
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem., 07 April 2023
Sec. Medicinal and Pharmaceutical Chemistry
Volume 11 - 2023 | https://doi.org/10.3389/fchem.2023.1160164
This article is part of the Research TopicFrontiers in Chemistry: Rising Stars 2022View all 13 articles
 Camilla Scarpellini1Sophie Valembois1Kenneth Goossens1
Camilla Scarpellini1Sophie Valembois1Kenneth Goossens1 Mike Vadi2,3
Mike Vadi2,3 Caroline Lanthier1
Caroline Lanthier1 Greta Klejborowska1
Greta Klejborowska1 Pieter Van Der Veken1
Pieter Van Der Veken1 Hans De Winter1
Hans De Winter1 Mathieu J. M. Bertrand2,3†
Mathieu J. M. Bertrand2,3† Koen Augustyns1*†
Koen Augustyns1*†Receptor-Interacting serine/threonine-Protein Kinase 1 (RIPK1) emerged as an important driver of inflammation and, consequently, inflammatory pathologies. The enzymatic activity of RIPK1 is known to indirectly promote inflammation by triggering cell death, in the form of apoptosis, necroptosis and pyroptosis. Small molecule Receptor-Interacting serine/threonine-Protein Kinase 1 inhibitors have therefore recently entered clinical trials for the treatment of a subset of inflammatory pathologies. We previously identified GSK2656157 (GSK’157), a supposedly specific inhibitor of protein kinase R (PKR)-like ER kinase (PERK), as a much more potent type II Receptor-Interacting serine/threonine-Protein Kinase 1 inhibitor. We now performed further structural optimisation on the GSK’157 scaffold in order to develop a novel class of more selective Receptor-Interacting serine/threonine-Protein Kinase 1 inhibitors. Based on a structure-activity relationship (SAR) reported in the literature, we anticipated that introducing a substituent on the para-position of the pyridinyl ring would decrease the interaction with PERK. Herein, we report a series of novel GSK’157 analogues with different para-substituents with increased selectivity for Receptor-Interacting serine/threonine-Protein Kinase 1. The optimisation led to UAMC-3861 as the best compound of this series in terms of activity and selectivity for Receptor-Interacting serine/threonine-Protein Kinase 1 over PERK. The most selective compounds were screened in vitro for their ability to inhibit RIPK1-dependent apoptosis and necroptosis. With this work, we successfully synthesised a novel series of potent and selective type II Receptor-Interacting serine/threonine-Protein Kinase 1 inhibitors based on the GSK’157 scaffold.
Cell death is increasingly recognised as an important driver of inflammation, and is consequently believed to be at the origin of various inflammatory diseases when improperly regulated (Newton et al., 2021). A subset of innate immune receptors can trigger the dismantlement of the cell, and the resulting cell death can promote the production of pro-inflammatory mediators by activating immune receptors on neighbouring effector cells. Next to apoptosis, which is generally considered immunologically silent, lytic forms of cell death, such as apoptosis-driven secondary necrosis, necroptosis and pyroptosis, release intracellular factors, known as danger-associated molecular patterns (DAMPs), that activate immune receptors and induce inflammatory responses. In addition, the death of epithelial cells can also affect the functionality of bodily barriers, and thereby trigger/exacerbate inflammation through the sensing of pathogen-associated molecular patterns (PAMPs) from microbes that have breached the barriers. Accumulating evidence indicates that genetic targeting of cell death can revert the inflammatory pathology state in various mouse models of acute and chronic inflammatory diseases. Hence, drugs that inhibit cell death are currently under investigation as potential therapies for human inflammatory and autoimmune diseases. Among these drugs, pharmacological inhibitors of the receptor-interacting serine/threonine protein kinase 1 (RIPK1) have entered clinical trials for the treatment of autoimmune diseases (psoriasis, ulcerative colitis and rheumatoid arthritis), as well as some cancers and neurological disorders (Mifflin et al., 2020).
RIPK1 functions as a central signalling node downstream of several immune receptors, where it can paradoxically function as a scaffold to promote cell survival or as an active kinase to trigger cell death (Delanghe et al., 2020). RIPK1 has been best studied in the context of tumour necrosis factor (TNF) signalling. Binding of TNF to TNFR1 induces recruitment of RIPK1 to the cytosolic tail of the receptor, an initial step in the assembly of the receptor signalling complex (Complex I). Within this complex, RIPK1 functions as a ubiquitylated scaffold that contributes to the activation of the MAPK and NF-κB signalling pathways, which collectively promote transcription of pro-inflammatory and pro-survival genes. Kinases from the MAPK and NF-κB pathway, including MK2 and IKKα/β, signal back to RIPK1 to maintain it in an inactive prosurvival state (Dondelinger et al., 2015; 2017; 2019; Jaco et al., 2017; Menon et al., 2017; Lafont et al., 2018; Xu et al., 2018). Consequently, conditions that affect proper activation of these kinases remove the brake on RIPK1, which can then autophosphorylate on Ser166, dissociate from complex I and promote assembly of the cytosolic caspase-8 activating complex (complex II) that triggers apoptosis induction. Caspase-8 activation by RIPK1 was also recently reported to induce cleavage and activation of the pore forming molecule gasdermin D (GSDMD) in macrophages, thereby promoting induction of a specific form of pyroptosis (Orning et al., 2018; Chen et al., 2021). Furthermore, conditions that prevent activation of caspase-8 in Complex II (e.g., by the use of the pan-caspase inhibitor zVAD.fmk) induce further recruitment of RIPK3 and MLKL to complex II, now called the necrosome, leading to RIPK3 activation, phosphorylation of MLKL by RIPK3, and subsequent necroptosis induction (Rodriguez et al., 2016; Mompea et al., 2018; Weber et al., 2018; Klöditz and Fadeel, 2019). The enzymatic activity of RIPK1 is therefore capable of inducing three distinct forms of cell death, which have all been linked to inflammation (Mifflin et al., 2020). Consequently, the importance of specific targeting RIPK1 to prevent chronic inflammatory processes has emerged.
The first RIPK1 inhibitor described in the literature was necrostatin-1 (Nec-1) in 2005 (Figure 1) (Degterev et al., 2005). It served as a valuable tool compound to study RIPK1 structure and to demonstrate the implication of RIPK1 kinase-dependent cell death in animal models of human diseases (Xie et al., 2013). Particularly, the co-crystallization of Nec-1s, a stable form of Nec-1, with the kinase domain of RIPK1 demonstrated the typical type III kinases inhibitor mode of interaction, with binding to the specific RIPK1 allosteric pocket formed by the DLG-out conformation and without interacting with the hinge region (Wang et al., 2006; Zheng et al., 2008; Takahashi et al., 2012). Since then, several additional RIPK1 inhibitors have been reported (Martens et al., 2020; Chen et al., 2022; Shi et al., 2022). Among them, a class of compounds featuring a benzoxazepinone core emerged with GSK2882481 (Clark et al., 2009; Harris et al., 2016), but interspecies differences were observed when comparing humans to non-primate RIPK1, probably due to the different amino acids featured by the enzyme affecting protein flexibility (Berger et al., 2015; Harris et al., 2017). This class of compound was further optimised aiming to develop a drug with suitable oral bioavailability. The lead optimisation led to the discovery of GSK2982772 (GSK’772) which is currently in phase IIb clinical trial for the treatment of ulcerative colitis, psoriasis, and rheumatoid arthritis (Weisel et al., 2017).
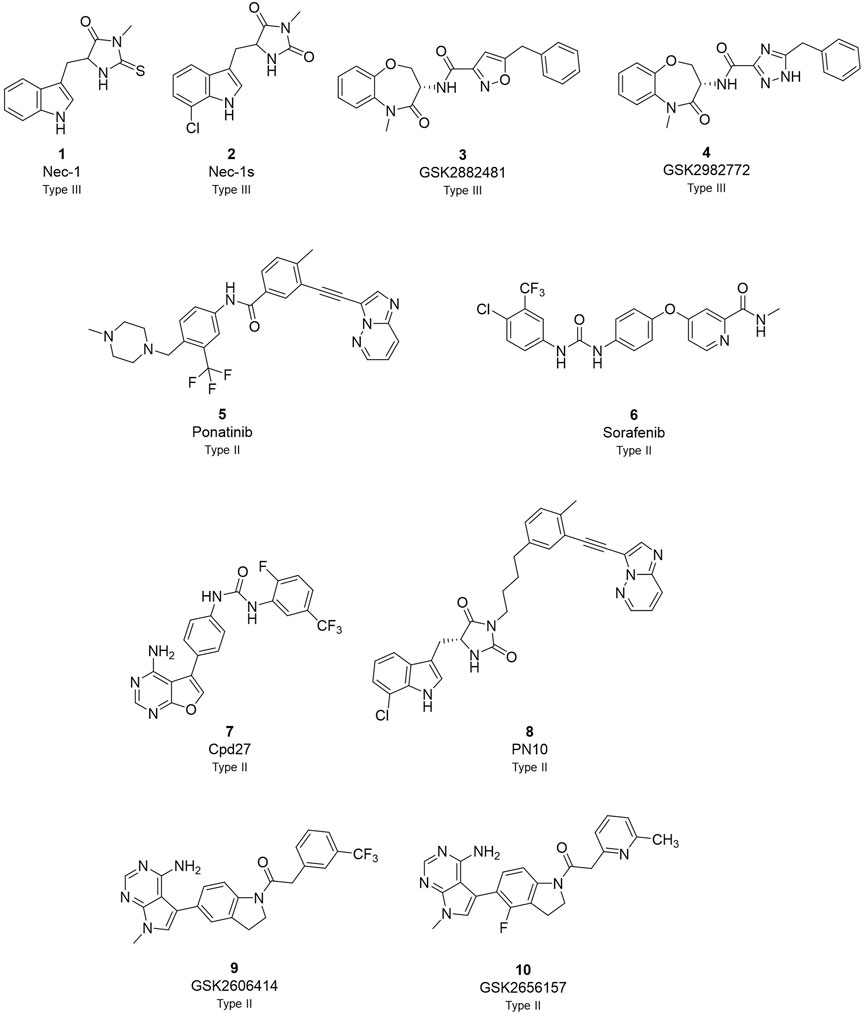
FIGURE 1. Overview of currently known RIPK1 inhibitors.
Among the different RIPK1-inhibitors available in the literature (Figure 1), the type II class has been less investigated in disease models and in clinic (Martens et al., 2020; Chen et al., 2022; Shi et al., 2022). In the type II RIPK1 inhibitors family, tyrosine kinase inhibitor (TKI) ponatinib and its analogues, and sorafenib are reported as dual inhibitors for both RIPK1 and RIPK3 (Fauster et al., 2015; Martens et al., 2017). Compound 27 (cpd27) showed promising pharmacokinetics properties but low kinase selectivity (Harris et al., 2013). PN10 is a hybrid compound which combines the structures of Nec-1 and ponatinib (Najjar et al., 2015). This strategy increased specificity and selectivity of the analogue retaining the interaction with the allosteric pocket of Nec-1 and the interaction with the hinge region of ponatinib as type II kinases inhibitors. Besides, GSK identified a class of dual inhibitors represented by GSK2593067 and GSK2593074 which completely blocked necroptosis in both human and murine cells as type II inhibitors (Zhou et al., 2019).
We previously reported that two commonly used Protein kinase R (PKR)-like ER kinase (PERK) inhibitors, GSK2606414 (GSK’414) and GSK2656157 (GSK’157), were actually potent inhibitors of RIPK1 (Rojas-Rivera et al., 2017). Interestingly, and in comparison to the above mentioned RIPK1 inhibitors, GSK’157 appeared to function as a type II kinases inhibitor of RIPK1 (Axten et al., 2013). A kinome scan perfomed for both, GSK’414 and GSK’157 suggested a good selectivity profile. GSK’414 was screened against a panel of 294 kinases at 10 µM with an inhibition of 20 kinases with more than 85%, while the optimised analogue GSK’157 inhibited 17 out of 300 kinases with more than 80% at the same concentration. The reported structure-activity relationship (SAR) suggested that derivatisation of the corresponding para-position of the pyridinyl ring could reduce affinity for PERK (Axten et al., 2012; 2013). Moreover, recent molecular modelling studies confirmed the different nature of the C-alpha helix domain in RIPK1 with the presence of an additional lipophilic pocket compared to PERK (Chintha et al., 2019). We investigated the structural differences between PERK and RIPK1 and we verified the presence of an extra pocket with additional space to place the para-substituent. We hypothesised that this para-substituent would reduce the affinity for PERK and at the same time, keep or enhance the affinity for RIPK1. Based on these assumptions, we herein report the design, synthesis and biological evaluation of novel GSK’157 analogues with increased selectivity for RIPK1 over PERK (Figure 2).
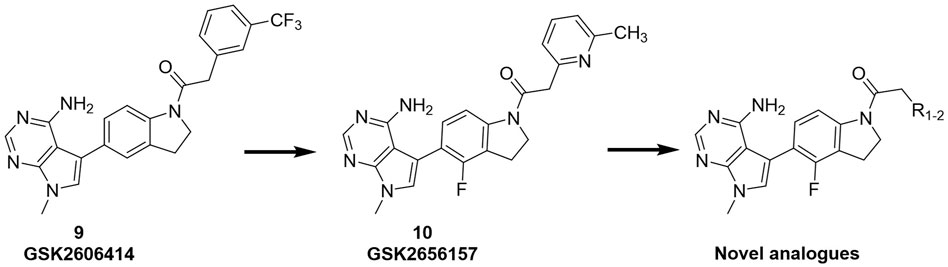
FIGURE 2. Overview of the novel GSK’157 analogues featuring a substituent on corresponding para-position or the pyridinyl or aryl moiety.
Unless otherwise stated, laboratory reagent grade solvents were used. Reagents were obtained from various commercial sources and were used without any prior purification. Characterisation of all compounds was done with 1H and 13C NMR and mass spectrometry. NMR spectra were recorded with a 400 MHz Bruker Avance III Nanobay spectrometer with Ultrashield. All obtained spectra were analysed using MestReNova analytical chemistry software. Chemical shifts are displayed in ppm and coupling constants are shown in hertz (Hz). ES mass spectra were obtained from an Esquire 3000plus Ion Trap Mass Spectrometer from Bruker Daltonics. The UPLC (ultra performance liquid chromatography), used to quantify the purity of the products, was an ACQUITY UPLC H-Class system with a TUV detector Waters coupled to an MS detector Waters Qda. Waters Acquity UPLC BEH C18 1.7 μm, 2.1 mm × 50 mm column was used. The eluent was composed of two different solvents. Solvent C consisted of water with 0.1% formic acid, solvent D was acetonitrile. For most of the experiments, unless stated otherwise, the general method was used. The column was first equilibrated for 0.15 min with a mixture of 95% solvent C and 5% solvent D. After that, solvent D was increased linearly to 100% over 1.75 min before being held constant for 0.25 min, followed by a mixture of 95% solvent C and 5% solvent D for 0.75 min (flow rate 0.7 mL/min). All mass spectra were recorded over a m/z range of 100–1000. The wavelength for UV detection was 254 nm. Method II starts with equilibration of column for 0.15 min with a mixture of 95% solvent C and 5% solvent D. After that, solvent D was increased linearly to 100% over 2.50 min before being held constant for 0.75 min, followed by a mixture of 95% solvent C and 5% solvent D for 0.75 min (flow rate 0.7 mL/min). Key target compounds for the activity were analyzedby high resolution mass spectrometry: 10 μL of each sample (concentration = 10−5 M) was injected using the CapLC system (Waters, Manchester, United Kingdom) and electrosprayed using a standard electrospray source. Samples were injected with an interval of 5 min. Positive ion mode accurate mass spectra (HRMS) were acquired usinga Q-TOF II instrument (Waters, Manchester, United Kingdom). The MS was calibrated prior to use with a 0.2% H3PO4 solution. The spectra were lock mass corrected using the knownmassof the nearest H3PO4 cluster. During the chemical synthesis, flash purification was performed when necessary on a Biotage ISOLERA One flash system equipped with an internal variable dual-wavelength diode array detector (200–400 nm). For the normal phase, purifications SNAP cartridges (10–340 g, flow rate of 10–100 mL/min) were used and reversed-phase purifications were done making use of KP-C18 or Buchi EcoFlex C18 cartridges. Dry sample loading was done by self-packing sample cartridges using Celite® 545. Gradients used varied for each purification. The following section comprises the synthetic procedures and analytical data for all compounds reported in this manuscript. Several synthesis procedures that were used in the preparation of intermediates and final products are summarized here as “General Procedures”. The purities of all final products were found to be >95%. Analyses indicated by the symbols of the elements or functions were within ±0.4% of the theoretical values.
Antibodies and reagents were purchased from the following companies: anti-PERK (Cell Signaling Technology; no. 3192), anti-RIPK1 (BD Biosciences Laboratories; no. 610459), pPERK (Cell Signaling Technology; no. #3179), pRIPK1 (Cell Signaling Technology; no. #31122 (for S166) or ThermoFischer, custom made (for S166/T169)), Tunicamicyn (Tm; Sigma-Aldrich, St Louis, MO, United States; no. T7765), Tubulin (Abacam, no. ab21058), 9-Epimer-11,12-dihydro-(5Z)-7- Oxozeanol ((5Z)-7-Oxozeaenol or TAK1 inhibitor (TAK1i); AnalytiCon Discovery GmbH, Potsdam, Germany; no. NP-0009245), Carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]-fluoromethylketone or zVAD.fmk (Bachem, Bubendorf, Switzerland; no. N-1510), GSK2656157 (ApexBio, Boston, MA, United States; no. B2175.). Recombinant human TNF-α is produced and purified to at least 99% homogeneity in the laboratory of VIB in Gent.
Mouse embryonic fibroblasts (MEFs) were isolated from C57Bl6/J E12.5 embryos according to standard protocol and immortalized by transfection of an SV40 large T-expressing construct. Ripk3+/+ and Ripk3−/− MEFs have been isolated from littermate embryos of Ripk3 ± pregnant females and have been described previously (Vanlangenakker et al., 2011; Dondelinger et al., 2013). The MEFs cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum, L-glutamine (200 mM), sodium pyruvate (400 mM), penicillin (100 IU/mL), and streptomycin (0.1 mg/mL) in normoxic conditions (5% CO2). HT-29 cells were cultured in McCoys modified medium supplemented with 10% fetal calf serum, L-glutamine (200 mM), sodium pyruvate (400 mM), penicillin (100 IU/mL), and streptomycin (0.1 mg/mL) in normoxic conditions (5% CO2).
The cell-death measurements were carried out using a Fluostar Omega fluorescence plate reader (BMG Labtech, Ortenberg, Germany) with temperature and atmosphere-controlled settings, as previously reported (Grootjans et al., 2016). In brief, the cells were seeded in duplicate at 10,000 cells per well in a 96-well adherent plate and incubated overnight at 37°C with 5% CO2. The next day, the cells were pretreated for 30 min with 1/10 dilution series of the indicated compounds ranging from 1 μM to 0.01 nM in the presence of 2.5 μM Sytox Green (Invitrogen, Waltham, MA, United States; no. S7020) and 50 µM zVAD.fmk. All the compound stock solutions were prepared in DMSO at 200 µM concentration. After stimulating the cells with human TNF at 20 ng/mL the plate was transferred to the incubator. Sytox Green intensity was measured in function of time at 0, 14 and 24 h with an excitation filter of 485 nm, an emission filter of 520 nm. The cell death was calculated by subtracting the background fluorescence from the induced Sytox Green fluorescence and by dividing the obtained result by the maximal fluorescence (minus the background fluorescence) obtained by permeabilization of the cells by using Triton X-100 at a final concentration of 0.1%. Curves were plotted in GraphPad 9 for the IC50 calculation.
RIPK1 (h) is incubated with 8 mM MOPS pH 7.0, 0.2 mM EDTA, 0.33 mg/mL myelin basic protein, 10 mM Magnesium acetate and [γ-33P-ATP] (specific activity and concentration as required). The reaction is initiated by the addition of the Mg/ATP mix. After incubation for 120 min at room temperature, the reaction is stopped by the addition of phosphoric acid to a concentration of 0.5%. An aliquot of the stopped reaction is spotted onto a filter and washed four times for 4 min in 0.425% phosphoric acid and once in methanol prior to drying and scintillation counting.
MEFs were seeded in 6-well plates at a density of 300.000 cells/well in 2 mL. The next day, the cells were suspended in 1 mL of medium and pre-treated for 30 min with the indicated compounds at 1/5 dilutions series for PERK ranging from 5 to 0.04 µM and 1 μg/mL of Tm followed by 4 h treatment with 20 ng/mL of hTNF. For RIPK1, the cells were pre-treated at 1/10 dilutions series ranging from 10 to 0.01 nM, 50 µM zVAD.fmk and 1 µM TAKi before stimulation with 20 ng/mL of hTNF for 2.5 h. Then, the cells were washed two times with 1 mL of ice-cold PBS before lysis in 0.2 mL of Laemmli buffer 1x (10% (w/v) SDS, 50% glycerol, 0.5% bromophenol blue, 250 mM Tris–HCl pH 6.8) and 250 mM of dithiothreitol (DTT). The cell lysate was then collected and boiled at 95 °C for 10min and stored at −20°C. Proteins expression was later monitored by immunoblotting.
Molecular docking was carried out using Glide (Chen et al., 2018). The input structures for PERK and RIPK1 were extracted from the Protein Data Bank (PDB ID: 4M7I and 4NEU, respectively) (Saur et al., 2021). Missing loops were modelled using MODELLER v10.2 (Fiser and Sali, 2003), and the structures were prepared using the Protein Preparation Wizard in Maestro (Madhavi Sastry et al., 2013). Ligands were prepared using the LigPrep module of Maestro, assuming a physiological pH of 7.4. Docking in Glide was performed with the OPLS2005 force field using the extra precision protocol. The binding site was defined based on the inhibitors present in the respective crystal structures. Docking was initially done unconstrained, but due to unrealistic poses being generated for PERK, core constraints were added to the adenylyl mimetic to ensure kinase-inhibitor-like docking poses by constraining its distance to the hinge region. Otherwise, default settings were kept.
According to the design strategy mentioned above, a series of 17 novel analogues presenting para-substituted pyridinyl (18–29) and aryl (30–34) groups were designed and synthesised. Different halogens together with small alkyls such as methyl and trifluoromethyl were introduced in both pyridinyl (18–22) and aryl (30–34) analogues. Specifically for the pyridinyl compounds, additional small groups were added with different polarity (23 with -O-CH3, 26 with -OH and the 4-pyridinyl analogue 27) and these were compared with compounds 24, 28 and 29 featuring bulkier moieties (5-cyclopropyl, 5-benzyl and 5-phenyl). The molecules were then tested for their selectivity profile against PERK and RIPK1 by Western blot. Afterwards, their cell death inhibitory activity was quantified by fluorometric cell death assay and finally compound UAMC-3861 (22) was selected for further biological characterisation (Grootjans et al., 2016).
The novel library was synthesised following the synthetic route reported for GSK’414 and GSK’157 (Axten et al., 2012; 2013). The common intermediate 17 was synthesised from the commercially available 4-fluoroindole as depicted in Figure 3. After the reduction of the 4-fluoroindole 11 to the corresponding indoline 12, the secondary amine group was Boc-protected (13) before allylic bromination with the N-bromosuccinimide (14). Finally, the reaction with the bis(pinacolato) diboron led to the corresponding boronic ester intermediate that was then used in a Suzuki−Miyaura coupling reaction with 5-Bromo-7-methyl-7H-pyrrolo [2,3-d]pyrimidin-4-amine (38) synthesised as reported in Figure 4. Briefly, the conditions for the methylation of the 4-chloro-7H-pyrrolo [2,3-d]pyrimidine (35) NH and the following bromination were modified compared to the route of Axten M. J. et al. As reported in the experimental section (Axten et al., 2013). After the Suzuki-Miyaura coupling, the intermediate 16 was deprotected under acidic conditions and the free indoline was coupled with different aryl acetic acids to obtain the desired final compounds (18–25 and 27–34). The corresponding acetic acids were either commercially available or synthesised as reported in the experimental section. To obtain the hydroxy 26, the methoxy group of compound 23 was demethylated in presence of borontribromide (BBr3). The corresponding R1-2 groups are reported in Table 1.
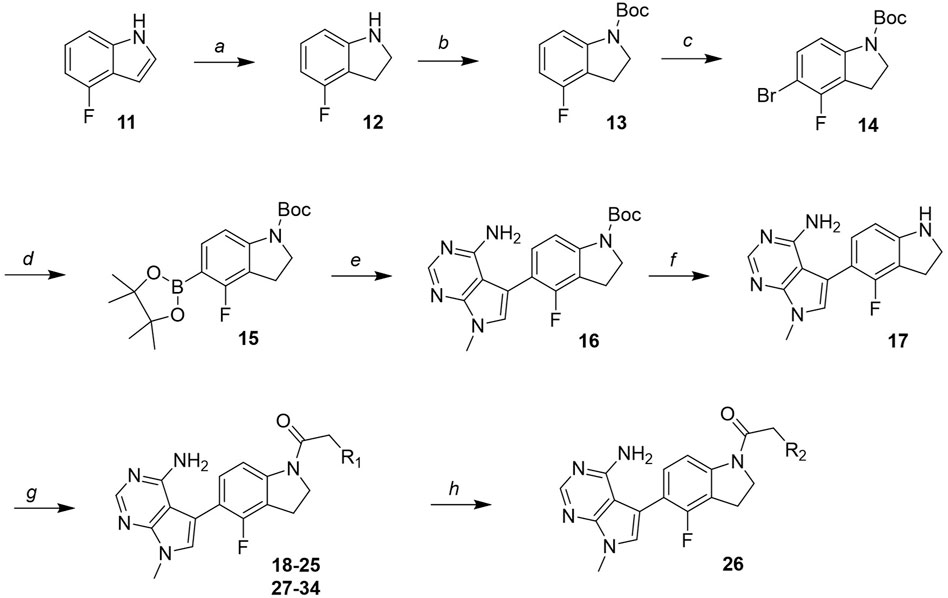
FIGURE 3. Synthesis of the common intermediate 17 based on GSK’157 synthetic route followed by amide coupling to obtain novel RIPK1 inhibitors 18–34. Reagents and conditions: (a) 4-fluoro-1H-indole, NaBH3CN, AcOH, 0°C, 10 min, rt, 1 h; (b) Boc2O, DMAP, DCM, rt, 48 h; (c) NBS, DCM, rt, 1 h; (d) bis(pinacolato) diboron, KOAc, 1,4-dioxane, water, 80°C for 16 h (e) (t-Bu)3PHBF4, Pd2 (dba)3, 90°C, 2 h; (f) HCl in 1,4-dioxane 4N, 1,4-dioxane, rt, 16 h; (g) HATU, corresponding carboxylic acid, DIPEA, DMF, rt, 16 h; (h) 23, BBr3, DCM, 0°C, 48 h.
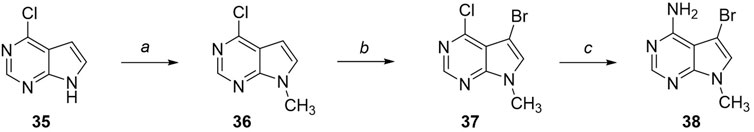
FIGURE 4. Synthesis of the coupling reagent 5-Bromo-7-methyl-7H-pyrrolo [2,3-d]pyrimidin-4-amine. Reagents and conditions: (a) 4-chloro-7H-pyrrolo [2,3-d]pyrimidine, Cs2CO3, CH3I, DMF, 1 h, rt; (b) NBS, DCM, 0°C, 1 h, rt, 3 h; (c) NH3 aq 30%, 120°C, 48 h.
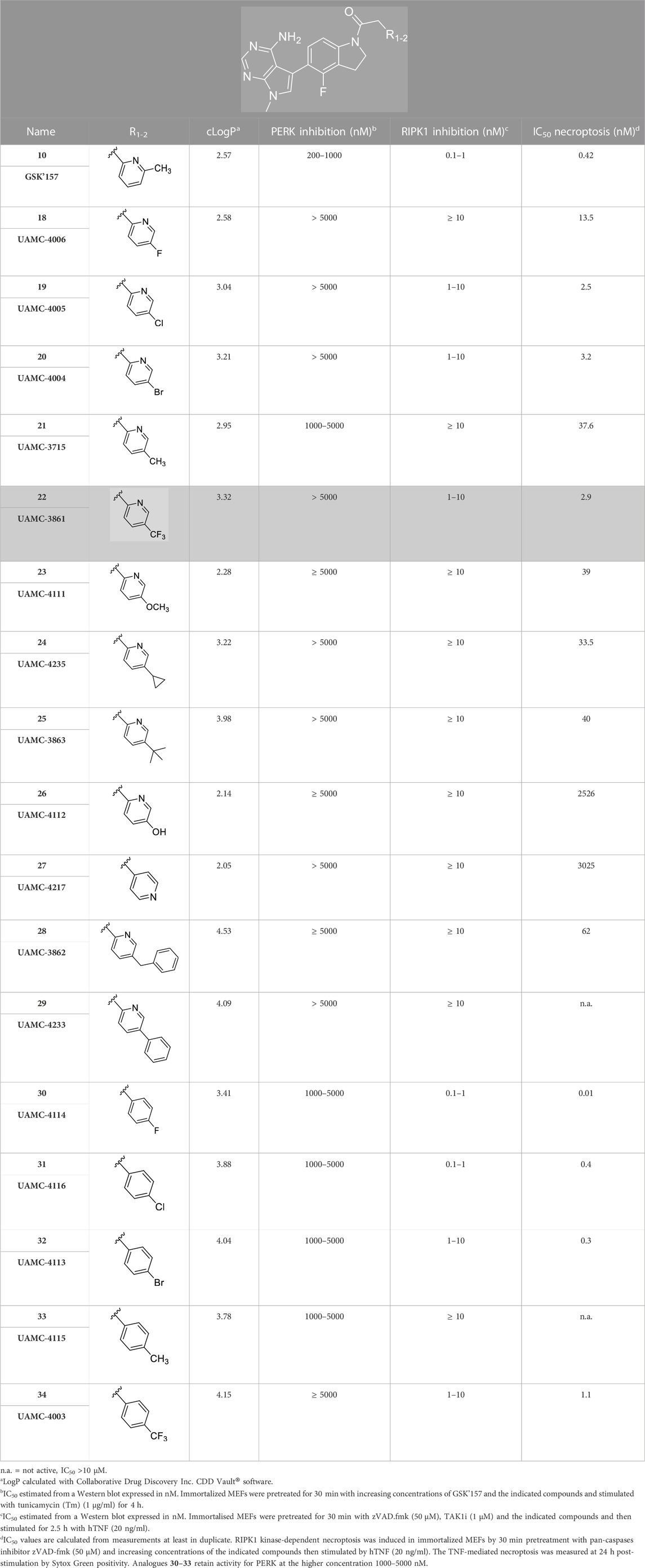
TABLE 1. Structures of the novel synthesised analogues and their corresponding IC50.
The compounds were first tested for their abilities to inhibit PERK and RIPK1 enzymatic activities in mouse embryonic fibroblasts (MEFs). To do so, we monitored, by immunoblot, the effects of the compounds on the autophosphorylation of PERK and RIPK1.
Activation of PERK was induced by stimulation of the cells with Tunicamycin, as previsouly reported (Bull and Thiede, 2012). More precisely, the immortalised MEFs were pre-incubated for 30 min with the analogues 18–34 at different concentrations (0.04, 0.2, 1, and 5 µM) prior to 4 h stimulation with Tunicamycin (1 μg/mL). As shown in Figures 5A–J, 12 out of the 17 compounds did not inhibit PERK at the tested concentrations while GSK’157 inhibited the autophosphorylation of PERK in a concentration range of 200–1000 nM. Their activities were evaluated by comparing the intensity of the signal to unstimulated MEFs (considered as zero phosphorylation) and Tunicamycin stimulated cells (considered as 100% PERK phosphorylation). The 12 compounds (18–20, 22–29 and 34) that had lost their inhibitory capacity on PERK were then tested for their ability to inhibit RIPK1 as shown in Figures 6A–H. Activation and autophosphorylation of RIPK1 was induced by stimulating the cells with TNF in the presence of TAK1 inhibitor and of the pan-caspases inhibitor zVAD.fmk, a well established necroptotic trigger (Vanlangenakker et al., 2011). The MEFs were pre-incubated for 30 min with the different compounds (0.01 nM–1000 nM) and then stimulated with human TNF (20 ng/mL) for 2.5 h. Similar to the PERK assessment, IC50 values for RIPK1 inhibition were estimated as a range of concentrations, and are reported in Table 1. These experiments provide an estimation of the specificity of our analogues against PERK and RIPK1 in comparison to the benchmark GSK’157 already reported as a potent PERK and RIPK1 inhibitor.
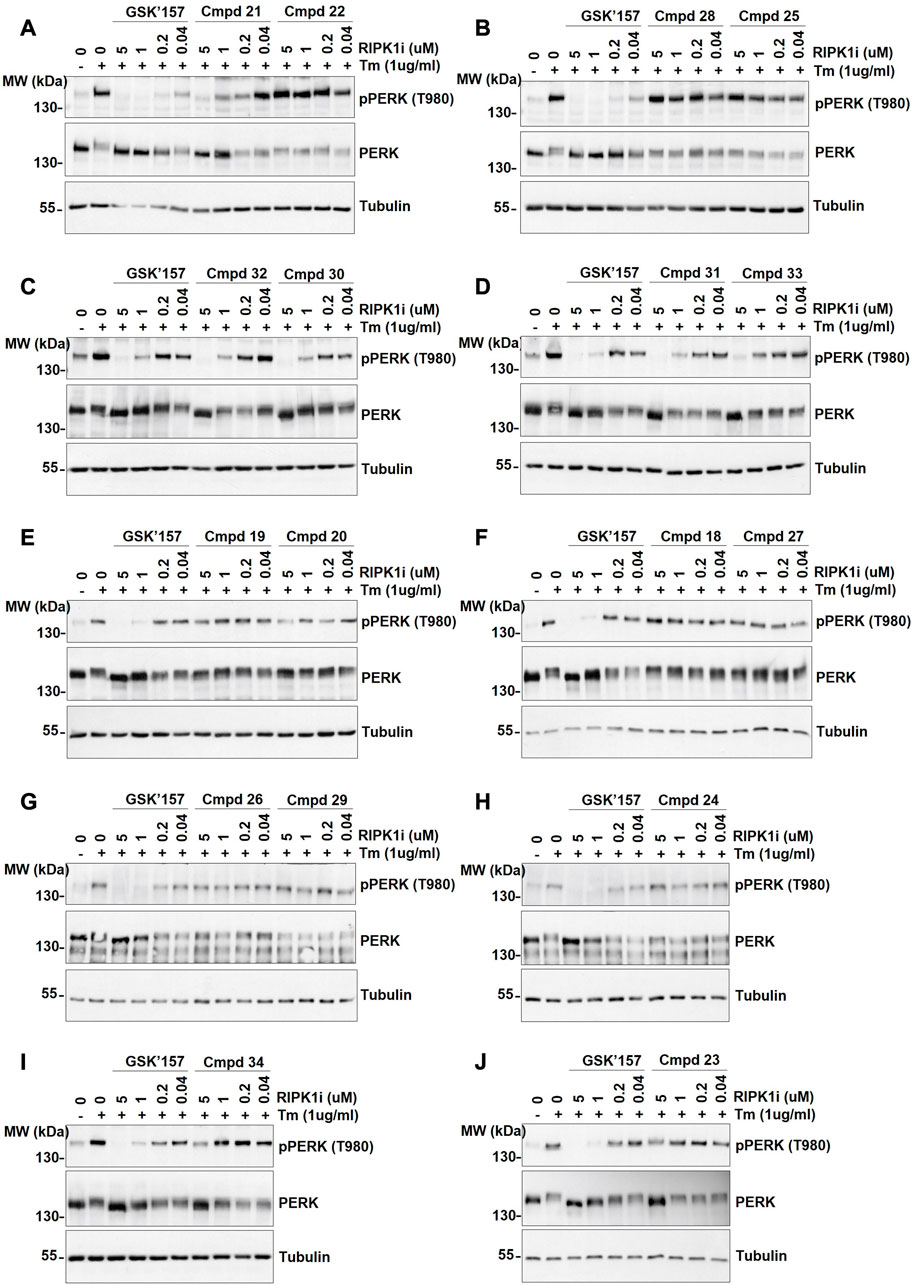
FIGURE 5. The effect of GSK’157 in comparison with compounds (cmpd) 18–34 on PERK autophosphorylation is reported in figures (A–J). Immortalized MEFs were pretreated for 30 min with increasing concentrations of GSK’157 and the indicated compounds and stimulated with tunicamycin (Tm) (1 μg/mL) for 4 h. The cell lysates were then immunoblotted as indicated.
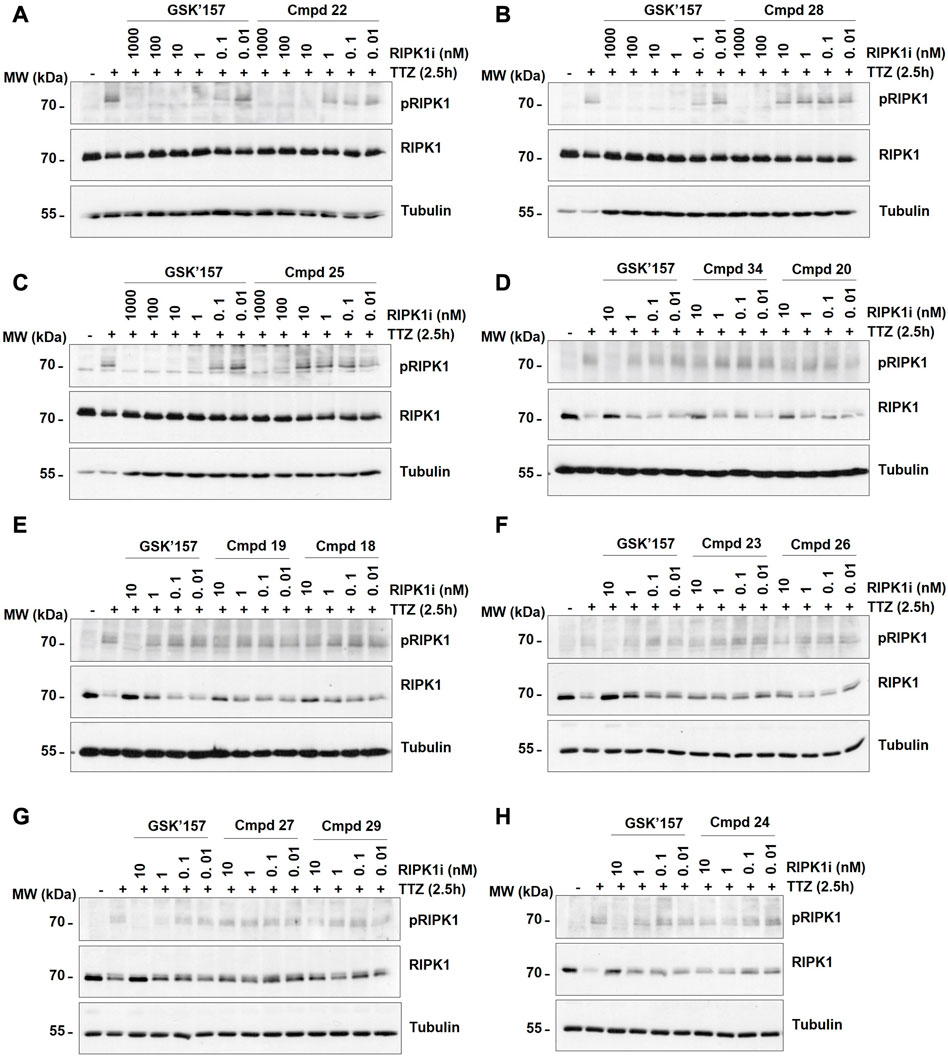
FIGURE 6. The effect of GSK’157 in comparison with compounds (cmpd) 18–20, 22–29 and 34 (not-active on PERK) on RIPK1 autophosphorylation (pS166) is reported in figures (A–H). Immortalized MEFs were pretreated for 30 min with zVAD.fmk (50 μM), TAK1i (1 μM) and the indicated compounds and then stimulated for 2.5 h with h TNF (20 ng/mL). TTZ = hTNF + TAK1i + zVAD.fmk. The cell lysates were then immunoblotted as indicated.
Consequently, the compounds were evaluated for their potential protection of RIPK1-mediated necroptosis following human TNF stimulation. The corresponding IC50 are reported in Table 1. The library was screened in MEFs cells due to their sensibility to TNF-induced necroptosis as described in the literature (Vanlangenakker et al., 2011). The IC50 values were calculated based on the cell response measured over 24 h by Sytox Green positivity.
The series of novel compounds were screened for their activity to selectively inhibit RIPK1 kinase-dependent necroptosis (Table 1) and their inhibitory effect on PERK and RIPK1 autophosporylation (Figure 5A–J and 6A-H). The novel analogues were compared with the benchmark GSK’157 which was confirmed as one of the most potent RIPK1 inhibitors currently available (IC50 = 0.42 nM). The compounds with a para-substituted-pyridinyl moiety (18–29) showed negligible PERK inhibition in contrast to GSK’157. Similar results were observed for the para-phenyl series (30–34), but to a lesser extent. The results confirmed the detrimental role of the para-substituent for PERK potency. In contrast, these compounds retained or even improved RIPK1 inhibition compared to GSK’157.
The IC50 value for necroptosis inhibition of the analogues substituted with halogen (F, Cl and Br) resulted in potent compounds. Compound 18 with the fluorine (IC50 = 13.5 nM) was less potent than the corresponding analogues 19 (IC50 = 2.5 nM) and 20 (IC50 = 3.2 nM), featuring a chlorine and bromide, respectively. The methyl analogue 21 was less potent (IC50 = 37 nM), whereas the corresponding trifluoromethyl (22, IC50 = 2.9 nM) is similar in potency to chlorine (19) and bromine (20). The introduction of methoxy (26), cyclopropyl (24) and tert-butyl (25) resulted in compounds that were equipotent to the methyl analogue 21. Polar substitutions such as the para-hydroxy (26) and a shift of the ring nitrogen of the pyridine ring (27) were detrimental for necroptosis inhibition (IC50 > 2 µM). This result confirms the high-hydrophobic nature of the RIPK1-allosteric pocket. Similarly, the insertion of bulky substituents such as benzyl and phenyl (28 and 29) negatively affects the activity of the analogues.
Furthermore, when a halogen was introduced on the corresponding para position of phenyl series, the activity increased notably with IC50 values <0.5 nM. Compound 30 with fluorine is the most potent compound of the entire series (IC50 = 0.01 nM), with 40-fold higher potency than GSK’157. To the best of our knowledge, this is the most potent necroptosis inhibitor ever reported (Martens et al., 2020; Chen et al., 2022; Shi et al., 2022). However, compared to the corresponding pyridinyl analogues (18–20), the phenyl analogues 30–32 retained some PERK inhibition, albeit at concentration higher than 1 µM which still results in excellent selectivity. Surprisingly, the introduction of a para-methyl substituent on the phenyl ring (33) resulted in a complete loss of necroptosis inhibition. The corresponding trifluoromethyl analogue (34, IC50 = 1.1 nM) was again a potent necroptosis inhibitor. This striking difference in potency between the methyl and trifluoromethyl analogue was also seen in the pyridinyl series (21 and 22), although less prominent.
Compound 22 (UAMC-3861) was preferably selected for further characterisation. We chose an analogue that showed no PERK inhibition at the highest concentration tested (5 µM) combined with a single-digit nanomolar necroptosis inhibition. Compounds 19 and 20 also meet these criteria. However, different studies suggested that the trifluoromethyl moiety improves the interaction with the hydrophobic allosteric pocket of RIPK1 (Liu and Gray, 2006; Harris et al., 2013).
The selected compound 22 together with GSK’157 and the clinically most advanced RIPK1 inhibitor GSK’772, were tested for necroptosis and apoptosis inhibition in murine MEFs and human HT-29 cells (Figures 7A–G; Table 2). Moreover, the three compounds were tested for inhibition of hRIPK1 (Table 2).
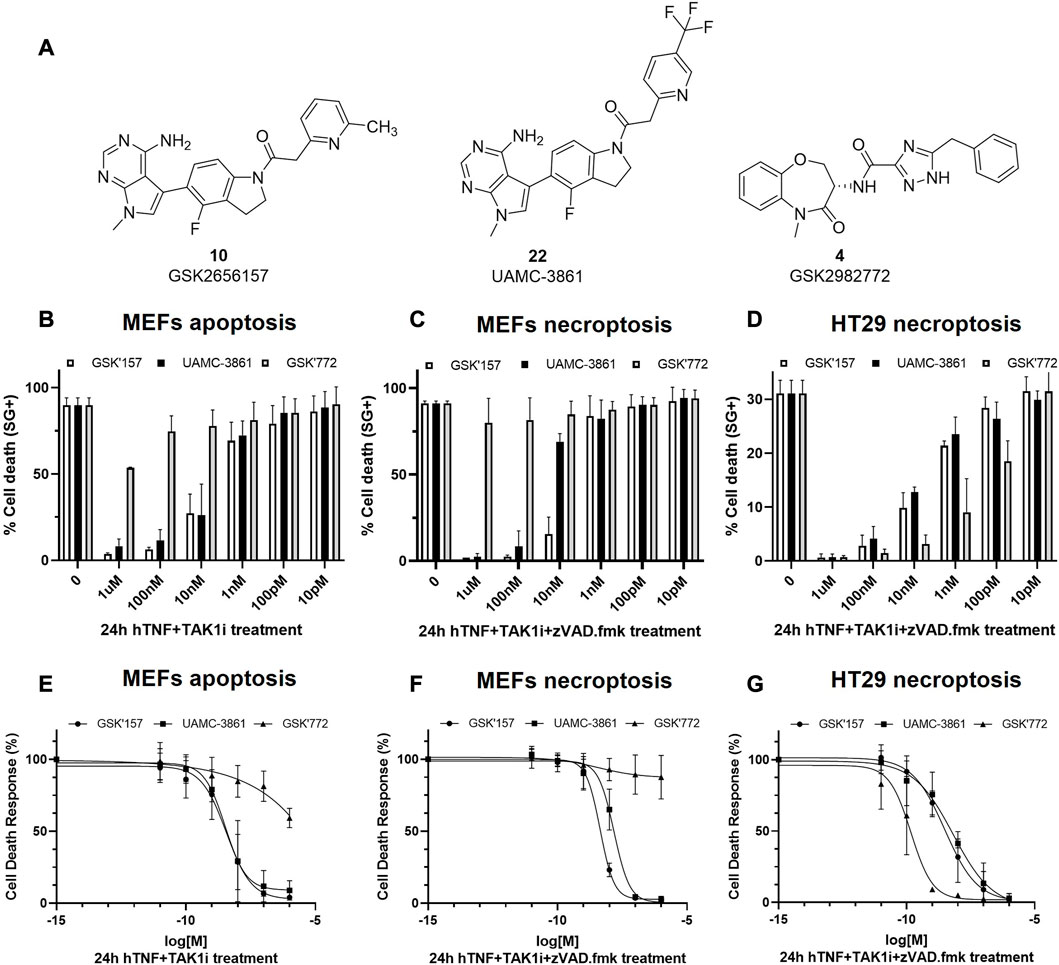
FIGURE 7. RIPK1-dependent cell death inhibition in mouse and human cells for GSK'157, UAMC-3861 and GSK'772. (A) Chemical structures of the compounds that were tested in vitro for RIPK1-dependent apoptosis and necroptosis. (B,C) MEFs and (D) HT-29 were pre-treated for 30 min with increasing concentrations of the indicated compounds. RIPK1 kinase-dependent apoptosis was induced by TAK1i + hTNF (100 pg/mL) and RIPK1 kinase-dependent necroptosis by zVAD.fmk + TAK1i + hTNF (100 pg/mL) and hTNF + zVAD.fmk (20 ng/mL). Cell death was measured over time by Sytox Green positivity (SG+) and the results are presented as mean ± SEM of three independent experiments. (E–G) MEFs and HT-29 were pre-treated for 30 min with increasing concentrations of the indicated compounds. In presence or absence of TAK1i (1 µM) and/or zVAD.
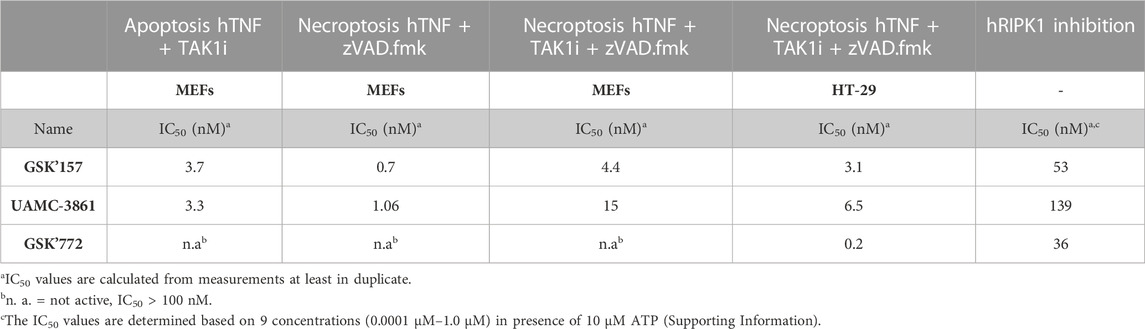
TABLE 2. Overview of the IC50 values of compounds GSK'157, UAMC-3861 and GSK'772 in mouse and human cells.
It was previously reported that the binding of hTNF to TNFR1 can induce RIPK1 kinase-dependent apoptosis and necroptosis (Jin and El-Deiry, 2006; Vanlangenakker et al., 2011). In MEFs, TNF together with the pan caspases inhibitor zVAD.fmk induces RIPK1-dependent necroptosis while TNF together with TAK1 inhibitor induces RIPK1 kinase-dependent apoptosis. We also included the conditions selected for the Western blot experiments, where necroptosis was induced by the combination of hTNF, zVAD.fmk and TAK1 inhibitor.
In RIPK1 kinase-dependent necroptosis induced with hTNF + zVAD.fmk in mouse cells (MEFs) GSK’157 and UAMC-3861 showed similar potency. As expected, when necroptosis was induced with the stronger condition in MEFs cells (hTNF + TAKi + zVAD.fmk), the IC50 values decreased from 6-fold (GSK’157) to 14-fold (UAMC-3861). As previously reported by Harris P. A. et al., GSK’772 is confirmed to be a species-specific inhibitor with no activity in mouse RIPK1-dependent necroptosis (Harris et al., 2017). To induce necroptosis in human colorectal adenocarcinoma cell line (HT-29) together with hTNF and the pan caspases inhibitor zVAD.fmk, TAK1 inhibitor is also needed. The high potency of the human necroptosis inhibitor GSK’772 (IC50 = 0.2 nM) is confirmed while GSK’157 (IC50 = 3.1 nM) and UAMC-3861 (IC50 = 6.5 nM) are less potent, but still in the same single-digit nanomolar range as observed in mouse cells. Only necroptosis conditions were tested in HT-29 since the cell line is not sensitive to RIPK1-dependent apoptosis as reported in the literature (Kong et al., 2018).
A similar trend in inhibitory potency for GSK772, GSK’157 and UAMC-3861 was also confirmed in the enzymatic assay against human RIPK1 (Table 2).
UAMC-3861 was further studied for its mode of interaction with both RIPK1 and PERK in a computational study using molecular docking (Figures 8A–F). This was done in comparison with the binding mode of GSK’157. In RIPK1, UAMC-3861 is able to form an extra methionine interaction with the trifluoromethyl-pyridinyl substituent. Moreover, the amide group of UAMC-3861 interacted with the backbone amine of Asp156 while the gatekeeper Met92 and Met67 formed methionine-aromatic interactions with the 4-fluoroindoline ring, and the trifluoromethyl-pyridinyl substituent, respectively. In addition, the docking study also confirms the high hydrophobicity of the RIPK1 allosteric pocket, since the CF3 moiety is surrounded by Met66, Met67 (C-helix), Leu70, Val75 (loop following C-helix), Leu129, Val134 (catalytic loop), and Leu159 (after DLG) as seen in Figure 8A. In PERK (Figure 8B), a stabilisation of the pyridinyl substituent was detected through the interaction between the pyridinyl ring and the backbone amine of Phe162. In addition, a methionine-aromatic interaction between the gatekeeper Met887 and the pyridinyl substituent was observed. When we compared the different poses of the substituent, in RIPK1 the pyridinyl is situated between the C-helix, the DLG-motif, and the activation loop; for PERK it is between the C-helix and the beta-sheet domain. The different interactions for GSK’157 methyl-pyridinyl and the corresponding trifluoromethyl-pyridinyl of UAMC-3861 are depicted in Figure 8C (RIPK1) and 8D (PERK). In Figures 8E,F, the different nature of the two allosteric pockets is illustrated. The allosteric pocket of PERK (green) is partly solvent-exposed, differently from the more hydrophobic one of RIPK1 (blue), which is buried inside the protein. Superposition of the docking poses in RIPK1 onto PERK and vice versa shows that neither docking pose is compatible with the other protein. In particular, the UAMC-3861 docking pose is incompatible with PERK due to steric clashes with the DFG-motif which impede the pyridinyl substituent from binding to the analogous RIPK1 allosteric binding pocket. Instead, the substituent is forced into a different, more solvent-exposed pocket, which is more unfavourable for binding of hydrophobic moieties. Concludingly, this docking study confirmed that the para-substituted pyridine ring is favourable for binding to RIPK1 whereas it is detrimental for binding to PERK.
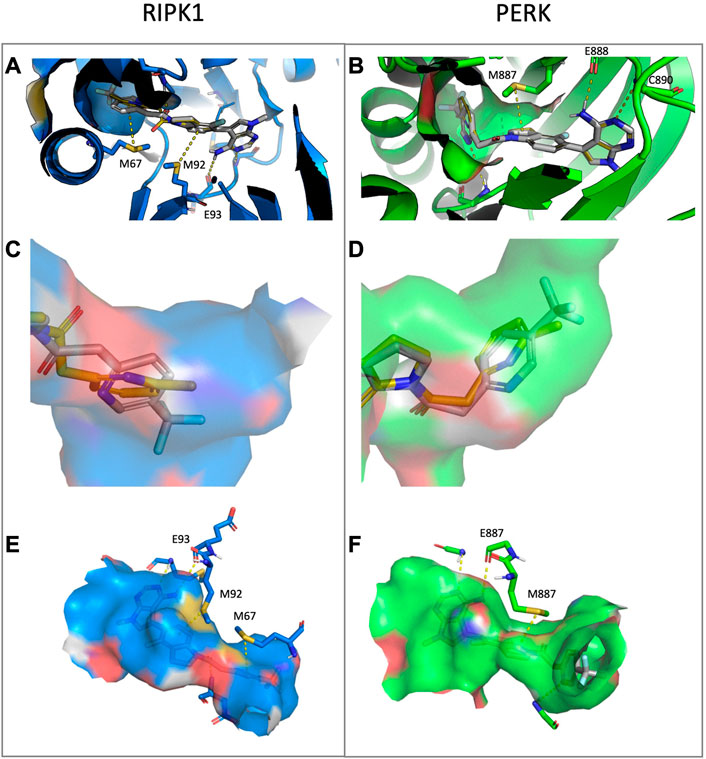
FIGURE 8. Docking of compound UAMC-3861 (grey sticks) and GSK’157 (yellow lines) in RIPK1 (blue) and PERK (green) (A–B), representation of the allosteric pockets of RIPK1 and PERK with GSK’157 and UAMC-3861 (C–D) and surface representation of the binding pocket for RIPK1 (blue) and PERK (green) and the different interactions with UAMC-3861 (E–F). (A) In RIPK1, UAMC-3861 is able to interacts with the gatekeeper Met92 and tend to form a methionine-aromatic interaction with 4-fluorindoline ring. (B) In PERK, the trifluoromethyl-pyridinyl substituent is surrounded by a less hydrophobic environment. The pyridinyl substituent forms a methionine-aromatic interaction with the gatekeeper Met887. (C) Zoom on the allosteric pocket of RIPK1 where the positions of the pyridinyl moieties of GSK’157 and UAMC-3861 are compared. (D) Zoom on the allosteric pocket of PERK where the different orientations of the pyridinyl rings are compared. (E,F) The trifluoromethyl-pyridinyl moiety occupied a different pocket in RIPK1 (E) and PERK (F). While for the latter the binding site is slightly solvent-exposed, for RIPK1 the pocket is buried, featuring a more hydrophobic profile due to the presence of three Leu, two Val, and Met (not shown in detail).
The role of TNF-mediated inflammation and cell death in different pathologies is widely recognised. Particularly, the TNF inflammatory cascade can induce inflammation and the release of cytokines through the activation of RIPK1-dependent apoptosis or necroptosis. The previously reported work in which GSK’157, a well-known PERK inhibitor, was revealed as a potent RIPK1 inhibitor opened the door to further optimisation of the chemical structure to enhance RIPK1 selectivity over PERK (Rojas-Rivera et al., 2017). Moreover, a kinome scan reported by Axten J.M. et al. For GSK’157 suggested a good selectivity profile of the compound, even though RIPK1 was not included in the panel. In this study we confirmed the hypothesis that the potency and selectivity for RIPK1 could be increased by derivatising the corresponding para-position of the GSK’157 pyridinyl ring (Axten et al., 2013; Chintha et al., 2019). In addition, based on the mode of interaction of GSK’157 this should result in a typical type II kinases inhibitor, a less explored class of RIPK1 inhibitors. We compared the potency of a para-substituted pyridinyl series (18–29) with the corresponding phenyl series (30–34). This demonstrates the importance of the pyridine ring to increase the selectivity for RIPK1 over PERK. Moreover, a smaller size of the para-substituent positively affected the potency for RIPK1. The hydrophobic nature of the RIPK1 allosteric pocket accommodating the para-substituted pyridine ring was confirmed, particularly considering the dramatic decrease of potency for the analogues having polar para substituents. To the best of our knowledge, the para-fluoro phenyl analogue (30) is the most potent necroptosis inhibitor described with an IC50 = 0.01 nM. However, since this compound retained some PERK inhibition at higher concentrations, we selected UAMC-3861 (22) for further evaluation in both murine and human cell lines. UAMC-3861 was confirmed as a highly potent compound with single digit nanomolar IC50 values for the inhibition of RIPK1-dependent necroptosis in both mouse and human cells as well as for the inhibition of RIPK1 dependent apoptosis in mouse cells. Compared with the clinical RIPK1 inhibitor GSK’772, UAMC-3861 is slightly less potent in human cells, but it has the advantage of potency in mouse cells in which GSK’772 is inactive. This suggests that UAMC-3861 will be an excellent tool compound to study RIPK1-dependent cell death in mouse models with the potential to translate to human cell lines.
The original contributions presented in the study are included in the article/Supplementary Materials, further inquiries can be directed to the corresponding author.
CS wrote the manuscript, synthesised the compounds and performed the biological characterisation. SV synthesised the compounds. KG performed the molecular docking. MV contribute to the biological characterisation. CL, GK, PV, and HW revised the manuscript. MB and KA conceived the topic, supervised the work and critically revised the manuscript.
This work was supported by the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No. 765608. This research was also funded by the University of Antwerp, BOF DOCPRO grant no.44874 and FWO-EOS Research project no. 36579. Research in the group of MB is financially supported by the Vlaams Instituut voor Biotechnologie (VIB), by Ghent University (iBOF ATLANTIS), by grants from the Fonds voor Wetenschappelijk Onderzoek Vlaanderen (FWO) (G035320N, G044518N, EOS G0G6618N, EOS G0I5722N) and from the Flemish Government (Methusalem BOF09/01M00709 and BOF16/MET_V/007 - attributed to P. Vandenabeele).
We would like to thank Frontiers in Chemistry for allowing us to share our research in Frontiers in Chemistry: Rising Stars 2022.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2023.1160164/full#supplementary-material
Axten, J. M., Medina, J. R., Feng, Y., Shu, A., Romeril, S. P., Grant, S. W., et al. (2012). Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro- 1H-indol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J. Med. Chem. 55, 7193–7207. doi:10.1021/jm300713s
Axten, J. M., Romeril, S. P., Shu, A., Ralph, J., Medina, J. R., Feng, Y., et al. (2013). Discovery of GSK2656157: An optimized PERK inhibitor selected for preclinical development. ACS Med. Chem. Lett. 4, 964–968. doi:10.1021/ml400228e
Berger, S., Harris, P., Nagilla, R., Kasparcova, V., Hoffman, S., Swift, B., et al. (2015). Characterization of GSK′963: A structurally distinct, potent and selective inhibitor of RIP1 kinase. Cell Death Discov. 1, 15009. doi:10.1038/cddiscovery.2015.9
Bull, V. H., and Thiede, B. (2012). Proteome analysis of tunicamycin-induced ER stress. Electrophoresis 33, 1814–1823. doi:10.1002/elps.201100565
Chen, K. W., Demarco, B., Ramos, S., Heilig, R., Goris, M., Grayczyk, J. P., et al. (2021). RIPK1 activates distinct gasdermins in macrophages and neutrophils upon pathogen blockade of innate immune signaling. Proc. Natl. Acad. Sci. U. S. A. 118, e2101189118–e2101189119. doi:10.1073/pnas.2101189118
Chen, L., Zhang, X., Ou, Y., Liu, M., Yu, D., Song, Z., et al. (2022). Advances in RIPK1 kinase inhibitors. Front. Pharmacol. 13, 1123234. doi:10.3389/fphar.2022.976435
Chen, Y., Ke, G., Ma, Y., Zhu, Z., Liu, M., Liu, Y., et al. (2018). A synthetic light-driven substrate channeling system for precise regulation of enzyme cascade activity based on DNA origami. J. Am. Chem. Soc. 140, 8990–8996. doi:10.1021/jacs.8b05429
Chintha, C., Carlesso, A., Gorman, A. M., Samali, A., and Eriksson, L. A. (2019). Molecular modeling provides a structural basis for PERK inhibitor selectivity towards RIPK1. RSC Adv. 10, 367–375. doi:10.1039/c9ra08047c
Clark, M. A., Acharya, R. A., Arico-muendel, C. C., Belyanskaya, S. L., Benjamin, D. R., Carlson, N. R., et al. (2009). Design, synthesis and selection of DNA-encoded small-molecule libraries. Nat. Chem. Biol. 5, 647–654. doi:10.1038/nchembio.211
Degterev, A., Huang, Z., Boyce, M., Li, Y., Jagtap, P., Mizushima, N., et al. (2005). Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 1, 112–119. doi:10.1038/nchembio711
Delanghe, T., Dondelinger, Y., and Bertrand, M. J. M. (2020). RIPK1 kinase-dependent death: A symphony of phosphorylation events. Trends Cell Biol. 30, 189–200. doi:10.1016/j.tcb.2019.12.009
Dondelinger, Y., Aguileta, M. A., Goossens, V., Dubuisson, C., Grootjans, S., Dejardin, E., et al. (2013). RIPK3 contributes to TNFR1-mediated RIPK1 kinase-dependent apoptosis in conditions of cIAP1/2 depletion or TAK1 kinase inhibition. Cell Death Differ. 20, 1381–1392. doi:10.1038/cdd.2013.94
Dondelinger, Y., Delanghe, T., Priem, D., Wynosky-Dolfi, M. A., Sorobetea, D., Rojas-Rivera, D., et al. (2019). Serine 25 phosphorylation inhibits RIPK1 kinase-dependent cell death in models of infection and inflammation. Nat. Commun. 10, 1729. doi:10.1038/s41467-019-09690-0
Dondelinger, Y., Delanghe, T., Rojas-Rivera, D., Priem, D., Delvaeye, T., Bruggeman, I., et al. (2017). MK2 phosphorylation of RIPK1 regulates TNF-mediated cell death. Nat. Cell Biol. 19, 1237–1247. doi:10.1038/ncb3608
Dondelinger, Y., Jouan-Lanhouet, S., Divert, T., Theatre, E., Bertin, J., Gough, P. J., et al. (2015). NF-κB-Independent role of ikkα/ikkβ in preventing RIPK1 kinase-dependent apoptotic and necroptotic cell death during TNF signaling. Mol. Cell 60, 63–76. doi:10.1016/j.molcel.2015.07.032
Fauster, A., Rebsamen, M., Huber, K. V. M., Bigenzahn, J. W., Stukalov, A., Lardeau, C. H., et al. (2015). A cellular screen identifies ponatinib and pazopanib as inhibitors of necroptosis. Cell Death Dis. 6, e1767. doi:10.1038/cddis.2015.130
Fiser, A., and Sali, A. (2003). ModLoop: Automated modeling of loops in protein structures. Bioinformatics 19, 2500–2501. doi:10.1093/bioinformatics/btg362
Grootjans, S., Hassannia, B., Delrue, I., Goossens, V., Wiernicki, B., Dondelinger, Y., et al. (2016). A real-time fluorometric method for the simultaneous detection of cell death type and rate. Nat. Protoc. 11, 1444–1454. doi:10.1038/nprot.2016.085
Harris, P. A., Bandyopadhyay, D., Berger, S. B., Campobasso, N., Capriotti, C. A., Cox, J. A., et al. (2013). Discovery of small molecule RIP1 kinase inhibitors for the treatment of pathologies associated with necroptosis. ACS Med. Chem. Lett. 4, 1238–1243. doi:10.1021/ml400382p
Harris, P. A., Berger, S. B., Jeong, J. U., Nagilla, R., Bandyopadhyay, D., Campobasso, N., et al. (2017). Discovery of a first-in-class receptor interacting protein 1 (RIP1) kinase specific clinical candidate (GSK2982772) for the treatment of inflammatory diseases. J. Med. Chem. 60, 1247–1261. doi:10.1021/acs.jmedchem.6b01751
Harris, P. A., King, B. W., Bandyopadhyay, D., Berger, S. B., Campobasso, N., Capriotti, C. A., et al. (2016). DNA-encoded library screening identifies benzo[b] [1,4]oxazepin-4-ones as highly potent and monoselective receptor interacting protein 1 kinase inhibitors. J. Med. Chem. 59, 2163–2178. doi:10.1021/acs.jmedchem.5b01898
Jaco, I., Annibaldi, A., Lalaoui, N., Wilson, R., Tenev, T., Laurien, L., et al. (2017). MK2 phosphorylates RIPK1 to prevent TNF-induced cell death. Mol. Cell 66, 698–710.e5. doi:10.1016/j.molcel.2017.05.003
Jin, Z., and El-Deiry, W. S. (2006). Distinct signaling pathways in TRAIL-versus tumor necrosis factor-induced apoptosis. Mol. Cell. Biol. 26, 8136–8148. doi:10.1128/mcb.00257-06
Klöditz, K., and Fadeel, B. (2019). Three cell deaths and a funeral: Macrophage clearance of cells undergoing distinct modes of cell death. Cell Death Discov. 5, 65. doi:10.1038/s41420-019-0146-x
Kong, Q., Lv, J., Yan, S., Chang, K., and Wang, G. (2018). A novel naphthyridine derivative, 3u, induces necroptosis at low concentrations and apoptosis at high concentrations in human melanoma A375 cells. doi:10.3390/ijms19102975
Lafont, E., Draber, P., Rieser, E., Reichert, M., Kupka, S., de Miguel, D., et al. (2018). TBK1 and IKKε prevent TNF-induced cell death by RIPK1 phosphorylation. Nat. Cell Biol. 20, 1389–1399. doi:10.1038/s41556-018-0229-6
Liu, Y., and Gray, N. S. (2006). Rational design of inhibitors that bind to inactive kinase conformations. Nat. Chem. Biol. 2, 358–364. doi:10.1038/nchembio799
Madhavi Sastry, G., Adzhigirey, M., Day, T., Annabhimoju, R., and Sherman, W. (2013). Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided. Mol. Des. 27, 221–234. doi:10.1007/s10822-013-9644-8
Martens, S., Hofmans, S., Declercq, W., Augustyns, K., and Vandenabeele, P. (2020). Inhibitors targeting RIPK1/RIPK3: Old and new drugs. Trends Pharmacol. Sci. 41, 209–224. doi:10.1016/j.tips.2020.01.002
Martens, S., Jeong, M., Tonnus, W., Feldmann, F., Hofmans, S., Goossens, V., et al. (2017). Sorafenib tosylate inhibits directly necrosome complex formation and protects in mouse models of inflammation and tissue injury. Cell Death Dis. 8, e2904. doi:10.1038/cddis.2017.298
Menon, M. B., Gropengießer, J., Fischer, J., Novikova, L., Deuretzbacher, A., Lafera, J., et al. (2017). P38 MAPK/MK2-dependent phosphorylation controls cytotoxic RIPK1 signalling in inflammation and infection. Nat. Cell Biol. 19, 1248–1259. doi:10.1038/ncb3614
Mifflin, L., Ofengeim, D., and Yuan, J. (2020). Receptor-interacting protein kinase 1 (RIPK1) as a therapeutic target. Nat. Rev. Drug Discov. 19, 553–571. doi:10.1038/s41573-020-0071-y
Mompea, M., Li, W., Li, J., Laage, S., Siemer, A. B., Bozkurt, G., et al. (2018). The structure of the necrosome RIPK1-RIPK3 core, a human hetero-amyloid signaling complex. Cell 173, 1244–1253.e10. doi:10.1016/j.cell.2018.03.032
Najjar, M., Suebsuwong, C., Ray, S. S., Thapa, R. J., Maki, J. L., Nogusa, S., et al. (2015). Structure guided design of potent and selective ponatinib-based hybrid inhibitors for RIPK1. Cell Rep. 10, 1850–1860. doi:10.1016/j.celrep.2015.02.052
Newton, K., Dixit, V. M., and Kayagaki, N. (2021). Dying cells fan the flames of inflammation. Sci. (80- 374, 1076–1080. doi:10.1126/SCIENCE.ABI5934
Orning, P., Weng, D., Starheim, K., Ratner, D., Best, Z., Lee, B., et al. (2018). Pathogen blockade of TAK1 triggers caspase-8–dependent cleavage of gasdermin D and cell death. Sci. (80-. ) 362, 1064–1069. doi:10.1126/science.aau2818
Rodriguez, D. A., Weinlich, R., Brown, S., Guy, C., Fitzgerald, P., Dillon, C. P., et al. (2016). Characterization of RIPK3-mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death Differ. 23, 76–88. doi:10.1038/cdd.2015.70
Rojas-Rivera, D., Delvaeye, T., Roelandt, R., Nerinckx, W., Augustyns, K., Vandenabeele, P., et al. (2017). When PERK inhibitors turn out to be new potent RIPK1 inhibitors: Critical issues on the specificity and use of GSK2606414 and GSK2656157. Cell Death Differ. 24, 1100–1110. doi:10.1038/cdd.2017.58
Saur, I. M. L., Panstruga, R., and Schulze-Lefert, P. (2021). NOD-Like receptor-mediated plant immunity: From structure to cell death. Nat. Rev. Immunol. 21, 305–318. doi:10.1038/s41577-020-00473-z
Shi, K., Zhang, J., Zhou, E., Wang, J., and Wang, Y. (2022). Small-molecule receptor-interacting protein 1 (RIP1) inhibitors as therapeutic agents for multifaceted diseases: Current medicinal chemistry insights and emerging opportunities. J. Med. Chem. 1, 14971–14999. doi:10.1021/acs.jmedchem.2c01518
Takahashi, N., Duprez, L., Grootjans, S., Cauwels, A., Nerinckx, W., Duhadaway, J. B., et al. (2012). Necrostatin-1 analogues: Critical issues on the specificity, activity and in vivo use in experimental disease models. Cell Death Dis. 3, e437. doi:10.1038/cddis.2012.176
Vanlangenakker, N., Bertrand, M. J. M., Bogaert, P., Vandenabeele, P., and Vanden Berghe, T. (2011). TNF-induced necroptosis in L929 cells is tightly regulated by multiple TNFR1 complex i and II members. Cell Death Dis. 2, e230. doi:10.1038/cddis.2011.111
Wang, K., Li, J., Degterev, A., Hsu, E., Yuan, J., and Yuan, C. (2006). Structure – activity relationship analysis of a novel necroptosis inhibitor, Necrostatin-5. Bioorg. Med. Chem. Lett. 17, 1455–1465. doi:10.1016/j.bmcl.2006.11.056
Weber, K., Roelandt, R., Bruggeman, I., Estornes, Y., and Vandenabeele, P. (2018). Nuclear RIPK3 and MLKL contribute to cytosolic necrosome formation and necroptosis. Commun. Biol. 1, 6–13. doi:10.1038/s42003-017-0007-1
Weisel, K., Scott, N. E., Tompson, D. J., Votta, B. J., Madhavan, S., Povey, K., et al. (2017). Randomized clinical study of safety, pharmacokinetics, and pharmacodynamics of RIPK1 inhibitor GSK2982772 in healthy volunteers. Pharmacol. Res. Perspect. 5, e00365. doi:10.1002/prp2.365
Xie, T., Peng, W., Liu, Y., Yan, C., Maki, J., Degterev, A., et al. (2013). Structural basis of RIP1 inhibition by necrostatins. Structure 21, 493–499. doi:10.1016/j.str.2013.01.016
Xu, D., Jin, T., Zhu, H., Chen, H., Ofengeim, D., Zou, C., et al. (2018). TBK1 suppresses RIPK1-driven apoptosis and inflammation during development and in aging. Cell 174, 1477–1491.e19. doi:10.1016/j.cell.2018.07.041
Zheng, W., Degterev, A., Hsu, E., Yuan, J., and Yuan, C. (2008). Structure–activity relationship study of a novel necroptosis inhibitor, necrostatin-7. Bioorg. Med. Chem. Lett. 18, 4932–4935. doi:10.1016/j.bmcl.2008.08.058
Keywords: RIPK1 inhibitor, necroptosis, regulated cell death, inflammation, type II kinase inhibitor, PERK (PKR-like endoplasmic reticulum kinase)
Citation: Scarpellini C, Valembois S, Goossens K, Vadi M, Lanthier C, Klejborowska G, Van Der Veken P, De Winter H, Bertrand MJM and Augustyns K (2023) From PERK to RIPK1: Design, synthesis and evaluation of novel potent and selective necroptosis inhibitors. Front. Chem. 11:1160164. doi: 10.3389/fchem.2023.1160164
Received: 06 February 2023; Accepted: 28 March 2023;
Published: 07 April 2023.
Edited by:
Clemens Zwergel, Sapienza University of Rome, ItalyReviewed by:
Piia Bartos, University of Eastern Finland, FinlandCopyright © 2023 Scarpellini, Valembois, Goossens, Vadi, Lanthier, Klejborowska, Van Der Veken, De Winter, Bertrand and Augustyns. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Koen Augustyns, a29lbi5hdWd1c3R5bnNAdWFudHdlcnBlbi5iZQ==
†These authors share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.