 Joyeeta Lodh1
Joyeeta Lodh1 Shounik Paul
Shounik Paul Wolfgang Schöfberger
Wolfgang Schöfberger Soumyajit Roy
Soumyajit Roy- 1Eco-Friendly Applied Materials Laboratory (EFAML), Materials Science Centre, Department of Chemical Sciences, Mohanpur Campus, Indian Institute of Science, Education and Research, Kolkata, West Bengal, India
- 2Institute of Organic Chemistry, Laboratory for Sustainable Chemistry and Catalysis (LSusCat), Johannes Kepler University (JKU), Linz, Austria
Although the core of electrochemistry involves simple oxidation and reduction reactions, it can be complicated in real electrochemical organic reactions. The principles used in electrochemical reactions have been derived using physical organic chemistry, which drives other organic/inorganic reactions. This review mainly comprises two themes: the first discusses the factors that help optimize an electrochemical reaction, including electrodes, supporting electrolytes, and electrochemical cell design, and the second outlines studies conducted in the field over a period of 10 years. Electrochemical reactions can be used as a versatile tool for synthetically important reactions by modifying the constant electrolysis current.
1 Introduction
Over the years, synthetic chemists have always faced the challenge of generating molecules with structures ranging from a few nanometers to increasingly bulky and complex structures in a sustainable way (Bryan et al., 2013; Kuttruff et al., 2014; Cernak et al., 2016). Owing to these challenges, the techniques and strategies are being constantly monitored to increase the chemical toolbox. These factors made photochemistry (Lodh et al., 2018; Mallick and Roy, 2018) and electrochemistry (Moeller, 2000; Sperry and Wright, 2006; Yoshida et al., 2008; Ogibin et al., 2009; Sequeira and Santos, 2009) re-emerge as an important route to synthesize industrially important precursor. The earliest mention of electrochemical methods could be dated back to the 19th century when the Hall–Heroult (Grjotheim et al., 1977) and Chloralkali processes (Lakshmanan and Murugesan, 2014) were developed to perform the electrolysis of aqueous sodium chloride and Al2O3, respectively. However, despite the huge popularity of electrochemical reactions to synthesize valuable chemicals on a metric-ton scale, the technique has rarely been used in organic chemistry for the synthesis of value-added chemicals. The electrochemical pathway is advantageous in many ways over the traditional pathway owing to its mild condition, tolerance for a functional group, sustainability, and easy scalability. The major challenges to adapting the electroorganic reactions include 1) separation of product, which is an issue for both conventional and electroorganic synthesis; 2) reaction setup, which is critical to the electroorganic reaction; and 3) the choice of the solvent, which influences the ionic conductivity in the reaction setup. The major challenges to adapting the electroorganic reactions include 1) separation of product, which is an issue for both conventional and electroorganic synthesis; 2) reaction setup, which is critical to the electroorganic reaction; and 3) the choice of the solvent, which influences the ionic conductivity in the reaction setup. It is important to realize that any chemical reaction will have its electrochemical counterpart. The reverse is also true, provided a distinct catalyst is known. However, one must realize that these two techniques are presently non-parallel and instead appear to be complementary. This could be changed with increased knowledge of factors influencing the chemical and electrochemical pathways. The advantages could be numerous once there are multiple parameters for the electrochemical pathway. The advantages could be 1) yield (in major cases adequate or ample); 2) negligible cost (excluding the cost of equipment, the chemical reagent and power are relatively cheap); and 3) easy work-up (the formation of side product is little, and in most cases, work-up involves the removal of only the electrolyte and solvent). Numerous review articles have been published in this field of organic electrochemistry that summarize the advances made over the years, such as the Kolbe reaction (Glasstone and Hickling, 1939; Vijh and Conway, 1967) and electrochemical reduction (Popp and Schultz, 1962; Elving and Pullman, 2009). The groups of Steckhan (Steckhan, 1986; Steckhan, 1987), Little (Francke and Little, 2014), and Nikishin (Ogibin et al., 2009) reviewed the advances in indirect electrolysis. The cathodic reduction and anodic oxidation processes were reviewed in detail by the groups of Yoshida (Yoshida et al., 2008), Schäfer (Schäfer, 1981; Schäfer, 2011), Wright (Sperry and Wright, 2006), and Tian (Chen et al., 2019). The anodic electrochemical process was also reviewed by the groups of Eberson (Eberson and Nyberg, 1973; Eberson and Nyberg, 1976), Shono (Shono, 1984), Adams (Adams, 1969), Schäfer (Schafer et al., 2007; Schafer, 2014), Chiba (Chiba and Kim, 2009), Moeller (Moeller, 1997; Moeller, 2000; Moeller, 2016; Feng et al., 2017), and Boydston (Ogawa and Boydston, 2015). The group of Waldvogel (Möhle et al., 2018; Waldvogel et al., 2018; Wiebe et al., 2018; Schulz and Waldvogel, 2019), Bao Guo Sun (Yang Q. L. et al., 2018), Tian-Sheng Mei (Ma et al., 2018), Zeng (Jiang et al., 2017), Zhang (Guo et al., 2015), and Daugulis (Daugulis et al., 2015) summarized the synthetic attempts at complex structures using electrochemistry. There were also detailed reviews on electrochemical alkyne functionalization by the Nisar Ahmed group (Martins et al., 2019). This review is divided into two major themes. In the first part, we focus on learning the basics of electrochemistry such that at any given point, for an electrochemical system, the relation between the current and voltage could be exploited to understand the behavior of an electrochemical cell and improve its catalytic performance. The second part focuses on synthetic electrochemical organic transformations on a regular laboratory scale with a special focus on the choice of electrodes. The examples have been chosen in a manner to provide exhaustive coverage of the electroorganic reactions (Table 1), at the same time eliminating an ambiguous example pertaining to selectivity, yield, and other parameters.
TABLE 1. Comparing different reaction strategies for electroorganic synthesis.
In this review, we not only attempt to present an overview of multiple parameters that govern the electrochemical pathway. We also focus a great deal on the types of organic reactions conducted over a period of 5 years. These reactions will be segregated according to the electrochemical methods employed. This review aims to help the researchers understand and adopt electrochemical pathways in a general synthetic organic reaction.
2 Basic requirements for electroorganic synthesis
In this section, we delve into the basics of electrochemistry, which might help a novice to foray into the field of electrochemistry. It is important to note the parameters which help distinguish any electrochemical organic reaction from a general organic reaction. The driving force for any redox reaction is potential, often referred to as voltage. One of the most important parameters would be a typical power source connected to three electrodes, namely, cathode, anode, and reference. When attached to an electrochemical cell, the power source controls the potential between the reference and working electrodes. The reaction of interest occurs at the working electrode, whereas the other could be referred to as the counter electrode. The anode and cathode can be designated as working electrodes. The potential difference between the cathode and anode would be driven by the power source in case a reference electrode is used. In order to create an oxidative environment at the anode and a reductive environment at the cathode, the power source would push electrons in the cathode from the anode. Another important aspect of any electrochemically driven reaction would be current, which is indicative of the rate of electron flow, is impossible to disentangle from voltage, and facilitates redox reaction. The fourth and final parameter would be the reaction medium containing an electrocatalyst, which acts as a circuit through which charged species move.
Before discussing the reaction types in an electrochemical cell, it is important to discuss what constitutes an electrochemical cell. The equipment for electrochemical cells could range from a simple beaker to elaborate instruments. However, in the interest of electroorganic synthesis, capacitors, redox-flow cells, and batteries would not be discussed.
2.1 Power supply
A simple method to ensure a supply of electricity would be attaching a suitable battery with a suitable potential, which helps overcome the resistance of an electrochemical cell. The power source could come in various forms. In electroorganic conversion, one might often encounter experiments conducted by potentiostats or galvanostats.
2.2 Electrodes
One of the most important constituents of an electrochemical organic synthesis is the choice of the electrode (Hilt, 2020) with regard to the surface area, reusability, and constituents. The major challenges in the field of electrochemistry would be the availability of the non-standardized nature of electrodes, including the myriad of semiconducting materials from the supermarket, such as aluminum foil. The choice of electrode material would significantly impact the fate of the reaction because the transfer of electrons occurs at the surface of the electrode. It is required to incorporate a “reference electrode.” However, the reference electrode is not a necessity for preparative constant potential experiments. It is important to gauge the adsorption of active species on the electrode surface and the diffusion of reactive species into the solution. The mode of operation could always be referred to from the literature.
2.3 Solvents and electrolytes
In an electrochemical cell, the reducing resistance or increasing conductivity is often dictated by solvents and electrolytes. Organic solvents employed in electroorganic conversions are often polar aprotic, which dissolves the substrates, electrolytes, and reactants. There have been numerous reviews on solvents and their properties for electrochemical reactions. This section briefly discusses solvents and their properties that make them relevant for electrochemical reactions (Mann and Grunwald, 1959; Sawyer et al., 1995; Lund and Hammerich, 2001; Janz, 2012; Kissinger and Heineman, 2018).
One of the important properties of an electrochemical solvent is its ability to dissolve and dissociate ionic salts to achieve high ionic conductivity. However, an exception to this rule is electrochemically induced precipitation or electrodeposition, wherein the solvation of solvents and products by the solvent is desired. Other properties of the electrochemical solvent include its polarity, oxidation/reduction property, acidity/basicity, and electrophilic/nucleophilic behavior. The susceptibility of solvent toward oxidation/reduction is a critical point because a solvent is often exposed to electrodes at high oxidizing or reducing potentials (Bockris et al., 1992; Conway and Bockris, 2012). In order to maintain the charge neutrality of the cell, the presence of charged species (electrolytes) is important. Electrolytes also help in the modification of the surface of the electrode during the reaction. The electrolytes can range from ionic liquids to polar solvents, which are conducive due to the presence of soluble organic salts, such as Bu4NF4.
2.4 Electrochemical cell design
The designing of an electrochemical cell depends entirely on the nature of the experiments. The materials for designing the electrochemical cell could vary from glass (e.g., quartz or Pyrex) to nylon or Teflon. (White, 2012). However, an electrochemical cell should be designed keeping in mind the following points: 1) it should be cost-effective and 2) it must be inert toward the electrochemical reaction. Additionally, it is important to note that the material used for making the electrochemical cell must not interfere during sensitive measurements. For example, systems involving hydrofluoride or high pH can lead to the corrosion of glass. Similarly, in the presence of an organic solvent, a cell made of plastic might decompose. This section provides a brief description of the two-electrode cell, three-electrode cell, and electrochemical cell used for specialized purposes.
2.4.1 Undivided cell
In order to assemble an undivided cell (Ma et al., 2021) as shown in Figures 1, 2, it is important to ensure that 1) the electrodes do not touch each other and 2) there is a distance between the electrode holders and electrodes. To create an inert atmosphere, the beaker-type cell could be sealed with a Teflon stopper. The bulk electrolysis at the undivided cell not only transforms substrate A but also could make a change in the material at the counter electrode. It is important to ensure that product A should not convert back to substrate A at the counter electrode causing the electrolysis to become inefficient (Figure 3).

FIGURE 1. Schematic for an undivided electrochemical cell.
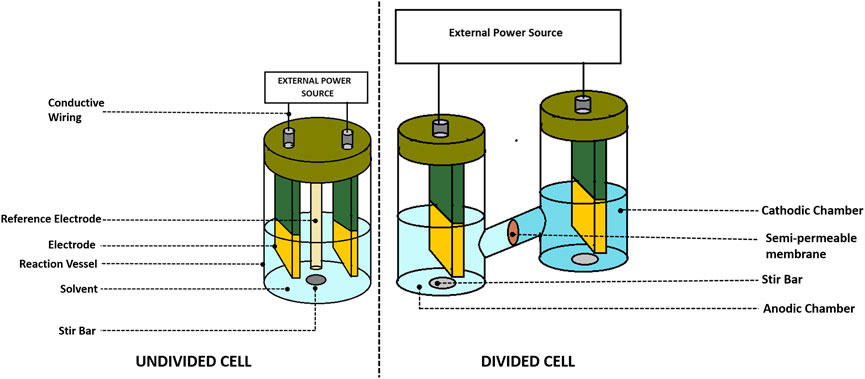
FIGURE 2. The undivided and divided cell configuration of electrochemical cell used in electroorganic synthesis.
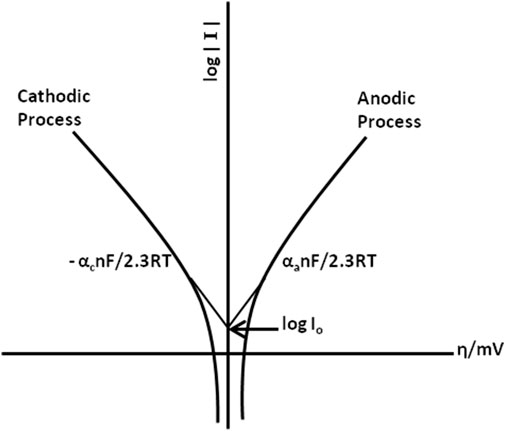
FIGURE 3. Schematic for a divided electrochemical cell.
2.4.2 Divided cell
The divided cells (Pollok and Waldvogel, 2020) are more elaborate than the undivided cells (see in Figures 2, 3). They are separated using a porous membrane, which allows conductivity. There is a necessity for a separator in a two-compartment cell to concur the further reaction of substrate A at the counter electrode due to byproduct electrolysis such as product B. It is also feasible that the productive electrolysis occurs at the counter electrode. The electric current could be used at the electrode (paired electrolysis) because there is no issue of separation between product A and product B.
2.4.2.1 Two-electrode electrochemical cell
The two-electrode electrochemical cell (Grimshaw, 2000; Frontana-Uribe et al., 2010) consists of a working electrode and a reference electrode wherein the potential of the reference electrode is monitored at a constant value and the potential of the working electrode is measured with respect to the reference electrode. The reference electrode can be considered an ideal non-polarized electrode as the current passes through the working and reference electrodes. The two-electrode electrochemical cells have been used in polarographic studies. In such studies, the dropping mercury electrode (DME) usually acts as the working electrode and a large mercury pool acts as the reference electrode. The area of the pool is substantially higher than that of the working electrode (DME). Thus, the pool can be regarded as unpolarized, making it a good reference for potential control. Solution resistance, Rs, and the iRs drop are important parameters that should be monitored. Non-aqueous mediums are usually more resistive as a system. In order to cope with it, ultramicroelectrodes can be used because they have a quite low current profile in the range of nA. This low current profile helps with solutions providing resistance in the scale of kΩ to MΩ.
2.4.2.2 Three-electrode electrochemical cell
The three-electrode electrochemical cells (Kingston et al., 2019) are majorly used in studies wherein the cell resistance is high. Although the potential of the working electrode is measured with respect to the reference electrode, the current over here passes through the working electrode and a counter electrode (auxiliary). The reference electrode, in this case, approaches ideal non-polarizability because no current passes through it. Hence, it is more reliable for monitoring the potential. During the experiments, the tip of the reference electrode is kept in the vicinity of the working electrode to minimize solution resistance. It is important to ensure that the tip of the reference electrode must not interfere with the mass transfer of electrolyte species. In most electrochemical experiments, three electrodes can be placed together in one compartment. However, under special conditions, it might be necessary to isolate the reference or auxiliary electrode from the solution containing the working electrode. We can choose a reference electrode based on the electrochemical reactions we perform (Inzelt et al., 2013). The incompatibility of the reference electrode with that of the system can lead to clogging of the reference electrode, precipitation of the charged species with poor solubility in the frit, and an increase in junction potential. Temperature and pressure are also important parameters while deciding on the reference electrode. The time of the experiment is also a limiting factor while deciding on the reference electrode because using a less robust reference electrode, in this case, can result in the stability of the reference electrode over a period of time. For the reference electrode to be flexible, a double junction or a salt bridge could be incorporated, which separates the reference electrode from the working electrode in case of fixed potential.
2.4.3 Quasi-divided cell
The quasi-divided cell merges the feature of the undivided and divided electrochemical cells. Usually, the quasi-divided cell (Senboku et al., 2022) constitutes electrodes with two different surface areas and galvanostatic electrolysis under a low current. The two different electrodes are subjected to different environments.
2.4.3.1 Small surface counter electrode
Owing to the small surface area of the electrode, the current density is high at the small surface counter wire electrode and flows in the opposite direction. The concentration for the starting material is low, as is the mass transfer to the electrode. Hence, the solvent gets electrolyzed due to a large amount of current.
2.4.3.2 Large surface working electrode
The low constant current at the working electrode causes low current density. As a result, the substrate with the lowest redox potential would get electrolyzed during the entire course of quasi-potentiostatic electrolysis. Usually, the undesired side reaction occurs only at the end.
2.4.4 Separator (for the divided cell)
The cell compartment for the divided cell must be separated using a conductive material. The porous membrane should allow the transport of ions. However, the transport of reactants and products must be restricted. The separator could be chosen from materials such as polymer membranes to ceramic frits.
2.5 Types of reactions in an electrochemical cell
Considering two completely stable and soluble species, A and B, in an electrolysis medium consisting of an excess of electrolyte (which is electroinactive), the simplest electrode interconversion that could occur at the inert surface is
For the above electrode reaction to occur, it is important to ensure the supply of reactants to the surface of the electrode. Additionally, it is essential to remove the product for the electron transfer reaction to occur. For example, the reduction of A to B would involve the following steps:
The cathodic current and the rate of reduction can be gauged by the rate of the above-mentioned overall sequence, which also must be dependent on the slowest step. However, the electrode reactions are rarely this simple. They involve electron transfer at multiple steps (Marcus, 1959; Marcus, 1964; Hammes-Schiffer and Soudackov, 2008). It could broadly be categorized into three types based on electrode processes.
2.5.1 Coupled chemical reactions
The chemical reactions, under favorable circumstances, can proceed via a single pathway generation of only one product. However, the organic reactions majorly follow competitive pathways leading to the formation of multiple products (Evans, 2008). For example, reducing p-iodonitrobenzene could lead to the generation of different products via different pathways.
2.5.1.1 Chemical reaction could follow two pathways
1) Homogenous: the reaction would occur as a result of electron transfer and species B is transported away from the surface. 2) Heterogenous: species R is adsorbed on the surface.
It is rarely possible that the electroactive species formed during a chemical reaction is not the major species in bulk solution or that the reaction at the electrodes disturbs the equilibrium in the homogenous solution. For example, acetic acid would dissociate before getting reduced via the electron transfer:
If the transport of acetic acid to the surface of the cathode is faster than the dissociation reaction, the current density will be limited due to the evolution of hydrogen.
2.5.2 Adsorption
Adsorption plays an important role in the field of electrochemistry. During electrocatalysis, it is important for the intermediates to get adsorbed on the surface because it leads to a lower energy pathway (Eqs 1–3). In the adsorption pathway, electron transfer occurs at the surface of the electrode. However, there might or might not be any interaction between the surface of the electrode and the reagent (A or B). It is feasible to slow down the electron transfer reaction, change the product, or modify the electrode reaction by adsorption of species that might not be directly involved in the electron transfer process.
2.5.3 Phase formation
There could also be a formation of a new phase through these electroreactions (Eq. 6), or one phase could get transformed into another. Eq. 6 includes steps such as diffusion, phase transformation, electron transfer, adsorption of intermediates, protonation, and hydration equilibria:
However, a phase formation would require a multi-step process involving nucleation and subsequent growth. It is important to consider that the growth of a new phase could lead to the incorporation of metal ion atoms at an appropriate position in the lattice or diffusion of metal atoms due to the reduction of metal ions in the solution.
2.6 Coulombic efficiency
For an electrochemical reaction, an ionically conductive reaction medium is required. The energy efficiency of an electroorganic synthesis could be determined using the electrolytic conductivity,
where Q is the amount of charge passed.
With the increase in the number of cycles, nC decreases.
2.7 Classical electrochemical techniques
This section discusses the classical electrochemical experiments to help the readers choose the techniques suitable for their experiments. The classical techniques have been chosen for their availability in commercial instruments. Electrochemical processes involve diffusion-controlled mass transport, facilitated by 1) applying controlled hydrodynamics that acts as a limiting factor to diffusion layer thickness; 2) using a large concentration of electrolyte to limit the migration of mobility of ions in the electric field (within the working and counter electrodes); 3) limiting the interference of natural convention by completing the experiment in the shortest time possible; 4) introducing a steady state diffusion controlled current by using microelectrodes before the interference of microelectrodes.
Electrochemical techniques are considered versatile, precise, and powerful owing to their large linear dynamic range and comparatively low-cost instruments. Most of the electrochemical techniques are based on the concept of measuring the resultant current due to the continuously changing applied potential at the electrode solution interface. Modern electrochemical techniques include stripping analysis which involves concentrating the analyte into or onto the surface of the electrode. There are different forms of stripping analysis (Kalvoda and Kopanica, 1989; Farghaly et al., 2014), such as anodic stripping voltammetry (ASV), cathodic stripping analysis (CSV), adsorptive stripping voltammetry (AdSV), and potentiometric stripping analysis (PSA). The modern variation of stripping analysis appeared first with fast linear sweep voltammetry, followed by the development of square wave and pulse polarography.
Another rapid voltage scan technique is cyclic voltammetry (Joseph, 2000), in which the direction of the voltage scan is reversed. The resulting current is recorded at the applied potential of the working electrode in forward and reverse directions. The parameters to consider in cyclic voltammetry are 1) anodic and cathodic peak current (Ipa and Ipc); 2), anodic and cathodic peak potential (Epa and Epc); and 3) half peak potential where cathodic and anodic currents reach their half value (Ep/2).
Pulsed voltammetry is a series of potential pulses with increasing amplitudes. The peak current is recorded at the end of each pulse. Different forms of pulsed voltammetry (Barker and Gardner, 1960) include normal pulse voltammetry (NPV) and differential pulse voltammetry (DPV).
Square wave voltammetry (SWV) (Barker et al., 1958; Borman, 1982) is another technique used, which was first introduced by Barker et al. The major advantage of SWV over DPV is its speed and higher current compared to analogous-differential pulse response. Several other electrochemical techniques include potentiometry (Joseph, 2000), which can provide information about the analyte using the potential between the two electrodes; Bulk electrolysis can be used for bulk synthesis of product, which requires large ration of surface area to solution volume.
3 Functionalization
3.1 C-N functionalization
Electrochemical reactions are important methods for C-H functionalization in a wide range of heterocyclic substrates. They have been proven to be very effective for substrates that are resistant to the conventional method of reaction initiation by peroxide radicals. It is important for us to study and understand the mechanistic approach of electrochemical C-H functionalization because it is of great interest in medicinal chemistry.
Electrochemical oxidation is an important method to functionalize the C-H bonds of aromatic and benzylic carbons involving single-electron transfer (SET). However, the limitation of the method lies in the fact that the aromatic and benzylic compounds in the presence of unprotected imidazole get electrochemically oxidized. There have been numerous reviews in this field. However, the ones by Zheng et al. (2020) and Kärkäs (2018) stand out. In this regard, work has been done by the Yoshida group wherein a new approach for C-N coupling of protected imidazoles was conducted based on the electrooxidative C-H functionalization of organic counterparts. This approach is applicable to aromatic and benzylic compounds. The group has demonstrated the electrooxidative C-N coupling of substrates such as N-methylimidazole and its aromatic and benzylic derivatives. The reaction proceeds with the formation of an electrochemically inactive imidazolium ion, which is transformed into N-benzylimidazoles or N-aryl with piperidine treatment. The reaction is highly chemoselective and straightforward and does not involve metal catalysts in the synthesis of N-substituted imidazoles (Morofuji et al., 2014).
The group also pioneered in developing several innovative methods for C-H amination, including metal-free benzylic amination via electrochemically generated benzylaminosulfonium ion (Hayashi et al., 2017). One of the methods introduces the intermediacy of electrooxidatively inactive cationic intermediates, which limits overoxidation. In order to address the problem of regioselectivity in some cases, an intermolecular approach was adopted. An organic transformation was designed using a pyridine ring instead of a pyrimidine ring, and 2-pyrimidyloxybenzene was synthesized using 2-bromopyrimidine and phenol. The oxidation of 2-pyrimidyloxybenzene at an anode generated a cyclized cationic intermediate. Furthermore, on treatment with piperidine, it gave 2-aminobenzoxazole, which is an important therapeutic molecule (Morofuji et al., 2015). One of the significant examples of C-H amination (Figure 4) was reported by the Mei group. They had worked on C-H amination of arenes with secondary amines electrochemically using Cu(OTf)2. n-Bu4NI was used as a redox mediator. The analysis of cyclic voltammogram, radical inhibition experiments, kinetic profiles, and isotopic effects are indicative of the fact that the reaction proceeds through an SET reaction, and it has been assumed that a Cu (III) species might be involved (Yang et al., 2018). The Ackermann group worked on electrochemical C-H amination of azole through cross-dehydrogenative N-H/C-H functionalization. The reaction could proceed easily without the involvement of a metal catalyst or any additional electrolyte. Instead, the acid additive was found to assist the electrochemical C-H amination (Sauermann et al., 2018). The group has also reported C-H amination via cobalt-catalyzed C-H Activation. During cyclic voltammetric studies, the oxidative potential of the cobalt catalyst (1.05 VSCE) in the presence of a base, KOAc, supported the idea of SET. In fact, a higher potential of 1.51 VSCE indicated the generation of Co (III) species during SET (Sauermann et al., 2018) using SCE as a reference electrode. The group also has similar interesting works in this regard (Tian et al., 2018).
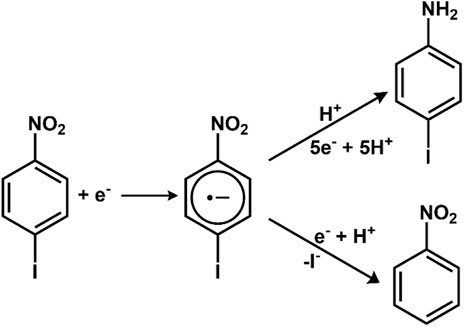
FIGURE 4. Different reaction pathways under coupled chemical process.
One of the noteworthy works on C-H amination was from the Aiwen Lei group, wherein they reported a simple method for the synthesis of N-aryl phenothiazines from phenothiazines and unprotected phenols. The reaction was performed in a CH3CN/CH3OH solvent in an undivided cell under a constant current of 7 mA for 100 min, and p-methoxyphenol reacted with phenothiazine in the presence of nBu4NBF4 as the electrolyte (Figure 3). The mechanistic studies have highlighted the fact that amine substrates get oxidized to radical cation and lead to the initiation of the reaction (Tang et al., 2018b). The group developed an approach for oxidative intramolecular C(sp3)-H amination of amides electrochemically in an undivided cell using platinum as a cathode and a carbon rod as an anode under constant current. While tetrabutylammonium acetate was incorporated as an electrolyte, it was also found to promote cleavage of the N-H bond and initiate hydrogen bond formation with amide. In fact, the intermolecular inert δ C(sp3)-H amination was found to occur through the 1,5-HAT process (Hu et al., 2018b). In 2019, the Lei group further reported intermolecular cross-coupling between aromatic substrates and sulphonimides (aromatic C-H imidation) induced by electrochemical oxidation (Hu et al., 2019). The reaction follows an N-radical addition pathway, and the cyclic voltammetry experiments showed that the N-centered imidyl radicals are produced as a result of proton-coupled electron transfer (PCET), which is mediated by anodic oxidation and nBu4NOAc (tetrabutylammonium acetate). The group also reported electrochemical dehydrogenative C(sp3)-H/N-H cross-coupling for developing a series of N-Mannich bases, wherein controlled experiments indicated that acetic acid played a major role in the formation of C(sp3)-N bond (Wang et al., 2019b).
Another work on oxidative C(sp3)-H/N-H cross-coupling of imidazopyridines with diarylamines was reported by the same group (Liu et al., 2019). There have been numerous reports on dehydrogenative C(sp3)-H/N-H cross-coupling (Wang and Stahl, 2019). The Lin group reported electrochemical diazidation of alkenes using an Mn-based catalyst (Fu et al., 2017). The group converted alkenes in the presence of sodium azide to 1,2-diazides. Further, the use of Mn-based catalysts and electrochemical energy under mild conditions of 1,2-diazides was reduced to vicinal diamines, which are important precursors for the natural product and molecular catalyst and as therapeutic agents. The formation of the second C-N bond is very challenging. Hence, to eliminate the problem, a redox active catalyst was introduced in the system through kinetic control to generate selectivity. Thus, the catalyst could very easily react with N3− to form a complex and ultimately form a metal adduct (M-N3), which would react with in situ generated carbon radical via oxidative addition or direct group transfer before reductive elimination to finish the cycle of diazidation.
The role of hydrogen atom transfer during electrochemical dehydrogenative C(sp3)-H amination was highlighted beautifully by the Magnus Rueping group (Nikolaienko et al., 2019). The electrooxidative amination of C(sp2)-H bonds was also reported by the group of Ian A. Nicholls, which involves cross-coupling of amines with aryl amides using Cu(OAc)2 as a catalyst due to the synergy between Cu catalyst and the electrocatalysis under mild conditions (Kathiravan et al., 2019). The Li-Zhu Wu group reported electrochemical ortho-amination of alkoxyl arene facilitated by trifluoroacetic acid. Trifluoroacetic acid, in this case, regulates the regioselectivity. The control experiments indicate that the arene radical cation intermediate is involved in the successful conversion. Increasing the current density during the experiments preserves the selectivity but decreases the yield (Wang et al., 2019a).
Another interesting work on the electrochemical oxidation of amino acids was reported by the Yahui Wang group. The group reported a decarboxylative C(sp3)-N coupling reaction that proceeded via anodic oxidative decarboxylation of carboxylic acid to form stabilized carbocations, which are trapped by amides or azoles to form C-N bonds. N-based nucleophiles need to be reactive and stable under electrolytic conditions to trap the stable iminium cations generated from the anodic oxidation of amino acids. Azoles, in this case, fulfill both criteria (Shao et al., 2019). Another attempt toward electrochemical oxidative C-N bond formation via Csp2-C sp3 bond cleavage was reported by the Zeng group and the Jiao group (Adeli et al., 2019).
3.2 C-C coupling
The Magnus Rueping group reported the first electrochemical approach for cross-electrophile coupling to generate 1,1-diarylalkane derivatives from alkyl and aryl halides using a nickel-based catalyst, (6,6′-dimethyl-2,2′-dipyridyl)NiBr2. The mechanistic studies reveal the generation of Ni0 species. Further, the Ni hydride species produced was found to facilitate the chain walking pathway to efficiently catalyze the electro-reductive process and hydroarylation (Kumar et al., 2020). The Antonchick group nicely summarized important works reported in the field of metal-free oxidative C-C bond formation through C-H bond functionalization (Narayan et al., 2015). The Waldvogel group had several methods for achieving homocoupling in phenolic substrates (Kirste et al., 2011a; Kirste et al., 2011b). In this regard, the group developed an active molybdenum-based electrode anode for performing dehydrogenative aryl coupling. This electrode in HFIP formed a high valent molybdenum species, which formed an active surface layer upon dissolving in the electrolyte (Beil et al., 2018).
The radical-cation-pool strategy could be used to synthesize asymmetrical biaryls from inactivated electron-rich compounds. Its origin could be traced back to the cation-pool method (Okajima et al., 2005; Nokami et al., 2008; Morofuji et al., 2012). The Yoshida group exploited the method to report C-C bond formation via C-H cross-coupling of two inactivated aromatic compounds. The process could be traced back to the generation of aromatic radical cations under an oxidative environment, followed by the coupling of the radical cation species with other aromatic substrates under a non-oxidative environment. The Waldvogel group reported metal-free electrochemical cross-coupling of phenols and arenes in fluorinated solvent using boron-doped diamond (BDE) anodes (Kirste et al., 2012). The group is also credited with electrochemical dehydrogenative cross-coupling to produce a series of 2,2′-diaminobiaryls (Schulz et al., 2017) and C-C cross-coupling of thiophenes with phenols (Wiebe et al., 2017). Cross-dehydrogenative coupling (Li, 2009) is an important strategy for the construction of C-C bond formation by activating two different C-H bonds. The classical method involves the use of a stoichiometric amount of oxidants to remove hydrogen and electrons, leading to the generation of intermediates that activate the C-H bond. However, the presence of a stoichiometric amount of oxidant would limit the application. In recent years, the focus has shifted toward a \ greener approach (Hu et al., 2018a).
3.3 C-H oxygenation
The Baran and Blackmond group reported an electrochemical initiation method, which generated increased yields during the C-H activation of complex pharmaceutical substrates (O’Brien et al., 2014). Usually, sulfinate radical sources are used to synthesize complex alkyl- and fluoroalkyl-substituted heterocycles. However, they display a low yield for a large number of substrates. The electrochemical C-H functionalization of carbamates using N-acyliminium ion is a very important method for synthesizing biologically important precursors, but the mechanism was not understood. One of the important works by the Baran group involves the activation of the inactivated C-H group through electrochemical oxidation via simple carbon and nickel electrodes and quinuclidine as a redox mediator. The electrochemical oxidation of sclareolide at C2 occurred at a lower potential owing to the presence of quinuclidine. Hence, the units, silyl ether, and ketones, among others, were well tolerated. The mechanism involves the formation of quinuclidine cation at the anode. It cleaves the inactivated C-H bond, followed by oxidation using molecular oxygen. The Baran group also reported phenomenal work on electrochemical allylic C-H oxidation (Horn et al., 2016). The allylic oxidation suffers from the use of toxic reagents, low yield, and expensive catalysts. The situation could be modified by introducing a co-oxidant, identifying a more effective electrochemical mediator, and designing an inexpensive reaction setup. Cl4NHPI was chosen as the electrochemical mediator instead of the conventional NHPI because it was found that attaching an electron-withdrawing group to the phthalimide moiety increased the activity of the catalyst. The mechanistic cycle of allylic oxidation was explained as follows: the pyridine deprotonated the Cl4NHPI followed by anodic oxidation, leading to the generation of tetra-chlorophthalimido N-oxyl radical species. The olefinic substrate would undergo hydrogen atom abstraction regenerating a stable allylic radical and Cl4NHPI. The tBuOO. radical would form allylic peroxide, which generates enone on the elimination of tBuOH.
Another work on electrochemical allylic oxidation was reported by the Waldvogel group (Edinger and Waldvogel, 2014), wherein they have further worked on the innovation introduced by Baran et al. The molecular oxygen as a co-oxidant was replaced by tert-butylhydroperoxide. The reactivity of the mediator was enhanced by substituting the tetrachloro-derivative. High-surface glassy carbon was used to lower the current density at the anode (Waldvogel and Selt, 2016). Another work by Baran involves C-H oxidation, wherein quinuclidine radical cation was generated through anodic oxidation, which led to the cleavage of the inactivated C-H bond and molecular oxygen producing the oxidation product. HFIP served as an electron acceptor leading to the generation of H2 during the cathodic process (Kawamata et al., 2017). The group has also worked significantly in the field of electrochemical oxidative dimerization (Rosen et al., 2014).
The Liu group reported electrochemical oxidation of the benzylic C-H bond. The reaction was performed at room temperature and in aqueous solvents. The anodic oxidation of tBuOOH resulted in the formation of tBuOO. radical, which leads to C-H abstraction at the benzylic position of the substrate, resulting in the formation of benzyl radical. This benzylic radical reacts with tBuOOH, generating corresponding oxygenated products. The KIE experiments suggested the
The Aiwen Lei group reported C-H oxygenation at ambient conditions at 23°C catalyzed by cobalt catalyst. The reaction proceeded via the oxidation of substrate by a cobalt (III) species generated by a mixture of catalyst Co(oAc)2 and NaOPiv in MeOH (Sauermann et al., 2017). An important work on C(sp2)-H acetoxylation of arenes via electrochemical oxidation was reported by the Zhang group (Li et al., 2017). The reaction does not employ any stoichiometric amount of oxidant and occurs at a mild temperature. The kinetic isotopic effect gave a mechanistic insight, which suggests Pd(II) atom coordinated with the N-atom of the oxime. It is followed by the rate-determining step of C-H activation and further undergoes N oxidation of Pd(II) to Pd(IV) and reductive elimination to give the desired product. The Little group worked on electrochemically in situ generated tris(p-bromophenyl) aminium radical cation initiated aromatic C-H bond functionalization. The solvents play a very important role in C-H Activation (Figure 5), protecting the product and generating highly active catalysts and, even sometimes, the air-regenerable oxidant (Hashiguchi et al., 2012).
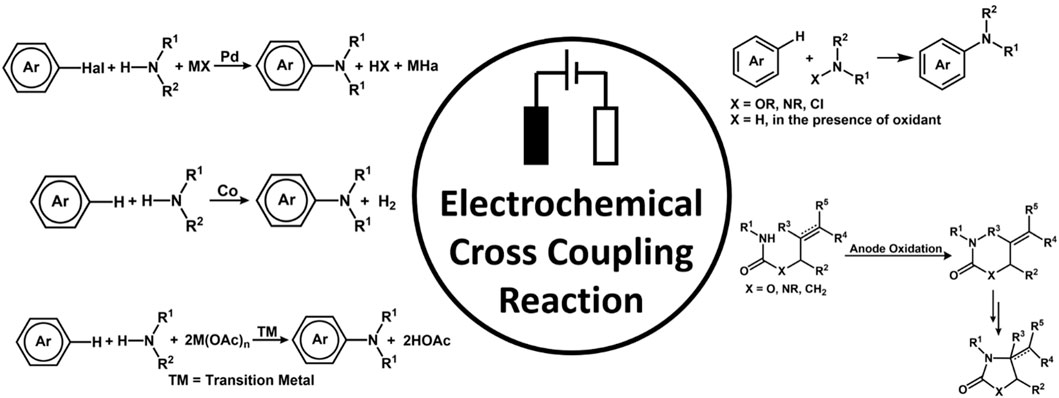
FIGURE 5. Strategies for different electrochemical cross-coupling, oxidative amination, C-N bond formation, and C-H imidation.
An effective electrochemical Friedel–Crafts alkylation protocol was developed for electron-rich aromatic substituents with N-vinyl amides (Li et al., 2015). The in situ-generated TBPA cation was used as an electron transfer reagent for initiating a cationic chain reaction. Polymeric ionic liquid-carbon black composite was used as a supporting electrolyte. Cyclic voltammetry and control experiments proved that cationic chain reaction proceeds through oxidative activation of the C-H bonds of aromatic compounds. The group also successfully achieved electrochemical aziridination of alkenes (Chen et al., 2015a). A number of structural variants of aziridines were synthesized using an undivided cell operating at a constant current and were mediated by n-Bu4NI in catalytic amounts. The reaction followed a radical pathway and occurred without the absence of additional conducting salt. The Xu group also reported electrochemically driven decarboxylative C-H functionalization of heteroarenes. The work dealt with C-H alkylation and carbamoylation of electron-deficient N-heterocycles with oxamic acid and carboxylic acid through H2 evolution (Lai et al., 2020).
An interesting piece of work was reported by the Ding group on direct and selective electrochemical conversion of benzyl C-H using a sole oxygen source and water as a co-solvent (Sun et al., 2020). The on-set potential of benzyl C-H bond activation could be reduced significantly due to the addition of water, which was enhanced further by oxygen vacancy-rich MnO2 as an electrocatalyst. The approach excluded the use of mediators. In recent studies, the focus has been more on finding green sources and overcoming anodic potential. The above section demonstrated how the electro-oxidation of the C-H bond gave rise to either carbon-centered cations or radicals, which upon the attack by oxygen-containing reagents, could offer myriad products, such as aldehydes, ketones, and esters with C-O bond formation. The electrochemical approach could be used to manipulate the oxidation state of the product by offering precise anodic potential, thus providing control over the selectivity of oxidation.
3.4 Sulphonation, selenation, and silylation
The Aiwen Lei group worked on direct arylsulfonylation of ynones with sulfinic acid through an electrooxidative pathway. The reaction was performed in an undivided cell using two platinum plates as a cathode and working electrode, TBAI as redox catalyst, CH3CN/DCE as a solvent, and LiClO4 as electrolytes while passing a constant current of 10 mA·cm−2. The reaction produced a variety of indenones in excellent yield. The group also developed a selective and highly efficient protocol for the synthesis of β-alkoxy and β-amino sulfides via electrochemical oxidative aminosulfenylation and oxysulfenylation of alkenes involving thiophenols/thiols as thiolating agents (Yuan et al., 2018).
Interesting work was reported by the Sun group on direct C(sp2)-H selenation of indoles without using an oxidant- and transition metal-free method. The control experiments led to the following observation: atmospheric air pressure led to a higher rate of selenation, protonic solvent interfered slightly with the yield, and the addition of iodine salt to the system enhanced the selenation process. Hence, KI was introduced into the system as a redox mediator and an electrolyte to increase the conductivity. The indoles and their substituents underwent C-H selenation electrocatalytically at the C3 position and generated selenyl indoles at a good yield (Zhang et al., 2018b).
An interesting piece by Huang reported electrochemically driven stereoselective synthesis of sulfur constituting β-enaminonitrile derivatives through oxidative C(sp3)-H functionalization of acetonitrile (He et al., 2019). The stereoselectivity was enhanced and used in the presence of a phosphine oxide catalyst. The activation of the C(sp3)-H bond produces cyanomethyl radicals in the presence of KI as a redox catalyst at a lower anodic potential leading to the formation of the C-S bond and enamines. Another example of C-S bond formation was reported by the Yi Pan group. The work was based on the Ullmann type of thiolation of aryl iodides catalyzed by NiCl2.glyme (Figure 6). The reaction was performed in an undivided cell in the presence of graphene/nickel foam electrodes because they help in enhancing the charge exchange. However, the reaction observed an increased yield due to the ratio of added electrolytes. The highest isolated yield was observed with the addition of three equivalent LiBr at Ecell of 3 V (Wang et al., 2019c).
FIGURE 6. The challenge of electrochemical C-H activation.
The Huo Cao group reported electrochemical diselenylation of indolizines using selenide, ketones, and pyridines in an external oxidant and transition metal catalyst-free condition in an undivided cell. The work dealt extensively with electrochemically driven intermolecular C-Se formation of α-bromoketones, 2-methylpyridines with diselenides producing diselenylated indolizines through direct C-H bond selenylation. A series of electrolytes were tested, such as KI, KBr, n-Bu4NBF4, n-Bu4NI, and LiClO4, and it was observed that halogen anions played a key role in the transformation. Subsequently, KI was chosen because it generated the highest yield (Li et al., 2020). An approach toward direct trifluoromethylsilylation and cyanosilylation of aldehydes via an electrochemically induced intramolecular pathway was reported by the Kedong Yuan group (Yang et al., 2020). The cathodic activation of trimethylsilyl reagents, such as SiMe3CF3 and SiMe3CN, allows the Si-O affinity to generate concerted anion intermediates, which further generates products with higher selectivity and yield via intramolecular -CF3 and -CN migration. The generation of concerted anion intermediate through direct cathodic activation of CF3-SiMe3 was energetically favorable (ΔG = −0.29 V) from DFT calculations.
3.5 Halogenation and trifluoromethylation
The Zeng group reported electrochemical Minisci-type trifluoromethylation of electron-deficient heterocycles wherein bromide ions are used as redox mediators (Dou et al., 2019). The efficiency of the process is maintained due to in situ formation of sulphonyl hypobromite intermediate, which regulates the concentration of the CF3 radical. The reaction proceeds in an undivided cell under galvanostatic conditions without the presence of supporting electrolytes. Another important work on electrochemical decarboxylative trifluoromethylation was reported by the Yubin Huang group (Hong et al., 2019). The group successfully performed decarboxylative trifluoromethylation of α,β-unsaturated carboxylic acid using Langlois reagent, which acts as a trifluoromethyl precursor. The reaction produces Cvinyl-CF3 compounds in good yield. The CF3 radical is generated at the anode by CF3SO2Na via SET while releasing SO2. The CF3 radical then attacks α,β-unsaturated carboxylic acid, which is oxidized to release oxygenated product and CO2. Over the years, several electrochemical approaches have been resorted to perform trifluoromethylation (Figure 7). Similarly, the Mo group devised an electrochemical strategy for trifluoromethylation using the Langlois reagent acting as a source of CF3. A simple Mn salt, MnBr2.4H2O, was used as a redox mediator, which is inclusive to a range of functionalized heterocycles, thus yielding a good amount of functionalized heterocycles bearing the CF3 moiety. The controlled experiments and mechanistic studies suggest the fact that there is a formation of CF3-bound high oxidation state Mn species, which assists in transition-metal-mediated CF3 transfer mechanism for C(sp2)-H functionalization. The electrochemical reaction between MnII and CF3SO2Na was studied using 19F NMR Spectroscopy. CF3SO2Na is known to display a singlet peak at −87.5 ppm, which disappeared on the application of the 1.1 F/mol current. Instead, a new peak appeared at −73.0 ppm, denoting that it was completely consumed on electrolysis generating a new CF3 species (Zhang et al., 2019).
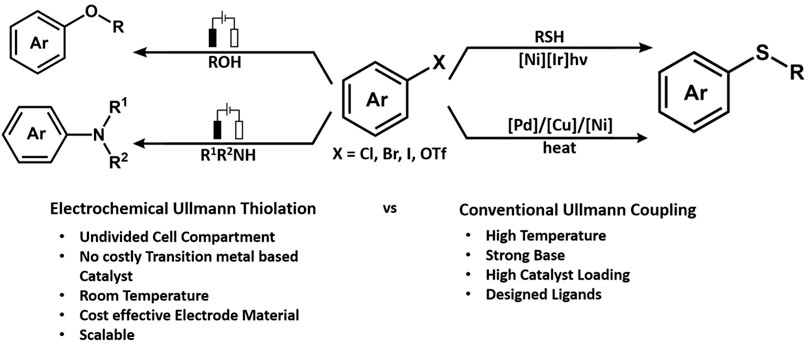
FIGURE 7. Schematic comparing electrochemical Ullmann thiolation with conventional Ullmann coupling.
An electrochemical approach toward intramolecular oxytrifluoromethylation of N-tethered alkenyl alcohol to generate CF3-containing morpholine derivative was reported by the Geraldine Masson group (Claraz et al., 2020). The approach includes direct anodic oxidation using the Langlois reagent as the trifluoromethylating reagent. The desired derivatives of morpholines were achieved by performing cyclization of alken-6-ol with the Langlois reagent using graphitic carbon anode and platinum cathode under constant current in the presence of LiClO4 as an electrolyte and a mixture of CH3CN and H2O as a solvent. The Lei group developed a protocol to perform oxytrifluoromethylation and aminotrifluoromethylation of alkenes under constant current electrolysis under the influence of the Lewis acid catalyst, Y(OTf)3. The method ensured the difunctionalization of the C-C bond (Zhang et al., 2018a). In this regard, the Ackermann group reported electrochemical di-/trifluoromethylation of N-substituted acrylamides under catalyst and oxidant-free conditions. The reaction condition displays high tolerance toward the functional group and provides judicious access to 3,4-dihydroquinolin-2(1H)-ones and fluoroalkylated oxindoles. It was observed that with the addition of BHT (7,2,4-di-tert-butyl-4-methylphenol), a radical scavenger to the medium, the trifluoromethylation got completely suppressed, which suggests that SET is involved in the mechanistic process.
Another work reported by the group involves di-/trifluoromethylation or cyclization of N-acrylamides under oxidant-free conditions. The noteworthy features of the work involve 1) effective fluoroalkylation without any involvement of metal catalyst or photoredox conditions; 2) catalyst-free, direct generation of radicals through electrolysis of sulfinate salts; 3) unprecedented mild conditions at 23°C; and 4) abundant scope for the formation of di- and trifluoromethylated oxindoles and 3,4-dihydroquinolin-2(1H)-ones (Ruan et al., 2019). The Yi Pan group reported electrochemical difunctionalization of olefin through the radical fluoroalkylation pathway. The method is advantageous because olefins can be modified easily via cascade dual anodic oxidations. The redox potential for the three fluoroalkylating agents—NaSO2CF2H, NaSO2CF3, and NaSO2C6F13—was 0.590, 0.814, and 0.742 V, respectively, using cyclic voltammetry. Thus, NaSO2CF2H can be easily oxidized at the anode. Thus, the order of reactivity of fluoroalkane sulphonates for oxidative radical fluoroalkylation was NaSO2CF3 > NaSO2C6F13 > NaSO2CF2H (Zou et al., 2019).
The Yulai Hu group reported electrochemical radical chlorination, bromination, and formyloxylation of various alkenes using NaCl, NaBr, and NaSO2CF3 as radical sources. (Sun et al., 2019). Electrochemical deconstructive chlorination of cycloalkanols catalyzed by MnCl2.4H2O was reported by the Morrill group. The reaction proceeded with the formation of alkoxy radical intermediates, leading to the formation of β- and γ-chlorinated ketones (Allen et al., 2019). The Shenlin Huang group was the first to develop an electrochemical approach toward oxydihalogenation of alkynes. The mechanistic scheme involved the generation of Cl− from CHCl3 through nucleophilic substitution of I−. Furthermore, the Cl− underwent oxidation at the anode to form chlorine radicals, which in addition to the alkyne, produced vinyl radicals. The vinyl radical on oxidation followed by nucleophilic addition of water generated enol moiety. Further enol on recombination with chlorine radical generated α,α-dihaloketone (Meng et al., 2020).
3.6 Annulation reaction
We will briefly discuss the annulation reaction wherein two bonds are generated in a single step. This reaction is an effective way to prepare cyclic compounds (Figure 8). The focus is particularly on dehydrogenative annulation reaction through X-H (heteroatom or X = C) functionalization. The Hai-Chao Xu group demonstrated, for the first time, the synthesis of saturated heterocycles using 1,4-diheteroatom via alkene annulations. The annulations of 1,1-diphenylethylene and ethylene glycol were performed in an undivided cell involving a platinum plate as cathode and RVC as anode wherein iPrCO2H was used as an acid additive and (2,4-Br2C6H3)3N (triarylamine) as redox catalyst (Ep/2 = 1.48 V vs. SCE). The 1,4-dioxane product displayed the highest yield of 91% (Cai and Xu, 2018).
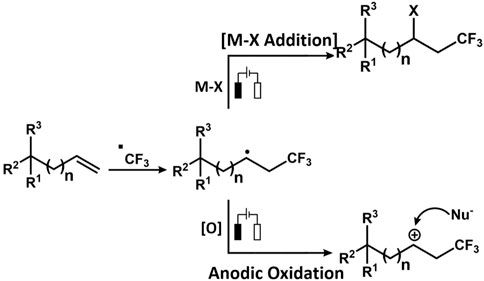
FIGURE 8. Electrochemical approaches for the generation of Trifluoromethyl radical.
The Aiwen Lei group also contributed significantly to the field of annulation reaction listed in Table 1. In 2018, the group reported electrochemical dehydrogenative C-H/N-H [4 + 2] annulations of aryl vinyl amide with alkene/alkyne using Co(acac)2 as a catalyst. The reaction exhibited high functional group tolerance. The controlled experiments for mechanistic studies determined that the key step for the process might be the electrochemical oxidation of the Co(II) complex at the anode (Tang et al., 2018a). The Ackermann group reported for the first time an electrochemical alkyne annulation reaction via C-H/N-H activation catalyzed by a cobalt-based catalyst, Co(OAc)2. The cyclic voltammetry experiments for C-H activation revealed that the oxidation potential of the catalyst (1.19 VSCE) was lower by 320 mV than that of the substrates (1.51 VSCE) using SCE as the reference electrode, indicating that the catalyst promotes SET. The group also reported for the first time the metalla-electrocatalyzed C-H activation through Rhoda-catalyzed alkyne annulations. A modular electro-flow cell was incorporated with porous graphite felt, which ensured efficient turnover number, leading to successful C-H/N-H activation with excellent regioselectivity and chemoselectivity. The catalyst Rh(III) carboxylate was regenerated via anodic oxidation, and H2 was the sole byproduct of cathodic reduction (Kong et al., 2019). The group also reported the electrochemical synthesis of aza-polycyclic aromatic hydrocarbon through rhodium-catalyzed cascade C-H activation and alkyne annulations (Kong et al., 2020). A multifunctional O-methylamidoxime was designed to sustain high regio- and chemoselectivity. The reaction was performed using [
3.7 Synthesis of cyclic carbamate
The Stahl group reported an electrochemical method for α-oxygenation of cyclic carbamates using water as a nucleophile, and bicyclic aminoxyl was used as a mediator. In the aminoxyl-mediated electrochemical process, the substrate oxygenation is observed to proceed at a potential lower by 1 V compared to the redox potential of the carbamate substrate, measured using an Ag/AgNO3 reference electrode, because we observe functional group compatibility, which is inaccessible in the case of conventional Shono-type reaction, which proceeds via direct electrochemical substrate oxidation. This was, in fact, one of the first examples of α-functionalization of non-activated cyclic carbamates using oxoammonium oxidants (Wang et al., 2018). The Urieta group performed electrochemical studies on the α-alkoxylation of carbamates, which led to the conclusion that it followed an ECEC mechanism, wherein the first two steps occur in a concerted manner. This was further confirmed by theoretical studies on absolute redox potential for stepwise and concerted pathways. The reaction shows anomalous regioselectivity, which was caused by the dipole moment vector of the asymmetric carbamates, which, in interaction with the positive charge of the anode, orients the molecule in a way that it favors the cleavage of the C-H bond for the less substituted site.
3.8 C-H functionalization of ethers and esters
A very important piece of work was reported by the Tristan H. Lambert group that reported C-H functionalization of ethers catalyzed by trisaminocyclopropenium (TAC) at a mild electrochemical potential under visible light irradiation (Huang et al., 2020). The reaction occurs under oxidant-free conditions in which ethers undergo coupling reaction with substrates such as purines, pyrazoles, alkynes, and alkenes with high regioselectivity toward less hindered α-position. The reaction proceeds via hydrogen atom transfer reaction from the substrates to the photoexcited TAC radical dication. The studies show a new direction toward combining photocatalysis with electrocatalysis. The Waldvogel group reported work on the electrochemically driven synthesis of 1,1,1,3,3,3- hexafluoroisopropanol aryl ether in the presence of BDE electrodes. A base such as triethylamine was used because it formed a highly conductive media with HFIP, resulting in the superfluous behavior of the additional electrolyte (Waldvogel et al., 2020) see in Table 1. The mechanistic study suggests that the reaction might proceed via ECEC-type mechanistic.
3.9 Alcohol oxidation
Recently, the focus has shifted to alcohol oxidations in fuel cells. For low-temperature fuel cells, methanol could be an alternative to hydrogen because methanol could easily be oxidized to CO2 without having to break the C-C bond. Organic aminoxyls, in the presence of chemical oxidants such as Br2, NaOCl, and PhI(OAc)2, act as widely accepted mediators for the transformation in both acidic and basic conditions (Tojo and Fernández, 2010b). Electrochemical alcohol oxidation mediated by aminoxyl was documented extensively in different comprehensive reviews (Tojo and Fernández, 2010a; Nutting et al., 2018). The Stahl group reported electrochemical alcohol oxidation using aminoxyl mediators, including TEMPO, its derivatives, and several bicyclic aminoxyl species, such as ABNO. 4-PhCO2-TEMPO and 4-AcNH-TEMPO (ACT) were observed to have the highest E1/2 leading to the conclusion that the driving force for the transformation is more powerful than the steric effect. In fact, this behavior contradicts the observation for the reaction catalyzed by organic aminoxyls in the presence of chemical oxidants such as Br2, NaOCl, and PhI(OAc)2, wherein sterically less hindered bicyclic aminoxyls prove to be an effective catalyst (Rafiee et al., 2015; Cardiel et al., 2019). Over the years, ACT has proven to be an effective electrocatalytic mediator for alcohol oxidation with broader substrate scope. ACT was reported to be effective at selective alcohol oxidation in lignin (Cardiel et al., 2019). However, their efficiency is limited by the diffusion of the catalyst to the surface of the electrode. The issue could be addressed by immobilizing the catalyst to the surface of the electrode.
Pyrene-TEMPO conjugate catalysts adsorbed on MWCNTs were far more superlative compared to ACT (Das and Stahl, 2017). The oxidation reaction of 4-methoxybenzyl alcohol had a TOF of approximately 4,000 h−1 and TON >1,800 (Rafiee et al., 2018). Cu-based aminoxyl systems were highly effective for the aerobic oxidation of alcohol with high functional group tolerance (Steves and Stahl, 2013; Ryland and Stahl, 2014). While aminoxyl-driven alcohol oxidation employing chemical oxidants was two-electron processes, the mechanistic studies have validated that the aminoxyl species and CuII serve as a one-electron oxidant (Hoover et al., 2013; Ryland et al., 2014; McCann and Stahl, 2015). The electrochemical alcohol oxidation using the Cu/TEMPO system operated at 0.5 V lower potential than the reaction based on only TEMPO (Badalyan and Stahl, 2016; Ryan et al., 2019).
In a report by the Whitesides group, Fe2+/Fe3+ redox couple was used as a mediator for methane oxidation with Pt black catalyst (Bergens et al., 1994). The Fe2+/Fe3+ as a redox couple mediator was also extended to ethylene glycol, peat, kraft lignin, and sub-bituminous coal as fuel without additional catalysts (Gorman et al., 1996; Weibel et al., 2005). These mediators could function using unconventional fuels. Polyoxometalates have been widely used as catalysts in the oxidation of alcohols, hydrogen, and biomass in fuel cells. In a report by the Deng group, H3PMo12O40 and polyol were irradiated with a metal halide lamp for 17.5 h, after which the solution was passed into an anodic compartment that employed H12P3Mo18V7O85 as the cathodic POM mediator, leading to enhanced power densities (Wu et al., 2016b). Sulphonated anthraquinone was also tested as a mediator on different kinds of fuel, such as alcohol, carbohydrates, and lignin (Hertl and Weetall, 1985b; Weetal et al., 1985). Thus, mediated electrochemistry provides enough opportunity for power generation in complex fuels. Low-potential polyoxometalates proved to be good anodic mediators with greater redox reversibility, stability, and low cost. The mediated cells, which use biomass, alcohol, and other unconventional fuels, produce lower power output than conventional fuel cells. However, the ability to replace the Pt and non-Pt group metal by deriving power through unconventional sources would further lead to new developments.
4 Electrochemical CO2 reduction
A selective and efficient method for electrochemical carbon dioxide reduction (Lu et al., 2014b) is one of the greatest challenges in the field of artificial photosynthesis to produce cleaner fuels. In 2014, the Salehi-Khojin group (Shen et al., 2016) reported molybdenum disulfide as a superior catalyst in an ionic medium for carbon dioxide reduction compared to noble metal owing to high current density and low overpotential of 54 mV, as measured against Ag wire as a reference electrode (Asadi et al., 2014). The superior activity of molybdenum disulfide is due to its molybdenum terminated edges, which are responsible for its high d-electron density and metallic character. It has been observed that vertically aligned MoS2 displays a high CO2 reduction current density of around 130 mA·cm−2 for both low- and high-applied potential owing to the presence of a high density of active sites, especially Mo atoms. The group also reported 2D nanoflakes of metal dichalcogenides for catalyzing CO2 reduction in an EMIM-BF4 medium. The nanoflakes exhibited a current density of 18.95 mA·cm−2 and a faradaic efficiency of 24%. The CO formation reported a turnover frequency of 0.28 with an overpotential as low as 54 mV. The Norskov group (Chen et al., 2016b; Shi et al., 2016) also reported molybdenum sulfides and selenides as probable electrocatalysts for CO2 reduction (Chan et al., 2014). There was also an investigation on the electrochemical reduction of CO2 to CH4 via eight electron transfer mechanisms using tungsten carbides (WC) and transition metal decorated WC (Wannakao et al., 2015). Silver has always been a traditional electrocatalyst in different kinds of reactions (Ma et al., 2014; Ma et al., 2016b; Singh et al., 2016). The Jiao group nanoporous silver catalyst that reduces CO2 to CO with 92% selectivity 3,000 times increased the rate of current compared to its polycrystalline counterpart at a moderate overpotential, which is <0.50 V (Lu et al., 2014a). Such high activity is owing to the greater stability of
The Hyungjun Kim group reported the covalency-aided electrochemical reaction (CAER) wherein the p-block dopants affect reaction energy by imposing partial covalency in the metal catalyst that enhances the catalytic activity beyond the modulations arising from d-block dopants (Lim et al., 2014). The Yang group explored the activity of gold-copper bimetallic nanoparticles, which are majorly governed by two factors, namely, the geometric and electronic effects, which help determine their catalytic activities (Kim et al., 2014). The Thomas J. Meyer group (Zhang et al., 2014b) synthesized tin nanocrystals through the hydrothermal method in which selective reduction of CO2 to formate occurs at an overpotential as low as 340 mV (Zhang et al., 2014a). The maximum faradaic efficiency for formate formation was calculated as 93%, under a current density of >10%, and high stability on graphene support. The increased reactivity toward carbon dioxide reduction is due to two factors: 1) the strength of interaction between the tin surface and
There have also been reports on the electrochemical methanation of carbon dioxide using a dispersible copper nanoscale catalyst. The Alivisatos group demonstrated how copper nanoparticles supported on glassy carbon (n-Cu/C) were almost four times more effective than high-purity copper foil electrodes (Manthiram et al., 2014). The Cu (core)/Cuo (shell) catalyst was reported to be effective for the electrochemical reduction of carbon dioxide to formic acid (Lan et al., 2014). With a catalyst loading of 1.0 mg·cm−2, the faradaic efficiency of CO and HCOOH was greater than other catalyst loading. One of the significant works on CO2 reduction to C3-C4 products, such as n-propanol and n-butane, was reported by the Jaeyoung Lee group. They had reported a Cl-induced biphasic electrode consisting of (Cu2O) and metallic copper (Cu). The synergistic effect between (Cu2O) and metallic copper (Cu) led to an abundance of Cu+1 and stabilization of the reaction intermediate (Lee et al., 2015). Copper nanoforms with hierarchical porosity have also been used for electrochemical CO2 reduction (Sen et al., 2014). The Collins group reported Cu nanoparticles supported on single-walled carbon nanotube (SWCNT), Ketjen black (KB), and carbon black (CB) for effective reduction of CO2 to hydrocarbons such as (CH4, C2H2, and C2H4) (Baturina et al., 2014). The size of Cu nanoparticles ranged from 10 to 30 nm. The supported nanoparticles were less susceptible to aerobic oxidation and agglomeration during the reductive treatment in H2. The Feng Jiao group (Rosen et al., 2015b) developed highly dense Cu nanowires (Ma et al., 2016a) for electrochemical CO2 reduction. It required a minimal overpotential of 0.3 V to reach a current density of 1 mA/cm2. Ag/AgCl was used as a reference electrode. It displayed an FE of ∼60% toward CO production. Another notable work reports Cu nanocubes as electrocatalysts with the greatest selectivity toward ethylene as a product for CO2 reduction, wherein the nanocube structure favors the formation of multicarbon products. It was observed that the local pH was much higher than the bulk pH during the reaction, which enhanced the selectivity of the product. The higher roughness of the nanostructured catalyst draws a higher current of −0.5 V (Roberts et al., 2015). There were numerous reports on CO2 reduction using copper-based electrocatalysts (Zhang et al., 2014c; Cheng et al., 2016) such as copper meso-/nanocrystals (Chen et al., 2015a; Wang et al., 2016), copper oxide (Kas et al., 2014; Ren et al., 2015; Mistry et al., 2016), copper electrodes (Ba et al., 2014; Xiao et al., 2016), and copper nanoparticles (Raciti et al., 2015; Kwon et al., 2016; Loiudice et al., 2016; Song et al., 2016). The Guido Mul group reported that the higher selectivity of ethylene for Cu nanoparticles was due to 1) a high local current density, 2) a low electrolyte concentration (i.e., buffer capacity), and 3) a high CO2 pressure. The sizes of nanoparticles affect their catalytic activity (Kas et al., 2015; Kas et al., 2016). The Bao group reported an electrochemical reduction of CO2 using different sizes of palladium nanoparticles in the range of 2.4–10.3 nm. The DFT studies have concluded that the edges and the corner sites of Pd nanoparticles were more effective in the reduction of CO2 than the terrace site. However, the competitive HER rate was similar for all three sites (Gao et al., 2015).
Numerous reports were made on electrochemical CO2 reduction using nanoparticles (Ding et al., 2016; Kim et al., 2016). Zn dendrites were used as electrocatalysts for CO conversion from CO2 by the Jiao group (Rosen et al., 2015a). Their catalytic activity in the aqueous bicarbonate electrolyte was threefold higher than the bulk Zn counterparts. The crystal structure of Zn was manipulated over the years to control product selectivity (Won et al., 2016). Sn dendrite electrodes were also reported as effective for the reduction of CO2 to formate (Won et al., 2015). The activity of the heat-treated electrode was superior and produced formate (228.6 μmol·h−1 cm−2 at −1.36 V). The reduced electrode SnOx/Sn with abundant O content could withstand highly reductive conditions and stabilize the
The Bouwan group reported electrocatalytic CO2 conversion to oxalate using a tetranuclear copper (II) complex (Angamuthu et al., 2010). Several works also reported effective CO evolution activity using bismuth-based catalysts (Ding et al., 2016; Sun et al., 2016; Zhang et al., 2016). The Wang group reported a molecular Cu–porphyrin complex (copper(II)-5,10,15,20-tetrakis-(2,6-dihydroxyphenyl)porphyrin) as an electrocatalyst for CO2 reduction to hydrocarbons (Weng et al., 2016). At the potential of −0.976 V vs. RHE, the catalyst produced methane and ethylene at the partial current densities of 13.2 and 8.4 mA·cm−2. Another notable work in this regard was reported by the Dyer group, in which 6,7-dimethyl-4-hydroxy-2-mercaptopteridine were used to catalyze the reduction of CO2 on a glassy carbon electrode (Xiang et al., 2014). The bulk electrolysis of the saturated CO2 solution using RVC as a working electrode produced methanol in the presence of PTE. A transient intermediate, PTE carbamate was characterized by FTIR, followed by 2e− reduction of CO2 to HCOOH, HCHO, and CH3OH. The Clifford P. Kubiak group (Machan et al., 2014b) reported electrocatalytic CO2 reduction using Re(I) bipyridine complex in which the methyl acetamidomethyl groups were added to the 4,4′-position of a 2,2′-bipyridyl ligand (Machan et al., 2014a). The complex catalyzed the reductive disproportionation of CO2 to CO at a lower potential, as low as 250 mV. The TOF and faradaic efficiency was low for the process. However, the reported work was a rare example of the use of hydrogen bonding and several other driving forces for molecular assembly of abiotic catalysis. [CoIIIN4H(Br)2]+ (N4H = 2,12-dimethyl-3,7,11,17-tetraazabicyclo-[11.3.1]-heptadeca-1(7),2,11, 13,15-pentaene) was reported as a catalyst in acetonitrile under glassy carbon working electrode for electrocatalytic CO2 reduction. The CO was generated as a product with a faradaic efficiency of 45%
The Bocarsly group (Detweiler et al., 2014) suggested MnBr(6-(2-hydroxyphenol)-2,2′-bipyridine) (CO)3 as an effective catalyst for the reduction of CO2 to CO, which bore a ligand structure with a phenolic proton in close proximity to the CO2 site, which allowed proton assisted C-O bond cleavage. The reduction occurred at an overpotential as low as 440 mV (Agarwal et al., 2015). Molecular manganese complexes, both immobilized and non-immobilized, have been a popular choice over the years. Ma et al. (2016a) presented a popular cAn interesting work that explored the electrochemical activity of pyridinium ion (pyrH+) and pyridine (py) on gold electrodes in both nitrogen and CO2 environment. The electrochemistry of pyridinium reduction depended on pH. The addition of CO2 saw an increased current density at the same potential displayed for pyridinium reduction (Lucio and Shaw, 2015). The Haywood group reported pyridine-catalyzed CO2 reduction to methanol. The bulk electrolysis in the galvanostatic and potentiostatic regimes occurred under pressure of 55-bar CO2, resulting in the formation of methanol with an FE of 10% as the charge of 5–10 C·cm−2 was passed (Haywood et al., 2016). The group 6 complexes of [M(CO)4 (bpy)] were also employed as efficient electrocatalysts by the Frantisek Hartl group (Tory et al., 2015). They could be used as electrocatalysts for the reduction of CO2 because the reduction potential of CO2 was less negative than that of radical anion [M(CO)4 (bpy)].−. The cyclic voltammetry studies revealed that five-coordinate [M(CO)3 (bpy)]2− was the active catalyst in the presence of Au as a working electrode. Similarly, there were also reports on tetracarbonyl complexes of low valent group 6 transition metals with diimine bidentate ligands, M(CO)4 (diamine), working as an efficient homogenous catalyst for electrochemical reduction of CO2 in non-aqueous media. The Walensky group reported a similar (α-diimine)M(CO)3Br (M = Mn, Re) complex for the electrochemical conversion of CO2 to CO (Vollmer et al., 2015). Notable work was reported by the Carter group on the influence of weak Bronsted acid (H2O, phenol, MeOH, and TFE) on homogenous electrocatalysts, such as [Re (bpy) (CO)3]− and [Mn (bpy) (CO)3] −. The TOF varied for different Brønsted acids. Moreover, for the same Brønsted acids, the TOF was higher for Re catalysts than for Mn catalysts. The order of Brønsted acid based on the TOF was TFE > MeOH > H2O (Riplinger and Carter, 2015). The Surendranath group (Hall et al., 2015) reported the synthesis of active graphitic surface for catalysis through condensation of fac-Re(5,6-diamino-1,10-phenanthroline)(CO)3Cl complexes with surface o-quinone moieties as an electrocatalyst for CO2 reduction to CO. The catalyst surface (GCC-Re surfaces) consisted of a uniform arrangement of Re centers wherein local coordination sites act as local coordination site (Oh et al., 2016).
Interestingly, the Grubbs and Gray group reported brush polymer ion gels comprising 1-butyl-3-methyl-imidazolium, PS-PEO-PS triblock brush polymer, bis-(trifluoromethylsulfonyl) imide (BMIM-TFSI), Re(bpy)(CO)3Cl, ferrocene (Fc), and cobaltocenium (Co
Another popular class of catalysts for electrochemical CO2 reduction was carbine-based complexes. The Wolf and Patrick group reported a range of lutidine- and pyridine-based bis-N-heterocyclic carbene (NHC) palladium pincer (Osadchuk et al., 2016; Stanton et al., 2016) complexes (Therrien et al., 2014; Therrien et al., 2015). The lutidine-based complex reduced CO2 to CO at a potential of −1.6 V vs. Ag/AgNO3 in the presence of 2,2,2-trifluoroacetic acid (TFA) as the proton source. The DFT studies have beautifully predicted the requirement of the proton source in the electrochemical system with respect to the degree of activation of CO2 and threw some light on the charge transfer dynamics of the electrocatalyst with respect to CO2.
The Hupp, Farha, and Kubiak group reported Fe_MOF-525, a metal-organic framework for the electrochemical reduction of CO2 to CO (Hod et al., 2015). The MOF was reportedly stable during the CV experiments, and the CO2-saturated solution exhibited a catalytic wave at and before the potential of the catalytically active Fe(I/0) couple. The controlled potential electrolysis (CPE) at an overpotential of ∼650 mV yielded a current density of a few to several milliamperes. Ag/AgCl/KCl (saturated) was used as a reference electrode. A highly selective electrochemical reduction of CO2 to CO was reported at a pH of 4.3 with an overpotential of 480 mV using a pyrene appended iron triphenyl porphyrin ring carrying six pendant OH groups on the phenyl ring in all ortho and ortho’ positions immobilized on carbon nanotubes (Lu et al., 2016) through non-covalent interactions (Maurin and Robert, 2016). Thus, it could be deduced that Fe-porphyrin was a quite popular catalyst for electrochemical CO2 reduction (Choi et al., 2016a; Wu et al., 2016a). Co-based MOF, Al2(OH)2TCPP-Co, was reported as an electrocatalyst for CO2 reduction to CO by the Yaghi group. The MOF-integrated catalytic system (Kang et al., 2016) generated greater turnover with stability for a greater duration. The spectrochemical studies showed the reduction of Co (II) to Co (I) during catalysis (Kornienko et al., 2015). The Jean-Michel Savéant group developed an excellent catalyst for the electrochemical conversion of CO2 to CO using electrogenerated Fe0 porphyrin synthesized by substituting two opposite phenyl rings in tetraphenylporphyrin with ortho, ortho’- phenol groups, whereas the other two were perfluorinated (Costentin et al., 2014). The Ott group developed a protocol for CO formation using a methyl group in the ruthenium–bipyridine catalyst [Ru(tBu3tpy)(bpy)(NCCH3)]2+ to enable CO2 existing at a one-electron reduced state to enter an inaccessible cycle (Johnson et al., 2016). Other works investigated the energy of electrochemical CO2 reduction on metal-porphyrin/bipyridine motifs (Cheng et al., 2015; Ding et al., 2016; Larrazábal et al., 2016; Shen et al., 2016).
One of the notable works on electrochemical CO2 reduction was conducted by the Yasuaki Einaga group, in which they reduced CO2 in seawater using boron doped-diamond electrodes (BDD) (Nakata et al., 2014). The method was advantageous because it overcame the problem of low yield for higher-order products and reduced the generation of H2. The BDD electrons were used owing to their high faradaic efficiency for the production of formaldehyde (around 74%). The high faradaic efficiency is due to the sp3-bonded carbon of BDD. There were also reports on CO2 reduction in EMIM–BF4 ionic liquid medium at BDD modified by Cu nanoparticles (Roy et al., 2016). The size of Cu nanoparticles deposited at the BDD electrodes was 30 nm. The Hongtao Yu group reported a non-metallic catalyst and an N-doped nanodiamond/Si rod array (NDD/Si RA) for CO2 reduction. The catalyst led to the formation of formate from CO2 at an onset potential of −0.36 V (vs. RHE). The FE was approximately 91% at −0.8 V to −1.0 V (Liu et al., 2015). The Purkait group reported electrochemical carbon dioxide reduction coupled with the removal of dye (oxidation) using a Co3O4 electrode (Gao et al., 2016) (anode) and Sn and Zn electrocatalysts (cathode) (Yadav and Purkait, 2015). The reaction was performed in the presence of NaHCO3 and KHCO3 as electrolytes. The cyclic voltammetry data were collected over a range of applied potentials (2–3.8 V). This applied potential helped not only in the formation of HCOOH but also in the removal of the dye.
Graphenes have immense potential as electrocatalysts for electrochemical CO2 reduction (Wu et al., 2016a). The Hashimoto and Kamiya group reported Ni-N-modified graphene in which the Ni-N structure exhibited high catalytic activity toward CO2 reduction to CO (Su et al., 2016). In fact, the competitive HER rate for Ni-N-Gr was less compared to the nickel electrode. The EXAFS and XPS revealed that the introduction of Ni atoms coordinated to N into the sp2 network of graphene needed heat treatment and Ni-N was the active center. A high selectivity toward ethanol during CO2 reduction was obtained for Cu nanoparticle/N-doped graphene with an FE of 63% and selectivity of 84%. Bimetallic nanoparticles (Sarfraz et al., 2016), Pd-Cu, were dispersed on the graphene surface to catalyze the reduction of CO2 (Liu et al., 2016). The samples with 1 wt% Pd–2wt% Cu/graphene displayed the lowest overpotential and higher current density. Clearly, graphene or catalyst-supported graphenes were a popular choice for electrochemical carbon dioxide reduction (Poh et al., 2014). The Peter Strasser group metal-doped the N-carbon material as an efficient catalyst for the conversion of CO2 to CO. The metal-doped N-carbon material was observed to exceed the mass activity of the carbon-supported Au catalyst by 80% at −0.5 VRHE (Varela et al., 2015). Along similar lines, the Xiao Dong Zhou group (Wu et al., 2014) reported an article on understanding the defect, defect density, and selectivity for N-doped carbon nanotubes in the field of electrochemical CO2 reduction (Sharma et al., 2015). In fact, the presence of pyridinic- and graphitic-nitrogen led to a significant decrease in overpotential (−0.18 V) and an increase in selectivity toward CO. The Lou and Ajayan group synthesized heavily nitrogen-doped carbon nanotubes using the CVD method that ensured total N-content to be 5 atom%. The maximum FE for NCNT was calculated as 80% at an overpotential of −0.26 V (Wu et al., 2015). Solvents and ionic liquids also modulated the course of electrochemical CO2 reduction (Grills et al., 2014; Oh et al., 2014; Sun et al., 2014; Gurkan et al., 2015; Chen et al., 2016a).
Recently, scientists discovered that to solve the CO2 emission problem and attain sustainability, one must convert it back to fuels. Now, this process requires energy and a good catalyst. The search for good catalysts resulted in metallic copper being a suitable system that can accomplish this job. However, there are certain limitations: they lack selectivity, H2 evolution is a trivial competitor, and sometimes they are operational at a higher overpotential (Hahn et al., 2017). Moreover, modeling the 2D/3D Cu/Cu2O surfaces is computationally costly, causing serious scientific concerns regarding the development of the catalysts (Garza et al., 2018). Here, the family of molecular metal complexes came into play with a facile synthetic methodology, control over the catalyst design, spectroscopic signatures, and ease of computational modeling (Hiroto et al., 2017). Thus, we focused our research attention on a well-known class of catalysts: corroles.
Molecular catalysts have been in development for a long time, but they have suffered from two major drawbacks: 1) C-C step-up chemistry fails (Cope et al., 2017) and 2) difficult catalyst recyclability and product recovery (Costentin et al., 2015; Rao et al., 2017). Thus, we focus on nanoscale heterogenized molecular systems such as cobalt phthalocyanine immobilized over multi-walled carbon nanotubes where remarkable activity for CO2 reduction to CO was observed at an overpotential of 0.52 V with a FECO >95%, and the nanoscale heterogenization effect resulted in obtaining a high current density of 15 mAcm−2 in the neutral aqueous medium. All the potentials were measured against Ag/AgCl reference electrode (Zhang et al., 2017). Similarly, the M. T. M. Koper group studied the immobilization of Co protoporphyrin on a pyrolytic graphite electrode. With CO2 electroreduction, they not only achieved volumes of CO but also minor products such as 6e− reduced methanol and 8e− reduced methane at a relatively low overpotential of 0.5 V. The potential was measured using RHE as the reference electrode. Their work also provided insights into controlling the selectivity of CO2 reduction by suppressing H2 evolution and the pH effect in controlling CO formation over H2 evolution, indicating the formation of Brønsted base type intermediate (carboxyhydroxyl intermediate) (Shen et al., 2015). The Hailiang Wang group explored Cu(II) porphyrin immobilized over the carbon paper electrode, which showed increased CO2 reducibility to hydrocarbons, such as methane and ethylene at −0.976 V vs. RHE (Weng et al., 2016). Corroles with structural similarity to porphyrins were a suitable candidate for electrochemical CO2 reduction, which was first explored by the Zeev Gross and E. Fujita group. Co and Fe corroles were investigated under homogeneous conditions, resulting in CO formation. Their studies revealed that the Co(I) and Fe(I) states of the catalysts formed under electrochemical bias are responsible for CO2 reduction (Grodkowski et al., 2002).
Our collaboration and the synthetic excellence of Professor Wolfgang Schӧfberger resulted in a (–S-PEG(7)-OMe)3 modified Co(III)-corrole, which led to a 12e reduction of CO2 to ethanol electrochemically at a low potential of −0.8 V vs. RHE (Gonglach et al., 2019). Systematic spectroscopic investigation and DFT calculation led to the proposition that shuttling back and forth between Co(III) and Co(I) states under electrochemical bias follows two divergent reaction pathways: one leading to the 6e reduced product methanol and the other 12e reduced product ethanol as shown in Figure 9. The low operational potential of the catalysts and formation of highly reduced products with C-C step-up was due to the formation of easily reducible glyoxal type intermediate (as shown in Figure 10). Now, to understand the mode of action of the metal center in the catalysis, Mn(III)-corrole was designed with the help of (–S-PEG(7)-OMe)3 modification (De et al., 2020). The previously supported Mn metallocomplexes showed excellent CO2 reducibility as in the case of Mn(bipyridine)-pyrene complexes where [MnBr(2,2′-bipyridine)(CO)3] was anchored over carbon nanotubes using pyrene moieties. High surface loading of the Mn complex led to the formation of Mn(0) dimer under electrochemical bias, reducing CO2 to CO. However, for low surface loading, Mn hydride formation shifted the reaction to a formic acid pathway (Reuillard et al., 2017). The A. J. Cowan group studied the same with different Mn-bipyridine carbonyl complexes immobilized over multi-walled carbon nanotubes (MWCNT) (Walsh et al., 2014; Walsh et al., 2015). The S. Sato group studied the Mn complex immobilized MWCNT and established a relationship between heterogeneous electrochemical CO2 reduction and the promotional effect of K+ ions. They found excellent stability of the electrodes under electrochemical conditions for at least 48 h with a current density of 2.0 mAcm−2 at −0.39 V vs. RHE. The significant lowering of the overpotential was a result of the electron storage properties of the MWCNTs and surface adsorption of K+, promoting the adsorption of CO2 over the catalysts (Sato et al., 2018). Based on the above experimental and theoretical support, our group conducted CO2 electroreduction over Mn(III)-corrole (–S-PEG (7)-OMe)3 immobilized over carbon paper electrodes. The 8e reduction of CO2 to acetate resulted from a faradaic efficiency of 63% and TOF of 8.25 h−1 (De et al., 2020). Comparing both the CO2 reduction results (Co(III) and Mn(III)-corrole) and tallying the chemical reduction study with density functional theory calculations, we reached the conclusion that the catalysis was metal-centered with the corrole ligand stabilizing multiple redox intermediates.
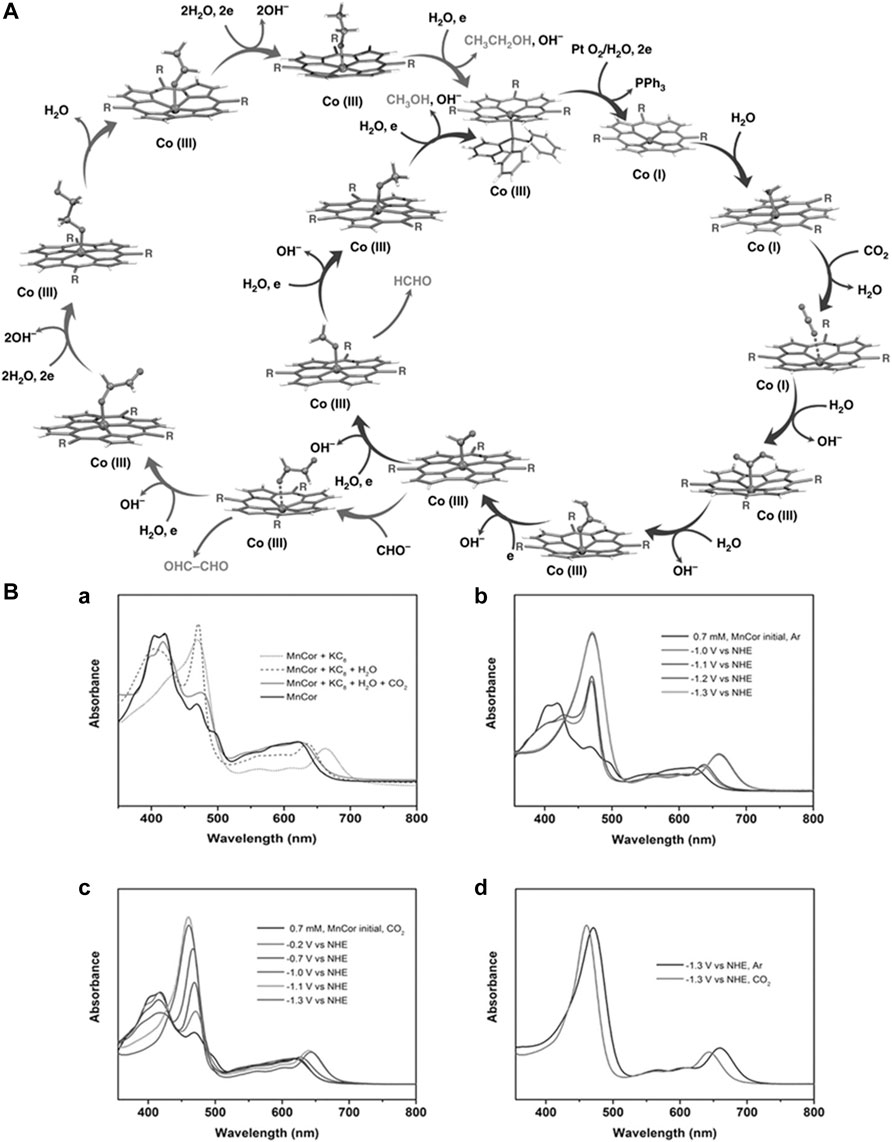
FIGURE 9. (A) Proposed single site mechanism of CO2 reduction using Co corrole. (B) (a) EAS of Mn corrole in acetonitrile under argon (black solid line), the chemically reduced form in the presence of KC8 (red dotted line), after the addition of water (blue, dashed line), and subsequent dosage of CO2 (green solid line). (b–d) Potential dependent SEC-UV/Vis of 0.7 mm Mn corrole in acetonitrile with 2% water and 0.2 M TBAPF6 as electrolyte after 2 min CPE (b) under argon, (c) under CO2, and (d) comparison of UV/Vis spectra observed at −1.3 V vs. NHE under argon (black) and CO2 dosage (red). SEC-UV/V measurements were recorded with a light transparent platinum mini-grid as working, as counter, and an Ag-microwire as a pseudo-reference electrode.
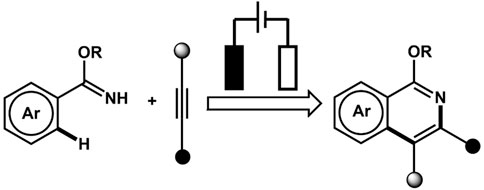
FIGURE 10. A representative diagram for electro-catalyzed C-H annulations.
5 Future perspective
The use of electricity to achieve chemical transformations, which traditionally involve cumbersome steps, including a stoichiometric amount of reagents, has opened up a whole new avenue in the field of catalysis. The field of electrosynthesis is evolving with the adoption of new concepts and strategies from other fields, such as magneto-electrochemistry (Dong et al., 2014; Vock et al., 2015; Clausmeyer et al., 2016) and ultrasonication (Atobe, 2014). Innovation in electrolyte and electrode systems is needed constantly to circumvent the limitation of the current electrode materials, such as mercury lead. The modern electrode system should be resistant to corrosion and should provide a large overpotential for undesired side reactions. BDE electrodes and synthetic carbon allotropes with modified surfaces have proven to be a step in a similar direction. There should also be enough emphasis on investigating solvents not only as a reaction medium but as a tool to moderate selectivity in the electrosynthetic pathway. There is also a vast scope for development in other aspects of electrochemical synthesis such as supporting electrolytes and electrolysis cells. The sustainability of electrochemical reactions might popularize it as a common synthetic method rather than a niche technology.
6 Conclusion
In this review, we have discussed different factors driving electrochemical reactions and have summarized different endeavors of electrochemical organic reaction and electrochemical CO2 reduction by several groups across the world. In order to perform a successful electrochemical reaction, the major challenges have been the generation and trapping of radical cations, current optimization, and the development of site-selective reactions on a microelectrode array. Over the years, we have been able to oxidize and reduce an array of substrates through these electrochemical reactions. This approach has been utilized over the years to drive reactions in a simple photovoltaic cell to a simple battery to preparative electrolysis setups to a microelectrode array. It is difficult for any other approach to explore such a varied scope of chemistry, which has been explored by electrochemical studies.
Author contributions
All authors contributed to the conception and design of the review article. JL, SP, HS, and LS wrote the first draft of the manuscript. JL, SP, HS, and LS wrote sections of the manuscript. WS and SR corrected the review and wrote additional sections of the review manuscript. All authors contributed to the manuscript revision and read and approved the submitted version.
Acknowledgments
The authors thank the Indian Institute of Science, Education and Research (IISER-K), Kolkata, for the funding. This is a short text to acknowledge the contributions of specific colleagues, institutions, or agencies that aided the efforts of the authors.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adams, R. N. (1969). Anodic oxidation pathways of aromatic hydrocarbons and amines. Acc. Chem. Res. 2, 175–180. doi:10.1021/ar50018a003
Adeli, Y., Huang, K., Liang, Y., Jiang, Y., Liu, J., Song, S., et al. (2019). Electrochemically oxidative C–C bond cleavage of alkylarenes for anilines synthesis. ACS Catal. 9, 2063–2067. doi:10.1021/acscatal.8b04351
Agarwal, J., Shaw, T. W., Schaefer, H. F., and Bocarsly, A. B. (2015). Design of a catalytic active site for electrochemical CO2 reduction with Mn (I)-tricarbonyl species. Inorg. Chem. 54, 5285–5294. doi:10.1021/acs.inorgchem.5b00233
Aljabour, A., Apaydin, D. H., Coskun, H., Ozel, F., Ersoz, M., Stadler, P., et al. (2016). Improvement of catalytic activity by nanofibrous CuInS2 for electrochemical CO2 reduction. ACS Appl. Mat. Interfaces 8, 31695–31701. doi:10.1021/acsami.6b11151
Allen, B. D., Hareram, M. D., Seastram, A. C., Mcbride, T., Wirth, T., Browne, D. L., et al. (2019). Manganese-catalyzed electrochemical deconstructive chlorination of cycloalkanols via alkoxy radicals. Org. Lett. 21, 9241–9246. doi:10.1021/acs.orglett.9b03652
Angamuthu, R., Byers, P., Lutz, M., Spek, A. L., and Bouwman, E. (2010). Electrocatalytic CO2 conversion to oxalate by a copper complex. Science 327 (5963), 313–315.
Asadi, M., Kumar, B., Behranginia, A., Rosen, B. A., Baskin, A., Repnin, N., et al. (2014). Robust carbon dioxide reduction on molybdenum disulphide edges. Nat. Commun. 5, 4470–4478. doi:10.1038/ncomms5470
Atobe, M. (2014). “Electrosynthesis under ultrasound and centrifugal fields,” in Encyclopedia of applied electrochemistry (Berlin, Germany: Springer).
Ba, X., Yan, L.-L., Huang, S., Yu, J., Xia, X.-J., and Yu, Y. (2014). New way for CO2 reduction under visible light by a combination of a Cu electrode and semiconductor thin film: Cu2O conduction type and morphology effect. J. Phys. Chem. C 118, 24467–24478. doi:10.1021/jp5063397
Badalyan, A., and Stahl, S. S. (2016). Cooperative electrocatalytic alcohol oxidation with electron-proton-transfer mediators. Nature 535, 406–410. doi:10.1038/nature18008
Barker, G., and Gardner, A. (1960). Pulse polarography. Z. Anal. Chem. 173, 79–83. doi:10.1007/bf00448718
Barker, G., Gonsalves, M., Macpherson, J., Slevin, C., and Unwin, P. (1958). Square wave polarography and some related techniques. Anal. Chim. Acta 18, 118–131. doi:10.1016/s0003-2670(00)87111-1
Baturina, O. A., Lu, Q., Padilla, M. A., Xin, L., Li, W., Serov, A., et al. (2014). CO2 electroreduction to hydrocarbons on carbon-supported Cu nanoparticles. ACS Catal. 4, 3682–3695. doi:10.1021/cs500537y
Beil, S. B., Müller, T., Sillart, S. B., Franzmann, P., Bomm, A., Holtkamp, M., et al. (2018). Active molybdenum-based anode for dehydrogenative coupling reactions. Angew. Chem. Int. Ed. 57, 2450–2454. doi:10.1002/anie.201712718
Bergens, S. H., Gorman, C. B., Palmore, G. T. R., and Whitesides, G. M. (1994). A redox fuel cell that operates with methane as fuel at 120 C. Science 265, 1418–1420. doi:10.1126/science.265.5177.1418
Bockris, J. O. M., Conway, B. E., and White, R. E. (1992). Modern aspects of electrochemistry. Berlin, Germany: Springer Science & Business Media.
Bryan, M. C., Dillon, B., Hamann, L. G., Hughes, G. J., Kopach, M. E., Peterson, E. A., et al. (2013). Sustainable practices in medicinal chemistry: Current state and future directions. J. Med. Chem. 56, 6007–6021. doi:10.1021/jm400250p
Cai, C.-Y., and Xu, H.-C. (2018). Dehydrogenative reagent-free annulation of alkenes with diols for the synthesis of saturated O-heterocycles. Nat. Commun. 9, 3551–3557. doi:10.1038/s41467-018-06020-8
Cardiel, A. C., Taitt, B. J., and Choi, K.-S. (2019). Stabilities, regeneration pathways, and electrocatalytic properties of nitroxyl radicals for the electrochemical oxidation of 5-hydroxymethylfurfural. ACS Sustain. Chem. Eng. 7, 11138–11149. doi:10.1021/acssuschemeng.9b00203
Cernak, T., Dykstra, K. D., Tyagarajan, S., Vachal, P., and Krska, S. W. (2016). The medicinal chemist's toolbox for late stage functionalization of drug-like molecules. Chem. Soc. Rev. 45, 546–576. doi:10.1039/c5cs00628g
Chai, G.-L., and Guo, Z.-X. (2016). Highly effective sites and selectivity of nitrogen-doped graphene/CNT catalysts for CO 2 electrochemical reduction. Chem. Sci. 7, 1268–1275. doi:10.1039/c5sc03695j
Chan, K., Tsai, C., Hansen, H. A., and Nørskov, J. K. (2014). Molybdenum sulfides and selenides as possible electrocatalysts for CO2 reduction. ChemCatChem 6, 1899–1905. doi:10.1002/cctc.201402128
Chapovetsky, A., Do, T. H., Haiges, R., Takase, M. K., and Marinescu, S. C. (2016). Proton-assisted reduction of CO2 by cobalt aminopyridine macrocycles. J. Am. Chem. Soc. 138, 5765–5768. doi:10.1021/jacs.6b01980
Chen, C. S., Handoko, A. D., Wan, J. H., Ma, L., Ren, D., and Yeo, B. S. (2015a). Stable and selective electrochemical reduction of carbon dioxide to ethylene on copper mesocrystals. Catal. Sci. Technol. 5, 161–168. doi:10.1039/c4cy00906a
Chen, J., Lv, S., and Tian, S. (2019). Electrochemical transition-metal-catalyzed C− H bond functionalization: Electricity as clean surrogates of chemical oxidants. ChemSusChem 12, 115–132. doi:10.1002/cssc.201801946
Chen, J., Yan, W.-Q., Lam, C. M., Zeng, C.-C., Hu, L.-M., and Little, R. D. (2015b). Electrocatalytic aziridination of alkenes mediated by n-bu4NI: A radical pathway. Org. Lett. 17, 986–989. doi:10.1021/acs.orglett.5b00083
Chen, L. D., Urushihara, M., Chan, K., and Nørskov, J. K. (2016b). Electric field effects in electrochemical CO2 reduction. ACS Catal. 6, 7133–7139. doi:10.1021/acscatal.6b02299
Chen, L., Guo, S. X., Li, F., Bentley, C., Horne, M., Bond, A. M., et al. (2016a). Electrochemical reduction of CO2 at metal electrodes in a distillable ionic liquid. ChemSusChem 9, 1271–1278. doi:10.1002/cssc.201600359
Cheng, M.-J., Kwon, Y., Head-Gordon, M., and Bell, A. T. (2015). Tailoring metal-porphyrin-like active sites on graphene to improve the efficiency and selectivity of electrochemical CO2 reduction. J. Phys. Chem. C 119, 21345–21352. doi:10.1021/acs.jpcc.5b05518
Cheng, T., Xiao, H., and Goddard, W. A. (2016). Reaction mechanisms for the electrochemical reduction of CO2 to CO and formate on the Cu (100) surface at 298 K from quantum mechanics free energy calculations with explicit water. J. Am. Chem. Soc. 138, 13802–13805. doi:10.1021/jacs.6b08534
Chiba, K., and Kim, S. (2009). Anodic carbon-carbon bond formation in lithium perchlorate/nitromethane electrolyte solution. Electrochemistry 77, 21–29. doi:10.5796/electrochemistry.77.21
Choi, J., Benedetti, T. M., Jalili, R., Walker, A., Wallace, G. G., and Officer, D. L. (2016a). High performance Fe porphyrin/ionic liquid Co-catalyst for electrochemical CO2 reduction. Chem. Eur. J. 22, 14158–14161. doi:10.1002/chem.201603359
Choi, S. Y., Jeong, S. K., Kim, H. J., Baek, I.-H., and Park, K. T. (2016b). Electrochemical reduction of carbon dioxide to formate on tin–lead alloys. ACS Sustain. Chem. Eng. 4, 1311–1318. doi:10.1021/acssuschemeng.5b01336
Claraz, A. L., Courant, T., and Masson, G. R. (2020). Electrochemical intramolecular oxytrifluoromethylation of N-tethered alkenyl alcohols: Synthesis of functionalized morpholines. Org. Lett. 22, 1580–1584. doi:10.1021/acs.orglett.0c00176
Clausmeyer, J., Wilde, P., Löffler, T., Ventosa, E., Tschulik, K., and Schuhmann, W. (2016). Detection of individual nanoparticle impacts using etched carbon nanoelectrodes. Electrochem. Commun. 73, 67–70. doi:10.1016/j.elecom.2016.11.003
Conway, B. E., and Bockris, J. M. (2012). Modern aspects of electrochemistry: No. 9. Berlin, Germany: Springer Science & Business Media.
Cope, J. D., Liyanage, N. P., Kelley, P. J., Denny, J. A., Valente, E. J., Webster, C. E., et al. (2017). Electrocatalytic reduction of CO 2 with CCC-NHC pincer nickel complexes. Chem. Commun. 53, 9442–9445. doi:10.1039/c6cc06537f
Costentin, C., Passard, G., Robert, M., and Savéant, J.-M. (2014). Ultraefficient homogeneous catalyst for the CO2-to-CO electrochemical conversion. Proc. Natl. Acad. Sci. U. S. A. 111, 14990–14994. doi:10.1073/pnas.1416697111
Costentin, C., Robert, M., and SavéAnt, J.-M. (2015). Current issues in molecular catalysis illustrated by iron porphyrins as catalysts of the CO2-to-CO electrochemical conversion. Acc. Chem. Res. 48, 2996–3006. doi:10.1021/acs.accounts.5b00262
Das, A., Nutting, J. E., and Stahl, S. S. (2019). Electrochemical C–H oxygenation and alcohol dehydrogenation involving Fe-oxo species using water as the oxygen source. Chem. Sci. 10, 7542–7548. doi:10.1039/c9sc02609f
Das, A., and Stahl, S. S. (2017). Noncovalent immobilization of molecular electrocatalysts for chemical synthesis: Efficient electrochemical alcohol oxidation with a pyrene–TEMPO conjugate. Angew. Chem. Int. Ed. Engl. 56, 9018–9023. doi:10.1002/ange.201704921
Daugulis, O., Roane, J., and Tran, L. D. (2015). Bidentate, monoanionic auxiliary-directed functionalization of carbon–hydrogen bonds. Acc. Chem. Res. 48, 1053–1064. doi:10.1021/ar5004626
De, R., Gonglach, S., Paul, S., Haas, M., Sreejith, S., Gerschel, P., et al. (2020). Frontispiz: Electrocatalytic reduction of CO 2 to acetic acid by a molecular manganese corrole complex. Angew. Chem. Int. Ed. Engl. 132, 202082662. doi:10.1002/ange.202082662
Detweiler, Z. M., White, J. L., Bernasek, S. L., and Bocarsly, A. B. (2014). Anodized indium metal electrodes for enhanced carbon dioxide reduction in aqueous electrolyte. Langmuir 30, 7593–7600. doi:10.1021/la501245p
Ding, C., Li, A., Lu, S.-M., Zhang, H., and Li, C. (2016). In situ electrodeposited indium nanocrystals for efficient CO2 reduction to CO with low overpotential. ACS Catal. 6, 6438–6443. doi:10.1021/acscatal.6b01795
Dong, J., Huang, Y., Jin, L., Lin, H., and Yang, H. (2014). Thermal optimization of a high-speed permanent magnet motor. IEEE Trans. Magn. 50, 749–752. doi:10.1109/tmag.2013.2285017
Dou, G.-Y., Jiang, Y.-Y., Xu, K., and Zeng, C.-C. (2019). Electrochemical Minisci-type trifluoromethylation of electron-deficient heterocycles mediated by bromide ions. Org. Chem. Front. 6, 2392–2397. doi:10.1039/c9qo00552h
Du, D., Lan, R., Humphreys, J., Sengodan, S., Xie, K., Wang, H., et al. (2016). Achieving both high selectivity and current density for CO2 reduction to formate on nanoporous tin foam electrocatalysts. ChemistrySelect 1, 1711–1715. doi:10.1002/slct.201600451
Eberson, L., and Nyberg, K. (1973). Anodic aromatic substitution. Acc. Chem. Res. 6, 106–112. doi:10.1021/ar50063a004
Eberson, L., and Nyberg, K. (1976). Synthetic uses of anodic substitution reactions. Tetrahedron 32, 2185–2206. doi:10.1016/0040-4020(76)85132-0
Edinger, C., and Waldvogel, S. R. (2014). Electrochemical deoxygenation of aromatic amides and sulfoxides. Eur. J. Org. Chem. 2014, 5144–5148. doi:10.1002/ejoc.201402714
Elving, P. J., and Pullman, B. (2009). Mechanisms of organic electrode reactions. Adv. Chem. Phys. 3, 1–31. doi:10.1002/9780470143490.ch1
Evans, D. H. (2008). One-electron and two-electron transfers in electrochemistry and homogeneous solution reactions. Chem. Rev. 108, 2113–2144. doi:10.1021/cr068066l
Farghaly, O. A., Hameed, R. A., and Abu-Nawwas, A.-a. H. (2014). Analytical application using modern electrochemical techniques. Int. J. Electrochem. Sci. 9, 3287–3318.
Feng, R., Smith, J. A., and Moeller, K. D. (2017). Anodic cyclization reactions and the mechanistic strategies that enable optimization. Acc. Chem. Res. 50, 2346–2352. doi:10.1021/acs.accounts.7b00287
Francke, R., and Little, R. D. (2014). Redox catalysis in organic electrosynthesis: Basic principles and recent developments. Chem. Soc. Rev. 43, 2492–2521. doi:10.1039/c3cs60464k
Frontana-Uribe, B. A., Little, R. D., Ibanez, J. G., Palma, A., and Vasquez-Medrano, R. (2010). Organic electrosynthesis: A promising green methodology in organic chemistry. Green Chem. 12, 2099–2119. doi:10.1039/c0gc00382d
Fu, N., Sauer, G. S., Saha, A., Loo, A., and Lin, S. (2017). Metal-catalyzed electrochemical diazidation of alkenes. Science 357, 575–579. doi:10.1126/science.aan6206
Gao, D., Zhou, H., Wang, J., Miao, S., Yang, F., Wang, G., et al. (2015). Size-dependent electrocatalytic reduction of CO2 over Pd nanoparticles. J. Am. Chem. Soc. 137, 4288–4291. doi:10.1021/jacs.5b00046
Gao, S., Jiao, X., Sun, Z., Zhang, W., Sun, Y., Wang, C., et al. (2016). Ultrathin Co3O4 layers realizing optimized CO2 electroreduction to formate. Angew. Chem. Int. Ed. Engl. 55, 708–712. doi:10.1002/ange.201509800
Garza, A. J., Bell, A. T., and Head-Gordon, M. (2018). Mechanism of CO2 reduction at copper surfaces: Pathways to C2 products. ACS Catal. 8, 1490–1499. doi:10.1021/acscatal.7b03477
Glasstone, S., and Hickling, A. (1939). The hydrogen peroxide theory of electrolytic oxidation. Chem. Rev. 25, 407–441. doi:10.1021/cr60082a003
Gonglach, S., Paul, S., Haas, M., Pillwein, F., Sreejith, S. S., Barman, S., et al. (2019). Molecular cobalt corrole complex for the heterogeneous electrocatalytic reduction of carbon dioxide. Nat. Commun. 10, 3864–3873. doi:10.1038/s41467-019-11868-5
Gorman, C. B., Bergens, S. H., and Whitesides, G. M. (1996). Platinum-catalyzed oxidations of organic compounds by ferric sulfate: Use of a redox fuel cell to mediate complete oxidation of ethylene glycol by dioxygen at 80° C. J. Catal. 158, 92–96. doi:10.1006/jcat.1996.0009
Grills, D. C., Matsubara, Y., Kuwahara, Y., Golisz, S. R., Kurtz, D. A., and Mello, B. A. (2014). Electrocatalytic CO2 reduction with a homogeneous catalyst in ionic liquid: High catalytic activity at low overpotential. J. Phys. Chem. Lett. 5, 2033–2038. doi:10.1021/jz500759x
Grimshaw, J. (2000). Electrochemical reactions and mechanisms in organic chemistry. Amsterdam, Netherlands: Elsevier.
Grjotheim, K., Krohn, C., Malinovsky, M., Matiasovsky, K., and Thonstad, J. (1977). “Aluminum electrolysis,” in The chemistry of the Hall-heroult process (Dusseldorf, Germany: Aluminium- Verlag GmbH), 350.
Grodkowski, J., Neta, P., Fujita, E., Mahammed, A., Simkhovich, L., and Gross, Z. (2002). Reduction of cobalt and iron corroles and catalyzed reduction of CO2. J. Phys. Chem. A 106, 4772–4778. doi:10.1021/jp013668o
Guo, X.-X., Gu, D.-W., Wu, Z., and Zhang, W. (2015). Copper-catalyzed C–H functionalization reactions: Efficient synthesis of heterocycles. Chem. Rev. 115, 1622–1651. doi:10.1021/cr500410y
Gurkan, B., Simeon, F., and Hatton, T. A. (2015). Quinone reduction in ionic liquids for electrochemical CO2 separation. ACS Sustain. Chem. Eng. 3, 1394–1405. doi:10.1021/acssuschemeng.5b00116
Hahn, C., Hatsukade, T., Kim, Y.-G., Vailionis, A., Baricuatro, J. H., Higgins, D. C., et al. (2017). Engineering Cu surfaces for the electrocatalytic conversion of CO2: Controlling selectivity toward oxygenates and hydrocarbons. Proc. Natl. Acad. Sci. U. S. A. 114, 5918–5923. doi:10.1073/pnas.1618935114
Hall, A. S., Yoon, Y., Wuttig, A., and Surendranath, Y. (2015). Mesostructure-induced selectivity in CO2 reduction catalysis. J. Am. Chem. Soc. 137, 14834–14837. doi:10.1021/jacs.5b08259
Hammes-Schiffer, S., and Soudackov, A. V. (2008). Proton-coupled electron transfer in solution, proteins, and electrochemistry. J. Phys. Chem. B 112, 14108–14123. doi:10.1021/jp805876e
Harnisch, F., and Schröder, U. (2019). Tapping renewables: A new dawn for organic electrosynthesis in aqueous reaction media. ChemElectroChem 6, 4126–4133. doi:10.1002/celc.201900456
Hashiguchi, B. G., Bischof, S. M., Konnick, M. M., and Periana, R. A. (2012). Designing catalysts for functionalization of unactivated C–H bonds based on the CH activation reaction. Acc. Chem. Res. 45, 885–898. doi:10.1021/ar200250r
Hayashi, R., Shimizu, A., Song, Y., Ashikari, Y., Nokami, T., and Yoshida, J. I. (2017). Metal-free benzylic C− H amination via electrochemically generated benzylaminosulfonium ions. Chem. Eur. J. 23, 61–64. doi:10.1002/chem.201604484
Haywood, S. K., Rybchenko, S. I., Touhami, D., and Wadhawan, J. D. (2016). Study of pyridine-mediated electrochemical reduction of CO2 to methanol at high CO2 pressure. ChemSusChem 9, 1660–1669. doi:10.1002/cssc.201600267
He, T.-J., Ye, Z., Ke, Z., and Huang, J.-M. (2019). Stereoselective synthesis of sulfur-containing β-enaminonitrile derivatives through electrochemical Csp 3–H bond oxidative functionalization of acetonitrile. Nat. Commun. 10, 833–839. doi:10.1038/s41467-019-08762-5
Hertl, W., and Weetall, H. (1985a). A photo-chemical electrical fuel cell: Part I. Alcohol fuels. Bioelectrochemistry Bioenergetics 14, 357–366. doi:10.1016/0302-4598(85)80008-8
Hertl, W., and Weetall, H. (1985b). A photo-chemical electrical fuel cell: Part II. Carbohydrate fuels. Bioelectrochemistry Bioenergetics 14, 367–373. doi:10.1016/0302-4598(85)80009-x
Hilt, G. (2020). Basic strategies and types of applications in organic electrochemistry. ChemElectroChem 7, 395–405. doi:10.1002/celc.201901799
Hiroto, S., Miyake, Y., and Shinokubo, H. (2017). Synthesis and functionalization of porphyrins through organometallic methodologies. Chem. Rev. 117, 2910–3043. doi:10.1021/acs.chemrev.6b00427
Hod, I., Sampson, M. D., Deria, P., Kubiak, C. P., Farha, O. K., and Hupp, J. T. (2015). Fe-porphyrin-based metal–organic framework films as high-surface concentration, heterogeneous catalysts for electrochemical reduction of CO2. ACS Catal. 5, 6302–6309. doi:10.1021/acscatal.5b01767
Hong, H., Li, Y., Chen, L., Li, B., Zhu, Z., Chen, X., et al. (2019). Electrochemical synthesis strategy for cvinyl-CF3 compounds through decarboxylative trifluoromethylation. J. Org. Chem. 84, 5980–5986. doi:10.1021/acs.joc.9b00766
Hoover, J. M., Ryland, B. L., and Stahl, S. S. (2013). Mechanism of copper (I)/TEMPO-catalyzed aerobic alcohol oxidation. J. Am. Chem. Soc. 135, 2357–2367. doi:10.1021/ja3117203
Horn, E. J., Rosen, B. R., Chen, Y., Tang, J., Chen, K., Eastgate, M. D., et al. (2016). Scalable and sustainable electrochemical allylic C–H oxidation. Nature 533, 77–81. doi:10.1038/nature17431
Hu, X., Zhang, G., Bu, F., Luo, X., Yi, K., Zhang, H., et al. (2018a). Photoinduced oxidative activation of electron-rich arenes: alkenylation with H 2 evolution under external oxidant-free conditions. Chem. Sci. 9, 1521–1526. doi:10.1039/c7sc04634k
Hu, X., Zhang, G., Bu, F., Nie, L., and Lei, A. (2018b). Electrochemical-oxidation-induced site-selective intramolecular C (sp3)–H amination. ACS Catal. 8, 9370–9375. doi:10.1021/acscatal.8b02847
Hu, X., Zhang, G., Nie, L., Kong, T., and Lei, A. (2019). Electrochemical oxidation induced intermolecular aromatic CH imidation. Nat. Commun. 10, 5467–5476. doi:10.1038/s41467-019-13524-4
Huan, T. N., Prakash, P., Simon, P., Rousse, G., Xu, X., Artero, V., et al. (2016). CO2 reduction to CO in water: Carbon nanotube–gold nanohybrid as a selective and efficient electrocatalyst. ChemSusChem 9, 2317–2320. doi:10.1002/cssc.201600597
Huang, H., Strater, Z. M., and Lambert, T. H. (2020). Electrophotocatalytic C–H functionalization of ethers with high regioselectivity. J. Am. Chem. Soc. 142, 1698–1703. doi:10.1021/jacs.9b11472
Inzelt, G., Lewenstam, A., and Scholz, F. (2013). Handbook of reference electrodes. Berlin, Germany: Springer.
Jiang, Y., Xu, K., and Zeng, C. (2017). Use of electrochemistry in the synthesis of heterocyclic structures. Chem. Rev. 118, 4485–4540. doi:10.1021/acs.chemrev.7b00271
Johnson, B. A., Maji, S., Agarwala, H., White, T. A., Mijangos, E., and Ott, S. (2016). Activating a low overpotential CO2 reduction mechanism by a strategic ligand modification on a ruthenium polypyridyl catalyst. Angew. Chem. Int. Ed. Engl. 55, 1857–1861. doi:10.1002/ange.201508490
Kalvoda, R., and Kopanica, M. (1989). Adsorptive stripping voltammetry in trace analysis. Pure Appl. Chem. 61, 97–112. doi:10.1351/pac198961010097
Kang, X., Zhu, Q., Sun, X., Hu, J., Zhang, J., Liu, Z., et al. (2016). Highly efficient electrochemical reduction of CO 2 to CH 4 in an ionic liquid using a metal–organic framework cathode. Chem. Sci. 7, 266–273. doi:10.1039/c5sc03291a
Kärkäs, M. D. (2018). Electrochemical strategies for C–H functionalization and C–N bond formation. Chem. Soc. Rev. 47, 5786–5865. doi:10.1039/c7cs00619e
Kas, R., Hummadi, K. K., Kortlever, R., De Wit, P., Milbrat, A., Luiten-Olieman, M. W., et al. (2016). Three-dimensional porous hollow fibre copper electrodes for efficient and high-rate electrochemical carbon dioxide reduction. Nat. Commun. 7, 10748–10757. doi:10.1038/ncomms10748
Kas, R., Kortlever, R., Milbrat, A., Koper, M. T., Mul, G., and Baltrusaitis, J. (2014). Electrochemical CO 2 reduction on Cu 2 O-derived copper nanoparticles: Controlling the catalytic selectivity of hydrocarbons. Phys. Chem. Chem. Phys. 16, 12194–12201. doi:10.1039/c4cp01520g
Kas, R., Kortlever, R., Yılmaz, H., Koper, M. T., and Mul, G. (2015). Manipulating the hydrocarbon selectivity of copper nanoparticles in CO2 electroreduction by process conditions. ChemElectroChem 2, 354–358. doi:10.1002/celc.201402373
Kathiravan, S., Suriyanarayanan, S., and Nicholls, I. A. (2019). Electrooxidative amination of sp2 C–H bonds: Coupling of amines with aryl amides via copper catalysis. Org. Lett. 21, 1968–1972. doi:10.1021/acs.orglett.9b00003
Kawamata, Y., Yan, M., Liu, Z., Bao, D.-H., Chen, J., Starr, J. T., et al. (2017). Scalable, electrochemical oxidation of unactivated C–H bonds. J. Am. Chem. Soc. 139, 7448–7451. doi:10.1021/jacs.7b03539
Kim, C., Jeon, H. S., Eom, T., Jee, M. S., Kim, H., Friend, C. M., et al. (2015). Achieving selective and efficient electrocatalytic activity for CO2 reduction using immobilized silver nanoparticles. J. Am. Chem. Soc. 137, 13844–13850. doi:10.1021/jacs.5b06568
Kim, D., Resasco, J., Yu, Y., Asiri, A. M., and Yang, P. (2014). Synergistic geometric and electronic effects for electrochemical reduction of carbon dioxide using gold–copper bimetallic nanoparticles. Nat. Commun. 5, 4948–8. doi:10.1038/ncomms5948
Kim, K.-S., Kim, W. J., Lim, H.-K., Lee, E. K., and Kim, H. (2016). Tuned chemical bonding ability of Au at grain boundaries for enhanced electrochemical CO2 reduction. ACS Catal. 6, 4443–4448. doi:10.1021/acscatal.6b00412
Kingston, C., Palkowitz, M. D., Takahira, Y., Vantourout, J. C., Peters, B. K., Kawamata, Y., et al. (2019). A survival guide for the “electro-curious”. Acc. Chem. Res. 53, 72–83. doi:10.1021/acs.accounts.9b00539
Kirste, A., Elsler, B., Schnakenburg, G., and Waldvogel, S. R. (2012). Efficient anodic and direct phenol-arene C, C cross-coupling: The benign role of water or methanol. J. Am. Chem. Soc. 134, 3571–3576. doi:10.1021/ja211005g
Kirste, A., Hayashi, S., Schnakenburg, G., Malkowsky, I. M., Stecker, F., Fischer, A., et al. (2011a). Highly selective electrosynthesis of biphenols on graphite electrodes in fluorinated media. Chem. Eur. J. 17, 14164–14169. doi:10.1002/chem.201102182
Kirste, A., Schnakenburg, G., and Waldvogel, S. R. (2011b). Anodic coupling of guaiacol derivatives on boron-doped diamond electrodes. Org. Lett. 13, 3126–3129. doi:10.1021/ol201030g
Kissinger, P., and Heineman, W. R. (2018). Laboratory techniques in electroanalytical chemistry, revised and expanded. United States: CRC Press.
Kong, W.-J., Finger, L. H., Messinis, A. M., Kuniyil, R., Oliveira, J. C., and Ackermann, L. (2019). Flow rhodaelectro-catalyzed alkyne annulations by versatile C–H activation: Mechanistic support for rhodium (III/IV). J. Am. Chem. Soc. 141, 17198–17206. doi:10.1021/jacs.9b07763
Kong, W. J., Shen, Z., Finger, L. H., and Ackermann, L. (2020). Electrochemical access to aza-polycyclic aromatic hydrocarbons: Rhoda-electrocatalyzed domino alkyne annulations. Angew. Chem. Int. Ed. 59, 5551–5556. doi:10.1002/anie.201914775
Kornienko, N., Zhao, Y., Kley, C. S., Zhu, C., Kim, D., Lin, S., et al. (2015). Metal–organic frameworks for electrocatalytic reduction of carbon dioxide. J. Am. Chem. Soc. 137, 14129–14135. doi:10.1021/jacs.5b08212
Kortlever, R., Peters, I., Balemans, C., Kas, R., Kwon, Y., Mul, G., et al. (2016). Palladium–gold catalyst for the electrochemical reduction of CO 2 to C 1–C 5 hydrocarbons. Chem. Commun. 52, 10229–10232. doi:10.1039/c6cc03717h
Kortlever, R., Peters, I., Koper, S., and Koper, M. T. (2015). Electrochemical CO2 reduction to formic acid at low overpotential and with high faradaic efficiency on carbon-supported bimetallic Pd–Pt nanoparticles. ACS Catal. 5, 3916–3923. doi:10.1021/acscatal.5b00602
Kumar, G. S., Peshkov, A., Brzozowska, A., Nikolaienko, P., Zhu, C., and Rueping, M. (2020). Nickel-catalyzed chain-walking cross-electrophile coupling of alkyl and aryl halides and olefin hydroarylation enabled by electrochemical reduction. Angew. Chem. Int. Ed. Engl. 59, 6575–6581. doi:10.1002/ange.201915418
Kuttruff, C. A., Eastgate, M. D., and Baran, P. S. (2014). Natural product synthesis in the age of scalability. Nat. Prod. Rep. 31, 419–432. doi:10.1039/c3np70090a
Kwon, Y., Lum, Y., Clark, E. L., Ager, J. W., and Bell, A. T. (2016). CO2 electroreduction with enhanced ethylene and ethanol selectivity by nanostructuring polycrystalline copper. ChemElectroChem 3, 1012–1019. doi:10.1002/celc.201600068
Lacy, D. C., Mccrory, C. C., and Peters, J. C. (2014). Studies of cobalt-mediated electrocatalytic CO2 reduction using a redox-active ligand. Inorg. Chem. 53, 4980–4988. doi:10.1021/ic403122j
Lai, X. L., Shu, X. M., Song, J., and Xu, H. C. (2020). Electrophotocatalytic decarboxylative C− H functionalization of heteroarenes. Angew. Chem. Int. Ed. 59, 10626. doi:10.1002/anie.202002900
Lakshmanan, S., and Murugesan, T. (2014). The chlor-alkali process: Work in progress. Clean. Technol. Environ. Policy 16, 225–234. doi:10.1007/s10098-013-0630-6
Lan, Y., Gai, C., Kenis, P. J., and Lu, J. (2014). Electrochemical reduction of carbon dioxide on Cu/CuO core/shell catalysts. ChemElectroChem 1, 1577–1582. doi:10.1002/celc.201402182
LarrazáBal, G. N. O., Martín, A. J., Mitchell, S., Hauert, R., and PéRez-Ramírez, J. (2016). Enhanced reduction of CO2 to CO over Cu–in electrocatalysts: Catalyst evolution is the key. ACS Catal. 6, 6265–6274. doi:10.1021/acscatal.6b02067
Lee, S., Kim, D., and Lee, J. (2015). Electrocatalytic production of C3-C4 compounds by conversion of CO2 on a chloride-induced bi-phasic Cu2O-Cu catalyst. Angew. Chem. Int. Ed. Engl. 127, 14914–14918. doi:10.1002/ange.201505730
Li, C.-J. (2009). Cross-dehydrogenative coupling (CDC): Exploring C− C bond formations beyond functional group transformations. Acc. Chem. Res. 42, 335–344. doi:10.1021/ar800164n
Li, J., Liu, X., Deng, J., Huang, Y., Pan, Z., Yu, Y., et al. (2020). Electrochemical diselenylation of indolizines via intermolecular C–Se formation with 2-methylpyridines, α-bromoketones and diselenides. Chem. Commun. 56, 735–738. doi:10.1039/c9cc08784b
Li, L.-J., Jiang, Y.-Y., Lam, C. M., Zeng, C.-C., Hu, L.-M., and Little, R. D. (2015). Aromatic C–H bond functionalization induced by electrochemically in situ generated tris (p-bromophenyl) aminium radical cation: Cationic chain reactions of electron-rich aromatics with enamides. J. Org. Chem. 80, 11021–11030. doi:10.1021/acs.joc.5b02222
Li, Y.-Q., Yang, Q.-L., Fang, P., Mei, T.-S., and Zhang, D. (2017). Palladium-catalyzed C (sp2)–H acetoxylation via electrochemical oxidation. Org. Lett. 19, 2905–2908. doi:10.1021/acs.orglett.7b01138
Lim, H.-K., Shin, H., Goddard, W. A., Hwang, Y. J., Min, B. K., and Kim, H. (2014). Embedding covalency into metal catalysts for efficient electrochemical conversion of CO2. J. Am. Chem. Soc. 136, 11355–11361. doi:10.1021/ja503782w
Liu, K., Wu, J., Deng, Y., Song, C., Song, W., and Lei, A. (2019). Electrochemical C− H/N− H oxidative cross coupling of imidazopyridines with diarylamines to synthesize triarylamine derivatives. ChemElectroChem 6, 4173–4176. doi:10.1002/celc.201900138
Liu, X., Zhu, L., Wang, H., He, G., and Bian, Z. (2016). Catalysis performance comparison for electrochemical reduction of CO 2 on Pd–Cu/graphene catalyst. RSC Adv. 6, 38380–38387. doi:10.1039/c6ra03160a
Liu, Y., Chen, S., Quan, X., and Yu, H. (2015). Efficient electrochemical reduction of carbon dioxide to acetate on nitrogen-doped nanodiamond. J. Am. Chem. Soc. 137, 11631–11636. doi:10.1021/jacs.5b02975
Lodh, J., Mallick, A., and Roy, S. (2018). Light-driven carbon dioxide reduction coupled with conversion of acetylenic group to ketone by a functional Janus catalyst based on keplerate {Mo132}. J. Mat. Chem. A Mat. 6, 20844–20851. doi:10.1039/c8ta06243a
Loiudice, A., Lobaccaro, P., Kamali, E. A., Thao, T., Huang, B. H., Ager, J. W., et al. (2016). Tailoring copper nanocrystals towards C2 products in electrochemical CO2 reduction. Angew. Chem. Int. Ed. 55, 5789–5792. doi:10.1002/anie.201601582
Lu, Q., Rosen, J., Zhou, Y., Hutchings, G. S., Kimmel, Y. C., Chen, J. G., et al. (2014a). A selective and efficient electrocatalyst for carbon dioxide reduction. Nat. Commun. 5, 3242–3246. doi:10.1038/ncomms4242
Lu, X., Leung, D. Y., Wang, H., Leung, M. K., and Xuan, J. (2014b). Electrochemical reduction of carbon dioxide to formic acid. ChemElectroChem 1, 836–849. doi:10.1002/celc.201300206
Lu, X., Tan, T. H., Ng, Y. H., and Amal, R. (2016). Highly selective and stable reduction of CO2 to CO by a graphitic carbon nitride/carbon nanotube composite electrocatalyst. Chem. Eur. J. 22, 11991–11996. doi:10.1002/chem.201601674
Lucio, A. J., and Shaw, S. K. (2015). Pyridine and pyridinium electrochemistry on polycrystalline gold electrodes and implications for CO2 reduction. J. Phys. Chem. C 119, 12523–12530. doi:10.1021/acs.jpcc.5b03355
Lund, H., and Hammerich, O. (2001). Organic electrochemistry: Revised and expanded. New York: Marcel Dekker.
Ma, C., Fang, P., Liu, D., Jiao, K.-J., Gao, P.-S., Qiu, H., et al. (2021). Transition metal-catalyzed organic reactions in undivided electrochemical cells. Chem. Sci. 12, 12866–12873. doi:10.1039/d1sc04011a
Ma, C., Fang, P., and Mei, T.-S. (2018). Recent advances in C–H functionalization using electrochemical transition metal catalysis. ACS Catal. 8, 7179–7189. doi:10.1021/acscatal.8b01697
Ma, M., Djanashvili, K., and Smith, W. A. (2016a). Controllable hydrocarbon formation from the electrochemical reduction of CO2 over Cu nanowire arrays. Angew. Chem. Int. Ed. Engl. 55, 6792–6796. doi:10.1002/ange.201601282
Ma, M., Trześniewski, B. J., Xie, J., and Smith, W. A. (2016b). Selective and efficient reduction of carbon dioxide to carbon monoxide on oxide-derived nanostructured silver electrocatalysts. Angew. Chem. Int. Ed. Engl. 128, 9900–9904. doi:10.1002/ange.201604654
Ma, S., Lan, Y., Perez, G. M., Moniri, S., and Kenis, P. J. (2014). Silver supported on titania as an active catalyst for electrochemical carbon dioxide reduction. ChemSusChem 7, 866–874. doi:10.1002/cssc.201300934
Machan, C. W., Chabolla, S. A., Yin, J., Gilson, M. K., Tezcan, F. A., and Kubiak, C. P. (2014a). Supramolecular assembly promotes the electrocatalytic reduction of carbon dioxide by Re (I) bipyridine catalysts at a lower overpotential. J. Am. Chem. Soc. 136, 14598–14607. doi:10.1021/ja5085282
Machan, C. W., Sampson, M. D., Chabolla, S. A., Dang, T., and Kubiak, C. P. (2014b). Developing a mechanistic understanding of molecular electrocatalysts for CO2 reduction using infrared spectroelectrochemistry. Organometallics 33, 4550–4559. doi:10.1021/om500044a
Mallick, A., and Roy, S. (2018). Visible light driven catalytic gold decorated soft-oxometalate (SOM) based nanomotors for organic pollutant remediation. Nanoscale 10, 12713–12722. doi:10.1039/c8nr03534b
Mann, C. K., and Grunwald, E. (1959). Electroanalytical chemistry. J. Am. Chem. Soc. 81, 5266. doi:10.1021/ja01528a068
Manthiram, K., Beberwyck, B. J., and Alivisatos, A. P. (2014). Enhanced electrochemical methanation of carbon dioxide with a dispersible nanoscale copper catalyst. J. Am. Chem. Soc. 136, 13319–13325. doi:10.1021/ja5065284
Marcus, R. A. (1964). Chemical and electrochemical electron-transfer theory. Annu. Rev. Phys. Chem. 15, 155–196. doi:10.1146/annurev.pc.15.100164.001103
Marcus, R. A. (1959). On the theory of electrochemical and chemical electron transfer processes. Can. J. Chem. 37, 155–163. doi:10.1139/v59-022
Marko, J. A., Durgham, A., Bretz, S. L., and Liu, W. (2019). Electrochemical benzylic oxidation of C–H bonds. Chem. Commun. 55, 937–940. doi:10.1039/c8cc08768g
Martins, G. M., Shirinfar, B., Hardwick, T., Murtaza, A., and Ahmed, N. (2019). Organic electrosynthesis: Electrochemical alkyne functionalization. Catal. Sci. Technol. 9, 5868–5881. doi:10.1039/c9cy01312a
Maurin, A., and Robert, M. (2016). Noncovalent immobilization of a molecular iron-based electrocatalyst on carbon electrodes for selective, efficient CO2-to-CO conversion in water. J. Am. Chem. Soc. 138, 2492–2495. doi:10.1021/jacs.5b12652
Mccann, S. D., and Stahl, S. S. (2015). Copper-catalyzed aerobic oxidations of organic molecules: Pathways for two-electron oxidation with a four-electron oxidant and a one-electron redox-active catalyst. Acc. Chem. Res. 48, 1756–1766. doi:10.1021/acs.accounts.5b00060
Mcnicholas, B. J., Blakemore, J. D., Chang, A. B., Bates, C. M., Kramer, W. W., Grubbs, R. H., et al. (2016). Electrocatalysis of CO2 reduction in brush polymer ion gels. J. Am. Chem. Soc. 138, 11160–11163. doi:10.1021/jacs.6b08795
Meng, X., Zhang, Y., Luo, J., Wang, F., Cao, X., and Huang, S. (2020). Electrochemical oxidative oxydihalogenation of alkynes for the synthesis of α, α-dihaloketones. Org. Lett. 22, 1169–1174. doi:10.1021/acs.orglett.0c00052
Mistry, H., Varela, A. S., Bonifacio, C. S., Zegkinoglou, I., Sinev, I., Choi, Y.-W., et al. (2016). Highly selective plasma-activated copper catalysts for carbon dioxide reduction to ethylene. Nat. Commun. 7, 12123–12129. doi:10.1038/ncomms12123
Moeller, K. D. (2016). Anodic olefin coupling reactions: A mechanism driven approach to the development of new synthetic tools. Interface Mag. 25, 53–59. doi:10.1149/2.f07162if
Moeller, K. D. (1997). “Intramolecular carbon-carbon bond forming reactions at the anode,” in Electrochemistry VI electroorganic synthesis: Bond formation at anode and cathode (Germany: Springer), 49–86.
Moeller, K. D. (2000). Synthetic applications of anodic electrochemistry. Tetrahedron 49, 9527–9554. doi:10.1016/s0040-4020(00)00840-1
Möhle, S., Zirbes, M., Rodrigo, E., Gieshoff, T., Wiebe, A., and Waldvogel, S. R. (2018). Modern electrochemical aspects for the synthesis of value-added organic products. Angew. Chem. Int. Ed. 57, 6018–6041. doi:10.1002/anie.201712732
MorlanéS, N., Takanabe, K., and Rodionov, V. (2016). Simultaneous reduction of CO2 and splitting of H2O by a single immobilized cobalt phthalocyanine electrocatalyst. ACS Catal. 6, 3092–3095. doi:10.1021/acscatal.6b00543
Morofuji, T., Shimizu, A., and Yoshida, J.-I. (2014). Direct C–N coupling of imidazoles with aromatic and benzylic compounds via electrooxidative C–H functionalization. J. Am. Chem. Soc. 136, 4496–4499. doi:10.1021/ja501093m
Morofuji, T., Shimizu, A., and Yoshida, J.-I. (2015). Electrochemical intramolecular C-H amination: Synthesis of benzoxazoles and benzothiazoles. Chem. Eur. J. 21, 3211–3214. doi:10.1002/chem.201406398
Morofuji, T., Shimizu, A., and Yoshida, J. I. (2012). Metal-and chemical-oxidant-free C H/C H cross-coupling of aromatic compounds: The use of radical-cation pools. Angew. Chem. Int. Ed. Engl. 124, 7371–7374. doi:10.1002/ange.201202788
Nakata, K., Ozaki, T., Terashima, C., Fujishima, A., and Einaga, Y. (2014). High-yield electrochemical production of formaldehyde from CO2 and seawater. Angew. Chem. Int. Ed. 53, 871–874. doi:10.1002/anie.201308657
Narayan, R., Matcha, K., and Antonchick, A. P. (2015). Metal-free oxidative C-C bond formation through C-H bond functionalization H bond functionalization. Chem. Eur. J. 21, 14678–14693. doi:10.1002/chem.201502005-
Nikolaienko, P., Jentsch, M., Kale, A. P., Cai, Y., and Rueping, M. (2019). Electrochemical and scalable dehydrogenative C (sp 3)− H amination via remote hydrogen atom transfer in batch and continuous flow. Chem. Eur. J. 25, 7177–7184. doi:10.1002/chem.201806092
Nokami, T., Ohata, K., Inoue, M., Tsuyama, H., Shibuya, A., Soga, K., et al. (2008). Iterative molecular assembly based on the cation-pool method. Convergent synthesis of dendritic molecules. J. Am. Chem. Soc. 130, 10864–10865. doi:10.1021/ja803487q
Nutting, J. E., Rafiee, M., and Stahl, S. S. (2018). Tetramethylpiperidine N-oxyl (TEMPO), phthalimide N-oxyl (PINO), and related N-oxyl species: Electrochemical properties and their use in electrocatalytic reactions. Chem. Rev. 118, 4834–4885. doi:10.1021/acs.chemrev.7b00763
O'brien, A. G., Maruyama, A., Inokuma, Y., Fujita, M., Baran, P. S., and Blackmond, D. G. (2014). Radical C-H functionalization of heteroarenes under electrochemical control H functionalization of heteroarenes under electrochemical control. Angew. Chem. Int. Ed. 53, 11868–11871. doi:10.1002/anie.201407948-
Ogawa, K. A., and Boydston, A. J. (2015). Recent developments in organocatalyzed electroorganic chemistry. Chem. Lett. 44, 10–16. doi:10.1246/cl.140915
Ogibin, Y. N., Elinson, M. N., and Nikishin, G. I. (2009). Mediator oxidation systems in organic electrosynthesis. Russ. Chem. Rev. 78, 89–140. doi:10.1070/rc2009v078n02abeh003886
Oh, S., Gallagher, J. R., Miller, J. T., and Surendranath, Y. (2016). Graphite-conjugated rhenium catalysts for carbon dioxide reduction. J. Am. Chem. Soc. 138, 1820–1823. doi:10.1021/jacs.5b13080
Oh, Y., Vrubel, H., Guidoux, S., and Hu, X. (2014). Electrochemical reduction of CO 2 in organic solvents catalyzed by MoO 2. Chem. Commun. 50, 3878–3881. doi:10.1039/c3cc49262a
Okajima, M., Suga, S., Itami, K., and Yoshida, J.-I. (2005). Cation pool” method based on C− C bond dissociation. Effective generation of monocations and dications. J. Am. Chem. Soc. 127, 6930–6931. doi:10.1021/ja050414y
Osadchuk, I., Tamm, T., and Ahlquist, M. R. S. (2016). Reduced state of iridium PCP pincer complexes in electrochemical CO2 hydrogenation. ACS Catal. 6, 3834–3839. doi:10.1021/acscatal.6b01233
Poh, H. L., Sofer, Z., Luxa, J., and Pumera, M. (2014). Transition metal-depleted graphenes for electrochemical applications via reduction of CO2 by lithium. Small 10, 1529–1535. doi:10.1002/smll.201303002
Pollok, D., and Waldvogel, S. R. (2020). Electro-organic synthesis–a 21 st century technique. Chem. Sci. 11, 12386–12400. doi:10.1039/d0sc01848a
Popp, F. D., and Schultz, H. P. (1962). Electrolytic reduction of organic compounds. Chem. Rev. 62, 19–40. doi:10.1021/cr60215a002
Raciti, D., Livi, K. J., and Wang, C. (2015). Highly dense Cu nanowires for low-overpotential CO2 reduction. Nano Lett. 15, 6829–6835. doi:10.1021/acs.nanolett.5b03298
Rafiee, M., Konz, Z. M., Graaf, M. D., Koolman, H. F., and Stahl, S. S. (2018). Electrochemical oxidation of alcohols and aldehydes to carboxylic acids catalyzed by 4-acetamido-TEMPO: An alternative to “Anelli” and “Pinnick” oxidations. ACS Catal. 8, 6738–6744. doi:10.1021/acscatal.8b01640
Rafiee, M., Miles, K. C., and Stahl, S. S. (2015). Electrocatalytic alcohol oxidation with TEMPO and bicyclic nitroxyl derivatives: Driving force trumps steric effects. J. Am. Chem. Soc. 137, 14751–14757. doi:10.1021/jacs.5b09672
Rao, H., Schmidt, L. C., Bonin, J., and Robert, M. (2017). Visible-light-driven methane formation from CO 2 with a molecular iron catalyst. Nature 548, 74–77. doi:10.1038/nature23016
Rasul, S., Anjum, D. H., Jedidi, A., Minenkov, Y., Cavallo, L., and Takanabe, K. (2015). A highly selective copper–indium bimetallic electrocatalyst for the electrochemical reduction of aqueous CO2 to CO. Angew. Chem. Int. Ed. Engl. 127, 2174–2178. doi:10.1002/ange.201410233
Ren, D., Deng, Y., Handoko, A. D., Chen, C. S., Malkhandi, S., and Yeo, B. S. (2015). Selective electrochemical reduction of carbon dioxide to ethylene and ethanol on copper (I) oxide catalysts. ACS Catal. 5, 2814–2821. doi:10.1021/cs502128q
Reuillard, B., Ly, K. H., Rosser, T. E., Kuehnel, M. F., Zebger, I., and Reisner, E. (2017). Tuning product selectivity for aqueous CO2 reduction with a Mn (bipyridine)-pyrene catalyst immobilized on a carbon nanotube electrode. J. Am. Chem. Soc. 139, 14425–14435. doi:10.1021/jacs.7b06269
Riplinger, C., and Carter, E. A. (2015). Influence of weak Brønsted acids on electrocatalytic CO2 reduction by manganese and rhenium bipyridine catalysts. ACS Catal. 5, 900–908. doi:10.1021/cs501687n
Roberts, F. S., Kuhl, K. P., and Nilsson, A. (2015). High selectivity for ethylene from carbon dioxide reduction over copper nanocube electrocatalysts. Angew. Chem. Int. Ed. Engl. 127, 5179–5182. doi:10.1002/anie.201412214
Rosen, B. R., Werner, E. W., O’brien, A. G., and Baran, P. S. (2014). Total synthesis of dixiamycin B by electrochemical oxidation. J. Am. Chem. Soc. 136, 5571–5574. doi:10.1021/ja5013323
Rosen, J., Hutchings, G. S., Lu, Q., Forest, R. V., Moore, A., and Jiao, F. (2015a). Electrodeposited Zn dendrites with enhanced CO selectivity for electrocatalytic CO2 reduction. ACS Catal. 5, 4586–4591. doi:10.1021/acscatal.5b00922
Rosen, J., Hutchings, G. S., Lu, Q., Rivera, S., Zhou, Y., Vlachos, D. G., et al. (2015b). Mechanistic insights into the electrochemical reduction of CO2 to CO on nanostructured Ag surfaces. ACS Catal. 5, 4293–4299. doi:10.1021/acscatal.5b00840
Roy, N., Shibano, Y., Terashima, C., Katsumata, K. I., Nakata, K., Kondo, T., et al. (2016). Ionic liquid assisted selective and controlled electrochemical CO2 reduction at cu-modified boron-doped diamond electrode. ChemElectroChem 3, 1044–1047. doi:10.1002/celc.201600105
Ruan, Z., Huang, Z., Xu, Z., Mo, G., Tian, X., Yu, X.-Y., et al. (2019). Catalyst-free, direct electrochemical tri-and difluoroalkylation/cyclization: Access to functionalized oxindoles and quinolinones. Org. Lett. 21, 1237–1240. doi:10.1021/acs.orglett.9b00361
Ryan, M. C., Whitmire, L. D., Mccann, S. D., and Stahl, S. S. (2019). Copper/TEMPO redox redux: Analysis of PCET oxidation of TEMPOH by copper (II) and the reaction of TEMPO with copper (I). Inorg. Chem. 58, 10194–10200. doi:10.1021/acs.inorgchem.9b01326
Ryland, B. L., Mccann, S. D., Brunold, T. C., and Stahl, S. S. (2014). Mechanism of alcohol oxidation mediated by copper (II) and nitroxyl radicals. J. Am. Chem. Soc. 136, 12166–12173. doi:10.1021/ja5070137
Ryland, B. L., and Stahl, S. S. (2014). Practical aerobic oxidations of alcohols and amines with homogeneous copper/TEMPO and related catalyst systems. Angew. Chem. Int. Ed. 53, 8824–8838. doi:10.1002/anie.201403110
Sarfraz, S., Garcia-Esparza, A. T., Jedidi, A., Cavallo, L., and Takanabe, K. (2016). Cu–Sn bimetallic catalyst for selective aqueous electroreduction of CO2 to CO. ACS Catal. 6, 2842–2851. doi:10.1021/acscatal.6b00269
Sato, S., Saita, K., Sekizawa, K., Maeda, S., and Morikawa, T. (2018). Low-energy electrocatalytic CO2 reduction in water over Mn-complex catalyst electrode aided by a nanocarbon support and K+ cations. ACS Catal. 8, 4452–4458. doi:10.1021/acscatal.8b01068
Sauermann, N., Mei, R., and Ackermann, L. (2018). Electrochemical C− H amination by cobalt catalysis in a renewable solvent. Angew. Chem. Int. Ed. Engl. 57, 5184–5188. doi:10.1002/ange.201802206
Sauermann, N., Meyer, T. H., Tian, C., and Ackermann, L. (2017). Electrochemical cobalt-catalyzed C–H oxygenation at room temperature. J. Am. Chem. Soc. 139, 18452–18455. doi:10.1021/jacs.7b11025
Sawyer, D. T., Sobkowiak, A., and Roberts, J. L. (1995). Electrochemistry for chemists. United States: Wiley.
Schäfer, H. J. (1981). Anodic and cathodic CC-bond formation. Angew. Chem. Int. Ed. Engl. 20, 911–934. doi:10.1002/anie.198109111
Schäfer, H. J. (2014). Carbon–carbon bond formation via electron transfer: Anodic coupling. ChemCatChem 6, 2792–2795. doi:10.1002/cctc.201402366
Schäfer, H. J. (2011). Contributions of organic electrosynthesis to green chemistry. Comptes Rendus Chim. 14, 745–765. doi:10.1016/j.crci.2011.01.002
Schäfer, H. J., Harenbrock, M., Klocke, E., Plate, M., and Weiper-Idelmann, A. (2007). Electrolysis for the benign conversion of renewable feedstocks. Pure Appl. Chem. 79, 2047–2057. doi:10.1351/pac200779112047
Schulz, L., Enders, M., Elsler, B., Schollmeyer, D., Dyballa, K. M., Franke, R., et al. (2017). Reagent-and metal-free anodic C− C cross-coupling of aniline derivatives. Angew. Chem. Int. Ed. 56, 4877–4881. doi:10.1002/anie.201612613
Schulz, L., and Waldvogel, S. R. (2019). Solvent control in electro-organic synthesis. Synlett 30, 275–286. doi:10.1055/s-0037-1610303
Sen, S., Liu, D., and Palmore, G. T. R. (2014). Electrochemical reduction of CO2 at copper nanofoams. ACS Catal. 4, 3091–3095. doi:10.1021/cs500522g
Senboku, H., Hayama, M., and Matsuno, H. (2022). Electrochemical Friedel–Crafts-type amidomethylation of arenes by a novel electrochemical oxidation system using a quasi-divided cell and trialkylammonium tetrafluoroborate. Beilstein J. Org. Chem. 18, 1040–1046. doi:10.3762/bjoc.18.105
Sequeira, C., and Santos, D. (2009). Electrochemical routes for industrial synthesis. J. Braz. Chem. Soc. 20, 387–406. doi:10.1590/s0103-50532009000300002
Shao, X., Zheng, Y., Tian, L., Martín-Torres, I., Echavarren, A. M., and Wang, Y. (2019). Decarboxylative csp<sup>3</sup>–N bond formation by electrochemical oxidation of amino acids. Org. Lett. 21, 9262–9267. doi:10.1021/acs.orglett.9b03696
Sharma, P. P., Wu, J., Yadav, R. M., Liu, M., Wright, C. J., Tiwary, C. S., et al. (2015). Nitrogen-doped carbon nanotube arrays for high-efficiency electrochemical reduction of CO2: On the understanding of defects, defect density, and selectivity. Angew. Chem. Int. Ed. Engl. 127, 13905–13909. doi:10.1002/ange.201506062
Shen, J., Kolb, M. J., Gottle, A. J., and Koper, M. T. (2016). DFT study on the mechanism of the electrochemical reduction of CO2 catalyzed by cobalt porphyrins. J. Phys. Chem. C 120, 15714–15721. doi:10.1021/acs.jpcc.5b10763
Shen, J., Kortlever, R., Kas, R., Birdja, Y. Y., Diaz-Morales, O., Kwon, Y., et al. (2015). Electrocatalytic reduction of carbon dioxide to carbon monoxide and methane at an immobilized cobalt protoporphyrin. Nat. Commun. 6, 8177–8178. doi:10.1038/ncomms9177
Shi, C., Chan, K., Yoo, J. S., and Nørskov, J. K. (2016). Barriers of electrochemical CO2 reduction on transition metals. Org. Process Res. Dev. 20, 1424–1430. doi:10.1021/acs.oprd.6b00103
Shono, T. (1984). Electroorganic chemistry in organic synthesis. Tetrahedron 40, 811–850. doi:10.1016/s0040-4020(01)91472-3
Singh, M. R., Kwon, Y., Lum, Y., Ager, J. W., and Bell, A. T. (2016). Hydrolysis of electrolyte cations enhances the electrochemical reduction of CO2 over Ag and Cu. J. Am. Chem. Soc. 138, 13006–13012. doi:10.1021/jacs.6b07612
Song, Y., Peng, R., Hensley, D. K., Bonnesen, P. V., Liang, L., Wu, Z., et al. (2016). High-selectivity electrochemical conversion of CO2 to ethanol using a copper nanoparticle/N-doped graphene electrode. ChemistrySelect 1, 6055–6061. doi:10.1002/slct.201601169
Sperry, J. B., and Wright, D. L. (2006). The application of cathodic reductions and anodic oxidations in the synthesis of complex molecules. Chem. Soc. Rev. 35, 605–621. doi:10.1039/b512308a
Stanton, C. J., Vandezande, J. E., Majetich, G. F., Schaefer, H. F., and Agarwal, J. (2016). Mn-NHC electrocatalysts: Increasing π acidity lowers the reduction potential and increases the turnover frequency for CO2 reduction. Inorg. Chem. 55, 9509–9512. doi:10.1021/acs.inorgchem.6b01657
Steckhan, E. (1986). Indirect electroorganic syntheses—a modern chapter of organic electrochemistry [new synthetic methods (59)]. Angew. Chem. Int. Ed. Engl. 25, 683–701. doi:10.1002/anie.198606831
Steckhan, E. (1987). “Organic syntheses with electrochemically regenerable redox systems,” in Electrochemistry I (Germany: Springer), 1–69.
Steves, J. E., and Stahl, S. S. (2013). Copper (I)/ABNO-catalyzed aerobic alcohol oxidation: Alleviating steric and electronic constraints of Cu/TEMPO catalyst systems. J. Am. Chem. Soc. 135, 15742–15745. doi:10.1021/ja409241h
Studt, F., Sharafutdinov, I., Abild-Pedersen, F., Elkjær, C. F., Hummelshøj, J. S., Dahl, S., et al. (2014). Discovery of a Ni-Ga catalyst for carbon dioxide reduction to methanol. Nat. Chem. 6, 320–324. doi:10.1038/nchem.1873
Su, P., Iwase, K., Nakanishi, S., Hashimoto, K., and Kamiya, K. (2016). Nickel-nitrogen-modified graphene: An efficient electrocatalyst for the reduction of carbon dioxide to carbon monoxide. Small 12, 6083–6089. doi:10.1002/smll.201602158
Sun, L., Ramesha, G. K., Kamat, P. V., and Brennecke, J. F. (2014). Switching the reaction course of electrochemical CO2 reduction with ionic liquids. Langmuir 30, 6302–6308. doi:10.1021/la5009076
Sun, X., Ma, H.-X., Mei, T.-S., Fang, P., and Hu, Y. (2019). Electrochemical radical formyloxylation–bromination, − chlorination, and− trifluoromethylation of alkenes. Org. Lett. 21, 3167–3171. doi:10.1021/acs.orglett.9b00867
Sun, X., Zhu, Q., Kang, X., Liu, H., Qian, Q., Zhang, Z., et al. (2016). Molybdenum–bismuth bimetallic chalcogenide nanosheets for highly efficient electrocatalytic reduction of carbon dioxide to methanol. Angew. Chem. Int. Ed. Engl. 128, 6883–6887. doi:10.1002/ange.201603034
Sun, Y., Li, X., Yang, M., Xu, W., Xie, J., and Ding, M. (2020). Highly selective electrocatalytic oxidation of benzyl C–H using water as safe and sustainable oxygen source. Green Chem. 22, 7543–7551. doi:10.1039/d0gc01871f
Tang, S., Wang, D., Liu, Y., Zeng, L., and Lei, A. (2018a). Cobalt-catalyzed electrooxidative CH/NH [4+ 2] annulation with ethylene or ethyne. Nat. Commun. 9, 798–804. doi:10.1038/s41467-018-03246-4
Tang, S., Wang, S., Liu, Y., Cong, H., and Lei, A. (2018b). Electrochemical oxidative C− H amination of phenols: Access to triarylamine derivatives. Angew. Chem. Int. Ed. Engl. 130, 4827–4831. doi:10.1002/ange.201800240
Therrien, J. A., Wolf, M. O., and Patrick, B. O. (2014). Electrocatalytic reduction of CO2 with palladium bis-N-heterocyclic carbene pincer complexes. Inorg. Chem. 53, 12962–12972. doi:10.1021/ic502056w
Therrien, J. A., Wolf, M. O., and Patrick, B. O. (2015). Polyannulated bis (N-heterocyclic carbene) palladium pincer complexes for electrocatalytic CO2 reduction. Inorg. Chem. 54, 11721–11732. doi:10.1021/acs.inorgchem.5b01698
Tian, C., Massignan, L., Meyer, T. H., and Ackermann, L. (2018). Electrochemical C− H/N− H activation by water-tolerant cobalt catalysis at room temperature. Angew. Chem. Int. Ed. Engl. 130, 2407–2411. doi:10.1002/ange.201712647
Tojo, G., and Fernández, M. (2010a). Basic reactions in organic synthesis. Oxidation of primary Alcohols to carboxylic acids. New York, NY: Springer.
Tojo, G., and Fernández, M. (2010b). Oxidation of primary alcohols to carboxylic acids basic reactions in organic synthesis. New York: Springer.
Torelli, D. A., Francis, S. A., Crompton, J. C., Javier, A., Thompson, J. R., Brunschwig, B. S., et al. (2016). Nickel–gallium-catalyzed electrochemical reduction of CO2 to highly reduced products at low overpotentials. ACS Catal. 6, 2100–2104. doi:10.1021/acscatal.5b02888
Tory, J., Setterfield-Price, B., Dryfe, R. A., and Hartl, F. (2015). [M(CO)4(2, 2′-bipyridine)] (M=Cr, Mo, W) complexes as efficient catalysts for electrochemical reduction of CO2 at a gold electrode. ChemElectroChem 2, 213–217. doi:10.1002/celc.201402282
Varela, A. S., Ranjbar Sahraie, N., Steinberg, J., Ju, W., Oh, H. S., and Strasser, P. (2015). Metal-doped nitrogenated carbon as an efficient catalyst for direct CO2 electroreduction to CO and hydrocarbons. Angew. Chem. Int. Ed. Engl. 127, 10758–10762. doi:10.1002/anie.201502099
Vijh, A., and Conway, B. (1967). Electrode kinetic aspects of the Kolbe reaction. Chem. Rev. 67, 623–664. doi:10.1021/cr60250a003
Vock, S., Tschulik, K., Uhlemann, M., Hengst, C., Fähler, S., Schultz, L., et al. (2015). Magnetostatic nearest neighbor interactions in a Co48Fe52 nanowire array probed by in-field magnetic force microscopy. J. Appl. Phys. 118, 233901. doi:10.1063/1.4937275
Vollmer, M. V., Machan, C. W., Clark, M. L., Antholine, W. E., Agarwal, J., Schaefer, H. F., et al. (2015). Synthesis, spectroscopy, and electrochemistry of (α-Diimine) M (CO) 3Br, M= Mn, Re, complexes: Ligands isoelectronic to bipyridyl show differences in CO2 reduction. Organometallics 34, 3–12. doi:10.1021/om500838z
Waldvogel, S. R., Dörr, M., Röckl, J., Rein, J., and Schollmeyer, D. (2020). Electrochemical C-H functionalization of (hetero) arenes–optimized by DoE. Chem. Eur. J. 26, 10195–10198. doi:10.1002/chem.202001171
Waldvogel, S. R., Lips, S., Selt, M., Riehl, B., and Kampf, C. J. (2018). Electrochemical arylation reaction. Chem. Rev. 118, 6706–6765. doi:10.1021/acs.chemrev.8b00233
Waldvogel, S. R., and Selt, M. (2016). Electrochemical allylic oxidation of olefins: Sustainable and safe. Angew. Chem. Int. Ed. 55, 12578–12580. doi:10.1002/anie.201606727
Walsh, J. J., Neri, G., Smith, C. L., and Cowan, A. J. (2014). Electrocatalytic CO 2 reduction with a membrane supported manganese catalyst in aqueous solution. Chem. Commun. 50, 12698–12701. doi:10.1039/c4cc06404f
Walsh, J. J., Smith, C. L., Neri, G., Whitehead, G. F., Robertson, C. M., and Cowan, A. J. (2015). Improving the efficiency of electrochemical CO2 reduction using immobilized manganese complexes. Faraday Discuss. 183, 147–160. doi:10.1039/c5fd00071h
Wang, F., Rafiee, M., and Stahl, S. S. (2018). Electrochemical functional-group-tolerant shono-type oxidation of cyclic carbamates enabled by aminoxyl mediators. Angew. Chem. Int. Ed. Engl. 130, 6796–6800. doi:10.1002/ange.201803539
Wang, F., and Stahl, S. S. (2019). Merging photochemistry with electrochemistry: Functional-group tolerant electrochemical amination of C (sp3)− H bonds. Angew. Chem. Int. Ed. Engl. 58, 6451–6456. doi:10.1002/ange.201813960
Wang, J.-H., Lei, T., Nan, X.-L., Wu, H.-L., Li, X.-B., Chen, B., et al. (2019a). Regioselective ortho amination of an aromatic C–H bond by trifluoroacetic acid via electrochemistry. Org. Lett. 21, 5581–5585. doi:10.1021/acs.orglett.9b01910
Wang, P., Yang, Z., Wu, T., Xu, C., Wang, Z., and Lei, A. (2019b). Electrochemical oxidative C (sp3)− H/N− H cross-coupling for N-mannich bases with hydrogen evolution. ChemSusChem 12, 3073–3077. doi:10.1002/cssc.201802676
Wang, Y., Deng, L., Wang, X., Wu, Z., Wang, Y., and Pan, Y. (2019c). Electrochemically promoted nickel-catalyzed carbon–sulfur bond formation. ACS Catal. 9, 1630–1634. doi:10.1021/acscatal.8b04633
Wang, Z., Yang, G., Zhang, Z., Jin, M., and Yin, Y. (2016). Selectivity on etching: Creation of high-energy facets on copper nanocrystals for CO2 electrochemical reduction. ACS Nano 10, 4559–4564. doi:10.1021/acsnano.6b00602
Wannakao, S., Artrith, N., Limtrakul, J., and Kolpak, A. M. (2015). Engineering transition-metal-coated tungsten carbides for efficient and selective electrochemical reduction of CO2 to methane. ChemSusChem 8, 2745–2751. doi:10.1002/cssc.201500245
Weetal, H. H., Forsyth, B. D., and Hertl, W. (1985). A direct fuel cell for the production of electricity from lignin. Biotechnol. Bioeng. 27, 972–979. doi:10.1002/bit.260270707
Weibel, D. B., Boulatov, R., Lee, A., Ferrigno, R., and Whitesides, G. M. (2005). Modeling the anodic half-cell of a low-temperature coal fuel cell. Angew. Chem. Int. Ed. Engl. 117, 5682–5686. doi:10.1002/anie.200501192
Weng, Z., Jiang, J., Wu, Y., Wu, Z., Guo, X., Materna, K. L., et al. (2016). Electrochemical CO2 reduction to hydrocarbons on a heterogeneous molecular Cu catalyst in aqueous solution. J. Am. Chem. Soc. 138, 8076–8079. doi:10.1021/jacs.6b04746
White, R. E. (2012). Electrochemical cell design. Berlin, Germany: Springer Science & Business Media.
Wiebe, A., Gieshoff, T., Möhle, S., Rodrigo, E., Zirbes, M., and Waldvogel, S. R. (2018). Electrifying organic synthesis. Angew. Chem. Int. Ed. 57, 5594–5619. doi:10.1002/anie.201711060
Wiebe, A., Lips, S., Schollmeyer, D., Franke, R., and Waldvogel, S. R. (2017). Single and twofold metal-and reagent-free anodic C− C cross-coupling of phenols with thiophenes. Angew. Chem. Int. Ed. 56, 14727–14731. doi:10.1002/anie.201708946
Won, D. H., Choi, C. H., Chung, J., Chung, M. W., Kim, E. H., and Woo, S. I. (2015). Rational design of a hierarchical tin dendrite electrode for efficient electrochemical reduction of CO2. ChemSusChem 8, 3092–3098. doi:10.1002/cssc.201500694
Won, D. H., Shin, H., Koh, J., Chung, J., Lee, H. S., Kim, H., et al. (2016). Highly efficient, selective, and stable CO2 electroreduction on a hexagonal Zn catalyst. Angew. Chem. Int. Ed. Engl. 55, 9443–9446. doi:10.1002/ange.201602888
Wu, J., Liu, M., Sharma, P. P., Yadav, R. M., Ma, L., Yang, Y., et al. (2016a). Incorporation of nitrogen defects for efficient reduction of CO2 via two-electron pathway on three-dimensional graphene foam. Nano Lett. 16, 466–470. doi:10.1021/acs.nanolett.5b04123
Wu, J., Risalvato, F. G., Ma, S., and Zhou, X.-D. (2014). Electrochemical reduction of carbon dioxide III. The role of oxide layer thickness on the performance of Sn electrode in a full electrochemical cell. J. Mat. Chem. A 2, 1647–1651. doi:10.1039/c3ta13544f
Wu, J., Yadav, R. M., Liu, M., Sharma, P. P., Tiwary, C. S., Ma, L., et al. (2015). Achieving highly efficient, selective, and stable CO2 reduction on nitrogen-doped carbon nanotubes. ACS Nano 9, 5364–5371. doi:10.1021/acsnano.5b01079
Wu, W., Liu, W., Mu, W., and Deng, Y. (2016b). Polyoxymetalate liquid-catalyzed polyol fuel cell and the related photoelectrochemical reaction mechanism study. J. Power Sources 318, 86–92. doi:10.1016/j.jpowsour.2016.03.074
Xiang, D., Magana, D., and Dyer, R. B. (2014). CO2 reduction catalyzed by mercaptopteridine on glassy carbon. J. Am. Chem. Soc. 136, 14007–14010. doi:10.1021/ja5081103
Xiao, H., Cheng, T., Goddard, W. A., and Sundararaman, R. (2016). Mechanistic explanation of the pH dependence and onset potentials for hydrocarbon products from electrochemical reduction of CO on Cu (111). J. Am. Chem. Soc. 138, 483–486. doi:10.1021/jacs.5b11390
Xu, F., Li, Y.-J., Huang, C., and Xu, H.-C. (2018). Ruthenium-catalyzed electrochemical dehydrogenative alkyne annulation. ACS Catal. 8, 3820–3824. doi:10.1021/acscatal.8b00373
Yadav, V., and Purkait, M. (2015). Electrochemical studies for CO2 reduction using synthesized Co3O4 (Anode) and Cu2O (Cathode) as electrocatalysts. Energy fuels. 29, 6670–6677. doi:10.1021/acs.energyfuels.5b01656
Yang, H., Shen, Y., Xiao, Z., Liu, C., Yuan, K., and Ding, Y. (2020). The direct trifluoromethylsilylation and cyanosilylation of aldehydes via an electrochemically induced intramolecular pathway. Chem. Commun. 56, 2435–2438. doi:10.1039/c9cc08975f
Yang, Q.-L., Wang, X.-Y., Lu, J.-Y., Zhang, L.-P., Fang, P., and Mei, T.-S. (2018a). Copper-catalyzed electrochemical C–H amination of arenes with secondary amines. J. Am. Chem. Soc. 140, 11487–11494. doi:10.1021/jacs.8b07380
Yang, Q.-L., Xing, Y.-K., Wang, X.-Y., Ma, H.-X., Weng, X.-J., Yang, X., et al. (2019). Electrochemistry-enabled Ir-catalyzed vinylic C–H functionalization. J. Am. Chem. Soc. 141, 18970–18976. doi:10.1021/jacs.9b11915
Yang, Q. L., Fang, P., and Mei, T. S. (2018b). Recent advances in organic electrochemical C—H functionalization. Chin. J. Chem. 36, 338–352. doi:10.1002/cjoc.201700740
Yoshida, J.-I., Kataoka, K., Horcajada, R., and Nagaki, A. (2008). Modern strategies in electroorganic synthesis. Chem. Rev. 108, 2265–2299. doi:10.1021/cr0680843
Yuan, Y., Chen, Y., Tang, S., Huang, Z., and Lei, A. (2018). Electrochemical oxidative oxysulfenylation and aminosulfenylation of alkenes with hydrogen evolution. Sci. Adv. 4, eaat5312. doi:10.1126/sciadv.aat5312
Zhang, L., Zhang, G., Wang, P., Li, Y., and Lei, A. (2018a). Electrochemical oxidation with lewis-acid catalysis leads to trifluoromethylative difunctionalization of alkenes using CF3SO2Na. Org. Lett. 20, 7396–7399. doi:10.1021/acs.orglett.8b03081
Zhang, S., Kang, P., and Meyer, T. J. (2014a). Nanostructured tin catalysts for selective electrochemical reduction of carbon dioxide to formate. J. Am. Chem. Soc. 136, 1734–1737. doi:10.1021/ja4113885
Zhang, S., Kang, P., Ubnoske, S., Brennaman, M. K., Song, N., House, R. L., et al. (2014b). Polyethylenimine-enhanced electrocatalytic reduction of CO2 to formate at nitrogen-doped carbon nanomaterials. J. Am. Chem. Soc. 136, 7845–7848. doi:10.1021/ja5031529
Zhang, X., Wang, C., Jiang, H., and Sun, L. (2018b). Convenient synthesis of selenyl-indoles via iodide ion-catalyzed electrochemical C–H selenation. Chem. Commun. 54, 8781–8784. doi:10.1039/c8cc04543g
Zhang, X., Wu, Z., Zhang, X., Li, L., Li, Y., Xu, H., et al. (2017). Highly selective and active CO 2 reduction electrocatalysts based on cobalt phthalocyanine/carbon nanotube hybrid structures. Nat. Commun. 8, 14675–14678. doi:10.1038/ncomms14675
Zhang, Y.-J., Sethuraman, V., Michalsky, R., and Peterson, A. A. (2014c). Competition between CO2 reduction and H2 evolution on transition-metal electrocatalysts. ACS Catal. 4, 3742–3748. doi:10.1021/cs5012298
Zhang, Z., Chi, M., Veith, G. M., Zhang, P., Lutterman, D. A., Rosenthal, J., et al. (2016). Rational design of Bi nanoparticles for efficient electrochemical CO2 reduction: The elucidation of size and surface condition effects. ACS Catal. 6, 6255–6264. doi:10.1021/acscatal.6b01297
Zhang, Z., Zhang, L., Cao, Y., Li, F., Bai, G., Liu, G., et al. (2019). Mn-mediated electrochemical trifluoromethylation/C (sp2)–H functionalization cascade for the synthesis of azaheterocycles. Org. Lett. 21, 762–766. doi:10.1021/acs.orglett.8b04010
Zheng, Q., Liu, C. F., Chen, J., and Rao, G. W. (2020). C–H functionalization of aromatic amides. Adv. Synth. Catal. 362, 1406–1446. doi:10.1002/adsc.201901158
Zhu, W., Zhang, Y.-J., Zhang, H., Lv, H., Li, Q., Michalsky, R., et al. (2014). Active and selective conversion of CO2 to CO on ultrathin Au nanowires. J. Am. Chem. Soc. 136, 16132–16135. doi:10.1021/ja5095099
Keywords: electrocatalysis, organic reaction, CO2 reduction, kinetics, electrochemical techniques and methods
Citation: Lodh J, Paul S, Sun H, Song L, Schöfberger W and Roy S (2023) Electrochemical organic reactions: A tutorial review. Front. Chem. 10:956502. doi: 10.3389/fchem.2022.956502
Received: 30 May 2022; Accepted: 07 December 2022;
Published: 10 January 2023.
Edited by:
Yuvaraj Hunge, Daegu Gyeongbuk Institute of Science and Technology (DGIST), South KoreaReviewed by:
Bidhan Pandit, Universidad Carlos III de Madrid, SpainHani Nasser Abdelhamid, Assiut University, Egypt
Copyright © 2023 Lodh, Paul, Sun, Song, Schöfberger and Roy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wolfgang Schöfberger, d29sZmdhbmcuc2Nob2ZiZXJnZXJAamt1LmF0; Soumyajit Roy, cy5yb3lAaWlzZXJrb2wuYWMuaW4=