 Qing Lang
Qing Lang Yuangang Xu
Yuangang Xu Ming Lu
Ming Lu- School of Chemical Engineering, Nanjing University of Science and Technology, Nanjing, Jiangsu, China
All-nitrogen materials, as a unique branch of energetic materials, have gained huge attentions, of which cyclo-N5− derivatives are the representative synthetically reported materials. However, the energetic performance of cyclo-N5− compounds has certain limitations and cannot go beyond that of CL-20. In order to reach the higher energy, in this work, we presented two kinds of polynitrogen species, N4 and N8. Two isomers of N4 and four isomers of N8 were fully calculated by using density functional theory (DFT). Theoretical results show that all these polynitrogen materials exhibit excellent heats of formation (7.92–16.60 kJ g−1), desirable detonation performance (D: 9766–11620 m s−1; p: 36.8–61.1 GPa), as well as the remarkable specific impulses (330.1–436.2 s), which are much superior to CL-20. Among them, N4-2 (tetraazahedrane) (D: 10037 m s−1; p: 40.1 GPa; Isp: 409.7 s) and cube N8-4 (D: 11620 m s−1; p: 61.1 GPa; Isp: 436.2 s) have the highest energetic properties, which are expected to become promising high-energy-density-materials. Moreover, electrostatic surface potentials, Frontier molecular orbitals, infrared spectra, natural bond orbital charges, and weak interactions were also investigated to further understand their relationship between structure and performance.
Introduction
In modern society, energetic materials (EMs) have played a significant role in both military and civilian fields, such as gas generating agents, propellants, and explosives for bombs and mines (Yin and Shreeve, 2015; Bu et al., 2019). With the continuous innovation and development of EMs, high-energy-density materials (HEDMs) have attracted great interests due to the desirable energy reserve (Zhang et al., 2015; Zhang et al., 2016; Wang et al., 2019). 2,4,6,8,10,12-hexanitro-2,4,6,8,10,12-hexaazaisowurtzitane (CL-20) and octanitrocubane (ONC) are two typical HEDMs with excellent energetic performance (CL-20: ρ: 2.04 g cm−3, D: 9445 m s−1, p: 46.7 GPa; ONC: ρ: 1.98 g cm−3, D: 10100 m s−1, p: 50.0 GPa) (Eaton et al., 2000; Lang et al., 2020). Unfortunately, few of the reported CHON high-energy-density materials so far have higher energy than CL-20, let alone ONC.
In terms of the above issues, all-nitrogen energetic materials have become a research hotspot, which are able to release huge energy by breaking the N-N (159.9 kJ mol−1), N = N (418.2 kJ mol−1) bonds in structure and forming N≡N (946 kJ mol−1) (Bondarchuk, 2020; Lang et al., 2021). However, the synthesis of all-nitrogen compounds is very difficult, resulting in extremely slow development. To date, only azido anion (N3−), pentanitrogen cation (N5+) and pentazolate anion (cyclo-N5-) are successfully synthesized (Christe et al., 1999; Christe, 2017; Wang et al., 2018). N5+ was first reported in 1995, and was proven to have a polyline structure. All N5+-based compounds were found to be very sensitive towards external stimuli and their synthesis needs harsh conditions like anhydrous, oxygen-free environment and ultra-low temperatures (Vij et al., 2001; Wilson et al., 2003). Hence, these materials are difficult to get further applications. On the contrary, cyclo-N5- can exist stably at normal pressure and temperature, and the first cyclo-N5- salt (N5)6(H3O)3(NH4)4Cl, was reported in 2017 (Zhang et al., 2017). Subsequently, a large number of cyclo-N5- energetic materials have been prepared and their energetic properties have also been fully estimated (Yang et al., 2018; Xu et al., 2019; Luo et al., 2020; Xu et al., 2022). It is disappointing that cyclo-N5- derivatives do not exhibit the expected high density and high detonation performance, whose energy is merely comparable (or even poorer) to RDX, and could not surpass CL-20 (Lin et al., 2020; Wozniak and Piercey, 2020). Main reason for this lies in the fact that all current pentazolate derivatives have the non-energetic or low-energetic cations (i.e., NH4+, N2H5+, etc.) in the structures, which affect the whole performance (Xu et al., 2020a). Therefore, to bring energy to a higher level, the transformation from all-nitrogen ions to pure all-nitrogen materials is essential, but also a great challenge (Scheme 1A).
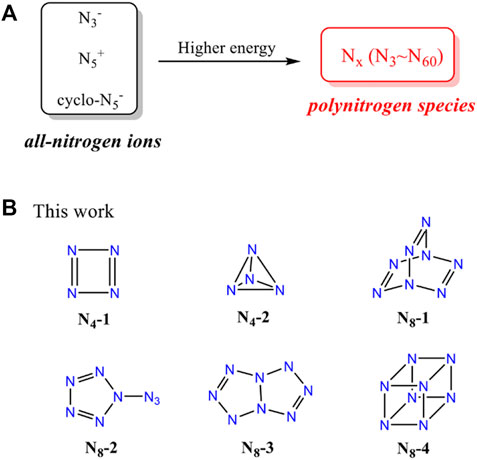
SCHEME 1. (A) The transformation from all-nitrogen ions to pure all-nitrogen materials. (B) The structures of N4 and N8 molecules in this work.
As computational chemistry matures, polynitrogen species (Nx, x from 3 to 60) are particularly attractive due to the extremely high heats of formation arising from abundant N-N and N = N high-energy bonds (Greschner et al., 2016; Türker, 2019). Meanwhile, the decomposition products of these materials are mostly N2, making them become green HEDMs (Wu et al., 2014). For the developments of Nx, density functional theory (DFT) calculations are helpful methods and widely used by many researches (Chen et al 1999; Arhangelskis et al., 2018). According to the theoretical evaluations, the heats of formation of polynitrogen species reach up to 2–5 kcal g−1, and detonation velocities even breakthrough the level of 10000 m s−1, which are much superior to CL-20 and ONC (Zarko, 2010; Türker, 2018). Moreover, calculations show that these materials have excellent specific impulses (300–500 s), allowing solid propellants equipped with high energetic efficiency to compete with liquid propellants (Türker, 2019). Thus, they exhibit great prospects and are expected to be next-generation high-energy-density materials.
In this work, we devoted to presenting a systematic study on the structures and properties of N4 and N8 species. Apart from the chain N4 and N8 (Samartzis and Wodtke, 2010; Hirshberg et al., 2014), two cyclo-N4 isomers and four cyclo-N8 isomers are calculated by using DFT methods, including optimized structures, molecular electrostatic potentials, Frontier molecular orbitals, non-covalent interaction, natural bond orbital, as well as energetic properties (Scheme 1B). Our study on the structure-property relationships of N4 and N8 conformation aims to a better understanding of polynitrogen materials.
Calculation methods
The molecular optimization and frequency calculations were performed by Gaussian 09 (Frisch et al., 2009) package under DFT-B3LYP method with 6–31++G(d,p) basis set. All of the optimized structures were characterized to be true local energy minimum on potential energy surfaces without imaginary frequencies. The geometrical configurations were optimized with no constraints imposed under default convergence criteria. Based on the optimized structures, the highest occupied molecular orbital (HOMO), the lowest unoccupied molecular orbital (LUMO), and infrared (IR) spectrum were calculated. And by using Multiwfn program (Lu and Chen, 2012), the electrostatic surface potential (ESP) and interaction region indicator (IRI) analysis were obtained at the same level of theory based on the optimized structures.
For covalent compounds, the intermolecular interactions within the crystals were considered to improve the accuracy of the density of the molecule (cm3 per molecule). So, an equation suggested by Politzer et al., (2009) was used.
Here M is the molecular mass, and Vm is the volume of the isolated gas phase molecule, ν is the balance of charges between positive potential and negative potential on molecular surface, and σtot2 is strengths and variabilities of the overall surface potentials. The coefficients α, β, γ are 0.9183, 0.0028, 0.0443, respectively.
The calculation of the detonation performance including detonation velocity (D) and detonation pressure (p) was performed with K-J equation proposed by Kamlet and Jacobs (1968).
where Q is heat of detonation, N is the number of moles of the gas generated per gram, M̅ is the average molecular weight of gaseous product, and ρ is the calculated density.In the cases of this study, all the molecules have the characteristic of c ≥ 2a + b/2, so the calculation method of Q, N, and M̅ are:
Where a, b, c and d stand for the number of C, H, O, N atoms in the explosive molecule, respectively; ΔHf is the standard heat of formation of the explosive. In addition, the detonation performances of these compounds were also evaluated by the EXPLO5 V6.05.04 program (Sućeska, 2020).The value of Isp was performed using the NASA Chemical Equilibrium with Applications (CEA) thermochemical code at 6.86 MPa with an expansion ratio 70:1, and the supersonic section ratio is considered to be Ae/At = 10, whereas the initial temperature is 298.15 K (Gordon and McBride, 1994; McBride, 1996).
Results and discussion
Molecular structure
The optimized structures of two N4 isomers and four N8 isomers are presented in Figure 1. The bond lengths of rectangle-shaped N4-1 are 1.256 Å (N2-N3) and 1.536 Å (N1-N2), and the bond angles are uniform 90°. N4-1 is a flat molecule, which can be evident from the dihedral angles of N4-N1-N2-N3, 0.021 and N1-N2-N3-N4, -0.017. Compound N4-2 exhibits a regular tetrahedron structure, with the bond length of 1.452 Å and bond angle of 60, and therefore is known as tetraazahedrane (TdN4). As for four N8 allotropes, N8-1shows a shape similar to a windmill, of which the bond lengths of double bonds (N1 = N2) are 1.220 Å and those of single bonds (N2-N3) are 1.509 Å. While N8-2 and N8-3 exhibit good planarity, with the dihedral angles of -179.9 (N8-2, N4-N1-N2-N3), -0.02 (N8-3, N5-N4-N3-N2) and 0.06 (N8-3, N5-N4-N8-N7). The N-N bonds distance in N8-2 range from 1.133 to 1.388 Å, where N2-N3 of azido group has the shortest value of 1.133 Å. The bond lengths of pentazole ring are 1.345 Å (N7-N8) and 1.304 Å (N6-N7), which are close to those of N8-3 (N1-N2, 1.323 Å; N3-N4, 1.337 Å). Similarly, the bond angles of pentazole ring in N8-2 and N8-3 are both in the range of 103.7°–114.1°. N8-4 shows a special cube structure, possessing the bond length of 1.521 Å and bond angle of 90.
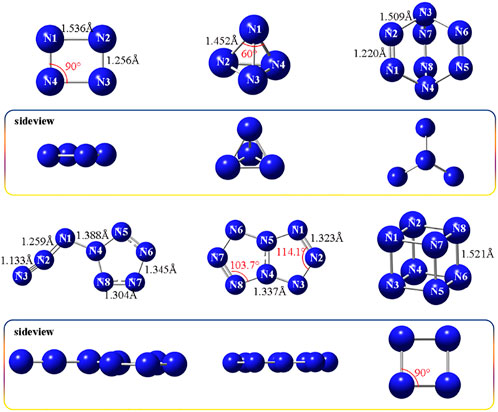
FIGURE 1. Molecular structures of N4-1, N4-2, N8-1, N8-2, N8-3, and N8-4, respectively.
In general, the longer bond length may mean the lower stability. For N4 isomers, tetraazahedrane (N4-2) has the shorter N-N bonds than rectangle N4-1, suggesting that the three-dimensional tetrahedral structure is conducive to enhancing the molecular stability. In addition to chain N4, tetrahedral form of N4 has already received growing concerns (Cacace et al., 2002; Nguyen, 2003; Samartzis and Wodtke, 2010). Among the N8 isomers, cube N8 (N8-4) has the longest bond distance of 1.521 Å. The structure of this material is the analogue of cubane (CH)8, which is the skeleton of high-energy-density material ONC. The internuclear N–N–N angles of 90° are far from the standard angle, which is considered to store powerful energy. ONC with remarkable energetic performance is a representative example. However, the longer bond lengths and the unusual bond angles of N8-4 may generate the possibility of instability. During subsequent calculations, we found that N8-4 has the highest total energy (-437.44 au) among four N8 isomers (Supplementary Table S1), and the result is in agreement with the structural features.
Electrostatic surface potential analysis
Electrostatic surface potentials (ESP) of six N4 and N8 materials were analyzed by Multiwfn program (Manzetti and Lu, 2013). As shown in Figure 2, The minima and maxima of ESP are labeled by blue and red, respectively. The negative electronic potentials in blue indicating the strongest attraction are scattered on the edge of the molecules, while the positive potentials in red representing the strongest repulsion are located at the center of the molecules. Overall, the values of electronic potentials of N4 isomers are smaller than those of N8 isomers, and N4-2 has the lower maximum of ESP with the value of 18.74 kcal mol−1 than N4-1 (33.23 kcal mol−1). As for N8 compounds, N8-3 and N8-4 exhibit the largest maximum surface of ESP with the values of 48.46 kcal mol−1 and 41.42 kcal mol−1, respectively. The surface minima and maximum of ESP also illustrate the main electrophilic and nucleophile reaction sites, respectively. Meanwhile, molecular surface area in different electrostatic potential intervals was obtained to help us to better understand quantitative distribution of ESP (Figure 3). Among six materials, the distribution of ESP for N8-2 is most uniform, while the distributions of ESP for others show the characteristics of “high middle and low sides”.

FIGURE 2. Electrostatic surface potentials mapped molecular vdW surface of N4-1, N4-2, N8-1, N8-2, N8-3, and N8-4. Significant surface local minima and maxima of ESP are represented as orange and cyan spheres, and labelled by dark blue and red texts, respectively. The unit is in kcal/mol.
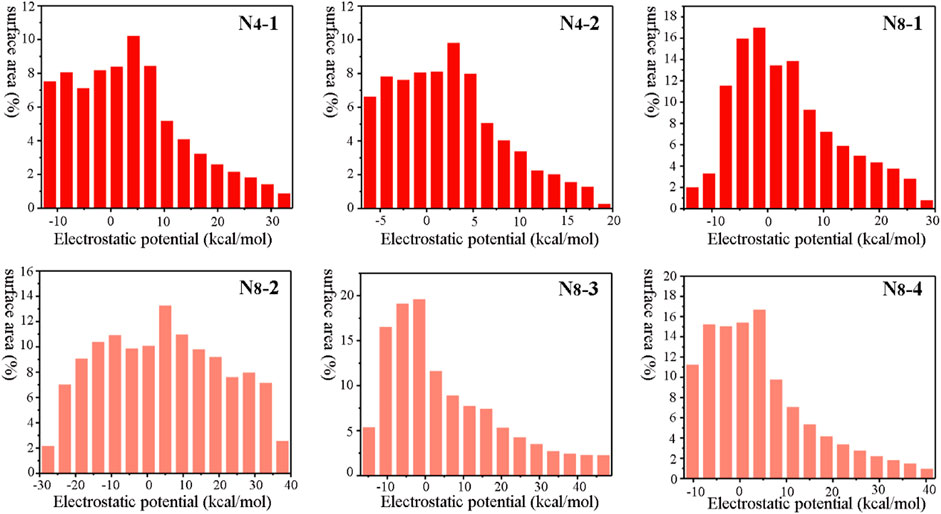
FIGURE 3. Molecular surface area of N4-1, N4-2, N8-1, N8-2, N8-3, and N8-4, respectively.
Vibration analysis
The IR spectra of the all-nitrogen compounds were calculated and the results are presented in Figure 4. Both N4 isomers show only one typical peak, as well as the cube N8, of which the peaks are at 561, 980, 792 cm−1, respectively. For other N8 isomers, the spectra are more complex. The strong peak at 1527 cm−1 of N8-1 is assigned to stretching vibration modes of the three N=N bonds in structure, while other two weak peaks at 467 and 1093 cm−1 arise from the stretching vibrations of single bonds. As for N8-2, the peaks at 980, 1204, 1408 cm−1 result from the stretching vibration modes of pentazole ring, and the strongest absorption at 2262 cm−1 arises from the stretching vibrations of N1–N2 and N2–N3 bonds in azide group. Another weak absorption at 784 cm−1 is assigned to stretching vibration of N1-N4 bond connecting pentazole and -N3. The peak at 235 cm−1 in the spectrum of N8-3 arises from the torsion vibration of fused N4-N5 bond. And others at 965, 1027, 1316 cm−1 belong to the stretching vibrations of two five-membered rings, of which the strongest absorption at 1027 cm−1 is stretching vibrations of N1-N2, N7-N8.
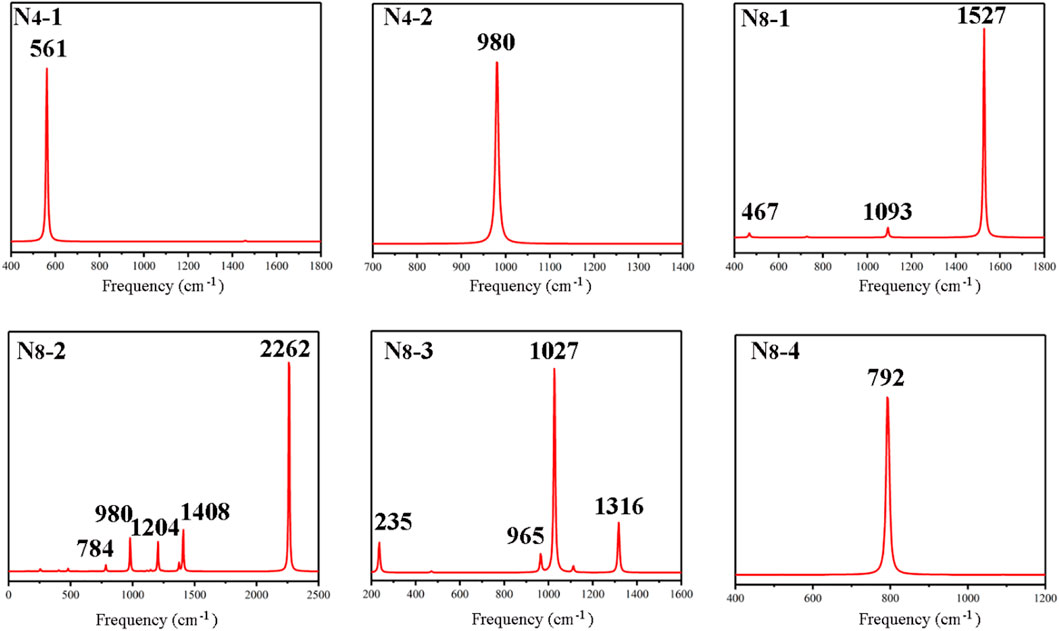
FIGURE 4. Calculated IR spectra for N4-1, N4-2, N8-1, N8-2, N8-3, and N8-4, respectively.
Weak interactions and orbitals analysis
Exploring the noncovalent interactions of a molecule is beneficial to help us understand the structural characteristics and stabilities of energetic materials. In this work, interaction region indicator (IRI) analysis as an effective method were adopted and the results are shown in Figure 5 (Lu and Chen, 2021). There are two kinds of interactions in each molecule, which are the chemical bonding interactions in blue isosurface and the steric effects in red. For all nitrogen allotropes, the repulsing interactions are concentrated in the center of the rings. Additionally, the scatter maps between IRI and sign(λ2)ρ of all compounds are also obtained. Several spikes in the range of -0.4–-0.25 au of all compounds correspond to the chemical bonding interactions, while the spikes ranging in 0–0.1 au mean the repulsive forces. It can be seen that there is no spike in the scatter plot of two N4 isomers, suggesting their weaker repulsions than N8 isomers. Among the N8 allotropes, the plot of cube N8 (N8-4) has two spikes with the value of 0.030 and 0.075 au, and others only respectively have one spike. According to the above analysis, cube N8 seems to exhibit the stronger steric effects and thus the poorer stability than other N8 conformations.
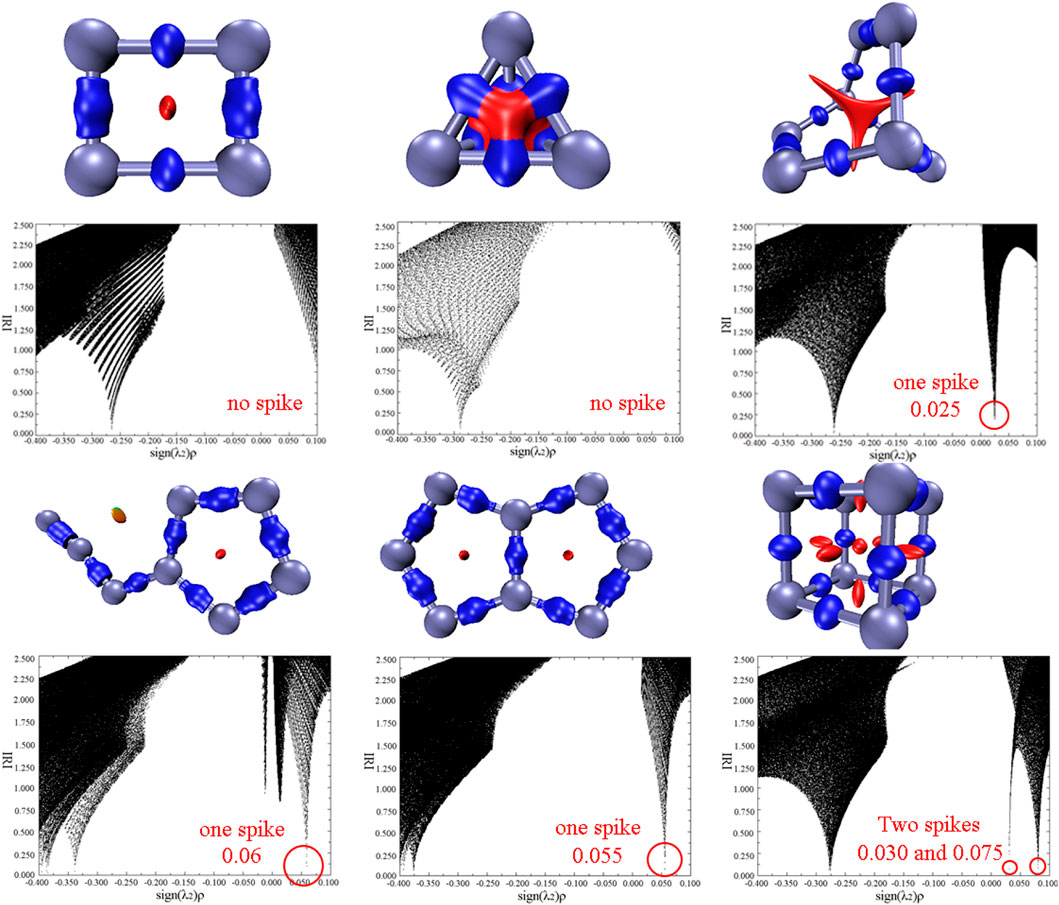
FIGURE 5. Isosurface maps of IRI= 1.0, and scatter map between IRI and sign(λ2)ρ for N4-1, N4-2, N8-1, N8-2, N8-3, and N8-4, respectively. λ2 represents the second largest eigenvalue of Hessian of the electron density, and ρ is the electron density.
In order to further investigate the molecular stability, the FMOs including highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) of all N4 and N8 isomers were explored. The HOMO and LUMO orbitals and the energy gaps are presented in Figure 6. The red parts represent the positive phase and the green represent the negative phase. The HOMOs and LUMOs are concentrated on each N atom in molecules, except for N8-3, whose HOMOs and LUMOs mainly occupy the unfused N atoms. The HOMOs-LUMOs distributions of these N4 and N8 materials exhibit good symmetrical features, apart from N8-2. The energy gaps for all materials are 2.74, 11.30, 5.36, 5.24, 6.25, 5.06 eV, respectively. Previous researches have demonstrated that a molecule with better stability may possess higher energy gap (Kosar and Albayrak, 2011; He et al., 2019). Hence, it can be preliminarily judged that tetrahedral-formed N4 has superior molecular stability than rectangle one due to the higher energy gaps. For N8 isomers, N8-3 with fused-ring structure has the largest energy gap and cube-shaped N8-4 has the lowest value, revealing the better stability of fused structures and the poor stability of caged materials.

FIGURE 6. HOMO-LUMO energy levels and energy gaps of N4-1, N4-2, N8-1, N8-2, N8-3, and N8-4, respectively.
Natural bond orbital analysis
Natural bond orbital charges (Glendening et al., 2011) of four N8 isomers were calculated to understand their charge, bond order, and bond type, and the results are presented in Figure 7. Interestingly, two N atoms in all N8 isomers exhibit positive charges, whereas the other six N atoms exhibit negative charges. N8-2 shows the most positive charge with the value of 1.519 (N2). For N8-1 and N8-3, their distributions of NBO charges are axisymmetric, which are consistent with the characteristics of their structures. And the N atoms in N8-3 has the more negative charges than those in N8-1. The negative charges in N8-4 are -0.005, which is the lowest among the four N8 isomers.
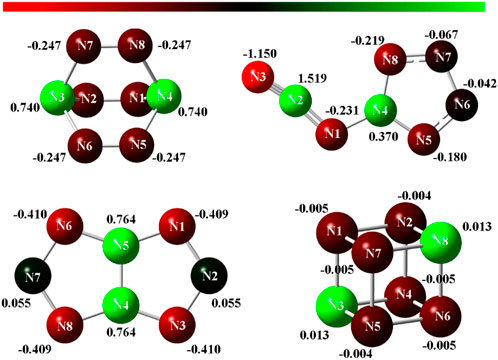
FIGURE 7. Natural bond orbital charges of N4-1, N4-2, N8-1, N8-2, N8-3, and N8-4, respectively.
Energetic properties
In order to accurately predict the detonation properties of all N4 and N8 isomers, the density and the heat of formation as two important factors should be obtained in advanced. In this study, the densities of all materials were calculated using Equation. 1 proposed by Politzer et, al, and the gas phase heats of formation (ΔfH298K(g)) were estimated by the atomization approach (Table 2 and Supplementary Table S2). Subsequently, based on the densities and enthalpies, the detonation performance is calculated by both K-J equation and EXPL O 5 program, which are currently the most widely used computational methods.
As presented in Table 1, because of the more N atoms in structures, N8 isomers have much higher densities than N4 ones. The density of tetrahedral N4 (N4-2) and cube N8 (N8-4) are highest, with the values of 1.502 g cm−3 and 1.749 g cm−3. The densities of other three N8 molecules are relatively similar in the range of 1.664–1.697 g cm−3. Unfortunately, the densities of these all-nitrogen materials seem no advantages compared to traditional CHON energetic materials, like RDX (1.80 g cm−3), HMX (1.91 g cm−3) and CL-20 (2.04 g cm−3).

TABLE 1. B3LYP-6-311++G(d,p) computed properties and calculated densities for N4 and N8 allotropes.
The heats of formation (details in Supporting Information) and detonation performance of all compounds are shown in Table 2. It is worth noting that these N4 and N8 allotropes has very excellent heats of formation of 7.92–16.60 kJ g−1, of which cube N8 has the highest value of 16.60 kJ g−1. This may arise from the numerous N-N and N=N bonds in the structure. The excellent enthalpies of these all-nitrogen materials are much superior to representative CHON explosives, such as RDX (0.36 kJ g−1), HMX (0.35 kJ g−1) and CL-20 (0.90 kJ g−1). In addition, the six poly-nitrogen materials exhibit prominent detonation performance. The values of detonation velocities fall in the range of 9747–11620 m s−1, while the detonation pressures are in the range of 36.80–61.10 GPa (calculated by EXPL O 5). Among them, N8 isomers show higher detonation performance than N4 ones due to the higher density. There is no doute that N8-4 has the best detonation performance (D: 11620 m s−1; p: 61.1 GPa). All the all-nitrogen materials presented in this work has much higher energy density than CL-20 (D: 9445 m s−1, p: 46.7 GPa). And compared to N5+N3− in our previous work (Xu et al., 2020b), N8-2 (ρ: 1.669 g cm−3, D: 9747 m s−1, p: 38.0 GPa) exhibits higher density and higher detonation performance based on the same structural composition of pentazole ring and azide (N5+N3−: ρ: 1.55 g cm−3, D: 9290 m s−1, p: 34.67 GPa). The above discussion not only confirms that all-nitrogen materials have the great prospects because of the huge energy, but also that covalent poly-nitrogen compounds would be more powerful than all-nitrogen salts.
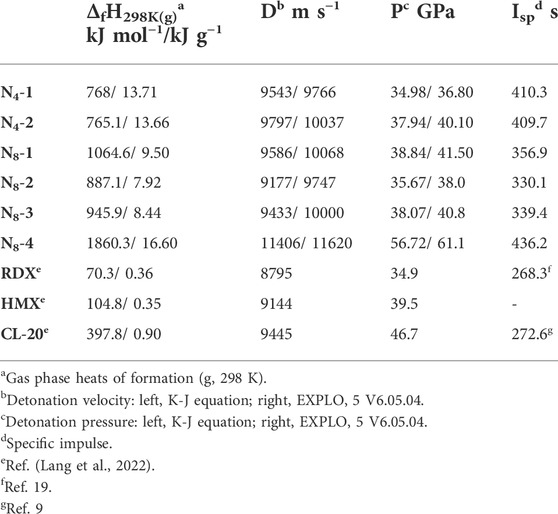
TABLE 2. Physico-chemical properties of N4 and N8 allotropes compared with RDX, HMX and CL-20.
In addition to the excellent detonation performance, N4 and N8 isomers also possess exceptional theoretical specific impulse (Isp). The values of Isp for two N4 isomers are 410.3 s and 409.7 s, respectively, which are higher than those of N8-1 (356.9 s), N8-2 (330.1 s), N8-3 (339.4 s), but poorer than N8-4 (436.2 s). Cube N8 in this work combines the best detonation performance and best specific impulse, suggesting the unique advantages of cage structure. And the specific impulses (330.1–436.2 s) of six poly-nitrogen molecules are much superior to that of CL-20 (272.6 s). It is believed that these ploy-nitrogen materials are expected to become the promising candidates as solid rocket propellants.
Conclusion
In this study, two structures of cyclo-N4 (N4-1, N4-2) and four structures of cyclo-N8 (N8-1, N8-2, N8-3, N8-4) were designed and calculated through density functional theory methods. The optimized structures, molecular electrostatic potentials, Frontier molecular orbitals, non-covalent interaction, natural bond orbital, and most importantly, the energetic properties of six poly-nitrogen materials were fully investigated. According to the theoretical calculation, N4-2 with tetrahedron structure possesses shorter N-N bond lengths than rectangle-shaped N4-1, and thus the higher molecular stability. Cube N8-4 has the longest bond lengths and unusual bond angle among N8 isomers, which may lead to the poor stability. The results from Frontier molecular orbitals and non-covalent interaction are also consistent with structure analysis. The densities of six compounds are 1.441–1.749 g cm−3, and the heats of formation are 7.92–16.60 kJ g−1. With such remarkable enthalpies, these N4 and N8 isomers exhibit excellent detonation performance (D: 9747–11620 m s−1; p: 36.8–61.6 GPa) and high specific impulses (Isp: 330.1–436.2 s), which are superior to CL-20 (D: 9445 m s−1; p: 46.7 GPa, Isp: 272.6 s). In particular, N4-2 and N8-4 have highest energy density among N4 and N8 species, respectively, revealing their huge development potential as next-generation HEDMs beyond CL-20.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding authors.
Author contributions
ML, PW, QLI, and YX designed the study QLA and YX conducted the DFT calculation QLA. prepared the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (No. 22105102, 22135003 and 21975127), Young Elite Scientist Sponsorship Program by CAST (No. YESS20210074), and the Fundamental Research Funds for the Central Universities (No. 30921011204).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2022.993036/full#supplementary-material
References
Arhangelskis, M., Katsenis, A. D., Morris, A. J., and Friscic, T. (2018). Computational evaluation of metal pentazolate frameworks: Inorganic analogues of azolate metal-organic frameworks. Chem. Sci. 9 (13), 3367–3375. doi:10.1039/c7sc05020h
Bondarchuk, S. V. (2020). Bipentazole (N10): A low-energy molecular nitrogen allotrope with high intrinsic stability. J. Phys. Chem. Lett. 11 (14), 5544–5548. doi:10.1021/acs.jpclett.0c01542
Bu, R., Xiong, Y., Wei, X., Li, H., and Zhang, C. (2019). Hydrogen bonding in CHON-containing energetic crystals: A review. Cryst. Growth Des. 19 (10), 5981–5997. doi:10.1021/acs.cgd.9b00853
Cacace, F., Petris, G., and Troiani, A. (2002). Experimental detection of tetranitrogen. science 295, 480–481. doi:10.1126/science.1067681
Chen, C., Sun, K., and Shyu, S. (1999). Theoretical study of various N10 structures. J. Mol. Struct. THEOCHEM 459, 113–122. doi:10.1016/S0166-1280(98)00260-7
Christe, K. O. (2017). Polynitrogen chemistry enters the ring. Science 355 (6323), 351. doi:10.1126/science.aal5057
Christe, K. O., Wilson, W. W., Sheehy, J. A., and Boatz, J. A. (1999). N5+: A novel homoleptic polynitrogen ion as a high energy density. Mater. Angew. Chem. Int. Ed. 38 (13), 2180–2184. doi:10.1002/1521-3773(20010817)40:16<2947::AID-ANIE11112947>3.0.CO;2-C
Eaton, P. E., Gilardi, R. L., and Zhang, M. (2000). Polynitrocubanes: Advanced high-density, high-energy materials. Adv. Mat. 12 (15), 1143–1148. doi:10.1002/1521-4095(200008)12:15<1143::AID-ADMA1143>3.0
Frisch, M. J., Trucks, G. W., and Schlegel, H. B. (2009). Gaussian 09, revision A.02. Wallingford, CT: Gaussian, Inc.
Glendening, E. D., Landis, C. R., and Weinhold, F. (2011). Natural bond orbital methods. WIREs Comput. Mol. Sci. 2 (1), 1–42. doi:10.1002/wcms.51
Gordon, S., and McBride, B. J. (1994). Computer program for calculation of complex chemical equilibrium compositions and applications, Part 1: Analysis. Cleveland, Ohio: NASA Lewis Research Center.
Greschner, M. J., Zhang, M., Majumdar, A., Liu, H., Peng, F., Tse, J. S., et al. (2016). A new allotrope of nitrogen as high-energy density material. J. Phys. Chem. A 120 (18), 2920–2925. doi:10.1021/acs.jpca.6b01655
He, P., Zhang, J., and Wu, J. (2019). DFT studies on new family of high‐energy density energetic bis(trinitromethyl) azo tetrazoles and triazoles. J. Phys. Org. Chem. 32, e3953. doi:10.1002/poc.3953
Hirshberg, B., Gerber, R. B., and Krylov, A. I. (2014). Calculations predict a stable molecular crystal of N8. Nat. Chem. 6 (1), 52–56. doi:10.1038/nchem.1818
Kamlet, M. J., and Jacobs, S. J. (1968). Chemistry of detonations. I. A simple method for calculating detonation properties of C–H–N–O explosives. J. Chem. Phys. 48 (1), 23–35. doi:10.1063/1.1667908
Kosar, B., and Albayrak, C. (2011). Spectroscopic investigations and quantum chemical computational study of (E)-4-methoxy-2-[(p-tolylimino)methyl]phenol. Spectrochimica Acta Part A Mol. Biomol. Spectrosc. 78, 160–167. doi:10.1016/j.saa.2010.09.016
Lang, Q., Jiang, S., Xu, Y., and Lu, M. (2022). Theoretical study on N-oxide pentazolate high-energy-density materials: Toward excellent energetic performance and good stability. J. Phys. Org. Chem. 35 (7). doi:10.1002/poc.4342
Lang, Q., Sun, Q., Wang, Q., Lin, Q., and Lu, M. (2020). Embellishing bis-1, 2, 4-triazole with four nitroamino groups: Advanced high-energy-density materials with remarkable performance and good stability. J. Mat. Chem. A 8 (23), 11752–11760. doi:10.1039/d0ta03008b
Lang, Q., Sun, Q., Xu, Y., Wang, P., Lin, Q., and Lu, M. (2021). From mono-rings to bridged bi-rings to caged bi-rings: A promising design strategy for all-nitrogen high-energy-density materials N10 and N12. New J. Chem. 45 (14), 6379–6385. doi:10.1039/d1nj00522g
Lin, Q., Wang, P., Xu, Y., and Lu, M. (2020). Pentazolate anion cyclo-N5−: Development of a new energetic. Mater. Eng. 6 (9), 964–966. doi:10.1016/j.eng.2020.04.011
Lu, T., and Chen, F. (2012). Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 33 (5), 580–592. doi:10.1002/jcc.22885
Lu, T., and Chen, Q. (2021). Interaction region indicator: A simple real space function clearly revealing both chemical bonds and weak interactions. Chem. Methods. 1 (5), 231–239. doi:10.1002/cmtd.202100007
Luo, J., Xia, H., Zhang, W., Song, S., and Zhang, Q. (2020). A promising hydrogen peroxide adduct of ammonium cyclopentazolate as a green propellant component. J. Mat. Chem. A 8 (25), 12334–12338. doi:10.1039/d0ta03010d
Manzetti, S., and Lu, T. (2013). The geometry and electronic structure of aristolochic acid: Possible implications for a frozen resonance. J. Phys. Org. Chem. 26 (6), 473–483. doi:10.1002/poc.3111
McBride, B. J. (1996). Computer program for calculation of complexchemical equilibrium compositions and applications. Cleveland, Ohio: NASA Lewis Research Center.
Nguyen, M. T. (2003). Polynitrogen compounds. Coord. Chem. Rev. 244, 93–113. doi:10.1016/S0010-8545(03)00101-2
Politzer, P., Martinez, J., Murray, J. S., Concha, M. C., and Toro-Labbé, A. (2009). An electrostatic interaction correction for improved crystal density prediction. Mol. Phys. 107 (19), 2095–2101. doi:10.1080/00268970903156306
Samartzis, P. C., and Wodtke, A. M. (2010). All-nitrogen chemistry: How far are we from N60? Int. Rev. Phys. Chem. 25 (4), 527–552. doi:10.1080/01442350600879319
Türker, L. (2019). A density functional study on some cyclic N10 isomers. Def. Technol. 15 (2), 154–161. doi:10.1016/j.dt.2018.08.005
Türker, L. (2018). Contemplation on some cyclic N8 isomers-A DFT treatment. Def. Technol. 14 (1), 19–27. doi:10.1016/j.dt.2017.04.009
Vij, A., Wilson, W. W., Vij, V., Tham, F. S., Sheehy, J. A., and Christe, K. O. (2001). Polynitrogen chemistry. Synthesis, characterization, and crystal structure of surprisingly stable fluoroantimonate salts of N5+. J. Am. Chem. Soc. 123, 6308–6313. doi:10.1021/ja010141g
Wang, P., Xu, Y., Lin, Q., and Lu, M. (2018). Recent advances in the syntheses and properties of polynitrogen pentazolate anion cyclo-N5- and its derivatives. Chem. Soc. Rev. 47 (20), 7522–7538. doi:10.1039/c8cs00372f
Wang, Y., Song, S., Huang, C., Qi, X., Wang, K., Liu, Y., et al. (2019). Hunting for advanced high-energy-density materials with well-balanced energy and safety through an energetic host–guest inclusion strategy. J. Mat. Chem. A 7 (33), 19248–19257. doi:10.1039/c9ta04677a
Wilson, W. W., Vij, A., Vij, V., Bernhardt, E., and Christe, K. O. (2003). Polynitrogen chemistry: Preparation and characterization of (N5)2SnF6, N5SnF5, and N5B(CF3)4. Chem. Eur. J. 9 (12), 2840–2844. doi:10.1002/chem.200304973
Wozniak, D. R., and Piercey, D. G. (2020). Review of the current synthesis and properties of energetic pentazolate and derivatives thereof. Engineering 6 (9), 981–991. doi:10.1016/j.eng.2020.05.019
Wu, Z., Benchafia el, M., Iqbal, Z., and Wang, X. (2014). N8(-) polynitrogen stabilized on multi-wall carbon nanotubes for oxygen-reduction reactions at ambient conditions. Angew. Chem. Int. Ed. Engl. 53 (46), 12555–12559. doi:10.1002/anie.201403060
Xu, Y., Ding, L., Yang, F., Li, D., Wang, P., Lin, Q., et al. (2022). LiN5: A novel pentazolate salt with high nitrogen content. Chem. Eng. J. 429, 132399. doi:10.1016/j.cej.2021.132399
Xu, Y., Li, D., Tian, L., Jiang, Z., Wang, P., and Lu, M. (2020a2020). Prediction of the energetic performance of pentazolate salts. Chin. J. Energetic Mater. 28 (8), 718–723. doi:10.11943/CJEM2019208
Xu, Y., Tian, L., Li, D., Wang, P., and Lu, M. (2019). A series of energetic cyclo-pentazolate salts: Rapid synthesis, characterization, and promising performance. J. Mat. Chem. A 7 (20), 12468–12479. doi:10.1039/c9ta01077g
Xu, Y., Wang, P., Lin, Q., and Lu, M. (2020b). Density functional theory studies on two novel poly-nitrogen compounds: N5+N3− and N5+N5−. J. Phys. Org. Chem. 34 (2). doi:10.1002/poc.4135
Yang, C., Zhang, C., Zheng, Z., Jiang, C., Luo, J., Du, Y., et al. (2018). Synthesis and characterization of cyclo-pentazolate salts of NH4(+), NH3OH(+), N2H5(+), C(NH2)3(+), and N(CH3)4(+). J. Am. Chem. Soc. 140 (48), 16488–16494. doi:10.1021/jacs.8b05106
Yin, P., and Shreeve, J. M. (2015). From N-nitro to N-nitroamino: Preparation of high-performance energetic materials by introducing nitrogen-containing ions. Angew. Chem. Int. Ed. 54 (48), 14513–14517. doi:10.1002/anie.201507456
Zarko, V. E. (2010). Searching for ways to create energetic materials based on polynitrogen compounds (review). Combust. Explos. Shock Waves 46 (2), 121–131. doi:10.1007/s10573-010-0020-x
Zhang, C., Sun, C., Hu, B., Yu, C., and Lu, M. (2017). Synthesis and characterization of the pentazolate anion cyclo-N5ˉ in (N5)6(H3O)3(NH4)4Cl. Science 355, 374–376. doi:10.1126/science.aah3840
Zhang, J., Dharavath, S., Mitchell, L. A., Parrish, D. A., and Shreeve, J. M. (2016). Energetic salts based on 3, 5-Bis(dinitromethyl)-1, 2, 4-triazole monoanion and dianion: Controllable preparation, characterization, and high performance. J. Am. Chem. Soc. 138 (24), 7500–7503. doi:10.1021/jacs.6b03819
Keywords: ploynitrogen, high-energy-density materials, DFT calculation, energetic performance, propellant
Citation: Lang Q, Lin Q, Wang P, Xu Y and Lu M (2022) Density functional theory studies on N4 and N8 species: Focusing on various structures and excellent energetic properties. Front. Chem. 10:993036. doi: 10.3389/fchem.2022.993036
Received: 13 July 2022; Accepted: 18 August 2022;
Published: 08 September 2022.
Edited by:
Alexander Shtukenberg, New York University, United StatesReviewed by:
Venkataramanan N. S, K. Ramakrishnan College of Engineering, IndiaPing Yin, Beijing Institute of Technology, China
Copyright © 2022 Lang, Lin, Wang, Xu and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuangang Xu, eXVhbmdhbmd4dUAxNjMuY29t; Ming Lu, bHVtaW5nQG5qdXN0LmVkdS5jbg==