
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. , 19 July 2022
Sec. Catalytic Reactions and Chemistry
Volume 10 - 2022 | https://doi.org/10.3389/fchem.2022.957412
This article is part of the Research Topic Catalysis Rising Stars in China View all 10 articles
 Shengliang Zhai†Ling Zhang†Jikai Sun†
Shengliang Zhai†Ling Zhang†Jikai Sun† Lei SunShuchao Jiang
Lei SunShuchao Jiang Tie Yu*
Tie Yu* Dong ZhaiChengcheng LiuZhen Li
Dong ZhaiChengcheng LiuZhen Li Guoqing Ren*
Guoqing Ren*Single-atom catalysts (SACs) as the new frontier in heterogeneous catalysis have attracted increasing attention. However, the rational design of SACs with high catalytic activities for specified reactions still remains challenging. Herein, we report the rational design of a Pd1-PdNPs synergistic structure on 2,6-pyridinedicarbonitrile-derived covalent triazine framework (CTF) as an efficient active site for CO2 hydrogenation to formate under ambient conditions. Compared with the catalysts mainly comprising Pd1 and PdNPs, this hybrid catalyst presented significantly improved catalytic activity. By regulating the ratio of Pd1 to PdNPs, we obtained the optimal catalytic activity with a formate formation rate of 3.66 molHCOOM·molPd−1·h−1 under ambient conditions (30°C, 0.1 MPa). Moreover, as a heterogeneous catalyst, this hybrid catalyst is easily recovered and exhibits about a 20% decrease in the catalytic activity after five cycles. These findings are significant in elucidating new rational design principles for CO2 hydrogenation catalysts with superior activity and may open up the possibilities of converting CO2 under ambient conditions.
In recent years, single-atom catalysts (SACs) have attracted significant interest in heterogeneous catalysis for their advantages of 100% metal atom use, single active site structure, and unexpected high activity and selectivity for various reactions (Yang et al., 2013; Wang et al., 2018). Since the concept of SACs was proposed in 2011 (Qiao et al., 2011), it has solved the problems of low metal loadings (Liu et al., 2016; Li et al., 2018), poor thermal stability (Jones et al., 2016; Liu et al., 2020a; Liu et al., 2020b), and difficulties in large-scale production (Liu et al., 2020b; He et al., 2020) after nearly 10 years of vigorous development. However, although SACs are presented as homogeneous active centers, their catalytic activity is often difficult to compare with that of homogeneous catalysts and enzyme catalysts, and even lower than that of the corresponding nanocatalyst system in some cases (Ding et al., 2015). One of the reasons is that it is often difficult for a single metal atom center to activate multiple reactants with different properties or a single polyatomic molecule. The catalytic activity of monatomic catalysts does not depend on their single active center, but also on their surrounding chemical environment. The design and controllable construction of the synergistic structure between the single-atom center and its surrounding active sites is the key to achieving the high-efficiency catalytic performance of SACs.
Synergistic catalysis has been widely studied in the field of nanocatalyst and has shown obvious advantages in improving the activity and product selectivity of catalysts. For example, Liu et al. (2017) designed a Schiff-base-mediated gold catalyst for hydrogenation to formate reaction. The Schiff-base functional group grafted on a SiO2 support helps CO2 activation by the formation of a weak carbamate zwitterionic intermediate, and Au is for H2 dissociation. We previously introduced a CeO2 promoter with excellent dissociation ability to water molecules and oxygen molecules into an Au/MgGa2O4 catalyst to establish the synergistic catalytic effect between nano Au and the CeO2 promoter, which significantly improved the catalytic activity for the water gas shift and catalytic combustion reaction (Ren et al., 2019). As for SACs, although the concept of an atomic scaled synergistic effect has not been clearly proposed and systematically studied, there are some research examples of synergistic effects. For example, we recently reported a highly active dual single-Pd-atom catalyst, which could catalyze the hydrogenation of CO2 to formate under ambient conditions (Ren et al., 2022). It was found that the pore enrichment effect of microporous structures and the ternary synergetic effect among two neighboring Pd atoms and a rich nitrogen environment were the main reasons for this extraordinary catalytic activity. Liu et al. (2018) prepared a Ni-N-C SAC with metal loading up to 7.5 wt% by using N-doped C as the support, which exhibited excellent catalytic activity and cyclic stability for the hydrogenolysis of cellulose to ethylene glycol. The theoretical calculation results showed that the H2 molecule was activated by assistance of the nearest uncoordinated pyridine N atom of Ni. Therefore, the design of the synergistic catalytic structure based on the properties of reactants provides a good idea for the rational design of highly effective SACs.
In this work, we focused on the synergistic effect between single atoms and nanoparticles for CO2 hydrogenation to formate, an attractive reaction to achieve CO2 emission reduction and safe hydrogen storage (Alvarez et al., 2017; Eppinger and Huang, 2017; Su et al., 2019). Through theoretical calculations, we found that Pd1 and PdNPs were the preferred active sites for CO2 activation and hydrogen dissociation, respectively. Experimentally, we synthesized a Pd1-PdNPs synergistic structure on 2,6-pyridinedicarbonitrile-derived covalent triazine framework (CTF) by modulating the Pd loadings and reduction time, which exhibited high efficiency for the ambient hydrogenation of CO2 to formate. The optimal catalytic performance of the catalyst was obtained by regulating the ratio of Pd1 to PdNPs. Furthermore, the recycling stability was also investigated. This work provides a new strategy for the rational design of highly active SACs and is very advantageous with regard to putting forward the conversion CO2 into practical applications.
All the chemicals used in this study were of analytical grade and were used without further purification unless otherwise noted. Anhydrous zinc chloride (ZnCl2, ≥99%), 2,6-pyridinedicarbonitrile (2,6-DCP, ≥ 97%), and palladium trifluoroacetate (Pd(O2CCF3)2, ≥98%) were purchased from Shanghai Macklin Biochemical Co., Ltd. Hydrochloric acid (HCl, 36.0–38.0%) and sodium bicarbonate (NaHCO3, ≥99.5%) were purchased from Sinopharm Chemical Reagent Co., Ltd. High purity compressed N2, 10% H2/N2, and 50% CO2/50% H2 gases were obtained from the Deyi Gas Products Co., Ltd. (Qingdao, China).
The CTF-400: 2,6-DCP-derived CTF was synthesized as described elsewhere (Kuhn et al., 2008). In detail, 2,6-pyridinedicarbonitrile and anhydrous ZnCl2 were mixed at the ratio of 1:5 (w/w) and ground under the glove box. The mixed powder was transferred into a quartz ampoule tube, and then evacuated, sealed, and heated to 400°C for 40 h. After the reaction, the mixture was subsequently ground and washed with large amounts of water and diluted HCl (2 M) to remove the residual ZnCl2. After that, the resulting black powder was dried in a vacuum at 150°C for 12 h, and the resulting product was denoted to be “CTF-400.”
Pd/[CTF-400]: Pd/[CTF-400] catalysts were prepared by soaking the CTF-400 support powder in the aqueous solution of palladium trifluoroacetate with a Pd nominal weight loading of 10 wt% and 0.5 wt% for 12 h with magnetic stirring in a N2 atmosphere. The suspensions were then filtrated and washed with deionized water. The resulting filter cake was dried at 80°C for 12 h under a vacuum. The samples were denoted as 10Pd/[CTF-400] and 0.5Pd/[CTF-400], respectively, and the Pd actual weight loadings were detected to be 6.48 wt% and 0.28 wt% from ICP-AES analysis, respectively. 10Pd/[CTF-400]-R-t samples were achieved by hydrogen reduction at 300°C for a different time. Specifically, the sample was heated to 300°C with the programming of 5°C/min under a constant flow of nitrogen. Then, the sample was kept at 300°C for 5, 10, 15, 30, 60, and 180 min under a constant flow of 10% H2/90% N2 at 10 ml/min, respectively. Subsequently, the furnace was cooled down to room temperature under the protection of nitrogen.
Fourier-transform infrared spectra (FT-IR) were performed in transmission mode on a Bruker VERTEX 70v spectrometer equipped with a DLATGS detector. The sample was diluted with KBr powder.
Surface areas and pore size distribution analyses were measured on Quantachrome Autosorb-iQ. N2 was used as the adsorbate and surface areas were calculated using the BET analysis method. All of the samples were degassed under a vacuum at 260°C for 8 h before measurement.
Elemental analyses were performed on a Vario El elemental analyzer.
X-ray photoelectron spectroscopy (XPS) data were analyzed on a Thermofisher ESCALAB 250Xi spectrometer using a monochromatized Al Kα X-ray source (1,486.6 eV). All the XPS data were calibrated by using C 1s binding energy at 284.8 eV.
Transmission electron microscopy (TEM) analysis was performed on FEI Tecnai G2 F20 at 200 keV. Aberration-corrected scanning transmission electron microscopy (AC-STEM) and EDX mapping analysis were performed on a JEOL JEM-ARM200F.
CO2 hydrogenation to formate reaction was carried out in a base solution under ambient conditions (30°C and 1 bar). In general, 20 mg catalysts were added into 5 ml NaHCO3 (1 mol/L) in a three-necked bottle connected to a balloon. Then, the feed gas comprising CO2 (50% vol%) and H2 (50% vol%) was introduced after purging the residual air. After stirring for 12 h under ambient conditions, formate in the reaction mixture was determined by high-performance liquid chromatography (HPLC). In recycling experiments, the catalyst was recovered by filtration, washed with water, and dried under a vacuum. The reaction rate was calculated according to the following equation:
The calculation was performed by using the M06l/6-31G* method for nonmetal elements whereas the M06l/LANL2DZ method was used for metal atoms (Hehre et al., 1972; Hay and Wadt, 1985; Zhao and Truhlar, 2006). The metals were augmented with the corresponding LANL2DZ pseudo-potential, which was both acceptable in precision and time-consuming. Vibrational frequencies of the optimized configurations were analyzed to validate that these configurations correspond to the local minima or transition state (TS). The TS with one imaginary frequency was found and verified by the intrinsic reaction coordinate (IRC) method (Fukui, 1981).
The DFT calculations were performed on the Vienna ab initio simulation package (VASP) (Kresse and Furthmuller, 1996b; Kresse and Furthmuller, 1996a) to investigate the CO2 hydrogenation process on a Pd bulk surface and Pd1/CTF. The optB88-vdW was used to describe the exchange-correlation functional, which described the van der Waals forces appropriately (Dion et al., 2004; Lee et al., 2010). The projector augmented wave (PAW) potentials (Blochl, 1994) were used for electron–ion interactions, with a plane-wave kinetic energy cutoff of 400 eV. The geometry structures were relaxed until the forces on all atoms were less than 0.05 eV/Å. The transition states were searched using the Climbing Image Nudged Elastic Band (CI-NEB) method (Henkelman et al., 2000). Each transition state was relaxed until the forces on all atoms were less than 0.05 eV/Å.
The Pd (1 1 1) surface was modeled by a three-layer slab with a (4 × 4) surface unit cell and a vacuum thickness of 20 Å. The bottom two atomic layers of Pd (1 1 1) were fixed while the remaining layer, together with the adsorbates, were fully relaxed during relaxation. The lattice constant for Pd1/CTF is the same as for Pd, with a size of 13.79 Å × 13.79 Å × 24.51 Å. The Brillouin zone was sampled using a (2 × 2×1) k-point grid based on the Monkhorst-Pack (1976) scheme.
To achieve the most active catalyst structure, two different Pd species, Pd1 and PdNPs, were investigated to carry out the CO2 activation and H2 dissociation process. We found that 2,6-pyridinedicarbonitrile-derived CTF-coordinated Pd1 exhibited much higher adsorption energy for the CO2 molecule than that of the Pd (111) surface (−1.15 vs. −0.28 eV), indicating that Pd1 could be served as the active site for CO2 activation (Figures 1A,B). However, further calculation showed that the dissociation of the hydrogen molecule on Pd1 was difficult in thermodynamics. In contrast, the dissociation of the hydrogen molecule on the Pd surface is thermodynamically feasible (Supplementary Table S1). Moreover, metallic Pd has also been proved to have high activity in H2 dissociation with a nearly zero activation barrier (Ni and Zeng, 2009; Lozano et al., 2010). Therefore, we infer that Pd1 and PdNPs catalyze CO2 hydrogenation in collaboration through the hydrogen molecule dissociation on PdNPs and CO2 hydrogenation on Pd1. The potential energy surface (PES) of CO2 hydrogenation to formic acid on this hybrid catalyst is shown in Figure 1C. The first hydrogenation step is the rate-determining step with a barrier energy of 1.37 eV. The second step of hydrogenation is exothermic, with an energy of 2.90 eV. The energy barrier of this step is only 0.12 eV. The theoretical calculations confirm that the hydrogenation of CO2 catalyzed by the Pd1-PdNPs synergistic structure has a low barrier and can occur under ambient conditions.

FIGURE 1. (A,B) CO2 adsorption energy on Pd1/CTF and the Pd surface; (C) the potential energy surface (PES) of CO2 hydrogenation on Pd1/CTF.
The 2,6-pyridinedicarbonitrile-derived covalent triazine framework is formed by the trimerization of 2,6-pyridinedicarbonitrile in molten ZnCl2 at 400°C for 40 h, and is labeled CTF-400 (Figure 2A). FT-IR, shown in Supplementary Figure S1, confirmed the formation of the corresponding covalent triazine rings. In addition, the prepared CTF-400 possessed a surface area of 418 m2 g−1, a pore size of 0.53 nm, and a total pore volume of 0.21 m3 g−1 (Supplementary Table S2). The nitrogen content was estimated to be 19.51 wt% (Supplementary Table S3), which is high enough to load and stabilize the supported Pd species. The Pd/[CTF-400] catalyst was prepared by soaking the support powders in aqueous palladium trifluoroacetate followed by filtrating and washing after stirring for 12 h under a N2 atmosphere. By varying the initial Pd loading percent to be 0.5 wt% and 10 wt%, the single-atomic dispersed sample and the coexistence of Pd1 and PdNPs samples were prepared. The samples were denoted to be 0.5Pd/[CTF-400] and 10Pd/[CTF-400], respectively. After further reduction of 10Pd/[CTF-400] at 300°C for 3 h, the PdNPs-dominated sample was also achieved as the reference, which was denoted to be 10Pd/[CTF-400]-R-3h.
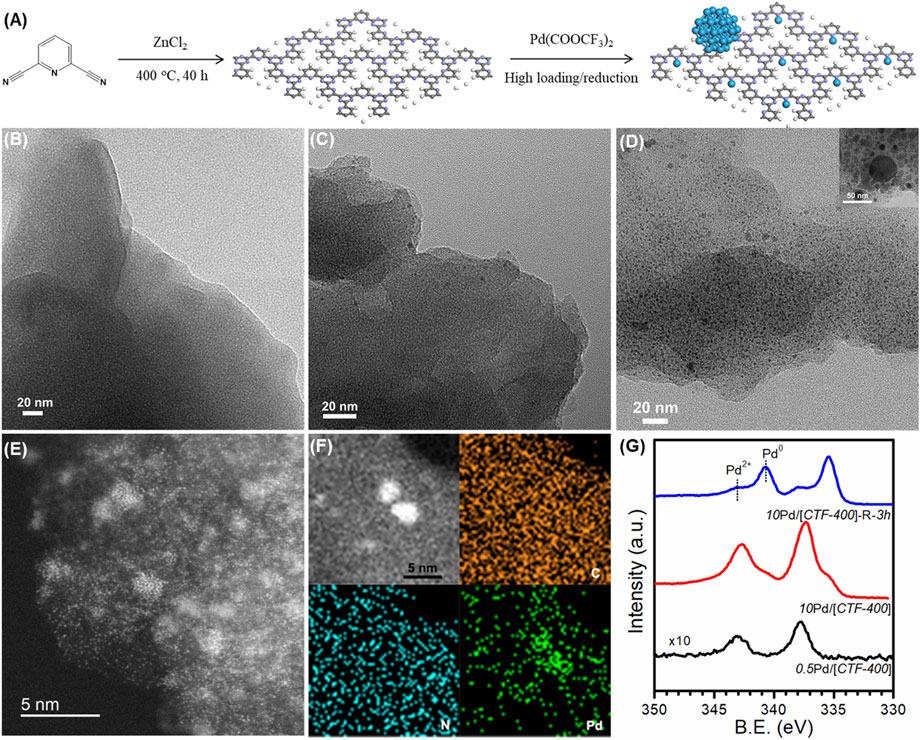
FIGURE2. Structural characterizations of Pd/[CTF-400]. (A) Schematic illustration of the fabrication procedures of Pd/[CTF-400]. (B–D) HRTEM images of the characterization of 0.5Pd/[CTF-400], 10Pd/[CTF-400], and 10Pd/[CTF-400]-R-3h. (E,F) Aberration-corrected STEM and EDS mapping of 10Pd/[CTF-400]. (G) XPS profiles for 0.5Pd/[CTF-400], 10Pd/[CTF-400], and 10Pd/[CTF-400]-R-3h samples.
Figures 2B–D display the TEM images at low magnification for 0.5Pd/[CTF-400], 10Pd/[CTF-400], and 10Pd/[CTF-400]-R-3h, respectively. It is obvious that 0.5Pd/[CTF-400] presents the notable character of CTF-400 without any Pd nanoparticles, which indicates that Pd is atomically dispersed on CTF-400 (Figure 2B). Scattered Pd nanoparticles appear on the 10Pd/[CTF-400] sample, which illustrates that Pd atoms on CTF-400 have partially aggregated to nanoparticles (Figure 2C). Further observation of this sample using AC-STEM confirms that Pd single atoms and nanoparticles coexist with good contact (Figures 2E,F). However, for the 10Pd/[CTF-400]-R-3h sample, a large amount of Pd nanoparticles with a major size of ∼3 nm was presented. The formation of PdNPs was due to the aggregation of highly dispersed Pd atoms. For the as-prepared catalyst, most of the Pd species are well dispersed as single-atoms and sub-nanoparticles (as shown in Figures 2E,F), which have high surface energy and are unstable at high temperatures and reductive atmospheres. Therefore, the percentage of PdNPs in the 10Pd/[CTF-400]-R-3h sample increased after reduction for 3 h. In addition, electronic properties for both samples were also estimated by XPS. As shown in Figure 2G, the binding energy of Pd 3d5/2 in 0.5Pd/[CTF-400] is 337.3 eV, which can be attributed to that for Pd2+ presented as Pd1 in geometry. In addition to Pd2+, 10Pd/[CTF-400] also shows the characteristic of metallic Pd corresponding to PdNPs in geometry with a binding energy of Pd 3d5/2 at 335.2 eV, and the ratio of Pd2+ to Pd0 is estimated to be about 90%. Different from that of 10Pd/[CTF-400], Pd0 is dominant in the 10Pd/[CTF-400]-R-3h sample with the ratio of Pd0 to Pd2+ being more than 70%. On the whole, the Pd1, Pd1-PdNPs hybrid, and PdNPs-dominated catalysts were successfully prepared, which can be used as good model catalysts to study the synergistic effect between different Pd active sites.
The catalytic performances of the as-prepared Pd catalysts for CO2 hydrogenation were studied at 30°C in a H2/CO2 mixture (0.1 MPa) with NaHCO3 (1 mol/L) as an additive in the liquid phase. After a reaction of 12 h, the formate was detected by using HPLC. As shown in Figure 3A and Table 1, compared with the Pd1 and PdNPs nanoparticle-dominated catalysts, the CO2 hydrogenation activity of the 10Pd/[CTF-400] catalyst was significantly enhanced (entries 1, 2 and 8 in Table 1). In detail, 10Pd/[CTF-400] exhibited a formate formation rate of 2.60 molHCOOM·molPd−1·h−1, which was obviously higher than that of 0.5Pd/[CTF-400] (0.026 molHCOOM·molPd−1·h−1) and 10Pd/[CTF-400]-R-3h (1.42 molHCOOM·molPd−1·h−1). Moreover, the heterogeneous nature of this catalyst allows it to be easily recovered by centrifugation, and the recycling tests indicated that there was around a 20% decrease in the catalytic activity after five uses (Figure 3B). This result demonstrates that the catalyst can be reused after a simple separation process, which is very advantageous with regard to practical applications.
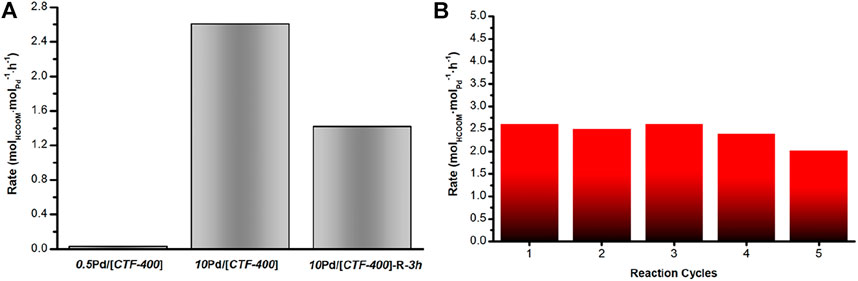
FIGURE 3. Catalytic performance of Pd/[CTF-400] for the hydrogenation of carbon dioxide to formate. (A) Formate formation rate of 0.5Pd/[CTF-400], 10Pd/[CTF-400], and 10Pd/[CTF-400]-R-3h. (B) Reuse test of 10Pd/[CTF-400]. Reaction conditions: 20 mg catalyst, 30°C, 1bar (CO2/H2 = 1:1), and 5 ml NaHCO3 (1 M), 12 h.
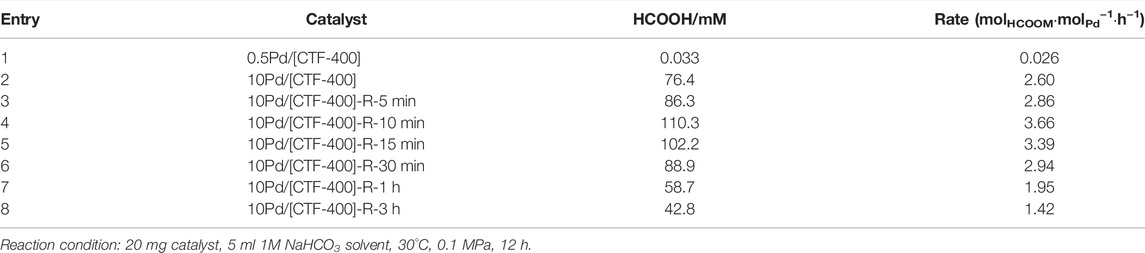
TABLE 1. Performance of Pd/[CTF-400] for CO2 hydrogenation to formate.
The aforementioned result confirms the theoretical predictions that the coexistence of Pd1 and PdNPs is necessary for the activation of H2 and CO2 at the same time. To obtain the optimized catalytic activity, the ratio of Pd1 to PdNPs was optimized. We attempted to get the sample with optimal Pd1/PdNPs by lowering the 10Pd/[CTF-400] sample into a tubular furnace at 300°C and reducing it in 10 vol% H2 for 5, 10, 15, 30, and 60 min, respectively. The corresponding samples are denoted as 10Pd/[CTF-400]-R-t, where “t” represents the reduction time. X-ray photoelectron spectroscopy (XPS) was performed to get the ratios between Pd single atoms and nanoparticles (Figure 4A and Supplementary Figures S3–S7). With careful deconvolution from the overlapped peaks of Pd2+ 3d and Pd0 3d, the ratios of Pd1 ion to the total palladium were estimated to be 89.9, 46.7, 43.1, 38.7, 34.3, and 33.7% with the continuous extension of hydrogen reduction time from 0 to 60 min (Figure 4A). Figure 4B shows the catalytic performances of 10Pd/[CTF-400]-R-t under ambient conditions (30°C, 0.1 MPa). It can be seen that the catalytic activities exhibit a volcano-type curve with a decrease in the Pd1 ratio. 10Pd/[CTF-400]-R-10 min with a Pd1 ratio of 43.1% performs the best catalytic activity, and the formate formation rate reaches 3.66 molHCOOM·molPd−1·h−1. The existence of the optimal Pd1 ratio is due to the rate equilibrium of hydrogen dissociation on Pd0 nanoparticles and carbon dioxide activation on Pd1.
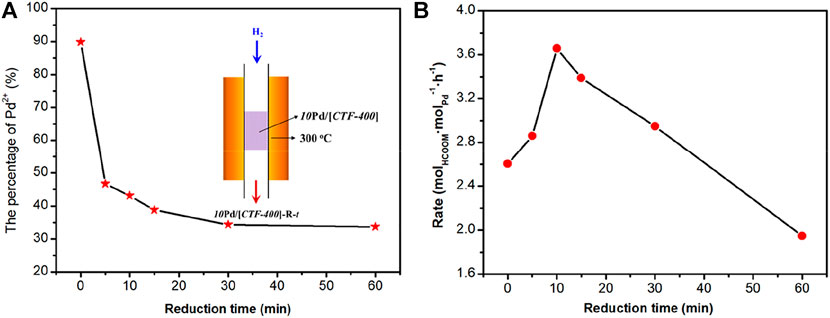
FIGURE 4. Optimization of the ratio of Pd single-atoms to nanoparticles for the hydrogenation of carbon dioxide to formate. (A) Percentage of Pd1 in 10Pd/[CTF-400]-R-t samples. (B) Formate formation rate of 10Pd/[CTF-400]-R-t samples (t = 0, 5, 10, 15, 30, and 60 min).
Based on the aforementioned analysis, the synergistic effect mechanism of 10Pd/[CTF-400] for the hydrogenation of CO2 to formate is shown in Figure 5. The 10Pd/[CTF-400] catalyst integrates both PdNPs and Pd1 into one catalyst system, where the PdNPs boosts the dissociation of H2 whereas Pd1 ions undertake the activation task of CO2. Through the atom diffusion process, H atoms generated at PdNPs move to the adsorbed CO2 on Pd1 for high-efficiency hydrogenation. Theoretical calculations shown in Figure 1 confirmed the rationality of the tasks over each active site, that is, hydrogen dissociation occurs more easily on PdNP, and carbon dioxide hydrogenation activation prefers to occur on Pd1. Experiments further verified the synergistic effect between Pd1 and PdNPs, and the 10Pd/[CTF-400] catalyst performed nearly two orders of magnitude higher activity than 0.5Pd/[CTF-400] and twice the reactivity of 10Pd/[CTF-400]-R-3 h.
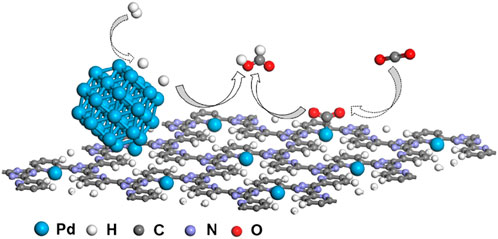
FIGURE 5. Proposed synergistic effect mechanism of Pd/[CTF-400] for the hydrogenation of carbon dioxide to formate.
In summary, we have rationally designed a highly efficient catalytic system for the hydrogenation of carbon dioxide to formate under ambient conditions based on theoretical predictions. Through modeling the CO2 adsorption and hydrogen dissociation process on both Pd1 and PdNPs, it was found that Pd1 performed the higher adsorption energy for CO2 and could be a potential candidate for CO2 activation. Compared with Pd1, hydrogen dissociation occurred more easily on Pd nanoparticles. Based on this prediction, the Pd/[CTF-400] catalyst integrating both Pd1 and PdNPs on one catalyst system was synthesized and realized the hydrogenation of CO2 to formate with a formate formation rate of 3.66 molHCOOM·molPd−1·h−1 under ambient conditions (30°C, 1 bar). This hybrid catalyst presented nearly two orders of magnitude higher than the catalyst containing bare Pd1 and twice the reactivity of that containing bare Pd0 nanoparticles. These discoveries may pave the way for the construction of active SACs with synergistic effects and open up the possibilities of converting CO2 under ambient conditions.
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding author.
GR conceived the concept and design research. SZ, LZ, and GR designed and carried out the catalyst synthesis, characterization, and catalytic test. LS and JS executed the theoretical calculations. TY carried out the TEM characterization. All authors were involved in the writing of the manuscript. The authors declare no competing interests.
This work was supported by the National Key Research and Development Program of China (No. 2017YFA0204800), the National Natural Science Foundation of China (No. 21525315), the Natural Science Foundation of Shandong Province (ZR2020QB056), and the Fundamental Research Funds of Shandong University (2019GN021 to G. R, 2019GN111 to T. Y., and 2019HW016 to L.S.), Shandong University Future Program for Young Scholars (Nos. 62460082164128 and 62460082064083).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2022.957412/full#supplementary-material
Álvarez, A., Bansode, A., Urakawa, A., Bavykina, A. V., Wezendonk, T. A., Makkee, M., et al. (2017). Challenges in the Greener Production of Formates/Formic Acid, Methanol, and DME by Heterogeneously Catalyzed CO2 Hydrogenation Processes. Chem. Rev. 117 (14), 9804–9838. doi:10.1021/acs.chemrev.6b00816
Blöchl, P. E. (1994). Projector Augmented-Wave Method. Phys. Rev. B 50 (24), 17953–17979. doi:10.1103/PhysRevB.50.17953
Ding, K., Gulec, A., Johnson, A. M., Schweitzer, N. M., Stucky, G. D., Marks, L. D., et al. (2015). Identification of Active Sites in CO Oxidation and Water-Gas Shift over Supported Pt Catalysts. Science 350 (6257), 189–192. doi:10.1126/science.aac6368
Dion, M., Rydberg, H., Schröder, E., Langreth, D. C., and Lundqvist, B. I. (2004). Van Der Waals Density Functional for General Geometries. Phys. Rev. Lett. 92 (24), 246401. doi:10.1103/Physrevlett.92.246401
Eppinger, J., and Huang, K.-W. (2017). Formic Acid as a Hydrogen Energy Carrier. ACS Energy Lett. 2 (1), 188–195. doi:10.1021/acsenergylett.6b00574
Fukui, K. (1981). The Path of Chemical Reactions - the IRC Approach. Acc. Chem. Res. 14 (12), 363–368. doi:10.1021/Ar00072a001
Hay, P. J., and Wadt, W. R. (1985). Ab Initio effective Core Potentials for Molecular Calculations. Potentials for K to Au Including the Outermost Core Orbitals. J. Chem. Phys. 82 (1), 299–310. doi:10.1063/1.448975
He, X., Deng, Y., Zhang, Y., He, Q., Xiao, D., Peng, M., et al. (2020). Mechanochemical Kilogram-Scale Synthesis of Noble Metal Single-Atom Catalysts. Cell. Rep. Phys. Sci. 1 (1), 100004. doi:10.1016/j.xcrp.2019.100004
Hehre, W. J., Ditchfield, R., and Pople, J. A. (1972). Self-Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian-type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 56(5), 2257, 2261. doi:10.1063/1.1677527
Henkelman, G., Uberuaga, B. P., and Jónsson, H. (2000). A Climbing Image Nudged Elastic Band Method for Finding Saddle Points and Minimum Energy Paths. J. Chem. Phys. 113 (22), 9901–9904. doi:10.1063/1.1329672
Jones, J., Xiong, H., Delariva, A. T., Peterson, E. J., Pham, H., Challa, S. R., et al. (2016). Thermally Stable Single-Atom Platinum-On-Ceria Catalysts via Atom Trapping. Science 353 (6295), 150–154. doi:10.1126/science.aaf8800
Kresse, G., and Furthmüller, J. (1996a). Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 6 (1), 15–50. doi:10.1016/0927-0256(96)00008-0
Kresse, G., and Furthmüller, J. (1996b). Efficient Iterative Schemes Forab Initiototal-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 54 (16), 11169–11186. doi:10.1103/PhysRevB.54.11169
Kuhn, P., Antonietti, M., and Thomas, A. (2008). Porous, Covalent Triazine-Based Frameworks Prepared by Ionothermal Synthesis. Angew. Chem. Int. Ed. 47 (18), 3450–3453. doi:10.1002/anie.200705710
Lee, K., Murray, É. D., Kong, L., Lundqvist, B. I., and Langreth, D. C. (2010). Higher-accuracy van der Waals density functional. Phys. Rev. B 82 (8), 081101. doi:10.1103/Physrevb.82.081101
Li, H., Wang, L., Dai, Y., Pu, Z., Lao, Z., Chen, Y., et al. (2018). Synergetic Interaction between Neighbouring Platinum Monomers in CO2 Hydrogenation. Nat. Nanotech 13 (5), 411–417. doi:10.1038/s41565-018-0089-z
Liu, K., Tang, Y., Yu, Z., Ge, B., Ren, G., Ren, Y., et al. (2020a). High-loading and Thermally Stable Pt1/MgAl1.2Fe0.8O4 Single-Atom Catalysts for High-Temperature Applications. Sci. China Mat. 63 (6), 949–958. doi:10.1007/s40843-020-1267-2
Liu, K., Zhao, X., Ren, G., Yang, T., Ren, Y., Lee, A. F., et al. (2020b). Strong Metal-Support Interaction Promoted Scalable Production of Thermally Stable Single-Atom Catalysts. Nat. Commun. 11 (1), 1263. doi:10.1038/S41467-020-14984-9
Liu, P., Zhao, Y., Qin, R., Mo, S., Chen, G., Gu, L., et al. (2016). Photochemical Route for Synthesizing Atomically Dispersed Palladium Catalysts. Science 352 (6287), 797–800. doi:10.1126/science.aaf5251
Liu, Q., Yang, X., Li, L., Miao, S., Li, Y., Li, Y., et al. (2017). Direct Catalytic Hydrogenation of CO2 to Formate over a Schiff-Base-Mediated Gold Nanocatalyst. Nat. Commun. 8 (1), 1407. doi:10.1038/s41467-017-01673-3
Liu, W., Chen, Y., Qi, H., Zhang, L., Yan, W., Liu, X., et al. (2018). A Durable Nickel Single-Atom Catalyst for Hydrogenation Reactions and Cellulose Valorization under Harsh Conditions. Angew. Chem. Int. Ed. 57 (24), 7071–7075. doi:10.1002/anie.201802231
Lozano, A., Gross, A., and Busnengo, H. F. (2010). Molecular Dynamics Study of H2 Dissociation on H-Covered Pd(100). Phys. Rev. B 81 (12), 121402. doi:10.1103/Physrevb.81.121402
Monkhorst, H. J., and Pack, J. D. (1976). Special Points for Brillouin-Zone Integrations. Phys. Rev. B 13 (12), 5188–5192. doi:10.1103/PhysRevB.13.5188
Ni, M., and Zeng, Z. (2009). Density Functional Study of Hydrogen Adsorption and Dissociation on Small Pdn (N=1-7) Clusters. J. Mol. Struct. THEOCHEM 910 (1-3), 14–19. doi:10.1016/j.theochem.2009.06.008
Qiao, B., Wang, A., Yang, X., Allard, L. F., Jiang, Z., Cui, Y., et al. (2011). Single-atom Catalysis of CO Oxidation Using Pt1/FeOx. Nat. Chem. 3 (8), 634–641. doi:10.1038/Nchem.1095
Ren, G.-Q., Pei, G.-X., Zhang, J.-C., and Li, W.-Z. (2019). Activity Promotion of Anti-sintering Au MgGa2O4 Using Ceria in the Water Gas Shift Reaction and Catalytic Combustion Reactions. Chin. J. Catal. 40 (4), 600–608. doi:10.1016/S1872-2067(19)63295-X
Ren, G., Sun, J., Zhai, S., Yang, L., Yu, T., Sun, L., et al. (2022). Ambient Hydrogenation of Carbon Dioxide into Liquid Fuel by a Heterogeneous Synergetic Dual Single-Atom Catalyst. Cell. Rep. Phys. Sci. 3 (1), 100705. doi:10.1016/j.xcrp.2021.100705
Su, X., Yang, X.-F., Huang, Y., Liu, B., and Zhang, T. (2019). Single-Atom Catalysis toward Efficient CO2 Conversion to CO and Formate Products. Acc. Chem. Res. 52 (3), 656–664. doi:10.1021/acs.accounts.8b00478
Wang, A., Li, J., and Zhang, T. (2018). Heterogeneous Single-Atom Catalysis. Nat. Rev. Chem. 2 (6), 65–81. doi:10.1038/s41570-018-0010-1
Yang, X.-F., Wang, A., Qiao, B., Li, J., Liu, J., and Zhang, T. (2013). Single-Atom Catalysts: A New Frontier in Heterogeneous Catalysis. Acc. Chem. Res. 46 (8), 1740–1748. doi:10.1021/ar300361m
Keywords: synergistic effect, single-atom catalysts (SACs), CO2 hydrogenation, formic acid, ambient conditions
Citation: Zhai S, Zhang L, Sun J, Sun L, Jiang S, Yu T, Zhai D, Liu C, Li Z and Ren G (2022) Rational Design of Synergistic Structure Between Single-Atoms and Nanoparticles for CO2 Hydrogenation to Formate Under Ambient Conditions. Front. Chem. 10:957412. doi: 10.3389/fchem.2022.957412
Received: 31 May 2022; Accepted: 24 June 2022;
Published: 19 July 2022.
Edited by:
Haifeng Xiong, Xiamen University, ChinaCopyright © 2022 Zhai, Zhang, Sun, Sun, Jiang, Yu, Zhai, Liu, Li and Ren. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tie Yu, eXV0aWVAc2R1LmVkdS5jbg==; Guoqing Ren, cmVuZ3VvcWluZ0BzZHUuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.