4762 µs-1). Estimation of the potential energy transfer rate constant kET can be calculated as the difference between the rate radiative decay from EuIII in the presence and absence of a TbIII transfer partner (kET = kEuTb–kEu), using data from [Eu2Tb] and [Eu]2DTPA, respectively. Calculation with τavg values for these compounds yield a value of kET = 3582 µs-1. Analogous changes in lifetime are not observed in complexes where either metal is paired with YbIII. Additionally, DO3A-bound YbIII and EuIII complexes show minimal variations in lifetime when paired with a spectroscopically silent LuIII in the central DTPA pocket ([Yb2Lu] [Eu2Lu], Supplementary Table S6).
Indeed, the 5DJ energy levels of TbIII (J = 4) and EuIII (J = 2, 1, 0) exhibit appreciable energetic overlap. The 395 nm excitation feature in [Eu2Tb] populates a EuIII 5L6 state which is energetically higher than the TbIII 5D4 level (ΔE = 4947 cm-1) and can populate the latter via non-radiative decay to 5D2 and subsequent ET (Figure 6) (Bispo et al., 2018). Additionally, there is the possibility of phonon-assisted ET facilitated by proximate X-H oscillators. The manifold provided by vibrational overtones of O-H and O-D oscillators can provide an alternate energetic pathway to populate 7FJ states of TbIII from direct EuIII 5L6 population. Irrespective of these pathways, the emissive states of both TbIII and EuIII are higher in energy than the 7FJ states of either metal (Supplementary Figure S2.33) and have been reported to communicate in similar systems (Zaïm et al., 2014). The simultaneous reduction in lifetime of both EuIII and TbIII is exclusive to [Eu2Tb] and is not present in the parent complexes. The ability of each to act as both donor and acceptor when paired together appears to facilitate a series of ET and possibly back energy transfer (BET) processes between the metal centers.
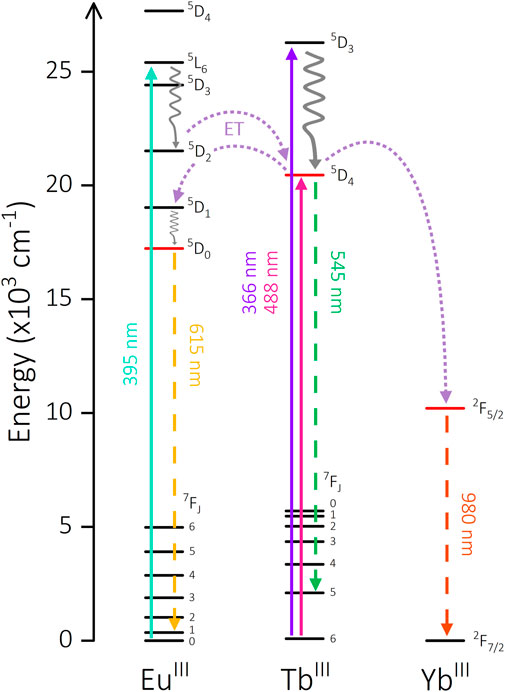
FIGURE 6. Energy level diagram showing potential intermetallic energy transfer mechanisms in [Ln2Ln’] complexes. Solid arrow: excitation, waved arrow: non-radiative decay, dashed arrow: emission, dotted arrow: energy transfer pathway.
Despite evidence of solvent interactions and consequent vibrational quenching, solution lifetimes are comparable or often longer than those in the solid state. The additional components in solid-state data likely arise from a combination of SAP
5 Conclusion
A series of three trinuclear bimetallic lanthanide complexes have been synthesized with selective introduction of metals into specific DO3A and DTPA binding sites. All three species exhibit strong sensitized emission when excited via a phenyl linker and represent a broad spectral range, from visible to NIR depending on the metal combination selected. Extensive photophysical measurements investigating the effects of temperature and excitation wavelength revealed communication between EuIII and TbIII centers when closely bound within the same complex, highlighting the potential for energy transfer between metals. The absence of evidence of intermetallic communication in the YbIII heterometallics emphasizes the impact of solid-state quenching and solution-state molecular motion on multimetallic lanthanide scaffolds that could facilitate energy transfer. Further work on optimizing the distance between metal centers and varying LnIII selection will be investigated in the future, with the aim of observing energy transfer and therefore accessing more intricate photophysics and applications.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
MET conducted all synthesis, characterization, measurements and analysis for compounds [Yb2Tb] and [Yb2Eu]. Initial synthesis and characterization of [Eu2Tb] was completed by JH and photophysical measurements finished by MET. LSN, SH, and PP secured funding and provided supervision and discussion. LSN and SF initiated the research and facilitated creation of the project. All authors contributed to the article and approved the submitted version.
Funding
The BBSRC Doctoral Training Partnership 2 BB/M011208/1 for studentship funding. PP is funded under the UKRI Future Leaders Fellowship scheme [MR/T021519/1].
Acknowledgments
MET and LSN thank the University of Manchester for access to facilities including the Centre for Radiochemistry Research National Nuclear User’s Facility (NNUF, EP/T011289/1).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2023.1232690/full#supplementary-material
References
Abad Galán, L., Aguilà, D., Guyot, Y., Velasco, V., Roubeau, O., Teat, S. J., et al. (2021). Accessing lanthanide-to-lanthanide energy transfer in a family of site-resolved [LnIIILnIII′] heterodimetallic complexes. Chem. Eur. J. 27, 7288–7299. doi:10.1002/chem.202005327
Aboshyan-Sorgho, L., Besnard, C., Pattison, P., Kittilstved, K. R., Aebischer, A., Bünzli, J.-C. G., et al. (2011). Near-Infrared→Visible light upconversion in a molecular trinuclear d–f–d complex. Angew. Chem. Int. Ed. 50, 4108–4112. doi:10.1002/anie.201100095
Aboshyan-Sorgho, L., Nozary, H., Aebischer, A., Bu, J.-C. G., Morgantini, P.-Y., Kittilstved, K. R., et al. (2012). Optimizing millisecond time scale near-infrared emission in polynuclear chrome(III)−Lanthanide(III) complexes. J. Am. Chem. Soc. 134 (30), 12675–12684. doi:10.1021/ja304009b
Beeby, A., Clarkson, I. M., Dickins, R. S., Faulkner, S., Parker, D., Royle, L., et al. (1999). Non-radiative deactivation of the excited states of europium, terbium and ytterbium complexes by proximate energy-matched OH, NH and CH oscillators: An improved luminescence method for establishing solution hydration states. J. Chem. Soc. Perkin Trans. 2 (3), 493–504. doi:10.1039/a808692c
Binnemans, K. (2015). Interpretation of europium(III) spectra. Coord. Chem. Rev. 295, 1–45. doi:10.1016/j.ccr.2015.02.015
Bispo, A. G., Lima, S. A. M., and Pires, A. M. (2018). Energy transfer between terbium and europium ions in barium orthosilicate phosphors obtained from sol-gel route. J. Lumin. 199, 372–378. doi:10.1016/j.jlumin.2018.03.057
Bünzli, J. C. G. (2010). Lanthanide luminescence for biomedical analyses and imaging. Chem. Rev. 110 (5), 2729–2755. doi:10.1021/cr900362e
Bünzli, J. C. G., and Piguet, C. (2005). Taking advantage of luminescent lanthanide ions. Chem. Soc. Rev. 34, 1048–1077. doi:10.1039/b406082m
Carnall, W. T., Fields, P. R., and Rajnak, K. (1968a). Electronic energy levels of the trivalent lanthanide aquo ions. III. Tb3+. J. Chem. Phys. 49, 4447–4449. doi:10.1063/1.1669895
Carnall, W. T., Fields, P. R., and Rajnak, K. (1968b). Electronic energy levels of the trivalent lanthanide aquo ions. IV. Eu3+. J. Chem. Phys. 49, 4450–4455. doi:10.1063/1.1669896
Doffek, C., Alzakhem, N., Bischof, C., Wahsner, J., Güden-Silber, T., Lügger, J., et al. (2012). Understanding the quenching effects of aromatic C-H- and C-D-oscillators in near-IR lanthanoid luminescence. J. Am. Chem. Soc. 134, 16413–16423. doi:10.1021/ja307339f
Dutta, S., Kim, S. K., Eun, J. L., Kim, T. J., Kang, D. S., Chang, Y., et al. (2006). Synthesis and magnetic relaxation properties of paramagnetic Gd-complexes of new DTPA-bis-amides. The X-ray crystal structure of [Gd(L)(H2O)]·3H2O (L = DTPA-bis(4-carboxylicphenyl)amide). Bull. Korean Chem. Soc. 27, 1038–1042. doi:10.5012/bkcs.2006.27.7.1038
Fan, Y., and Zhang, F. (2019). A new generation of NIR-II probes: Lanthanide-based nanocrystals for bioimaging and biosensing. Adv. Opt. Mater. 7, 1801417. doi:10.1002/adom.201801417
Faulkner, S., and Pope, S. J. A. (2003). Lanthanide-sensitized lanthanide luminescence: Terbium-sensitized ytterbium luminescence in a trinuclear complex. J. Am. Chem. Soc. 125, 10526–10527. doi:10.1021/ja035634v
Foucault-Collet, A., Gogick, K. A., White, K. A., Villette, S., Pallier, A., Collet, G., et al. (2013). Lanthanide near infrared imaging in living cells with Yb3+ nano metal organic frameworks. Proc. Natl. Acad. Sci. U. S. A. 110, 17199–17204. doi:10.1073/pnas.1305910110
Hemmer, E., Benayas, A., Légaré, F., and Vetrone, F. (2016). Exploiting the biological windows: Current perspectives on fluorescent bioprobes emitting above 1000 nm. Nanoscale Horiz. 1, 168–184. doi:10.1039/c5nh00073d
Idée, J. M., Port, M., Robic, C., Medina, C., Sabatou, M., and Corot, C. (2009). Role of thermodynamic and kinetic parameters in gadolinium chelate stability. J. Mag. Res. Imag. 30, 1249–1258. doi:10.1002/jmri.21967
Jin, G. Q., Sun, D., Xia, X., Jiang, Z. F., Cheng, B., Ning, Y., et al. (2022). Bioorthogonal lanthanide molecular probes for near-infrared fluorescence and mass spectrometry imaging. Angew. Chem. Int. Ed. 61, 202208707. doi:10.1002/anie.202208707
Maniaki, D., Sickinger, A., Barrios Moreno, L. A., Aguilà, D., Roubeau, O., Settineri, N. S., et al. (2023). Distributive Nd-to-Yb energy transfer within pure [YbNdYb] heterometallic molecules. Inorg. Chem. 62, 3106–3115. doi:10.1021/acs.inorgchem.2c03940
Miller, K. J., Saherwala, A. A., Webber, B. C., Wu, Y., Sherry, A. D., and Woods, M. (2010). The population of SAP and TSAP isomers in cyclen-based lanthanide(III) chelates is substantially affected by solvent. Inorg. Chem. 49, 8662–8664. doi:10.1021/ic101489t
Monteiro, J. H. S. K. (2020). Recent advances in luminescence imaging of biological systems using lanthanide(III) luminescent complexes. Molecules 25 (9), 2089. doi:10.3390/molecules25092089
Natrajan, L. S., Villaraza, A. J. L., Kenwright, A. M., and Faulkner, S. (2009). Controlled preparation of a heterometallic lanthanide complex containing different lanthanides in symmetrical binding pockets. Chem. Commun. 40, 6020–6022. doi:10.1039/b913702e
Nielsen, L. G., and Sørensen, T. J. (2019). Including and declaring structural fluctuations in the study of lanthanide(III) coordination Chemistry in solution. Inorg. Chem. 59 (1), 94–105. doi:10.1021/acs.inorgchem.9b01571
Nonat, A., Bahamyirou, S., Lecointre, A., Przybilla, F., Mély, Y., Platas-Iglesias, C., et al. (2019). Molecular upconversion in water in heteropolynuclear supramolecular Tb/Yb assemblies. J. Am. Chem. Soc. 141, 1568–1576. doi:10.1021/jacs.8b10932
Pujales-Paradela, R., Savić, T., Pérez-Lourido, P., Esteban-Gómez, D., Angelovski, G., Botta, M., et al. (2019). Lanthanide complexes with 1H paraCEST and 19F response for magnetic resonance imaging applications. Inorg. Chem. 58, 7571–7583. doi:10.1021/acs.inorgchem.9b00869
Rivas, C., Stasiuk, G. J., Gallo, J., Minuzzi, F., Rutter, G. A., and Long, N. J. (2013). Lanthanide(III) complexes of rhodamine-DO3A conjugates as agents for dual-modal imaging. Inorg. Chem. 52, 14284–14293. doi:10.1021/ic402233g
Sastri, V. S., Bünzli, J.-C., Rao, V. R., Rayudu, G. V. S., and Perumareddi, J. R. (2003). “Spectroscopy of lanthanide complexes,” in Modern aspects of rare earths and their complexes (United States: American Chemical Society), 569–731. doi:10.1016/B978-044451010-5/50022-5
Sørensen, T. J., and Faulkner, S. (2018). Multimetallic lanthanide complexes: Using kinetic control to define complex multimetallic arrays. Acc. Chem. Res. 51, 2493–2501. doi:10.1021/acs.accounts.8b00205
Sørensen, T. J., Tropiano, M., Kenwright, A. M., and Faulkner, S. (2017). Triheterometallic lanthanide complexes prepared from kinetically inert lanthanide building blocks. Eur. J. Inorg. Chem. 2017, 2165–2172. doi:10.1002/ejic.201700027
Tircso, G., Webber, B. C., Kucera, B. E., Young, V. G., and Woods, M. (2011). Analysis of the conformational behavior and stability of the SAP and TSAP isomers of lanthanide(III) NB-DOTA-type chelates. Inorg. Chem. 50, 7966–7979. doi:10.1021/ic2012843
Tropiano, M., Blackburn, O. A., Tilney, J. A., Hill, L. R., Just Sørensen, T., and Faulkner, S. (2015). Exploring the effect of remote substituents and solution structure on the luminescence of three lanthanide complexes. J. Lumin. 167, 296–304. doi:10.1016/j.jlumin.2015.06.035
Xu, W., Alam Bony, B., Rong Kim, C., Su Baeck, J., Chang, Y., Eun Bae, J., et al. (2013). Mixed lanthanide oxide nanoparticles as dual imaging agent in biomedicine. Sci. Rep. 3, 3210. doi:10.1038/srep03210
Keywords: Lanthanide, luminescence, energy transfer, NIR emission, Macrocycle
Citation: Thornton ME, Hemsworth J, Hay S, Parkinson P, Faulkner S and Natrajan LS (2023) Heterometallic lanthanide complexes with site-specific binding that enable simultaneous visible and NIR-emission. Front. Chem. 11:1232690. doi: 10.3389/fchem.2023.1232690
Received: 01 June 2023; Accepted: 17 July 2023;
Published: 31 July 2023.
Edited by:
Aleksandar Kondinski, University of Cambridge, United KingdomReviewed by:
Carlos D. S. Brites, University of Aveiro, PortugalDimitrije Mara, Institute of General and Physical Chemistry, Serbia
Copyright © 2023 Thornton, Hemsworth, Hay, Parkinson, Faulkner and Natrajan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Louise S. Natrajan, bG91aXNlLm5hdHJhamFuQG1hbmNoZXN0ZXIuYWMudWs=