 Yousaf Khan1Shahid Iqbal2*Mazloom Shah3Aneela Maalik1Rafaqat Hussain4
Yousaf Khan1Shahid Iqbal2*Mazloom Shah3Aneela Maalik1Rafaqat Hussain4 Shoaib Khan4*Imran Khan4Rami Adel Pashameah5Eman Alzahrani6Abd-ElAziem Farouk7Mohammed Issa Alahmdi8Hisham S. M. Abd-Rabboh9,10
Shoaib Khan4*Imran Khan4Rami Adel Pashameah5Eman Alzahrani6Abd-ElAziem Farouk7Mohammed Issa Alahmdi8Hisham S. M. Abd-Rabboh9,10- 1Department of Chemistry, COMSATS Universityislamabad Campus, Islamabad, Pakistan
- 2Department of Chemistry, School of Natural Sciences (SNS), National University of Science and Technology (NUST), Islamabad, Pakistan
- 3Department of Chemistry, Abbottabad University of Science and Technology (AUST), Abbottabad, Pakistan
- 4Department of Chemistry, Hazara University, Mansehra, Pakistan
- 5Department of Chemistry, Faculty of Applied Science, Umm Al-Qura University, Makkah, Saudi Arabia
- 6Department of Chemistry, College of Science, Taif University, Taif, Saudi Arabia
- 7Department of Biotechnology College of Science, Taif University, Taif, Saudi Arabia
- 8Department of Chemistry, Faculty of Science, University of Tabuk, Tabuk, Saudi Arabia
- 9Chemistry Department, Faculty of Science, King Khalid University, Abha, Saudi Arabia
- 10Department of Chemistry, Faculty of Science, Ain Shams University, Cairo, Egypt
The 7-quinolinyl-bearing triazole analogs were synthesized (1d–19d) and further assessed in vitro for their inhibitory profile against α-amylase andα-glucosidase. The entire analogs showed a diverse range of activities having IC50 values between 0.80 ± 0.05 µM to 40.20 ± 0.70 µM (α-amylase) and 1.20 ± 0.10 µM to 43.30 ± 0.80 µM (α-glucosidase) under the positive control of acarbose (IC50 = 10.30 ± 0.20 µM) (IC50 = 9.80 ± 0.20 µM) as the standard drug. Among the synthesized scaffolds, seven scaffolds 12d, 10d, 8d, 9d, 11d, 5d, and 14d showed excellent α-amylase and α-glucosidase inhibitory potentials with IC50 values of 4.30 ± 0.10, 2.10 ± 0.10, 1.80 ± 0.10, 1.50 ± 0.10, 0.80 ± 0.05, 5.30 ± 0.20, and 6.40 ± 0.30 µM (against α-amylase) and 3.30 ± 0.10, 2.40 ± 0.10, 1.20 ± 0.10, 1.90 ± 0.10, 8.80 ± 0.20, 7.30 ± 0.40, and 5.50 ± 0.10 µM (against α-glucosidase), respectively, while the remaining 12 scaffolds 19d, 8d, 17d, 16d, 15d, 7d, 4d, 3d, 1d, 2d, 13d and 6 d showed less α-amylase and α-glucosidase inhibitory potentials than standard acarbose but still found to be active. Structure–activity connection studies also showed that scaffolds with electron-withdrawing groups like -Cl, -NO2, and -F linked to the phenyl ring had higher inhibitory potentials for -amylase and -glucosidase than scaffolds with -OCH3, -Br, and -CH3 moieties. In order to better understand their binding sites, the powerful scaffolds 11d and 9d were also subjected to molecular docking studies. The results showed that these powerful analogs provide a number of important interactions with the active sites of both of these targeted enzymes, including conventional hydrogen bonding, pi–pi stacking, pi–sulfur, pi–anion, pi–pi, pi–sigma, T-shaped, and halogen (fluorine). Furthermore, various techniques (spectroscopic), including 1H, 13C-NMR, and HREI-MS mass, were used to explore the correct structure of newly afforded hybrid scaffolds based on quinoline-bearing triazole ring.
1 Introduction
In the past few decades, DM (diabetes mellitus) was known to be the most familiar epidemic disorder, which adversely affects the metabolic functions performed by the endocrine system. It is growing and spreading rapidly all over the world. Islets of Langerhans are well-known specialized cells that are responsible for producing insulin in the human body when a diabetic patient does not either produce insulin or is incapable of consuming insulin properly. Due to this dysfunction, the glucose (sugar) level of blood increases in the body, resulting in hyperglycemia. DM is categorized into two types, insulin-reliant, which is due to a lack of production of insulin in the human body termed type-1; however, in type-2 diabetes, either insulin is not produced by the human body or is incapable of utilizing insulin properly. After cardiovascular and cancer diseases, which result in mortality, DM is among the most frequent and rapidly growing chronic diseases (Li et al., 2004; Chauhan et al., 2010). However, inhibition of α-amylase and α-glucosidase, as being carbolytic enzymes, is one of the common approaches to minimizing or managing these metabolic disorders (Zaharudin et al., 2019). Lipase, protease, and amylase are the most used enzymes in food biotechnology and industry (Asgher et al., 2007). α-amylase, which belongs to the class of metalloenzymes, catalyzes the formation of glucose and maltose through hydrolysis of starch (polysaccharides) (Sundarram and Murthy, 2014; Taha et al., 2017). The human body absorbs starch through the sequential catalytic action of α-glucosidase (intestinal) andα-amylase (Asgher et al., 2007). Inhibitors of α-amylase and α-glucosidase play a crucial role in modulating the glucose level of blood after taking a meal. Therefore, the α-amylase and α-glucosidase inhibitory potential can be clinically utilized as chemotherapeutic agents and finds applications in the treatment of DM, in addition to obesity (Gunawan-Puteri et al., 2012; Taha et al., 2017).
In addition to the usage of several approaches, the inhibitors of α-glucosidase are used as a therapeutic approach for the treatment of DM (Jong-Anurakkun et al., 2007). The α-glucosidase inhibitors slow down the absorption of glucose, therefore resulting in a decrease in the digestion process. Miglitol and acarbose are important inhibitors of the α-glucosidase enzyme commonly used to decrease the glucose level of postprandial blood. These synthesized inhibitors inhibit glucose formation in the small intestine through inhibition of the α-glucosidase enzyme. Acarbose, an antidiabetic drug with α-amylase and α-glucosidase inhibitory potentials, is currently used in type-2 DM treatment (Salar et al., 2017). Nonetheless, different side effects, such as abdominal pain and flatulence, are associated with acarbose. The use of only drugs is often limited by these side effects; consequently, the efficiency was improved by using in combination with other antidiabetic drugs. Therefore, designing and constructing more active scaffolds having fewer side effects to treat DM is time-consuming (Meneilly et al., 2000).
Quinoline scaffolds find application in numerous pharmaceutically potent drugs and received much attention in the field of medicinal chemistry. Quinoline scaffolds were reported to show a broad range of biological activities such as antifungal (Tabassum et al., 2021), antibacterial (Ghosh et al., 2020), antileishmanial (Gupta et al., 2021), anticonvulsant (Al-Ostoot et al., 2021), anti-Zaka virus (Taha et al., 2019), antimalarial (Vinindwa et al., 2021), proton pump inhibitors (Cernicchi et al., 2021), antiproliferative (Ammar et al., 2021), antiulcer (Atukuri et al., 2020), antidiabetic (Taha et al., 2019), antimicrobial, anti-inflammatory (Douadi et al., 2020), antitrypanosomal (Zhang et al., 2018), antitubercular (Nesaragi et al., 2021), antioxidant agent (Velmurugan et al., 2021), and antihypertensive (Kumari et al., 2019).
It is well known that scaffolds harboring the 1,2,4-triazole motif have a variety of biological and pharmacological properties, including anticonvulsant, anti-inflammatory, antiproliferative, and antifungal activities (Fadaly et al., 2020; Jahani et al., 2020; Joshi et al., 2020). Furthermore, 1,2,4-triazole is a pharmacologically active scaffold that is crucial for therapeutic candidates such as anastrozole, vorozole, and letrozole, all of which have been shown to be efficient aromatase inhibitors and have applications in the treatment of breast cancer (Ammazzalorso et al., 2021). Additionally, it was shown that 1,2,4-triazole containing mercapto scaffolds have antitubercular, antibacterial, antifungal, and anticancer effects (Mallikarjuna et al., 2009; Pitucha et al., 2020).
In the recent past, researchers had investigated numerous heterocyclic moieties of different categories and classes to explore their inhibitory profile in search of lead molecules. Keeping in view the α-amylase and α-glucosidase profile of quinoline and triazole-3-thione analogs, herein in this study, we have incorporated two heterocyclic rings in the same molecule as7-quinolinyl-bearing triazole-3-thione analogs (1d–19d) with the hope of further expanding their inhibitory potentials as dual α-amylase and α-glucosidase inhibitors (Figure 1).
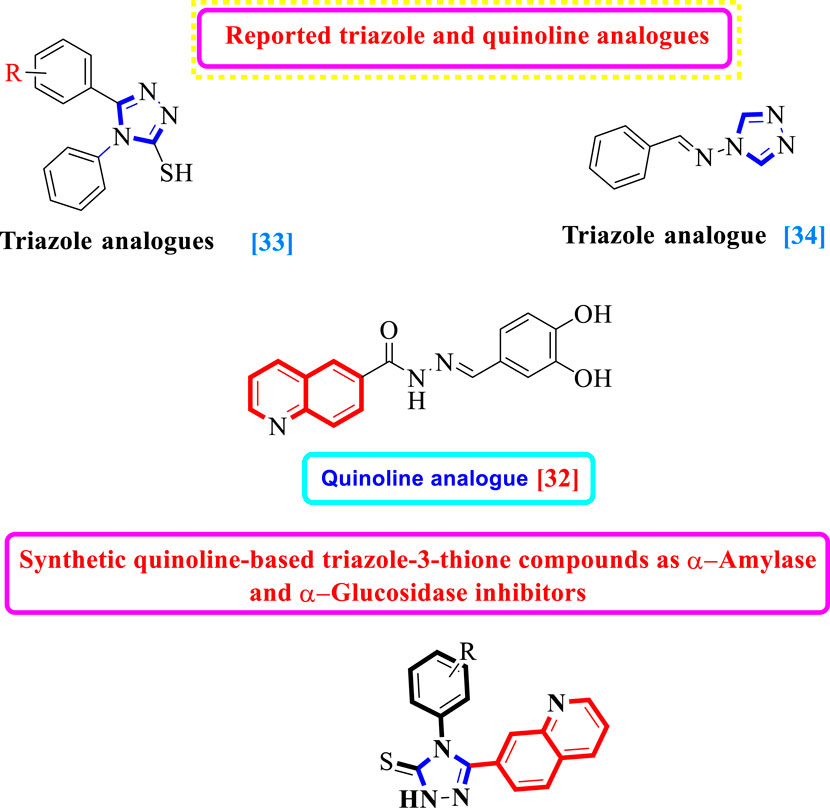
FIGURE 1. Hybridization of the quinoline moiety with a triazole ring.
2 Results and discussion
2.1 Chemistry
To afford new hybrid scaffolds (1d–19d) based on the quinoline-bearing triazole skeleton, methyl quinoline-7-carboxylate (0.5 mmol) (a) solution stirred in EtOH (10 ml) was reacted and refluxed with hydrazine hydrate (1 equivalent) for 5 h. As the reaction was completed, a solid residue (0.5 mmol) was obtained by evaporation of the solvent and then washed with cold water and dried to afford quinoline-7-carbohydrazide (b). In the second step, the mixture of quinoline-7-carbohydrazide (0.5 mmol), the corresponding phenyl isothiocyanates (1 equivalent), and Et3N (1 ml) in THF (10 ml) was refluxed for 12 h and then quenched with water (10 ml) to form a precipitate, which was further filtered and using EtOH (5 ml) recrystallized to access an intermediate, which further underwent cyclization with 2% NaOH (10 ml) followed by neutralization with dilute HCl (5 ml) to afford the targeted quinoline-based triazole (1d–19d) scaffolds in the appropriate yield (Scheme 1) Table 1.
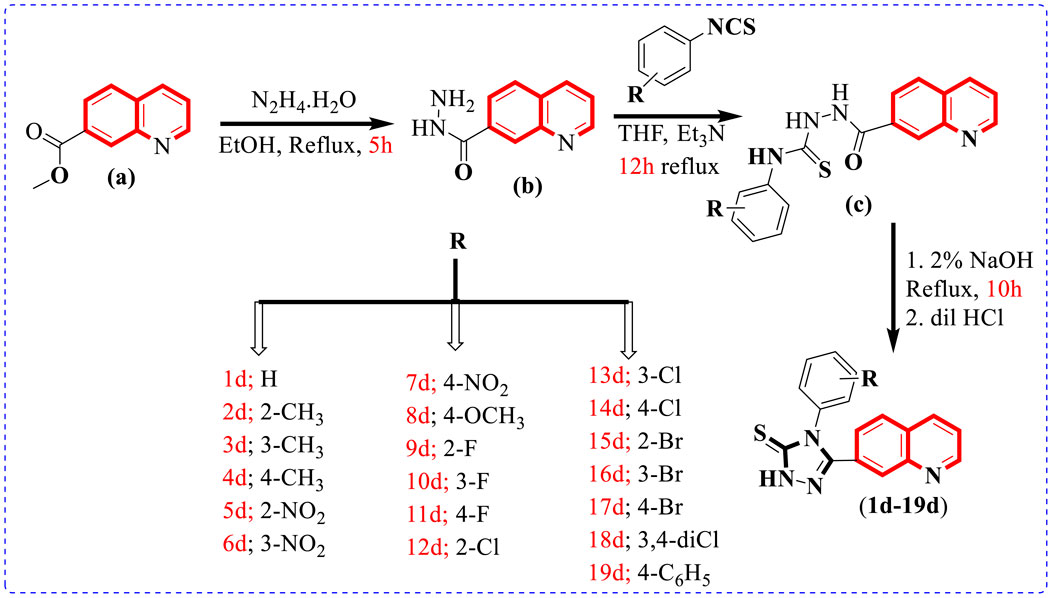
SCHEME 1. Preparation of 7-quinolinyl-based triazole-3-thione derivatives (1d–19d).
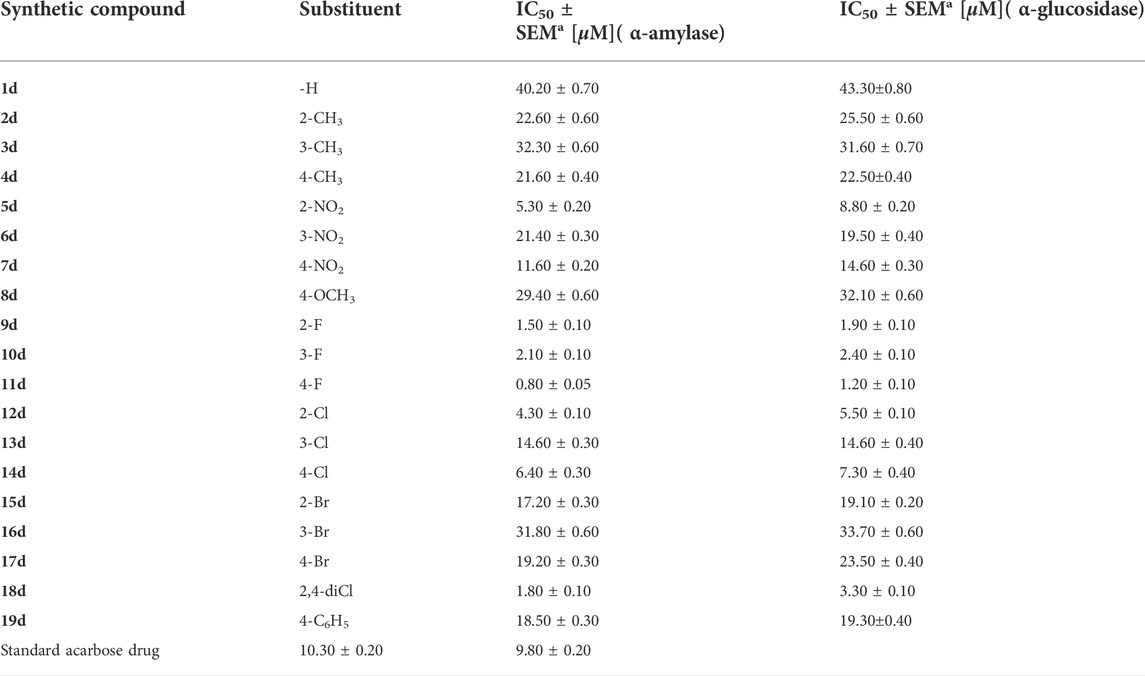
TABLE 1. Inhibition profile (in vitro) of targeted α-amylase and α-glucosidase enzymes of hybrid scaffolds of quinoline-based triazole (1d–19d).
2.2 Biological activity
2.2.1 Inhibition study of α-amylase and α-glucosidase
In this study, nineteen scaffolds based on quinoline-bearing triazole were afforded and then assessed against α-amylase and α-glucosidase to explore their inhibition profile. The entire analogs showed a diverse range of activities having IC50 values between 0.80 ± 0.05 µM and 40.20 ± 0.70 µM (α-amylase) and 1.20 ± 0.10 µM and 43.30 ± 0.80 µM (α-glucosidase) under the positive control of acarbose [(IC50 = 10.30 ± 0.20 µM) (IC50 = 9.80 ± 0.20 µM)] as the standard drug. Among the synthesized scaffolds, seven scaffolds 12d, 10d, 18d, 9d, 11d, 5d and 14d showed superior α-amylase and α-glucosidase inhibitory potentials than standard acarbose drugs with IC50 values of 4.30 ± 0.10, 2.10 ± 0.10, 1.80 ± 0.10, 1.50 ± 0.10, 0.80 ± 0.05, 5.30 ± 0.20, and 6.40 ± 0.30 µM (against α-amylase) and 3.30 ± 0.10, 2.40 ± 0.10, 1.20 ± 0.10, 1.90 ± 0.10, 8.80 ± 0.20, 7.30 ± 0.40, and 5.50 ± 0.10 µM (against α-glucosidase), respectively, while the remaining 12 scaffolds 19d, 8d, 17d, 16d, 15d, 7d, 4d, 3d, 1d, 2d, 13d and 6d showed less α-amylase and α-glucosidase inhibitory potentials than standard acarbose but still found to be active. SAR studies suggest that the entire part of synthetic scaffolds, including quinoline, triazole ring, and N-phenyl ring, actively participates in the activity, and any change observed in the activity was due to alteration in position, number/s, and nature of substituents around phenyl ring B.
2.2.1.1 Structure–activity relationship study for α-amylase enzyme
It was noteworthy from SAR studies that quinoline-based triazole scaffolds that hold the fluoro moiety as being the electron-withdrawing entity attached to various positions of the aryl ring show a significant role in the inhibition of the α-amylase enzyme, and hence hybrid scaffolds based on the quinoline-bearing triazole moiety showed better inhibition than -CH3, -OCH3, and–Br groups bearing scaffolds. This enhanced inhibitory potential of fluoro moiety-bearing analogs was due to greater electron withdrawing affinity of the –F moiety, which makes the ph-ring more effective for interactions with α-amylase active pocket. Moreover, it also seemed that the activity was altered by bringing variation in the position of the –F moiety around aryl ring B. In current newly afforded scaffolds, the analog 11d that holds the –F moiety at 4-position of aryl ring B linked to the N-triazole skeleton was found to show ten-fold enhanced potency than standard acarbose drug. However, analogs 9d with ortho-F and 10d having meta-F substitutions displayed less potency than scaffold 11d, although these analogs resemble in their structure with analog 9d but have different positions around phenyl ring B. This variation in potency of these–F moiety-bearing scaffolds was due to different positions of the -F moiety at the phenyl ring, indicating that alteration in the position of the –F moiety may result in diverse potency.
Similarly, the alteration in the position of the attached substitute also affects other–Cl moiety-bearing scaffolds 14d, 12d, and 13d. By comparing scaffold 12d that holds ortho–Cl moiety with scaffolds 14d bearing para-Cl and 13d having meta-Cl substitutions, the ortho-Cl moiety holding analog 12d showed enhanced potency than its counterparts 14d and 13d, although these scaffolds are structurally similar. This indicates that the activity was diminished by shifting ortho-Cl moiety either to para- or meta-position as in scaffolds 14d and 13d. In addition, compound 14d bearing para-Cl substitution showed 2-folds more potency than scaffold 7d bearing para-NO2 moieties on the aryl ring, although the position of substituents around the aryl ring is the same. The activity was different in the case of both these compounds, which may be owing to the diverse nature of attached substituents (-Cl and–NO2). Moreover, by comparing the ortho-Cl moiety bearing scaffold 12d with compound 5d that hold the ortho-NO2 group on the aryl ring of the triazole skeleton, the scaffold 12d has emerged as a better inhibitor of α-amylase than scaffold 5d. This difference in potency between these analogs was caused by the unique properties of both the -Cl and -NO2 moieties. If we compared compound 14d (IC50 = 6.40 ± 0.30 µM) bearing mono-chloro substitution on 4-position of the attached N-ph-ring with its counterpart 2 bearing di-Cl substitution linked to 3-and 4-position of the phenyl ring, the compound 18d showed better activity than compound 14d due to the attached di-Cl substitutions that offered stronger electron-withdrawing effect. Analog 1d having un-substituted attached ph-ring is recognized as the least potent inhibitor among the newly constructed scaffolds. However, the activity may be enhanced when substituents of either electron-withdrawing/electron-donating properties are attached to it at various positions of the ph-ring. Therefore, compounds 8d (having para–OCH3), 4d (having para–CH3), 14d (having (having para–Cl), and 18d (having 3,4–di-Cl) moieties showed enhanced α–amylase inhibitory potentials when compared to compound 1d with un-substituted ph-ring, indicating that the activity was largely dependent on attached electron-withdrawing/electron-donating groups (Figure 2).
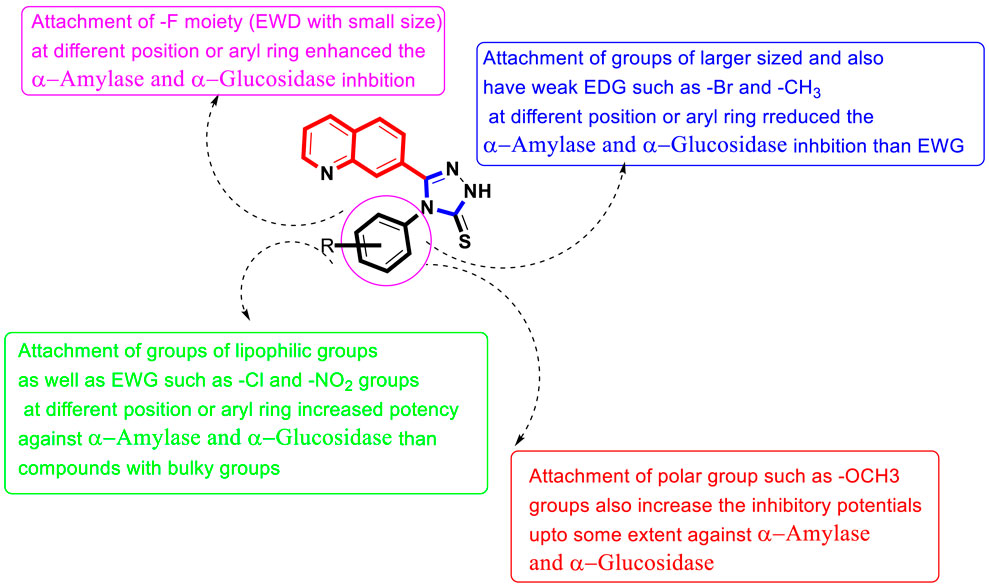
FIGURE 2. Summary of SAR studies of quinoline-based triazole against α-amylase and α-glucosidase enzymes.
2.2.1.2 Structure–activity relationship study for α-glucosidase enzyme
Among halogen-substituted analogs, fluoro-substituted analogs (10d, 11d and 9d) showed enhanced α–glucosidase inhibitory potentials when compared to Cl-substituted (14d, 12d and 13d) and bromo-substituted analogs (17d, 16d and 15d). Similarly, Cl-substituted scaffolds display better activity than bromo-substituted analogs. This shows that the α–glucosidase activity was enhanced by increasing the electron-withdrawing nature of the attached substituent; therefore, halogen-substituted scaffolds showed activity in order of–F > Cl > Br. Moreover, by comparing the para-F moiety bearing analog 11d with compounds 10d bearing meta-F and 9d having ortho-F group, compound 11d showed superior potentials than its counterparts 10d and 9d, which are similar in structure to compound 11d. This difference in activity found in these fluoro-substituted analogs was due to the different positions of the attached electron-withdrawing fluoro moiety around the ph-ring. If we compare analog 14d and 12d with mono-chloro substitution with compound 18d bearing 3, 4-di Cl chloro groups, the di-Cl moieties bearing scaffold 18d showed superior activity than a mono-Cl moiety containing scaffolds 14d and 12d. This enhanced activity of compound 18d may be due to the stronger electron-withdrawing effect offered by the two chloro groups as compared to compound 14d and 12d bearing mono-chloro substitution. Similarly, by comparing scaffold 5d bearing ortho-nitro substitution with its structurally similar analog 7d that holds the nitro group at the para-position of the phenyl ring, the compound 5d showed enhanced activity than its counterpart 7d. This shows that alteration in the position of substitution around the phenyl ring greatly affects the enzymatic activity (Figure 2).
Based on the aforementioned observation, it was concluded that the inhibitory potentials of both α-amylase and α-glucosidase were largely dependent on substitution patterns around the ph-ring attached to the hybrid triazole-based quinoline skeleton. Moreover, it was also noted that the activities changed by bringing alterations in nature, position, and number/s of attached substituents.
2.2 Experimental
The experimental data (general procedure, spectral analysis, α-amylase, and α-glucosidase inhibition, and docking protocols (Li et al., 2015; Mrabti, 2018; Rao et al., 2021) were included in the supporting documentation.
3 Molecular docking
To explore the active part in newly constructed quinoline-based triazole scaffolds against α-amylase and α-glucosidase, molecular docking was carried out. Among newly afforded scaffolds being tested, the analogs that hold fluoro moiety showed enhanced inhibitory potentials (in vitro), and hence these fluoro-substituted analogs were subjected to the molecular docking study to explore their binding site and results obtained from molecular docking show that the most potent analogs 11d and 9d not only display better potency (in vitro) but also furnish numerous important interactions (in silico) with the active site of the amino acid.
It was shown by the PLI profile of potent analog 11d bearing–F moiety that analog 11d against α-amylase displays various important interactions (Figure 3) such as TYR-62 (π–π stacked), HIS-299 (donor interactions), ASP-3002 (H-B), GLU-233 (H-B), LEU-162 (π–R), HIS-305 (donor interactions), and TRP-59 (π–π T-shaped). Similarly, analog 9d in α-amylase showed various interactive residues from a certain distance, while the residues are TRP-59 (π–π stacked), GLN-63 (H-B), LEU-165 (π–sigma), and TRY-62 (H-B) as shown in Figure 3.
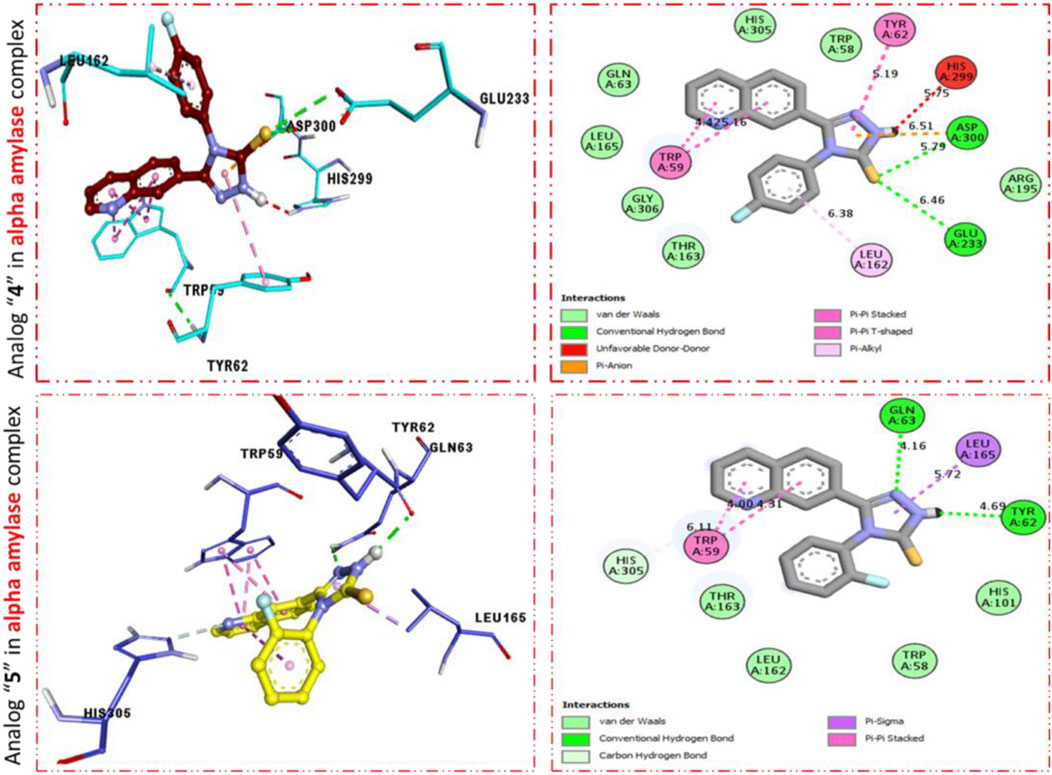
FIGURE 3. PLI profile indicates the corresponding surface of α-amylase enzyme and its interactions with fluoro-substituted scaffolds 11 d and 9d.
Furthermore, scaffold 4 also displays better interactions against the α–glucosidase enzyme. This analog 11d having para-F moiety on the extended aryl ring interacts with α-glucosidase via several interactions, including MET-470 (π–S), ALA-234 (π–R), ASP-568 (π–anion), TRP-329 (π–π T-shaped), TRP-432 (π-sigma), ARG-552 (H-B), and ASP-232 (π–anion) as shown in Figure 4, while analog 9d exhibited the following interactions against alpha glucosidase such as TRP-432 (π–π T-shaped), ASP-232 (H-F), MET-470 (π–S), ALA-234 (π–R), ASP-568 (H-F), TRP-329 (π–π T-Stacked), and ARG-552 (H-B) along with van der Waals interaction (Figure 4).
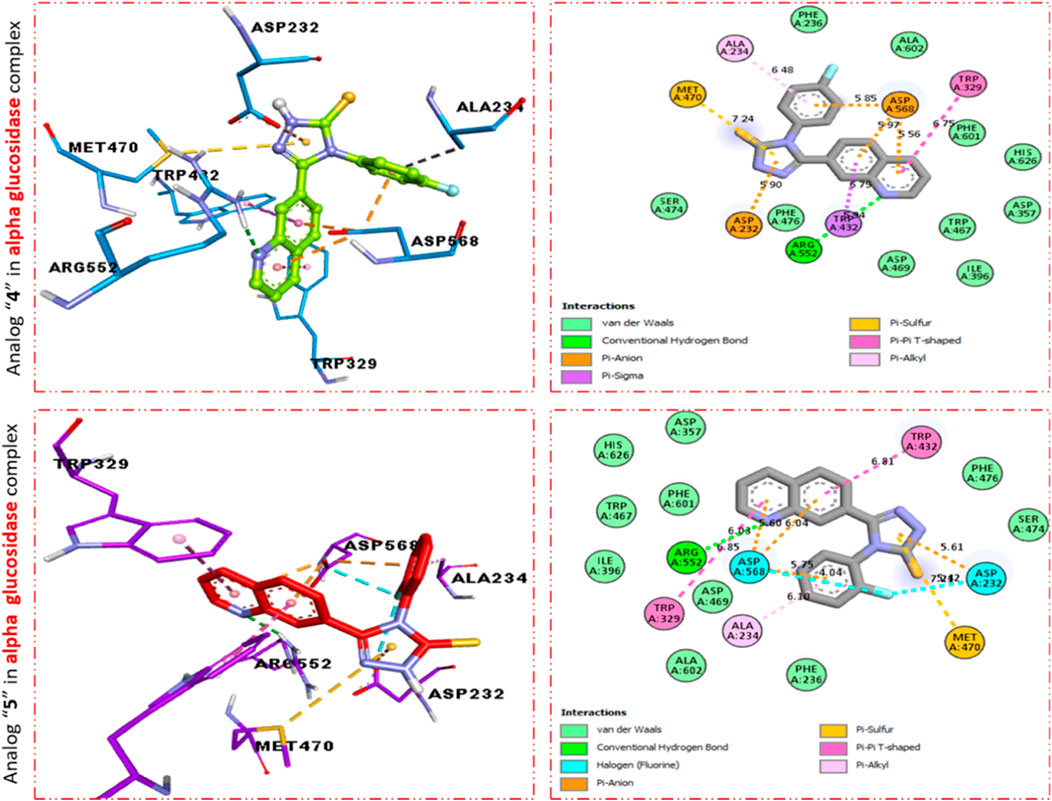
FIGURE 4. PLI profile indicates the corresponding surface of α-glucosidase enzyme and its interactions with fluoro-substituted scaffolds 11 d and 9d.
It was observed by comparison of the protein–ligand interaction profile of most active scaffolds 11d and 9d with protein–ligand interaction of standard acarbose drugs against α-amylase and α-glucosidase enzymes that the active scaffolds 11d and 9d established some additional interactions such as halogen (fluorine), π–π T-shaped, π–sulfur, π–alkyl, π–sigma, π–π stacking, and π–anion interactions with the active part of the amino acid of α-amylase and α-glucosidase than standard acarbose drug which furnish only conventional HB van der Waals and carbon HB interactions. Therefore, scaffolds 11d and 9d were found to be more potent against α-amylase and α-glucosidase than acarbose standard drugs.
The most potent scaffolds at 11d and 9d have different potency (in vitro) for both enzymes being targeted and hence established a diverse range of interactions (molecular docking) with active sites of these enzymes (Tables 2–4). This difference in activity and different interactions (in silico) possessed by these scaffolds toward enzymes being targeted was due to different positions of the attached fluoro moiety around the aryl part of the quinoline-triazole skeleton. It was shown (Figure 5) from docking results that these potent analogs 11d and 9d (possess the–F at a different position) furnish a better binding mode of interactions with the active part of enzymes being targeted (α-amylase and α-glucosidase) than standard acarbose drug which established only conventional HB, van der Waals, and carbon HB interactions with the active part of targeted enzymes (α-amylase and α-glucosidase).
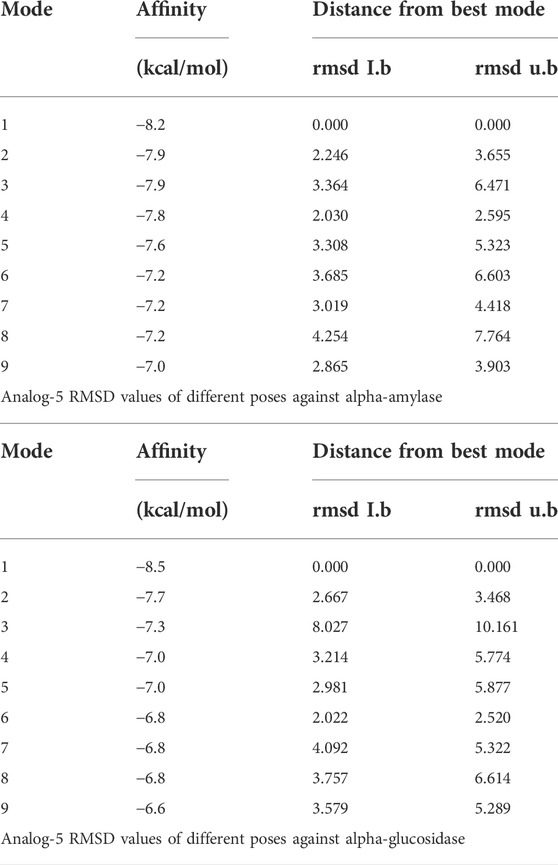
TABLE 2. RMSD values of different poses of analog 11d against α-amylase and α-glucosidase enzymes.
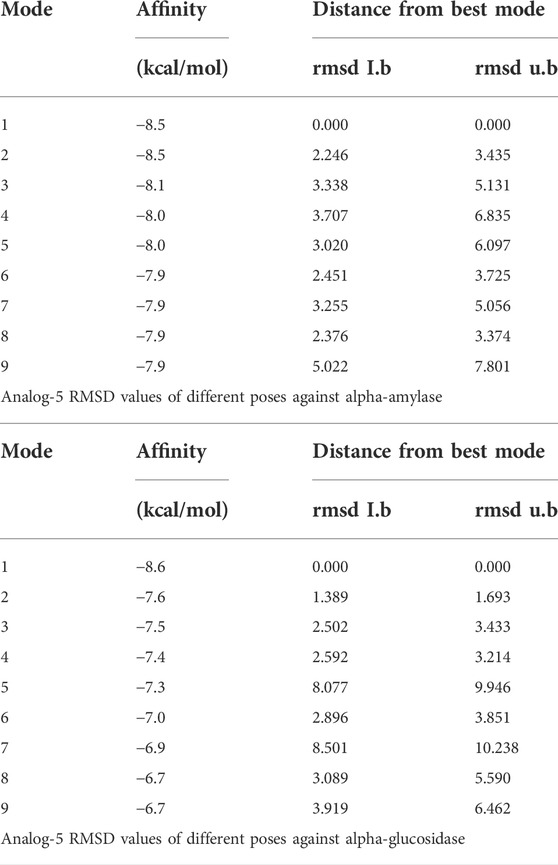
TABLE 3. RMSD values of different poses of analog 9d against α-amylase and α-glucosidase enzymes.
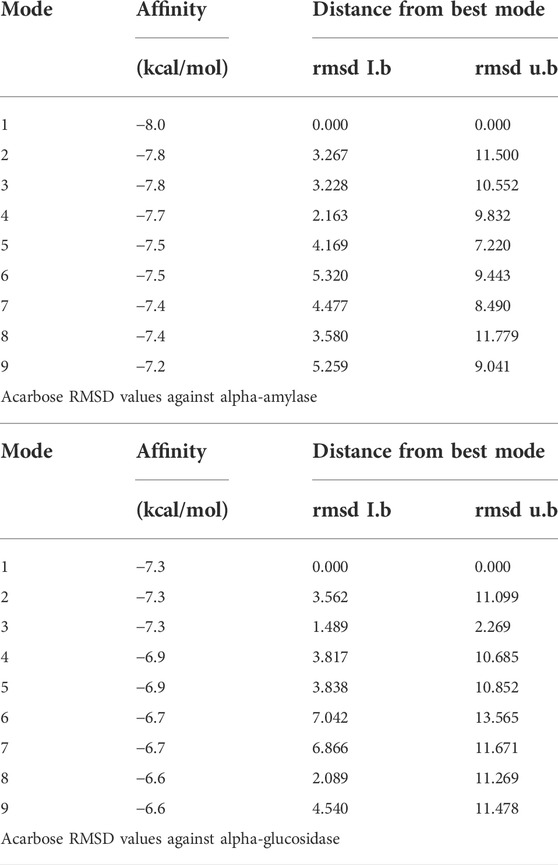
TABLE 4. RMSD values of different poses of standard acarbose drug against α-amylase and α-glucosidase enzymes.
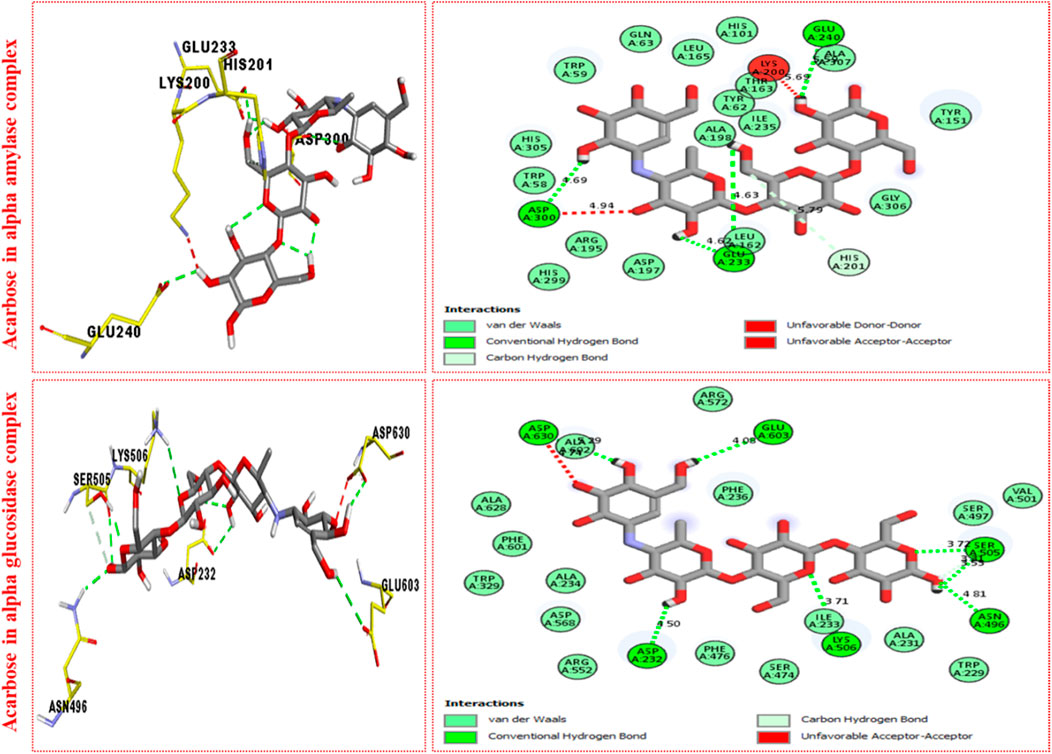
FIGURE 5. PLI profile indicates the corresponding surface of α-amylase and α-glucosidase enzymes and their interactions with the standard acarbose drug.
4 Conclusion
In conclusion, the present study synthesized novel hybrid analogs (1d–19d) based on a quinoline-bearing triazole moiety in their core structure. The entire constructed analogs were also additionally assessed for α-amylase and α-glucosidase (in vitro) inhibitory activities. All scaffolds being synthesized were found to display a diverse range of inhibitory potentials between 0.80 ± 0.05µM and 40.20 ± 0.70 µM (against α-amylase) and 1.20 ± 0.10 µM and 43.30 ± 0.80 µM (against α-glucosidase). Compound 11d (IC50 = 0.80 ± 0.05 µM) (IC50 = 1.20 ± 0.10 µM) holding para-fluoro substitution on the phenyl ring emerged as the most active analog among the synthesized series. Moreover, compound 9d with fluoro substitution at the ortho-position of the phenyl ring was identified as the second most active analog against α-amylase and α-glucosidase enzymes. Therefore, it was concluded based on SAR studies that scaffolds bearing electron-withdrawing groups such as–NO2, -Cl, and –F attached to the phenyl ring, resulting in enhanced inhibitory potentials. When these active scaffolds 11d and 9d underwent molecular docking analysis, they created a number of significant contacts with the active sites of the two targeted enzymes. The protein–ligand contact profile for compound 11d against α-amylase shows that it established numerous important interactions such as His-305 (donor interactions), Tyr-62 (π–π stacked), His-299 (donor interactions), Asp-3002 (H-B), Glu-233 (H-B), Leu-162 (π–R), and Trp-59 (π–π T-shaped), while against α-glucosidase it also furnishes numerous important interactions, including Met-470 (π-S), Ala-234 (π–R), Asp-568 (π–anion), Trp-329 (π–π T-shaped), Trp-432 (π–sigma), Arg-552 (H-B), and Asp-232 (π–anion). Furthermore, the structures of all the newly synthesized quinoline-based triazole analogs were confirmed by employing different techniques, including 1H NMR, HREI-MS, and 13C-NMRspectroscopy.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding authors.
Author contributions
The manuscript was written with the contributions of all authors. All authors have approved the final version of the manuscript.
Acknowledgments
The authors extend their appreciation to the Deanship of Scientific Research at King Khalid University for funding this work through the General Research Project under grant number (RGP.2/183/43). The authors would like to thank the Deanship of Scientific Research at Umm Al-Qura University for supporting this work by grant code: (22UQU4320141DSR21). Email: cmFwYXNoYUB1cXUuZWR1LnNh.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2022.995820/full#supplementary-material
References
Al-Ostoot, F. H., Salah, S., and Khanum, S. A. (2021). Recent investigations into synthesis and pharmacological activities of phenoxy acetamide and its derivatives (chalcone, indole and quinoline) as possible therapeutic candidates. J. Iran. Chem. Soc. 18 (8), 1839–1875. doi:10.1007/s13738-021-02172-5 |
Ammar, Y. A., Elhagali, G. A., Abusaif, M. S., Selim, M. R., Zahran, M. A., Naser, T., et al. (2021). Carboxamide appended quinoline moieties as potential anti-proliferative agents, apoptotic inducers and Pim-1 kinase inhibitors. Med. Chem. Res. 30 (9), 1649–1668. doi:10.1007/s00044-021-02765-y |
Ammazzalorso, A., Gallorini, M., Fantacuzzi, M., Gambacorta, N., De Filippis, B., Giampietro, L., et al. (2021). Design, synthesis and biological evaluation of imidazole and triazole-based carbamates as novel aromatase inhibitors. Eur. J. Med. Chem. 211, 113115. doi:10.1016/j.ejmech.2020.113115 | |
Asgher, M., Asad, M. J., Rahman, S. U., and Legge, R. L. (2007). A thermostable α-amylase from a moderately thermophilic Bacillus subtilis strain for starch processing. J. food Eng. 79 (3), 950–955. doi:10.1016/j.jfoodeng.2005.12.053 |
Atukuri, D., Vijayalaxmi, S., Sanjeevamurthy, R., Vidya, L., Prasannakumar, R., and Raghavendra, M. M. (2020). Identification of quinoline-chalcones and heterocyclic chalcone-appended quinolines as broad-spectrum pharmacological agents. Bioorg. Chem. 105, 104419. doi:10.1016/j.bioorg.2020.104419 | |
Cernicchi, G., Felicetti, T., and Sabatini, S. (2021). Microbial efflux pump inhibitors: A journey around quinoline and indole derivatives. Molecules 26 (22), 6996. doi:10.3390/molecules26226996 | |
Chauhan, A., Sharma, P. K., Srivastava, P., Kumar, N., and Dudhe, R. (2010). Plants having potential antidiabetic activity: A review. Der Pharm. Lett. 2 (3), 369–387.
Douadi, K., Chafaa, S., Douadi, T., Al-Noaimi, M., and Kaabi, I. (2020). Azoimine quinoline derivatives: Synthesis, classical and electrochemical evaluation of antioxidant, anti-inflammatory, antimicrobial activities and the DNA/BSA binding. J. Mol. Struct. 1217, 128305. doi:10.1016/j.molstruc.2020.128305 |
Fadaly, W. A., Elshaier, Y. A., Hassanein, E. H., and Abdellatif, K. R. (2020). New 1, 2, 4-triazole/pyrazole hybrids linked to oxime moiety as nitric oxide donor celecoxib analogs: Synthesis, cyclooxygenase inhibition anti-inflammatory, ulcerogenicity, anti-proliferative activities, apoptosis, molecular modeling and nitric oxide release studies. Bioorg. Chem. 98, 103752. doi:10.1016/j.bioorg.2020.103752 | |
Ghosh, S., Pal, S., Praveena, K. S. S., and Mareddy, J. (2020). 2-Chloro-7-methyl-3-((4-((p-tolyloxy) methyl)-1H-1, 2, 3-triazol-1-yl) methyl) quinoline: Crystal structure, hydrogen bonding and anti-bacterial activity. J. Mol. Struct. 1212, 128137. doi:10.1016/j.molstruc.2020.128137 |
Gunawan-Puteri, M. D., Kato, E., and Kawabata, J. (2012). α-Amylase inhibitors from an Indonesian medicinal herb, Phyllanthus urinaria. J. Sci. Food Agric. 92 (3), 606–609. doi:10.1002/jsfa.4615 | |
Gupta, O., Pradhan, T., Bhatia, R., and Monga, V. (2021). Recent advancements in anti-leishmanial research: Synthetic strategies and structural activity relationships. Eur. J. Med. Chem. 223, 113606. doi:10.1016/j.ejmech.2021.113606 | |
Jahani, R., Abtahi, S. R., Nematpour, M., Dastjerdi, H. F., Chamanara, M., Hami, Z., et al. (2020). Design, synthesis, and pharmacological evaluation of novel 1, 2, 4-triazol-3-amine derivatives as potential agonists of GABAA subtype receptors with anticonvulsant and hypnotic effects. Bioorg. Chem. 104, 104212. doi:10.1016/j.bioorg.2020.104212 | |
Jong-Anurakkun, N., Bhandari, M. R., and Kawabata, J. (2007). α-Glucosidase inhibitors from Devil tree (Alstoniascholaris). Food Chem. 103 (4), 1319–1323. doi:10.1016/j.foodchem.2006.10.043 |
Joshi, R., Kumari, A., Singh, K., Mishra, H., and Pokharia, S. (2020). Triorganotin (IV) complexes of Schiff base derived from 1, 2, 4-triazole moiety: Synthesis, spectroscopic investigation, DFT studies, antifungal activity and molecular docking studies. J. Mol. Struct. 1206, 127639. doi:10.1016/j.molstruc.2019.127639 |
Kumari, L., Mazumder, A., Pandey, D., Yar, M. S., Kumar, R., Mazumder, R., et al. (2019). Synthesis and biological potentials of quinoline analogues: A review of literature. Mini. Rev. Org. Chem. 16 (7), 653–688. doi:10.2174/1570193x16666190213105146 |
Li, W. L., Zheng, H. C., Bukuru, J., and De Kimpe, N. (2004). Natural medicines used in the traditional Chinese medical system for therapy of diabetes mellitus. J. Ethnopharmacol. 92 (1), 1–21. doi:10.1016/j.jep.2003.12.031 | |
Li, Z., Gu, J., Zhuang, H., Kang, L., Zhao, X., and Guo, Q. (2015). Adaptive molecular docking method based on information entropy genetic algorithm. Appl. Soft Comput. 26, 299–302. doi:10.1016/j.asoc.2014.10.008 |
Mallikarjuna, B. P., Sastry, B. S., Kumar, G. S., Rajendraprasad, Y., Chandrashekar, S. M., and Sathisha, K. (2009). Synthesis of new 4-isopropylthiazole hydrazide analogs and some derived clubbed triazole, oxadiazole ring systems–A novel class of potential antibacterial, antifungal and antitubercular agents. Eur. J. Med. Chem. 44 (11), 4739–4746. doi:10.1016/j.ejmech.2009.06.008 | |
Meneilly, G. S., Ryan, E. A., Radziuk, J., Lau, D. C., Yale, J. F., Morais, J., et al. (2000). Effect of acarbose on insulin sensitivity in elderly patients with diabetes. Diabetes care 23 (8), 1162–1167. doi:10.2337/diacare.23.8.1162 | |
Mrabti, N. N. (2018). QSAR study and molecular docking of benzimidazole derivatives inhibitors of p38 kinase. Moroc. J. Chem. 6 (3), 6–3. doi:10.48317/IMIST.PRSM/morjchem-v6i3.9273 |
Nesaragi, A. R., Kamble, R. R., Bayannavar, P. K., Shaikh, S. K. J., Hoolageri, S. R., Kodasi, B., et al. (2021). Microwave assisted regioselective synthesis of quinoline appended triazoles as potent anti-tubercular and antifungal agents via copper (I) catalyzed cycloaddition. Bioorg. Med. Chem. Lett. 41, 127984. doi:10.1016/j.bmcl.2021.127984 | |
Pitucha, M., Janeczko, M., Klimek, K., Fornal, E., Wos, M., Pachuta-Stec, A., et al. (2020). 1, 2, 4-Triazolin-5-thione derivatives with anticancer activity as CK1γ kinase inhibitors. Bioorg. Chem. 99, 103806. doi:10.1016/j.bioorg.2020.103806 | |
Rao, C. M. M. P., Naidu, N., Priya, J., Rao, K. P. C., Ranjith, K., Shobha, S., et al. (2021). Molecular docking and dynamic simulations of benzimidazoles with beta-tubulins. Bioinformation 17 (3), 404. doi:10.6026/97320630017404 | |
Salar, U., Khan, K. M., Chigurupati, S., Taha, M., Wadood, A., Vijayabalan, S., et al. (2017). New hybrid hydrazinyl thiazole substituted chromones: As potential α-amylase inhibitors and radical (DPPH & ABTS) scavengers. Sci. Rep. 7 (1), 16980–17017. doi:10.1038/s41598-017-17261-w | |
Sundarram, A., and Murthy, T. P. K. (2014). α-Amylase production and applications: A review. J. Appl. Environ. Microbiol. 2 (4), 166–175. doi:10.12691/jaem-2-4-10 |
Tabassum, R., Ashfaq, M., and Oku, H. (2021). Current pharmaceutical aspects of synthetic quinoline derivatives. Mini Rev. Med. Chem. 21 (10), 1152–1172. doi:10.2174/1389557520999201214234735 | |
Taha, M., Javid, M. T., Imran, S., Selvaraj, M., Chigurupati, S., Ullah, H., et al. (2017). Synthesis and study of the α-amylase inhibitory potential of thiadiazole quinoline derivatives. Bioorg. Chem. 74, 179–186. doi:10.1016/j.bioorg.2017.08.003 | |
Taha, M., Sultan, S., Imran, S., Rahim, F., Zaman, K., Wadood, A., et al. (2019). Synthesis of quinoline derivatives as diabetic II inhibitors and molecular docking studies. Bioorg. Med. Chem. 27 (18), 4081–4088. doi:10.1016/j.bmc.2019.07.035 | |
Velmurugan, K., Don, D., Kannan, R., Selvaraj, C., VishnuPriya, S., Selvaraj, G., et al. (2021). Synthesis, antibacterial, anti-oxidant and molecular docking studies of imidazoquinolines. Heliyon 7 (7), e07484. doi:10.1016/j.heliyon.2021.e07484 | |
Vinindwa, B., Dziwornu, G. A., and Masamba, W. (2021). Synthesis and evaluation of Chalcone-Quinoline based molecular hybrids as potential anti-malarial agents. Molecules 26 (13), 4093. doi:10.3390/molecules26134093 | |
Zaharudin, N., Staerk, D., and Dragsted, L. O. (2019). Inhibition of α-glucosidase activity by selected edible seaweeds and fucoxanthin. Food Chem. 270, 481–486. doi:10.1016/j.foodchem.2018.07.142 | |
Keywords: quinoline, triazole, α-amylase enzymes, α-glucosidase enzymes, molecular docking
Citation: Khan Y, Iqbal S, Shah M, Maalik A, Hussain R, Khan S, Khan I, Pashameah RA, Alzahrani E, Farouk A-E, Alahmdi MI and Abd-Rabboh HSM (2022) New quinoline-based triazole hybrid analogs as effective inhibitors of α-amylase and α-glucosidase: Preparation, in vitro evaluation, and molecular docking along with in silico studies. Front. Chem. 10:995820. doi: 10.3389/fchem.2022.995820
Received: 19 July 2022; Accepted: 17 August 2022;
Published: 15 September 2022.
Edited by:
Wagdy Mohamed Eldehna, Kafrelsheikh University, EgyptCopyright © 2022 Khan, Iqbal, Shah, Maalik, Hussain, Khan, Khan, Pashameah, Alzahrani, Farouk, Alahmdi and Abd-Rabboh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shahid Iqbal, c2hhaGlkZ2NzMTBAeWFob28uY29t; Shoaib Khan, c2hvYWlia2hhbnN3YXRpQGdtYWlsLmNvbQ==