
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem., 28 April 2023
Sec. Medicinal and Pharmaceutical Chemistry
Volume 11 - 2023 | https://doi.org/10.3389/fchem.2023.1179948
This article is part of the Research TopicDevelopment of Novel Physiologically Active Compounds: Hit-to-Lead Strategies in Drug DiscoveryView all 3 articles
 Xinye Bao1†‡Jian Wei2‡Cheng Tao3
Xinye Bao1†‡Jian Wei2‡Cheng Tao3 Muhammad Adnan Bashir1Hai-Jun Zhang1Bian Bao1
Muhammad Adnan Bashir1Hai-Jun Zhang1Bian Bao1 Jian Chen4*
Jian Chen4* Hongbin Zhai1*
Hongbin Zhai1*During the past decades, rheumatoid arthritis had become a serious problem, torturing millions of patients because of unclear pathogenesis and no ideal therapies. Natural products remain an important source of medicines to treat various major diseases such as rheumatoid arthritis (RA) given their excellent biocompatibility and structural diversity. Herein, we have developed a versatile synthetic method for constructing various skeletons of akuammiline alkaloid analogs based on our previous research on the total synthesis of the related indole alkaloids. We have also evaluated the effect of these analogs on the proliferation of RA fibroblast-like synoviocytes (FLSs) in vitro and analyzed the corresponding structure-activity relationship (SAR). Among these analogs, compounds 9 and 17c have demonstrated a promising inhibitory effect on the proliferation of RA-FLSs, with IC50 values of 3.22 ± 0.29 μM and 3.21 ± 0.31 μM, respectively. Our findings provide a solid foundation for future pharmacological studies on akuammiline alkaloid derivatives and inspiration for the development of anti-RA small molecule drugs derived from natural products.
It is widely acknowledged that human beings are affected by a range of chronic illnesses, including the potentially debilitating rheumatoid arthritis (RA). If patients with RA are not treated in a timely manner, it could lead to irreversible joint damage and further deterioration over time. During the past decades, chemotherapy that utilizes chemical drugs for RA treatment has held a dominant position in RA therapy. However, emergent drug resistance and undesirable side effects still exist and impede the treatment of RA. (Smolen et al., 2018). Hence, the development of small molecule anti-RA drugs is still highly desirable.
Natural products, due to their good biocompatibility and structural diversity, have acted as an important source of medicines in treating major diseases such as RA. (Yu et al., 2022). Many natural products, such as flavonoids, polyphenols, terpenoids, and alkaloids possessing anti-inflammatory properties, have been used clinically for this purpose. (Ma and Jiang, 2016; Wang et al., 2021; Guo et al., 2022). Multiple component extracts from plants (including herbs), such as Melodinus henryi extracts, Betelvine (Piper betle L.) extracts, and Fuzi lipid-soluble alkaloids, have been reported for RA treatment. (Li et al., 2019; Li et al., 2020; Murugesan et al., 2020; Yang et al., 2020). Scientists continue to research natural products, aiming to find more effective and viable treatments for chronic diseases. For instance, Shen et al. (2020) reported the mechanism of berberine treatment, which involved the inhibition of cell proliferation of fibroblast-like synoviocytes (FLSs), the effector cells of synovial hyperplasia of RA, via immunomodulatory effects. Moreover, Zhang and coworkers found that an indole alkaloid, Tabersonine (Tab), decreased the expression of inflammatory factors and reduced the proliferation of FLSs, in a manner similar to berberine. (Zhang et al., 2022a). However, akuammiline alkaloid-based small molecules for RA treatment have been investigated less. The structurally complex akuammiline alkaloids have been challenging to synthesize and thus create an abundant synthetic supply for biological investigations, which has hampered their potential to be developed into innovative drugs.
Since the unique indole heterocycle moiety of akuammiline alkaloids present in many FDA-approved marketed anti-inflammatory medicines generally correlates to the biological activities of the compounds, (Estevao et al., 2010; Guerra et al., 2011; Kumari and Singh, 2019; Sharma et al., 2021; Grau et al., 2023), it is reasonable to simplify the structure of the original akuammiline alkaloids by preserving the key indole heterocycle moiety for novel small molecule anti-inflammatory drug development.
Azido is a common functional group found in many small molecule drugs, including the classic antiviral drug zidovudine (AZT). Also, the presence of the azido moiety was found to possess a bone-targeting effect. (Zhang et al., 2021a). Moreover, triazoles and sulfonamides are important fragments present in the structure of marketed small molecule drugs with a wide range of bioactivities such as antibacterial, analgesic, anti-inflammatory, anti-tumor, and antiviral activities, and thus possess excellent pharmacological applications. (Haider et al., 2013; Abdellatif et al., 2016; Bonandi et al., 2017; Akgul et al., 2018; Pandit et al., 2018; Bozorov et al., 2019; Zhang, 2019; Shaker et al., 2020; Zhang et al., 2021b; Dai et al., 2022; Du et al., 2023). Therefore, we hypothesized that the introduction of azido and sulfonyl groups to the simplified structure of akuammiline alkaloid picrinine might result in improved and complementary anti-RA effects. According to the above hypothesis, we designed a series of akuammiline alkaloid derivatives (Scheme 1).
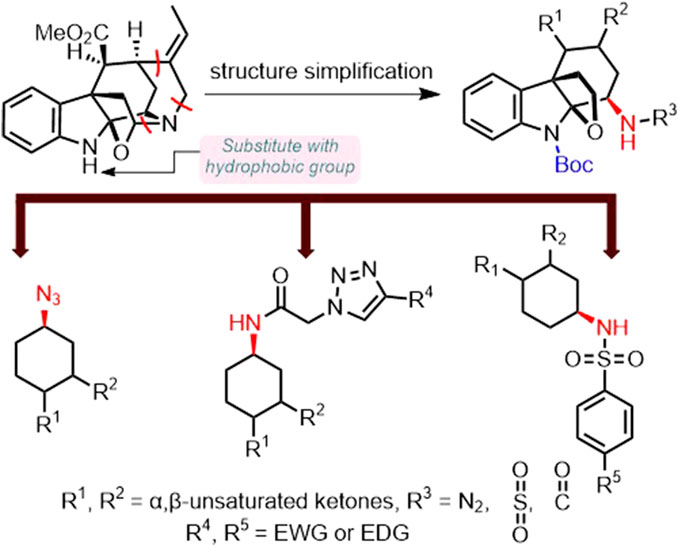
SCHEME 1. The primary design of akuammiline alkaloid derivatives.
Based on our prior research on the total synthesis of the akuammiline alkaloids, (Wang et al., 2017; Zou et al., 2021), a flexible synthetic process was established for creating the analogs of the akuammiline alkaloids with various skeletons and the effect of these compounds on the proliferation of RA-FLSs in vitro was investigated.
Our work commenced with the synthesis of two known intermediates 2 and 3 (Scheme 2). (Wang et al., 2017; Zou et al., 2021) According to our previous work, we prepared the intermediates with two different polycyclic skeletons through an eight-step protocol featuring a gold(I)-catalyzed cascade cyclization for the construction of the α,β-acetylenic ketone moiety as well as a stereoselective introduction of azido groups via the azidoalkoxylation reaction. (Wang et al., 2017; Zou et al., 2021).
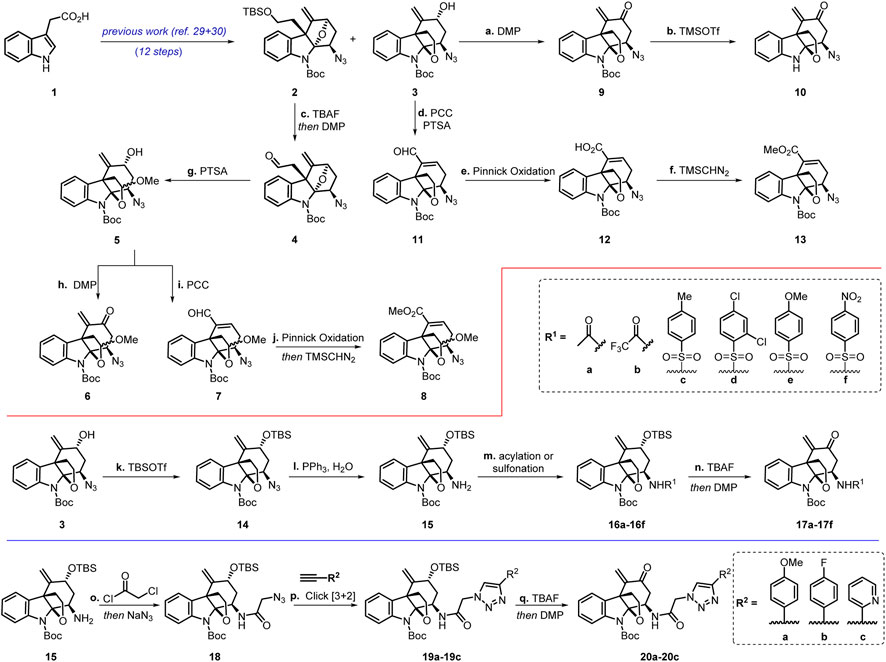
SCHEME 2. Chemical synthesis of the Akuammiline alkaloid derivatives. Reagents and conditions: (A) DMP (1.5 equiv), NaHCO3 (2 equiv), DCM, 0°C, 85%; (B) TMSOTf (3 equiv), DCM, 0°C, 40%; (C) TBAF (3 equiv), THF, 50°C; then DMP (1.5 equiv), NaHCO3 (2 equiv), DCM, 0°C, 82%; (D) PTSA (0.1 equiv), MeOH, reflux; then remove MeOH, PCC (2 equiv), PTSA (1 equiv), DCM, reflux, 63%; (E) NaClO2 (10 equiv), NaH2PO4·2H2O (10 equiv), 2-methyl-2-butene (10 equiv), tBuOH/H2O = (1:1), 0 °C to rt, 89%; (F) TMSCHN2 (2 equiv), PhMeOH/MeOH = (10:1), 0 °C to rt, 78%; (G) PTSA (0.1 equiv), MeOH, reflux, 88%; (H) DMP (1.5 equiv), NaHCO3 (2 equiv), DCM, 0°C, 86%; (I) PCC (2 equiv), PTSA (1 equiv), DCM, reflux, 75%; (J) NaClO2 (10 equiv), NaH2PO4.2H2O (10 equiv), 2-methyl-2-butene (10 equiv), tBuOH/H2O = (1:1), 0 °C to rt; then TMSCHN2 (2 equiv), PhMeOH/MeOH = (10:1), 0 °C to rt, 59%; (K) TBSOTf (1.2 equiv), 2,6-lutidine (2 equiv), DCM, 0°C, 80%; (L) PPh3 (3 equiv), H2O, THF, 50°C, 66%; (m) (para-substituted) benzenesulfonyl chlorides (1.1 equiv), Et3N (2 equiv), DCM, 0°C, 84–93%; (n) TBAF (5 equiv), THF, 60°C; then DMP (1.5 equiv), NaHCO3 (2 equiv), DCM, 0°C, 62–78%; (o) chloroacetyl chloride (1.1 equiv), Et3N (2 equiv), DCM, 0°C; then NaN3 (3 equiv), DMF/H2O = (3:1), rt, 64%; (p) three (para-substituted) benzene acetylene (1.1 equiv), CuSO4·H2O (0.2 equiv), sodium ascorbate (0.2 equiv), tBuOH/H2O = (1:1), rt, 65–72%; (q) TBAF (5 equiv), THF, 60°C; then DMP (1.5 equiv), NaHCO3 (2 equiv), DCM, 0°C, 60–71%.
DCM = dichloromethane, DMF = N, N-Dimethylformamide, DMP = Dess−Martin periodinane, THF = tetrahydrofuran, TBAF = tetra-n-butylammonium fluoride, PCC = pyridinium chlorochromate, PTSA = p-toluenesulfonic acid, TMSOTf = trimethylsilyl triflate, TBSOTf = tert-butyldimethylsilyl triflate.
Since both 2 and 3 contain the common azido moiety, we undertook structural derivatization by introducing other functional groups to these compounds. From compound 2, we explored the ring opening of the tetrahydrofuran unit to form the cyclic acetal moiety. Desilylation of compound 2 with TBAF followed by oxidation with DMP afforded aldehyde 4. Treatment of 4 with PTSA affected the ring opening reaction of the tetrahydrofuran moiety and the cyclization to afford acetals 5 as a mixture of two inseparable diastereomers in a 63% combined yield (d.r. = 2:1). Compound 6 (also containing two inseparable diastereomers) was obtained from 5 by oxidation with DMP. On the contrary, compound 7 was delivered upon treatment of 5 with PCC, presumably through an oxidative transposition of the secondary allyl alcohol. Aldehyde 7 was further oxidized with NaClO2 to furnish a carboxylic acid, the esterification of which with TMSCHN2 generated methyl esters 8. Meanwhile, intermediate 3 was oxidized with DMP to give enone 9, which was transformed into compound 10 upon Boc deprotection with TMSOTf. Furthermore, starting from allylic alcohol 3, α,β-unsaturated aldehyde 11, acid 12, and ester 13 were synthesized in a fashion analogous to that for the preparation of compounds 7 and 8.
In an endeavor to introduce a triazole or a sulfonyl group, the hydroxyl in intermediate 3 was protected as a TBS ether to lead to 14 in a 66% yield. Reduction of the azido group fraction of 14 under the Staudinger conditions produced the corresponding primary amine 15. Compound 15 was acylated or sulfonylated to afford a series of amides or sulfonamide intermediates (16a-16f). For sulfonamides, there is an electron-donating or electron-withdrawing group at the para position. Subsequently, desilylation of 16a-16f with TBAF followed by oxidation with DMP afforded compounds 17a-17f.
The azido group in 14 failed to undergo the [3 + 2] cycloaddition reaction, presumably due to the steric hindrance. Upon treatment with chloroacetyl chloride followed by an SN2 reaction with NaN3, amine 15 was converted into azide 18 containing a more accessible azido group compared to that in 14. Click reaction of 18 with three different terminal alkynes formed 1,2,3-triazoles 19a-19c in 65–72% yields. Desilylation of 19a-19c with TBAF followed by oxidation with DMP furnished enones 20a-20c in 60–71% yields.
As such, three types of desirable akuammiline alkaloid derivatives were obtained, including azides 2-14, amides/sulfonamides 16a-16f and 17a-17f, and triazoles 19a-19c and 20a-20c. Biological studies of these compounds were subsequently conducted.
Considering that FLSs are effector cells of synovial hyperplasia of RA, the effect of the compounds on the viability of RA-FLSs was assessed after stimulation for 24 h in the current study. As shown in Table 1, the 24 h-half maximal inhibition concentrations (IC50) of 6, 9, 17a, 17c, 17d, and 17f on MH7A cells were less than 10 μM, among which 9 and 17c exhibited the optimal inhibitory effect on the proliferation of MH7A cells with an IC50 of 3.22 ± 0.29 μM and 3.21 ± 0.31 μM, respectively. These results suggested that 9 and 17c may have the potential to be applied to treating RA.
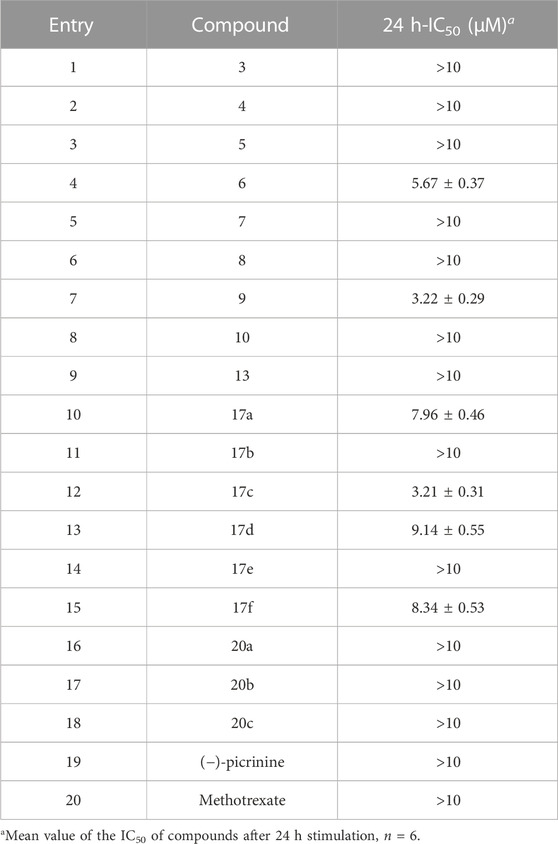
TABLE 1. 24h-IC50 of compounds on MH7A cells.
From a structural perspective, the six alkaloid derivatives that exhibited significant biological activity can be divided into three categories (Scheme 3). All these compounds have a unique tetracyclic core and an enone moiety.
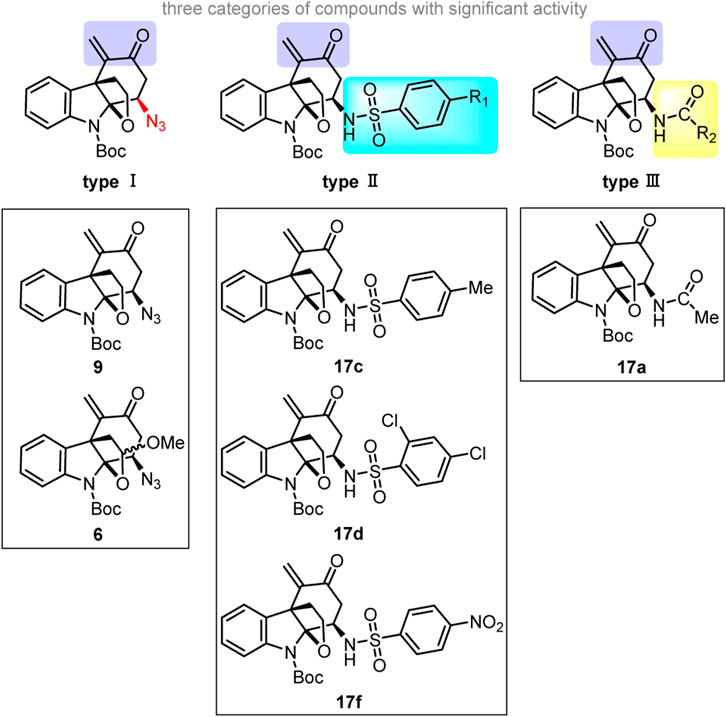
SCHEME 3. The skeletons of three types of effective derivatives and six specific lead compounds.
As for type I derivatives, the difference in their inhibitory effect on the proliferation of RA-FLSs may correlate with the azido substituent on the cyclohexane ring in addition to the conjugated enone moiety. Compounds 6 and 9 showed significant inhibition of proliferation in MH7A cells, whereas azides 7, 8, 11, and 13 with an α,β-unsaturated aldehyde or ester moiety had no obvious inhibition effect, suggesting that the combination of the azido group and the α,β-unsaturated ketone is critical to the enhancement of the inhibitory effect on the proliferation of RA-FLSs. (Arshad et al., 2017; Zhang et al., 2022b; Zhou et al., 2022). Among the amides/sulfonamides (see the structure of 17), compound 17c, a para-toluenesulfonamide, displayed the strongest inhibitory effect on the proliferation of RA-FLSs. (Zhao et al., 2016; Zhao et al., 2019). Moreover, the presence of one strong electron-donating group (17e) or one or more electron-withdrawing groups (17d, 17f) on the benzene ring within the sulfonamide moiety greatly decreased the inhibitory effect on the proliferation of RA-FLSs.
After a solution of 3 (108 mg, 0.28 mmol) was dissolved in dry DCM (5.0 mL), DMP (196.1 mg, 0.42 mmol) and NaHCO3 (47.1 mg, 0.56 mmol) were added to it at 0°C. The resultant mixture was stirred for 2 h at the same temperature. Then, the saturated aqueous Na2S2O3 (10 mL) was added to the mixture. The mixture was stirred for 0.5 h at room temperature and extracted with DCM (3 × 10 mL). The combined organic layer was washed with brine and then dried over Na2SO4. The solvent was removed under vacuum, and the residue was purified by column chromatography (petroleum ether/ethyl acetate = 4:1) to afford 9 (91.3 mg, 85%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.72–7.65 (m, 1H), 7.20 (ddd, J = 8.5, 6.2, 2.6 Hz, 1H), 7.05–6.95 (m, 2H), 6.21 (s, 1H), 5.64 (s, 1H), 5.34–5.29 (m, 1H), 4.32 (ddd, J = 8.6, 7.4, 1.1 Hz, 1H), 3.76 (ddd, J = 11.9, 9.1, 4.9 Hz, 1H), 2.66–2.51 (m, 2H), 2.48 (d, J = 4.7 Hz, 1H), 2.35 (dd, J = 17.4, 2.4 Hz, 1H), 1.62 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 195.5, 151.8, 146.9, 142.4, 130.5, 129.1, 124.1, 123.9, 120.5, 115.0, 105.2, 82.9, 67.8, 60.0, 58.0, 42.3, 39.1, 28.3. HRMS (ESI): C20H22N4NaO4+ [(M + Na)+]: calcd: 405.1533; found: 405.1531.
After a solution of 9 (91 mg, 0.24 mmol) was dissolved in dry DCM (5.0 mL), TMSOTf (125 μL, 0.72 mmol) was added at 0°C. After being stirred for 20 min, the reaction mixture was quenched with saturated NaHCO3 and extracted with EtOAc. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated. The residue was purified by flash column chromatography (petroleum ether/ethyl acetate = 4:1) to give 10 (26.8 mg, 40%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.09 (td, J = 7.6, 1.3 Hz, 1H), 7.01 (dd, J = 7.6, 1.3 Hz, 1H), 6.80 (td, J = 7.5, 1.0 Hz, 1H), 6.61 (d, J = 7.8 Hz, 1H), 6.23 (d, J = 0.8 Hz, 1H), 5.68 (s, 1H), 4.42 (s, 1H), 4.29 (ddd, J = 8.8, 4.9, 3.0 Hz, 1H), 4.17 (dd, J = 4.9, 2.8 Hz, 1H), 3.82–3.68 (m, 1H), 2.77 (dd, J = 17.3, 2.9 Hz, 1H), 2.62 (dd, J = 17.3, 4.9 Hz, 1H), 2.55–2.48 (m, 2H).13C NMR (101 MHz, CDCl3) δ 196.4, 148.0, 146.9, 129.7, 129.1, 124.2, 121.5, 120.28, 108.7, 103.0, 68.0, 60.5, 60.0, 43.0, 40.0. HRMS (ESI): C15H15N4O2+ [(M + H)+]: calcd: 283.1190; found: 283.1189.
PTSA (104.6 mg, 0.55 mmol) was added to a solution of 3 (2.1 g, 5.5 mmol) in MeOH (20 mL). The resultant mixture was heated to reflux in an oil bath for 15 min, and then the solvent was removed in vacuo and the residue was dissolved in DCM (20 mL). PCC (2.4 g, 11 mmol), PTSA (1.0 g, 5.5 mmol), and silica gel (2.4 g) were added to the above mixture and then heated to reflux in an oil bath for 15 min. The mixture was filtered, and the filtrate was concentrated in vacuo. The residue was purified by column chromatography (petroleum ether/ethyl acetate = 15:1) to afford 11 (1.5 g, 63%, dr = 2:1, determined by crude 1H NMR) as a white foam.
After a solution of unsaturated aldehyde 11 (1.3 g, 3.4 mmol) was dissolved in t-BuOH (10 mL) and 2-methyl-2-butene (6 mL) at 0°C before a solution of sodium chlorite (3.1 g, 34.0 mmol) and monobasic sodium phosphate (4.1 g, 34.0 mmol) in H2O (10 mL) was added to it. The reaction was warmed to room temperature. After vigorous stirring for 4 h, the reaction mixture was poured into water (30 mL) and the resulting mixture was diluted with EtOAc (3 × 20 mL). The layers were separated, and the aqueous layer was extracted with EtOAc. The organic layers were combined and dried over Na2SO4, and concentrated in vacuo to produce a crude oil, which would be used for the next step without further purification.
After a solution of the acid 12 obtained from above in PhMe/MeOH (30 mL/3 mL) was cooled to 0°C, a solution of TMSCHN2 in hexanes (2.1 M, 3.4 mL) was added dropwise. The mixture was warmed to room temperature and stirred for 0.5 h before being quenched with silica gel. The solvent was directly removed in vacuo and the residue was then purified by flash column chromatography on silica gel (petroleum ether/ethyl acetate = 30:1) to give 13 (1.40 g, 69% over two steps) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.70 (d, J = 8.3 Hz, 1H), 7.61 (ddd, J = 7.7, 1.5, 0.6 Hz, 1H), 7.20 (ddd, J = 8.2, 7.4, 1.5 Hz, 1H), 6.98 (td, J = 7.5, 1.1 Hz, 1H), 6.87 (td, J = 4.7, 0.8 Hz, 1H), 5.46 (s, 1H), 3.69 (ddd, J = 12.3, 8.6, 4.9 Hz, 1H), 3.07 (dd, J = 12.6, 4.9 Hz, 1H), 2.56 (td, J = 12.5, 7.9 Hz, 1H), 2.43 (dd, J = 4.6, 3.1 Hz, 2H), 1.63 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 165.7, 152.3, 142.2, 135.3, 134.5, 132.0, 128.6, 124.8, 123.4, 115.4, 105.2, 82.8, 68.8, 57.7, 57.6, 51.7, 39.2, 28.5, 28.3. HRMS (ESI): C21H24N4NaO5+ [(M + Na)+]: calcd: 435.1639; found: 435.1642.
After a solution of 5 (50.2 mg, 0.12 mmol) was dissolved in dry DCM (4.0 mL), DMP (84 mg, 0.18 mmol) and NaHCO3 (20.1 mg, 0.24 mmol) were added to it at 0 °C. The resultant mixture was stirred for 2h at the same temperature. Then, the saturated aqueous Na2S2O3 (15 mL) was added to the mixture. The mixture was stirred for 0.5 h at room temperature and extracted with DCM (3 × 10 mL). Then, the combined organic layers were washed with brine and then dried over Na2SO4. The solvent was removed under vacuum and the residue was purified by column chromatography (petroleum ether/ethyl acetate = 4:1) to afford 6 (43 mg, 86%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.66 (s, 1H), 7.18 (ddd, J = 8.5, 4.9, 3.8 Hz, 1H), 6.99 (dd, J = 4.0, 0.9 Hz, 2H), 6.18 (s, 1H), 5.58 (s, 1H), 5.42 (dd, J = 3.3, 2.2 Hz, 1H), 5.37 (s, 1H), 3.23 (s, 3H), 2.72–2.66 (m, 2H), 2.58 (dd, J = 17.6, 4.3 Hz, 1H), 2.40 (dd, J = 17.6, 2.4 Hz, 1H), 1.65 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 195.1, 151.7, 147.3, 132.0, 128.9, 123.8, 123.7, 120.3, 115.4, 106.8, 105.3, 82.8, 55.0, 48.1, 38.9, 28.4. HRMS (ESI): C21H24N4NaO5+ [(M + Na)+]: calcd: 435.1639; found: 435.1640.
After a solution of compound 3 (210 mg, 0.55 mmol) was dissolved in DCM (5 mL), 2,6-lutidine (128 uL, 1.10 mmol) and TBSOTf (150 uL, 0.66 mmol) were added to it at 0 °C and stirred for 0.5 h. Then the mixture was diluted with DCM (3 × 10 mL), washed with H2O three times, dried over Na2SO4, filtered, and concentrated under a vacuum. The crude product was dissolved in THF (30 mL) and then Ph3P (430 mg, 1.65 mmol) and H2O (0.1 mL, 5.5 mmol) were added. The reaction mixture was heated at 50 °C for 12 h. The solvent was directly removed in vacuo and the residue was then purified by flash column chromatography (DCM/MeOH = 50:1). The crude product was directly dissolved in DCM (3 mL), and then triethylamine (160 uL, 1.1 mmol) and 4-Methylbenzenesulfonyl chloride (116 mg, 0.61 mmol) were added sequentially at 0°C. The resulting solution was warmed to room temperature and stirred for 2 h and then was quenched with NH4Cl (aq.). The aqueous phase was extracted with DCM. The combined organic layers were washed with brine and then dried over Na2SO4. The solvent was removed in vacuo and the residue was then purified by flash column chromatography (petroleum ether/ethyl acetate = 5:1) to give 16c (134.5 mg, 39% over three steps) as a white solid.
After a solution of 16c (134 mg, 0.21 mmol) was dissolved in THF (3 mL), Tetrabutylammonium fluoride (1.0 M in THF, 1 mL, 1.05 mmol) was added. The resulting mixture was stirred for 24 h at 60°C and then was quenched with NH4Cl (aq.). After the reaction was terminated, the aqueous layer was extracted with EtOAc. The combined organic layers were washed with H2O and brine, dried over anhydrous Na2SO4, and concentrated in vacuo to produce a crude oil, which was purified by column chromatography (petroleum ether/ethyl acetate = 2:1). The crude product was directly dissolved in DCM (3 mL), and then DMP (147 mg, 0.32 mmol) and NaHCO3 (35.3 mg, 0.42 mmol) were added sequentially at 0°C. The resultant mixture was stirred for 2 h at the same temperature. Then, the saturated aqueous Na2S2O3 (10 mL) was added to the mixture. The mixture was stirred for 0.5 h at room temperature and extracted with DCM (3 × 5 mL). The combined organic layer was washed with brine, then dried over Na2SO4. The solvent was removed under vacuum, and the residue was purified by column chromatography (petroleum ether/ethyl acetate = 4:1) to afford 17c (83.6 mg, 78% over two steps) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.74 (d, J = 8.0 Hz, 2H), 7.65 (d, J = 8.2 Hz, 1H), 7.31 (d, J = 8.0 Hz, 2H), 7.16 (ddd, J = 8.5, 6.5, 2.3 Hz, 1H), 7.00–6.90 (m, 2H), 6.15 (s, 1H), 5.56 (s, 1H), 5.24 (s, 1H), 4.74 (dt, J = 3.9, 1.8 Hz, 1H), 4.16 (dd, J = 9.2, 7.3 Hz, 1H), 3.65 (ddd, J = 12.1, 9.2, 4.8 Hz, 1H), 2.95 (dd, J = 17.9, 4.3 Hz, 1H), 2.49 (dt, J = 12.1, 6.0 Hz, 1H), 2.43 (s, 3H), 2.33 (dd, J = 11.9, 4.7 Hz, 1H), 2.25 (dt, J = 17.9, 1.7 Hz, 1H), 1.41 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 195.5, 151.4, 146.6, 143.8, 142.6, 136.2, 130.2, 129.7, 129.1, 127.3, 124.2, 123.9, 120.2, 114.9, 103.5, 82.7, 67.6, 60.2, 51.1, 42.0, 38.2, 28.0, 21.5. HRMS (ESI): C27H30N2NaO6S+ [(M + H)+]: calcd: 533.1717; found: 533.1720.
The product of 15 (260 mg, 0.55 mmol) was directly dissolved in DCM (3 mL), and then triethylamine (160 uL, 1.1 mmol) and acetyl chloride (43.5 μL, 0.61 mmol) were added sequentially at 0°C. The resulting solution was warmed to room temperature and stirred for 2 h and then was quenched with NH4Cl (aq.). The aqueous phase was extracted with DCM. The combined organic layers were washed with brine and then dried over Na2SO4. The solvent was removed in vacuo and the residue was then purified by flash column chromatography (petroleum ether/ethyl acetate = 5:1) to give 16a (249 mg, 88%) as a white solid.
After a solution of 16a (108 mg, 0.21 mmol) was dissolved in THF (3 mL), Tetrabutylammonium fluoride (1.0 M in THF, 1 mL, 1.05 mmol) was added. The resulting mixture was stirred for 24 h at 60°C and then was quenched with NH4Cl (aq.). After the reaction was terminated, the aqueous layer was extracted with EtOAc. The combined organic layers were washed with H2O and brine, dried over anhydrous Na2SO4, and concentrated in vacuo to produce a crude oil, which was purified by column chromatography (petroleum ether/ethyl acetate = 2:1). The crude product was directly dissolved in DCM (3 mL), and then DMP (147 mg, 0.32 mmol) and NaHCO3 (35.3 mg, 0.42 mmol) were added sequentially at 0°C. The resultant mixture was stirred for 2 h at the same temperature. Then, the saturated aqueous Na2S2O3 (10 mL) was added to the mixture. The mixture was stirred for 0.5 h at room temperature and extracted with DCM (3 × 5 mL). The combined organic layer was washed with brine, then dried over Na2SO4. The solvent was removed under vacuum, and the residue was purified by column chromatography (petroleum ether/ethyl acetate = 4:1) to afford 17a (54.5 mg, 65% over two steps) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.70 (d, J = 8.3 Hz, 1H), 7.20 (ddd, J = 8.4, 6.5, 2.3 Hz, 1H), 7.02–6.92 (m, 2H), 6.29 (s, 1H), 5.67 (d, J = 5.8 Hz, 2H), 5.39 (q, J = 2.0 Hz, 1H), 4.30–4.21 (m, 1H), 3.75 (ddd, J = 11.8, 9.4, 5.2 Hz, 1H), 3.07 (dd, J = 17.6, 4.4 Hz, 1H), 2.53–2.35 (m, 2H), 2.32 (dd, J = 17.6, 2.6 Hz, 1H), 1.93 (s, 3H), 1.59 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 196.5, 170.2, 151.4, 146.6, 142.8, 130.1, 129.3, 124.1, 123.7, 121.3, 115.0, 104.1, 82.9, 67.2, 60.2, 48.6, 42.4, 38.9, 28.3, 23.5. HRMS (ESI): C22H26N2NaO5+ [(M + Na)+]: calcd: 421.1734; found: 421.1737.
The product of 15 (260 mg, 0.55 mmol) was directly dissolved in DCM (3 mL), and then triethylamine (160 uL, 1.1 mmol) and 2,2,2-trifluoroacetyl chloride (54 μL, 0.61 mmol) were added sequentially at 0°C. The resulting solution was warmed to room temperature and stirred for 2 h and then was quenched with NH4Cl (aq.). The aqueous phase was extracted with DCM. The combined organic layers were washed with brine and then dried over Na2SO4. The solvent was removed in vacuo and the residue was then purified by flash column chromatography (petroleum ether/ethyl acetate = 5:1) to give 16b (263 mg, 84%) as a white solid.
After a solution of 16b (119.5 mg, 0.21 mmol) was dissolved in THF (3 mL), Tetrabutylammonium fluoride (1.0 M in THF, 1 mL, 1.05 mmol) was added. The resulting mixture was stirred for 24 h at 60°C and then was quenched with NH4Cl (aq.). After the reaction was terminated, the aqueous layer was extracted with EtOAc. The combined organic layers were washed with H2O and brine, dried over anhydrous Na2SO4, and concentrated in vacuo to produce a crude oil, which was purified by column chromatography (petroleum ether/ethyl acetate = 2:1). The crude product was directly dissolved in DCM (3 mL), and then DMP (147 mg, 0.32 mmol) and NaHCO3 (35.3 mg, 0.42 mmol) were added sequentially at 0°C. The resultant mixture was stirred for 2 h at the same temperature. Then, the saturated aqueous Na2S2O3 (10 mL) was added to the mixture. The mixture was stirred for 0.5 h at room temperature and extracted with DCM (3 × 5 mL). The combined organic layer was washed with brine, then dried over Na2SO4. The solvent was removed under vacuum, and the residue was purified by column chromatography (petroleum ether/ethyl acetate = 4:1) to afford 17b (59 mg, 62% over two steps) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.68 (d, J = 8.2 Hz, 1H), 7.22 (ddd, J = 8.5, 6.5, 2.3 Hz, 1H), 7.05–6.94 (m, 2H), 6.70 (s, 1H), 6.29 (s, 1H), 5.71 (s, 1H), 5.40 (td, J = 4.9, 2.9 Hz, 1H), 4.35–4.24 (m, 1H), 3.84–3.72 (m, 1H), 3.08 (dd, J = 17.9, 4.7 Hz, 1H), 2.51–2.39 (m, 3H), 1.59 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 195.3, 157.5, 157.1, 156.8, 156.4, 151.4, 146.1, 142.4, 129.6, 129.4, 124.1, 124.0, 122.3, 119.7, 116.8, 115.1, 113.9, 111.1, 103.4, 83.4, 67.5, 60.3, 49.2, 42.3, 37.9, 28.2. 19F NMR (376 MHz, CDCl3) δ -76.0. HRMS (ESI): C22H23F3N2NaO5+ [(M + Na)+]: calcd: 475.1451; found: 475.1452.
The product of 15 (260 mg, 0.55 mmol) was directly dissolved in DCM (3 mL), and then triethylamine (160 uL, 1.1 mmol) and 2,4-dichlorobenzenesulfonyl chloride (150 mg, 0.61 mmol) were added sequentially at 0°C. The resulting solution was warmed to room temperature and stirred for 2 h and then was quenched with NH4Cl (aq.). The aqueous phase was extracted with DCM. The combined organic layers were washed with brine and then dried over Na2SO4. The solvent was removed in vacuo and the residue was then purified by flash column chromatography (petroleum ether/ethyl acetate = 5:1) to give 16d (326 mg, 87%) as a white solid.
After a solution of 16d (143 mg, 0.21 mmol) was dissolved in THF (3 mL), Tetrabutylammonium fluoride (1.0 M in THF, 1 mL, 1.05 mmol) was added. The resulting mixture was stirred for 24 h at 60°C and then was quenched with NH4Cl (aq.). After the reaction was terminated, the aqueous layer was extracted with EtOAc. The combined organic layers were washed with H2O and brine, dried over anhydrous Na2SO4, and concentrated in vacuo to produce a crude oil, which was purified by column chromatography (petroleum ether/ethyl acetate = 2:1). The crude product was directly dissolved in DCM (3 mL), and then DMP (147 mg, 0.32 mmol) and NaHCO3 (35.3 mg, 0.42 mmol) were added sequentially at 0°C. The resultant mixture was stirred for 2 h at the same temperature. Then, the saturated aqueous Na2S2O3 (10 mL) was added to the mixture. The mixture was stirred for 0.5 h at room temperature and extracted with DCM (3 × 5 mL). The combined organic layer was washed with brine, then dried over Na2SO4. The solvent was removed under vacuum, and the residue was purified by column chromatography (petroleum ether/ethyl acetate = 4:1) to afford 17d (89 mg, 75% over two steps) as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.01 (d, J = 8.5 Hz, 1H), 7.65 (d, J = 8.3 Hz, 1H), 7.53 (d, J = 2.0 Hz, 1H), 7.42 (dd, J = 8.5, 2.0 Hz, 1H), 7.17 (ddd, J = 8.5, 6.9, 1.9 Hz, 1H), 7.02–6.90 (m, 2H), 6.17–6.12 (m, 1H), 5.63 (s, 1H), 5.50 (d, J = 2.7 Hz, 1H), 4.97–4.90 (m, 1H), 4.10 (dd, J = 9.3, 7.3 Hz, 1H), 3.67 (ddd, J = 12.2, 9.3, 4.8 Hz, 1H), 2.85 (dd, J = 18.0, 4.1 Hz, 1H), 2.50 (td, J = 12.0, 7.4 Hz, 1H), 2.38 (dd, J = 11.9, 4.7 Hz, 1H), 2.34–2.24 (m, 1H), 1.49 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 195.4, 151.3, 146.4, 142.6, 139.7, 135.9, 132.8, 132.0, 131.3, 129.9, 129.3, 127.5, 124.2, 124.0, 120.9, 115.1, 103.4, 83.0, 67.7, 60.2, 51.5, 42.1, 38.7, 28.2. HRMS (ESI): C26H26Cl2N2NaO6S+ [(M + Na)+]: calcd: 587.0781; found: 587.0781.
The product of 15 (260 mg, 0.55 mmol) was directly dissolved in DCM (3 mL), and then triethylamine (160 uL, 1.1 mmol) and 4-Methoxybenzenesulfonyl chloride (126 mg, 0.61 mmol) were added sequentially at 0°C. The resulting solution was warmed to room temperature and stirred for 2 h and then was quenched with NH4Cl (aq.). The aqueous phase was extracted with DCM. The combined organic layers were washed with brine and then were dried over Na2SO4. The solvent was removed in vacuo and the residue was then purified by flash column chromatography (petroleum ether/ethyl acetate = 5:1) to give 16e (328 mg, 93%) as a white solid.
After a solution of 16e (135 mg, 0.21 mmol) was dissolved in THF (3 mL), Tetrabutylammonium fluoride (1.0 M in THF, 1 mL, 1.05 mmol) was added. The resulting mixture was stirred for 24 h at 60°C and then was quenched with NH4Cl (aq.). After the reaction was terminated, the aqueous layer was extracted with EtOAc. The combined organic layers were washed with H2O and brine, dried over anhydrous Na2SO4, and concentrated in vacuo to produce a crude oil, which was purified by column chromatography (petroleum ether/ethyl acetate = 2:1). The crude product was directly dissolved in DCM (3 mL), and then DMP (147 mg, 0.32 mmol) and NaHCO3 (35.3 mg, 0.42 mmol) were added sequentially at 0°C. The resultant mixture was stirred for 2 h at the same temperature. Then, the saturated aqueous Na2S2O3 (10 mL) was added to the mixture. The mixture was stirred for 0.5 h at room temperature and extracted with DCM (3 × 5 mL). The combined organic layer was washed with brine, then dried over Na2SO4. The solvent was removed under vacuum, and the residue was purified by column chromatography (petroleum ether/ethyl acetate = 4:1) to afford 17e (80 mg, 72% over two steps) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.83–7.74 (m, 2H), 7.66 (d, J = 8.2 Hz, 1H), 7.17 (ddd, J = 8.5, 6.6, 2.2 Hz, 1H), 7.02–6.91 (m, 4H), 6.16 (s, 0H), 5.57 (s, 1H), 5.11 (s, 1H), 4.77 (dt, J = 3.8, 1.7 Hz, 1H), 4.19 (dd, J = 9.3, 7.3 Hz, 1H), 3.88 (s, 3H), 3.68 (ddd, J = 12.2, 9.3, 4.8 Hz, 1H), 2.94 (dd, J = 17.9, 4.2 Hz, 1H), 2.49 (td, J = 12.1, 7.4 Hz, 1H), 2.36 (dd, J = 11.9, 4.7 Hz, 1H), 2.25 (ddd, J = 17.8, 2.1, 1.2 Hz, 1H), 1.45 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 195.4, 163.2, 151.4, 146.7, 142.6, 130.8, 130.2, 129.5, 129.2, 124.2, 124.0, 120.2, 115.0, 114.4, 103.5, 82.8, 67.6, 60.2, 55.6, 51.2, 42.2, 38.1, 28.1. HRMS (ESI): C27H30N2NaO7S+ [(M + Na)+]: calcd: 549.1666; found: 549.1669.
The product of 15 (260 mg, 0.55 mmol) was directly dissolved in DCM (3 mL), and then triethylamine (160 uL, 1.1 mmol) and 4-nitrobenzenesulfonyl chloride (135 mg, 0.61 mmol) were added sequentially at 0 °C. The resulting solution was warmed to room temperature and stirred for 2 h and then was quenched with NH4Cl (aq.). The aqueous phase was extracted with DCM. The combined organic layers were washed with brine and then dried over Na2SO4. The solvent was removed in vacuo and the residue was then purified by flash column chromatography (petroleum ether/ethyl acetate = 5:1) to give 16f (326 mg, 90%) as a white solid.
After a solution of 16f (138 mg, 0.21 mmol) was dissolved in THF (3 mL), Tetrabutylammonium fluoride (1.0 M in THF, 1 mL, 1.05 mmol) was added. The resulting mixture was stirred for 24 h at 60 °C and then was quenched with NH4Cl (aq.). After the reaction was terminated, the aqueous layer was extracted with EtOAc. The combined organic layers were washed with H2O and brine, dried over anhydrous Na2SO4, and concentrated in vacuo to produce a crude oil, which was purified by column chromatography (petroleum ether/ethyl acetate = 2:1). The crude product was directly dissolved in DCM (3 mL), and then DMP (147 mg, 0.32 mmol) and NaHCO3 (35.3 mg, 0.42 mmol) were added sequentially at 0 °C. The resultant mixture was stirred for 2 h at the same temperature. Then, the saturated aqueous Na2S2O3 (10 mL) was added to the mixture. The mixture was stirred for 0.5 h at room temperature and extracted with DCM (3 × 5 mL). The combined organic layer was washed with brine, then dried over Na2SO4. The solvent was removed under vacuum, and the residue was purified by column chromatography (petroleum ether/ethyl acetate = 4:1) to afford 17f (80 mg, 70% over two steps) as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.38 (d, J = 8.9 Hz, 2H), 8.06 (d, J = 8.9 Hz, 2H), 7.60 (d, J = 8.3 Hz, 1H), 7.19 (ddd, J = 8.5, 7.1, 1.7 Hz, 1H), 7.03–6.91 (m, 2H), 6.18 (s, 1H), 5.63 (s, 1H), 5.38–5.33 (m, 1H), 5.00 (dt, J = 4.4, 2.2 Hz, 1H), 4.20–4.11 (m, 1H), 3.69 (ddd, J = 12.0, 9.3, 5.0 Hz, 1H), 2.90 (dd, J = 17.8, 4.2 Hz, 1H), 2.47 (td, J = 12.0, 7.4 Hz, 1H), 2.40–2.35 (m, 1H), 2.32 (ddd, J = 17.8, 2.3, 1.0 Hz, 1H), 1.52 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 195.0, 151.2, 150.2, 146.2, 145.2, 142.3, 129.9, 129.4, 128.7, 124.3, 124.2, 124.1, 121.3, 115.0, 103.3, 83.3, 67.6, 60.1, 51.3, 42.3, 38.8, 28.2. HRMS (ESI): C26H27N3NaO8S+ [(M + Na)+]: calcd: 567.1411; found: 567.1412.
After a solution of compound 15 (1.3 g, 2.75 mmol) was dissolved in DCM (20 mL), triethylamine (0.8 mL, 5.5 mmol) and chloroacetyl chloride (239 uL, 3.0 mmol) were added at 0°C. Having been stirred for 2 h, the mixture was diluted with DCM (3 × 20 mL), washed with H2O three times, dried over Na2SO4, filtered, and concentrated under a vacuum. The crude product was dissolved in DMF (12 mL) and H2O (4 mL) and then NaN3 (536 mg, 8.25 mmol) was added. The reaction mixture was heated at room temperature for 24 h. The solvent was directly removed in vacuo and the residue was then purified by flash column chromatography (DCM/MeOH = 100:1) to give 18 (978 mg, 64%, over two steps) as a white solid.
After a solution of 18 (86 mg, 0.15 mmol) was dissolved in t-BuOH/H2O (2 mL/2 mL), sodium ascorbate (6.0 mg, 0.03 mmol), CuSO4·H2O (5.4 mg, 0.03 mmol) and 1-ethynyl-4-methoxybenzene (22 mg, 0.17 mmol) were added at room temperature. After a vigorous stirring for 3 h, the resulting mixture was diluted with EtOAc (3 × 5 mL). The layers were separated, and the aqueous layers were extracted with EtOAc. The organic layers were combined and dried over Na2SO4, concentrated in vacuo, and then purified by flash column chromatography (petroleum ether/ethyl acetate = 5:1) to give 19a (72 mg, 68%, over two steps) as a white solid.
After a solution of 19a (72 mg, 0.105 mmol) was dissolved in THF (3 mL), tetrabutylammonium fluoride (1.0 M in THF, 0.5 mL, 0.53 mmol) was added, and the resulting mixture was stirred for 24 h at 60°C, and then was quenched with NH4Cl (aq.). After the reaction was terminated, the aqueous layer was extracted with EtOAc. The combined organic layers were washed with H2O and brine, dried over anhydrous Na2SO4, and concentrated in vacuo to produce a crude oil, which was purified by column chromatography (petroleum ether/ethyl acetate = 2:1). The crude product was directly dissolved in DCM (3 mL), and then DMP (74 mg, 0.16 mmol) and NaHCO3 (18 mg, 0.21 mmol) were added sequentially at 0°C. The resultant mixture was stirred for 2 h at the same temperature. Then, the saturated aqueous Na2S2O3 (10 mL) was added to the mixture. The mixture was stirred for 0.5 h at room temperature and extracted with DCM (3 × 5 mL). The combined organic layers were washed with brine and then dried over Na2SO4. The solvent was removed under vacuum, and the residue was purified by column chromatography (petroleum ether/ethyl acetate = 4:1) to afford 20a (39 mg, 65% over two steps) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.81–7.72 (m, 3H), 7.66 (d, J = 8.3 Hz, 1H), 7.17 (ddd, J = 8.4, 7.3, 1.4 Hz, 1H), 7.04–6.98 (m, 2H), 6.94 (td, J = 7.5, 1.0 Hz, 1H), 6.86 (dd, J = 7.7, 1.5 Hz, 1H), 6.23 (d, J = 5.4 Hz, 1H), 5.97 (s, 1H), 5.53–5.45 (m, 1H), 5.40 (s, 1H), 5.12 (d, J = 16.5 Hz, 1H), 4.98 (d, J = 16.5 Hz, 1H), 4.14 (dd, J = 9.0, 7.3 Hz, 1H), 3.87 (s, 3H), 3.63 (ddd, J = 11.8, 9.2, 5.0 Hz, 1H), 3.01 (dd, J = 17.8, 4.1 Hz, 1H), 2.34 (dd, J = 17.8, 2.5 Hz, 1H), 2.28–2.13 (m, 2H), 1.60 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 196.2, 165.2, 160.0, 151.4, 148.6, 146.3, 142.7, 129.9, 129.3, 127.0, 124.1, 123.9, 122.4, 121.7, 120.3, 115.0, 114.5, 103.6, 83.1, 67.4, 60.1, 55.3, 53.4, 48.3, 41.9, 38.4, 29.7, 28.3. HRMS (ESI): C31H34N5O6+ [(M + H)+]: calcd: 572.2504; found: 572.2505.
After a solution of 18 (86 mg, 0.15 mmol) was dissolved in t-BuOH/H2O (2 mL/2 mL), sodium ascorbate (6.0 mg, 0.03 mmol), CuSO4·H2O (5.4 mg, 0.03 mmol) and 1-ethynyl-4-fluorobenzene (20.5 mg, 0.17 mmol) were added at room temperature. After a vigorous stirring for 3 h, the resulting mixture was diluted with EtOAc (3 × 5 mL). The layers were separated, and the aqueous layer was extracted with EtOAc. The organic layer was combined and dried over Na2SO4, concentrated in vacuo, and then purified by flash column chromatography (petroleum ether/ethyl acetate = 5:1) to give 19b (73 mg, 72%, over two steps) as a white solid.
After a solution of 19b (72 mg, 0.107 mmol) was dissolved in THF (3 mL), tetrabutylammonium fluoride (1.0 M in THF, 0.5 mL, 0.53 mmol) was added, and the resulting mixture was stirred for 24 h at 60°C, and then was quenched with NH4Cl (aq.). After the reaction was terminated, the aqueous layer was extracted with EtOAc. The combined organic layers were washed with H2O and brine, dried over anhydrous Na2SO4, and concentrated in vacuo to produce a crude oil, which was purified by column chromatography (petroleum ether/ethyl acetate = 2:1). The crude product was directly dissolved in DCM (3 mL), and then DMP (74 mg, 0.16 mmol) and NaHCO3 (18 mg, 0.21 mmol) were added sequentially at 0 °C. The resultant mixture was stirred for 2 h at the same temperature. Then, the saturated aqueous Na2S2O3 (10 mL) was added to the mixture. The mixture was stirred for 0.5 h at room temperature and extracted with DCM (3 × 5 mL). The combined organic layers were washed with brine, then dried over Na2SO4. The solvent was removed under vacuum, and the residue was purified by column chromatography (petroleum ether/ethyl acetate = 4:1) to afford 20b (42 mg, 71% over two steps) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.87–7.76 (m, 3H), 7.64 (d, J = 8.3 Hz, 1H), 7.16 (dtd, J = 8.8, 7.2, 1.8 Hz, 3H), 6.94 (td, J = 7.5, 1.0 Hz, 1H), 6.86 (dd, J = 7.7, 1.4 Hz, 1H), 6.30 (d, J = 5.3 Hz, 1H), 5.97 (s, 1H), 5.48 (td, J = 4.5, 2.4 Hz, 1H), 5.41 (s, 1H), 5.11 (d, J = 16.4 Hz, 1H), 4.99 (d, J = 16.4 Hz, 1H), 4.17–4.06 (m, 1H), 3.63 (ddd, J = 11.7, 9.2, 5.1 Hz, 1H), 3.01 (dd, J = 17.7, 4.2 Hz, 1H), 2.33 (dd, J = 17.8, 2.5 Hz, 2H), 2.29–2.15 (m, 1H), 1.58 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 196.1, 165.0, 164.1, 161.6, 151.4, 147.7, 146.3, 142.6, 129.8, 129.3, 127.5, 127.4, 126.1, 126.0, 124.1, 123.9, 121.6, 121.0, 116.2, 116.0, 115.0, 103.6, 83.1, 67.4, 60.0, 53.4, 48.4, 42.0, 38.4, 28.3. 19F NMR (376 MHz, CDCl3) δ -112.5. HRMS (ESI): C30H31FN5O5+ [(M + H)+]: calcd: 560.2304; found: 560.2308.
After a solution of 18 (86 mg, 0.15 mmol) was dissolved in t-BuOH/H2O (2 mL/2 mL), sodium ascorbate (6.0 mg, 0.03 mmol), CuSO4·H2O (5.4 mg, 0.03 mmol) and 2-ethynylpyridine (17.5 mg, 0.17 mmol) were added at room temperature. After a vigorous stirring for 3 h, the resulting mixture was diluted with EtOAc (3 × 5 mL). The layers were separated, and the aqueous layer was extracted with EtOAc. The organic layer was combined and dried over Na2SO4, concentrated in vacuo, and then purified by flash column chromatography (petroleum ether/ethyl acetate = 4:1) to give 19c (64 mg, 65%, over two steps) as a white solid.
After a solution of 19c (64 mg, 0.098 mmol) was dissolved in THF (3 mL), tetrabutylammonium fluoride (1.0 M in THF, 0.5 mL, 0.53 mmol) was added, and the resulting mixture was stirred for 24 h at 60°C, and then was quenched with NH4Cl (aq.). After the reaction was terminated, the aqueous layer was extracted with EtOAc. The combined organic layers were washed with H2O and brine, dried over anhydrous Na2SO4, and concentrated in vacuo to produce a crude oil, which was purified by column chromatography (petroleum ether/ethyl acetate = 2:1). The crude product was directly dissolved in DCM (3 mL), and then DMP (69 mg, 0.15 mmol) and NaHCO3 (17 mg, 0.20 mmol) were added sequentially at 0 °C. The resultant mixture was stirred for 2 h at the same temperature. Then, the saturated aqueous Na2S2O3 (10 mL) was added to the mixture. The mixture was stirred for 0.5 h at room temperature and extracted with DCM (3 × 5 mL). The combined organic layers were washed with brine, then dried over Na2SO4. The solvent was removed under vacuum, and the residue was purified by column chromatography (petroleum ether/ethyl acetate = 4:1) to afford 20c (32 mg, 60% over two steps) as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.62 (ddd, J = 4.9, 1.8, 1.0 Hz, 1H), 8.19 (s, 1H), 8.17–8.13 (m, 1H), 7.82 (td, J = 7.8, 1.8 Hz, 1H), 7.67 (d, J = 8.3 Hz, 1H), 7.28 (ddd, J = 7.7, 4.9, 1.2 Hz, 1H), 7.17 (ddd, J = 8.4, 7.3, 1.5 Hz, 1H), 6.94 (td, J = 7.4, 1.0 Hz, 1H), 6.86 (dd, J = 7.7, 1.4 Hz, 1H), 6.26 (d, J = 5.6 Hz, 1H), 6.01 (s, 1H), 5.51 (ddd, J = 6.2, 4.1, 2.5 Hz, 1H), 5.42 (s, 1H), 5.13 (d, J = 16.6 Hz, 1H), 5.04 (d, J = 16.6 Hz, 1H), 4.18–4.10 (m, 1H), 3.63 (ddd, J = 10.9, 9.2, 5.6 Hz, 1H), 2.99 (dd, J = 18.0, 4.1 Hz, 1H), 2.35 (dd, J = 17.9, 2.6 Hz, 1H), 2.29–2.15 (m, 2H), 2.10 (d, J = 9.9 Hz, 1H), 1.59 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 196.2, 164.8, 151.4, 149.7, 149.4, 149.2, 146.4, 142.7, 137.0, 129.8, 129.3, 124.1, 123.8, 123.7, 123.3, 121.6, 120.1, 115.0, 103.7, 83.1, 67.4, 60.1, 53.4, 48.3, 41.8, 38.4, 28.3. HRMS (ESI): C29H31N6O5+ [(M + H)+]: calcd: 543.2350; found: 543.2352.
MH7A cells (Jennio Biotech Co., Ltd, China) were cultured as we reported previously. (Chen et al., 2022). For cell viability detection, MH7A cells were seeded in 96-well plates overnight before being stimulated with different dosages of compounds (2.5–20 μM) for 24 h. Subsequently, the treatment of the resulting cells was co-incubated with CCK-8 solution (10 μL/well; APExBIO Corporation) for 1 h. Finally, the absorbance of each well was tested through a microplate reader (450 nm; BioTek, United States).
In this study, we synthesized a series of novel akuammiline alkaloid derivatives, among which 9 and 17c exhibited the optimal inhibitory effect on the proliferation of RA-FLSs with an IC50 of (3.22 ± 0.29 μM) and (3.21 ± 0.31 μM), respectively. These results suggested that 9 and 17c may have potential activity in treating RA as lead compounds.
In summary, we presented the flexible synthetic process for creating analogs of akuammiline alkaloids with various skeletons and analyzed their effects on the proliferation of RA-FLSs in vitro from their structure. We also demonstrated that 9 and 17c elicited a strong inhibitory effect on the proliferation of RA-FLSs. Our study is certain to lay a good foundation for further pharmacology studies of akuammiline alkaloid derivatives and provides a good example for the development of anti-RA small molecule drugs that originate from natural products.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
JW, CT, XB, and JC designed the experiments. XB, JW, HZ, and BB conducted the experiments. XB, CT, and MB wrote the manuscript. All authors have approved the final version of the manuscript.
We thank the National Natural Science Foundation of China (22231003, 22271008), Shenzhen Science and Technology Innovation Committee (JCYJ20180504165454447, GXWD20201231165807007-20200812100115001), Shenzhen Science and Technology Program (Grant No. KQTD20190929174023858), Guangdong Science and Technology Program (2017B030314002), Industry and Information Technology Bureau of Shenzhen Municipality (201806151622209330), Shenzhen-Hong Kong Institute of Brain Science-Shenzhen Fundamental Research Institutions (2023SHIBS0004), the third foster plan in 2019 “Molecular Imaging Probe Preparation and Characterization of Key Technologies and Equipment” (ZDKJ20190305003) for the development of key technologies and equipment in major science and technology infrastructure in Shenzhen, the National Ten Thousand Talent Program (the Leading Talent Tier), and the Shenzhen Bay Laboratory Open Research Program (SZBL2019062801010), for the financial support.
We are grateful to Prof. Zhiqiang Ma of South China University of Technology for enlightening discussions.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2023.1179948/full#supplementary-material
Abdellatif, K. R., Lamie, P. F., and Omar, H. A. (2016). 3-Methyl-2-phenyl-1-substituted-indole derivatives as indomethacin analogs: Design, synthesis and biological evaluation as potential anti-inflammatory and analgesic agents. J. Enzyme Inhib. Med. Chem. 31, 318–324. doi:10.3109/14756366.2015.1022174
Akgul, O., Di Cesare Mannelli, L., Vullo, D., Angeli, A., Ghelardini, C., Bartolucci, G., et al. (2018). Discovery of novel nonsteroidal anti-inflammatory drugs and carbonic anhydrase inhibitors hybrids (NSAIDs–CAIs) for the management of rheumatoid arthritis. J. Med. Chem. 61, 4961–4977. doi:10.1021/acs.jmedchem.8b00420
Arshad, L., Jantan, I., Bukhari, S. N., and Haque, M. A. (2017). Immunosuppressive effects of natural α,β-unsaturated carbonyl-based compounds, and their analogs and derivatives, on immune cells: A review. Front. Pharmacol. 8, 22. doi:10.3389/fphar.2017.00022
Bonandi, E., Christodoulou, M. S., Fumagalli, G., Perdicchia, D., Rastelli, G., and Passarella, D. (2017). The 1,2,3-triazole ring as a bioisostere in medicinal chemistry. Drug Discov. Today 22, 1572–1581. doi:10.1016/j.drudis.2017.05.014
Bozorov, K., Zhao, J., and Aisa, H. A. (2019). 1,2,3-Triazole-containing hybrids as leads in medicinal chemistry: A recent overview. Bioorg Med. Chem. 27, 3511–3531. doi:10.1016/j.bmc.2019.07.005
Chen, J., Lin, X., He, J., Liu, J., He, J., Tao, C., et al. (2022). Novel isatin-based hybrids as potential anti-rheumatoid arthritis drug candidates: Synthesis and biological evaluation. Bioorg Chem. 128, 106063. doi:10.1016/j.bioorg.2022.106063
Dai, J., Tian, S., Yang, X., and Liu, Z. (2022). Synthesis methods of 1,2,3-/1,2,4-triazoles: A review. Front. Chem., 10. doi:10.3389/fchem.2022.891484
Du, J., Liu, P., Zhu, Y., Wang, G., Xing, S., Liu, T., et al. (2023). Novel tryptanthrin derivatives with benzenesulfonamide substituents: Design, synthesis, and anti-inflammatory evaluation. Eur. J. Med. Chem. 246, 114956. doi:10.1016/j.ejmech.2022.114956
Estevao, M. S., Carvalho, L. C., Ribeiro, D., Couto, D., Freitas, M., Gomes, A., et al. (2010). Antioxidant activity of unexplored indole derivatives: Synthesis and screening. Eur. J. Med. Chem. 45, 4869–4878. doi:10.1016/j.ejmech.2010.07.059
Grau, L., Soucek, R., and Pujol, M. D. (2023). Resveratrol derivatives: Synthesis and their biological activities. Eur. J. Med. Chem. 246, 114962. doi:10.1016/j.ejmech.2022.114962
Guerra, A. S., Malta, D. J., Laranjeira, L. P., Maia, M. B., Colaco, N. C., de Lima Mdo, C., et al. (2011). Anti-inflammatory and antinociceptive activities of indole–imidazolidine derivatives. Int. Immunopharmacol. 11, 1816–1822. doi:10.1016/j.intimp.2011.07.010
Guo, C., He, L., Hu, N., Zhao, X., Gong, L., Wang, C., et al. (2022). Aconiti Lateralis Radix Praeparata lipid-soluble alkaloids alleviates IL-1β-induced inflammation of human fibroblast-like synoviocytes in rheumatoid arthritis by inhibiting NF-κB and MAPKs signaling pathways and inducing apoptosis. Cytokine 151, 155809. doi:10.1016/j.cyto.2022.155809
Haider, S., Alam, M. S., Hamid, H., Shafi, S., Nargotra, A., Mahajan, P., et al. (2013). Synthesis of novel 1,2,3-triazole based benzoxazolinones: Their TNF-α based molecular docking with in-vivo anti-inflammatory, antinociceptive activities and ulcerogenic risk evaluation. Eur. J. Med. Chem. 70, 579–588. doi:10.1016/j.ejmech.2013.10.032
Kumari, A., and Singh, R. K. (2019). Medicinal chemistry of indole derivatives: Current to future therapeutic prospectives. Bioorg Chem. 89, 103021. doi:10.1016/j.bioorg.2019.103021
Li, S., Liu, X., Chen, X., and Bi, L. (2020). Research progress on anti-inflammatory effects and mechanisms of alkaloids from Chinese medical herbs. Evid. Based Complement. Altern. Med. 2020, 1303524. doi:10.1155/2020/1303524
Li, T. P., Zhang, A. H., Miao, J. H., Sun, H., Yan, G. L., Wu, F. F., et al. (2019). Applications and potential mechanisms of herbal medicines for rheumatoid arthritis treatment: A systematic review. RSC Adv. 9, 26381–26392. doi:10.1039/c9ra04737a
Ma, Q., and Jiang, J. G. (2016). Functional components from nature-derived drugs for the treatment of rheumatoid arthritis. Curr. Drug Targets 17, 1673–1686. doi:10.2174/1389450117666160527122233
Murugesan, S., Ravichandran, D., Lakshmanan, D. K., Ravichandran, G., Arumugam, V., Raju, K., et al. (2020). Evaluation of anti rheumatic activity of Piper betle L. (Betelvine) extract using in silico, in vitro and in vivo approaches. Bioorg Chem. 103, 104227. doi:10.1016/j.bioorg.2020.104227
Pandit, S. S., Kulkarni, M. R., Pandit, Y. B., Lad, N. P., and Khedkar, V. M. (2018). Synthesis and in vitro evaluations of 6-(hetero)-aryl-imidazo[1,2-b]pyridazine-3-sulfonamide’s as an inhibitor of TNF-α production. Bioorg Med. Chem. Lett. 28, 24–30. doi:10.1016/j.bmcl.2017.11.026
Shaker, A. M. M., Abdelall, E. K. A., Abdellatif, K. R. A., and Abdel-Rahman, H. M. (2020). Synthesis and biological evaluation of 2-(4-methylsulfonyl phenyl) indole derivatives: Multi-target compounds with dual antimicrobial and anti-inflammatory activities. BMC Chem. 14, 23. doi:10.1186/s13065-020-00675-5
Sharma, D., Chaubey, P., and Suvarna, V. (2021). Role of natural products in alleviation of rheumatoid arthritis—A review. J. Food Biochem. 45, e13673. doi:10.1111/jfbc.13673
Shen, P., Jiao, Y., Miao, L., Chen, J. H., and Momtazi-Borojeni, A. A. (2020). Immunomodulatory effects of berberine on the inflamed joint reveal new therapeutic targets for rheumatoid arthritis management. J. Cell Mol. Med. 24, 12234–12245. doi:10.1111/jcmm.15803
Smolen, J. S., Aletaha, D., Barton, A., Burmester, G. R., Emery, P., Firestein, G. S., et al. (2018). Rheumatoid arthritis. Nat. Rev. Dis. Prim. 4, 18001. doi:10.1038/nrdp.2018.1
Wang, T., Duan, X., Zhao, H., Zhai, S., Tao, C., Wang, H., et al. (2017). Asymmetric total synthesis of (−)-Aspidophylline A. Org. Lett. 19, 1650–1653. doi:10.1021/acs.orglett.7b00448
Wang, Y., Chen, S., Du, K., Liang, C., Wang, S., Owusu Boadi, E., et al. (2021). Traditional herbal medicine: Therapeutic potential in rheumatoid arthritis. J. Ethnopharmacol. 279, 114368. doi:10.1016/j.jep.2021.114368
Yang, M., Wang, Y., Fan, Z., Xue, Q., Njateng, G. S. S., Liu, Y., et al. (2020). Acute and sub-acute toxicological evaluations of bioactive alkaloidal extract from Melodinus henryi and their main chemical constituents. Nat. Prod. Bioprospect 10, 227–241. doi:10.1007/s13659-020-00252-2
Yu, L., Li, S., Pu, L., Yang, C., Shi, Q., Zhao, Q., et al. (2022). Traditional Tibetan medicine: therapeutic potential in rheumatoid arthritis. Front. Pharmacol. 13, 938915. doi:10.3389/fphar.2022.938915
Zhang, B. (2019). Comprehensive review on the anti-bacterial activity of 1,2,3-triazole hybrids. Eur. J. Med. Chem. 168, 357–372. doi:10.1016/j.ejmech.2019.02.055
Zhang, C., Zhang, S., Liao, J., Gong, Z., Chai, X., and Lyu, H. (2022). Towards better sinomenine-type drugs to treat rheumatoid arthritis: Molecular mechanisms and structural modification. Molecules 27, 8645. doi:10.3390/molecules27248645
Zhang, R.-J., Li, S.-F., Shi, H.-J., Zhao, J.-L., Geng, Y., Fan, H.-C., et al. (2022). J. Explor Res. Pharmacol. 7, 125–132.
Zhang, T.-Y., Li, C.-S., Cao, L.-T., Bai, X.-Q., Zhao, D.-H., and Sun, S.-M. (2021). New ursolic acid derivatives bearing 1,2,3-triazole moieties: Design, synthesis and anti-inflammatory activity in vitro and in vivo. Mol. Divers 26, 1129–1139. doi:10.1007/s11030-021-10236-0
Zhang, X., Ji, A., Wang, Z., Lou, H., Li, J., Zheng, L., et al. (2021). Azide-dye unexpected bone targeting for near-infrared window II osteoporosis imaging. J. Med. Chem. 64, 11543–11553. doi:10.1021/acs.jmedchem.1c00839
Zhao, C., Rakesh, K. P., Ravidar, L., Fang, W. Y., and Qin, H. L. (2019). Pharmaceutical and medicinal significance of sulfur (SVI)-Containing motifs for drug discovery: A critical review. Eur. J. Med. Chem. 162, 679–734. doi:10.1016/j.ejmech.2018.11.017
Zhao, F., Wang, J., Ding, X., Shu, S., and Liu, H. (2016). Application of sulfonyl in drug design. Chin. J. Org. Chem. 36, 490. doi:10.6023/cjoc201510006
Zhou, S., Jiang, W., Chen, G., and Huang, G. (2022). Design and synthesis of novel double-ring conjugated enones as potent anti-rheumatoid arthritis agents. ACS Omega 7, 44065–44077. doi:10.1021/acsomega.2c05492
Keywords: alkaloid derivates, rheumatoid arthritis, fibroblast-like synoviocytes, structure activity relationships, anti-inflammatory
Citation: Bao X, Wei J, Tao C, Bashir MA, Zhang H-J, Bao B, Chen J and Zhai H (2023) Akuammiline alkaloid derivatives: divergent synthesis and effect on the proliferation of rheumatoid arthritis fibroblast-like synoviocytes. Front. Chem. 11:1179948. doi: 10.3389/fchem.2023.1179948
Received: 05 March 2023; Accepted: 17 April 2023;
Published: 28 April 2023.
Edited by:
Syeda Abida Ejaz, Islamia University of Bahawalpur, PakistanReviewed by:
Saira Afzal, University of Lahore, PakistanCopyright © 2023 Bao, Wei, Tao, Bashir, Zhang, Bao, Chen and Zhai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jian Chen, Y2hlbmppYW5AcGt1c3poLmNvbQ==; Hongbin Zhai, emhhaWhiQHBrdS5lZHUuY24=
†ORCID: Xinye Bao, orcid.org/0009-0009-4752-2395
‡These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.