 Shibo Lin
Shibo Lin Xiaohong Gan
Xiaohong Gan
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem., 22 July 2022
Sec. Organic Chemistry
Volume 10 - 2022 | https://doi.org/10.3389/fchem.2022.962355
This article is part of the Research TopicSynthesis and Application of Organophosphorus CompoundsView all 6 articles
The palladium-catalyzed decarboxylative reactions of phenols and vinyl ethylene carbonate to produce allylic aryl ethers under mild conditions have been established. Adopting an inexpensive PdCl2(dppf) catalyst promotes the efficient conversion of phenols to the corresponding allylic aryl ethers via the formation of a new C-O bond in good isolated yields with complete regioselectivities, acceptable functional group tolerance and operational simplicity. The robust procedure could be completed smoothly by conducting a scaled-up reaction with comparable efficiency to afford the target product.
Aryl ethers are common structural elements found in physiologically important molecules as well as complicated natural substances. (Trost and Toste 1998; Cao et al., 2002; Tanaka et al., 2003; Trost et al., 2003; Sawayama et al., 2004; Trost 2004; Trost et al., 2004; Pochetti et al., 2010). Among the many different varieties, allylic aryl ethers have gotten a lot of interest because of their wide range of uses in academia and industry (Trost and Tang 2002; Parker and Fokas 2006; Varghese and Hudlicky 2014; Zhang et al., 2019a; Zhang et al., 2019b), and selective functionalization of the motif has acted as a foundation for the synthesis of novel bioactive compounds (Figure 1). The divergent synthesis of allylic aryl ethers from simple substrates, on the other hand, remains a difficult process that has yet to be fully explored. As a result, developing effective synthetic techniques and strategies to produce such therapeutically relevant compounds is of tremendous synthetic and applied importance (Consiglio and Waymouth 1989; Malkov et al., 1999; Trost et al., 2002; López et al., 2003; Mbaye et al., 2003; Ashfeld et al., 2004; Miyabe et al., 2005; Helmchen et al., 2007; Kawatsura et al., 2007; Trost and Brennan 2007; Bandini and Eichholzer 2009).
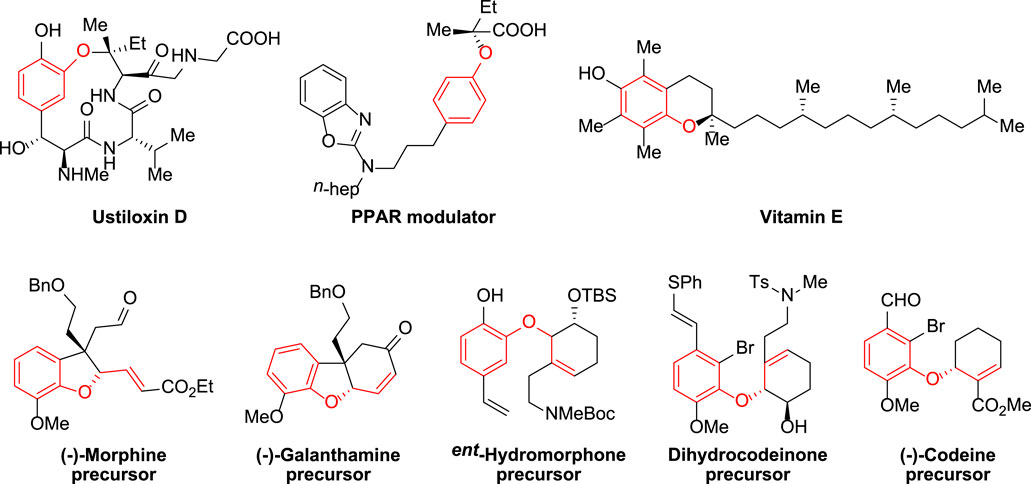
FIGURE 1. Representative important molecules containing aryl ether fragments.
Wandless and Trost, respectively, pioneered Pd-catalyzed allylic alkylation of phenol with π-allyl precursors (Sawayama et al., 2004) and intramolecular cyclization of phenol allyl carbonates (Trost and Toste 1998), which give effective methodologies for the synthesis of allylic C-O bonds. Nonetheless, the response yield is frequently inadequate. Trost (Trost et al., 2003) reported a viable decarboxylative allyl etherification process that employed vinyl epoxides as allylic donors and 4-methoxybenzyl alcohol as an interceptor to produce tertiary-alcohol derivatives in high yields (Figure 2A). For vinyl epoxides, methyl-substituted vinyl epoxides can be readily prepared by isoprene oxidation. Other 2-vinyl oxiranes with various 2-substituents, on the other hand, are difficult to obtain and rather unstable, limiting the reaction substrate’s extension. Recently, vinyl ethylene carbonates (VECs) have been created as adaptable allylic donors with more stable five-membered ring topologies and easier preparation techniques (Khan et al., 2017; Khan et al., 2019). The decarboxylative transformation of vinyl ethylene carbonates catalyzed by transition metals has shown to be a flexible approach for the creation of diverse allylic scaffolds (Shang and Liu 2011; Zhao and Szostak 2019). Kleij’s group reported the palladium-catalyzed decarboxylative allylation of VEC with phenol-based nucleophilic reagents (Xie et al., 2017), which afforded the branched product in high yields in the strong base condition (Figure 2B). It was shown that Cs2CO3 was critical for the formation of the branched products, and the presence of metal cations induced nucleophilic attack to occur at the central carbon of the palladium intermediate, thus generating the target compounds. This mild protocol is characterized by a fair scope in reaction partners, overall good yields and appreciable enantioinduction. In the same year, Zhang’s group reported the formation of tertiary alcohols or ethers from the reaction of water or alcohol as oxygen donors with VECs (Khan et al., 2017). The experimental process allowed smooth access in high yields, and the mechanism study indicated that the boron reagent played a key part in the reaction (Figure 2C). The process allowed rapid access to valuable tertiary alcohols and ethers in high yields with complete regioselectivities and high enantioselectivities. Work done by the Trost group (Trost and Toste 1999) had established that Pd π-allyl species undergo nucleophilic attack at the secondary carbon in preference to the primary carbon to deliver tertiary centers over secondary ones. This selectivity presumably arises from the greater carbocation-like character of the more highly substituted carbon center following ionization by the Pd (0) metal (Sawayama et al., 2004).
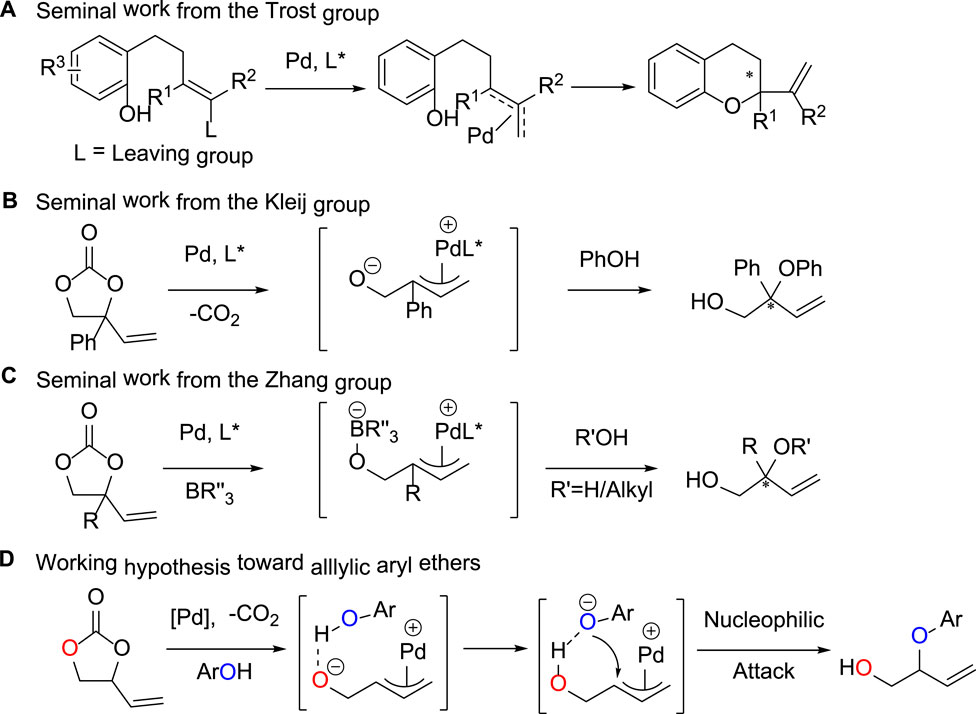
FIGURE 2. Strategy for Pd-catalyzed decarboxylative reactions of VEC with phenols.
Inspired by the above research results, we envisioned that, in the presence of an appropriate transition-metal catalyst, allylic aryl ethers could be constructed through decarboxylative etherification of VEC with activated phenolic nucleophilic reagents. As illustrated in Figure 2D, we hypothesized that the palladium-catalyzed decarboxylation of VEC could afford reactive amphiphilic π-allyl palladium intermediate, which subsequently forms the desired allylic ethers upon nucleophilic attack by phenols. Herein, we report the palladium-catalyzed conversion of VEC with phenols to construct functionalized allylic aryl ethers in high yields with excellent efficiencies and complete regioselectivities.
We commenced our studies by examining the decarboxylative etherification of an easily accessible 1-naphthol 1aa and VEC 2 as standard reaction partners with using palladium catalysis. The results are summarized in Table 1. Initially, a mixture of 1aa, 2, PdCl2(dppf) as the catalyst, and Cs2CO3 as the additive was heated in acetonitrile solvent at 80°C. We were pleased to isolate the desired product, 3aa, in 72% yield after 15 h (Table 1, entry 1). Subsequently, different palladium sources such as PdCl2(dppf)·CH2Cl2, Pd2 (dba)3, and PdCl2·(bipy) were screened for the reaction under similar conditions, respectively, but most of them obtained no target compound (Table 1, entries 2–4). In addition, no reaction was observed in the absence of palladium catalyst (Table 1, entry 5). With the efficient PdCl2(dppf) catalysis in hand, we then tested various additives to evaluate their effects on the reaction outcome. Fortunately, with the help of additives, the reaction efficiency could be promoted to different degrees. Cs2CO3 achieved the most excellent results in facilitating the reaction, while other additives, including CsOAc, DBU, K2CO3, and KOH, led to lower isolated yields (Table 1, entries 6–9). Note that the inferior result would be obtained without the help of additives (Table 1, entry 10). Furthermore, solvents such as DMF, DMSO, PhMe, 1,4-dioxane, DCM, DCE, and THF were screened based on the established catalytic system, and MeCN was determined to be the optimal solvent (Table 1, entries 11–17). To our delight, reducing the reaction temperature to 70°C increased the yield of 3aa from 72 to 81% (Table 1, entry 18). However, turning the temperature down continuously resulted in the remaining unreacted substrates, which was detrimental to the reaction yield (Table 1, entries 19–20). Therefore, the established optimized reaction conditions were 1aa (0.20 mmol) and 2 (0.30 mmol) as the substrates, PdCl2(dppf) (5 mol%) as the catalyst, and Cs2CO3 as the additive in acetonitrile (2 ml) for 15 h at 70°C.
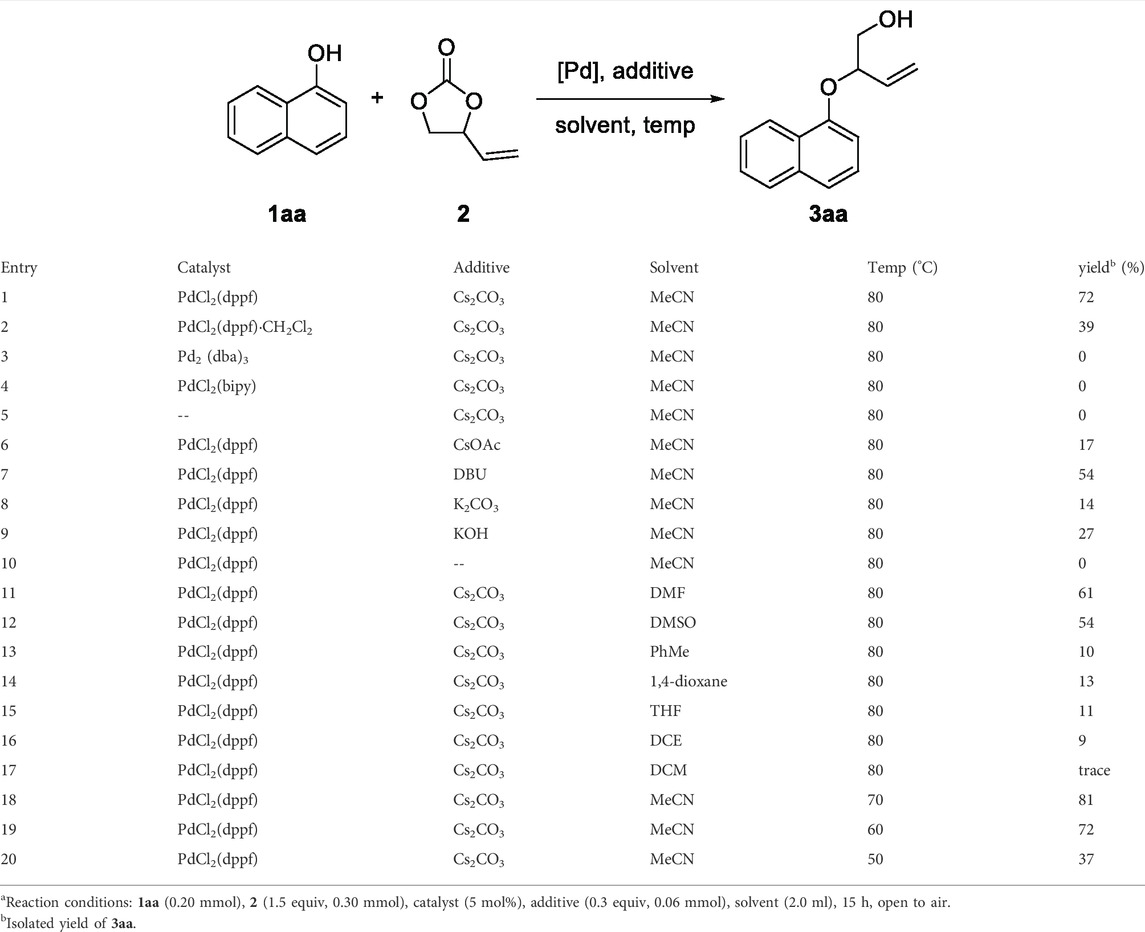
TABLE 1. Optimization of the reaction conditionsa.
As depicted in Figure 3, we investigated the substrate scope of the allylic etherifications under the optimal conditions. The reaction exhibited good functional group tolerance and was a generalized method to construct aryl allyl ether fragments facilely. First, we explored the effect of diverse monosubstituted phenol derivatives as substrates on the reaction. Halogens such as fluoro-, chloro-, and bromo-substituted phenols were well tolerated under identical conditions, affording the corresponding products in good to moderate yield (3b-3f). Further exploration found that the substitution position of halogen groups on the benzene ring greatly influences the reaction. For instance, halogen substitutions at the ortho-position of the phenyl ring (3b, 3c) generated much higher yields than did substitutions at the para and meta-positions (3d-3f). Notably, 2-Br substitution on the benzene ring afforded the target compound with an excellent yield of 90% (3b). Besides halogen groups, monosubstituted functional groups bearing electron-donating groups (-Me, -Et, -Ph) and electron-withdrawing groups (-CN, -NO2, -CF3, -COMe) on the phenol ring underwent the reaction smoothly to transform into designed products in moderate yields (3ab, 3g–3o). The reaction was also available to achieve phenols with multiple substituents, giving the etherified derivatives satisfyingly (3p–3t). Unfortunately, pyridyl ethers could not be prepared from hydroxy pyridine through the standard reaction conditions. Subsequently, we were prompted to check the feasibility of allylic etherification of quinolinol and isoquinolinol, which were demonstrated to be appropriate substrates, affording the desired product in 62 and 71% yields, respectively (3u, 3v). In addition, differently substituted naphthol and dibenzofuranol substrates performed high reactivity, and the corresponding naphthol allyl ethers were satisfyingly prepared (3w–3z). When both ortho-positions of the phenol ring are occupied, the reaction will not take place owing to the presence of higher magnitude of steric hindrance.
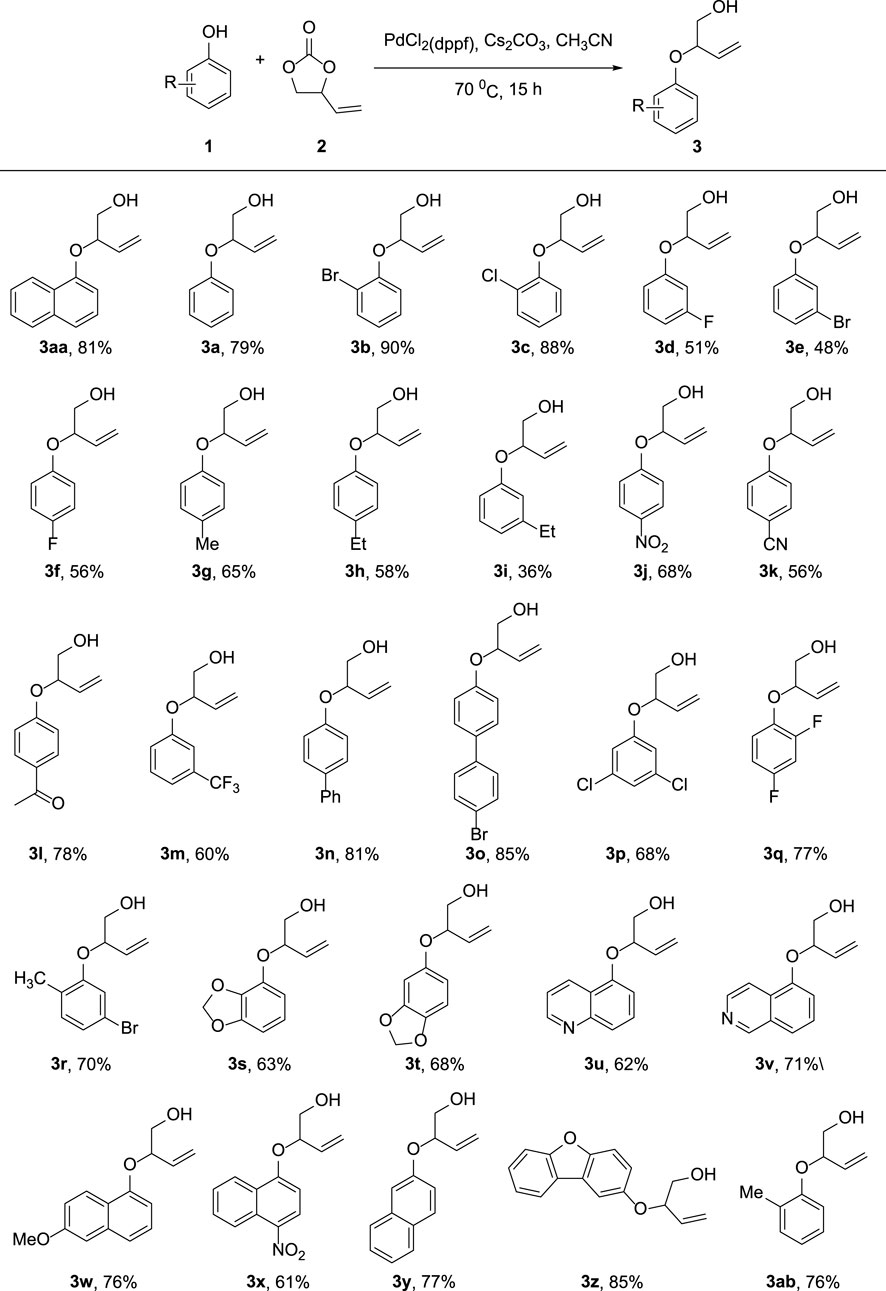
FIGURE 3. Substrate Scope of phenols. Reaction conditions: Phenol 1 (0.20 mmol), 2 (0.30 mmol), PdCl2(dppf) (5 mol%), Cs2CO3 (0.06 mmol), MeCN (2 ml), 70°C, 15 h. Isolated yields are reported.
The synthetic utility of the transformation was explored by conducting a gram-scale reaction of 1-naphthol 1aa and VEC 2 under the ideal condition, which provided the target compound 3aa with an isolated yield of 52%. Furthermore, competition experiments between differently substituted phenols revealed the preferential conversion of electron-deficient arenes (Figure 4).
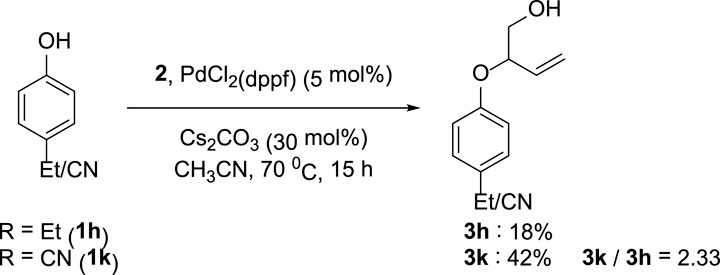
FIGURE 4. Competition experiments between 1h and 1k.
In summary, we have disclosed a general strategy for the synthesis of allylic aryl ethers through palladium-catalyzed O-H activation followed by decarboxylative reactions of aryl phenols and vinyl ethylene carbonate. Under mild conditions, allylic aryl ethers tolerating a broad scope of substitution types and functional groups could be obtained efficiently in good to excellent yield with complete regioselectivities. Notably, a scaled-up reaction could be conducted smoothly via this protocol.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
YY and XG designed the project. SL, XZ, LH, XL, QJ, and LX performed the experiments. SL analyzed the data. SL wrote the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2022.962355/full#supplementary-material
Ashfeld, B. L., Miller, K. A., and Martin, S. F. (2004). Direct, stereoselective substitution in [Rh(CO)2Cl]2-Catalyzed allylic alkylations of unsymmetrical substrates. Org. Lett. 6, 1321–1324. doi:10.1021/ol0496529
Bandini, M., and Eichholzer, A. (2009). Enantioselective gold-catalyzed allylic alkylation of indoles with alcohols: an efficient route to functionalized tetrahydrocarbazoles. Angew. Chem. Int. Ed. 48, 9533–9537. doi:10.1002/anie.200904388
Cao, B., Park, H., and Joullié, M. M. (2002). Total synthesis of ustiloxin D. J. Am. Chem. Soc. 124, 520–521. doi:10.1021/ja017277z
Consiglio, G., and Waymouth, R. M. (1989). Enantioselective homogeneous catalysis involving transition-metal-allyl intermediates. Chem. Rev. 89, 257–276. doi:10.1021/cr00091a007
Helmchen, G., Dahnz, A., Dübon, P., Schelwies, M., and Weihofen, R. (2007). Iridium-catalysed asymmetric allylic substitutions. Chem. Commun., 675–691. doi:10.1039/b614169b
Kawatsura, M., Ata, F., Wada, S., Hayase, S., Uno, H., Itoh, T., et al. (2007). Ruthenium-catalysed linear-selective allylic alkylation of allyl acetates. Chem. Commun., 298–300. doi:10.1039/b614015g
Khan, A., Khan, S., Khan, I., Zhao, C., Mao, Y., Chen, Y., et al. (2017). Enantioselective construction of tertiary C-O bond via allylic substitution of vinylethylene carbonates with water and alcohols. J. Am. Chem. Soc. 139, 10733–10741. doi:10.1021/jacs.7b04759
Khan, S., Li, H., Zhao, C., Wu, X., and Zhang, Y. J. (2019). Asymmetric allylic etherification of vinylethylene carbonates with diols via Pd/B cooperative catalysis: A route to chiral hemi-crown ethers. Org. Lett. 21, 9457–9462. doi:10.1021/acs.orglett.9b03663
López, F., Ohmura, T., and Hartwig, J. F. (2003). Regio- and enantioselective iridium-catalyzed intermolecular allylic etherification of achiral allylic carbonates with phenoxides. J. Am. Chem. Soc. 125, 3426–3427. doi:10.1021/ja029790y
Malkov, A. V., Davis, S. L., Baxendale, I. R., Mitchell, W. L., and Kocovský, P. (1999). Molybdenum(II)-Catalyzed allylation of electron-rich aromatics and heteroaromatics. J. Org. Chem. 64, 2751–2764. doi:10.1021/jo982178y
Mbaye, M. D., Demerseman, B., Renaud, J. L., Toupet, L., and Bruneau, C. (2003). [Cp*(η2-bipy)(MeCN)RuII] [PF6] catalysts for regioselective allylic substitution and characterization of dicationic [Cp*(η2-bipy)(η3-allyl)RuIV] [PF6]2 intermediates. Angew. Chem. Int. Ed. 42, 5066–5068. doi:10.1002/anie.200352257
Miyabe, H., Yoshida, K., Yamauchi, M., and Takemoto, Y. (2005). Hydroxylamines as oxygen atom nucleophiles in transition-metal-catalyzed allylic substitution. J. Org. Chem. 70, 2148–2153. doi:10.1021/jo047897t
Parker, K. A., and Fokas, D. (2006). Enantioselective synthesis of (−)-Dihydrocodeinone: A short formal synthesis of (−)-Morphine1,. J. Org. Chem. 71, 449–455. doi:10.1021/jo0513008
Pochetti, G., Mitro, N., Lavecchia, A., Gilardi, F., Besker, N., Scotti, E., et al. (2010). Structural insight into peroxisome proliferator-activated receptor gamma binding of two ureidofibrate-like enantiomers by molecular dynamics, cofactor interaction analysis, and site-directed mutagenesis. J. Med. Chem. 53, 4354–4366. doi:10.1021/jm9013899
Sawayama, A. M., Tanaka, H., and Wandless, T. J. (2004). Total synthesis of ustiloxin D and considerations on the origin of selectivity of the asymmetric allylic alkylation. J. Org. Chem. 69, 8810–8820. doi:10.1021/jo048854f
Shang, R., and Liu, L. (2011). Transition metal-catalyzed decarboxylative cross-coupling reactions. Sci. China Chem. 54, 1670–1687. doi:10.1007/s11426-011-4381-0
Tanaka, H., Sawayama, A. M., and Wandless, T. J. (2003). Enantioselective total synthesis of ustiloxin D. J. Am. Chem. Soc. 125, 6864–6865. doi:10.1021/ja035429f
Trost, B. M. (2004). Asymmetric allylic alkylation, an enabling methodology. J. Org. Chem. 69, 5813–5837. doi:10.1021/jo0491004
Trost, B. M., and Brennan, M. K. (2007). Palladium-catalyzed regio- and enantioselective allylic alkylation of bis allylic carbonates derived from Morita-Baylis-Hillman adducts. Org. Lett. 9, 3961–3964. doi:10.1021/ol701585b
Trost, B. M., Dogra, K., Hachiya, I., Emura, T., Hughes, D. L., Krska, S., et al. (2002). Designed ligands as probes for the catalytic binding mode in Mo-catalyzed asymmetric allylic alkylation we thank the national science foundation and the national institutes of health for their generous support of the work carried out at stanford. I.H. thanks the Japan society for the promotion of science for a postdoctoral fellowship. Mass spectra were provided by the mass spectrometry facility at the university of California, san francisco, which is supported by the NIH division of research resources. Angew. Chem. Int. Ed. 41, 1929. doi:10.1002/1521-3773(20020603)41:11<1929:aid-anie1929>3.0.co;2-o
Trost, B. M., Shen, H. C., Dong, L., Surivet, J-P., and Sylvain, C. (2004). Synthesis of chiral chromans by the Pd-catalyzed asymmetric allylic alkylation (AAA): Scope, mechanism, and applications. J. Am. Chem. Soc. 126, 11966–11983. doi:10.1021/ja048078t
Trost, B. M., Shen, H. C., Dong, L., and Surivet, J. P. (2003). Unusual effects in the pd-catalyzed asymmetric allylic alkylations: synthesis of chiral chromans. J. Am. Chem. Soc. 125, 9276–9277. doi:10.1021/ja036052g
Trost, B. M., and Tang, W. (2002). Enantioselective synthesis of (-)-codeine and (-)-morphine. J. Am. Chem. Soc. 124, 14542–14543. doi:10.1021/ja0283394
Trost, B. M., and Toste, F. D. (1998). A catalytic enantioselective approach to chromans and chromanols. A total synthesis of (−)-Calanolides A and B and the vitamin E nucleus. J. Am. Chem. Soc. 120, 9074–9075. doi:10.1021/ja981142k
Trost, B. M., and Toste, F. D. (1999). Regio- and enantioselective allylic alkylation of an unsymmetrical substrate: A working model. J. Am. Chem. Soc. 121, 4545–4554. doi:10.1021/ja9828713
Varghese, V., and Hudlicky, T. (2014). Short chemoenzymatic total synthesis of ent-hydromorphone: an oxidative dearomatization/intramolecular [4+2] cycloaddition/amination sequence. Angew. Chem. Int. Ed. 53, 4355–4358. doi:10.1002/anie.201400286
Xie, J., Guo, W., Cai, A., Escudero-Adán, E. C., and Kleij, A. W. (2017). Pd-catalyzed enantio- and regioselective formation of allylic aryl ethers. Org. Lett. 19, 6388–6391. doi:10.1021/acs.orglett.7b03247
Zhang, Q., Zhang, F. M., Zhang, C. S., Liu, S. Z., Tian, J. M., Wang, S. H., et al. (2019a). Catalytic asymmetric total syntheses of (-)-Galanthamine and (-)-Lycoramine. J. Org. Chem. 84, 12664–12671. doi:10.1021/acs.joc.9b01971
Zhang, Q., Zhang, F. M., Zhang, C. S., Liu, S. Z., Tian, J. M., Wang, S. H., et al. (2019b). Enantioselective synthesis of cis-hydrobenzofurans bearing all-carbon quaternary stereocenters and application to total synthesis of (‒)-morphine. Nat. Commun. 10, 2507. doi:10.1038/s41467-019-10398-4
Keywords: palladium-catalyzed, allylic etherification, phenols, vinyl ethylene carbonate, C-O bond formation
Citation: Lin S, Zhao X, He L, Li X, Jiang Q, Xiang L, Ye Y and Gan X (2022) Palladium-catalyzed allylic etherification of phenols with vinyl ethylene carbonate. Front. Chem. 10:962355. doi: 10.3389/fchem.2022.962355
Received: 06 June 2022; Accepted: 06 July 2022;
Published: 22 July 2022.
Edited by:
Jamal Rafique, Federal University of Mato Grosso do Sul, BrazilReviewed by:
Rui Shang, The University of Tokyo, JapanCopyright © 2022 Lin, Zhao, He, Li, Jiang, Xiang, Ye and Gan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongqin Ye, eWV5b25ncWluQGNkc2Rlcm15eS53ZWNvbS53b3Jr; Xiaohong Gan, bGluMDc0MTAyOEB5ZWFoLm5ldA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.