
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Chem. , 27 September 2022
Sec. Electrochemistry
Volume 10 - 2022 | https://doi.org/10.3389/fchem.2022.996383
This article is part of the Research Topic Interfacial Engineering of Carbon-Based Materials for Efficient Energy Conversion View all 6 articles
 Lei Wang*
Lei Wang* Lijuan Wang
Lijuan WangArtificial photosynthesis (AP) has been proved to be a promising way of alleviating global climate change and energy crisis. Among various materials for AP, molecular complexes play an important role due to their favorable efficiency, stability, and activity. As a result of its importance, the topic has been extensively reviewed, however, most of them paid attention to the designs and preparations of complexes and their water splitting mechanisms. In fact, ligands design and preparation also play an important role in metal complexes’ properties and catalysis performance. In this review, we focus on the ligands that are suitable for designing mononuclear catalysts for water splitting, providing a coherent discussion at the strategic level because of the availability of various activity studies for the selected complexes. Two main designing strategies for ligands in molecular catalysts, substituents modification and backbone construction, are discussed in detail in terms of their potentials for water splitting catalysts.
To address the problems of climate change and energy crisis, solar energy technologies have been developing and applying for decades thanks to the abundant, renewable energy sources of Sun light. Although photovoltaic technologies can convert solar energy into electrical energy, the energy conversion and utilization are highly dependent on the weather and time of a day. The intermittent and diffuse nature of solar energy and the need for taking full advantages of Sun light promote the development of more efficient storage technologies for solar energy (Akbari et al., 2019; Liu et al., 2019; Palacios et al., 2020).
Inspiring from the natural photosynthesis process, during which one oxygen, four protons and four electrons are liberated in water oxidation phase then the protons and electrons contributed to carbon dioxide fixation in the photosystem II (PSII), artificial photosynthesis has been extensively studied and is considered as an attractive technology to produce green and sustainable energy (Whang and Apaydin, 2018; Ye et al., 2018; Dogutan and Nocera, 2019; Zhang and Reisner, 2020). In artificial photosynthesis, water splitting including oxygen-evolving reaction (OER), Eq. 1, and hydrogen-evolving reaction (HER), Eq. 2, is attractive for the solar energy utilization and storage, where OER, due to the thermodynamical and kinetical barriers, is the bottleneck of this process.
There are two major mechanistic classifications for each water splitting process: 1) OER reaction: I2M, WNA, and 2) HER reaction: ECEC, EECC, as shown in Figure 1 (Blakemore et al., 2015; Shaffer et al., 2017; Wang et al., 2019). The general two types of OER mechanisms both involve O-O bond formation, where an O-O radical coupling interaction of two metallo-oxy radicals are involved in the coupling (I2M mechanism) or a water molecular attack on an electrophilic metal-oxo or metal-oxyl (WNA mechanism) (Shaffer et al., 2017). The two types of HER mechanisms are distinguished by the initial reduction: 1) one electron and two H-M(n−1)+ react with each other to generate H2 (ECEC mechanism), 2) two electrons and the resultant M(n−2)+ can be protonated to H-Mn+, which then can react with an external proton to yield H2 (EECC mechanism) (Wang et al., 2019).
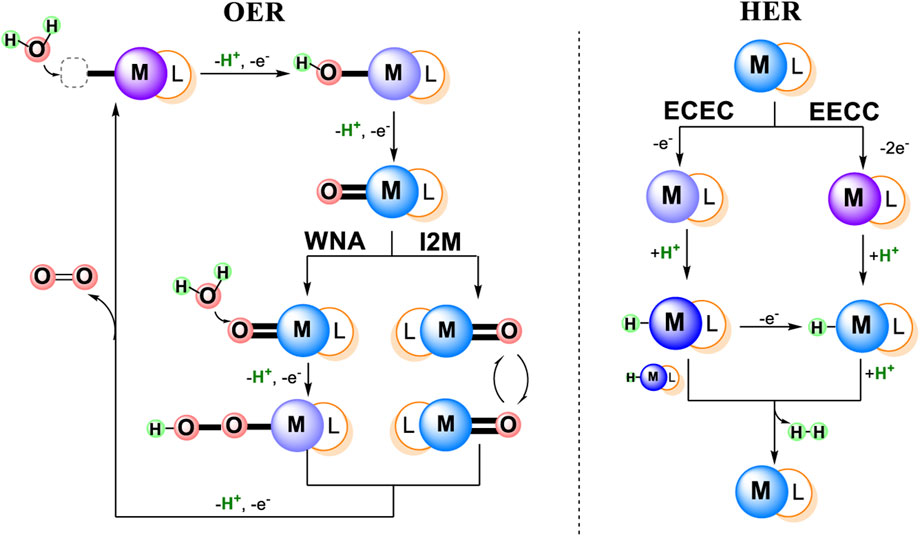
FIGURE 1. Schematic representation of general mechanisms for Water Oxidation and Water Reduction catalyzed by metal complexes.
Among catalysts for water splitting, molecular metal complexes have been paid tremendous attention due to the following advantages: 1) designable steric configuration and electronic structure; 2) tunable intrinsic activity; 3) clear catalytic mechanisms; 4) high selectivity of products 5) high atomic economy; 6) compatible with the development of various spectroscopic instruments. Over the last few decades, many efficient molecular water oxidation catalysts (WOCs) and water reduction catalysts (WRCs) were developed, such as the ruthenium catalysts (Concepcion et al., 2009b), iridium catalysts (Moore et al., 2011), manganese catalysts (Najafpour and Allakhverdiev, 2012), cobalt catalysts (Eckenhoff et al., 2013), platinum catalysts (Wang et al., 2015), iron catalysts (Mehrabani et al., 2020), copper catalysts (Liu et al., 2018), and nickel catalysts (Shaffer et al., 2017; Wang et al., 2019) etc. Thereby, many reviews are available in the literature, comparing various kinds of molecular catalysts comprehensively and summarizing catalytic mechanisms for water splitting (Meyer et al., 2017; Stolarczyk et al., 2018; Zhang and Sun, 2019a).
Several procedures are involved in developing molecular catalysts for water splitting: 1) ligand design, synthesis, and characterization; 2) metal complexes synthesis and performance characterization; 3) catalytic mechanism studies. In fact, the flourish of various ligands designed for WOCs and WRCs, as well as active metal site, establish the foundation of molecular catalysts performance. The combination of different metals and ligands will create thousands of molecular catalysts. Therefore, the modification strategies of ligands are recognized as one of the challenges to improve the intrinsic catalytic activity and stability of water splitting catalysts (WSCs).
In this review, we emphasize the modification strategies of ligands and their effect on the properties and performance of WSCs. We address here the two major designing strategies of ligands for mononuclear water splitting outlined in Figure 2B, including 1) substituents modification, and 2) backbone construction. We consider four strategies for substituents modification, including electronic effect, intermolecular interactions, steric hindrance, and anchoring groups, with providing corresponding examples for each of them. The backbone construction refers to the parent configurations that are organized by coordination number, including monodentate, bidentate, and polydentate ligands. In the end, we summarize the ligands’ designing strategies and highlight their prospects in future research of molecular complexes for artificial photosynthesis.
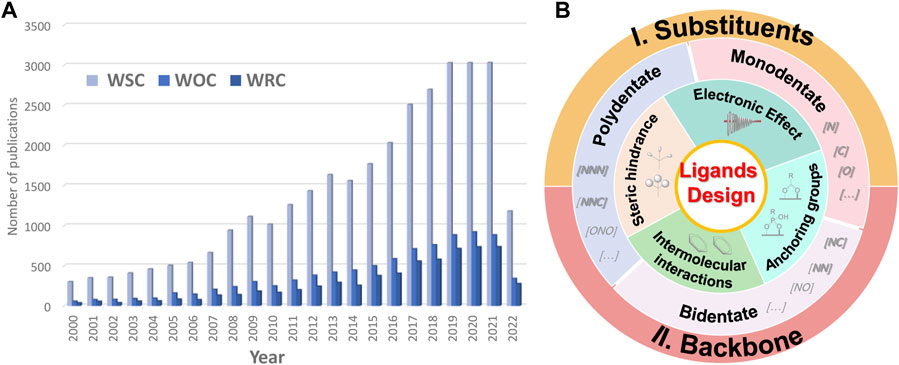
FIGURE 2. (A) Number of published papers related to molecular catalysts for water splitting from 2000 to 2022 (data obtained on 30 July 2022, from a search performed in the Web of Science for “molecular water splitting*“, “molecular water oxidation*“, and “molecular water reduction*” topics). (B) Designing strategies of ligands for molecular water splitting catalysts.
The large variety of organic substituents as well as the straightforward synthesis of both ligands and metal complexes open a large new window for the design of ligand-based WSCs. Though the same type of ligands may have different effect on WSCs’ performance depending on the mechanism of catalysts and the central active metal site, the major substituents modification strategies can be briefly summarized as: electronic effect, intramolecular interactions, steric hindrance, and anchoring groups.
Over the last decade, the exploration of electronic effect on the properties of metal complexes has been dramatically increased owning to its easy-monitored nature by various methods (Allen and Cook, 1963; Lanznaster et al., 2006; Kieltsch et al., 2010; Feng et al., 2014; Chen et al., 2015; Bellows et al., 2016; Matheu et al., 2019a; Meza-Chincha et al., 2020). It is worth to note that the electronic effect can usually be reflected by the Hammette parameter σ (σm or σp, depending on the position of substituent), which increases with the increasing of electron-withdrawing ability. Hansch et al. (1991) summarized σ values of various substituents, reporting that electron-withdrawing groups such as -CF3 and -Br possess positive values while the electron-donating groups such as -NH2 and -OEt have negative values, and σ of hydrogen (H) equals to zero. As a result, a plenty of works studied the relationships between σ and redox potential, λ, or reactivity, etc (Clark et al., 2018; Zhang et al., 2018; Suresh et al., 2022).
The electronic changes have notable effects on the electron density over metal center, resulting in changes of NMR spectra and electrochemical properties. NMR analysis from previous studies (Figures 3A,B) demonstrated that an electron-donating group causes significant up-shifting of protons in ligand(s), and an electron-withdrawing group has the opposite effect, i.e., lower the field chemical shifts of protons in NMR spectra (An et al., 2012; Duan et al., 2013; Sato et al., 2015). In general, electron-withdrawing groups decrease electronic density and stabilize the metal’s lower oxidation state, leading to more positive redox potentials as well as the overpotentials and less back-bonding into coordinated ligands. On the contrary, electron-donating groups can increase the stability of the higher oxidation state via increasing the electronic density over the metal center, resulting in more back-bonding interactions with coordinated ligands (Yoshida et al., 2010; Garrido-Barros et al., 2015). Typical examples of this case are WO catalysts [Ru (bda)L2] (complex 1a) (Duan et al., 2013; Sato et al., 2015), HER catalyst [NiP2PhN2C6H4X]2 (complex 4) (Kilgore et al., 2011a) and Pt (bpy-R2)Cl2 (complex 10) (Mcinnes et al., 1999), shown in Figure 4. The electrochemical investigation illustrated that the overpotential is drastically reduced as the electron-donating ability increases, and the redox potentials become more positive with introducing a more electron-withdrawing substituent, following a positive linear relationship between E1/2 and σ, shown in Figures 3C,D (Kilgore et al., 2011a; An et al., 2012; Duan et al., 2013; Duan et al., 2015; Mognon et al., 2015; Sato et al., 2015).
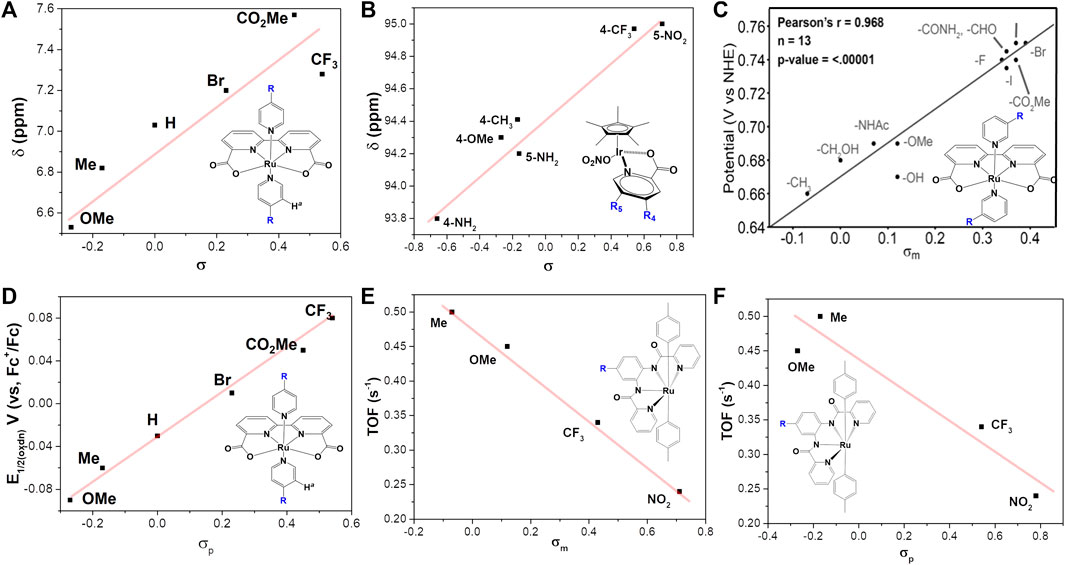
FIGURE 3. Plot of σ-Hammett parameter versus (A) 1H NMR chemical shift of ortho-proton on the pyridine ligands, data from Sato et al. (2015), (B) 13C NMR chemical shift of quaternary carbon atom on the Cp, data from Rodriguez et al. (2021), (C) Redox couples RuIII/RuII, adapt with permission from Angew. Chem. 2021,133,14625–14632. Copyright 2021 Angewandte Chemie published by Wiley-VCHGmbH, (D) The oxidation potential of [Ru (bda)-(pyR)2], Sato et al. (2015), (E,F) Turnover frequency (TOF) for complex 3, data from Abdel-Magied et al. (2017).
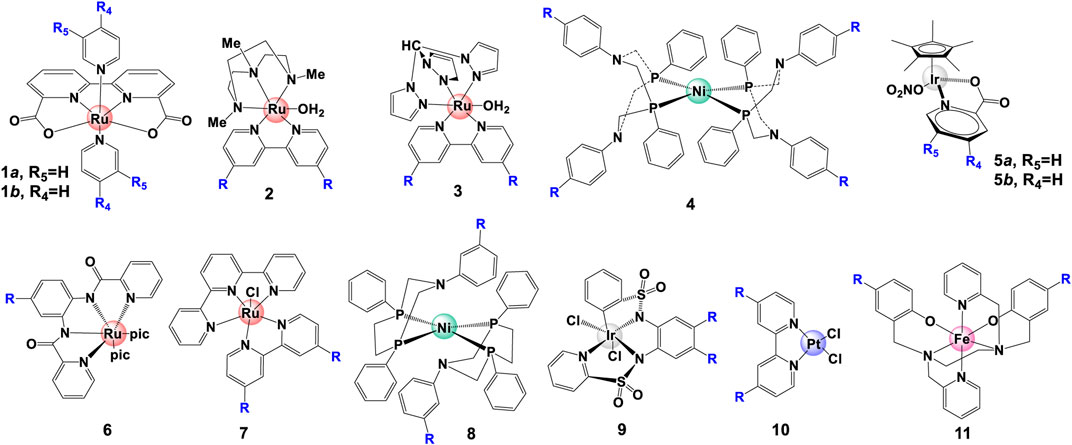
FIGURE 4. Selected water splitting catalysts with electron-withdrawing/-donating substituents.
Though the catalytic activity is determined by multiple factors, for the same series of catalysts with various electronic power, the introducing of electron donating groups usually tends to increase the catalytic activity of WOCs (Figures 3E,F), as predicted from the linear free energy relationships (Mognon et al., 2015; Abdel-Magied et al., 2017; Timmer et al., 2020). In their systematic study, Sun et al. reported how electron density affects catalytic performance of Ru-bda complex 1 (An et al., 2012; Duan et al., 2013; Sato et al., 2015; Zhang and Sun, 2019b; Corbucci et al., 2019). Proceeding by a I2M pathway, an electron-withdrawing group on 1 causes destabilization of the RuV = O species, favoring the O-O bond formation. For catalysts following WNA pathway, Rodriguez et al. (2021) reported that increasing electron-withdrawing ability can facilitate the nucleophilic attack at the IrV intermediate (a rate-determining step), therefore enhance the catalytic activity of [Cp*Ir (Xpic)NO3], as indicated by the correlations between σ and the measured TOFmax.
Yoshida et al. (2010) and Abdel-Magied et al. (2017) reported that for single-site Ru complex, a more electron-donating substituent affords a smaller oxidation potential of Ru center and enhances its catalytic activity. Besides the O2 evolution mechanism, in their case, the influence of substituents on deactivation pathway is important in changing catalytic efficiency. In another example, a HER catalyst [NiP2PhN2C6H4X]2 with electron-withdrawing -Br substituent shows a higher catalytic activity (TOF = 740 s−1) than its stronger competitor -CF3 (TOF = 95 s−1) because the reduced species can’t be protonated by the most electron-withdrawing groups in this family (Kilgore et al., 2011a).
Electron-donating/withdrawing substituents can also impact the UV-vis spectra. It is proposed that increasing the electronic power of substituents can improve the ligand field, hence affect the UV-vis absorptions. Take [Ru (bda) (py-4-R)] (complex 1a) as an example, the metal-to-ligand charge-transfer (1MLCT) band can be largely shifted to longer wavelength when a more electron-withdrawing substituent is modified on ligand (Sato et al., 2015). In fact, a linear relationship between the energy of the lowest LMCT band of complexes [Fe (bbpen-R)]ClO4 (complex 11 in Figure 4) and the Hammett parameter σ was found by Lanznaster et al. (2006). The properties of complexes with different electronic effect have been summarized in Table 1.
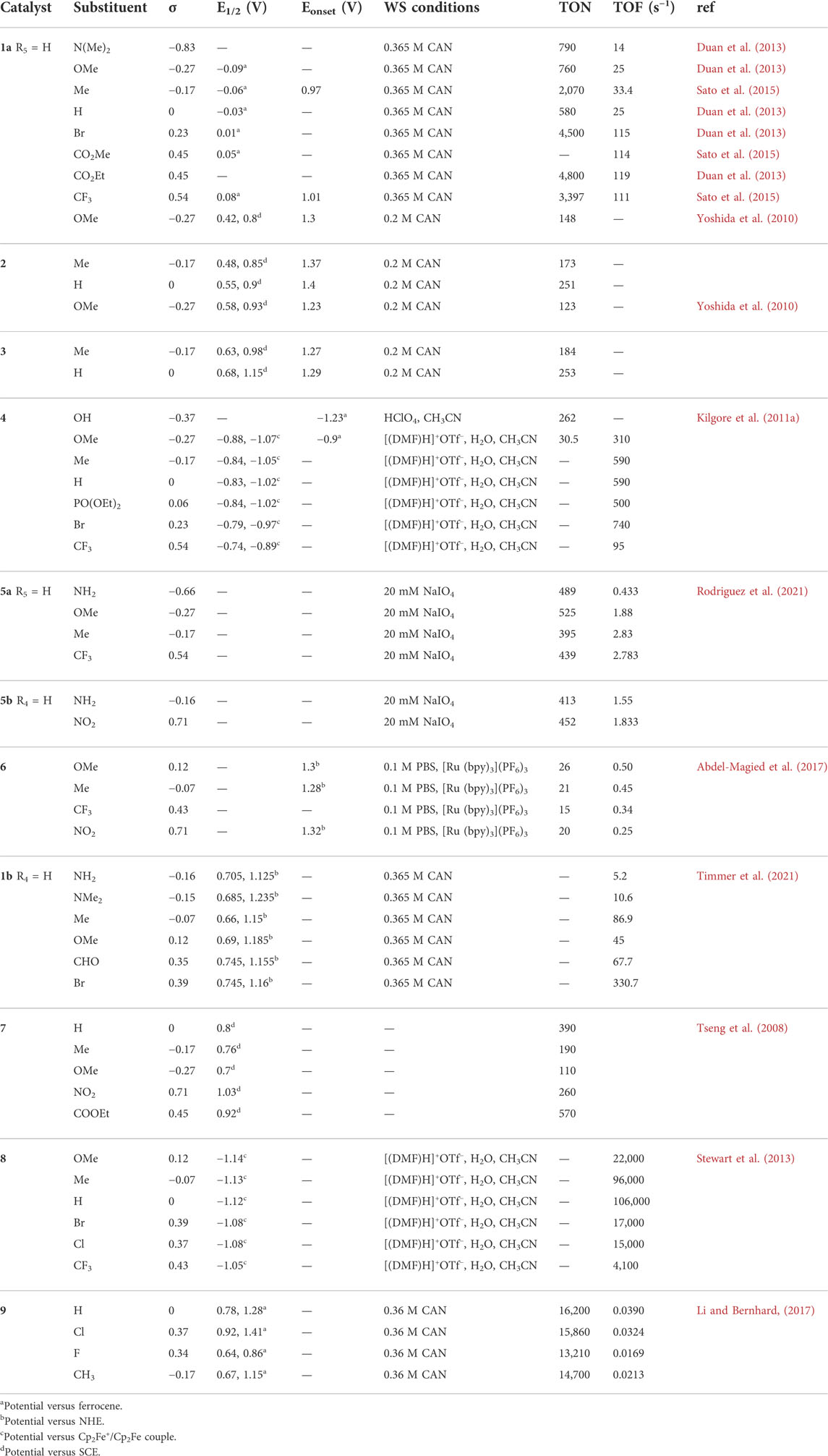
TABLE 1. Properties of metal complexes with different electron-donating/-withdrawing substituents. σ Data from ref (Hansch et al., 1991).
Intermolecular interactions are ubiquitous and often being used in pre-organizing molecular structures (Wang et al., 2019) or constructing dye-catalyst model for photocatalysis (Wang et al., 2022). In fact, non-covalent interactions in inter-catalyst coupling, such as hydrophobic effects, π—π stacking, halogen aromatic interactions, electrostatic interactions, off-set interaction etc., have shown impacts on the properties and catalytic activities of metal complexes in many studies (Zhang and Sun, 2019b).
The hydrophobic effect is often introduced by using lipophilic substituents. Generally, the hydrophobicity modification of ligands can pre-organize complexes and boost the association of two metal centers, thus improving water splitting catalytic activity. Complex 1a, complex 12, and complex 13 WO catalysts are good examples of this phenomenon. The hydrophobic modification on ligands improves the WO catalytic activity of Ru-bda from 22 s−1 to 146 s−1 (Liu et al., 2021). The octyl substituent in the carbene ligand of triazolylidene Cp*Ir-complexes induces the association of the iridium species, leading to a ∼10-fold increase of TOF (101 min−1) comparing with their methyl counterparts (TOF = 9.9 min−1), shown in Figure 5A (Corbucci et al., 2015). By exploiting the extreme hydrophobicity of semi-fluorinated side chains (Figure 5B), Chen et al. (2016) reported an enhanced efficient WOC, Co-(BimC3F8), with a TOF of 1.83 s−1, a 15-fold increase, at neutral pH without soluble cobalt, salts.
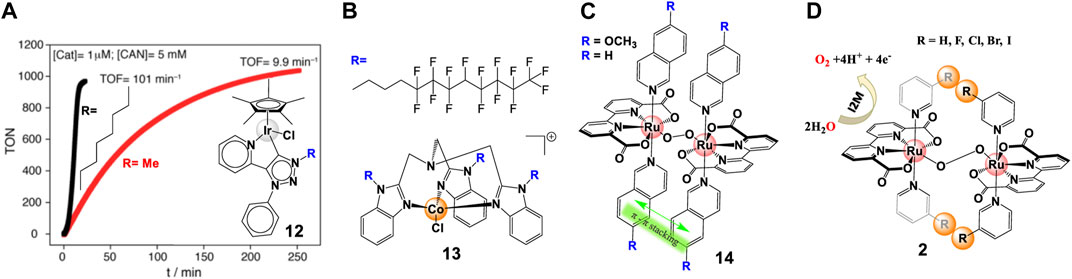
FIGURE 5. (A) TON of complex 12 at pH 1 measured by UV−vis spectroscopy, adapt with permission from ACS Catal. 2015, 5, 5, 2,714–2,718. Copyright 2015 American Chemical Society. (B) Chemical structure of complex 13. (C,D) Schematic diagram for noncovalent interactions between the axial ligands for complex 14 and complex 2.
π—π interactions were extensively used in multi-components photocatalytic system, especially in promoting the electron transfer between photosensitizers and catalysts (Wang et al., 2022). Neel et al. (2017) reviewed how to exploit non-covalent π interactions for catalyst design. In chemical-driven water splitting process, π—π stacking is employed to facilitate the intermolecular interactions and accelerate bimolecular coupling. For example, complex 14 with isoquinoline (isoq) displays an order of magnitude higher TOF of 303 s−1 than that of 32 s−1 for complex 1a with picoline (pic), as shown in Figure 5C (Duan et al., 2012b). A faster catalysis was observed by introducing MeO-isoq, which causes a more favorable π—π stacking effects in water (Richmond et al., 2014). Correlated ab initio calculations demonstrated that modulation of π—π stacking dispersion interactions can lower activation barrier, therefore a smaller driving force for the catalysis is obtained (Johansson et al., 2021).
Attempts to improve the performance of WSCs also include the introduction of halogen substituents. Xie et al. (2018) studied in detail the influence of halogen substituents on the performance of complex 1b (Figure 5D). A 10-fold enhancement of TOF (330 s−1) was found for R = I compare to R = H, which revealed that iodine can accelerate the O-O bond formation by facilitating the intermolecular interactions i.e. bimolecular coupling, due to its easy-polarization.
Electrostatic interactions were found to be another effective strategy to regulate binuclear catalysis. While attractive electrostatic interactions can facilitate inter-catalyst coupling and promote the catalytic performance, repulsive electrostatic interactions have a negative effect on the catalytic activity (Sampson et al., 2014; Pye and Mankad, 2017). In their recent study, Yi et al. (2021) prepared a family of Ru-bda catalysts (complex 15, Figure 6), functionalized with positively charged Me-bpy+ (Nmethyl-4,4′-bipyridinium) and/or negatively charged p-SO3-py- (pyridine-4-sulfonate) group, and identified the intermolecular electrostatic interactions by various methods. Complex 15c and the mixture M ([15a]: [15b] = 1:1) present 8–20 times higher TOF than complex 15a and 15b with repulsive effects. It was proved that electrostatic interactions are benefit to the formation of pre-reactive dimers, which were key intermediates in improving the catalytic activities. In fact, Richmond et al. (2014) had discovered the electrostatic effect in their earlier report, but they attributed the lowered catalytic activity to the high steric hindrance between the Me-bpy+ ligands.
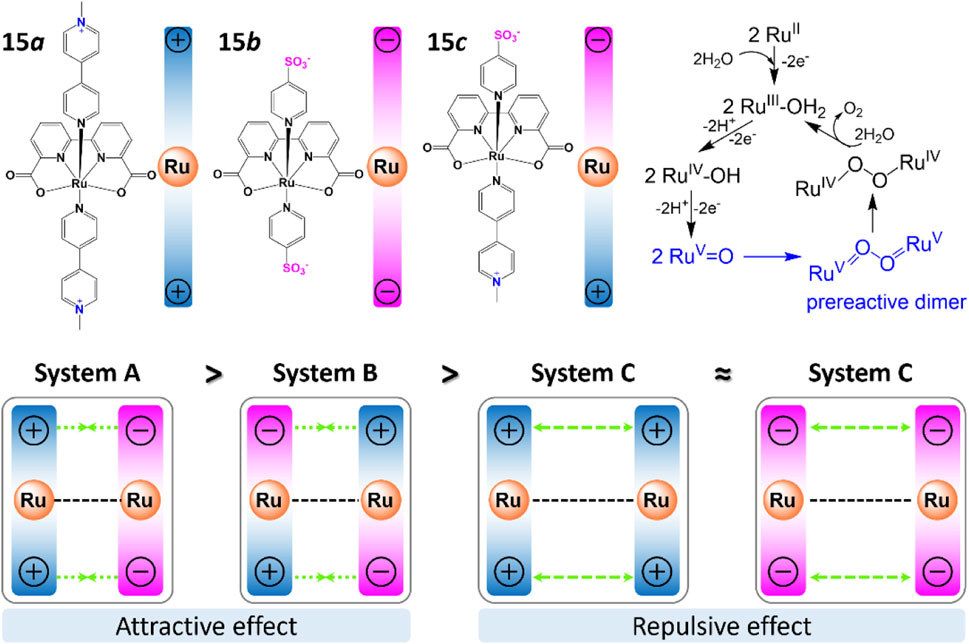
FIGURE 6. Effect of electrostatic interactions on Ru-bda catalysts, adapt with permission from J. Am. Chem. Soc. 2021, 143, 6, 2,484–2,490. Copyright 2021 American Chemical Society.
In addition to aforementioned intermolecular interactions, Timmer et al. (2021) discovered that off-set interactions that introduced by de-symmetrization of the axial ligands in complex 16 and 17 (Figure 7A) can provide enough space for the O-O bond formation and reduce reaction barrier. DFT calculations suggested that reduced kinetic barrier of the second-order O-O bond formation ensures high catalytic performance especially at low catalyst concentrations. For Ru-bda catalysts with isoq in the axis, the position of pyridine substituents is crucial for stacking. Instead of direct π—π interactions, the off-set interaction brought by bromide shorten the distance of Ru-Ru in the pre-reactive dimer.
FIGURE 7. (A) The chemical structures of complex 16 and 17, and the off-set interactions involved in Ru-bda catalyst, adapt with permission from Angew. Chem. Int. Ed. 2021, 60, 14504–14511. Copyright 2021 Angewandte Chemie International Edition published by Wiley-VCHGmbH. (B–G) Chemical structure of (B) copper complex 18 [Cu(bpy)(OH)2](Zhang et al., 2014), (C) Mn corrole complexes 19 (Gao et al., 2007), (D) nickel hangman complex 20 (Bediako et al., 2014), (E) cobalt hangman complex 21 (Dogutan et al., 2011; Neuman et al., 2020; Zhang et al., 2021), (F) cobalt corroles complex 22 (Sun et al., 2017), (G) iron porphyrins complex 23 (Graham and Nocera, 2014).
Another attractive way to facilitate the catalytic activity is to govern proton-coupled electron transfer (PCET) process by introducing proton acceptors/donators on ligands (Young et al., 2009; Wang et al., 2019; Zhang et al., 2021). Zhang et al. (2014) proposed ligand assisted PCET process for complex 18, where H on the 6 and 6′positions of bpy is replaced with hydroxyl groups, an internal base for proton transfer (Figure 7B). This directly lower the potential of complex about 200 mV, consequently resulting in its capability of driving WO to peroxide at a relatively low potential. Gao et al. (2007) designed bio-inspired manganese complex 19 with corrole ligands. The electrochemical data show that Mn corrole complexes 19 can catalyze oxidation of water to produce oxygen at quite low oxidation potentials, as indicated by its easy oxidation to high-valent states. Nocera et al. systematically studied the electrocatalytic behavior of Co/Ni hangman porphyrins complexes. They reported that owing to the pre-organization of water within the hangman cleft, the catalytic performance of these complexes (20, 21) can be dramatically boosted by employing carboxyl acid as a proton acceptor (Dogutan et al., 2011; Bediako et al., 2014; Neuman et al., 2020). Later, Sun et al. (2017) synthesized complex 22 and proved that the pendant hangman carboxyl moiety can act as intramolecular base to accelerate the APT process during the O-O bond formation. In the following up study, Nocera et al. found that the rate of catalysis of hangman iron porphyrins complexes 23 can be affected by nearly 3 orders of magnitude by improving the hanging group’s proton-donating ability (Graham and Nocera, 2014). Recently, the impact of carboxylate unites on electrocatalyzed WO process were deeply discussed by Das et al. (2021) Same as previous studies, the free carboxylic acid/carboxylate units can provide proton donor/acceptor sites through a chemically non-innocent way, hence can improve the overpotential and activity of the WO reaction dramatically.
Clearly, these intermolecular interactions offer inspirations for future design of WSCs. The comparison between modified and parent complexes have been summarized in Table 2.
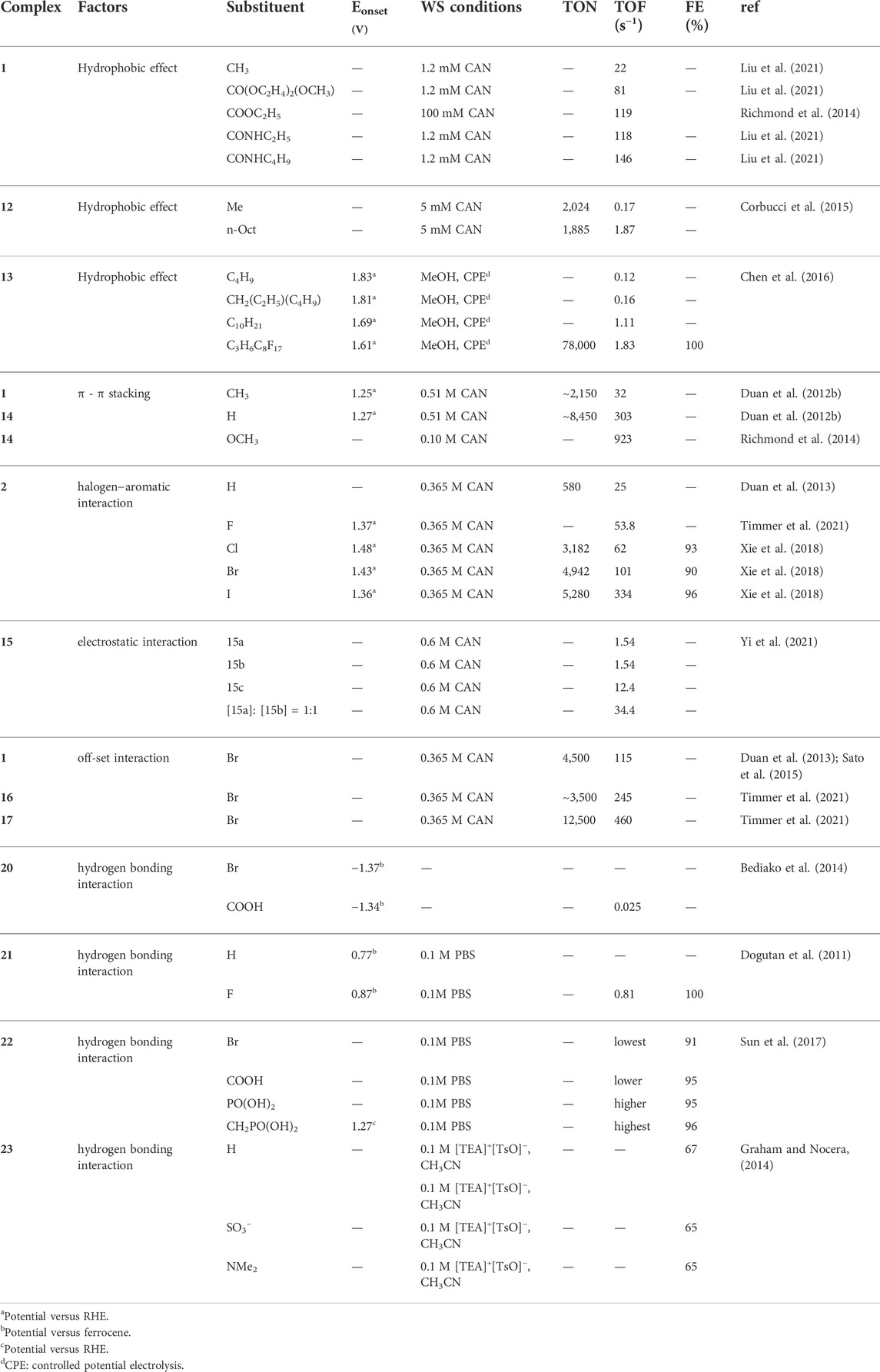
TABLE 2. Properties of metal complexes with different intermolecular interactions.
It has been proposed that steric hindrance plays an key role in the overall catalytic rate (Richmond et al., 2019). Steric hindrance can bring changes in the properties and catalytic performance for water splitting catalysts by changing the geometry and conformation (such as bond angles and bond lengths) of metal complexes. It is interesting to note that bulky group can also switch the catalytic reactivity. Smith et al. reported that with small substituents (R = H, Me), complexes 24 ([(Py2NR2)Mn(H2O)2]2+) in Figure 8 catalytically disproportionate H2O2 in aqueous solution while this reaction is shut down with a bulkier substituent (R = tBu), but becomes active for aqueous electrocatalytic H2O oxidation (Lee et al., 2014; Crandell et al., 2017).
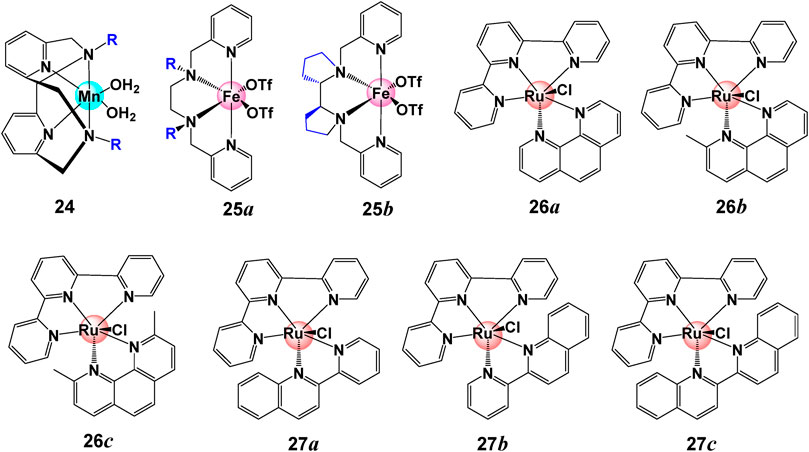
FIGURE 8. Chemical structures of complexes 24–27.
Generally, a bulky ligand can raise the activation barrier and slow down the reaction rates through steric tension in transition states or intermediates. Large substituents can shield the formation of active intermediate, hence lower the catalytic activity. Iron coordinating complex 25 with sharing a common structural topology but different geometry present different WO activity under the same condition. As indicated by L-Fe-L angle around 95–100°, the bulkier group inhibit the formation of the FeIV(O)-(μ-O)CeIV(OH) species (Fillol et al., 2011; Panchbhai et al., 2016). The steric hindrance is also observed for Ru-type complexes 26. When replacing H in the phen ligand (26a) with one (26b) or two methyl groups (26c), the activity and stability of complex is prohibited by the methyl group. The lowest TOF of 0.005 s−1 and TON of 60 were observed for complex 26c with two methyl groups, and a moderate TOF of 0.008 s−1 and TON of 155 were found for complex 26b (Kaveevivitchai et al., 2012). Similar observations were also reported for complex 27. When the benzene is proximal to Cl ligand (27b), the TOF is reduced as compared to complex 27a. The activity is fully suppressed when the bpy ligand is extended by two benzene rings, as shown in complex 27c (Kaveevivitchai et al., 2012).
Steric hindrance can also play its role by affecting the protonation reaction which, as we mentioned before, can influence water splitting activity. For example, for complex 28, the protonation reaction may occur in either 2-endo or 2-exo positions as shown in Figure 9. Clearly, the 2-endo protonation site of the Ni(I) intermediate 28–1 is favored for complex 28 to enter the catalytic cycle because the strong hydride donor abilities of the metal center can accelerate the rate of H2 elimination from 28–1. However, the bulky phosphine substituent can hinder the endo protonation of amines in these intermediates and also influence the hydride donor ability of [HNi (P2RNPh2)2]+ derivatives (Kilgore et al., 2011b; Wiese et al., 2012).
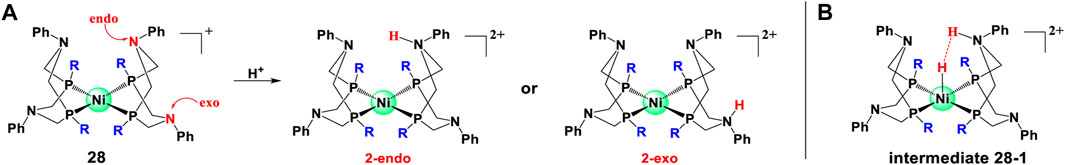
FIGURE 9. 2- endo or 2-exo protonation of pendant amines in complex 28 and important intermediate for hydrogen production, adapt with permission from Inorg. Chem. 2011, 50, 21, 10908–10918. Copyright 2011 American Chemical Society.
Ir-based WO complex 12 bearing pyridine triazolylidene ligands with hydrophobic octyl substituent has shown an enhanced activity as a consequence of the association of the iridium species. However, with studying the same complex with variable steric hindrance, Corbucci et al. (2019) discovered that a discontinuity of activity when change R from Me to Et, showing that steric group can inhibit the transfer of hydroperoxo or peroxo moiety from Ir intermediate to cerium, a process that slows oxygen evolution in cerium-driven WO process (Bucci et al., 2016).
In contrast to the halogen interaction that improves the catalytic WO activity for [Ru (bda)(R-py)2], heavy halogen atoms such as iodine (I) decreases the catalytic activity of [Ru (bda)(R-isoq)2] due to the steric hindrance of π−π overlap, demonstrating the importance of balance between polarizability and favorable π−π interactions (Xie et al., 2018). The complicated effect of steric effect was also found on Ru(II) complexes 29–31, show in Figure 10. Cyclic voltammetry and water oxidation studies illustrated that the complexes with tri-butyl-tpy ligand are easier to be oxidized because of donor attribute, hence showed boosted activity. However, these WOCs must involve a water molecule in the coordination sphere of metal to make the catalysis happen. The steric hindrance could severely inhibit the binding of water, resulting in a weakened catalytic activity when more t-butyl groups were introduced (Kaveevivitchai et al., 2012).
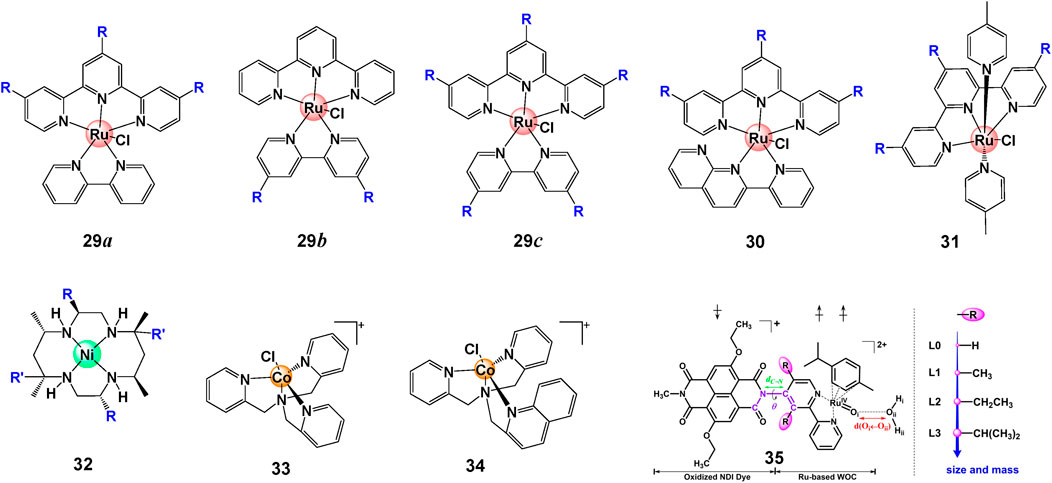
FIGURE 10. Chemical structures of complexes 29–34 and schematic structure of complexes 35, adapt with permission from ChemSusChem 2021, 14, 479–486. Copyright 2020 Published by Wiley-VCHGmbH.
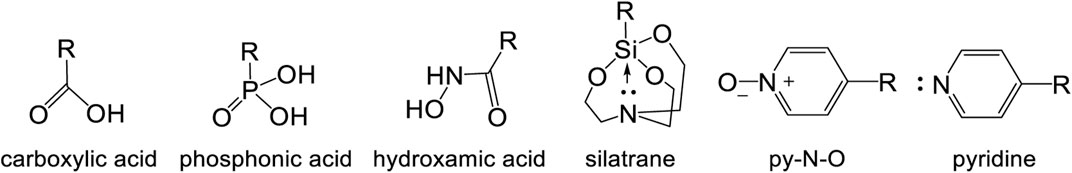
FIGURE 11. Commonly used anchoring groups for molecular water splitting process.
The steric hindrance can sometimes benefit catalysis when the isolation of transient intermediates is enabled (Chen et al., 2015). Lu et al. prepared three Ni complexes 32 with different number methyl group and studied their catalytic performance in aqueous buffer at pH 7.0. The catalytic activity increases with increasing the number of methyl group, suggesting that both suppressed axial coordination of phosphate anions with the NiIII center and increased oxidation potentials can promote catalytic performance (Wang et al., 2017; Hessels et al., 2020).
The steric hindrance can also be built up between photosensitizers (PS) and catalysts in photocatalytic systems. In comparison with complex 33, a dramatically diminished photocatalytic activity of 34 was observed when a quinoline is involved, indicating a steric hindrance effect (Figure 10). The steric hindrance inhibits the acceptance of electrons from PS− and impedes the formation of Co(III)–H, a pivotal intermediate for H2 evolution, from Co(I) (Guo et al., 2021). De Groot and Buda (2021) performed constrained ab initio computational simulations on catalyst-dye supramolecular complex 35 and proved that efficient high-performance dye-sensitized photoelectrochemical cells can be engineered by introducing steric substituents. The properties of partial complexes with different steric hindrance effect are summarized in Table 3.

TABLE 3. Properties of metal complexes with different steric hindrance effect.
Anchoring groups play an important role in heterogenizing molecular catalysts. By modifying anchoring groups, molecular catalysts are able to be stably loaded on semiconductors, which is beneficial to heterogenation of molecular catalysts. Zhang and Cole (2015) have conducted an extensive survey of anchoring groups used in DSSCs. Materna et al. (2017) have also thoroughly reviewed the anchoring groups for photocatalytic WO on metal oxide surfaces, including types and synthesis of surface anchors that used in DSPECs and their incorporations into molecules.
In addition to above mentioned anchoring groups as summarized by Materna et al. (2017), pyridine-N-oxide and pyridine was found to be effective anchoring groups as well (Lu et al., 2013; Mai et al., 2015). Wang et al. (2013) reported that pyridine-N-oxide can effectively bind on TiO2 surfaces. This guarantees the injection and adsorption of the dye molecules as indicated by an excellent IPCE of 95% and the best photon-to-electron conversion efficiency of 3.72%. Recently, (Zhu et al., 2020) found that although phosphonic acid leads to well-defined surfaces in DSPEC (dye-sensitized photoelectrosynthesis cells) assemblies, the on-surface dimerization leads to a diminished reactivity toward water oxidation compared to related monomers in solution. By contrast, the 4,4′-dipyridyl anchoring ligand of Ru-bda can maintain the monomeric structure of catalyst, affording stable photoanodes with high photocurrents and photon-to-current efficiency of 1.5% (Zhu et al., 2020).
In addition to substituent’s modification, changing backbone such as from [NN] to [NC] is another effective way of enhancing catalytic performance of metal complexes. The ligands for molecular complexes can be categorized by chelating numbers: monodentate, bidentate, tridentate, tetradentate, and polydentate ligands. In most cases, one catalyst contains more than one type of ligands, but herein we focus on structures of ligands and how they affect the overall performance of WSCs. In this section, we only consider the parent ligands without extending the discussions to substituent modification strategies which have been discussed above. For simplicity consideration, we discuss tridentate, tetradentate, and polydentate ligands together in “3.3 Multidentate ligands for WSCs” section. It should be noted that all the ligands we present below are the commonly used ligands for mononuclear WSCs and they are not comprehensive lists.
A monodentate ligand has only one atom that coordinates with a metal center. Some usual atoms are nitrogen (N), carbon (C), halogen (Cl, Br, I), oxygen (O), sulphury (S) and phosphorus (P).
Pyridine, a [N] type monodentate ligand, is one of the earliest developed monodentate chelating ligands where a ruthenium WOC was developed and is still widely used today (Carlin, 1961; Gersten et al., 1982). It can form coordinating bond with many transition metals such as Ru (Daniel et al., 2018; Kamdar and Grotjahn, 2019), Co, (Khademi et al., 2022; Navarro et al., 2022), Ni (Wang et al., 2019), and Cu (Lee et al., 2020), etc. And be used to prepare WSCs. Pal (2018) has well explained the coordinating mechanisms and emphasized its broad application in the fifth chapter of book “Pyridine”. Recently, pyridine was noticed again by Li et al. (2021), Zhu et al. (2022) as additives retarding the back-electron transfer or electron transfer between TiO2 photoanode and the oxidized dye in solar water oxidation, or between an organic light absorber and a molecular WOC on a photoanode.
Based on the coordinating mechanisms of pyridine, Duan et al. (2009) reported the first case of Ru-bda WO catalyst. DFT prediction implied that complex with higher HOMO energy has higher durability, i.e., stability and lifetime (Duan et al., 2012a). When change the pyridine ligand to phthalazine (pzt) ligands, an extraordinary TON of 55400 and high TOF of 286 s−1 were obtained. On the basis of computational analysis, a series of [N] ligands (Figure 12) with two benzene methine groups are occupied by “N” in ring, such as pyrimidine (pmd), pyrazine (prz), pyridazine (pdz), cinnoline, and phthalazine (ptz). This family of monodentate ligands shed light on the development of other types of WSCs.
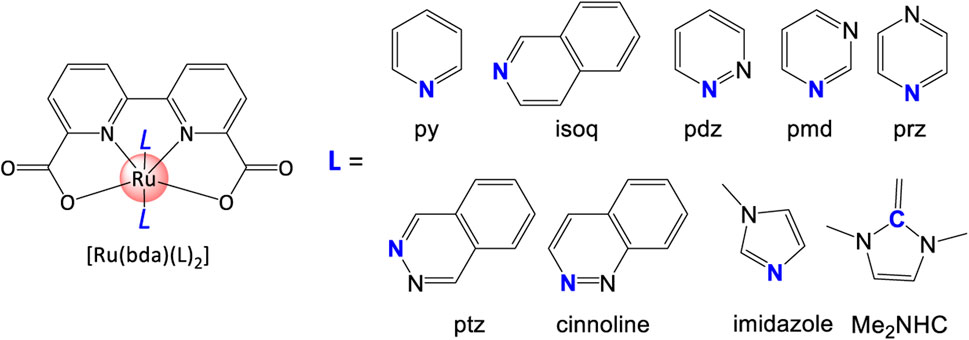
FIGURE 12. [N] and [C] ligands examples in this review.
Similar to pyridine-based [N] ligands in terms of metal-coordination properties and adjustable structures, a more electron-donating ligand, imidazole drew much attention. Imidazole has also been applied in preparing Ru-bda catalysts. The imidazole ligand can in situ form an active complex with the RuII center under the catalytic conditions (Wang et al., 2012). The key factor of catalytic performance in this case is that bulky ligand changes coupling between terminal oxygen atoms, and electronic properties.
In addition to [N] ligand, [C] ligand is another usual type of monodentate ligands such as carbene. Hetterscheid and Reek, 2011 studied [IrCp*(Me2NHC)(OH)2] (Me2NHC = N-dimethylimidazolin-2-ylidene) complex with a large TON of 2000. DFT calculations show that the oxidant potential of this WOC can heavily influence its catalytic water oxidation via various competing channels (Diaz-Morales et al., 2014; Venturini et al., 2014). Iglesias and Oro (2018) reviewed iridium–NHC catalysts, and more carbene-containing complexes will be discussed in detail in the following sections.
Other monodentate ligands such as halogen ions (F, Cl, Br, I) and aqua (OH2) were also extensively explored. When comparing the performance of complexes with different halogen ligands, two factors are usually considered: 1) steric effect coming from the different size of halogen atoms (I > Br > Cl), 2) the O–H∙∙∙halogen hydrogen bond intensity that may affect the water/proton exchange rate (Brammer et al., 2001). Besides these two factors, however, (Kaveevivitchai et al., 2012) proposed the third possibility: halogen atoms difference may change the water splitting pathway even if the complexes have the same chemical structure but different halogen atoms. Cyclic voltammetric data of Ru catalysts 29 d and 31 show that the aqua complexes are more difficult to oxidize than the analogous halide complexes. The WO data show that the aqua complexes performed better than the chloride and bromide complexes because the later ones require initial exchange of water for the Cl or Br ligand to produce the active intermediate species (Tseng et al., 2008; Masaoka and Sakai, 2009). However, the iodo-complexe presents unusual behavior where it catalyze considerably accelerate production of oxygen than the aqua-complex, as shown in Figures 13A,B. The unusually high initial rate for I-containing complex indicates the seven-coordinate rather than the six-coordinate intermediate pathway (Tseng et al., 2008).
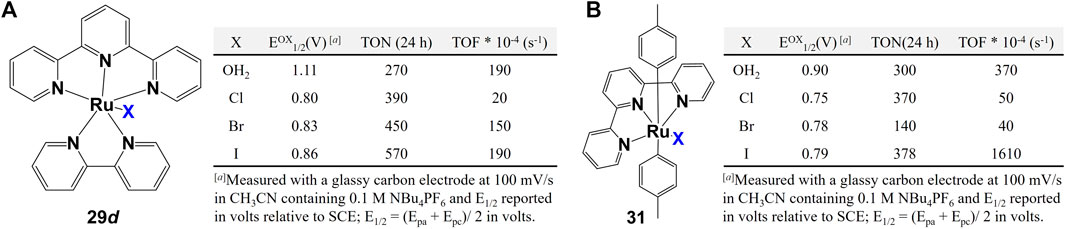
FIGURE 13. Complexes with halogen ions and aqua as monodentate ligands and their water oxidation. Data from ref (Kaveevivitchai et al., 2012).
In the second class of ligands, there are two coordinating atoms in each ligand. Three types of ligands are discussed according to their coordinating atoms: [NN] ligands, C-containing ligands, and O-containing ligands.
[NN] ligand is the most studied type of chelating ligands and hundreds of derivatives have been developed so far. It is difficult to get an absolute conclusion of which ligand is better than the other since the catalytic performance is usually mechanism-dependent, and the same factor may have different effects but there are some general rules that can be followed, such as changing the steric geometry, electronic density around metal center, pKa, its ability of accepting/donating protons or transferring electrons, or building up PCET pathway, etc.
2,2′-bipyrimidine (bpm) and 2,2′-bipyrazine (bpz) are stronger π-acceptors and less electron-donating groups with respect to 2,2′-bipyridine (bpy), hence usually result in higher oxidation potentials but lower TON values. For example, Eo (RuIII/II) of [Ru(tpy)(bpm)(OH2)]2+ and [Ru(tpy)(bpz)(OH2)]2+ catalysts are increased relative to that of [Ru(tpy)(bpy)(OH2)]2+ by metal-ligand back-bonding to bpm or bpz in Ru(II). However, rate constants for the rate limiting step of catalysts with bpm or bpz is larger than that with bpy, demonstrating that a less energy is required for the O-O bond formation for WOCs with bpm or bpz (Concepcion et al., 2008; Concepcion et al., 2009b; Concepcion et al., 2010). Similar observations were also reported for the family of [Cp*Ir(NN)Cl] catalysts (36) where Cp* is pentamethylcyclopentadienyl and [M(NNN)(NN)(OH2)]2+ catalysts (37) where M is Ir, Ru, or Os (Mcdaniel et al., 2008; Concepcion et al., 2009b; Blakemore et al., 2010). DFT calculation indicates that more nitrogen atoms in ligand result in less electron density at the reactant Ru-O bond, further explicating the slightly smaller TOF of catalyst with bpy than that with bpm or bpz (Jarvis et al., 2013). Moreover, the nitrogen atoms in the chelating ligands can also change the pKa and proton-electron transfer pathway of catalytic reactions. The potential-pH diagram for bpm complex and its comparison with bpy complex revealed that PCET avoiding charge buildup leads to the thermodynamical instability of Ru(III) and leads to its poor TON performance (Concepcion et al., 2009a).
Inspired by the high performance of Mn-ligating His332 of PS II, where deprotonation process improves the catalytic activity, imidazole-containing ligands such as 2,2′-diimidazole (H2bim), 2-(2′-pyridyl)-imidazole (pimH), and 2-(2′-pyridyl)-benzoimidazole (pybim) were designed. An imidazole ring can not only provide more nitrogen atoms to tune electron density at metal center but also serve as a proton donor to tune proton-electron transfer pathways. In addition, the deprotonation of imidazole moiety on ligand could lower catalytic onset potential. Stott et al. (2017) studied Cu-based catalysts Cu(NN)(OH2)2 with introducing an imidazole ring into the bpy ligand. The experimental results showed that deprotonation of an ionizable imidazole ring can lower the metal reduction potentials and catalytic overpotentials of catalysts. Similarly, Okamura et al. (2012) modified [Ru(tpy)(bpy)(OH2)]2+ by replacing bpy with H2bim, a lower water oxidation potential was observed compare to analogous Ru WOCs due to the high donor power and multi-electron storage ability.
In addition to changing nitrogen atoms and proton donors, steric hindrance can also influence the catalytic performance. As discussed in the preceding text, the steric impact on the activity and stability of complexes depends on water splitting conditions and mechanisms hence we can’t conclude its positive or negative effect. However, the as-needed enhanced steric effect of backbone can be generally designed either by bond fixation (to hinder rotation) such as replacing bpy with phen or by increasing the size of backbone such as modifying an extra benzene ring on ligand, as shown in Figure 14. For instance, the catalytic activity of [Ru(tpy)(NN)Cl]2+ decreased with fusing an extra ring onto the bpy ligand (pyqn) and the activity is fully suppressed when there are two phenyl rings (bqn) (Tseng et al., 2008; Zeng et al., 2015).
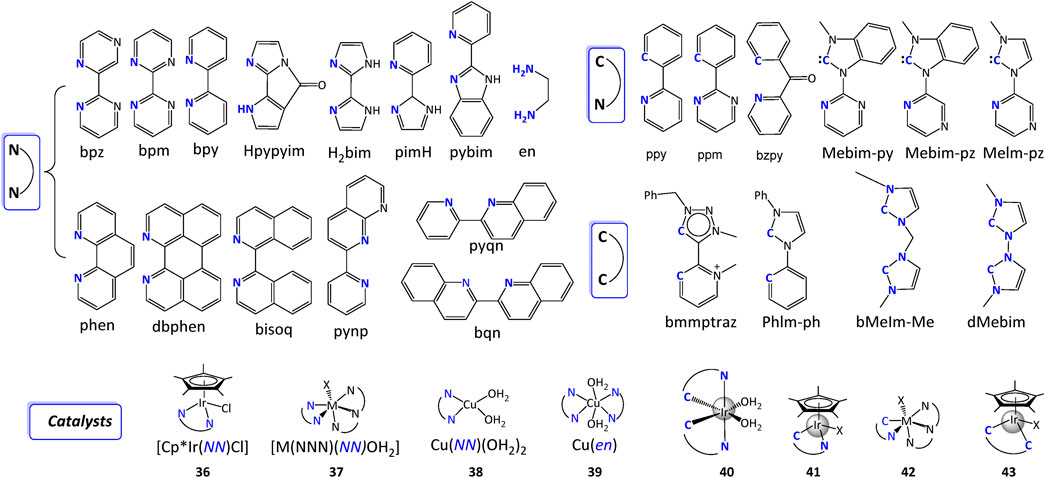
FIGURE 14. Selected [NN], [CN], and [CC] ligands and exampled water splitting catalysts discussed in this review. Coordinating atoms are labeled in blue.
Besides the commonly used bipyridine type bidentate [NN] ligands, amine-contained ligands were also developed in recent years especially in designing earth-abundant transition-metal complexes for molecular WSCs. Amine-type ligands are advantageous as 1) their simple characterizing conditions (Lu et al., 2016), 2) the easy active conformation during OER (Lee et al., 2020) in comparison with bpy ligands. For example, by mixing a Cu(II) salt and 1,2-ethylenediamine (en), Lu et al. (2016) synthesized Cu(en) catalyst with high WO activity over a wide pH range. WO catalysis occurs in solutions from pH 7 to 10 for [CuII(en)2(OH2)2]2+ complexes. At higher pH, a catalytically active layer of CuO/Cu(OH)2 formed on the electrode surface, producing O2 in a high Faradaic yield and at an overpotential superior to other Cu-based surface catalysts.
Notably, the [NN] ligands generally determine the electrochemical tunability by virtue of the substituents on ligands or the intrinsic structure changes. Despite their decent catalytic activity, however, the complexity of ligands’ designs and synthesis upgrade the difficulties of obtaining homogeneous WSCs that are simultaneously simple, robust, and effective. 2-Phenylpyridine (ppy) ligand (Figure 14), where a nitrogen in bpy is replaced by carbon, is presented as an efficient ligand for WSCs owing to the formation of strong carbon-metal bond that extremely robust under typical conditions. Since it was first reported by Schmid et al. (1994) in preparing a WO catalyst [Ir (ppy)2(H2O)2]+ (40), ppy was proven versatile enough to generate aquo complexes with various types of cyclometalating ligands by Mcdaniel et al. (2008). In the following work, Vilella et al. (2011) reported that [Ir(O)(X)(ppy)2]n (X = OH2, OH− or O2−, depending on the pH) was the active catalytic species and X = O specie has the most basic internal base, hence demonstrated the lowest energy barrier for O–O bond formation.
Crabtree et al. reported a series of highly active and robust cyclopentadiene (Cp*)-containing Ir complexes and compared their water splitting behaviors (Hull et al., 2009; Blakemore et al., 2010). Apparently, the robust carbon-metal bond contributes to an increase of TON for [(Cp*)Ir(ppy)Cl]+ (41), but a slight decrease of TOF in comparison with [(Cp*)Ir(bpy)Cl]+ (36), where a [NN] ligand was used. Subsequently, mechanism studies showed that unlike ppy-contained Cp* iridium complex, where water molecules directly interacted with the IrV = O and formed O-O bond, in the bpy analogue, [CeIV(NO3)3(OH)] complex bridged the IrIV-O• species and a water molecule, as shown in Figure 15 (Bucci et al., 2016). Furthermore, (Savini et al., 2010) studied a water soluble complex [(Cp*)Ir(bzpy)Cl]+ (bzpy = 2-benzoylpyridine) with long-term activity 2 times higher than [(Cp*)Ir(ppy)Cl]+ by creatively introducing a –C(O)– bridging between the two aryl rings. Obviously, the presence of an electron withdrawing group stabilizes the complex by enhancing the π-back donation from the metal. However, adding ketone group increased the flexibility of ligand and made them less robust than their ppy counterpart (Savini et al., 2011).
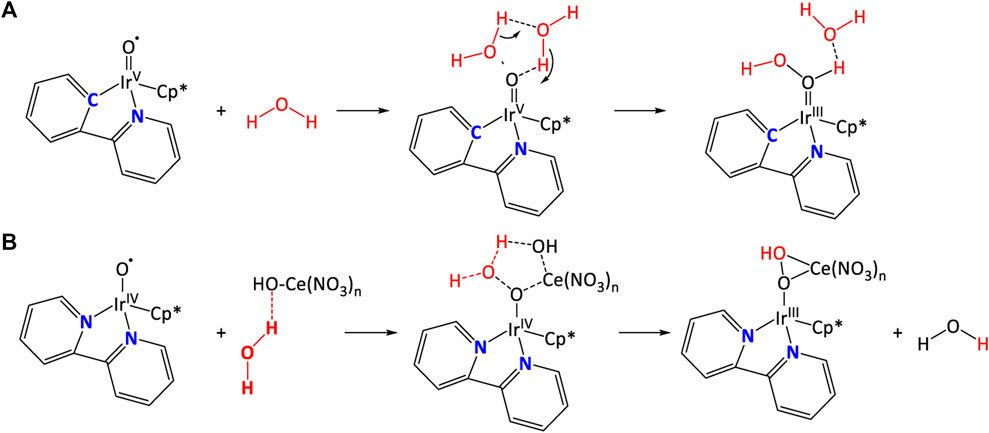
FIGURE 15. Proposed mechanism of binding water molecule for (A) (Cp*)Ir (ppy)Cl]+ (Blakemore et al., 2010), and (B) [(Cp*)Ir (bpy)Cl]+ (Bucci et al., 2016).
Another type of [CN] ligand is N-heterocyclic carbene (NHC)-pyridine ligands, as shown in Figure 14. Vaquer et al. (2013) synthesized aqua-Ru complexes with different number of carbene ligands and revealed a linear relationship between the number of carbene ligands and Δ E1/2, where Δ E1/2 = E1/2 (RuIV/III) - E1/2 (RuIII/II). The enhanced Δ E1/2 increased the stability of Ru(III) oxidation state and therefore electrocatalytic driving force for WO. Vivancos et al. (2018) have recently reviewed the N-heterocyclic carbenes complexes in details in terms of their synthesis, catalysis, and other applications.
The strong coordination bonds between NHC ligands and transition metal centers can also significantly increase the stability of complexes containing [CC] ligands, such as bmmptraz. The influence of NHC on the catalytic water splitting activity was obvious. Albrecht et al. compared the catalytic performance of [(Cp*)Ir (bmmptraz)(MeCN)]2+ containing [CC] ligand (43) and its [CN] type counterpart (41) (Lalrempuia et al., 2010). Both the TON and TOF of 43 were larger than 41. Moreover, by comparing the electrochemical behavior of [(Cp*)Ir (ppy)Cl]+ (41) and [(Cp*)Ir(PhIm-ph)Cl]+ (43), Crabtree et al. demonstrated that the NHC ligand on high-valent iridium has a stabilizing effect (Brewster et al., 2011).
Some unusual bidentate ligands were also developed in the past decade. An oxidation- and dissociation-resistant [NO] ligand, pyalkH, has been proved useful in stabilizing unusually high oxidation states, such as Rh (Sinha et al., 2015), Ir (Shopov et al., 2015; Shopov et al., 2017), and Mn (Michaelos et al., 2016). Fisher et al. (2017) prepared Cu(pyalk)2 WO electrocatalyst with high activity and stability. The oxhydryl group in pyalkH provided a deprotonated site for producing alkoxide form. The complex showed a decent TOF of 0.7 s−1 under basic conditions (pH > 10.4). Mechanism studies suggested that only the cis form of Cu(pyalk)2 could convert H2O to O2. Based on the same principle, various [OO] ligands were reported (Figure 16) (Concepcion et al., 2010; Lippert et al., 2010), as reviewed by Limburg et al. (2012).
FIGURE 16. Selected O-containing ligands discussed in this review. Coordinating atoms are labeled in blue.
Enormous multidentate ligands for WSCs have been reported. Therefore, many reviews are available in the literature covering ligands for molecular WSCs, including some focused on WOCs (Matheu et al., 2019b; Ye et al., 2019), WRCs (Huo et al., 2019), ruthenium-based catalysts (Yu et al., 2019), nickel-based catalysts (Wang et al., 2019), etc. The designing methods for these ligands follow similar rules to bidentate ligands from the strategy point of view, including steric geometry, electronic consideration, pKa modification, etc. Considering the large number of possibilities of permutations and combinations for the coordinating atoms, here we provide a few commonplace remarks and take some common ligands as examples, shown in Figure 17.
FIGURE 17. Some common multidentate ligands. Coordinating atoms are labeled in blue.
As discussed previously, an extended steric tension in ligand raises the activation barrier and hinder the reaction. Kang et al. (2014) compared the I2M barriers of complexes 44–46 (Figure 18), a larger increase of 31.2 kcal/mol was found from 45 to 46 than from 44 to 45 (4.8 kcal/mol). The O−O bond lengths of the three TS structures and their corresponding Ru−Ru distances prove the side effect of steric hindrance on the O−O bond formation. Therefore, for this series of complexes, a bulky ligand with large steric hindrance can severely discount the catalytic reaction rates.
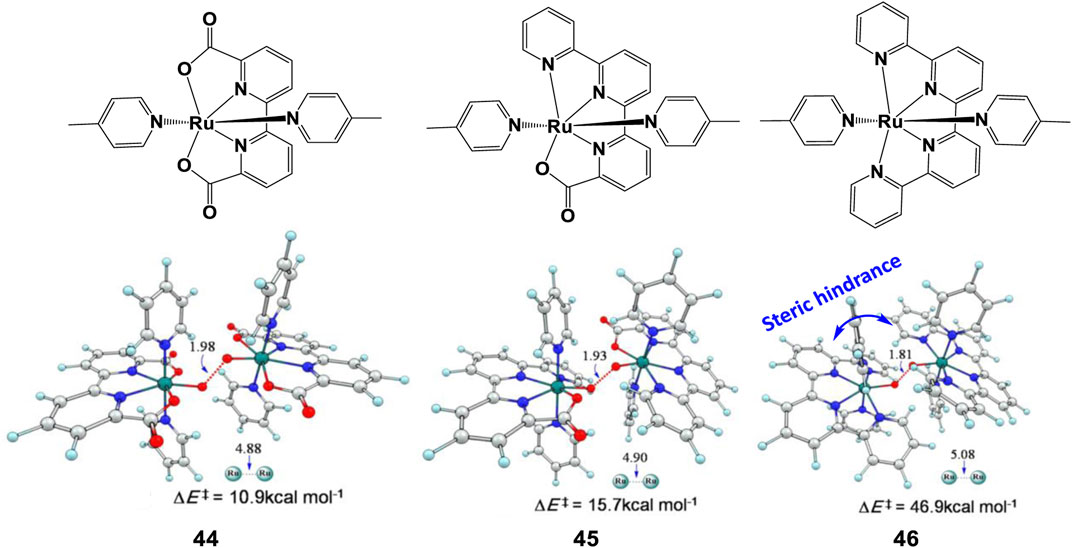
FIGURE 18. Chemical structures of complexes 44–46 and transition-state structures of O−O bond formation catalyzed with corresponding complexes following the I2M mechanism, adapt with permission from Inorg. Chem. 2014, 53, 7,130–7,136. Copyright 2014 American Chemical Society.
In addition to replacing coordinating site with bulky ligands, spatial configuration provides another attractive way of affecting the reactivity. Panchbhai et al. (2016) compared the catalytic (TON, TOF) and structural data for different iron-based WOCs with different geometry (47 and 48, Figure 19). It worth noting that the solid-state complex with smaller L–Fe–L angle (86° versus 95°) presented a lower TOF (0.007 versus 0.28) and TON (8 versus 360), illustrating the satirical interference on the activity.
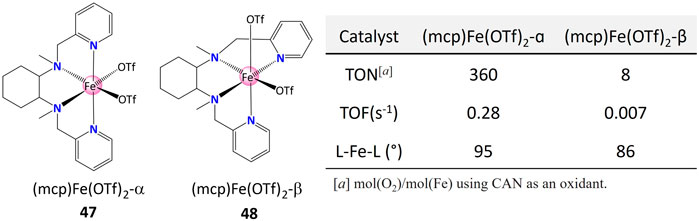
FIGURE 19. Comparison of catalytic and structural data for Fe WOCs 47 and 48. Data from ref (Panchbhai et al., 2016).
FIGURE 20. Chemical structures of (A) complexes containing ligand carboxylates, and (B) various cobalt molecular catalysts with different number and position of nitrogen in ligands.
Over the past decade, the impact of carboxylates in the ligand backbone on water splitting performance has been extensively studied and demonstrated its important role in decreasing the overpotential and increasing the WO rate. The carboxylate group(s) in ligand backbone contribute to: 1) the stability improvement of structure and photophysical properties via the formation of negatively charged ligands (Kärkäs et al., 2012); 2) the decreased potentials for the formation of higher valent intermediates (active species for water splitting) (Laine et al., 2015); 3) the buildup of PCET or APT pathways that can dramatically improve WO activity (Shatskiy et al., 2019; Liu et al., 2020; Das et al., 2021). In their pioneering study, Matheu et al. (2015) introduced a series of Ru-tda (tda = [2,2′:6′,2″-terpyridine]-6,6″-dicarboxylate) complex 49 with a large TOF of 8,000 s−1 at pH 7.0 and 50,000 s−1 at pH 10.0, which is 3–4 orders of magnitude better than Ru-bda at the same pH. DFT calculations manifested the key role of carboxylate as a proton acceptor in decreasing the activation energies for O–O bond formation (Matheu et al., 2015; Liu et al., 2020). This impact was also confirmed by replacing OH substituent with a -COOH group, when a 20-fold increase in TON of 4,000 was observed for complex 50 in comparison with complex 51 with TON of 180 (Kärkäs et al., 2012).
On the foundation of carboxylate ligands, Xie et al. (2016) developed a novel WO complex 53 containing phosphonate ligands which demonstrated multifunction in WO process: 1) provide effective pathways for electron donation and charge compensation; 2) increase the water solubility and stability of the complexes; 3) lower the redox-potential of high-valent metal–oxo species by charge compensation and σ-donation effects; 4) transfer protons in/out of the catalytic site to lower the activation energy for O-O bond-formation through PCET pathway. Moreover, these compounds retain the molecular activity when binding on metal-oxide surfaces, providing a desirable property for the incorporation of these catalysts in dye–catalyst assemblies (Xie et al., 2016).
Other attempts to improve the performance of WSCs were also achieved by changing the number or position of nitrogen atom in molecular cobalt catalysts, as reported by Kohler et al. (2021). They prepared five molecular Co(II) tetrapyridyl complexes 54–58 with different number and location of pyrazine functional groups and compared their redox potentials as well as catalytic activity. Complex 56 presented excellent activity (TOF = 3419 H2/Co/h, TON =1569 H2/Co) compared to complex 54 (TOF = 1017 H2/Co/h, TON =1268 H2/Co) while others showed inferior activity. It was explained that in the H2 photocatalysis process, the electron transfer from [Ru(bpy)2(bpy·−)]+ to Co(II) promotes activity for catalysts 56, while for 55, 57, and 58, the protons transfer promotes the overall activity. These results provided a novel option of facilitating the catalytic activity for aqueous H2 generation.
This review summarizes works on ligand designing strategies for molecular complexes aimed at achieving structure-containable molecular complexes for water splitting. From the perspective of modification position, two major strategies, substituents modification and backbone construction, are discussed. Detailed principle of how ligand modification affect catalytic performance is emphasized. The discussions are centered on electron density distribution, proton/electron-acceptance ability, bond angle, bond length, etc. Based on these consideration, various efforts including changing electron-donating/withdrawing ability, introducing intermolecular interactions, adding steric hindrance, etc. have targeted development of highly effective and stable WSCs. Comparing to the backbone construction strategy, substituents modification are more used in fine-tuning for the properties and performance of molecular catalysts.
Although some common strategies for tuning properties and performance of metal complexes are summarized in this review, as Reek Hessels et al. (2020) concluded that catalysts design rules are not universal among different transition metals and need comprehensive considerations of structure changes, mechanisms differences, and influence trends, etc. Multiple effects can be integrated in one system, and which one gains more advantages than the other needs to be analyzed case by case. In addition, the synthetic feasibility and complexity should also be considered when designing the metal complexes.
The manuscript was written through contributions of LeW. The figures in the manuscript were collected or drawn by LeW and LiW. All authors have given approval to the final version of the manuscript.
We acknowledge support from Shanghai Outstanding Academic Leaders Program.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abdel-Magied, A. F., Arafa, W. A., Laine, T. M., Shatskiy, A., Kärkäs, M. D., Åkermark, B., et al. (2017). Substituent effects in molecular ruthenium water oxidation catalysts based on amide ligands. ChemCatChem 9, 1583–1587. doi:10.1002/cctc.201601382
Akbari, H., Browne, M. C., Ortega, A., Huang, M. J., Hewitt, N. J., Norton, B., et al. (2019). Efficient energy storage technologies for photovoltaic systems. Sol. Energy 192, 144–168. doi:10.1016/j.solener.2018.03.052
Allen, A., and Cook, C. (1963). The effect of substituents on the stabilities of complexes of platinum with aryl acetylenes and phosphines. Can. J. Chem. 41, 1235–1238. doi:10.1139/v63-173
An, J., Duan, L., and Sun, L. (2012). Ru complexes containing pyridine dicarboxylate ligands: Electronic effects on their catalytic activity toward water oxidation. Faraday Discuss. 155, 267–275. doi:10.1039/c1fd00101a
Bediako, D. K., Solis, B. H., Dogutan, D. K., Roubelakis, M. M., Maher, A. G., Lee, C. H., et al. (2014). Role of pendant proton relays and proton-coupled electron transfer on the hydrogen evolution reaction by nickel hangman porphyrins. Proc. Natl. Acad. Sci. U. S. A. 111, 15001–15006. doi:10.1073/pnas.1414908111
Bellows, S. M., Brennessel, W. W., and Holland, P. L. (2016). Effects of ligand halogenation on the electron localization, geometry and spin state of low‐coordinate (β‐Diketiminato) iron complexes. Eur. J. Inorg. Chem. 20, 3344–3355. doi:10.1002/ejic.201600112
Blakemore, J. D., Schley, N. D., Balcells, D., Hull, J. F., Olack, G. W., Incarvito, C. D., et al. (2010). Half-sandwich iridium complexes for homogeneous water-oxidation catalysis. J. Am. Chem. Soc. 132, 16017–16029. doi:10.1021/ja104775j
Blakemore, J. D., Crabtree, R. H., and Brudvig, G. W. (2015). Molecular catalysts for water oxidation. Chem. Rev. 115, 12974–13005. doi:10.1021/acs.chemrev.5b00122
Brammer, L., Bruton, E. A., and Sherwood, P. (2001). Understanding the behavior of halogens as hydrogen bond acceptors. Cryst. Growth Des. 1, 277–290. doi:10.1021/cg015522k
Brewster, T. P., Blakemore, J. D., Schley, N. D., Incarvito, C. D., Hazari, N., Brudvig, G. W., et al. (2011). An iridium (IV) species, [Cp*Ir(NHC)Cl]+, related to a water-oxidation catalyst. Organometallics 30, 965–973. doi:10.1021/om101016s
Bucci, A., Menendez Rodriguez, G., Bellachioma, G., Zuccaccia, C., Poater, A., Cavallo, L., et al. (2016). An alternative reaction pathway for iridium-catalyzed water oxidation driven by cerium ammonium nitrate (can). ACS Catal. 6, 4559–4563. doi:10.1021/acscatal.6b01325
Carlin, R. L. (1961). Transition metal complexes of pyridine N-oxide. J. Am. Chem. Soc. 83, 3773–3775. doi:10.1021/ja01479a010
Chen, C., Bellows, S. M., and Holland, P. L. (2015). Tuning steric and electronic effects in transition-metal β-diketiminate complexes. Dalton Trans. 44, 16654–16670. doi:10.1039/c5dt02215k
Chen, B.-T., Morlanes, N., Adogla, E., Takanabe, K., and Rodionov, V. O. (2016). An efficient and stable hydrophobic molecular cobalt catalyst for water electro-oxidation at neutral pH. ACS Catal. 6, 4647–4652. doi:10.1021/acscatal.6b01237
Clark, M. L., Cheung, P. L., Lessio, M., Carter, E. A., and Kubiak, C. P. (2018). Kinetic and mechanistic effects of bipyridine (bpy) substituent, labile ligand, and brønsted acid on electrocatalytic CO2 reduction by Re (bpy) complexes. ACS Catal. 8, 2021–2029. doi:10.1021/acscatal.7b03971
Concepcion, J. J., Jurss, J. W., Templeton, J. L., and Meyer, T. J. (2008). One site is enough. Catalytic water oxidation by [Ru(tpy)(bpm)(OH2)]2+ and [Ru (tpy)(bpz)(OH2)]2+. J. Am. Chem. Soc. 130, 16462–16463. doi:10.1021/ja8059649
Concepcion, J. J., Jurss, J., Brennaman, M., Hoertz, P., Murakami Iha, N. Y., Patrocinio, A. O. v. T., et al. (2009a). Making oxygen with ruthenium complexes. Acc. Chem. Res. 42, 1954–1965. doi:10.1021/ar9001526
Concepcion, J. J., Jurss, J. W., Brennaman, M. K., Hoertz, P. G., Patrocinio, A. O. T., Murakami Iha, N. Y., et al. (2009b). Making oxygen with ruthenium complexes. Acc. Chem. Res. 42, 1954–1965. doi:10.1021/ar9001526
Concepcion, J. J., Jurss, J. W., Norris, M. R., Chen, Z., Templeton, J. L., and Meyer, T. J. (2010). Catalytic water oxidation by single-site ruthenium catalysts. Inorg. Chem. 49, 1277–1279. doi:10.1021/ic901437e
Corbucci, I., Petronilho, A., MüLler-Bunz, H., Rocchigiani, L., Albrecht, M., and Macchioni, A. (2015). Substantial improvement of pyridine-carbene iridium water oxidation catalysts by a simple methyl-to-octyl substitution. ACS Catal. 5, 2714–2718. doi:10.1021/acscatal.5b00319
Corbucci, I., Zaccaria, F., Heath, R., Gatto, G., Zuccaccia, C., Albrecht, M., et al. (2019). Iridium water oxidation catalysts based on pyridine‐carbene alkyl‐substituted ligands. ChemCatChem 11, 5353–5361. doi:10.1002/cctc.201901092
Crandell, D. W., Xu, S., Smith, J. M., and Baik, M.-H. (2017). Intramolecular oxyl radical coupling promotes O–O bond formation in a homogeneous mononuclear mn-based water oxidation catalyst: A computational mechanistic investigation. Inorg. Chem. 56, 4435–4445. doi:10.1021/acs.inorgchem.6b03144
Daniel, Q., Duan, L., Timmer, B. J., Chen, H., Luo, X., Ambre, R., et al. (2018). Water oxidation initiated by in situ dimerization of the molecular Ru(pdc) catalyst. ACS Catal. 8, 4375–4382. doi:10.1021/acscatal.7b03768
Das, B., Rahaman, A., Shatskiy, A., Verho, O., Karkas, M. D., and Åkermark, B. R. (2021). The impact of ligand carboxylates on electrocatalyzed water oxidation. Acc. Chem. Res. 54, 3326–3337. doi:10.1021/acs.accounts.1c00298
De Groot, H. J., and Buda, F. (2021). Tuning the Proton‐Coupled Electron‐Transfer rate by ligand modification in catalyst–dye supramolecular complexes for photocatalytic water splitting. ChemSusChem 14, 479–486. doi:10.1002/cssc.202001863
Diaz-Morales, O., Hersbach, T. J., Hetterscheid, D. G., Reek, J. N., and Koper, M. T. (2014). Electrochemical and spectroelectrochemical characterization of an iridium-based molecular catalyst for water splitting: Turnover frequencies, stability, and electrolyte effects. J. Am. Chem. Soc. 136, 10432–10439. doi:10.1021/ja504460w
Dogutan, D. K., and Nocera, D. G. (2019). Artificial photosynthesis at efficiencies greatly exceeding that of natural photosynthesis. Acc. Chem. Res. 52, 3143–3148. doi:10.1021/acs.accounts.9b00380
Dogutan, D. K., Mcguire, R., and Nocera, D. G. (2011). Electocatalytic water oxidation by cobalt (III) hangman β-octafluoro corroles. J. Am. Chem. Soc. 133, 9178–9180. doi:10.1021/ja202138m
Duan, L., Araujo, C. M., Ahlquist, M. S., and Sun, L. (2012a). Highly efficient and robust molecular ruthenium catalysts for water oxidation. Proc. Natl. Acad. Sci. U. S. A. 109, 15584–15588. doi:10.1073/pnas.1118347109
Duan, L., Bozoglian, F., Mandal, S., Stewart, B., Privalov, T., Llobet, A., et al. (2012b). A molecular ruthenium catalyst with water-oxidation activity comparable to that of photosystem II. Nat. Chem. 4, 418–423. doi:10.1038/nchem.1301
Duan, L., Wang, L., Inge, A. K., Fischer, A., Zou, X., and Sun, L. (2013). Insights into Ru-based molecular water oxidation catalysts: Electronic and noncovalent-interaction effects on their catalytic activities. Inorg. Chem. 52, 7844–7852. doi:10.1021/ic302687d
Duan, L., Wang, L., Li, F., Li, F., and Sun, L. (2015). Highly efficient bioinspired molecular Ru water oxidation catalysts with negatively charged backbone ligands. Acc. Chem. Res. 48, 2084–2096. doi:10.1021/acs.accounts.5b00149
Duan, L., Araujo, C. M., Xu, Y., and Sun, L. (2009). Isolated seven-coordinate Ru(IV) dimer complex with [HOHOH]– bridging ligand as an intermediate for catalytic water oxidation. J. Am. Chem. Soc. 131, 10397–10399. doi:10.1021/ja9034686
Eckenhoff, W. T., Mcnamara, W. R., Du, P., and Eisenberg, R. (2013). Cobalt complexes as artificial hydrogenases for the reductive side of water splitting. Biochimica Biophysica Acta - Bioenergetics 1827, 958–973. doi:10.1016/j.bbabio.2013.05.003
Feng, Z.-Q., Yang, X.-L., Ye, Y.-F., and Hao, L.-Y. (2014). The effect of electron-withdrawing group functionalization on antibacterial and catalytic activity of palladium (II) complexes. Bull. Korean Chem. Soc. 35, 1121–1127. doi:10.5012/bkcs.2014.35.4.1121
Fillol, J. L., Codolà, Z., Garcia-Bosch, I., Gómez, L., Pla, J. J., and Costas, M. (2011). Efficient water oxidation catalysts based on readily available iron coordination complexes. Nat. Chem. 3, 807–813. doi:10.1038/nchem.1140
Fisher, K. J., Materna, K. L., Mercado, B. Q., Crabtree, R. H., and Brudvig, G. W. (2017). Electrocatalytic water oxidation by a copper (II) complex of an oxidation-resistant ligand. ACS Catal. 7, 3384–3387. doi:10.1021/acscatal.7b00494
Gao, Y., Liu, J., Wang, M., Na, Y., Åkermark, B., and Sun, L. (2007). Synthesis and characterization of manganese and copper corrole xanthene complexes as catalysts for water oxidation. Tetrahedron 63, 1987–1994. doi:10.1016/j.tet.2006.12.060
Garrido-Barros, P., Funes-Ardoiz, I., Drouet, S., Benet-Buchholz, J., Maseras, F., and Llobet, A. (2015). Redox non-innocent ligand controls water oxidation overpotential in a new family of mononuclear Cu-based efficient catalysts. J. Am. Chem. Soc. 137, 6758–6761. doi:10.1021/jacs.5b03977
Gersten, S. W., Samuels, G. J., and Meyer, T. J. (1982). Catalytic oxidation of water by an oxo-bridged ruthenium dimer. J. Am. Chem. Soc. 104, 4029–4030. doi:10.1021/ja00378a053
Graham, D. J., and Nocera, D. G. (2014). Electrocatalytic H2 evolution by proton-gated hangman iron porphyrins. Organometallics 33, 4994–5001. doi:10.1021/om500300e
Guo, X., Li, C., Wang, W., Hou, Y., Zhang, B., Wang, X., et al. (2021). Polypyridyl Co complex-based water reduction catalysts: Why replace a pyridine group with isoquinoline rather than quinoline? Dalton Trans. 50, 2042–2049. doi:10.1039/c9dt04767k
Hansch, C., Leo, A., and Taft, R. (1991). A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 91, 165–195. doi:10.1021/cr00002a004
Hessels, J., Masferrer‐Rius, E., Yu, F., Detz, R. J., Klein Gebbink, R. J., and Reek, J. N. (2020). Nickel is a different pickle: Trends in water oxidation catalysis for molecular nickel complexes. ChemSusChem 13, 6629–6634. doi:10.1002/cssc.202002164
Hetterscheid, D. G., and Reek, J. N. (2011). Me2–NHC based robust Ir catalyst for efficient water oxidation. Chem. Commun. 47, 2712–2714. doi:10.1039/c0cc05108j
Hull, J. F., Balcells, D., Blakemore, J. D., Incarvito, C. D., Eisenstein, O., Brudvig, G. W., et al. (2009). Highly active and robust Cp* iridium complexes for catalytic water oxidation. J. Am. Chem. Soc. 131, 8730–8731. doi:10.1021/ja901270f
Huo, J., Zhang, Y.-B., Zou, W.-Y., Hu, X., Deng, Q., and Chen, D. (2019). Mini-review on an engineering approach towards the selection of transition metal complex-based catalysts for photocatalytic H2 production. Catal. Sci. Technol. 9, 2716–2727. doi:10.1039/c8cy02581a
Iglesias, M., and Oro, L. A. (2018). A leap forward in iridium–NHC catalysis: New horizons and mechanistic insights. Chem. Soc. Rev. 47, 2772–2808. doi:10.1039/c7cs00743d
Jarvis, E. A., Lee, B., Neddenriep, B., and Shoemaker, W. (2013). Computational comparison of stepwise oxidation and O–O bond formation in mononuclear ruthenium water oxidation catalysts. Chem. Phys. 417, 8–16. doi:10.1016/j.chemphys.2013.03.007
Johansson, M. P., Niederegger, L., Rauhalahti, M., Hess, C. R., and Kaila, V. R. (2021). Dispersion forces drive water oxidation in molecular ruthenium catalysts. RSC Adv. 11, 425–432. doi:10.1039/d0ra09004b
Kamdar, J. M., and Grotjahn, D. B. (2019). An overview of significant achievements in ruthenium-based molecular water oxidation catalysis. Molecules 24, 494. doi:10.3390/molecules24030494
Kang, R., Chen, K., Yao, J., Shaik, S., and Chen, H. (2014). Probing ligand effects on O–O bond formation of Ru-catalyzed water oxidation: A computational survey. Inorg. Chem. 53, 7130–7136. doi:10.1021/ic500008c
Kärkäs, M. D., Åkermark, T., Johnston, E. V., Karim, S. R., Laine, T. M., Lee, B. L., et al. (2012). Water oxidation by single‐site ruthenium complexes: Using ligands as redox and proton transfer mediators. Angew. Chem. Int. Ed. Engl. 51, 11757–11761. doi:10.1002/ange.201205018
Kaveevivitchai, N., Zong, R., Tseng, H.-W., Chitta, R., and Thummel, R. P. (2012). Further observations on water oxidation catalyzed by mononuclear Ru (II) complexes. Inorg. Chem. 51, 2930–2939. doi:10.1021/ic202174j
Khademi, A., Amiri, A., Tirani, F. F., and Schenk-Joß, K. (2022). Co (III) carboxamide complexes as electrocatalysts for water splitting. Int. J. Hydrog. Energy.
Kieltsch, I., Dubinina, G. G., Hamacher, C., Kaiser, A., Torres-Nieto, J., Hutchison, J. M., et al. (2010). Magnitudes of electron-withdrawing effects of the trifluoromethyl ligand in organometallic complexes of copper and nickel. Organometallics 29, 1451–1456. doi:10.1021/om901122z
Kilgore, U. J., Roberts, J. A., Pool, D. H., Appel, A. M., Stewart, M. P., Dubois, M. R., et al. (2011a). [Ni (PPh2NC6H4X2)2]2+ complexes as electrocatalysts for H2 production: Effect of substituents, acids, and water on catalytic rates. J. Am. Chem. Soc. 133, 5861–5872. doi:10.1021/ja109755f
Kilgore, U. J., Stewart, M. P., Helm, M. L., Dougherty, W. G., Kassel, W. S., Dubois, M. R., et al. (2011b). Studies of a series of [Ni(PR2NPh2)2(CH3CN)]2+ complexes as electrocatalysts for H2 production: Substituent variation at the phosphorus atom of the P2N2 ligand. Inorg. Chem. 50, 10908–10918. doi:10.1021/ic201461a
Kohler, L., Potocny, A. M., Niklas, J., Zeller, M., Poluektov, O. G., and Mulfort, K. L. (2021). Replacing pyridine with pyrazine in molecular cobalt catalysts: Effects on electrochemical properties and aqueous H2 generation. Catalysts 11, 75. doi:10.3390/catal11010075
Laine, T. M., Kärkäs, M. D., Liao, R.-Z., Åkermark, T., Lee, B.-L., Karlsson, E. A., et al. (2015). Efficient photochemical water oxidation by a dinuclear molecular ruthenium complex. Chem. Commun. 51, 1862–1865. doi:10.1039/c4cc08606f
Lalrempuia, R., Mcdaniel, N. D., Müller‐Bunz, H., Bernhard, S., and Albrecht, M. (2010). Water oxidation catalyzed by strong carbene‐type donor‐ligand complexes of iridium. Angew. Chem. Int. Ed. 49, 9765–9768. doi:10.1002/anie.201005260
Lanznaster, M., Neves, A., Bortoluzzi, A. J., Assumpção, A. M., Vencato, I., Machado, S. P., et al. (2006). Electronic effects of electron-donating and-withdrawing groups in model complexes for iron-tyrosine-containing metalloenzymes. Inorg. Chem. 45, 1005–1011. doi:10.1021/ic050869o
Lee, W. T., Muñoz, S. B., Dickie, D. A., and Smith, J. M. (2014). Ligand modification transforms a catalase mimic into a water oxidation catalyst. Angew. Chem. Int. Ed. 53, 9856–9859. doi:10.1002/anie.201402407
Lee, H., Wu, X., and Sun, L. (2020). Copper-based homogeneous and heterogeneous catalysts for electrochemical water oxidation. Nanoscale 12, 4187–4218. doi:10.1039/c9nr10437b
Li, M., and Bernhard, S. (2017). Synthetically tunable iridium (III) bis-pyridine-2-sulfonamide complexes as efficient and durable water oxidation catalysts. Catal. Today 290, 19–27. doi:10.1016/j.cattod.2016.11.027
Li, J., Zhu, Y., Li, F., Liu, G., Xu, S., and Sun, L. (2021). Dye-sensitized photoanode decorated with pyridine additives for efficient solar water oxidation. Chin. J. Catal. 42, 1352–1359. doi:10.1016/s1872-2067(20)63683-x
Limburg, B., Bouwman, E., and Bonnet, S. (2012). Molecular water oxidation catalysts based on transition metals and their decomposition pathways. Coord. Chem. Rev. 256, 1451–1467. doi:10.1016/j.ccr.2012.02.021
Lippert, C. A., Arnstein, S. A., Sherrill, C. D., and Soper, J. D. (2010). Redox-active ligands facilitate bimetallic O2 homolysis at five-coordinate oxorhenium (V) centers. J. Am. Chem. Soc. 132, 3879–3892. doi:10.1021/ja910500a
Liu, S., Lei, Y.-J., Xin, Z.-J., Lu, Y.-B., and Wang, H.-Y. (2018). Water splitting based on homogeneous copper molecular catalysts. J. Photochem. Photobiol. A Chem. 355, 141–151. doi:10.1016/j.jphotochem.2017.09.060
Liu, J., Chen, X., Cao, S., and Yang, H. (2019). Overview on hybrid solar photovoltaic-electrical energy storage technologies for power supply to buildings. Energy Convers. Manag. 187, 103–121. doi:10.1016/j.enconman.2019.02.080
Liu, Y., Su, X., Guan, W., and Yan, L. (2020). Ruthenium-based catalysts for water oxidation: The key role of carboxyl groups as proton acceptors. Phys. Chem. Chem. Phys. 22, 5249–5254. doi:10.1039/c9cp05893a
Liu, T., Li, G., Shen, N., Ahlquist, M. S., and Sun, L. (2021). Hydrophobic interactions of Ru-bda-type catalysts for promoting water oxidation activity. Energy fuels. 35, 19096–19103. doi:10.1021/acs.energyfuels.1c02097
Lu, J., Xu, X., Li, Z., Cao, K., Cui, J., Zhang, Y., et al. (2013). Zinc porphyrins with a pyridine‐ring‐anchoring group for dye‐sensitized solar cells. Chem. Asian J. 8, 956–962. doi:10.1002/asia.201201136
Lu, C., Du, J., Su, X.-J., Zhang, M.-T., Xu, X., Meyer, T. J., et al. (2016). Cu (II) aliphatic diamine complexes for both heterogeneous and homogeneous water oxidation catalysis in basic and neutral solutions. ACS Catal. 6, 77–83. doi:10.1021/acscatal.5b02173
Mai, C.-L., Moehl, T., Hsieh, C.-H., DéCoppet, J.-D., Zakeeruddin, S. M., GräTzel, M., et al. (2015). Porphyrin sensitizers bearing a pyridine-type anchoring group for dye-sensitized solar cells. ACS Appl. Mat. Interfaces 7, 14975–14982. doi:10.1021/acsami.5b03783
Masaoka, S., and Sakai, K. (2009). Clear evidence showing the robustness of a highly active oxygen-evolving mononuclear ruthenium complex with an aqua ligand. Chem. Lett. 38, 182–183. doi:10.1246/cl.2009.182
Materna, K. L., Crabtree, R. H., and Brudvig, G. W. (2017). Anchoring groups for photocatalytic water oxidation on metal oxide surfaces. Chem. Soc. Rev. 46, 6099–6110. doi:10.1039/c7cs00314e
Matheu, R., Ertem, M. Z., Benet-Buchholz, J., Coronado, E., Batista, V. S., Sala, X., et al. (2015). Intramolecular proton transfer boosts water oxidation catalyzed by a Ru complex. J. Am. Chem. Soc. 137, 10786–10795. doi:10.1021/jacs.5b06541
Matheu, R., Ertem, M. Z., Gimbert-Suriñach, C., Sala, X., and Llobet, A. (2019a). Seven coordinated molecular ruthenium–water oxidation catalysts: A coordination chemistry journey: Focus review. Chem. Rev. 119, 3453–3471. doi:10.1021/acs.chemrev.8b00537
Matheu, R., Garrido-Barros, P., Gil-Sepulcre, M., Ertem, M. Z., Sala, X., Gimbert-Suriñach, C., et al. (2019b). The development of molecular water oxidation catalysts. Nat. Rev. Chem. 3, 331–341. doi:10.1038/s41570-019-0096-0
Mcdaniel, N. D., Coughlin, F. J., Tinker, L. L., and Bernhard, S. (2008). Cyclometalated iridium (III) aquo complexes: Efficient and tunable catalysts for the homogeneous oxidation of water. J. Am. Chem. Soc. 130, 210–217. doi:10.1021/ja074478f
Mcinnes, E. L., Farley, R., Rowlands, C., Welch, A., and Yellowlees, L. (1999). On the electronic structure of [Pt (4, 4′-X2-bipy) Cl2]0/–/2–: An electrochemical and spectroscopic (UV/Vis, EPR, ENDOR) study. J. Chem. Soc. Dalton Trans., 4203–4208. doi:10.1039/a904658e
Mehrabani, S., Bikas, R., Zand, Z., Mousazade, Y., Allakhverdiev, S. I., and Najafpour, M. M. (2020). Water splitting by a pentanuclear iron complex. Int. J. Hydrogen Energy 45, 17434–17443. doi:10.1016/j.ijhydene.2020.04.249
Meyer, T. J., Sheridan, M. V., and Sherman, B. D. (2017). Mechanisms of molecular water oxidation in solution and on oxide surfaces. Chem. Soc. Rev. 46, 6148–6169. doi:10.1039/c7cs00465f
Meza-Chincha, A.-L., Lindner, J. O., Schindler, D., Schmidt, D., Krause, A.-M., Röhr, M. I., et al. (2020). Impact of substituents on molecular properties and catalytic activities of trinuclear Ru macrocycles in water oxidation. Chem. Sci. 11, 7654–7664. doi:10.1039/d0sc01097a
Michaelos, T. K., Lant, H. M., Sharninghausen, L. S., Craig, S. M., Menges, F. S., Mercado, B. Q., et al. (2016). Catalytic oxygen evolution from manganese complexes with an oxidation‐resistant N, N, O‐donor ligand. ChemPlusChem 81, 1129–1132. doi:10.1002/cplu.201600353
Mognon, L., Benet-Buchholz, J., and Llobet, A. (2015). Single site isomeric Ru WOCs with an electron-withdrawing group: Synthesis, electrochemical characterization, and reactivity. Inorg. Chem. 54, 11948–11957. doi:10.1021/acs.inorgchem.5b02260
Moore, G. F., Blakemore, J. D., Milot, R. L., Hull, J. F., Song, H.-E., Cai, L., et al. (2011). A visible light water-splitting cell with a photoanode formed by codeposition of a high-potential porphyrin and an iridium water-oxidation catalyst. Energy Environ. Sci. 4, 2389–2392. doi:10.1039/c1ee01037a
Najafpour, M. M., and Allakhverdiev, S. I. (2012). Manganese compounds as water oxidizing catalysts for hydrogen production via water splitting: From manganese complexes to nano-sized manganese oxides. Int. J. Hydrogen Energy 37, 8753–8764. doi:10.1016/j.ijhydene.2012.02.075
Navarro, M. Á., Cosano, D., Bhunia, A., Simonelli, L., Martin-Diaconescu, V., Romero-Salguero, F. J., et al. (2022). Cobaloxime tethered pyridine-functionalized ethylene-bridged periodic mesoporous organosilica as an efficient HER catalyst. Sustain. Energy Fuels 6, 398–407. doi:10.1039/d1se01437d
Neel, A. J., Hilton, M. J., Sigman, M. S., and Toste, F. D. (2017). Exploiting non-covalent π interactions for catalyst design. Nature 543, 637–646. doi:10.1038/nature21701
Neuman, N. S. I., Albold, U., Ferretti, E., Chandra, S., Steinhauer, S., Rößner, P., et al. (2020). Cobalt corroles as electrocatalysts for water oxidation: Strong effect of substituents on catalytic activity. Inorg. Chem. 59, 16622–16634. doi:10.1021/acs.inorgchem.0c02550
Okamura, M., Yoshida, M., Kuga, R., Sakai, K., Kondo, M., and Masaoka, S. (2012). A mononuclear ruthenium complex showing multiple proton-coupled electron transfer toward multi-electron transfer reactions. Dalton Trans. 41, 13081–13089. doi:10.1039/c2dt30773a
Pal, S. (2018). Pyridine: A useful ligand in transition metal complexes. Pyridine. 57, 57–74. doi:10.5772/intechopen.76986
Palacios, A., Barreneche, C., Navarro, M., and Ding, Y. (2020). Thermal energy storage technologies for concentrated solar power–A review from a materials perspective. Renew. Energy 156, 1244–1265. doi:10.1016/j.renene.2019.10.127
Panchbhai, G., Singh, W. M., Das, B., Jane, R. T., and Thapper, A. (2016). Mononuclear iron complexes with tetraazadentate ligands as water oxidation catalysts. Eur. J. Inorg. Chem. 20, 3262–3268. doi:10.1002/ejic.201600165
Papadakis, M., Barrozo, A., Straistari, T., Queyriaux, N., Putri, A., Fize, J., et al. (2020). Ligand-based electronic effects on the electrocatalytic hydrogen production by thiosemicarbazone nickel complexes. Dalton Trans. 49, 5064–5073. doi:10.1039/c9dt04775a
Pye, D. R., and Mankad, N. P. (2017). Bimetallic catalysis for C–C and C–X coupling reactions. Chem. Sci. 8, 1705–1718. doi:10.1039/c6sc05556g
Richmond, C. J., Matheu, R., Poater, A., Falivene, L., Benet‐Buchholz, J., Sala, X., et al. (2014). Supramolecular water oxidation with Ru–bda‐based catalysts. Chem. Eur. J. 20, 17282–17286. doi:10.1002/chem.201405144
Richmond, C. J., Escayola, S., and Poater, A. (2019). Axial ligand effects of Ru‐bda complexes in the O–O bond formation via the I2M bimolecular mechanism in water oxidation catalysis. Eur. J. Inorg. Chem., 2101–2108. doi:10.1002/ejic.201801450
Rodriguez, G. M., Zaccaria, F., Van Dijk, S., Zuccaccia, C., and Macchioni, A. (2021). Substituent effects on the activity of Cp* Ir (pyridine-carboxylate) water oxidation catalysts: Which ligand fragments remain coordinated to the active Ir centers? Organometallics 40, 3445–3453. doi:10.1021/acs.organomet.1c00464
Sampson, M. D., Nguyen, A. D., Grice, K. A., Moore, C. E., Rheingold, A. L., and Kubiak, C. P. (2014). Manganese catalysts with bulky bipyridine ligands for the electrocatalytic reduction of carbon dioxide: Eliminating dimerization and altering catalysis. J. Am. Chem. Soc. 136, 5460–5471. doi:10.1021/ja501252f
Sato, Y., Takizawa, S. Y., and Murata, S. (2015). Substituent effects on physical properties and catalytic activities toward water oxidation in mononuclear ruthenium complexes. Eur. J. Inorg. Chem., 5495–5502. doi:10.1002/ejic.201500958
Savini, A., Bellachioma, G., Ciancaleoni, G., Zuccaccia, C., Zuccaccia, D., and Macchioni, A. (2010). Iridium (III) molecular catalysts for water oxidation: The simpler the faster. Chem. Commun. 46, 9218–9219. doi:10.1039/c0cc03801f
Savini, A., Belanzoni, P., Bellachioma, G., Zuccaccia, C., Zuccaccia, D., and Macchioni, A. (2011). Activity and degradation pathways of pentamethyl-cyclopentadienyl-iridium catalysts for water oxidation. Green Chem. 13, 3360–3374. doi:10.1039/c1gc15899f
Schmid, B., Garces, F., and Watts, R. (1994). Synthesis and characterizations of cyclometalated iridium (III) solvento complexes. Inorg. Chem. 33, 9–14. doi:10.1021/ic00079a005
Shaffer, D. W., Xie, Y., and Concepcion, J. (2017). O–O bond formation in ruthenium-catalyzed water oxidation: Single-site nucleophilic attack vs. O–O radical coupling. Chem. Soc. Rev. 46, 6170–6193. doi:10.1039/c7cs00542c
Shatskiy, A., Bardin, A. A., Oschmann, M., Matheu, R., Benet‐Buchholz, J., Eriksson, L., et al. (2019). Electrochemically driven water oxidation by a highly active ruthenium‐based catalyst. ChemSusChem 12, 2251–2262. doi:10.1002/cssc.201900097
Shopov, D. Y., Rudshteyn, B., Campos, J. S., Batista, V. S., Crabtree, R. H., and Brudvig, G. W. (2015). Stable iridium (IV) complexes of an oxidation-resistant pyridine-alkoxide ligand: Highly divergent redox properties depending on the isomeric form adopted. J. Am. Chem. Soc. 137, 7243–7250. doi:10.1021/jacs.5b04185
Shopov, D. Y., Rudshteyn, B., Campos, J., Vinyard, D. J., Batista, V. S., Brudvig, G. W., et al. (2017). A full set of iridium (IV) pyridine-alkoxide stereoisomers: Highly geometry-dependent redox properties. Chem. Sci. 8, 1642–1652. doi:10.1039/c6sc03758e
Sinha, S. B., Shopov, D. Y., Sharninghausen, L. S., Vinyard, D. J., Mercado, B. Q., Brudvig, G. W., et al. (2015). A stable coordination complex of Rh (IV) in an N, O-donor environment. J. Am. Chem. Soc. 137, 15692–15695. doi:10.1021/jacs.5b12148
Stewart, M. P., Ho, M.-H., Wiese, S., Lindstrom, M. L., Thogerson, C. E., Raugei, S., et al. (2013). High catalytic rates for hydrogen production using nickel electrocatalysts with seven-membered cyclic diphosphine ligands containing one pendant amine. J. Am. Chem. Soc. 135, 6033–6046. doi:10.1021/ja400181a
Stolarczyk, J. K., Bhattacharyya, S., Polavarapu, L., and Feldmann, J. (2018). Challenges and prospects in solar water splitting and CO2 reduction with inorganic and hybrid nanostructures. ACS Catal. 8, 3602–3635. doi:10.1021/acscatal.8b00791
Stott, L. A., Prosser, K. E., Berdichevsky, E. K., Walsby, C. J., and Warren, J. J. (2017). Lowering water oxidation overpotentials using the ionisable imidazole of copper (2-(2′-pyridyl) imidazole). Chem. Commun. 53, 651–654. doi:10.1039/c6cc09208j
Sun, H., Han, Y., Lei, H., Chen, M., and Cao, R. (2017). Cobalt corroles with phosphonic acid pendants as catalysts for oxygen and hydrogen evolution from neutral aqueous solution. Chem. Commun. 53, 6195–6198. doi:10.1039/c7cc02400b
Suresh, C. H., Remya, G. S., and Anjalikrishna, P. K. (2022). Molecular electrostatic potential analysis: A powerful tool to interpret and predict chemical reactivity. Wiley Interdiscip. Rev. Comput. Mol. Sci., e1601.
Timmer, B. J., Kravchenko, O., Zhang, B., Liu, T., and Sun, L. (2020). Electronic influence of the 2, 2′-bipyridine-6, 6′-dicarboxylate ligand in Ru-based molecular water oxidation catalysts. Inorg. Chem. 60, 1202–1207. doi:10.1021/acs.inorgchem.0c03339
Timmer, B. J., Kravchenko, O., Liu, T., Zhang, B., and Sun, L. (2021). Off‐set interactions of ruthenium–bda type catalysts for promoting water‐splitting performance. Angew. Chem. Int. Ed. Engl. 133, 14625–14632. doi:10.1002/ange.202101931
Tseng, H.-W., Zong, R., Muckerman, J. T., and Thummel, R. (2008). Mononuclear ruthenium (II) complexes that catalyze water oxidation. Inorg. Chem. 47, 11763–11773. doi:10.1021/ic8014817
Vaquer, L., Miró, P., Sala, X., Bozoglian, F., Masllorens, E., Benet‐Buchholz, J., et al. (2013). Understanding electronic ligand perturbation over successive metal‐based redox potentials in mononuclear ruthenium–aqua complexes. ChemPlusChem 78, 235–243. doi:10.1002/cplu.201200268
Venturini, A., Barbieri, A., Reek, J. N., and Hetterscheid, D. G. (2014). Catalytic water splitting with an iridium carbene complex: A theoretical study. Chem. Eur. J. 20, 5358–5368. doi:10.1002/chem.201303796
Vilella, L., Vidossich, P., Balcells, D., and Lledós, A. (2011). Basic ancillary ligands promote O–O bond formation in iridium-catalyzed water oxidation: A DFT study. Dalton Trans. 40, 11241–11247. doi:10.1039/c1dt10660k
Vivancos, A. N., Segarra, C., and Albrecht, M. (2018). Mesoionic and related less heteroatom-stabilized N-heterocyclic carbene complexes: Synthesis, catalysis, and other applications. Chem. Rev. 118, 9493–9586. doi:10.1021/acs.chemrev.8b00148
Wang, L., Duan, L., Stewart, B., Pu, M., Liu, J., Privalov, T., et al. (2012). Toward controlling water oxidation catalysis: Tunable activity of ruthenium complexes with axial imidazole/DMSO ligands. J. Am. Chem. Soc. 134, 18868–18880. doi:10.1021/ja309805m
Wang, L., Yang, X., Li, S., Cheng, M., and Sun, L. (2013). A new type of organic sensitizers with pyridine-N-oxide as the anchoring group for dye-sensitized solar cells. RSC Adv. 3, 13677–13680. doi:10.1039/c3ra41182f
Wang, C., Chen, Y., and Fu, W.-F. (2015). New platinum and ruthenium Schiff base complexes for water splitting reactions. Dalton Trans. 44, 14483–14493. doi:10.1039/c5dt01055a
Wang, J.-W., Hou, C., Huang, H.-H., Liu, W.-J., Ke, Z.-F., and Lu, T.-B. (2017). Further insight into the electrocatalytic water oxidation by macrocyclic nickel (II) complexes: The influence of steric effect on catalytic activity. Catal. Sci. Technol. 7, 5585–5593. doi:10.1039/c7cy01527e
Wang, N., Zheng, H., Zhang, W., and Cao, R. (2018). Mononuclear first-row transition-metal complexes as molecular catalysts for water oxidation. Chin. J. Catal. 39, 228–244. doi:10.1016/s1872-2067(17)63001-8
Wang, J.-W., Liu, W.-J., Zhong, D.-C., and Lu, T.-B. (2019). Nickel complexes as molecular catalysts for water splitting and CO2 reduction. Coord. Chem. Rev. 378, 237–261. doi:10.1016/j.ccr.2017.12.009
Wang, L., Polyansky, D. E., and Concepcion, J. J. (2019). Self-assembled bilayers as an anchoring strategy: Catalysts, chromophores, and chromophore-catalyst assemblies. J. Am. Chem. Soc. 141, 8020–8024. doi:10.1021/jacs.9b01044
Wang, J.-W., Huang, H.-H., Wang, P., Yang, G., Kupfer, S., Huang, Y., et al. (2022). Co-facial π–π interaction expedites sensitizer-to-catalyst electron transfer for high-performance CO2 photoreduction. American Chemical Society, 1359–1374. doi:10.1021/jacsau.2c00073
Whang, D. R., and Apaydin, D. H. (2018). Artificial photosynthesis: Learning from nature. ChemPhotoChem 2, 109–160. doi:10.1002/cptc.201800045
Wiese, S., Kilgore, U. J., Dubois, D. L., and Bullock, R. M. (2012). [Ni (PMe2NPh2)2](BF4)2 as an electrocatalyst for H2 production. ACS Catal. 2, 720–727. doi:10.1021/cs300019h
Xie, Y., Shaffer, D. W., Lewandowska‐Andralojc, A., Szalda, D. J., and Concepcion, J. (2016). Water oxidation by ruthenium complexes incorporating multifunctional bipyridyl diphosphonate ligands. Angew. Chem. Int. Ed. Engl. 128, 8199–8203. doi:10.1002/ange.201601943
Xie, Y., Shaffer, D. W., and Concepcion, J. J. (2018). O–O radical coupling: From detailed mechanistic understanding to enhanced water oxidation catalysis. Inorg. Chem. 57, 10533–10542. doi:10.1021/acs.inorgchem.8b00329
Ye, S., Ding, C., Chen, R., Fan, F., Fu, P., Yin, H., et al. (2018). Mimicking the key functions of photosystem II in artificial photosynthesis for photoelectrocatalytic water splitting. J. Am. Chem. Soc. 140, 3250–3256. doi:10.1021/jacs.7b10662
Ye, S., Ding, C., Liu, M., Wang, A., Huang, Q., and Li, C. (2019). Water oxidation catalysts for artificial photosynthesis. Adv. Mat. 31, 1902069. doi:10.1002/adma.201902069
Yi, J., Zhan, S., Chen, L., Tian, Q., Wang, N., Li, J., et al. (2021). Electrostatic interactions accelerating water oxidation catalysis via intercatalyst O–O coupling. J. Am. Chem. Soc. 143, 2484–2490. doi:10.1021/jacs.0c07103
Yoshida, M., Masaoka, S., Abe, J., and Sakai, K. (2010). Catalysis of mononuclear aquaruthenium complexes in oxygen evolution from water: A new radical coupling path using hydroxocerium (IV) species. Chem. Asian J. 5, 2369–2378. doi:10.1002/asia.201000323
Young, E. R., Rosenthal, J., Hodgkiss, J. M., and Nocera, D. G. (2009). Comparative PCET study of a donor− acceptor pair linked by ionized and nonionized asymmetric hydrogen-bonded interfaces. J. Am. Chem. Soc. 131, 7678–7684. doi:10.1021/ja809777j
Yu, J., He, Q., Yang, G., Zhou, W., Shao, Z., and Ni, M. (2019). Recent advances and prospective in ruthenium-based materials for electrochemical water splitting. ACS Catal. 9, 9973–10011. doi:10.1021/acscatal.9b02457
Zeng, Q., Lewis, F. W., Harwood, L. M., and Hartl, F. (2015). Role of ligands in catalytic water oxidation by mononuclear ruthenium complexes. Coord. Chem. Rev. 304, 88–101. doi:10.1016/j.ccr.2015.03.003
Zhang, L., and Cole, J. M. (2015). Anchoring groups for dye-sensitized solar cells. ACS Appl. Mat. Interfaces 7, 3427–3455. doi:10.1021/am507334m
Zhang, J. Z., and Reisner, E. (2020). Advancing photosystem II photoelectrochemistry for semi-artificial photosynthesis. Nat. Rev. Chem. 4, 6–21. doi:10.1038/s41570-019-0149-4
Zhang, B., and Sun, L. (2019a). Artificial photosynthesis: Opportunities and challenges of molecular catalysts. Chem. Soc. Rev. 48, 2216–2264. doi:10.1039/c8cs00897c
Zhang, B., and Sun, L. (2019b). Ru-bda: Unique molecular water-oxidation catalysts with distortion induced open site and negatively charged ligands. J. Am. Chem. Soc. 141, 5565–5580. doi:10.1021/jacs.8b12862
Zhang, T., Wang, C., Liu, S., Wang, J.-L., and Lin, W. (2014). A biomimetic copper water oxidation catalyst with low overpotential. J. Am. Chem. Soc. 136, 273–281. doi:10.1021/ja409267p
Zhang, W., Wu, F., Li, J., Yan, D., Tao, J., Ping, Y., et al. (2018). Unconventional relation between charge transport and photocurrent via boosting small polaron hopping for photoelectrochemical water splitting. ACS Energy Lett. 3, 2232–2239. doi:10.1021/acsenergylett.8b01445
Zhang, X.-P., Wang, H.-Y., Zheng, H., Zhang, W., and Cao, R. (2021). O–O bond formation mechanisms during the oxygen evolution reaction over synthetic molecular catalysts. Chin. J. Catal. 42, 1253–1268. doi:10.1016/s1872-2067(20)63681-6
Zhu, Y., Wang, D., Huang, Q., Du, J., Sun, L., Li, F., et al. (2020). Stabilization of a molecular water oxidation catalyst on a dye− sensitized photoanode by a pyridyl anchor. Nat. Commun. 11, 4610–4618. doi:10.1038/s41467-020-18417-5
Keywords: molecular catalysts, modification strategies, ligands design, mononuclear complexes, water splitting
Citation: Wang L and Wang L (2022) Ligands modification strategies for mononuclear water splitting catalysts. Front. Chem. 10:996383. doi: 10.3389/fchem.2022.996383
Received: 17 July 2022; Accepted: 03 August 2022;
Published: 27 September 2022.
Edited by:
Xiaolei Yuan, Nantong University, ChinaReviewed by:
Kun Jiang, Shanghai Jiao Tong University, ChinaCopyright © 2022 Wang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lei Wang, bGVpd2FuZ0B1c3N0LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.