 Faheem Jan1,2
Faheem Jan1,2 Bo Li
Bo Li- 1Shenyang National Laboratory for Materials Science, Institute of Metal Research, Chinese Academy of Sciences, Shenyang, Liaoning, China
- 2School of Materials Science and Engineering, University of Science and Technology of China, Shenyang, Liaoning, China
- 3Institute of Catalysis for Energy and Environment, College of Chemistry and Chemical Engineering, Shenyang Normal University, Shenyang, China
HCl-assisted propane dehydrogenation (PDH) is an attractive route for propene production with good selectivity. In this study, the doping of CeO2 with different transition metals, including V, Mn, Fe, Co, Ni, Pd, Pt, and Cu, in the presence of HCl was investigated for PDH. The dopants have a pronounced effect on the electronic structure of pristine ceria that significantly alters the catalytic capabilities. The calculations indicate the spontaneous dissociation of HCl on all surfaces with a facile abstraction of the first hydrogen atom except on V- and Mn-doped surfaces. The lowest energy barrier of 0.50 and 0.51eV was found for Pd- and Ni-doped CeO2 surfaces. The surface oxygen is responsible for hydrogen abstraction, and its activity is described by the p-band center. Microkinetics simulation is performed on all doped surfaces. The increase in the turnover frequency (TOF) is directly linked with the partial pressure of propane. The adsorption energy of reactants aligned with the observed performance. The reaction follows first-order kinetics to C3H8. Furthermore, on all surfaces, the formation of C3H7 is found as the rate-determining step confirmed by the degree of rate control (DRC) analysis. This study provides a decisive description of catalyst modification for HCl-assisted PDH.
1 Introduction
Propylene is a building block of chemical industries used to produce many chemicals such as polypropylene, propylene oxide, acrylonitrile, and acrylic acid. Propylene is conventionally obtained by steam and fluid catalytic cracking of naphtha and light diesel (Sattler et al., 2014). However, these methods are thermodynamically limited, give low yield, and have complex separation of the desired products (Che-Galicia et al., 2014). Hence, searching for an alternative method is an urgent task. The large reservoir of shale gas worldwide opens a new channel to provide abundant light alkanes such as propane (Abedin et al., 2020). Under this circumstance, propane direct dehydrogenation (PDH) becomes an alternative and profitable catalytic method of producing propene.
There are two major routes of PDH: non-oxidative and oxidative dehydrogenation. The difference between these two routes is whether an oxidant, such as oxygen, is employed in the reaction. The non-oxidative route suffers from two inherent drawbacks, i) the endothermic nature makes it economically unfavorable in that it demands high energy input, and ii) the fast deactivation of the catalyst due to coke formation (Larsson et al., 1996; Lian et al., 2021). The use of oxidants in PDH overcomes the thermodynamics restraint and lowers the reaction temperature (Atanga et al., 2018). More importantly, coke formation is inhibited under oxidizing conditions (Carrero et al., 2014). However, the strong oxidizing ability of oxygen molecules induces the deep reaction of propene, which consequently reduces selectivity. The overoxidation issue is significantly lessened by using “soft oxidants,” including CO2, N2O, sulfur, and halogens, in propane dehydrogenation (Jiang et al., 2021; Zhi et al., 2022). Compared to an oxygen molecule, soft oxidants possess a much-reduced oxidizing ability that is detrimental to side reactions.
Among the soft oxidants, halogens and their halides are the most promising oxidants for propane dehydrogenation. Halogens (X) can assist the PDH pathways i) by molecular halogens (X2), ii) oxyhalogenation (X2, O2), iii) oxyhalogenation via halides (HX, O2), and iv) oxyhalogenation via molten salts of metal halides (LiX, O2). Halogens as an oxidant not only suppress the reactivity toward side reactions but also give a new active site for C-H bond activation. Therefore, the use of halogens over metal oxides and phosphate surfaces (CeO2, EuOX, CrPO4, and FePO4) shows a remarkable propene selectivity of up to 95% with 70% propane conversion at 500 °C compared to conventional catalysts (Xie et al., 2018; Zichittella et al., 2019a; Zichittella et al., 2020a).
CeO2 is extensively studied due to its activity toward HX oxidation, alkane, and alkene oxyhalogenation (Scharfe et al., 2020). Xie et al. reported the different transitions and rare metal oxides for PDH in the presence of chlorine. The study shows the instability of Fe2O3, CuO, and NiO, whereas the high reactivity of oxygen leads to COx as a major product on the RuO2 surface. Among them, a high propylene selectivity of 55% with 38% propane conversion was observed on the CeO2 surface in the presence of a halogen (Xie et al., 2018). The high conversion of oxygen is also found on the ceria surface, which leads to increased CO2 selectivity of up to 20%. The formation of CO2 can be reduced by the addition of dopants to the ceria surface. The nickel-doped ceria decreased the selectivity of CO2 to 15%. Furthermore, the nickel-doped ceria shows the highest selectivity of propylene at 80%, while the propane conversion reached 69%. In comparison, the undoped CeO2 gives only 55% selectivity with 38% propane conversion (Xie et al., 2018). The surface modification of CeO2 with doped metals makes a significant change in the catalytic performance (Su et al., 2018; Yang et al., 2018). However, an understanding of this enhancement induced by dopants is still absent, even though doped ceria has attracted much attention in oxidative dehydrogenation (Yang et al., 2018). Ceria doped with transition metals is an attractive approach that gives a balanced activity and selectivity for propane dehydrogenation. Our previous work identified the role of HBr in PDH for pristine CeO2 catalysts. This study revealed that the presence of HBr facilitated the C-H bond activation and altered the conventional reaction pathway. The apparent activation barrier is reduced with the presence of HBr (Jan et al., 2022).
In the current work, we further deepen the understanding of HCl-assisted propane dehydrogenation on metal-doped ceria. The dopant candidates include V, Mn, Fe, Co, Ni, Pd, Pt, and Cu. The geometry and electronic properties of doped ceria are carefully examined and compared. The electronic structure analysis indicated the pronounced effects induced by dopants and their implications on reactivities. The adsorption of reactants and reaction pathways are carefully examined. The thermodynamic and kinetic parameters from DFT calculations are streamed into the microkinetic simulation, and the most promising dopant candidates are proposed and verified. The current work provides a practical optimization method to further improve performance and reveal the origin of enhancement from the doping strategy.
2 Computational details
The reported work was performed using the spin-polarized density functional theory (DFT) method with the on-site Coulomb interaction (DFT + U) using the Vienna ab initio simulation package (VASP) (Kresse and Hafner, 1993; Kresse and Furthmuller, 1996). The DFT calculations were performed on the most stable CeO2 (111) surface according to the literature (Wang et al., 2013; Wang et al., 2015). A plane-wave basis set was used for valance electrons with a cutoff energy of 400 eV. The projector augmented-wave (PAW) method was used to describe ionic cores (Blöchl, 1994; Kresse and Joubert, 1999). The revised Perdew–Burke–Ernzerhof (RPBE) function was used as an exchange-correlation functional (Hammer et al., 1999). The Brillouin-zone integration was performed at a 1 × 2 × one Monkhorst–Pack k-point grid for transition-metal-modified M-CeO2 (111) surfaces (M = V, Mn, Fe, Co, Ni, Pd, Pt, and Cu). The van der Waals correction was performed by the DFT-D3 method (Grimme et al., 2010). For the description of cerium 4f, localized electrons were treated according to Dudarev et al. (1998). The value of U for this calculation was selected as 5.5 eV, as previously determined (Jan et al., 2022). The calculation was converged for all optimized structures to an accuracy of 1 × 10−5 eV, and force tolerance is 0.03 eV/Å.
A nine-atomic-layer slab with a 4 × 3 supercell in which the bottom six layers are kept fixed is used to model the ceria surface. The thickness of the vacuum was taken as 15 Å in the z direction. One cerium atom is substituted by transition metal M (M = V, Mn, Fe, Co, Ni, Pd, Pt, and Cu), as shown in Figure 1. The adsorption energy Eads was calculated as
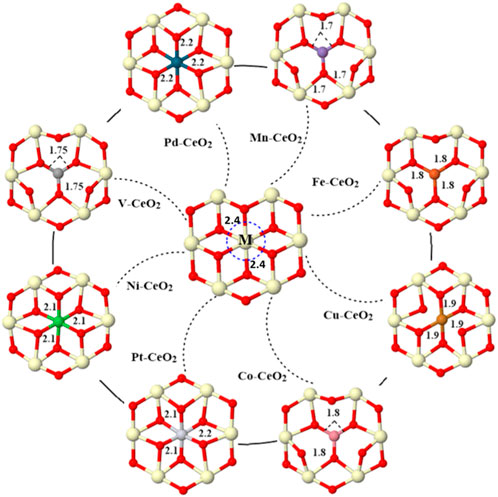
FIGURE 1. Optimized geometries of all doped surfaces. One of the surface cerium atoms is replaced with a dopant metal (Pd, Mn, Fe, Cu, Co, Pt, Ni, and V). The bond distance between the dopant and the first neighbor oxygen is indicated in Å. Cerium is light yellow, and oxygen is red.
in which Eadsorbate/slab represents the total energy of interaction species with the slab, Eslab is the individual energy of the CeO2/M-CeO2 surface, and the adsorbate represents the isolated molecules of C3H8, O2, and HCl, respectively. The climbing image nudged elastic band (CINEB) method was used to compute the reaction path (Henkelman et al., 2000). The true transition state was verified by frequency calculation on each surface.
The bonding strength is analyzed with crystal orbital Hamilton population (COHP) analysis (Dronskowski and Bloechl, 1993). The microkinetics analysis was performed by the MKMCXX simulation package (Filot et al., 2014). It is noted that microkinetic simulation is a mean-field method that discarded surface heterogeneity and dynamics evolution. Although there are some deficiencies, it is still a cost-effective way to mimic surface catalytic reactions. The Gibbs free energy with zero-point energy correction (ZPE) was calculated from DFT frequency calculations at 773 K. The ideal gas approximation was used for the entropy calculation of gas-phase species, while harmonic approximation was used for surface species. The reaction rates for adsorption/desorption species were calculated using Hertz–Knudsen kinetics. The reaction rate for elementary reactions is given by a set of ordinary differential equations as
where rj is the rate of the elementary reaction j, kj is the rate constant of j, and ci represents the concentration of species i in a given reaction. The rate constant for the adsorption step is calculated as
In the equation, P denotes the partial pressure of the gaseous molecule, A is the surface area on which the adsorbed molecule is present, m denotes the mass of the reactant, and T and kb are the temperature and Boltzmann’s constant, respectively. However, for the surface reaction, the rate constant of the forward/backward reactions is calculated as
where X represents the reacting species, ≠ shows the transition state, kr denotes the reaction rate for the forward/backward reaction, and Q and h represent the partition function and Planck’s constant, respectively.
3 Results and discussion
3.1 Structure and properties of doped ceria
The distance between the dopant and the first neighbor oxygen atom is shortened after doping, as shown in Figure 1. For pristine CeO2, the bond distance between Ce and O is 2.4 Å, while the distance between the metal dopant and oxygen is within a range of 1.7–2.2 Å after metal doping. Note also that the coordination number of the dopant is varied due to different valences. For pristine CeO2(111), Ce in the top layer has bonded with six oxygen atoms; three reside in the top layer, and the others are in the subsurface. Fe, Cu, Co, V, and Mn have less coordination, while Pt, Ni, and Pd have the same bonding oxygen atoms with a pristine surface. The introduction of dopants effectively modified the electronic structure as revealed from charge, PDOS, and COHP analysis. First, all dopant metals give electrons and become positively charged, as shown in Table 1.
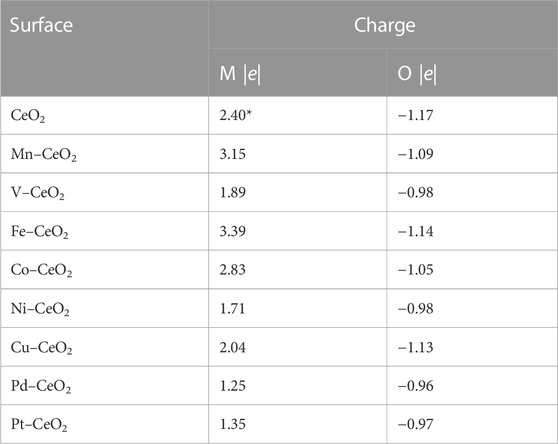
TABLE 1. Bader charge analysis of CeO2 and doped CeO2 surfaces. The * sign indicates the charge on the Ce atom in pristine CeO2.
Compared to Ce, Mn, Fe, and Co donated more electrons to surrounding atoms and became more positively charged, while the others have fewer transferred charges than Ce. PDOS analysis of surface oxygen shown in Figure 2 clearly indicated pronounced effects on the electronic structure caused by dopants. New states appeared within the gap between the top of the valence band and the bottom of the conduction band after metal doping that are not present for pristine CeO2. This observation is valid for all metal dopants except V and Mn. For the latter two, the conduction band is shifted toward the Fermi level. As surface oxygen will abstract hydrogen from propane, as discussed in the following, a doped surface will have a different behavior for C-H bond activation compared to pristine ceria, as PDOS suggests.
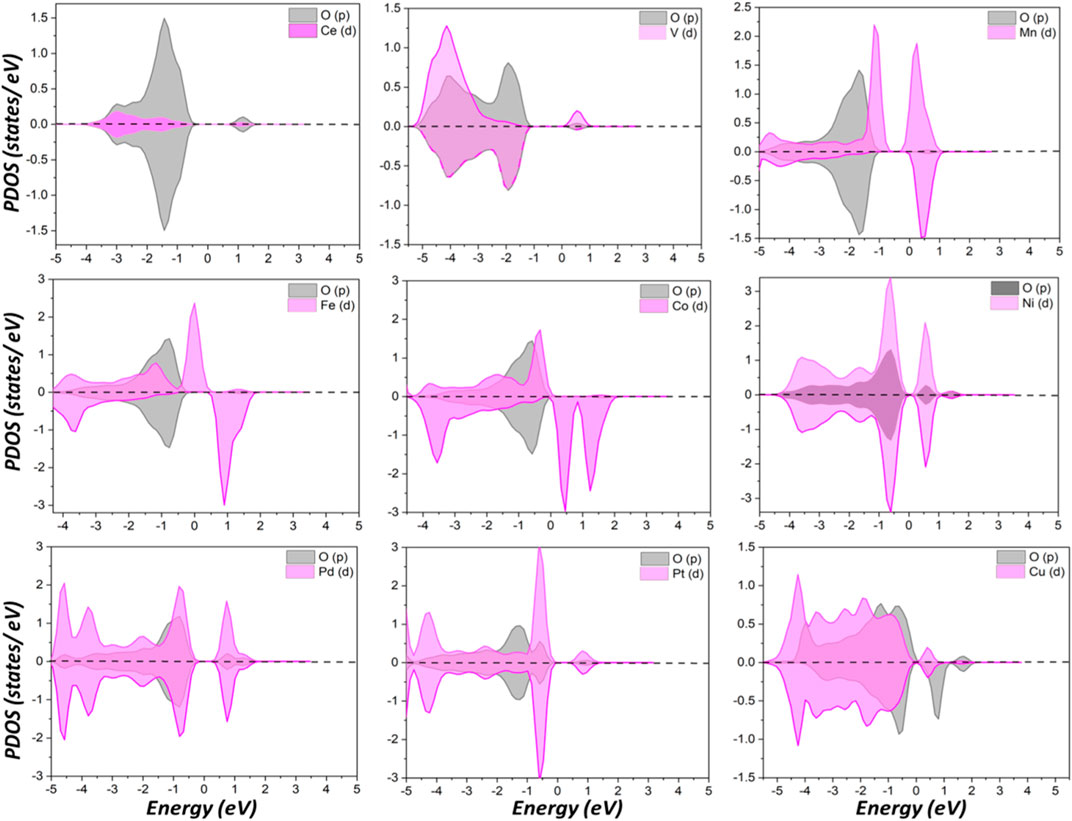
FIGURE 2. Partial density of states (PDOS) of surface oxygen p orbitals and doped metal d orbitals.
The activities of oxygen are also clearly indicated from the O p-band center analysis, as shown in Figure 3. Similar to the famous d-band center notation, the p-band center recently exhibited an ability to describe the activity of oxygen (Liu et al., 2019). As shown in Figure 3, the p-band centers of V and Mn are far from the Fermi level, which implies their inferior reactivities compared with others.
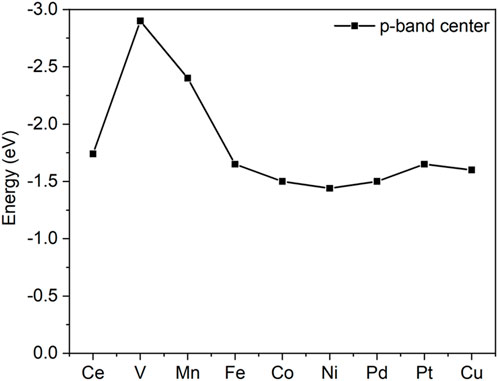
FIGURE 3. p-band centers of the surface oxygen of doped and pristine ceria.
3.2 HCl and propane adsorption
HCl is a polar molecule that is more active than a propane molecule. First, the adsorption of HCl is explored on doped and pristine CeO2. The structure optimization indicated that HCl has a spontaneous dissociation upon approaching the surface, as shown in Supplementary Figure S1. The dissociated Cl and H bind at metal and oxygen sites, respectively. HCl dissociation is a highly exothermic process, as indicated in Supplementary Table S1, and all doped surfaces have a stronger tendency for dissociation than pristine CeO2, which underscored the doping effects. The Ni-doped CeO2 has the most exothermic dissociation of −2.5 eV. The bond distance between Cl and the metal dopant is in the range of 2.2–2.76 Å, which falls into the sum of covalent radius. The strong bonding is also evident from the COHP analysis, as shown in Supplementary Figure S2, as the value of ICOHP is quite large. The addition of Cl also further engineered the electronic structure of the doped CeO2 surface due to its strong withdrawing electron ability. All metal dopants donated electrons to adsorbed Cl and became more positively charged. Hence, HCl dissociation induced a significant charge re-distribution that created more polar surface sites. The other fragment H* species became a surface hydroxyl (OH*) with a bond distance of 0.99 Å.
The adsorption of propane is also investigated, as shown in Figure 4B. As expected, the propane adsorption belongs to weak physisorption with adsorption energy in a range of −0.14 to −0.36 eV, except for Mn doping, which has a slight endothermic adsorption of 0.06 eV. Pt-doped ceria has the highest adsorption energy at −0.36 eV, and the distance between the adsorbed propane and the surface is in the range of 2.8–3.3 Å.
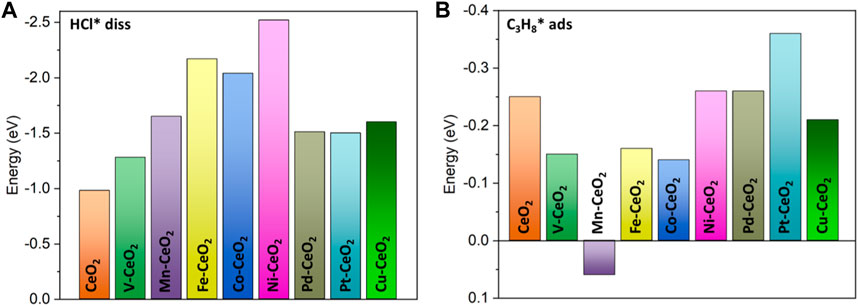
FIGURE 4. (A) Dissociation energy of the HCl molecule; (B) adsorption energy of the propane molecule.
3.3 C-H bond activation
The C-H bond activation is the core issue in propane dehydrogenation as two hydrogen abstractions are required to reach the desired product, propene. Due to its strong bonding energy, C-H activation in propane often is deemed as the descriptor for reactivity in dehydrogenation. On the chlorinated surface, the adsorbed propane proceeded via the first hydrogen abstraction from secondary carbon and formed the isopropyl species, as shown in Figure 5. The surface oxygen acts as an active site to attack the C-H bond. At the transition state, the C-H bond is stretched to be around 1.4 Å from its original bond distance of 1.1 Å. Pd-, Ni-, Pt-, and Co-doped ceria demonstrated relatively low barriers of 0.50, 0.51, 0.65, and 0.66 eV, respectively. In contrast, the energy barrier on the pristine CeO2 surface is calculated to be 0.88 eV. In contrast, V- and Mn-doped ceria possessed higher energy barriers of 1.11 and 1.20 eV that are larger than pristine CeO2. This observation is consistent with the previous oxygen p-band center analysis shown in Figure R5. The best dopants, Ni and Pd, have been suggested to have superior performance in propane dehydrogenation from experimental reports (Su et al., 2018; Xie et al., 2018).
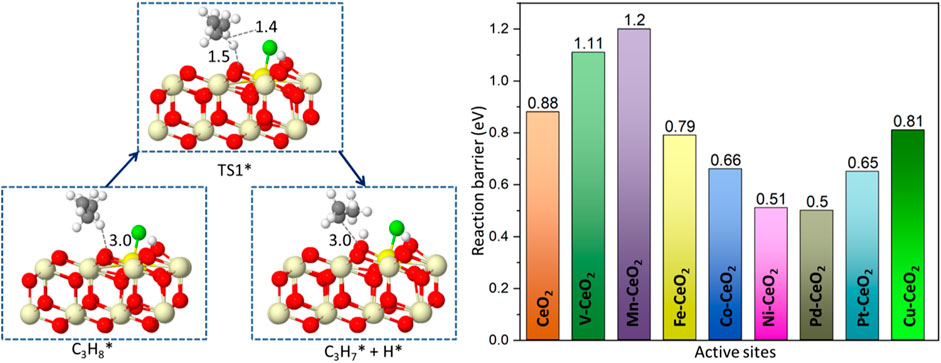
FIGURE 5. Reaction path and reaction barrier for the abstraction of a first hydrogen atom with a Gibbs free energy barrier (TS1) of all catalytic surfaces at 773 K. The yellow sphere represents the dopant position.
After the C-H bond was broken, the dissociated hydrogen moved toward the surface oxygen and formed surface hydroxyl. The other fragment, C3H7, is attracted by surface Cl* due to its electron affinity to form C3H7Cl*. This is an exothermic process, and the energy is decreased by over 1 eV for all doped ceria surfaces. C3H7Cl* is an important intermediate in the dehydrogenation process and opens a new pathway for propylene formation (Ding et al., 2013; Xie et al., 2018; Zichittella et al., 2020b). In contrast, the direct conversion of propane to propene without the involvement of C3H7Cl* is generally considered negligible in the oxyhalogenation process, which clearly indicates the importance of C3H7Cl*. (Zichittella et al., 2017; Xie et al., 2018; Zichittella et al., 2018; Zichittella et al., 2019b). The newly formed C3H7Cl* is located above the surface at a distance in a range of 2.5–3.3 Å, as shown in Supplementary Figure S4.
3.4 Propene formation
The generated C3H7Cl* will further undergo dehydrogenation, as shown in Figure 6. The C3H7Cl* dissociation leads to the formation of propylene, as shown in Figure 6. A two-step dissociation is calculated on all surfaces. The first step starts with the removal of a hydrogen atom from the primary carbon and leads to the formation of a C3H6Cl* species. This elementary step proceeds with an energy barrier, as indicated in Figure 6. Among examined doped ceria, Ni, Cu, Pd, and Co have the lowest barriers that are no more than 0.5 eV. It is noted that the barrier associated with the second hydrogen abstraction is smaller than that of the first hydrogen abstraction, as shown in Figure 6. It was also found that Ni-doped ceria possessed the lowest barriers for two successive hydrogen abstractions among investigated dopants. Compared to pristine CeO2, all doped catalysts decreased the second hydrogen abstraction barrier and showed a better catalytic ability. In the next step, C3H6Cl* species proceed via a spontaneous dissociation with no barrier, and the detached Cl* rebounded with the surface. This step is exothermic for all investigated doped ceria, as indicated in Supplementary Table S1. A catalytic cycle is completed after the targeted product C3H6 release, as shown in Scheme 1. Overall, the key intermediate, C3H7Cl, was a bridge between C-H bond activation in propane and propene formation, as illustrated in Scheme 1. The doped ceria shows a remarkable reduction of hydrogen abstraction barrier compared with the pristine sample.
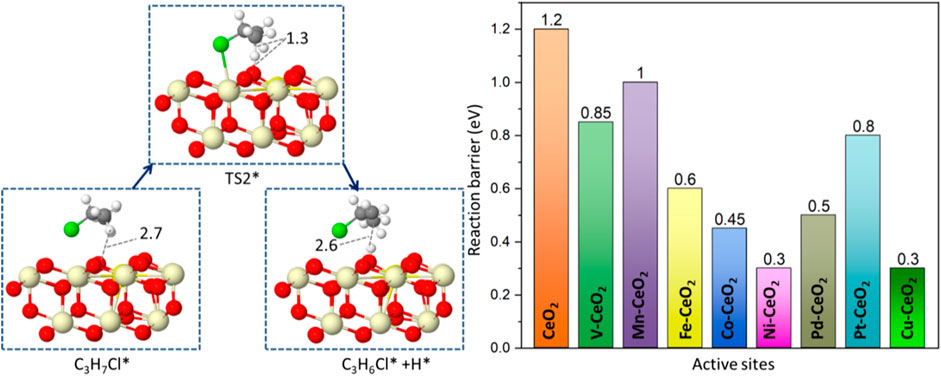
FIGURE 6. Reaction path and reaction barrier for the removal of the second H atom from C3H7Cl* with the Gibbs free energy barriers (TS2) of all catalytic surfaces.
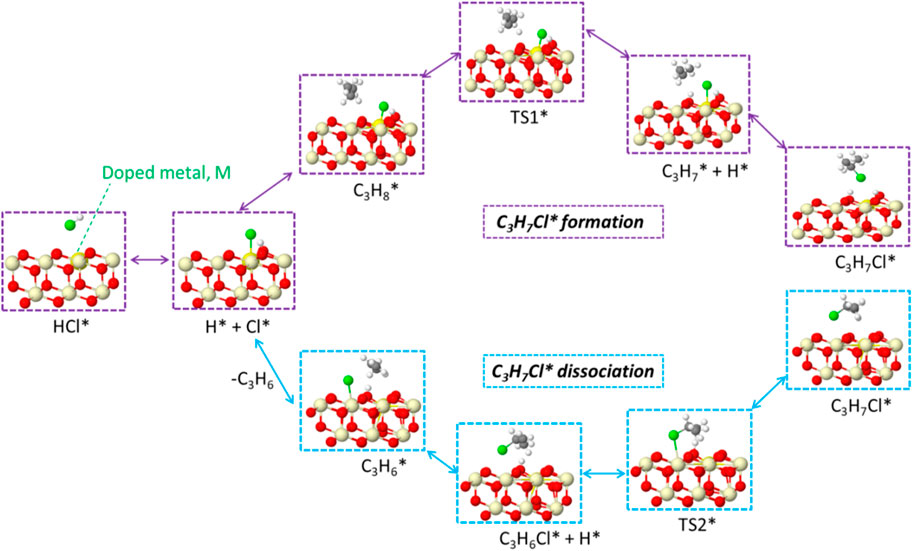
SCHEME 1. Schematic representation of the elementary steps for PDH.
The surface oxygen sites are covered with H-atoms after dehydrogenation. The interactions between surface oxygen and hydrogen lead to water formation, as shown in Supplementary Figure S7, with a low barrier of 0.1 eV. After water release, an oxygen vacancy site is formed that will be subsequently filled by oxygen molecule dissociation.
4 Microkinetics simulations
Microkinetics simulation was performed based on DFT calculations to determine the influence of different reaction conditions on the reaction kinetics and evaluate the most favorable catalytic surface. The main elementary steps are divided into adsorption, desorption, and surface reactions, as listed in Supplementary Table S2. From DFT calculations, the Gibbs free energy, rate constant, and reaction order are calculated at 773 K and 1 bar. For all elementary steps, the rate constants with pre-factors are listed in Supplementary Table S2 of the supporting information file.
The relationship between the reaction rates with the partial pressure of the reactant is shown in Figure 7. The investigated catalysts exhibited different dependencies on reactant pressure. Pd-, Ni-, Pt-, and Co-doped ceria have a strong positive correlation of TOF with pressure. TOF has a sharp increase with the pressure increase, while the other catalysts have a rather mild TOF increase.
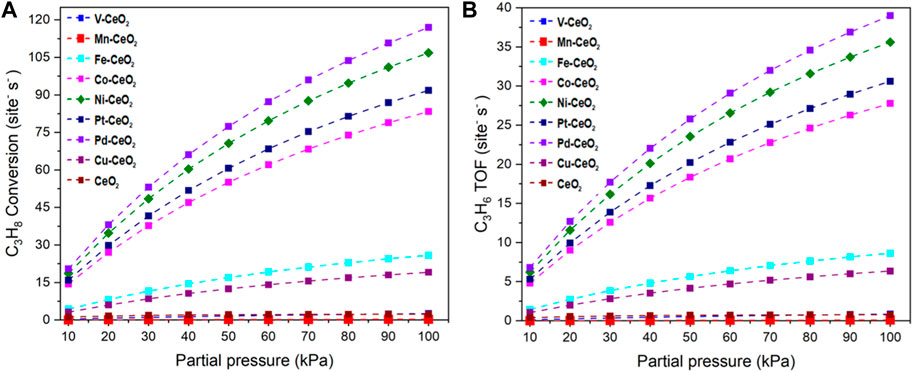
FIGURE 7. (A) C3H8* conversion rate. (B) C3H6 formation TOF under different partial pressures of propane.
The adsorption of propane and HCl regulates the performance of investigated catalysts, as shown in Figure 8. From the heat map, the modest binding energies of propane and HCl yield the best performance, which is around −0.3 eV for both cases. It is noted that Ni, Pt, and Pd possessed a bigger TOF than the other doped and pristine CeO2 samples. This is consistent with previous electronic structure and pathway analysis. DRC analysis was performed to identify the critical step among the elementary steps. The DRC coefficient for C3H7* formation was found to be unity for all investigated catalysts, as shown in Supplementary Table S3.
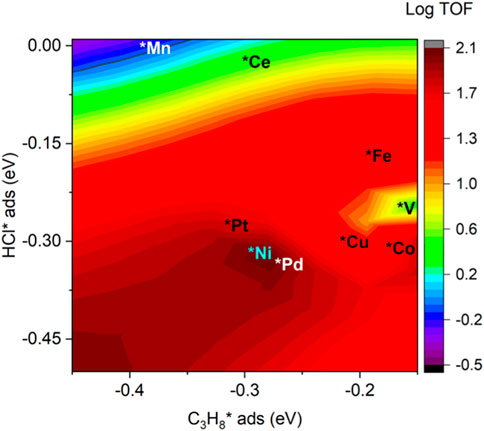
FIGURE 8. Relationship between TOF (C3H8 conversion) and the adsorption energies of HCl and C3H8.
Therefore, it is verified that the formation of C3H7* is the rate-determining step. C3H7* is formed by the abstraction of the first hydrogen from a secondary carbon atom. Further analysis indicates that the reaction follows the first-order kinetics to C3H8, which further confirms that the reaction kinetics are directly linked with the propane concentration.
5 Conclusion
In conclusion, the modification of ceria with metal dopants for PDH in the presence of HCl was studied with DFT-based microkinetics simulation. The charge analysis clearly indicated that dopants caused the charge transfer and created local polarization that increased the reactivities accordingly. The surface oxygen is identified as the active site to abstract hydrogen, and its activities are tuned by different dopants. The reaction starts with the spontaneous dissociation of the HCl molecule. The propyl species spontaneously attacks the adsorbed Cl* and gives the intermediate C3H7Cl* that, upon dissociation, results in the formation of propene. The catalytic performance of Ni- and Pd-CeO2 is found to be very promising for PDH. The TOF of propane conversion and propene formation is enhanced and directly related to the partial pressure of propane. DRC coefficient is found as unity for the formation of C3H7*, which confirmed that the first hydrogen removal is a rate-determining step on all catalytic surfaces. The reaction follows the first-order kinetics to C3H8. Overall, the current study provides unique insight into the reaction mechanism of HCl-assisted propane dehydrogenation and paves the way for future optimization.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding authors.
Author contributions
FJ: investigation and writing; MY: validation; NZ: formal analysis; XS: supervision; BL: conceptualization, writing, and supervision.
Funding
This work is supported by National Natural Science Foundation of China (22172100), the ShenYang Normal University (BS202208), and The Program for Excellent Talents in Shenyang Normal University. It is also supported by the Fundamental Research Funds for the Central Universities, China.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2023.1133865/full#supplementary-material
References
Abedin, M. A., Kanitkar, S., Bhattar, S., and Spivey, J. J. (2020). Mo oxide supported on sulfated hafnia: Novel solid acid catalyst for direct activation of ethane and propane. Appl. Catal. a-Gen 602, 117696. doi:10.1016/j.apcata.2020.117696
Atanga, M. A., Rezaei, F., Jawad, A., Fitch, M., and Rownaghi, A. A. (2018). Oxidative dehydrogenation of propane to propylene with carbon dioxide. Appl. Catal. 220, 429–445. doi:10.1016/j.apcatb.2017.08.052
Blöchl, P. E. (1994). Projector augmented-wave method. Phy. Rev. B 50 (24), 17953–17979. doi:10.1103/physrevb.50.17953
Carrero, C., Schlögl, R., Wachs, I., and Schomaecker, R. (2014). Critical literature review of the kinetics for the oxidative dehydrogenation of propane over well-defined supported vanadium oxide catalysts. Acs. Catal. 4 (10), 3357–3380. doi:10.1021/cs5003417
Che-Galicia, G., Quintana-Solórzano, R., Ruiz-Martínez, R. S., Valente, J. S., and Castillo-Araiza, C. O., (2014). Kinetic modeling of the oxidative dehydrogenation of ethane to ethylene over a MoVTeNbO catalytic system. Chem. Engr. 252, 75–88. doi:10.1016/j.cej.2014.04.042
Ding, K., Metiu, H., and Stucky, G. D. (2013). Interplay between bromine and iodine in oxidative dehydrogenation. ChemCatChem. 5 (7), 1906–1910. doi:10.1002/cctc.201200913
Dronskowski, R., and Bloechl, H. (1993). Crystal orbital Hamilton populations (COHP): Energy-resolved visualization of chemical bonding in solids based on density-functional calculations. J. Phys. Chem. 97 (33), 8617–8624. doi:10.1021/j100135a014
Dudarev, S. L., Botton, G. A., Savrasov, S. Y., Humphreys, C. J., and Sutton, A. P. (1998). Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys.l Rev. B 57 (3), 1505–1509. doi:10.1103/physrevb.57.1505
Filot, I. A., Santen, Van, and Hensen, E. J. (2014). The optimally performing Fischer–Tropsch catalyst. Angew. Chem. Int.Ed. 126 (47), 12960–12964. doi:10.1002/ange.201406521
Grimme, S., Antony, J., Ehrlich, S., and Krieg, H. (2010). A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132 (15), 154104. doi:10.1063/1.3382344
Hammer, B., Hansen, L. B., and Norskov, J. K. (1999). Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functionals. Phys. Chem. B Condens. Matter Matter. Phys. 59 (11), 7413–7421. doi:10.1103/physrevb.59.7413
Henkelman, G., Uberuaga, B. P., and Jónsson, H. J. T. (2000). A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113 (22), 9901–9904. doi:10.1063/1.1329672
Jan, F., Lian, Z., Zhi, S., Yang, M., Si, C., and Li, B. (2022). Revealing the role of HBr in propane dehydrogenation on CeO(2)(111) via DFT-based microkinetic simulation. Phys. Chem. Chem. Phys. 24, 9718–9726. doi:10.1039/d2cp00733a
Jiang, X., Sharma, L., Fung, V., Park, S. J., Jones, C. W., Sumpter, B. G., et al. (2021). Oxidative dehydrogenation of propane to propylene with soft oxidants via heterogeneous catalysis. Acs. Catal. 11 (4), 2182–2234. doi:10.1021/acscatal.0c03999
Kresse, G., and Furthmuller, J. (1996). Efficient iterative schemes forab initiototal-energy calculations using a plane-wave basis set. Phy. Rev. B 54 (16), 11169–11186. doi:10.1103/physrevb.54.11169
Kresse, G., and Hafner, J. (1993). Ab initio molecular dynamics for liquid metals. Phy. Rev. B 47 (1), 558–561. doi:10.1103/physrevb.47.558
Kresse, G., and Joubert, D. (1999). From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Chem. B Condens. Matter Matter. Phys. 59 (3), 1758–1775. doi:10.1103/physrevb.59.1758
Larsson, M., Hultén, M., Blekkan, E. A., and Andersson, B. (1996). The effect of reaction conditions and time on stream on the coke formed during propane dehydrogenation. Catal. 164 (1), 44–53. doi:10.1006/jcat.1996.0361
Lian, Z., Si, C., Jan, F., Zhi, S., and Li, B. (2021). Coke deposition on Pt-based catalysts in propane direct dehydrogenation: Kinetics, suppression, and elimination. Acs. Catal. 11 (15), 9279–9292. doi:10.1021/acscatal.1c00331
Liu, Z., Sun, Y., Wu, X., Hou, C., Geng, Z., Wu, J., et al. (2019). Charge transfer-induced O p-band center shift for an enhanced oer performance in LaCoO 3 film. Cryst. Eng. Comm. 21 (10), 1534–1538. doi:10.1039/c8ce01849a
Sattler, J. J., Ruiz-Martinez, J., Santillan-Jimenez, E., and Weckhuysen, B. M. (2014). Catalytic dehydrogenation of light alkanes on metals and metal oxides. Chem. Rev. 114 (20), 10613–10653. doi:10.1021/cr5002436
Scharfe, M., Zichittella, G., Paunović, V., and Perez-Ramirez, J. (2020). Ceria in halogen chemistry. Chin. J. Catal. 41 (6), 915–927. doi:10.1016/s1872-2067(19)63528-x
Su, Y.-Q., Filot, I. A., Liu, J.-X., and Hensen, E. (2018). Stable Pd-doped ceria structures for CH4 activation and CO oxidation. Acs. Catal. 8 (1), 75–80. doi:10.1021/acscatal.7b03295
Wang, Y.-G., Mei, D., Glezakou, V.-A., Li, J., and Rousseau, R. (2015). Dynamic formation of single-atom catalytic active sites on ceria-supported gold nanoparticles. Nat. Commun. 6 (1), 6511–6518. doi:10.1038/ncomms7511
Wang, Y.-G., Mei, D., Li, J., and Rousseau, R. J. (2013). DFT+ U study on the localized electronic states and their potential role during H2O dissociation and CO oxidation processes on CeO2(111) surface. J. Phy. Chem. C 117 (44), 23082–23089. doi:10.1021/jp409953u
Xie, Q., Zhang, H., Kang, J., Cheng, J., Zhang, Q., and Wang, Y. (2018). Oxidative dehydrogenation of propane to propylene in the presence of HCl catalyzed by CeO2 and NiO-modified CeO2 nanocrystals. Acs. Catal. 8 (6), 4902–4916. doi:10.1021/acscatal.8b00650
Yang, W., Li, C., Wang, H., Li, X., Zhang, W., and Li, H. (2018). Cobalt doped ceria for abundant storage of surface active oxygen and efficient elemental mercury oxidation in coal combustion flue gas. Appl. Catal. 239, 233–244. doi:10.1016/j.apcatb.2018.08.014
Zhi, S., Lian, Z., Si, C., Jan, F., Yang, M., and Li, B. (2022). A critical evaluation of the catalytic role of CO2 in propane dehydrogenation catalyzed by chromium oxide from a DFT-based microkinetic simulation. Phys. Chem. Chem. Phys. 24 (18), 11030–11038. doi:10.1039/d2cp00027j
Zichittella, G., Aellen, N., Paunović, V., Amrute, A. P., and Perez-Ramirez, J. (2017). Olefins from natural gas by oxychlorination. Angew. Chem. Int.Ed. 56 (44), 13670–13674. doi:10.1002/anie.201706624
Zichittella, G., Hemberger, P., Holzmeier, F., Bodi, A., and Perez-Ramirez, J. (2020). Operando photoelectron photoion coincidence spectroscopy unravels mechanistic fingerprints of propane activation by catalytic oxyhalogenation. J. Phys. Chem. Lett. 11 (3), 856–863. doi:10.1021/acs.jpclett.9b03836
Zichittella, G., Lüthi, J., Paunović, V., and Perez-Ramirez, J. (2020). Alkane functionalization via catalytic oxychlorination: Performance as a function of the carbon number. Energy. Tech. 8 (8), 1900622. doi:10.1002/ente.201900622
Zichittella, G., Puértolas, B., Paunović, V., Block, T., Pöttgen, R., and Perez-Ramirez, J. (2018). Halogen type as a selectivity switch in catalysed alkane oxyhalogenation. Catal. Sci. Techol. 8 (8), 2231–2243. doi:10.1039/c8cy00122g
Zichittella, G., Scharfe, M., Puértolas, B., Paunović, V., Hemberger, P., Bodi, A., et al. (2019). Halogen-dependent surface confinement governs selective alkane functionalization to olefins. Angew. Chem. Int.Ed. 58 (18), 5877–5881. doi:10.1002/anie.201811669
Keywords: propane dehydrogenation, microkinetic simulation, density functional theory, reaction mechanism, hydrogen chloride
Citation: Jan F, Yang M, Zhou N, Sun X and Li B (2023) Engineering the catalytic properties of CeO2 catalyst in HCl-assisted propane dehydrogenation by effective doping: A first-principles-based microkinetic simulation. Front. Chem. 11:1133865. doi: 10.3389/fchem.2023.1133865
Received: 29 December 2022; Accepted: 21 February 2023;
Published: 10 March 2023.
Edited by:
Jorge M. C. Marques, University of Coimbra, PortugalReviewed by:
M. Natalia D. S. Cordeiro, University of Porto, PortugalAlvaro Posada-Amarillas, University of Sonora, Mexico
Copyright © 2023 Jan, Yang, Zhou, Sun and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bo Li, Ym9saUBzeW51LmVkdS5jbg==; XiaoYing Sun, c3VueGlhb3lpbmc3OEAxNjMuY29t