
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. , 27 September 2022
Sec. Medicinal and Pharmaceutical Chemistry
Volume 10 - 2022 | https://doi.org/10.3389/fchem.2022.984069
This article is part of the Research Topic Frontiers in Chemistry: Editors Showcase 2021 View all 50 articles
 Rebecca Ferrisi1
Rebecca Ferrisi1 Francesca Gado1,2*†
Francesca Gado1,2*† Beatrice Polini3
Beatrice Polini3 Caterina Ricardi3Kawthar A. Mohamed4Lesley A. Stevenson5
Caterina Ricardi3Kawthar A. Mohamed4Lesley A. Stevenson5 Gabriella Ortore1
Gabriella Ortore1 Simona Rapposelli1Giuseppe Saccomanni1
Simona Rapposelli1Giuseppe Saccomanni1 Roger G. Pertwee5
Roger G. Pertwee5 Robert B. Laprairie4,6
Robert B. Laprairie4,6 Clementina Manera1,7*
Clementina Manera1,7* Grazia Chiellini3,7*
Grazia Chiellini3,7*It is well known that G protein–coupled receptors (GPCRs) assume multiple active states. Orthosteric ligands and/or allosteric modulators can preferentially stabilize specific conformations, giving rise to pathway-biased signaling. One of the most promising strategies to expand the repertoire of signaling-selective GPCR activators consists of dualsteric agents, which are hybrid compounds consisting of orthosteric and allosteric pharmacophoric units. This approach proved to be very promising showing several advantages over monovalent targeting strategies, including an increased affinity or selectivity, a bias in signaling pathway activation, reduced off-target activity and therapeutic resistance. Our study focused on the cannabinoid receptor type 2 (CB2R), considered a clinically promising target for the control of brain damage in neurodegenerative disorders. Indeed, CB2R was found highly expressed in microglial cells, astrocytes, and even in some neuron subpopulations. Here, we describe the design, synthesis, and biological evaluation of two new classes of potential dualsteric (bitopic) CB2R ligands. The new compounds were obtained by connecting, through different linkers, the pharmacophoric portion of the CB2R positive allosteric modulator (PAM), EC21a, with that of the CB2R selective orthosteric agonist LV62, both developed in our laboratories. A preliminary screening enabled us to identify compound JR64a as the most promising of the series. Indeed, functional examination highlighted a signaling ‘bias’ in favor of G protein activation over βarrestin2 recruitment, combined with high affinity for CB2R and the ability to efficiently prevent inflammation in human microglial cells (HMC3) exposed to LPS/TNFα stimulation, thus demonstrating great promise for the treatment of neurodegenerative diseases.
Several neurodegenerative disorders display alterations in components of the endocannabinoid system (ECS), and a cannabinoid-based approach has proven efficacious in the reversal of certain neurodegenerative events in pre-clinical models of neuroinflammation, oxidative stress, and neuronal loss, among others (Basavarajappa et al., 2009; Di Marzo 2009; Aymerich et al., 2018; Mastinu et al., 2018). The ECS is a complex signaling system consisting of cannabinoid receptors, their endogenous ligands (known as “endocannabinoids”), and the enzymes responsible for endocannabinoid biosynthesis, cellular uptake and catabolism (Bisogno et al., 2005). The effects of endocannabinoids are primarily mediated by CB1 and CB2 cannabinoid receptors (CB1Rs and CB2Rs), which are G protein-coupled receptors (GPCRs), predominantly associated with Gαi/o proteins (Howlett et al., 2002). Their activation inhibits adenylyl cyclase and certain voltage dependent calcium channels, and regulates mitogen-activated protein kinase (MAPK) and phosphatidylinositol-3-kinase (PI3K) pathways (Howlett et al., 2002). CB1Rs are highly expressed in the central nervous system (CNS) (Mackie 2005), where they play a well-established role in regulating neuronal excitability. In contrast, CB2Rs have reduced expression in the brain compared to CB1Rs. Indeed, the expression of CB2R in the brain has been mainly identified at the level of microglia and vascular elements (Walter et al., 2003; Ramirez et al., 2012), and their modulation is not accompanied by psychotropic side effects associated with the activation of CB1Rs.
Notably, activation of CB2Rs results in inhibition of neuroinflammatory signaling pathways (Bie et al., 2018); therefore, this receptor type may be a clinically promising target for the control of brain damage in neurodegenerative disorders, including neuropathic pain, Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, and multiple sclerosis (Benito et al., 2003; Benito et al., 2005; Han et al., 2013; Chung et al., 2016; Xu et al., 2016; Ferrisi et al., 2021). However, to date only a few synthetic CB2R agonists have reached an advanced stage of clinical trials (from ClinicalTrials.gov: GW842166X, S-777469, and JTE-907), probably because of the predominance of CB2Rs on immune cells, whose activation might cause immunosuppression (Oláh et al., 2017).
Recent observations indicate that GPCRs are dynamic proteins able to assume multiple active states providing an interaction surface for intracellular adaptor proteins (e.g., heterotrimeric G proteins, G protein-coupled receptor kinases, and βarrestins) each of them responsible for different signaling pathway. Some ligands (orthosteric ligands or allosteric modulators) may stabilize a unique receptor conformation inducing a particular signaling pathway at the expense of others. This results in differential coupling to the signal transduction cascade and a biased response, a scenario which is also referred to as biased signaling or stimulus bias. Ligand bias should generate a biased response relatively independent of the cell system tested (Gurevich and Gurevich, 2019; Wootten et al., 2013; Smith et al., 2018). Biased GPCR ligands have been shown to display beneficial biological responses in preclinical and clinical studies, which explains the growing interests of medicinal chemists in biased signaling (Bock and Bermudez 2021). One of the most promising strategies to expand the repertoire of signaling-selective GPCR activators consists of dualsteric/bitopic agents, which are hybrid compounds composed of orthosteric and allosteric pharmacophoric units (Kamal and Jockers 2009; Lane et al., 2013). They bridge two topographically distinct ligand-binding domains, joining both orthosteric and allosteric properties within a single therapeutic agent. This strategy could offer access to GPCR modulators with a unique receptor subtype and signaling selectivity profile by virtue of targeting an allosteric site, as well as greater affinity due to the concomitant engagement with the orthosteric site. It is noteworthy that the special pharmacological profile of a bitopic ligand may be reflected in its unique binding kinetics. Theoretically, dualsteric ligands may have higher affinities than the respective partners. This may be derived from the ability of the two counterparts to bind into their corresponding binding pockets in an ideal manner without inducing an unfavorable conformational rearrangement of the receptor. In this view, upon binding, each pharmacophore has its own binding kinetics that can induce synergistic effects (i.e., allosteric cooperativity) on the overall kinetics of the bitopic ligand, greater than simply combining two individual components, as reported, for instance, for the M2 muscarinic receptor ligand, THR-160209 (Steinfeld et al., 2007). However, it may be expected that bitopic ligands do not always display an improved binding affinity; for example, a compromised binding affinity of the bitopic ligand can also occur when the individual pharmacophores have different preferences on receptor states (Antony et al., 2009). Bitopic/dualsteric ligands, may prove to be particularly useful in situations where endogenous agonist tone is progressively lost, such as in neurodegenerative disorders, thanks to the co-presence of the orthosteric and the allosteric modulator (Gentry et al., 2015). Finally, this innovative approach may also provide novel bias ligands because the incorporation of two pharmacophores in one ligand can severely impact receptor flexibility and thus signaling output (Lane et al., 2013; Schrage and Kostenis 2017; Reinecke et al., 2019).
Here, we describe the design, synthesis, and biological evaluation of potential CB2R dualsteric ligands, characterized by general structures A and B (Figure 1). The two classes of compounds were obtained by connecting, through different linkers, the pharmacophoric portion of a previously identified CB2R positive allosteric modulator (PAM), namely EC21a (Gado et al., 2019; Shapiro et al., 2021), with that of the CB2R selective orthosteric agonist LV62, which belongs to the class of 1,8-naphthyridin-2(1H)-one-3-carboxamide derivatives, previously identified by us as potent CB2R orthosteric agonists (Lucchesi et al., 2014). The nitrogen atom in position 1 and the 4-methyl cyclohexyl group in position 3 of LV62 moiety were selected to connect in position N (1) of the central core of EC21a, obtaining respectively the A (JR22a, JR26a, JR58a, JR60a, JR61a, JR64a compounds) and B (JR14a, JR16a compounds) series. Previous structural activity relation (SAR) studies on 1,8-naphthyridin-2(1H)-one-3-carboxamide derivatives (Lucchesi et al., 2014; Cooper et al., 2018) and on EC21a analogues (Gado et al., 2019; Gado et al., 2021) indicated these positions as the most suitable to chemical modifications without significantly compromising activity. The choice of the correct linker plays a crucial role in allowing the two pharmacophores to interact correctly with the respective binding site. Obviously, a deep knowledge of the orthosteric and allosteric binding sites will make easier the choice (Newman et al., 2020). The orthosteric CB2R binding site is well known now (Li et al., 2019), on the contrary there are no reliable structural data about the allosteric binding site of the CB2R. For this reason, the linkers used to obtain our ligands have been chosen on the basis of what previously reported for the bivalent/bitopic ligand (Kamal and Jockers. 2009; Newman et al., 2020; Meijer et al., 2021; Obeng et al., 2021; Gado et al., 2022).
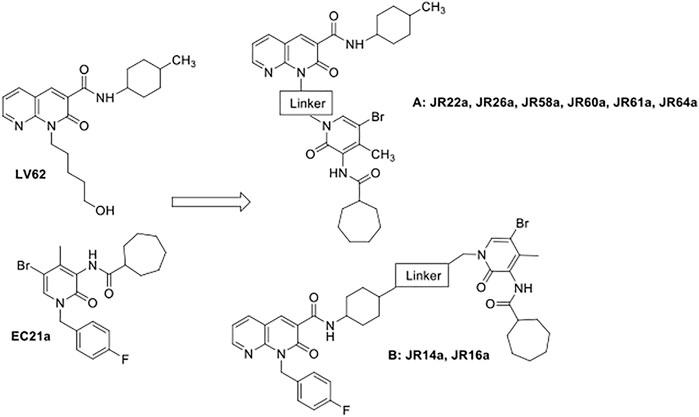
FIGURE 1. Design of potential CB2R dualsteric ligands (A) and (B).
A preliminary screening of the newly designed dualsteric CB2R ligands, led to the identification of compound JR64a as the most promising bitopic/dualsteric CB2R ligand of the series. Indeed, a signaling ‘bias’ in favor of G protein activation over βarrestin2 recruitment was observed, combined with high binding affinity for CB2R. Functional examination highlighted the ability of this compound to efficiently prevent inflammation in human microglial cells (HMC3) exposed to LPS/TNFα stimulation, as demonstrated by the reduced secretion of pro-inflammatory IL-6 and increased secretion of anti-inflammatory IL-10.
Commercially available reagents were purchased from Sigma Aldrich, Tokyo Chemical Industry or Fluorochem and used without purification. 1H NMR and 13C NMR were recorded at 400 and 100 MHz respectively, on a Bruker AVANCE IIITM 400 spectrometer. Chemical shift (δ) is reported in parts per million related to the residual solvent signal, while coupling constants (J) are expressed in Hertz (Hz). All compounds are >95% pure by HPLC analysis. The analytical HPLC system consisted of a Varian 9,012 Solvent Delivery System, coupled to a Varian ProStar 330 DAD detector with operating wavelengths in the range between 200 and 400 nm, and the Star LC Workstation version 6.41 software was used for instrument control, data acquisition, and data processing. Analyses were performed on a reverse phase C18 column (Luna C18 (2) 150 mm × 4.6 mm, 5 µm particle size, Phenomenex®). The mobile phase was constituted by H2O (eluent A) and ACN (eluent B) at a flow rate of 600 μL/min. A linear gradient starting from 40% of A, changing to 80% of A over 30 min, and returning to the initial conditions over 20 min was used. The target compounds are ≥97% pure by HPLC analysis (Supporting Information). High resolution mass spectra (HRMS) were recorded on a Q Exactive™ Plus Hybrid Quadrupole-Orbitrap™ Mass Spectrometer (Thermo Fisher Scientific), equipped with HESI source. The ESI-MS spectrum was recorded by direct injection at 5 μml min- 1 flow rate. Working conditions: positive polarity, spray voltage 3.5 kV, capillary temperature 300°C, S-lens RF level 55, sheath gas 20, auxiliary gas 3 (arbitrary units); negative polarity, spray voltage 3.4 kV, capillary temperature 270°C, S-lens RF level 55, sheath gas 35, auxiliary gas 8 (arbitrary units). Acquisition and analysis: Xcalibur 4.2 software (Thermo). For spectra acquisition a nominal resolution (at m/z 200) of 140,000 was used. Organic solutions were dried over anhydrous Na2SO4. Evaporation was carried out in vacuo using a rotating evaporator. Silica gel flash chromatography was performed using silica gel 60 Å (0.040–0.063 mm; Merck Life Science S. r.l.). Reactions were monitored by TLC on Merck aluminium silica gel (60 F254) plates that were visualized under a UV lamp (λ = 254 nm). Melting points were determined on a Kofler hot-stage apparatus and are uncorrected.
The synthesis of the precursors of JR compounds is reported in Supporting Information.
N-(4-(5-(4-(3-(5-bromo-3-(cycloheptanecarboxamido)-4- methyl -2-oxopyridin-1(2H)-yl)propyl)-1H-1,2,3-triazol-1-yl) pentanamido)cyclohexyl)-1-(4-fluorobenzyl)-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxamide (JR14a). To a solution of compounds 5 (40.0 mg, 0.08 mmol) and 15 (31.5 mg, 0.08 mmol) in DMF (1.71 ml) and water (0.85 ml), CuSO4.5H2O (20.0 mg, 0.08 mmol) and sodium ascorbate (47.6 mg, 0.24 mmol) were added. The reaction mixture was stirred at 80°C for 2 h. Subsequently, the solvent was removed under reduced pressure to give a residue which was dissolved in ethyl acetate and washed with saturated solution of NaHCO3. The organic phase was dried over Na2SO4, filtered, and evaporated under reduced pressure. The crude product obtained was purified by flash chromatography on silica gel using, firstly, ethyl acetate and 5% of methanol, and then ethyl acetate and 10% of methanol as eluent to afford compound JR14a (36.5 mg, 0.04 mmol) as a brownish solid. Yield: 50%. Mp: 108°C (dec.). 1H-NMR (CDCl3) δ (ppm) 9.90 (bd, 1H, J = 7.2 Hz, NH), 8.90 (s, 1H, H4), 8.72 (dd, 1H, J = 4.6 Hz; 1.8 Hz, H7), 8.10 (dd, 1H, J = 7.8 Hz; 1.8 Hz, H5), 7.55 (bs, 1H, NH), 7.43 (m, 3H, Ar-H + H6-Py), 7.37 (s, 1H, NCHC), 7.30 (dd, 1H, J = 7.8 Hz; 4.6 Hz, H6), 6.96 (AA’XX’ system, 2H, Ar-H), 5.78 (s, 2H, CH2), 5.63 (bd, 1H, J = 7.6 Hz, NH), 4.35 (t, 2H, J = 7.0 Hz, CH2-triazole), 4.20 (m, 1H, CHN), 3.99 (t, 2H, J = 7.0 Hz, CH2NCO), 3.93 (m, 1H, CH), 2.75 (t, 2H, J = 7.2 Hz, CH2-triazole), 2.51 (m, 1H, CHCO), 2.20 (t, 2H, J = 7.2 Hz, CH2CONH), 2.14 (s, 3H, CH3), 1.84 (m, 26H, cyclohexyl + cycloheptyl + 3xCH2). 13C-NMR (CDCl₃) δ (ppm) 176.11, 171.45, 162.87, 161.97, 162.22 (d, J = 245 Hz), 158.43, 152.30, 149.80, 146.50, 142.47, 142.23, 138.70, 133.10, 132.81 (d, J = 3 Hz), 130.29 (d, J = 8 Hz), 126.38, 123.31, 121.21, 119.56, 115.35 (d, J = 21 Hz), 115.10, 103.46, 49.92, 49.34, 47.85, 46.38, 45.54, 44.16, 35.78, 31.81, 29.72, 28.63, 28.55, 28.48, 28.24, 26.69, 22.56, 22.50, 20.43. HRMS-ESI: m/z calcd for C46H55BrFN9O5 [M-H]-, 910.34208; found 910.34450.
N-(4-(5-(4-((5-bromo-3-(cycloheptanecarboxamido)-4-methyl-2-oxopyridin-1(2H)-yl)methyl)-1H-1,2,3-triazol-1-yl)pentanamido)cyclohexyl)-1-(4-fluorobenzyl)-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxamide (JR16a). Compound JR16a was prepared from compounds 5 and 16 as described for compound JR14a and purified by flash column chromatography on a silica gel using ethyl acetate and 10% of methanol. Yield: 55%. Mp: 113 °C (dec.). 1H-NMR (CDCl3) δ (ppm) 9.90 (bd, 1H, J = 7.6 Hz, NH), 8.91 (s, 1H, H4), 8.72 (dd, 1H, J = 4.8 Hz; 2.0 Hz, H7), 8.10 (dd, 1H, J = 7.8 Hz; 2.0 Hz, H5), 7.67 (s, 1H, NCHC), 7.64 (bs, 1H, NH), 7.46 (m, 3H, Ar-H + H6-Py), 7.31 (dd, 1H, J = 7.8 Hz; 4.8 Hz, H6), 6.97 (AA’XX’ system, 2H, Ar-H), 5.79 (s, 2H, CH2), 5.56 (bd, 1H, J = 7.6 Hz, NH), 5.15 (s, 2H, CCH2N), 4.35 (t, 2H, J = 7.4 Hz, CH2-triazole), 4.21 (m, 1H, CHN), 3.95 (m, 1H, CH), 2.50 (m, 1H, CHCO), 2.19 (t, 2H, J = 7.6 Hz, CH2CONH), 2.13 (s, 3H, CH3), 1.84 (m, 24H, cyclohexyl + cycloheptyl + 2xCH2). 13C-NMR (CDCl₃) δ (ppm) 176.14, 171.45, 162.85, 162.17 (d, J = 245 Hz), 161.96, 158.25, 152.32, 149.75, 143.33, 142.53, 142.03, 138.72, 133.06, 132.75 (d, J = 3 Hz), 130.19 (d, J = 9 Hz), 126.17, 123.90, 123.23, 119.58, 115.32 (d, J = 22 Hz), 115.08, 103.91, 50.15, 47.73, 46.28, 45.54, 44.52, 44.14, 35.67, 31.77, 29.55, 28.58, 28.52, 28.19, 26.66, 22.54, 20.37. HRMS-ESI: m/z calcd for C44H51BrFN9O5 [M-H]-, 882.31075; found 882,31,397.
1-(5-(4-(3-(5-bromo-3-(cycloheptanecarboxamido)-4-methyl-2-oxopyridin-1(2H)-yl)propyl)-1H-1,2,3-triazol-1-yl)pentyl)-N-(4-methylcyclohexyl)-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxamide (JR22a). Compound JR22a was prepared from compounds 7 and 15 as described for compound JR14a and purified by flash column chromatography on a silica gel using ethyl acetate and 10% of methanol. Yield: 58%. Mp: 114 °C (dec.). 1H-NMR (CDCl3) δ (ppm) 9.96 and 9.60 (2d, 1H, J = 7.2 Hz, NH), 8.87 (s, 1H, H4), 8.70 (dd, 1H, J = 4.8 Hz; 2.0 Hz, H7), 8.08 (dd, 1H, J = 7.8 Hz; 2.0 Hz, H5), 7.47 (bs, 1H, NH), 7.44 (s, 1H, NCHC), 7.41 (s, 1H, H6-Py), 7.28 (dd, 1H, J = 7.8 Hz; 4.8 Hz, H6), 4.57 (2t, 2H, J = 7.6 Hz, CH2NCO), 4.38 (t, 2H, J = 7.2 Hz, CH2-triazole), 4.25 and 3.92 (2m, 1H, CH), 4.02 (t, 2H, J = 7.2 Hz, CH2NCO), 2.78 (t, 2H, J = 7.2 Hz, CH2-triazole), 2.50 (m, 1H, CHCO), 2.15 (s, 3H, CH3), 1.59 (m, 29H, cyclohexyl + cycloheptyl + 4xCH2), 0.97 and 0.92 (2d, 3H, J = 6.4 Hz, CH3). 13C-NMR (CDCl₃) δ (ppm) 176.10, 162.56, 161.96, 158.37, 152.12, 149.65, 146.46, 142.23, 142.00, 138.55, 133.09, 126.32, 123.14, 120.98, 119.15, 114.99, 103.44, 50.12, 49.36, 48.87, 47.77, 41.51, 33.94, 33.02, 32.04, 31.75, 29.96, 28.48, 28.18, 27.18, 26.64, 24.04, 22.46, 22.28, 20.42. HRMS-ESI: m/z calcd for C40H53BrN8O4 M-H]-, 787.33003; found 787.33307.
1-(5-(4-((5-bromo-3- (cycloheptanecarboxamido)-4-methyl-2-oxopyridin-1(2H)-yl)methyl)-1H-1,2,3-triazol-1-yl)pentyl)-N-(4-methylcyclohexyl) -2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxamide (JR26a). Compound JR26a was prepared from compounds 7 and 16 as described for compound JR14a and purified by flash column chromatography on a silica gel using ethyl acetate and 10% of methanol. Yield: 59%. Mp: 116 °C (dec.). 1H-NMR (CDCl3) δ (ppm) 9.98 and 9.59 (2d, 1H, J = 7.2 Hz, NH), 8.86 (s, 1H, H4), 8.69 (dd, 1H, J = 4.8 Hz; 2.0 Hz, H7), 8.07 (dd, 1H, J = 7.8 Hz; 2.0 Hz, H5), 7.66 (s, 1H, NCHC), 7.64 (bs, 1H, NH), 7.39 (s, 1H, H6-Py), 7.27 (dd, 1H, J = 7.8 Hz; 4.8 Hz, H6), 5.17 (s, 2H, CCH2N), 4.57 (2t, 2H, J = 7.6 Hz, CH2NCO), 4.35 (t, 2H, J = 7.4 Hz, CH2-triazole), 4.25 and 3.90 (2m, 1H, CH), 2.49 (m, 1H, CHCO), 2.14 (s, 3H, CH3), 1.56 (m, 27H, cyclohexyl + cycloheptyl + 3xCH2), 0.97 and 0.90 (2d, 3H, J = 6.6 Hz, CH3). 13C-NMR (CDCl₃) δ (ppm) 176.00, 162.62, 161.95, 158.16, 152.11, 149.73, 142.84, 142.00, 141.83, 138.51, 132.86, 126.13, 123.64, 123.28, 119.12, 114.99, 103.88, 50.41, 47.84, 45.74, 44.44, 41.42, 31.78, 31.09, 30.27, 29.87, 29.65, 28.17, 27.14, 26.66, 24.03, 21.58, 20.48. HRMS-ESI: m/z calcd for C38H49BrN8O4 M-H]-, 759.29873; found 759.29901.
1-(5-(5-bromo-3-(cycloheptanecarboxamido)-4- methyl-2-oxopyridin-1(2H)-yl)pentyl)-N- (4-methylcyclohexyl)-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxamide (JR61a). NaH (60% dispersion in mineral oil) (8.4 mg, 0.21 mmol) was added portion wise at 0°C to a solution of compound 14 (70 mg, 0.21 mmol) in DME (0.4 ml) and anhydrous DMF (0.1 ml). LiBr (36.5 mg, 0.42 mmol) was added 15 min later. The mixture was stirred for 20 min at room temperature. Compound 6 (182 mg, 0.42 mmol), as a mixture of 1:1 trans and cis isomers, was added dropwise, and the reaction was stirred at 65°C overnight. Solvents were removed under reduced pressure. The mixture obtained was dissolved in CHCl3 and washed three times with water. The organic phase was dried over Na2SO4, filtered, and evaporated under reduced pressure. The crude product obtained was purified by flash chromatography on silica gel using hexane/AcOEt 5:5 as eluent to afford compound JR61a (33.0 mg, 0.05 mmol) as a yellowish solid. Yield: 24%. Mp: 118 °C (dec.). 1H-NMR (CDCl3) δ (ppm) 9.98 and 9.62 (2d, 1H, J = 7.8 Hz, NH), 8.85 (s, 1H, H4), 8.69 (dd, 1H, J = 4.4 Hz; 1.6 Hz, H7), 8.06 (dd, 1H, J = 7.8 Hz; 1.6 Hz, H5), 7.56 (bs, 1H, NH), 7.34 (s, 1H, H6-Py), 7.26 (dd, 1H, J = 7.8 Hz; 4.4 Hz, H6), 4.57 (2t, 2H, J = 7.6 Hz, CH2NCO), 4.24 and 3.89 (2m, 1H, CH), 3.91 (t, 2H, J = 7.4 Hz, CH2NCO), 2.50 (m, 1H, CHCO), 2.15 (s, 3H, CH3), 1.58 (m, 27H, cyclohexyl + cycloheptyl + 3xCH2), 0.96 and 0.90 (2d, 3H, J = 6.4 Hz, CH3). 13C-NMR (CDCl₃) δ (ppm) 175.95, 162.44, 161.82, 158.08, 151.95, 149.58, 141.80, 141.63, 138.38, 132.54, 126.27, 123.09, 118.96, 114.83, 103.35, 49.97, 48.69, 47.63, 45.56, 41.36, 33.80, 32.87, 31.88, 31.59, 30.94, 30.10, 29.49, 28.68, 28.03, 27.14, 26.49, 23.90, 22.14, 21.41, 20.32. HRMS-ESI: m/z calcd for C35H46BrN5O4 M-H]-, 678.26604; found 678.26887.
1-(7-(5-bromo-3-(cycloheptanecarboxamido)-4-methyl-2-oxopyridin-1(2H)-yl)heptyl)-N-(4-methylcyclohexyl)-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxamide (JR64a). Compound JR64a was prepared from compounds 14 and 8 as described for compound JR61a and purified by flash column chromatography on a silica gel using hexane/AcOEt 5:5. Yield: 18%. Mp: 121 °C (dec.). 1H-NMR (CDCl3) δ (ppm) 10.01 and 9.65 (2d, 1H, J = 7.6 Hz, NH), 8.85 (s, 1H, H4), 8.69 (dd, 1H, J = 4.6 Hz; 1.8 Hz, H7), 8.06 (dd, 1H, J = 7.8 Hz; 1.8 Hz, H5), 7.49 (bs, 1H, NH), 7.31 (s, 1H, H6-Py), 7.26 (dd, 1H, J = 7.8 Hz; 4.6 Hz, H6), 4.54 (2t, 2H, J = 7.8 Hz, CH2NCO), 4.24 and 3.92 (2m, 1H, CH), 3.88 (t, 2H, J = 7.4 Hz, CH2NCO), 2.50 (m, 1H, CHCO), 2.15 (s, 3H, CH3), 1.60 (m, 31H, cyclohexyl + cycloheptyl + 5xCH2), 0.97 and 0.91 (2d, 3H, J = 6.4 Hz, CH3). 13C-NMR (CDCl₃) δ (ppm) 176.13, 162.68, 162.12, 158.25, 152.12, 149.84, 141.93, 141.75, 138.56, 132.65, 126.45, 123.33, 119.08, 115.05, 103.63, 50.29, 48.92, 47.93, 45.83, 42.03, 41.93, 34.03, 33.09, 32.12, 31.82, 30.32, 29.67, 29.30, 29.00, 28.25, 27.88, 26.99, 26.94, 26.71, 26.62, 22.36, 20.56. HRMS-ESI: m/z calcd for C37H50BrN5O4 M-H]-, 706.29734; found 706.29881.
1-(2-(2-(5-bromo-3-(cycloheptanecarboxamido)-4-methyl-2-oxopyridin-1(2H)-yl)ethoxy)ethyl)-N-(4-methylcyclohexyl)-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxamide (JR58a). Compound JR58a was prepared from compounds 14 and 9 as described for compound JR61a and purified by flash column chromatography on a silica gel using hexane/AcOEt 3:7. Yield: 59%. Mp: 110 °C (dec.). 1H-NMR (CDCl3) δ (ppm) 9.95 and 9.60 (2d, 1H, J = 7.6 Hz, NH), 8.84 (s, 1H, H4), 8.60 (dd, 1H, J = 4.6 Hz; 2.0 Hz, H7), 8.02 (dd, 1H, J = 7.6 Hz; 2.0 Hz, H5), 7.66 (bs, 1H, NH), 7.31 (s, 1H, H6-Py), 7.25 (dd, 1H, J = 7.6 Hz; 4.6 Hz, H6), 4.80 (m, 2H, CH2NCO), 4.20 and 3.85 (2m, 1H, CH), 4.00 (m, 2H, CH2NCO), 3.80 (m, 2H, OCH2), 3.73 (m, 2H, OCH2), 2.48 (m, 1H, CHCO), 2.14 (s, 3H, CH3), 1.56 (m, 21H, cyclohexyl + cycloheptyl), 0.92 and 0.87 (2d, 3H, J = 6.4 Hz, CH3). 13C-NMR (CDCl₃) δ (ppm) 175.99, 162.68, 161.75, 158.15, 151.80, 149.75, 142.11, 141.98, 138.36, 133.99, 125.84, 123.33, 119.16, 114.74, 102.73, 68.43, 67.99, 49.35, 48.74, 47.60, 45.83, 40.48, 40.42, 33.79, 32.88, 31.88, 31.61, 30.10, 29.52, 28.03, 26.49, 22.10, 21.41, 20.24. HRMS-ESI: m/z calcd for C34H44BrN5O5 M-H]-, 680,24,530; found 680.24717.
1-(4-(4-(5-bromo-3-(cycloheptanecarboxamido)-4-methyl-2-oxopyridin-1(2H)-yl)butoxy) butyl)-N-(4-methylcyclohexyl)-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxamide (JR60a). Compound JR60a was prepared from compounds 14 and 10 as described for compound JR61a and purified by flash column chromatography on a silica gel using hexane/AcOEt 3:7. Yield: 12%. Mp: 118 °C (dec.). 1H-NMR (CDCl3) δ (ppm) 10.01 and 9.65 (2d, 1H, J = 7.2 Hz, NH), 8.85 (s, 1H, H4), 8.66 (dd, 1H, J = 4.6 Hz; 1.4 Hz, H7), 8.05 (dd, 1H, J = 7.6 Hz; 1.4 Hz, H5), 7.51 (bs, 1H, NH), 7.34 (s, 1H, H6-Py), 7.24 (dd, 1H, J = 7.6 Hz; 4.6 Hz, H6), 4.61 (2t, 2H, J = 7.6 Hz, CH2NCO), 4.25 and 3.88 (2m, 1H, CH), 3.93 (t, 2H, J = 6.8 Hz, CH2NCO), 3.46 (m, 4H, 2xOCH2), 2.50 (m, 1H, CHCO), 2.14 (s, 3H, CH3), 1.58 (m, 29H, cyclohexyl + cycloheptyl, 4xCH2), 0.97 and 0.91 (2d, 3H, J = 6.4 Hz, CH3). 13C-NMR (CDCl₃) δ (ppm) 175.98, 162.50, 161.92, 158.15, 151.95, 149.67, 141.80, 141.62, 138.34, 132.70, 126.17, 123.07, 118.93, 114.84, 103.28, 70.42, 70.25, 49.83, 48.72, 47.66, 45.58, 44.91, 41.65, 33.81, 32.87, 31.90, 31.60, 30.09, 29.60, 28.03, 27.16, 27.12, 26.52, 26.49, 24.73, 22.60, 21.41, 20.24. HRMS-ESI: m/z calcd for C38H52BrN5O5 M-H]-, 736,30,788; found 736.30861.
CP55,940 was purchased from Cayman Chemicals (Ann Arbor, MI) [3H]CP55,940 at a concentration of 174.6 Ci/mmol was purchased from PerkinElmer (Guelph, ON). LPS (Escherichia coli 0111:B4) and TNFα were purchased from Sigma-Aldrich (Milan, Italy), whereas SR144528 was from Tocris (Bristol, United Kingdom). Chinese hamster ovary (CHO)-K1 cells originally obtained from ATCC (Catalog No. CCL-61, Manassas, VA) stably expressing human cannabinoid CB1R (hCB1R) or CB2R (hCB2R) described previously were maintained at 37°C, 5% CO2 in F-12 DMEM containing 1 mM l-glutamine, 10% FBS, and 1% Pen/Strep as well as hygromycin B (300 μg/ml) and G418 (600 μg/ml) for CHO-K1 hCB1R cells or G418 (400 μg/ml) for CHO-K1 hCB2R cells (Garai et al., 2020; Zagzoog et al., 2020). In the case of membrane collection for radioligand binding, cells were removed from flasks by scraping, centrifuged, and then frozen as a pellet at -80°C until required. Before use in a radioligand binding assay, cells were defrosted, diluted in Tris buffer (50 mM Tris [pH 7.4]) and homogenized with a 1 ml hand-held homogenizer. HitHunter® (cAMP) and PathHunter® (βarrestin2) CHO-K1 cells stably-expressing hCB2R from DiscoveRx (Eurofins, Fremont, CA) were maintained at 37 °C, 5% CO2 in F-12 DMEM containing 10% FBS and 1% Pen/Strep with 800 μg/ml geneticin (HitHunter®) or 800 μg/ml G418 and 300 μg/ml hygromycin B (PathHunter®), as previously reported (Garai et al., 2020; Zagzoog et al., 2020). The human microglial clone 3 cell line (HMC3) (ATCC® CRL-3304™) was cultured in high glucose DMEM supplemented with 10% FBS, streptomycin (100 g/ml) and penicillin (100 U/mL) (Sigma-Aldrich, Milan, Italy).
Inhibition of FSK-stimulated cAMP accumulation was measured using the DiscoveRx HitHunter assay. Twenty thousand cells/well were plated in low-volume 96-well plates and incubated overnight in Opti-MEM containing 1% FBS at 37°C and 5% CO2. Opti-MEM media was then removed and replaced with cell assay buffer (DiscoveRx) and cells were co-treated at 37°C with 10 µM FSK and ligands for 90 min cAMP antibody solution and cAMP working detection solutions were added to cells (DiscoveRx), and cells were incubated for 60 min at room temperature. cAMP solution A (DiscoveRx) was added, and cells were incubated for an additional 180 min at room temperature before chemiluminescence was measured on a Cytation5 plate reader (top read, gain 200, integration time 10,000 ms).
βarrestin2 recruitment was quantified using the DiscoveRx PathHunter® assay. Cells (20,000 cells/well in low-volume 96-well plates) were incubated overnight in Opti-MEM containing 1% FBS at 37 °C and 5% CO2. Cells were treated with ligands for 90 min at 37 °C. Detection solution was added to cells (DiscoveRx), and cells were incubated for 60 min at room temperature. Chemiluminescence was measured on a Cytation5 plate reader (top read, gain 200, integration time 10,000 ms).
CHO-K1 cells were thawed, diluted in Tris buffer (50 mM Tris [pH 7.4]) and homogenized in a 1 ml hand-held homogenizer. hCB1R and hCB2R CHO-K1 cell membranes were collected by cavitation in a pressure cell, and sedimented by ultracentrifugation. Pellets were resuspended in TME buffer (50 mM Tris, 5 mM MgCl2, 1 mM EDTA [pH 7.4]), and protein concentration was measured via the Bradford method according to the manufacturer’s directions (Bio-Rad Laboratories, Mississauga, ON). Competition binding experiments were conducted with 1 nM [3H]CP55,940 and Tris binding buffer (50 mM Tris, 0.1% BSA [pH 7.4], 2 ml). Radioligand binding began with the addition of CHO-K1 cell membranes (25 µg protein per sample). Assays were performed for 120 min at 37°C and stopped by the addition of ice-cold Tris binding buffer (pH 7.4), followed by vacuum filtration using a 24-well sampling manifold (Brandel Cell Harvester; Brandel Inc., Gaithersburg, MD, United States ). Brandel GF/B filter paper was soaked with wash buffer at 4 °C for at least 24 h. Each filter paper was washed 6 times with a 1.2 ml aliquot of Tris-binding buffer (pH 7.4), then air-dried and submerged in 5 ml of scintillation fluid (Ultima Gold XR, PerkinElmer). Liquid scintillation spectrometry was used to quantify radioactivity. For competition binding experiments, specific binding was equal to the difference in radioactivity with or without 1 µM unlabelled CP55,940.
The concentrations of pro-inflammatory (IL-6) and anti-inflammatory (IL-10) interleukins were determined by performing specific ELISA assays (MyBioSource, San Diego, CA, United States ) on collected culture media. Human microglial cells (HMC3) were exposed to pretreatment with test compounds for 1 h followed by stimulation with LPS (10 μg/ml)/TNFα (50 ng/ml) for 24 h. In competition experiments, the CB2R antagonist (SR144528, 1 μM) or the CB2R positive allosteric modulator (EC21a 1 or 10 μM) were administered 15 min before agonist administration.
The expression of CB2Rs was evaluated in human microglia cell lysates by Western blot experiments. Briefly, 50 μg of protein was diluted with Laemmli sample buffer 2×, boiled for 8 min at 96°C, separated on Criterion TGXTM gel (4–20%) and transferred into PVDF membranes. To avoid non-specific immunodetection, membranes were incubated for 45 min in T-TBS (20 mM Tris, 500 mM NaCl, 0.1% Tween-20, pH 8) containing 5% non-fat milk. Blots were then incubated overnight at 4°C with a rabbit anti- CB2R antibody (#ab3561, Abcam, Cambridge, United Kingdom). Next, the membranes were incubated with HRP-labeled secondary anti-rabbit (#MAB201P, Merck-Millipore, Darmstadt, Germany) for 2 h at room temperature. Detection of chemiluminescence signals and densitometric analysis of blots were performed using ImageQuant LAS 4000 (GE Healthcare, Milan, Italy) and ImageLab software (Bio-Rad, Hercules, CA, United States ), respectively.
Data for [3H]CP55,940 binding are expressed as % of maximum [3H]CP55,940 bound (i.e. 100%). HitHunter® cAMP, and PathHunter® βarrestin2 data are shown as % of maximal CP55,940 response (i.e. 100%). Concentration–response curves (CRC) were fit using non-linear regression (3 parameters) and used to calculate EC50, Emax, or IC50 (GraphPad, Prism, v. 9.0). Statistical analyses were conducted by one-way analysis of variance (ANOVA), as indicated in the figure legends, using GraphPad. Post-hoc analyses were performed using Tukey’s (one-way ANOVA) test. Homogeneity of variance was confirmed using Bartlett’s test. All results are reported as the mean ± the standard error of the mean (SEM) or 95% confidence interval (CI), as indicated. pvalues < 0.05 were considered significant.
The synthesis of novel compounds, JR14a, JR16a, JR22a, JR26a, JR58a, JR60a, JR61a and JR64a, along with their corresponding precursors, was accomplished as depicted in Schemes 1-5.
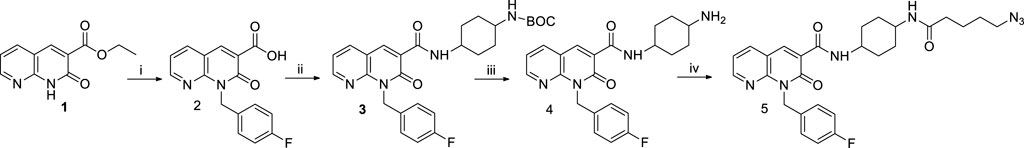
SCHEME 1. Synthetic pathway for the synthesis of the intermediate 5. Reagents and conditions: i) a) DMF, Cs₂CO₃, 1h, rt; b) p-fluorobenzyl bromide, 50°C, 12h; c) NaOH 10%, 100°C, 24 h; ii) a) DMF, NEt₃, TBTU, 0°C, 30 min.; b) cis-4-(Boc-amino) cyclohexylamine, 0°C, 30 min.; c) rt, 12 h; iii) CH₂Cl₂, CF₃COOH, −20°C, 3h; iv) a) 5-azidopentanoic acid, DMF, NEt₃, TBTU, 0°C, 30 min.; b) compound 4, 0°C, 30 min.; c) rt, 12h.
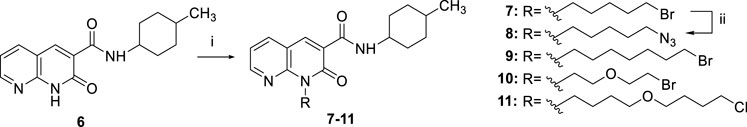
SCHEME 2. Synthetic pathway for the synthesis of the intermediates 7-11. Reagents and conditions: i) a) CsF, DMF, rt, 1h; b) suitable R-Br or R-Cl, 30 °C, 24 h; ii) NaN3, DMF, 60 °C, 12 h.
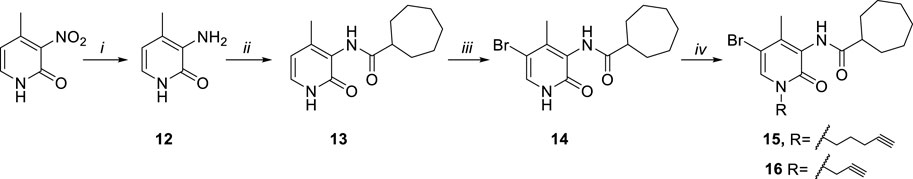
SCHEME 3. Synthetic pathway for the synthesis of the alkyne derivatives 15 and 16. Reagents and conditions: i) Fe, NH4Cl, H2O/EtOH 1:2, 80 °C, 3 h. ii) a) cycloheptanecarboxylic acid, C2O2Cl2, DMF, rt, 0.5 h b) NEt3, DCM, DMF, rt, 24 h. iii) Br2, CHCl3, rt, 12 h. iv) a) CsF, DMF, rt, 1 h b) R-bromide, 30°C, 12h.
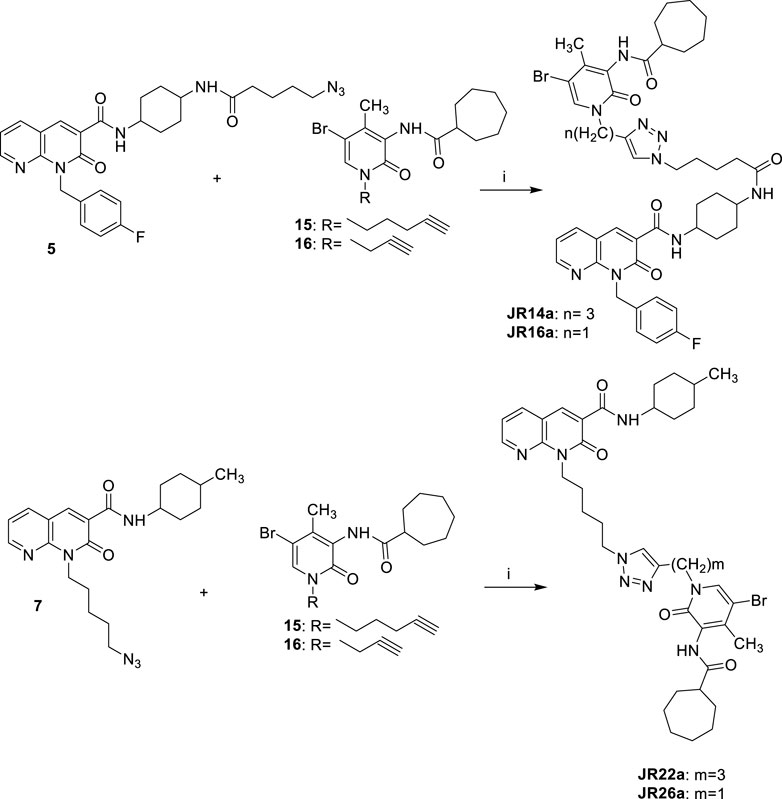
SCHEME 4. Synthetic pathway for the synthesis of compounds JR14a, JR16a and JR22a, JR26a Reagents and conditions: i) CuSO4·5H2O, sodium ascorbate, DMF/H2O 4:1, 80°C, 2 h.
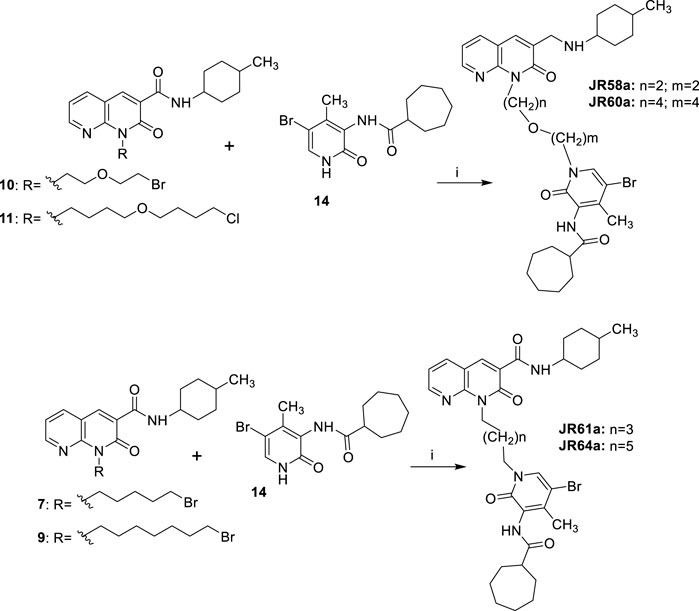
SCHEME 5. Synthetic pathway for the synthesis of compounds JR58a, JR60a, JR61a and JR64a. Reagents and conditions: i) a) NaH, DME, DMF, 0°C, 30 min; b) LiBr r.t. 20 min; suitable c) 65 °C, 12 h.
As described in Scheme 1, the ethyl ester 1 (Manera et al., 2009) was dissolved in anhydrous DMF with the p-fluorobenzyl bromide in the presence of cesium carbonate at 50 °C for 12 h. After this time and solvent removal, the crude product was immediately subjected to alkaline hydrolysis of ester group in the presence of NaOH al 10% at reflux for 24h, followed by acidification, affording the carboxylic acid 2. The amide 3 was obtained by reaction of derivative 2 with cis-4-(Boc-amino) cyclohexylamine in the presence of TBTU, NEt3, at room temperature for 12 h. The tert-butyl carbamate (BOC) deprotection of 3 was performed under acidic conditions by using CF₃COOH at −20°C for 3 h to yield the amino-derivative 4. Finally, the derivative 4 was reacted with 5-azidopentanoic acid (previously prepared by reacting 5-bromovaleric acid, with NaN₃ in DMF at 60°C for 12 h) in the presence of TBTU according to the procedure described above, to afford the desired compound 5 (Scheme 1).
As reported in Scheme 2, the N1-alkylation of 1,8-naphthyridine-3-carboxamide 6 [(Manera et al., 2009) in anhydrous DMF with the suitable halogenated reagent in the presence of cesium fluoride afforded the desired 1,8-naphthyridin-2-one derivatives 7, 9–11. Additionally, compound 7 was also used as starting material in the presence of sodium azide at 60°C for 12 h to give compound 8.
As reported in Scheme 3, the commercially available starting material 2-hydroxy-4-methyl-3-nitropyridine was treated with iron powder and ammonium chloride at 80°C for 3 h, to afford the amine compound 12. The reaction between the amine derivative 12 and the cycloheptanecarbonyl chloride in DMF and triethylamine initially at 0°C and then at room temperature for 24 h, gave the amides 13. The acyl chloride was prepared by reaction between cycloheptanecarboxylic acid and oxalyl chloride at room temperature for 30 min. The 5-bromo derivative 14 was obtained from compound 13 for treatment with Br2 in CHCl3 at room temperature for 12 h. Finally, compound 14 was subjected to a N-alkylation reaction by treatment with cesium fluoride in anhydrous DMF, at room temperature for 1 h and then with the suitable halogenated reagent at 50°C for 12 h, affording the desired alkyne derivatives 15 and 16.
As showed in Scheme 4, the final compounds JR14a and JR16a were easily obtained by click chemistry reaction of the azido derivative 5 with the alkyne derivatives 15 or 16 in DMF and water in the presence of CuSO4.5H2O and sodium ascorbate, at 80°C for 2 h. The same click reaction was also conducted between the azido derivative 7 with the alkyne derivatives 15 and 16 to afford the compounds JR22a and JR26a.
Finally, the synthesis of final compounds JR58a, JR60a, JR61a and JR64a, was accomplished as depicted in Scheme 5. The 5-bromo derivative 14 was subjected to a N-alkylation reaction by treatment with sodium hydride at 60% in anhydrous DME: DMF (4:1, v/v) mixture, at 0°C for 15 min, followed by the addition of lithium bromide, at room temperature for 20 min, and then of the appropriate N1-alkylated intermediate 7, 9–11 at 65 °C for 12 h, affording, after purification, the desired compounds JR58a, JR60a, JR61a and JR64a.
The new dualsteric CB2R ligands of general chemical structure A (JR22a, JR26a, JR58a, JR60a, JR61a, and JR64a) and B (JR14a, JR16a), along with their relative parent compounds EC21a and LV62, were screened to assay their ability to inhibit FSK-stimulated cAMP accumulation in CHO-K1 cells stably-expressing hCB2R (Table 1). The non-selective orthosteric CBR ligand CP55,490 was used as a reference compound. Cells were treated with 10 μM FSK and CP55,490 or compound for 90 min to assess ligand concentration-dependent activity. Regarding the parent compounds, for EC21a no response was detected in accordance with its allosteric nature. LV62 showed nanomolar potency (EC50 = 58 nM) and high relative efficacy (Emax = 110 ± 8.8) (Table 1; Figure 2A). The baseline for LV62 was higher than that of CP55,940 (Figure 2A), indicating that Emin and Emax may be poorly defined within this assay, which may be a product of the curve fit for LV62 or an artefact of the assay. In co-administration experiments, the activity of 10 nM LV62 was augmented by EC21a in a concentration-dependent manner (EC50 = 4.3 nM) (Table 1; Figure 2A), confirming the role of EC21a as a CB2R positive allosteric modulator (PAM) (Gado et al., 2019).
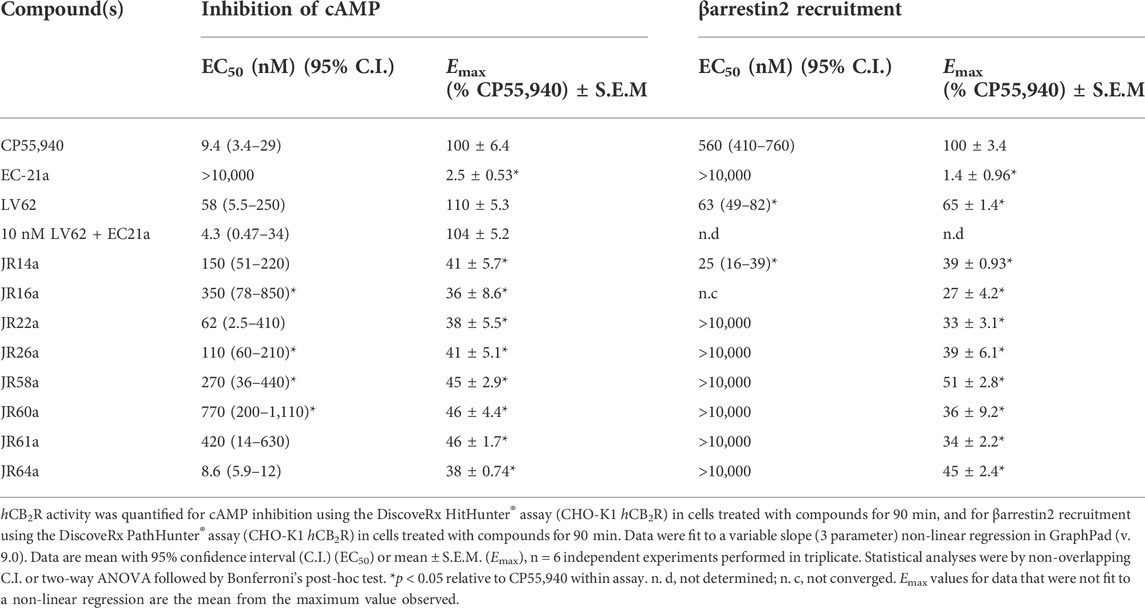
TABLE 1. Inhibition of forskolin-stimulated cAMP and βarrestin2 recruitment.
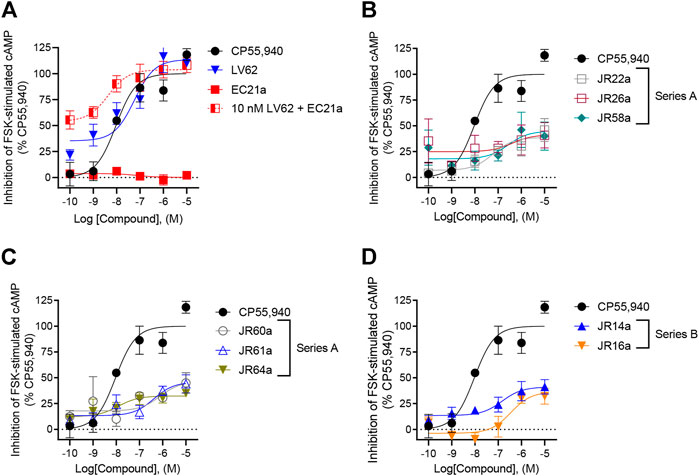
FIGURE 2. Evaluation of hCB2R-dependent inhibition of FSK-stimulated cAMP. CHO cells stably-expressing hCB2R were treated with 0.10 nM—10 μM compounds and 10 µM FSK for 90 min and cAMP inhibition was measured. (A) Parent compounds used for the design of the JR derivatives; (B,C) compounds of series A; (D) compounds of series B. CP55,940 data are the same in all four panels and presented for reference. Data are expressed as % CP55,940 response. Data were fitted to a non-linear regression (3 parameter model, GraphPad v. 9.0). Data are mean ± S.E.M. of six independent experiments performed in triplicate.
Among the compounds tested, JR64a and JR14a, were the most interesting of series A and B respectively (Table 1; Figure 2B). In particular JR64a was more potent than the orthosteric agonist LV62 (EC50(JR64a) = 8.6 nM; EC50(LV62) = 58 nM [not statistically significant]); and its potency was also almost comparable to that observed in EC21a and LV62 co-administration experiments (EC50(LV62+EC21a) = 4.3 nM). All other tested JR compounds displayed lower potency compared to the parent orthosteric agonist LV62 or the compounds JR64a (Table 1). Only a modest efficacy, ranging from 38 to 41%, and lower than that observed in EC21a and LV62 co-administration experiments (Emax(LV62+EC21a) = 104 ± 5.2), was observed for both JR14a and JR64a (Table 1). Therefore, the combination of EC21a (PAM) and LV62 (agonist) produced greater potency and efficacy than the resultant JR compounds, obtained conjoining these two ligands. Nevertheless, our results indicated that the ligands JR14a and JR64a displayed modest partial agonist activity in the inhibition of FSK-stimulated cAMP accumulation assay in hCB2R-expressing cells.
In addition to G protein-mediated signaling, GPCRs also interact with βarrestins, which facilitates receptor internalization, recycling, degradation, and signaling (Chen et al., 2014; Donthamsetti et al., 2020). Therefore, the complete panel of new orthosteric/allosteric hybrid CB2R ligands and their corresponding parent compounds were evaluated for their ability to enhance βarrestin2 recruitment in CHO cells stably-expressing hCB2R. Cells were treated with CP55,490 or compound for 90 min (Figure 3; Table 1). Regarding the parent compounds, no response was detected for EC21a alone, which is consistent with its activity as a CB2R PAM (Figure 3A; Table 1). The CB2R orthosteric agonist LV62 enhanced βarrestin2 recruitment with both potency and efficacy comparable to those previously observed in FSK-stimulated cAMP accumulation assays (Table 1). Interestingly, analogs JR14a and JR64a, previously shown to inhibit FSK-stimulated cAMP accumulation, exhibited different behaviors in enhancing βarrestin2 recruitment (Figure 3; Table 1). JR14a enhanced βarrestin2 recruitment with nanomolar potency (EC50(JR14a) = 25 nM), whereas JR64a, did not significantly enhance βarrestin2 recruitment at concentrations below 10,000 nM. Therefore, JR64a displayed greater potency in the inhibition of cAMP accumulation as compared to the recruitment of βarrestin2 (biased ligand). This result may be important for the design of new CB2R agonists devoid of the side effects related to the internalization and desensitization of CB2Rs. Indeed, it has been reported that CB2R agonists, by enhancing βarrestin2 recruitment, can induce internalization and desensitization of the receptor leading to a decrease in signaling and surface receptor levels (Chen et al., 2014).
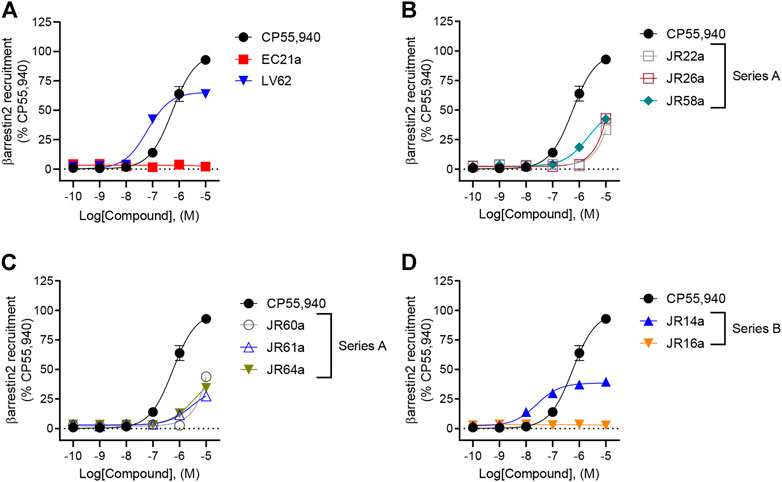
FIGURE 3. Evaluation of hCB2R-dependent recruitment of βarrestin2. CHO-K1 cells stably-expressing hCB2R were treated with 0.10 nM—10 μM compounds for 90 min and βarrestin2 recruitment was measured. (A) Parent compounds used for the design of the JR derivatives; (B,C) compounds of series A; (D) compounds of series B. CP55,940 data are the same in all four panels and presented for reference. Data are expressed as % CP55,940 response. Data were fitted to a non-linear regression (3 parameter model, GraphPad v. 9.0). Data are mean ± S.E.M. of six independent experiments performed in triplicate.
Following characterization of G protein-mediated cAMP inhibition and βarrestin2 recruitment, we assessed ligand affinity for LV62, EC21a, JR14a, and JR64a at hCB1R and hCB2R using a [3H]CP55,940 radioligand displacement assay on membranes derived from CHO-K1 cells stably-expressing either receptor. No affinity at hCB1R was detected for all compounds (Figure 4A; Table 2). At hCB2R, consistent with its CB2R PAM character, EC21a increased [3H]CP55,940 bound to hCB2R (Figure 4B; Table 2). LV62 fully displaced [3H]CP55,940 from hCB2R, as expected for an orthosteric compound (Figure 4B; Table 2). Remarkably, novel hCB2R ligands JR14a and JR64a displayed nanomolar relative affinities in the [3H]CP55,940 displacement assay with hCB2R CHO-K1 cell membranes (Figure 4B; Table 2; IC50(JR14a) = 1.8 nM; IC50(JR64a) = 0.60 nM).
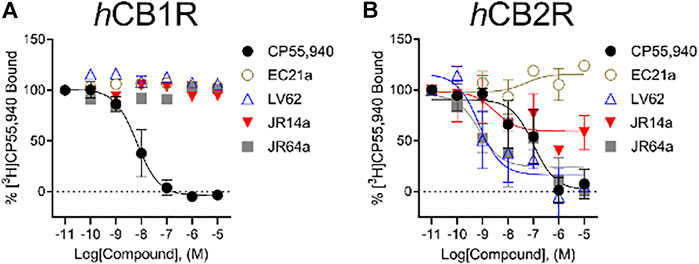
FIGURE 4. [3H]CP55,940 binding to hCB1R and hCB2R. Membranes from CHO-K1 cells stably-expressing hCB1R (A) or hCB2R (B) were incubated with 1 nM [3H]CP55,940 and 0.10 nM—10 μM compounds for 2 h. Data are expressed as % [3H]CP55,940 bound. Data were fitted to a non-linear regression (3 parameter model to estimate changes in displacement [IC50] which was substituted for EC50, GraphPad v. 9.0). Data are mean ± S.E.M. of at least three independent experiments performed in duplicate. Data from these graphs is presented in Table 2.

TABLE 2. [3H] CP55,940 binding.
Taking into account that the inflammatory process in microglial cells supports the onset and progression of several neurodegenerative and psychiatric disorders (Nakagawa and Chiba, 2014; Hansen et al., 2018; Haukedal and Freude 2019; Ho 2019), we decided to investigate the anti-inflammatory properties of novel CB2R ligand JR64a, which had emerged from a preliminary screening as the most promising of the series. At this aim, we first set up a human model of microglial inflammation, by exposing HMC3 cells to LPS/TNFα stimulus (Dello Russo et al., 2018). In agreement with our previous observations (Polini et al., 2020; Gado et al., 2022), exposure of microglial cells to LPS/TNFα resulted in a significant increase of the release of pro-inflammatory IL-6 as compared to control cells (Figure 5A), whereas no significant effects were observed on IL-10 release (Figure 5B). Expression of CB2R in HMC3 microglial cells was also assessed by WB analysis (Figure 5C). Then, dose-response experiments were carried out by exposing HMC3 cells to pretreatment with increasing concentrations (1, 10, and 25 μM) of test compound followed by LPS/TNFα treatment for 24 h (Figure 6). Notably, no relevant cytotoxic effects were detected in HMC3 cells after treatment with JR64a at 1 and 10 μM concentrations (Figure 7), whereas when used at 25 μM a modest decrease in cell viability (ca 10%) was observed (Figure 7). Measurements of pro- and anti-inflammatory interleukins levels (i.e. IL-6 and IL-10) in cell media by ELISA tests revealed that, when used at 10 or 25 μM concentration, analog JR64a displayed a marked anti-inflammatory activity, which was completely abolished after pretreatment with the CB2R selective antagonist SR144528 (1 μM, Figures 6A,B), suggesting a CB2R-mediated anti-inflammatory effect in HMC3 microglial cells. In parallel experiments, the anti-inflammatory effects of the orthosteric agonist LV62 administered at 1 or 10 μM concentration either individually or in equimolar combination with the CB2R-PAM EC21a were evaluated. As shown in Figures 6C,D, administration of EC21a at equimolar dosages enhanced LV62 anti-inflammatory activity, which resulted in a similar response to that observed on administration of equimolar concentrations of the hybrid compound JR64a (Figures 6A,B). Notably, co-administration of EC21a at equimolar or higher doses revealed no effects on JR64a anti-inflammatory activity in HMC3 cells (Figure 8), suggesting a potential ability for JR64a to interact with both orthosteric and allosteric sites of CB2R. However, additional investigation is needed to confirm the potential bitopic pharmacology of JR64a. In particular, computational studies using our recently proposed model of CB2R in complex with bitopic ligands (Gado et al., 2022) could certainly help to clarify the binding mode of JR64a and its ability of occupying both orthosteric and allosteric site of the CB2R, as well as to facilitate the rational design of novel and more effective CB2R bitopic ligands.
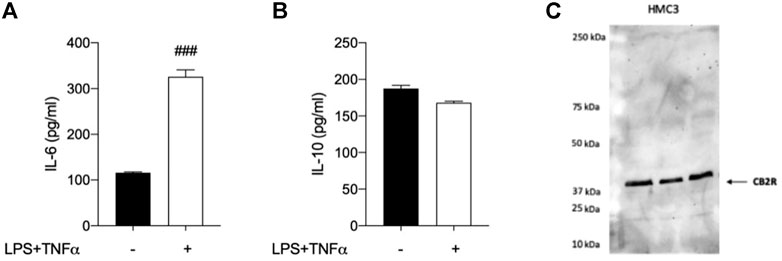
FIGURE 5. Analysis of HMC3 cell line response to inflammatory stimulus and expression of CB2R. (A) Pro-inflammatory interleukin-6 (IL-6) and (B) anti-inflammatory interleukin-10 (IL-10) levels were measured after exposure to LPS (10 μg/ml)/TNFα (50 ng/ml) stimulus for 24 h. (C) Western Blot (WB) analysis allowed the detection of CB2R in control HMC3 cell lysates. [A-B]: data represent means ± S.E.M. from n = 3 independent experiments, performed in duplicate. Statistical analysis was performed by ordinary one-way ANOVA followed by Tukey’s multiple comparison test. ###p < 0.005 compared to control cells.
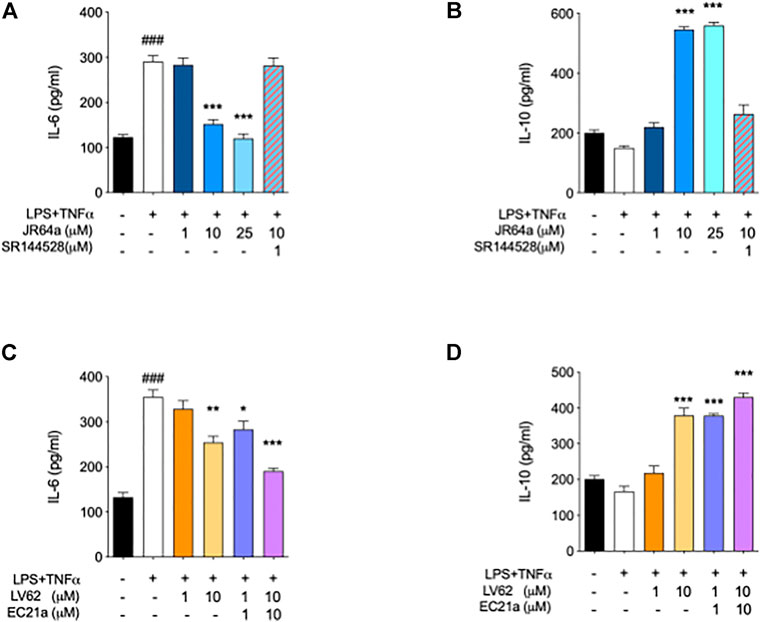
FIGURE 6. Ability of dualsteric CB2R ligand JR64a to decrease the inflammatory phenotype of LPS/TNFα stimulated HMC3 cells. Panels (A) and (B) show the effects of JR64a on the release of pro-and anti-inflammatory interleukines IL-6 and IL-10, respectively. Panels (C) and (D) show the effects produced in the same experimental setting by administering CB2R selective orthosteric agonist LV62 alone or in combination (1:1) with the CB2R PAM EC21a. Bars represent the release (pg/ml) of ILs in the presence of the compounds. Data represent mean ± SEM from n = 3 independent experiments performed in duplicate. Statistical analysis was performed by ordinary one-way ANOVA followed by Tukey’s multiple comparison test. ###p < 0.005 vs. control cells; *p < 0.05, **p < 0.01 and ***p < 0.005 vs. LPS/TNFα treated cells.
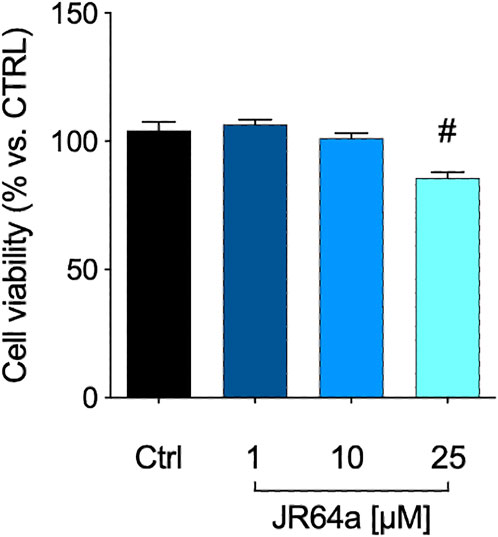
FIGURE 7. MTT assay performed with different concentrations of JR64a. Data represent means ± S.E.M. from n = 3 independent experiments, performed in triplicate. Statistical analysis was performed by ordinary one-way ANOVA followed by Tukey’s multiple comparison test. #p < 0.05 vs. control.
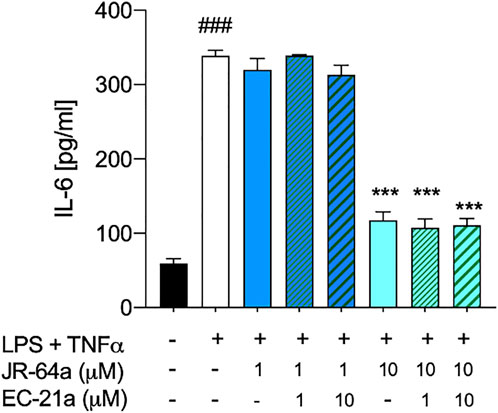
FIGURE 8. Co-administration of selective CB2R PAM EC21a doesn’t modify the effects of JR64a on the release of IL-6 in HMC3 cells exposed to LPS/TNFα stimulus. Bars represent the release (pg/ml) of IL-6 in the presence of the compounds. Data represent mean ± SEM from n = 3 independent experiments performed in duplicate. Statistical analysis was performed by ordinary one-way ANOVA followed by Tukey’s multiple comparison test. ###p < 0.005 vs. control cells; ***p < 0.005 vs. LPS/TNFα treated cells.
Recent progress made in the discovery and functional characterization of GPCRs ligands have enabled the design of bitopic/duasteric ligand as single chemical entities in which an orthosteric and an allosteric pharmacophore are chemically attached via a linker in a manner to allow simultaneous targeting of two binding sites (orthosteric and allosteric) within one receptor. This approach may offer several advantages over the classical ‘monovalent’ ligand, such as an increased affinity and selectivity for the target receptor, often accompanied with functional selectivity (stimulus bias) (Newman et al., 2020). However, the design of bitopic ligands needs deep knowledge of orthosteric and allosteric ligands for a given GPCR as well as its binding sites (orthosteric and allosteric.). For these reasons, bitopic ligands have so far been described predominantly for muscarinic receptors (Valant, et al., 2008; Keov, et al., 2014), adenosine receptors (Valant et al., 2014), and dopamine receptors (Draper-Joyce et al., 2018), for which detailed knowledge on allosteric binding sites and ligands is available.
In this study we have presented the synthesis and functional characterization of novel potential CB2R dualsteric agents, designed as hybrid compounds composed of two distinct pharmacophoric units, namely the CB2R PAM EC21a and the CB2R orthosteric agonist LV62, linked through different alkyl chains. After a preliminary screening, which included Gαi/o protein-dependent inhibition of FSK-stimulated cAMP accumulation, ligand induced recruitment of βarrestin2, and binding to hCB2R, analog JR64a was identified as the most promising dualsteric CB2R ligand. Indeed, a bias towards the modulation of the cAMP-pathway over the recruitment of βarrestin2 combined with high affinity for binding to hCB2R were observed. Subsequently, the lead compound was tested to evaluate its ability to decrease the inflammatory phenotype of LPS/TNFα-stimulated human microglial cells (HMC3). Our results indicated that analog JR64a displayed good anti-inflammatory properties when used at 10 μM concentration. Notably, as demonstrated by pharmacological antagonism, the anti-inflammatory effect displayed by JR64a revealed to be CB2R-mediated, and no relevant HMC3 cytotoxicity was also observed after administering JR64a at 10 μM concentration. Even though still at a preliminary level, functional assays indicated that the selective CB2R PAM analog EC21a was not able to increase the anti-inflammatory effects produced by JR64a in HMC3 cells, supporting the design of this compound as a potential CB2R dualsteric ligand. Certainly, the availability of dualsteric/bitopic ligands for the CB2R can provide a fundamental contribution to the clarification of the mechanism of action of this receptor. It is therefore essential to provide an unequivocal demonstration of a simultaneous allosteric/orthosteric mode of binding for new bitopic ligand candidates. The good news is that recent advances in CB2R structural biology might help in the construction of allosteric/bitopic ligands with a designed pharmacological profile. Therefore, future structural and functional studies will provide critical insights to confirm the actual CB2R-bitopic ligand behavior of our novel analog JR64a.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
GC, CM, RL, and RP conceived and designed the experiments. CM, RF, FG, GS, SR and GO performed the design, synthesis, and chemical characterization of all novel compounds. KM, LS, RF, RP, and RL carried out cAMP, βarrestin2, and radioligand displacement assays. BP, CR, FG and GC performed the interleukins release experiments. GC, CM, RP, and RL analysed the results. GC, CM and RL wrote the paper. All authors verified the data, contributed to the manuscript, and approved the final version.
This work was supported by University of Pisa Research Grant (PRA_2020_77), by Italian Ministry of Health–Ricerca Finalizzata 2016—NET-2016-02363765, by a CIHR-GSK partnership grant and an NSERC discovery grant to RL. KM is supported by an NSERC National graduate student award.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2022.984069/full#supplementary-material
Antony, J., Kellershohn, K., Mohr-Andrä, M., Kebig, A., Prilla, S., Muth, M., et al. (2009). Dualsteric GPCR targeting: A novel route to binding and signaling pathway selectivity. FASEB J. 23, 442–450. doi:10.1096/fj.08-114751
Aymerich, M. S., Aso, E., Abellanas, M. A., Tolon, R. M., Ramos, J. A., Ferrer, I., et al. (2018). Cannabinoid pharmacology/therapeutics in chronic degenerative disorders affecting the central nervous system. Biochem. Pharmacol. 157, 67–84. doi:10.1016/j.bcp.2018.08.016
Basavarajappa, B. S., Nixon, R. A., and Arancio, O. (2009). Endocannabinoid system: Emerging role from neurodevelopment to neurodegeneration. Mini Rev. Med. Chem. 9, 448–462. doi:10.2174/138955709787847921
Benito, C., Kim, W. K., Chavarria, I., Hillard, C. J., Mackie, K., Tolon, R. M., et al. (2005). A glial endogenous cannabinoid system is upregulated in the brains of macaques with simian immunodeficiency virus-induced encephalitis. J. Neurosci. 25, 2530–2536. doi:10.1523/JNEUROSCI.3923-04.2005
Benito, C., Nunez, E., Tolon, R. M., Carrier, E. J., Rabano, A., Hillard, C. J., et al. (2003). Cannabinoid CB2 receptors and fatty acid amide hydrolase are selectively overexpressed in neuritic plaque-associated glia in Alzheimer’s disease brains. J. Neurosci. 23, 11136–11141. doi:10.1523/JNEUROSCI.23-35-11136.2003
Bie, B., Wu, J., Foss, J.F., and Naguib, M. (2018). An overview of the cannabinoid type 2 receptor system and its therapeutic potential. Curr. Opin. Anaesthesiol. 31, 407–414. doi:10.1097/ACO.0000000000000616
Bisogno, T., Ligresti, A., and Di Marzo, V. (2005). The endocannabinoid signalling system: Biochemical aspects. Pharmacol. Biochem. Behav. 81, 224–238. doi:10.1016/j.pbb.2005.01.027
Bock, A., and Bermudez, M. (2021). Allosteric coupling and biased agonism in G protein-coupled receptors. FEBS J. 288, 2513–2528. doi:10.1111/febs.15783
Chen, X., Zheng, C., Qian, J., Sutton, S. W., Wang, Z., Lv, J., et al. (2014). Involvement of β-arrestin-2 and clathrin in agonist-mediated internalization of the human cannabinoid CB2 receptor. Curr. Mol. Pharmacol. 7, 67–80. doi:10.2174/1874467207666140714115824
Chung, Y. C., Shin, W. H., Baek, J. Y., Cho, E. J., Baik, H. H., Kim, S. R., et al. (2016). CB2 receptor activation prevents glial-derived neurotoxic mediator production, BBB leakage and peripheral immune cell infiltration and rescues dopamine neurons in the MPTP model of Parkinson’s disease. Exp. Mol. Med. 48, e205. doi:10.1038/emm.2015.100
Cooper, A. G., Oyagawa, C. R. M., Manning, J. J., Singh, S., Hook, S., Grimsey, N. L., et al. (2018). Development of selective, fluorescent cannabinoid type 2 receptor ligands based on a 1, 8-naphthyridin-2-(1H)-one-3-carboxamide scaffold. Medchemcomm 9, 2055–2067. doi:10.1039/c8md00448j
Dello Russo, C., Cappoli, N., Coletta, I., Mezzogori, D., Paciello, F., Pozzoli, G., et al. (2018). The human microglial HMC3 cell line: Where do we stand? A systematic literature review. J. Neuroinflammation 15, 259. doi:10.1186/s12974-018-1288-0
Di Marzo, V. (2009). The endocannabinoid system: Its general strategy of action, tools for its pharmacological manipulation and potential therapeutic exploitation. Pharmacol. Res. 60, 77–84. doi:10.1016/j.phrs.2009.02.010
Donthamsetti, P., Gallo, E. F., Buck, D. C., Stahl, E. L., Zhu, Y., Lane, J. R., et al. (2020). Arrestin recruitment to dopamine D2 receptor mediates locomotion but not incentive motivation. Mol. Psychiatry 25, 2086–2100. doi:10.1038/s41380-018-0212-4
Draper-Joyce, C. J., Michino, M., Verma, R. K., Klein Herenbrink, C., Shonberg, J., Kopinathan, A., et al. (2018). The structural determinants of the bitopic binding mode of a negative allosteric modulator of the dopamine D2 receptor. Biochem. Pharmacol. 148, 315–328. doi:10.1016/j.bcp.2018.01.002
Ferrisi, R., Ceni, C., Bertini, S., Macchia, M., Manera, C., and Gado, F. (2021). Medicinal Chemistry approach, pharmacology and neuroprotective benefits of CB2R modulators in neurodegenerative diseases. Pharmacol. Res. 170, 105607. doi:10.1016/j.phrs.2021.105607
Gado, F., Di Cesare Mannelli, L., Lucarini, E., Bertini, S., Cappelli, E., Digiacomo, M., et al. (2019). Identification of the first synthetic allosteric modulator of the CB2 receptors and evidence of its efficacy for neuropathic pain relief. J. Med. Chem. 62, 276–287. doi:10.1021/acs.jmedchem.8b00368
Gado, F., Ferrisi, R., Polini, B., Mohamed, K. A., Ricardi, C., Lucarini, E., et al. (2022). Design, synthesis, and biological activity of new CB2 receptor ligands: From orthosteric and allosteric modulators to dualsteric/bitopic ligands. J. Med. Chem. 65, 9918–9938. doi:10.1021/acs.jmedchem.2c00582
Gado, F., Mohamed, K. A., Meini, S., Ferrisi, R., Bertini, S., Digiacomo, M., et al. (2021). Variously substituted 2-oxopyridine derivatives: Extending the structure-activity relationships for allosteric modulation of the cannabinoid CB2 receptor. Eur. J. Med. Chem. 211, 113116. doi:10.1016/j.ejmech.2020.113116
Garai, S., Kulkarni, P. M., Schaffer, P. C., Leo, L. M., Brandt, A. L., Zagzoog, A., et al. (2020). Application of fluorine and nitrogen-walk approaches: Defining the structural and functional diversity of 2-phenylindole class of cannabinoid 1 receptor positive allosteric modulators. J. Med. Chem. 63, 542–568. doi:10.1021/acs.jmedchem.9b01142
Gentry, P. R., Sexton, P. M., and Christopoulos, A. (2015). Novel allosteric modulators of G protein- coupled receptors. J. Biol. Chem. 290, 19478–19488. doi:10.1074/jbc.R115.662759
Gurevich, V. V., and Gurevich, E. V. (2019). The structural basis of the arrestin binding to GPCRs. Mol. Cell. Endocrinol. 484, 34–41. doi:10.1016/j.mce.2019.01.019
Han, S., Jayant Thatte, J., Buzard, D. J., and Jones, R. M. (2013). Therapeutic utility of cannabinoid receptor type 2 (CB2) selective agonists. J. Med. Chem. 56, 8224–8256. doi:10.1021/jm4005626
Hansen, D. V., Hanson, J. E., and Sheng, M. (2018). Microglia in Alzheimer’s disease. J. Cell. Biol. 217, 459–472. doi:10.1083/jcb.201709069
Haukedal, H., and Freude, K. (2019). Implications of microglia in amyotrophic lateral sclerosis and frontotemporal dementia. J. Mol. Biol. 431, 1818–1829. doi:10.1016/j.jmb.2019.02.004
Ho, M. S. (2019). Microglia in Parkinson’s disease. Adv. Exp. Med. Biol. 1175, 335–353. doi:10.1007/978-981-13-9913-8_13
Howlett, A. C., Barth, F., Bonner, T. I., Cabral, G., Casellas, P., Devane, W. A., et al. (2002). International union of pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol. Rev. 54, 161–202. doi:10.1124/pr.54.2.161
Kamal, M., and Jockers, R. (2009). Bitopic ligands: All-in-one orthosteric and allosteric. F1000 Biol. Rep. 1, 77. doi:10.3410/B1-77
Keov, P., Lopez, L., Devine, S. M., Valant, C., Lane, J. R., Scammells, P. J., et al. (2014). Molecular mechanisms of bitopic ligand engagement with the M1 muscarinic acetylcholine receptor. J. Biol. Chem. 289, 23817–23837. doi:10.1074/jbc.M114.582874
Lane, J. R., Sexton, P. M., and Christopoulos, A. (2013). Bridging the gap: Bitopic ligands of G-protein-coupled receptors. Trends Pharmacol. Sci. 34, 59–66. doi:10.1016/j.tips.2012.10.003
Li, X., Hua, T., Vemuri, K., Ho, J. H., Wu, Y., Wu, L., et al. (2019). Crystal structure of the human cannabinoid receptor CB2. Cell. 76, 459–467.e13. e13. doi:10.1016/j.cell.2018.12.011
Lucchesi, V., Hurst, D. P., Shore, D. M., Bertini, S., Ehrmann, B. M., Allarà, M., et al. (2014). CB2-selective cannabinoid receptor ligands: Synthesis, pharmacological evaluation, and molecular modeling investigation of 1, 8-naphthyridin-2(1H)-one-3-carboxamides. J. Med. Chem. 21, 8777–8791. doi:10.1021/jm500807e
Mackie, K. (2005). Distribution of cannabinoid receptors in the central and peripheral nervous system. Handb. Exp. Pharmacol. 2005, 299–325. doi:10.1007/3-540-26573-2_10
Manera, C., Saccomanni, G., Adinolfi, B., Benetti, V., Ligresti, A., Cascio, M. G., et al. (2009). Rational design, synthesis, and pharmacological properties of new 1, 8-naphthyridin-2(1H)-on-3-carboxamide derivatives as highly selective cannabinoid-2 receptor agonists. J. Med. Chem. 52, 3644–3651. doi:10.1021/jm801563d
Mastinu, A., Premoli, M., Ferrari-Toninelli, G., Tambaro, S., Maccarinelli, G., Memo, M., et al. (2018). Cannabinoids in health and disease: Pharmacological potential in metabolic syndrome and neuroinflammation. Horm. Mol. Biol. Clin. Investig. 36,/j/hmbci.2018.36.issue-2/hmbci-2018-0013/hmbci-2018-0013.xml. doi:10.1515/hmbci-2018-0013
Meijer, F. A., Oerlemans, G. J. M., and Brunsveld, L. (2021). Orthosteric and allosteric dual targeting of the nuclear receptor RORγt with a bitopic ligand. ACS Chem. Biol. 16, 510–519. doi:10.1021/acschembio.0c00941
Nakagawa, Y., and Chiba, K. (2014). Role of microglial M1/M2 polarization in relapse and remission of psychiatric disorders and diseases. Pharmaceuticals 7, 1028–1048. doi:10.3390/ph7121028
Newman, A. H., Battiti, F. O,, and Bonifazi, A. (2020). 2016 philip S. Portoghese medicinal chemistry lectureship: Designing bivalent or bitopic molecules for G-protein coupled receptors. The whole is greater than the sum of its parts. J. Med. Chem. 63, 1779–1797. doi:10.1021/acs.jmedchem.9b01105
Obeng, S., Hiranita, T., León, F., McMahon, L. R., and McCurdy, C. R. (2021). Novel approaches, drug candidates, and targets in pain drug discovery. J. Med. Chem. 64, 6523–6548. doi:10.1021/acs.jmedchem.1c00028
Oláh, A., Szekanecz, Z., and Bíró, T. (2017). Targeting cannabinoid signaling in the immune system: “High”-ly exciting questions, possibilities, and challenges. Front. Immunol. 8, 1487. doi:10.3389/fimmu.2017.01487
Polini, B., Cervetto, C., Carpi, S., Pelassa, S., Gado, F., Ferrisi, R., et al. (2020). Positive allosteric modulation of CB1 and CB2 cannabinoid receptors enhances the neuroprotective activity of a dual CB1R/CB2R orthosteric agonist. Life 333, 333–414. doi:10.3390/life10120333
Ramirez, S. H., Hasko, J., Skuba, A., Fan, S., Dykstra, H., McCormick, R., et al. (2012). Activation of cannabinoid receptor 2 attenuates leukocyte-endothelial cell interactions and blood-brain barrier dysfunction under inflammatory conditions. J. Neurosci. 32, 4004–4016. doi:10.1523/JNEUROSCI.4628-11.2012
Reinecke, B. A., Wang, H., and Zhang, Y. (2019). Recent advances in the drug discovery and development of dualsteric/bitopic activators of G protein-coupled receptors. Curr. Top. Med. Chem. 19, 2378–2392. doi:10.2174/1568026619666191009164609
Schrage, R., and Kostenis, E. (2017). Functional selectivity and dualsteric/bitopic GPCR targeting. Curr. Opin. Pharmacol. 32, 85–90. doi:10.1016/j.coph.2016.12.001
Shapiro, L., Gado, F., Manera, C., and Escayg, A. (2021). Allosteric modulation of the cannabinoid 2 receptor confers seizure resistance in mice. Neuropharmacology 188, 108448. doi:10.1016/j.neuropharm.2021.108448
Smith, J. S., Lefkowitz, R. J., and Rajagopal, S. (2018). Biased signalling: From simple switches to allosteric microprocessors. Nat. Rev. Drug Discov. 17, 243–260. doi:10.1038/nrd.2017.229
Steinfeld, T., Mammen, M., Smith, J. A. M., Wilson, R. D., and Jasper, J. R. (2007). A novel multivalent ligand that bridges the allosteric and orthosteric binding sites of the M2 muscarinic receptor. Mol. Pharmacol. 72, 291–302. doi:10.1124/mol.106.033746
Valant, C., Gregory, K. J., Hall, N. E., Scammells, P. J., Lew, M. J., Sexton, P. M., et al. (2008). A novel mechanism of G protein-coupled receptor functional selectivity. J. Biol. Chem. 283, 29312–29321. doi:10.1074/jbc.M803801200
Valant, C., May, L. T., Aurelio, L., Chuo, C. H., White, P. J., Baltos, J. A., et al. (2014). Separation of on-target efficacy from adverse effects through rational design of a bitopic adenosine receptor agonist. Proc. Natl. Acad. Sci. U. S. A. 111, 4614–4619. doi:10.1073/pnas.1320962111
Walter, L., Franklin, A., Witting, A., Wade, C., Xie, Y., Kunos, G., et al. (2003). Nonpsychotropic cannabinoid receptors regulate microglial cell migration. J. Neurosci. 23, 1398–1405. doi:10.1523/JNEUROSCI.23-04-01398.2003
Wootten, D., Christopoulos, A., and Sexton, P. M. (2013). Emerging paradigms in GPCR allostery: Implications for drug discovery. Nat. Rev. Drug Discov. 12, 630–644. doi:10.1038/nrd4052
Xu, J., Tang, Y., Xie, M., Bie, B., Wu, J., Yang, H., et al. (2016). Activation of cannabinoid receptor 2 attenuates mechanical allodynia and neuroinflammatory responses in a chronic post-ischemic pain model of complex regional pain syndrome type I in rats. Eur. J. Neurosci. 44, 3046–3055. doi:10.1111/ejn.13414
Keywords: cannabinoid receptor type 2 (CB2R), CB2R allosteric modulators, dualsteric agents, antiinflammatory activity, human microglial cells
Citation: Ferrisi R, Gado F, Polini B, Ricardi C, Mohamed KA, Stevenson LA, Ortore G, Rapposelli S, Saccomanni G, Pertwee RG, Laprairie RB, Manera C and Chiellini G (2022) Design, synthesis and biological evaluation of novel orthosteric-allosteric ligands of the cannabinoid receptor type 2 (CB2R). Front. Chem. 10:984069. doi: 10.3389/fchem.2022.984069
Received: 01 July 2022; Accepted: 05 September 2022;
Published: 27 September 2022.
Edited by:
Frédéric Bihel Université de Strasbourg (CNRS), FranceReviewed by:
Malliga Iyer, National Institutes of Health (NIH), United StatesCopyright © 2022 Ferrisi, Gado, Polini, Ricardi, Mohamed, Stevenson, Ortore, Rapposelli, Saccomanni, Pertwee, Laprairie, Manera and Chiellini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesca Gado, ZnJhbmNlc2NhLmdhZG9AdW5pbWkuaXQ=; Clementina Manera, Y2xlbWVudGluYS5tYW5lcmFAdW5pcGkuaXQ=; Grazia Chiellini, Z3JhemlhLmNoaWVsbGluaUB1bmlwaS5pdA==
†Present address: Francesca Gado, Department of Pharmaceutical Sciences, University of Milan, Milan, Italy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.