 Xiaotian Zhao1†
Xiaotian Zhao1† Xuanhao Li
Xuanhao Li Guodong Ye
Guodong Ye- 1Department of Pharmacy, Chengdu Second Peoples Hospital, Chengdu, China
- 2Department of Dermatology, Chengdu Second Peoples Hospital, Chengdu, China
- 3Guangzhou Municipal and Guangdong Provincial Key Laboratory of Molecular Target & Clinical Pharmacology, the NMPA and State Key Laboratory of Respiratory Disease, School of Pharmaceutical Sciences and the Fifth Affiliated Hospital, Guangzhou Medical University, Guangzhou, China
The photodriven radical-mediated [3 + 2] cyclization reaction was found to yield polymers efficiently without being hindered by degradative chain transfer. The first reaction is a hydrogen abstraction process in which one hydrogen atom migrates from the α-methylene group of an allyl monomer to the triplet state (or fragments) of the photoinitiator, thus yielding primary allyl radicals as primary radicals and then begins chain propagation via a 3 + 2 cyclization reaction. Allyl ether monomers were found to be significantly higher than other allyl monomers even with the absence of amine-like synergists. In order to clarify the procedure of the hydrogen abstraction mechanism, we used four allyl-type monomers as hydrogen donors and three thioxanthone photoinitiators as hydrogen acceptors by the quantum chemistry method in terms of geometry and energy. The results were interpreted with transition-state theory and the interaction/deformation model. Then, the tunneling factors of hydrogen abstraction reactions were also investigated by Eckart’s correction. The results show allyl ether systems are more reactive than other allyl systems, and it would provide us with new insights into these hydrogen abstractions.
Introduction
The allyl-like compounds are characterized by the presence of the allyl group CH2 = CH-CH2-X-R (X = CH2, O, NH, OCO, S,… etc). Allyl monomers are also used in coating and copolymers based on them are used to improve the thermal stability and the resistance to wear of certain materials (Mark, 1978). Thiol-ene reactions are based on the CH2 = CH- with the help of initiators. Fenton reactions mainly focus on the adjacent CH2-applying cobalt driers (Yuan et al., 2004a; Yuan et al., 2004b). Polymers are also obtained by the insertion polymerization and copolymerization applications in engineering industries such as insulators, connecting sleeves, gas-tight seals, etc (Schildknecht, 1973). In our present work (Zhao et al., 2021a; Lun et al., 2022), we obtained the embolic microsphere based on allyl monomers, which has broad application prospects in embolization interventional therapy. In addition, the abovementioned wide range of applications of allyl polymers accounts for the interest in them in recent years (Herrero and Ullah, 2021).
In the past, allyl-like compounds polymerized with difficulty and gave polymers in low yields and with low molecular weights by thermopolymerization (Laible, 1958). However, in our early research, we used the photopolymerization of allyl ether monomers to obtain the embolic microsphere via a simple photodriven radical-mediated [3 + 2] cyclization reaction (PRMC) mechanism (Zhou et al., 2022). These mechanisms involve the cleavage of type I photoinitiators to yield radicals or the excited process of type II photoinitiators to yield triplet states in the first step. Subsequent abstraction of hydrogen from an allyl monomer promotes the formation of allyl radicals as primary radicals (Zhao et al., 2021b). Under irradiation, allyl radicals are excited and start a [3 + 2] cyclization reaction with the second allyl monomer and start chain-growth processes without being interrupted by degradative chain transfer (Curran, 2002). The degradative chain transfer is induced by the conjugated structure of three-center-three-electron in allyl radicals and causes a decrease in chain propagation. Compared to allyl ether monomer with other allyl-like monomers, it has extremely satisfied polymerization properties (Olivero et al., 2000).
Hydrogen abstraction (HAT) reactions are important initiation reactions generating primary allyl radicals. These HAT reactions could also be easily found in the free-radical polymerization of (meth)acrylates or autoxidation of alkyd resin using a drier (Dursun et al., 2003; Meereis et al., 2014), which also play a significant role in the first step. For example, the formation of the alkyl hydroperoxide is the “bottleneck” step in the autoxidation process, which reveals that polymerization requires a long time at the initial stage (Zhou et al., 2016). Now, this can be overcome by adding photoinitiators to increase the formation of the alkyl hydroperoxide. In addition, Yu et al. (2018) modified the combustion mechanism analysis of decalin developed by Dagaut et al. (2013), which indicated that hydrogen abstraction reactions are the important fuel consumption channels. Although hydrogen abstraction reactions are studied in many literature studies (Chen et al., 2021; Khiri et al., 2021), there are no available experimental and theoretical studies on the thermodynamic properties and kinetic parameters of hydrogen abstraction from allyl ether or other allyl monomers.
Aiming to provide accurate kinetic parameters and facilitate the modeling of allyl monomers, the high-precision quantum chemical method was used to study the hydrogen abstraction reactions. We used four allyl-like monomers as donors and TX series photoinitiators as acceptors. The molecular structure of our study is shown in Figure 1, and the four different monomers were selected to represent the allyl monomer, allyl ether monomer. Geometry, including the conformation of the donor and the geometry of the transition state (TS), and energy, including the transition state theory (TST) energy of donors and thermodynamic data (such as the activation energy, Ea) of the reaction in combination with kinetic descriptors (such as rate constants, k), are obtained.
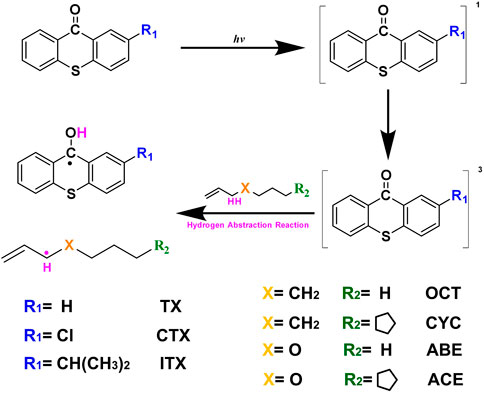
FIGURE 1. Mechanism of excited TXs as hydrogen acceptors reacting with C-H donors.
Computational details
The donors were 1-octene (OCT), allyl butyl ether (ABE), 4-cyclopentane olefin (CYC), and allyl cyclopentane ether (ACE). The acceptors were three thioxanthone photoinitiators, including thioxanthone (TX), 2-chloro-thioxanthone (CTX), and 2-isopropylthioxanthone (ITX). The density functional theory (DFT) method (Perdew and Ruzsinszky, 2010) of theoretical calculations was used to identify and quantify the interactions between different molecules. Three thioxanthone initiators are selected as abstracting agents (TX, CTX, and ITX), and the corresponding groups of twelve reactions were labeled with TX + OCT, TX + ABE, and so on. Geometry optimization calculations of all species are performed employing B3LYP/6-311++g(d,p) level of theory. The reaction energies and barrier heights were evaluated using several density functional theory methods at the M06-2X/6-311++g(d,p) level (Zhao and Truhlar, 2008) and were scaled with a zero-point energy (ZPE) scale factor of 0.97 (Alecu et al., 2010) and using Gaussian 16 package (Gaussian, 2016) in the gas phase. It involved reactants, products, and transition state (TS). All the structures of stable molecules were confirmed to have no imaginary frequency. Meanwhile, there was only one imaginary frequency of the transition state (TS). The intrinsic reaction coordinate (IRC) (Schlegel, 2011) calculations were implemented to verify that the transition state connects the two right stationary points at 298.15 K and 1 atm. Gibb’s free energy (ΔrG) and reaction enthalpies (ΔrH) are obtained from the difference of reactants and products. The Ea is obtained from the difference of reactants and TS based on the data of free energy. The bond dissociation energy (BDE) (Blanksby and Ellison, 2003) and electrostatic potential (ESP) (Murray and Politzer, 2011) were obtained by Multiwfn 3.6 (Lu and Chen, 2012) and VMD 1.9 (Humphrey et al., 1996). The k and tunneling factors (κ(T)) were computed with KiSThelP (Canneaux et al., 2014). We also performed the k of these groups were independent of pressure. The high-pressure-limit k of twelve reactions is calculated by using the TST method with the consideration of κ(T). The detailed coordinates of compounds are shown in the Supplementary Material.
Result and discussion
Donors’ descriptors
Two allyl ether monomers (ABE and ACE) combined with two allyl analogs (OCT and CYC) for comparison are performed with different electronic properties. It is a popular quantum mechanical descriptor for the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) energies (Zhou and Parr, 1990). They play a significant role in governing a wide range of chemical interactions. The HOMO–LUMO energy gaps of the four monomers are evaluated and listed in Figure 2. The OCT compounds are alpha molecular orbital level (32) and alpha molecular orbital level (33), which depend on the HOMO–LUMO gap, respectively. The energy values of the HOMO (32) orbital and LUMO (33) orbital were laying at an energy value of -8.53 and -0.02 eV, respectively. The detailed data can be found in Supplementary Table S1. In comparing these HOMO–LUMO energy gaps of different compounds, the ABE shows the highest value of 201.25 kcal/mol. The higher HOMO–LUMO energy gap implies the kinetic energy is higher and has high chemical reactivity (Saranya et al., 2018). The result indicates that the allyl ether monomers are more highly reactive than the allyl analog.
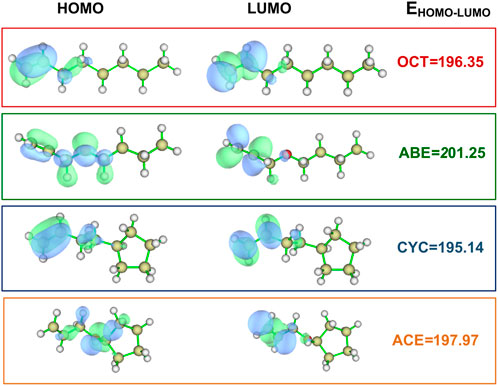
FIGURE 2. Values for HOMO and LUMO of the OCT, ABE, CYC, and ACE at the M06-2X/6-311++g(d,p) level (Unit: kcal/mol).
The BDE of C-H bonds in the donors is the key to hydrogen transfer (Zhou et al., 2016). As can be seen from the data in Figure 3, we found that ACE and ABE monomers have the lower BDE value at 78.67 and 79.42 kcal/mol, indicating two C-H bonds are more easily broken than others. The difference between the allyl and allyl ether was about 5 kcal/mol, implying that OCT and CYC do not actively participate in the HAT reaction.
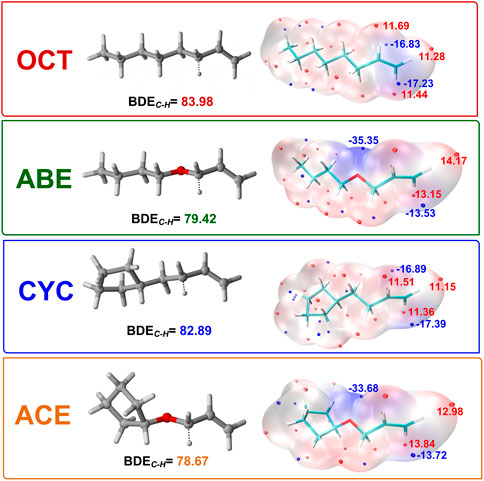
FIGURE 3. BDEs of C-H bonds in different donors and ESP-mapped molecular vdW surface of the four title monomers. Significant surface local minima and maxima of ESP are represented as red and blue spheres and labeled by red and blue texts, respectively (Unit: kcal/mol).
The oxygen atom adjacent to double bonds has strong effects than the methylene group to decrease BDE. The lower BDE value provides clear evidence for our hypothesis that hydrogen abstractions have more easily proceeded as the first step at the beginning of PRMC.
The ESP picture is portrayed with the aim of studying the donor’s electronic character. It is critical for understanding and predicting intermolecular interaction to the ESP value (Politzer and Murray, 2002), which depicts the molecular surface electronic density. Moreover, its value is also helpful for us to explore the reactivity of four monomers as shown in Figure 3. The surface area is also displayed in different colors. The negative and positive ESP were represented by blue and red spheres, respectively. It can be seen in the allyloxy region of the ABE monomer that the surface minima of ESP are present between -O-CH2 –CH = CH2 carbon atoms as shown by the blue area, and the vdW surface has a large negative value of ESP around -35 kcal/mol. The allyl ether monomers possessing more negative ESP had a stronger ability to attract electrophiles and thus are more likely to be the reactive site. This is identical to the value of BDE.
Transition state
In order to investigate the TS of HAT using four monomers as reactants, we calculated twelve HAT reactions as shown in Figure 4. Three thioxanthone initiators are selected as abstracting agents (TX, CTX, and ITX), and the corresponding twelve reactions are labeled with TX + OCT, TX + ABE, and so on.
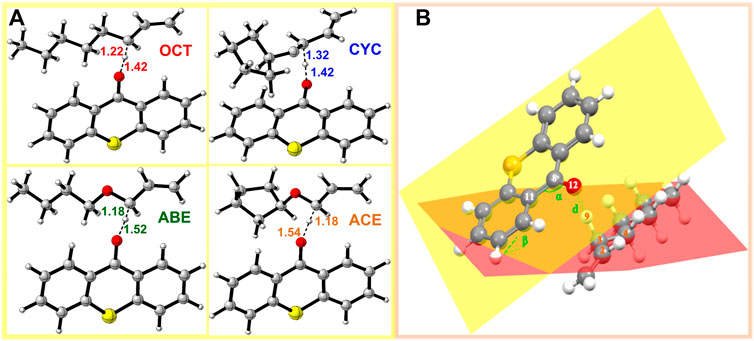
FIGURE 4. TS structure in HAT reactions: (A) structure of allyl monomers and allyl ether monomer complexes optimized at the level of B3LYP/6-311++G (d, p) (The dotted lines represent virtual σ bonds, and the unit is Å); (B) schematic view of the TS.
Geometries
In the four systems, as shown in Figure 4, the central H9 atom migrates from the carbon atoms in the methyl chain (C3) to the oxygen atom (O12) in the triplet states of thioxanthone. The comparative results of the bond angles (Δα and Δβ) are displayed in Table 1, which also contains the corresponding distances of atoms. These distances for the O12-C3 in the TS are significantly longer than those in the ground states, which indicate that they appear ready to be broken for old C3-H9 bonds and formed for new O12-H9 bonds, respectively. In addition, there are performing the different d values in different groups (More details are shown in Supplementary Table S2). As is shown previously in Figure 4B, the Δα and Δβ are variations in the bond angle of ∠O12 = C8-C11 and the depression angle change of red and yellow planes (including the angles of the O12 = C8-C11 and C2-C3-C4 planes). According to these changes in deformation, it leads to an increase in the potential energy surface, which is generally defined as the deformation energy (Ed). The interaction energy (Ei) is from the difference between Ea and Ed.
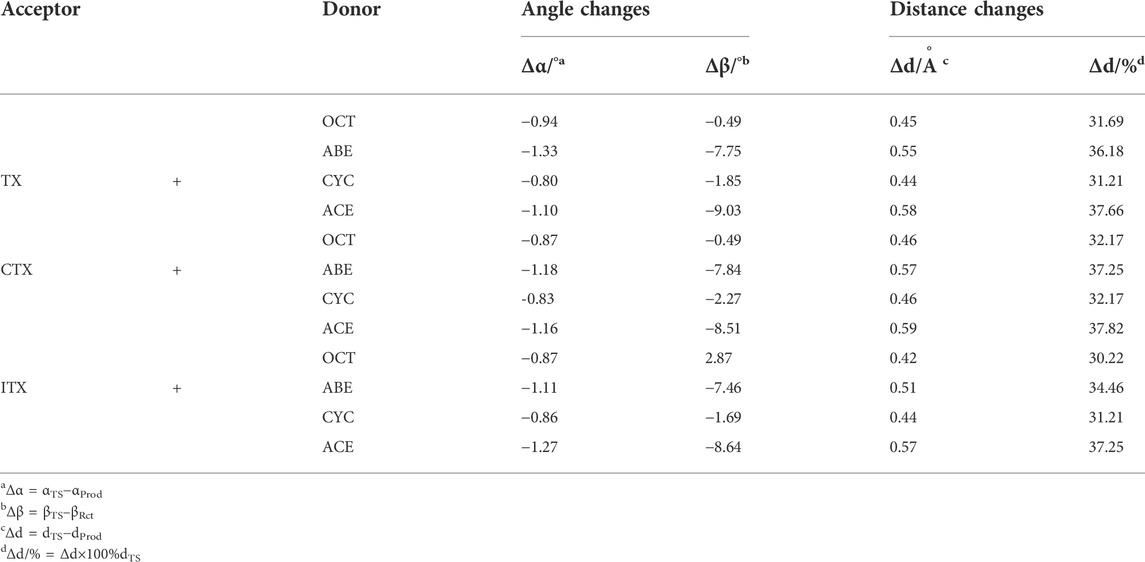
TABLE 1. Changes in bond angles and distances between the reaction complex (RC) and the transition state (TS).
As shown in Table 1, when allyl monomer reacted with triplet states of TX/CTX/ITX, almost no differences in Δα are present between the OCT and CYC, which values change below 0.1. It can be indicated that the deformations of the allyl monomers as reactants are nearly the same. However, the Δα value of ABE and ACE groups is greater, which shows that the deformation of allyl ether systems is larger than in allyl systems. These results were also validated by the Δβ values of four monomers, where the change in angle β with a value of 9.03o for TX + ACE was the greatest one in Table 1. The Δβ for OCT and CYC groups is much lower than that of allyl ether systems.
According to Table 2, the Ed of the ITX + CYC group would be found to have the highest value (12.65) of the four monomers. However, the results of the angle change Δβ show that the TX + ACE group has a higher value (9.03) than that of the ITX + CYC group. In other words, the Δβ for planes we have chosen does not have a positive correlation as we expected in Ed section. Finally, it can be seen that the Ed of allyl ether groups is lower than that of allyl groups in Table 2.
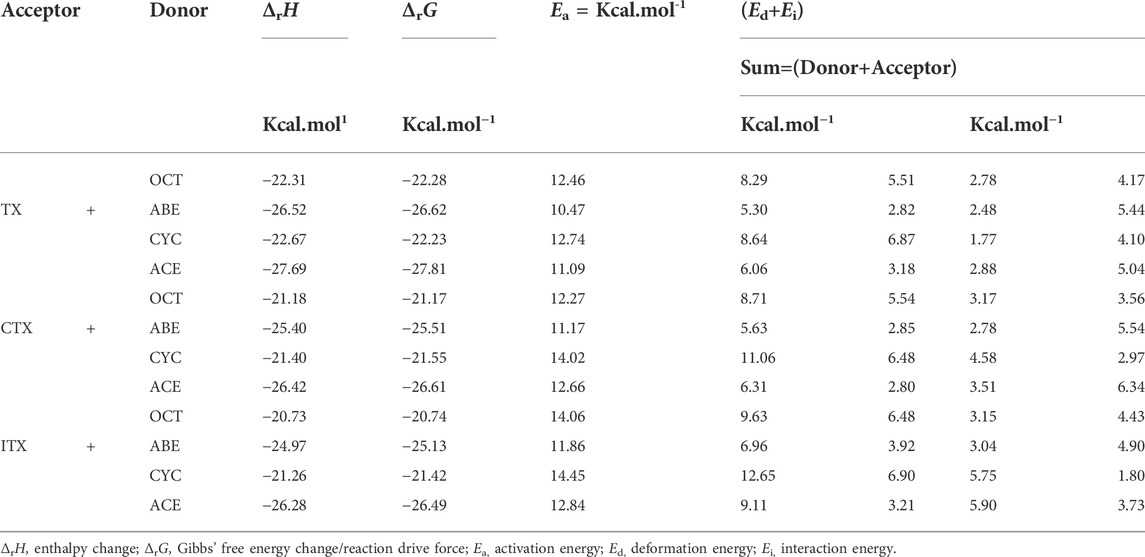
TABLE 2. Thermodynamic parameters of twelve hydrogen abstraction reactions.
We also obtained the bond length changes for the terminal atoms. Δd is the variation in bond length for O12-H9, and Δd% shows the magnitude of the change in the distance of the terminal oxygen. As is shown earlier, a comparison of the bond length reveals that ACE and ABE groups have a greater variation in bond length. In the CTX + ACE group, the bond length change reaches 37.82%, that is, 5% more than that in CYC groups. The values of allyl systems (OCT and CYC groups) are very close as above 31%. The greater Δd seems to indicate that the TS occurs earlier, and the earlier TS means that smaller activation energy (Ea) is required for the reaction. The Δd% of OCT and CYC is very close, and they might, therefore, have similar energy barriers. According to the value Ea in Table 2, it was also validated by the allyl ether systems for the lower Ea value than the allyl systems.
Energy
The thermodynamic properties of hydrogen abstraction reactions are obtained based on TST. According to the height difference in the potential energy surface, we can obtain Ea. The Ed is the energy accompanying the structural distortion from the reaction complex to the TS in the IRC calculation (Ess and Houk, 2008). Ed is the difference value of Ea and Ed according to the distortion/interaction model as mentioned earlier. The ΔrH, ΔrG, Ea, Ed, and Ei are summarized in Table 2.
The Ea values of allyl ether and allyl monomers are in the range of 10.47–12.84 and 12.27–14.06 kcal/mol, respectively. In the CYC groups, the Ea value of the ITX + CYC group is the largest, which reached 14.45 kcal/mol. However, the Ea for TX + ABE has taken the lowest value of 10.47 kcal/mol. For each group, the Ea height reactions are both in the order of CYC > OCT > ACE > ABE. Hence, one can expect that allyl ether monomers are more active among the HAT, and the corresponding reaction k is higher in Table 3.
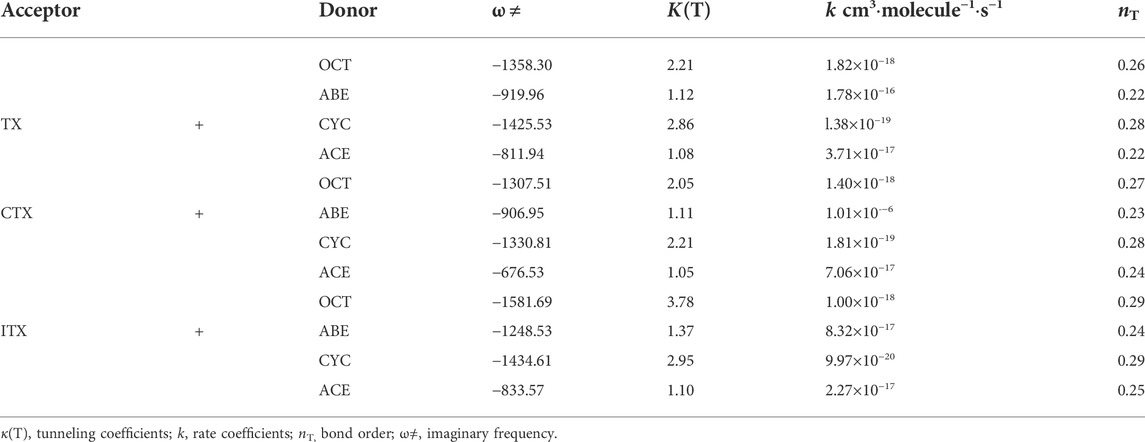
TABLE 3. Imaginary frequencies, tunneling factors, rate constants, and bond orders of the twelve reactions.
Moreover, it reveals that the ΔrG (−26.61 kcal/mol) in the CTX + ACE group is much more negative than that in the CTX + CYC group (−21.55 kcal/mol), providing a huge reaction driving force. Comparison of the ΔrG values shows that allyl ether systems have a greater negative value than allyl systems, indicating that the allyl ether systems have more reactivity. The results of ΔrG and Ea are entirely consistent with the observations obtained by gel permeation chromatography and thermogravimetry-derivative thermogravimetry analysis earlier. We found the molecular weights and polymeric yields of allyl ether monomers were significantly higher than those of other ones (Chen et al., 2022).
The Ed values were affected not only by the bond angle changes but also by the bond length changes (Grabowski, 2011). As shown in Figure 4, the distance between the TX + ACE group and O12-H is 1.54 Å, which is the greatest distance of the four monomers. After this step, the distance of the product is shortened to 0.96. According to Table 1, the Δd reaches 0.58 Å, which is larger than the other groups, 0.45 Å for OCT, 0.55 Å for ABE, and 0.44 Å for CYC. It could be seen that the HAT in the allyl ether systems crosses the barrier from a greater distance. Generally, a smaller Ed value will result in a smaller Ea, which favors the occurrence of the reaction.
Rate constant
The k could help us to understand the kinetic nature of the reaction processes. It is calculated from the formula of conventional TST (Hu et al., 2010):
where σ = 2 (for this reaction), which is the symmetry factor (degeneracy of the reaction path) that accounts for the two possible HAT from donors, kb is the Boltzmann constant, T is the absolute temperature, h is Planck’s constant, q≠, qA, and qB are, respectively, the partition functions of the reactants A and B and the transition state per unit volume, and E is the classical height of the barrier. The kinetic parameters of twelve H-abstract reactions, for example, Eckart’s correction, are shown in Table 3. Eckart’s tunneling factor, κ(T), is expressed by the following Eq. 2:
where ω≠ is the imaginary frequency of the transition state, T is the temperature, kb and h are the Boltzmann constant and Planck’s constant, respectively. Bond order, nT, is the criteria for ‘‘earliness” or ‘‘lateness” of the TS. The larger the value of nT, the later the TS would appear. According to the bond energy–bond order model (BEBO) (Blowers and Masel, 1998), nT is obtained from Eq. 3:
According to the value of nT, we found the appearance of TS in TX + ABE is earlier than the TX + OCT. Peculiarly, the ω ≠ of the TX + ABE group is lower than that of TX + OCT, so the corresponding κ(T) becomes very low. It indicated that including allyl ether group reached the TS state earlier than the allyl group in HAT. According to our observation, the κ(T) value induced by radicals is usually over 3, and it has a pronounced impact on the reactivity (Liang et al., 2018). However, in our study, κ(T) values were near to 1 for the allyl ether system. For the allyl system, the κ(T) value became higher. We found κ(T) value increases with increasing Ea. That means the low Ea is a significant factor in inducing this tunneling behavior. By comparing the k in twelve reactions, we found that the k value in the TX + ABE group reaction was 1.78 × 10−16 cm3∙molecule−1∙s−1 and is the highest. All the k values are reported in the form of a modified Arrhenius expression. The high-pressure limit k of these reactions for temperature varied from 500 to 2,500 K when treated with TST.
It can be seen from Figure 5 that the computational k for involved allyl ether (ABE/ACE) reactions are higher than those for allyl (OCT/CYC) reactions at the same temperature. It also shows a weaker negative temperature dependence for allyl ether reactions than the others. At 500 K, the k of ABE reactions is higher than that of ACE reactions since the former owns the lowest energy barriers in each group, but until 2,500 K, it can be seen that k in ACE reactions is similar to that in ABE. When ABE/ACE react with the same acceptor, the two reactions tend to possess similar k as a result of the small difference among their barrier heights. In addition, these reactions' k values with Eckart’s method (solid line) are relatively close to the without tunneling correction (dash line).
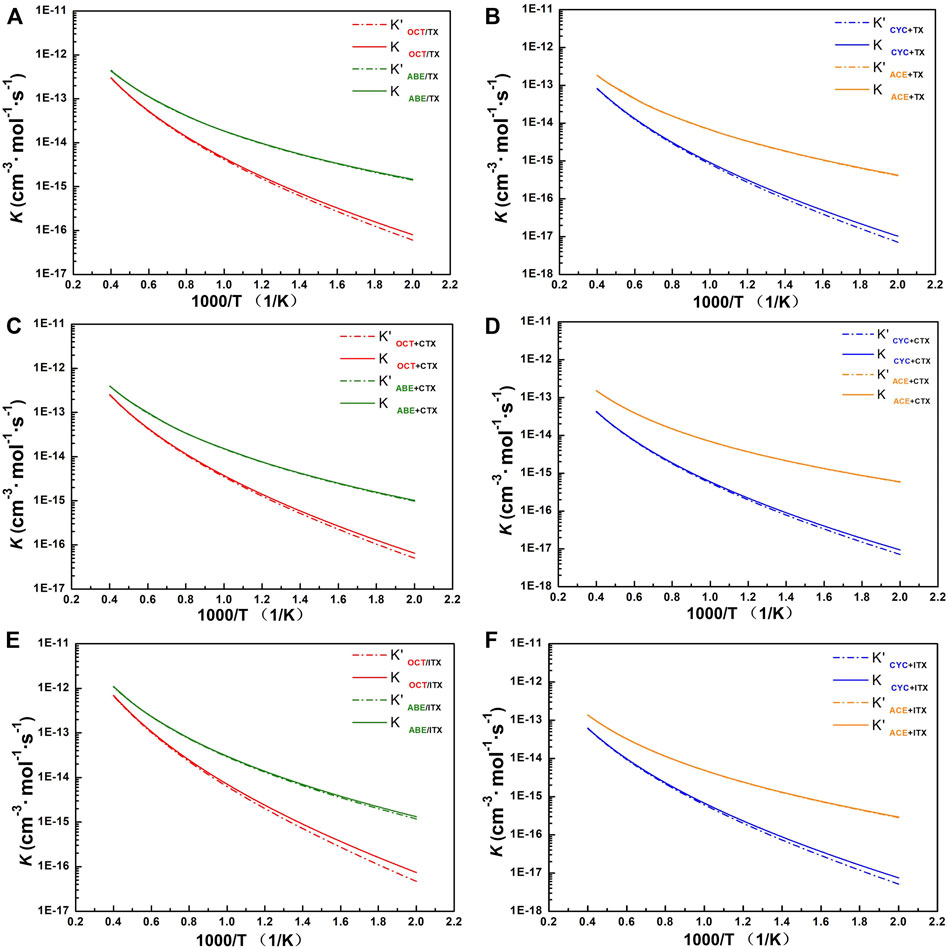
FIGURE 5. The profile of rate constant. (A) TX+OCT/ABE group. (B) TX+CYC/ACE group. (C) CTX+OCT/ABE group. (D) CTX+CYC/ACE group. (E) ITX+OCT/ABE group. (F) ITX+CYC/ACE group. k with and without tunneling correction as a function of temperature from 500 to 2,500 K. Dash line: rate constant without tunneling correction. Solid line: rate constant obtained with Eckart’s method.
For group TX + OCT/ABE, the change of our computed value for the OCT and ABE reactions’ constant rates is one to three orders of magnitude of the deviations in the temperature range of 500–1,000 K. Especially, the rate line of OCT reactions is sharper than that of ABE, and it indicates that reaction rate change of OCT is more sensitive at the beginning temperature-up. At the 500 K, the ABE reaction rate is 1.46 × 10−15, and the OCT reaction rate is 8.02 × 10−17. Until the 2,500 K, these values are similar to 4.35 × 10−13 and 3.02 × 10−13, respectively.
For group TX + CYC/ACE, the k of the TX + ACE reaction shows two orders of magnitude of the TX + CYC reaction at 500 K. While at the 2,500 K, the value of k for the TX + ACE reaction is one order of magnitude of the TX + CYC reaction. Comparing the reactions of the other acceptors (CTX and ITX), the tendency of values is similar to that of TX reactions, owing to these thioxanthone-based structures. More details can be found in Supplementary Table S3.
For the ITX + ABE reaction, our computational k of it is almost the highest over the entire temperature range, followed by reactions TX + ABE and CTX + ABE. Especially at 1,500 K, its value is one order of magnitude of the other. Moreover, by comparing the ABE structure to other donors, its computational rate constant value is found to be the highest between 500 and 2,500 K.
According to Supplementary Table S3, it revealed that for allyl ether monomers as donors, the k value is higher than that of allyl monomers, indicating that the allyl ether structure is more reaction effective. In addition, the tendency of all reaction rate values is in conformity with temperature.
Conclusion
Based on the result of the calculation, two types of monomers, allyl vs. allyl ether, have different chemical properties. By comparing the reactions of TSs with allyl or allyl ether, we found that the allyl ether system has more chemical reactivity in HAT. The allyl ether monomer processed not only higher electron density but also lower BDE than allyl systems. HAT in allyl ether monomer is more easily reacted according to lower Ea. Moreover, according to the k, the allyl ether system has a higher value and has a positive correlation with temperature. In our research earlier, we proposed the PRMC mechanism in the polymerization of allyl ether, and the hydrogen abstraction is the first step in the polymeric process. Meanwhile, our calculation works are of paramount values and fairly contribute to the building of the polymerization mechanism of allyl ether in the future.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material: further inquiries can be directed to the corresponding author.
Author contributions
GY conceived and conducted the methods including the explanation of [3 + 2] cyclization mechanism. XZ and WX wrote the manuscript and performed all the calculations. XC, SL, and XL helped XZ to provide guidance in the analysis of calculation data and put forward some beneficial advice for images and layouts. LH and XL assisted with the paperwork. All authors reviewed the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2022.967836/full#supplementary-material.
References
Alecu, I. M., Zheng, J., Zhao, Y., and Truhlar, D. G. (2010). Computational thermochemistry. Scale factor Databases and scale factors for vibrational frequencies obtained from electronic model Chemistries. J. Chem. Theory Comput. 6, 2872–2887. doi:10.1021/ct100326h
Blanksby, S. J., and Ellison, G. B. (2003). Bond dissociation energies of organic molecules. Acc. Chem. Res. 36, 255–263. doi:10.1021/ar020230d
Blowers, P., and Masel, R. I. (1998). Conservation of bond order during radical substitution reactions: Implications for the BEBO model. J. Phys. Chem. A 102, 9957–9964. doi:10.1021/jp9829243
Canneaux, S., Bohr, F., and Henon, E. (2014). KiSThelP: A program to predict thermodynamic properties and rate constants from quantum chemistry results. J. Comput. Chem. 35, 82–93. doi:10.1002/jcc.23470
Chen, P., Zhou, Y., Li, Q., Xiao, Q., Lun, Y., Huang, Y., et al. (2022). Study on the photopolymerization mechanism of allyl monomers. A photo-driven radical-mediated [3+2] cyclopolymerization mechanism to reduce degradation chain transfer. Polymer 255, 125153. doi:10.1016/j.polymer.2022.125153
Chen, W., Guo, X., Chen, L., Zhang, R., Li, Y., Feng, H., et al. (2021). A kinetics study on hydrogen abstraction reactions of cyclopentane by hydrogen, methyl, and ethyl radicals. Phys. Chem. Chem. Phys. 23, 7333–7342. doi:10.1039/d1cp00386k
Curran, H. (2002). A comprehensive modeling study of iso-octane oxidation. Combust. Flame 129, 253–280. doi:10.1016/S0010-2180(01)00373-X
Dagaut, P., Ristori, A., Frassoldati, A., Faravelli, T., Dayma, G., and Ranzi, E. (2013). Experimental and semi-detailed kinetic modeling study of decalin oxidation and pyrolysis over a wide range of conditions. Proc. Combust. Inst. 34, 289–296. doi:10.1016/j.proci.2012.05.099
Dursun, C., Degirmenci, M., Yagci, Y., Jockusch, S., and Turro, N. J. (2003). Free radical promoted cationic polymerization by using bisacylphosphine oxide photoinitiators: Substituent effect on the reactivity of phosphinoyl radicals. Polymer 44, 7389–7396. doi:10.1016/j.polymer.2003.09.020
Ess, D. H., and Houk, K. N. (2008). Theory of 1, 3-dipolar cycloadditions: Distortion/interaction and frontier molecular orbital models. J. Am. Chem. Soc. 130, 10187–10198. doi:10.1021/ja800009z
Grabowski, S. J. (2011). Halogen bond and its counterparts: Bent's rule explains the formation of nonbonding interactions. J. Phys. Chem. A 115, 12340–12347. doi:10.1021/jp205019s
Herrero, Y. R., and Ullah, A. (2021). Rapid, metal-free, catalytic conversion of glycerol to allyl monomers and polymers. ACS Sustain. Chem. Eng. 9, 9474–9485. doi:10.1021/acssuschemeng.1c03134
Hu, R., Zhang, Q., and Chen, Y. (2010). Reactions of C2(a 3Pi(u)) with selected saturated alkanes: A temperature dependence study. J. Chem. Phys. 132, 164312. doi:10.1063/1.3400070
Humphrey, W., Dalke, A., and Schulten, K. (1996). VMD: Visual molecular dynamics. J. Mol. Graph. 14, 33–38. doi:10.1016/0263-7855(96)00018-5
Khiri, D., Taamalli, S., Dao, D. Q., Nguyen, T.-B., Gasnot, L., Louis, F., et al. (2021). Thermochemical and kinetic studies of hydrogen abstraction reaction from C16H10 isomers by H atoms. Comput. Theor. Chem. 1201, 113257. doi:10.1016/j.comptc.2021.113257
Liang, Z., Liu, H., Su, N., Song, D., Zhang, Y., Huang, H., et al. (2018). Study of the deformation/interaction model: How interactions increase the reaction barrier. J. Chem. 2018, 1–8. doi:10.1155/2018/3106297
Lu, T., and Chen, F. (2012). Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580–592. doi:10.1002/jcc.22885
Lun, Y., Zhou, Y., Li, Q., Chen, P., Huang, Y., and Ye, G. (2022). Preparation and characterization of a magnetic microsphere synthesized from sucrose allyl ether for transcatheter arterial embolization. Mat. Today Chem. 24, 100772. doi:10.1016/j.mtchem.2022.100772
Mark, H. F. (1978). Encyclopedia of polymer science and technology; plastics, resins, rubbers, fibers. J. Chem. Educ. New York. doi:10.1021/ed055pA52.1
Meereis, C. T. W., Leal, F. B., Lima, G. S., Carvalho, R. V., Piva, E., and Ogliari, F. A. (2014). BAPO as an alternative photoinitiator for the radical polymerization of dental resins. Dent. Mat. 30, 945–953. doi:10.1016/j.dental.2014.05.020
Murray, J. S., and Politzer, P. (2011). The electrostatic potential: An overview. WIREs Comput. Mol. Sci. 1, 153–163. doi:10.1002/wcms.19
Olivero, S., Franco, D., Clinet, J. C., and Dunach, E. (2000). Electrochemical reduction of allyl ethers in the presence of nickel complexes: A review of synthetic applications. Collect. Czech. Chem. Commun. 65, 844–861. doi:10.1135/cccc20000844
Perdew, J. P., and Ruzsinszky, A. (2010). Fourteen easy lessons in density functional theory. Int. J. Quantum Chem. 110, 2801–2807. doi:10.1002/qua.22829
Politzer, P., and Murray, J. S. (2002). The fundamental nature and role of the electrostatic potential in atoms and molecules. Theor. Chem. Accounts Theory Comput. Model. 108, 134–142. doi:10.1007/s00214-002-0363-9
Saranya, M., Ayyappan, S., Nithya, R., Sangeetha, R. K., and Gokila, A. (2018). Molecular structure, NBO and HOMO-LUMO analysis of quercetin on single layer graphene by density functional theory. Dig. J. Nanomater. Bios. 13, 97–105.
Schlegel, H. B. (2011). Geometry optimization. WIREs Comput. Mol. Sci. 1, 790–809. doi:10.1002/wcms.34
Yu, L., Wu, Z., Qiu, Y., Qian, Y., Mao, Y., and Lu, X. (2018). Ignition delay times of decalin over low-to-intermediate temperature ranges: Rapid compression machine measurement and modeling study. Combust. Flame 196, 160–173. doi:10.1016/j.combustflame.2018.06.014
Yuan, H., Lu, X., Zeng, Z., Yang, J., and Chen, Y. (2004a). Allyl ether-modified unsaturated polyesters for UV/air dual-curable coatings. I: Synthesis and characterization of the oligomers and their cured films. J. Appl. Polym. Sci. 92, 2765–2770. doi:10.1002/app.20273
Yuan, H., Lu, X., Zeng, Z., Yang, J., and Chen, Y. (2004b). Allyl ether-modified unsaturated polyesters for UV/air dual-curable coatings. II: UV and air-curing behavior. J. Appl. Polym. Sci. 92, 2771–2776. doi:10.1002/app.20274
Zhao, X., Huang, W., Li, X., Lin, R., Li, Q., Wu, J., et al. (2021a). One-step preparation of photoclick method for embolic microsphere synthesis and assessment for transcatheter arterial embolization. Eur. J. Pharm. Biopharm. 166, 94–102. doi:10.1016/j.ejpb.2021.06.002
Zhao, X., Huang, W., Lin, S., Chen, X., Guo, X., Zou, D., et al. (2021b). Density functional theory guide for an allyl monomer polymerization mechanism: Photoinduced radical-mediated 3 + 2 cyclization. ACS omega 6, 15608–15616. doi:10.1021/acsomega.1c00165
Zhao, Y., and Truhlar, D. G. (2008). The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements. Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 120, 215–241. doi:10.1007/s00214-007-0310-x
Zhou, H., Huang, Y., Zhang, Y., Song, D., Huang, H., Zhong, C., et al. (2016). Hydrogen abstraction of carbon/phosphorus-containing radicals in photoassisted polymerization. RSC Adv. 6, 68952–68959. doi:10.1039/C6RA00156D
Zhou, Y., Chen, M., Chen, P., Zhao, X., Zhou, H., Wu, J., et al. (2022). Using a photodriven radical-mediated [3+2] cyclization reaction to prepare embolic microspheres from sucrose allyl ether without degradative chain transfer. Mat. Des. 218, 110694. doi:10.1016/j.matdes.2022.110694
Keywords: allyl ether, allyl, thioxanthone, density functional theory, hydrogen abstraction, transition state
Citation: Zhao X, Xu W, Chen X, Lin S, Li X, He L, Liao X and Ye G (2022) A comparison of hydrogen abstraction reaction between allyl-type monomers with thioxanthone-based photoinitiators without amine synergists. Front. Chem. 10:967836. doi: 10.3389/fchem.2022.967836
Received: 13 June 2022; Accepted: 25 July 2022;
Published: 02 September 2022.
Edited by:
Thomas S. Hofer, University of Innsbruck, AustriaCopyright © 2022 Zhao, Xu, Chen, Lin, Li, He, Liao and Ye. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guodong Ye, Z3poeWdkQGd6aG11LmVkdS5jbg==
†These authors have contributed equally to this work