 Mariya Novakova
Mariya Novakova Anupama Das
Anupama Das Catherine Alex2
Catherine Alex2 Alexei V. Demchenko
Alexei V. Demchenko- 1Department of Chemistry, Saint Louis University, St. Louis, MO, United States
- 2Department of Chemistry and Biochemistry, University of Missouri—St. Louis, One University Boulevard, St. Louis, MO, United States
Presented herein is a streamlined synthesis of building blocks of a rare sugar D-altrosamine. Also investigated was the glycosylation of different glycosyl acceptors with differentially protected altrosamine donors. High facial stereoselectivity was achieved with 3-O-picoloyl donors and reactive glycosyl acceptors via the H-bond-mediated aglycone delivery (HAD) pathway. In contrast, glycosidations of the altrosamine donor equipped with the 3-O-benzoyl group were poorly stereoselective.
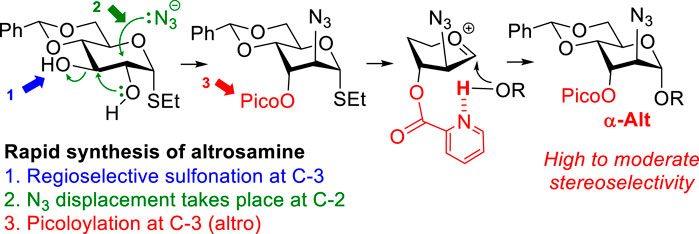
GRAPHICAL ABSTRACT
Introduction
Due to significant progress in recent years, many glycans can now be obtained by using chemical methods and automated platforms (Panza et al., 2018). However, the availability of selectively protected sugar building blocks remains scarce, which hampers the scientific progress in the area of carbohydrate synthesis. Despite general improvements in the application of protecting group strategies in the mainstream carbohydrate research (Polyakova et al., 2015; Jager and Minnaard, 2016; Ágoston et al., 2016; Kulkarni et al., 2018; Volbeda et al., 2019; Wang and Demchenko, 2019), building blocks for the introduction of uncommon (rare or unnatural) sugars remain largely underdeveloped or not available at all (Emmadi and Kulkarni, 2014; Sanapala and Kulkarni, 2016a; Sanapala and Kulkarni, 2016b; Behera and Kulkarni, 2018; Wang et al., 2020).
Early reports for the synthesis of rare sugar D-altrose relied on the degradation of heptuloses (Ritchmyer et al., 1939) or modification of fructose (Araujo et al., 2012) among others. The synthesis of D-altrosamine could be achieved from 2,3-anhydroaltrose that was obtained from D-glucose precursors via a multi-step protocol (Vega-Perez et al., 1995; Nilsson and Norberg, 2000; Chiu et al., 2007; Shrestha et al., 2022). These building blocks have previously been used as synthons to access derivatized rare sugars. Nevertheless, altrosamine remains prohibitively expensive to be used as the starting material both for laboratory and industrial applications. Reported herein is a streamlined and scalable procedure for the synthesis of D-altrosamine building blocks. Also investigated was the first glycosidation of altrosamine donors with standard glycosyl acceptors.
Results and discussion
Previously, we developed methods to obtain mannosamine building blocks from methyl 4,6-O-benzylidene-α-D-glucopyranoside 1 (Alex et al., 2020a; Alex et al., 2020b). High regioselectivity of sulfonation of diol 1 with triflic anhydride at C-2 (Knapp et al., 1990) was the key to success in obtaining 2-azido-2-deoxy-d-mannopyranoside 2 (Scheme 1A). This reaction proceeded via a stereospecific nucleophilic displacement at C-2 with sodium azide in DMF (A) (Knapp et al., 1992). Compound 2 was then subjected to sequential acetolysis and a leaving group introduction to afford thioglycoside 3. The latter was protected to afford 3-OH derivative 4, which was then picoloylated to afford donor 5 or benzoylated to afford donor 6. These donors were then used for stereoselective introduction of mannosides.
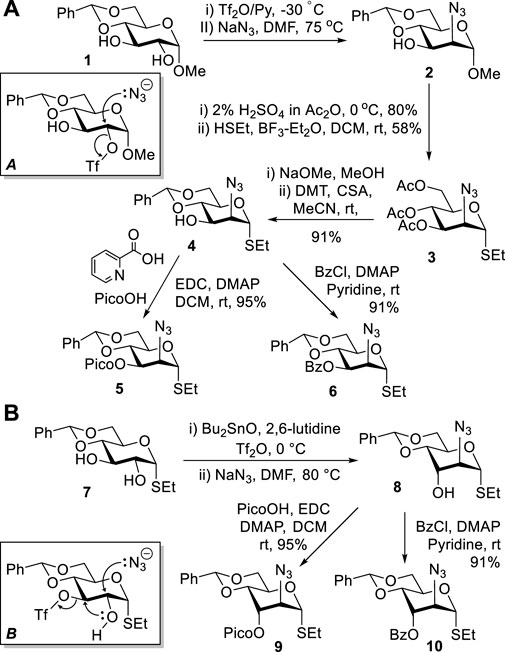
SCHEME 1. Previous synthesis of ManN3 building blocks 2–6 and the direct synthesis of D-altrosamine donors 9 and 10 from the D-gluco precursor 7.
However, the synthesis of 5 and 6 remained somewhat tedious and required a lengthy and multi-step process to arrive at the desired compounds. In an effort to streamline the approach, we attempted to carry out the synthesis from thioglycoside 7 (Takeo et al., 1993) instead of the previously employed methyl glycoside 1. All efforts to sulfonate thioglycoside 7 at position C-2 have failed, regardless of whether this reaction was performed in the presence of dibutyl tin oxide or not (Scheme 1B). Sulfonation was consistently directed to the C-3 position, and the subsequent nucleophilic displacement resulted in a cascade reaction with the anticipated pathway (B). Presumably, first, 2,3-cyclization would occur, and the resulting 2,3-epoxide (Walvoort et al., 2011) would then open upon the nucleophilic attack by N3 to afford D-altro-configured amino sugar 8. This discovery led to a straightforward one-pot protocol for the synthesis of this rare sugar series and derivatives thereof. To elaborate upon this finding, we protected the 3-OH derivative 8 with picoloyl or benzoyl groups to afford glycosyl donors 9 and 10 in good yields of 95 and 91%, respectively (Scheme 1B).
We have previously reported that picolinyl or picoloyl (Pico) protecting groups at remote C-3, C-4, or C-6 positions of pyranose sugars can provide high facial syn-stereoselectivity in glycosylations (Yasomanee and Demchenko, 2012). This is due to the H-bond-mediated aglycone delivery (HAD) reaction pathway (Mannino et al., 2021). For the HAD reaction to take place, the Pico nitrogen of the glycosyl donor has to establish a hydrogen bond with the hydroxyl group of the glycosyl acceptor. Upon activation of the glycosyl donor, the glycosyl acceptor forms the glycosidic bond, which is syn with respect to the Pico substituent.
As previously proposed by our group (Alex et al., 2020a; Alex and Demchenko, 2021; Alex et al., 2021), the mannosamine donor equipped with the 3-O-Pico group will favor formation of the β-linked mannoside, as shown in Scheme 2. In the case of 3-O-benzoylated (3-Bz) mannosamine donor, excellent α-stereoselectivity was obtained. This result was explained by the occurrence of a remote participation of the 3-O-benzoyl group. Our expectations for glycosyl donors of the D-altrosamine series were opposed to those observed with D-manno donors. This is because of the orientation of the substituent at C-3. Thus, we anticipated that 3-Pico donors will provide preferential α-stereoselectivity whereas 3-Bz donor will be β-altro stereoselective due to the remote participation effect (Scheme 2).
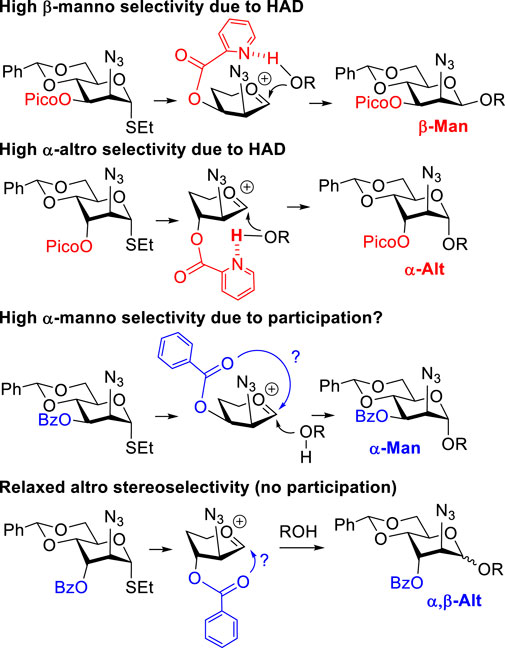
SCHEME 2. H-bond-mediated aglycone delivery and participation for mannosides and altrosides.
We hypothesized that the remote 3-O-Pico group in donor 9 will act as an H-bond acceptor for the incoming nucleophile (hydroxyl group of the glycosyl acceptor). As a result, the formation of α-altrosides was anticipated. With that, we set up a series of glycosylations with common sugar acceptors 11–14 (Ranade et al., 2010), as shown in Table 1. When glycosyl donor 9 was coupled with 6-OH acceptor 11 in the presence of NIS/TfOH in 1,2-dichloroethane (1,2-DCE), the expected disaccharide 15 was obtained in 98% and complete α-altro stereoselectivity (entry 1). Since the HAD reaction pathway is absent in 3-Bz donor 10, an opposing stereoselectivity was anticipated. However, the reaction between 3-Bz donor 10 and acceptor 11 yielded the corresponding disaccharide 16 in a high yield of 98%, albeit with modest stereoselectivity with α-anomer still favored (α/β = 3.0/1, entry 2). Both reactions were completed in 1 h. This result implies that the participation of the 3-Bz group seen for mannosides, (Crich et al., 2000) mannosamine glycosides, (Alex et al., 2020a; Alex et al., 2020b; Alex and Demchenko, 2021; Alex et al., 2021; Alex and Demchenko, 2022), and other sugar series, (Ustyuzhanina et al., 2006; Baek et al., 2009; Komarova et al., 2013; Komarova et al., 2014; Komarova et al., 2016) is practically ineffective in case of D-altro-configured sugars.

TABLE 1. Glycosidation of donors 9 and 10 with glycosyl acceptors 11–14.
When the reaction of 4-OH glycosyl acceptor 12 was conducted with glycosyl donor 9 under the promotion of NIS/TfOH, disaccharide 17 was obtained in 71% yield, albeit with poor stereoselectivity (α/β = 1.7/1, entry 3). When 3-Bz donor 10 was glycosidated with acceptor 12, disaccharide 18 was obtained in 84% yield with no stereoselectivity (α/β = 1.0/1, entry 4). A very similar trend was achieved in the reaction of 3-OH acceptor 13 with glycosyl donors 9 and 10. The corresponding disaccharides 19 and 20 were achieved in good yields of 63–80% but with modest stereoselectivity in both cases (α/β = 2.2–2.5/1, entries 5–6). When glycosyl donor 9 was glycosidated with 2-OH acceptor 14, disaccharide 21 was isolated in a good yield of 65% and with complete α-altro selectivity (entry 7). The reaction between 3-Bz glycosyl donor 10 and glycosyl acceptor 14 produced the corresponding disaccharide 22 in 66% yield, albeit with poor stereoselectivity (α/β = 1.5/1, entry 2). All glycosylations of secondary acceptors 12–14 were completed in 2 h. These results confirm the general trend previously seen in some HAD reactions wherein poorly nucleophilic acceptors provided lower stereoselectivity.
To confirm the stereoselectivity observed, we removed 3-ester groups from disaccharides 15 and 16 (Scheme 3). The comparison of the NMR data on the resulting disaccharide 23 ultimately confirmed the preferential α-altrosamine configuration of disaccharides obtained with 3-Bz donor 10.
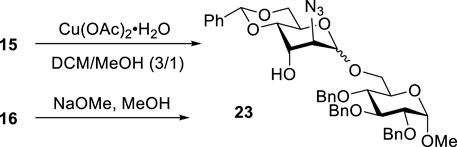
SCHEME 3. Deprotection of 15 and 16 to understand stereoselectivity.
In conclusion, a useful method for the synthesis of building blocks of D-altrosamine is reported. Also investigated was a stereoselective synthesis of α-glycosides of altrosamine. High facial stereoselectivity achieved with 3-O-picoloyl donors with reactive (6-OH and 2-OH) glycosyl acceptors was explained by the occurrence of the HAD reaction pathway. Stereoselectivity of reactions with 3-OH and 4-OH glycosyl acceptors was low because the HAD reaction was known to be less effective with poorly reactive acceptors (Yasomanee and Demchenko, 2012). Glycosidations of altrosamine donors equipped with the 3-O-benzoyl group were poorly stereoselective. Further investigation of these reactions is currently underway in our laboratory.
Experiment
In general, the reactions were performed using commercial reagents. The ACS grade solvents used for reactions were purified and dried in accordance with the standard procedures. Column chromatography was performed on silica gel 60 (70–230 mesh), and reactions were monitored by TLC on Kiesel gel 60 F254. The compounds were detected by examination under UV light and by charring with 10% sulfuric acid in methanol. The solvents were removed under reduced pressure at <40°C. ClCH2.CH2Cl (1,2-DCE) was distilled from CaH2 directly prior to application. Anhydrous DMF was used. Molecular sieves (4 Å), used for reactions, were crushed and activated in vacuo at 390°C during 8 h in the first instance and then for 2–3 h at 390°C directly prior to application. Optical rotations were measured by using a ‘Jasco P-2000’ polarimeter. 1H NMR spectra were recorded in CDCl3 at 400 MHz, and 13C NMR spectra were recorded in CDCl3 at 100 or 175 MHz. The 1H NMR chemical shifts are referenced to tetramethyl silane (TMS, δH = 0 ppm) or CDCl3 (δH = 7.26 ppm) for 1H NMR spectra for solutions in CDCl3. The 13C NMR chemical shifts are referenced to the central signal of CDCl3 (δC = 77.00 ppm) for solutions in CDCl3. Compound purity or compound ratios were accessed or calculated by comparing the integration intensities of the relevant signals in their 1H NMR spectra. Accurate mass spectrometry determinations were performed using the Agilent 6230 ESI-TOF LC/MS mass spectrometer.
Synthesis of glycosyl donors 9 and 10
Ethyl 2-azido-4,6-O-benzylidene-2-deoxy-1-thio-α-D-altropyranoside (8): lutidine (4.80 ml, 41.6 mmol), and Bu2SnO (0.10 g, 0.41 mmol) were added to a stirring solution of ethyl 4,6-O-benzylidene-1-thio-α-D-glucopyranoside (7, (Takeo et al., 1993) 2.60 g, 8.32 mmol) in CH2Cl2 (30 ml) under argon at room temperature. The resulting mixture was cooled to 0°C; Tf2O (4.90 ml, 29.1 mmol) was added dropwise. The mixture was then stirred for 1 h at 0°C. The reaction mixture was then diluted with CH2Cl2 (250 ml) and washed with H2O (5 × 50 ml). The organic phase was separated, dried with Na2SO4, concentrated under reduced pressure, and dried in vacuo. The crude residue was dissolved in DMF (50 ml), NaN3 (2.70 g, 41.6 mmol) was added, and the resulting mixture was stirred under argon for 16 h at 80°C. The reaction mixture was then diluted with CH2Cl2 (250 ml) and washed with H2O (5 × 50 ml). The organic phase was separated, dried with Na2SO4, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (ethyl acetate–hexane gradient elution) to afford the title compound as a white amorphous solid. Analytical data for 8: Rf = 0.50 (ethyl acetate/hexane, 3/7, v/v); [α]D24 +95.6 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ 7.52–7.35 (m, 5H, aromatic), 5.65 (s, 1H, >CHPh), 5.23 (s, 1H, H-1), 4.62 (m, 1H, J5,6a = 5.1, J5,6b = 10.1 Hz, H-5), 4.29 (dd, 1H, J6a,6b = 10.3 Hz, H-6a), 4.13 (dd, 1H, J3,4 = 2.8 Hz, H-3), 4.00 (d, 1H J2,3 = 3.2 Hz, H-2), 3.91 (dd, 1H, J4,5 = 9.8 Hz, H-4), 3.81 (m, 1H, H-6b), 2.88 (d, 1H, OH), 2.72–2.61 (m, 2H, SCH2), and 1.30 (t, 3H, SCH2CH3) ppm; 13C NMR (100 MHz, CDCl3): δ 137.0, 129.4, 128.4 (x2), 126.2 (x2), 102.3, 82.4, 76.3, 68.9, 67.6, 64.0, 58.8, 27.1, and 14.9 ppm; and HRMS [M + Na]+ calcd for C15H19N3O4SNa 360.0994; found 360.0995.
Ethyl 2-azido-4,6-O-benzylidene-2-deoxy-3-O-picoloyl-1-thio-α-D-altropyranoside (9): picolinic acid (0.39 g, 3.2 mmol), EDC (0.61 g, 3.2 mmol), and DMAP (51 mg, 0.42 mmol) were added to a solution of compound 8 (0.71 g, 2.1 mmol) in dry CH2Cl2 (10 ml), and the resulting mixture was stirred under argon for 3 h at room temperature. After that, the reaction mixture was diluted with CH2Cl2 (20 ml) and washed with water (10 ml), sat. aq. NaHCO3 (10 ml), and water (2 × 10 ml). The organic phase was separated, dried with Na2SO4, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (ethyl acetate–hexane gradient elution) to afford the title compound as a white amorphous solid in 95% yield (1.32 g, 2.98 mmol). Analytical data for 9: Rf = 0.50 (ethyl acetate/hexane, 1/1, v/v); [α]D24 + 57.4 (c = 1.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ 8.82 (dd, 1H, aromatic), 8.36 (d, 1H, J = 7.8 Hz, aromatic), 7.88 (m, 1H, aromatic), 7.30–7.55 (m, 6H, aromatic), 5.64 (s, 1H, >CHPh), 5.59 (dd, 1H, J3,4 = 3.2 Hz, H-3), 5.30 (s, 1H, H-1), 4.75 (m, 1H, J5,6a = 5.2, J5,6b = 10.1 Hz, H-5), 4.33 (dd, 1H, J6a,6b = 10.5 Hz, H-6a), 4.22 (d, 1H, J2,3 = 4.0 Hz, H-2), 4.14 (dd, 1H, J4,5 = 9.8 Hz, H-4), 3.86 (dd, 1H, H-6b), 2.69 (m, 2H, SCH2), and 1.31 (t, 3H, SCH2CH3) ppm; 13C NMR (100 MHz, CDCl3): δ 163.3, 150.4, 147.2, 137.1, 136.9, 129.1, 128.3 (x2), 127.1, 126.1 (x2), 125.9, 102.1, 82.5, 74.3, 68.9, 68.5, 62.4, 60.0, 27.2, and 15.1 ppm; and HRMS [M + H]+ calcd for C21H23N4O5S 443.1395; found 443.1384.
Ethyl 2-azido-3-O-benzoyl-4,6-O-benzylidene-2-deoxy-1-thio-α-D-altropyranoside (10): benzoyl chloride (0.25 ml, 2.0 mmol) and DMAP (24 mg, 0.20 mmol) were added to a solution of compound 8 (0.33 g, 1.0 mmol) in pyridine (15 ml), and the resulting mixture was stirred under argon for 2 h at room temperature. After that, the reaction was quenched with MeOH (5 ml), the volatiles were removed under reduced pressure, and the residue was co-evaporated with toluene. The resulting residue was diluted with CH2Cl2 (20 ml) and washed with water (10 ml), 1 N aq. HCl (10 ml), and water (2 × 10 ml). The organic phase was separated, dried with Na2SO4, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (ethyl acetate–hexane gradient elution) to give the title compound as an off-white amorphous solid in 91% yield (0.40 g, 0.91 mmol). Analytical data for 10: Rf = 0.50 (ethyl acetate/hexane, 2/3, v/v); [α]D24 +68.6 (c 1.0, CHCl3); 1H NMR (700 MHz, CDCl3): δ 8.28–8.25 (m, 2H, aromatic), 8.18–8.16 (m, 1H, aromatic), 7.66–7.61 (m, 1H, aromatic), 7.54–7.50 (m, 2H, aromatic), 7.46–7.42 (m, 2H, aromatic), 7.35–7.32 (m, 2H, aromatic), 5.67 (s, 1H, >CHPh), 5.56 (dd, 1H, J3,4 = 3.2 Hz, H-3), 5.34 (s, 1H, H-1), 4.83 (m, 1H, J5,6a = 5.2, J5,6b = 10.1 Hz, H-5), 4.38 (dd, 1H, J6a,6b = 10.5 Hz, H-6a), 4.26 (d, 1H, J2,3 = 3.6 Hz, H-2), 4.17 (dd, 1H, J4,5 = 9.8 Hz, H-4), 3.90 (dd, 1H, H-6b), 2.77–2.67 (m, 2H, SCH2), and 1.35 (t, 3H, SCH2CH3) ppm; 13C NMR (100 MHz, CDCl3): δ 165.5, 137.0, 133.7, 133.4, 130.2 (×2), 129.3, 129.1, 128.5 (x2), 128.2, 126.1 (×2), 102.2, 82.5, 74.4, 68.9, 67.9, 62.4, 60.0, 27.2, and 15.1 ppm; and HRMS [M + H]+ calcd for C22H24N3O5S 442.1436; found 442.1431.
Synthesis of disaccharides 15–22
General procedure for glycosylation in the presence of NIS/TfOH: a mixture of glycosyl donor (0.1 mmol), glycosyl acceptor (0.09 mmol), and freshly activated molecular sieves (4 Å, 100 mg) in 1,2-DCE (2.0 ml) was stirred under argon for 1 h at room temperature. The mixture was then cooled to −30°C, N-iodosuccinimide (NIS, 0.22 mmol) and trifluoromethanesulfonic acid (TfOH, 2.0 µl, 0.02 mmol) were added, and the resulting mixture was allowed to warm to ambient temperature and stirred for 1–2 h at room temperature. After that, the solids were filtered off and washed successively with CH2Cl2. The combined filtrate (30–40 ml) was washed with water (10 ml), 10% sodium thiosulfate (Na2S2O3, 10 ml), and water (2 × 10 ml). The organic phase was separated, dried with Na2SO4, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (acetone–toluene gradient elution) to afford corresponding disaccharide derivatives. Anomeric ratios (or anomeric purity) were determined by the comparison of the integral intensities of the relevant signals in 1H NMR spectra.
Methyl 6-O-(2-azido-4,6-O-benzylidene-2-deoxy-3-O-picoloyl-α-D-altropyranosyl)-2,3,4-tri-O-benzyl-α-D-glucopyranoside (15): the title compound was obtained as an off-white amorphous solid from glycosyl donor 9 and acceptor 1129 in 98% yield. Analytical data for 15: Rf = 0.40 (ethyl acetate/toluene, 3/7, v/v); [α]D24 +42.5 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ 8.64 (dd, 1H, aromatic), 8.04 (dd, 1H, aromatic), 7.50–7.00 (m, 22H, aromatic), 5.61 (s, 1H, >CHPh), 5.51 (dd, 1H, J3’,4’ = 3.0 Hz, H-3′), 4.95 (d, 1H, CHPh), 4.92 (s, 1H, H-1′), 4.69–4.84 (m, 3H, 3 x CHPh), 4.58 (d, 1H, CHPh), 4.46 (m, 2H, H-5′, CHPh), 4.45 (d, 1H, J1,2 = 3.5 Hz, H-1), 4.23 (d, 1H, J2′,3′ = 2.8 Hz, H-2′) 4.20 (dd, 1H, J6’a,6’b = 10.4 Hz, H-6′a), 4.09 (dd, 1H, J4’,5’ = 9.7 Hz, H-4′), 4.01 (dd, 1H, J6a,6b = 11.4 Hz, H-6a), 3.91 (dd, 1H, J3,4 = 9.3 Hz, H-3), 3.77 (dd, 1H, H-6b′), 3.71–3.67 (m, 1H, H-5), 3.63 (dd, 1H, H-6b), 3.48 (dd, 1H, J4,5 = 9.5 Hz, H-4), 3.27 (dd, 1H, J2,3 = 9.3 Hz, H-2), and 3.22 (s, 3H, CH3) ppm; 13C NMR (100 MHz, CDCl3): δ 164.2, 150.4, 147.4, 138.7, 138.2, 138.1, 137.0, 136.9, 129.1, 128.5 (x2), 128.4 (x2), 128.3 (x2), 128.2 (x4), 128.0 (x2), 127.9, 127.7 (x2), 127.5 (x2), 126.9, 126.1 (x2), 125.2, 102.1, 98.9, 98.0, 81.7, 80.1, 75.7, 74.8, 73.8, 73.4 (x2), 69.8, 69.0, 68.9, 67.2, 60.1, 59.3, and 55.2 ppm; and HRMS [M + Na]+ calcd for C47H48N4O11Na 867.3238; found 867.3212.
Methyl 6-O-(2-azido-3-O-benzoyl-4,6-O-benzylidene-2-deoxy-D-altropyranosyl)-2,3,4-tri-O-benzyl-α-D-glucopyranoside (16): the title compound was obtained as an off-white sticky semi-solid from glycosyl donor 10 and acceptor 1129 in 98% yield (α/β = 3.0/1). Selected analytical data for α-16: Rf = 0.40 (ethyl acetate/toluene, 3/7, v/v); 1H NMR (400 MHz, CDCl3): δ 8.11–7.98 (m, 3H, aromatic), 7.49 (m, 1H, aromatic), 7.43–7.09 (m, 21H, aromatic), 5.62 (s, 1H, >CHPh), 5.39 (dd, 1H, J3’,4’ = 3.1 Hz, H-3′), 4.91 (s, 1H, H-1′), 4.76–4.71 (m, 4H, H-5′, 3 x CHPh), 4.66–4.54 (m, 4H, H-6′a, 3 x CHPh), 4.41 (d, 1H, J1,2 = 3.5 Hz, H-1), 4.24 (d, 1H, J2,3 = 3.1 Hz, H-2′), 4.07 (dd, 1H, J4’,5’ = 9.7 Hz, H-4′), 3.90 (dd, 1H, J3,4 = 9.2 Hz, H-3), 3.79 (m, 2H, H-6′b), 3.71–3.63 (m, 2H, H-5, 6 b), 3.36–3.27 (m, 2H, H-2, 4), and 3.14 (s, 3H, CH3) ppm; 13C NMR (100 MHz, CDCl3): δ 165.9, 165.1, 138.7 (×2), 138.3, 138.1 (×2), 138.0, 137.1, 136.9, 133.7, 133.3, 129.8 (×2), 129.7, 129.6 (×2), 129.4, 129.2, 129.1, 129.0, 128.7, 128.6 (×2), 128.5 (×5), 128.4 (×4), 128.3 (×4), 128.2, 128.1 (×3), 128.0 (×6), 127.9 (×2), 127.8, 127.7, 127.6 (×6), 126.1 (×5), 102.3, 102.1, 99.1 (x2), 98.0, 97.9, 82.1, 81.7, 80.1, 79.8, 77.6, 77.1, 75.7, 74.9, 74.7, 74.0, 73.9, 73.5 (x2), 70.1, 69.7, 69.6, 69.1, 68.9, 68.7, 68.5 (x2), 67.3, 64.7, 61.0, 60.1, 59.4, 55.2, and 55.1 ppm; and HRMS [M + H]+ calcd for C48H50O11N3 844.3443; found 844.3440.
Methyl 4-O-(2-azido-4,6-O-benzylidene-2-deoxy-3-O-picoloyl-D-altropyranosyl)-2,3,6-tri-O-benzyl-α-D-glucopyranoside (17): the title compound was obtained as a white amorphous solid from glycosyl donor 9 and acceptor 1229 in 71% yield (α/β = 1.7/1). Selected analytical data for α-17: Rf = 0.40 (ethyl acetate/toluene, 3/7, v/v); 1H NMR (400 MHz, CDCl3): δ 8.77 (dd, 1H, aromatic), 8.07 (dd, 1H, aromatic), 7.73 (dd, 1H, aromatic), 7.47–7.23 (m, 21H, aromatic), 5.60 (s, 1H, >CHPh), 5.45 (dd, 1H, J3’,4’ = 3.0 Hz, H-3′), 5.20 (s, 1H, H-1′), 5.00 (d, IH, CHPh), 4.70 (d, 1H, CHPh), 4.65–4.54 (m, 3H, CHPh), 4.57 (d, 1H, J1,2 = 3.0 Hz, H-1) 4.50 (d, 1H, CHPh), 4.46–4.40 (m, 1H, J5’,6a’ = 10.1, J5’,6b’ = 5.2 Hz, H-5′), 4.17 (dd, 1H, J = 10.6 Hz, H-6a′), 4.08 (dd, 1H, J4’,5’ = 9.8 Hz, H-4′), 3.91 (d, 1H, J2’,3’ = 3.0 Hz, H-2′), 3.85 (dd, 1H, J6a,6b = 10.7 Hz, H-6a), 3.78–3.69 (m, 4H, H-3, 4, 6b, 6 b’), 3.57 (m, 1H, J5,6a = 5.4 Hz, H-5), 3.52 (dd, 1H, J2,3 = 9.4 Hz, H-2), and 3.33 (s, 3H, CH3) ppm; 13C NMR (100 MHz, CDCl3): δ 163.8, 150.3, 149.8, 147.7, 138.2, 138.1, 138.0, 137.9, 137.8, 137.7, 137.1 (×2), 137.0, 136.8, 129.2, 129.1, 128.6 (×4), 128.5 (×3), 128.4 (×5), 128.3, 128.2 (×7), 128.1 (×2), 127.9, 127.8 (×2), 127.7 (×5), 127.6 (×5), 127.0 (×2), 126.3, 126.2, 126.1 (×4), 126.0, 125.3, 125.2, 102.2, 102.1, 99.8, 99.7, 97.8 (×2), 81.5 (×2), 80.4, 80.3, 80.2, 76.5, 75.9, 75.4, 73.9, 73.7, 73.6, 73.2 (×2), 69.8, 69.3, 69.1, 69.0 (×2), 68.9, 68.7, 67.5, 61.7, 60.0, 59.9, 58.9, 55.3, 55.2, and 52.9 ppm; and HRMS [M + Na]+ calcd for C47H48N4O11Na 867.3238; found 867.3212.
Methyl 4-O-(2-azido-3-O-benzoyl-4,6-O-benzylidene-2-deoxy-D-altropyranosyl)-2,3,6-tri-O-benzyl-α-D-glucopyranoside (18): the title compound was obtained as a white amorphous solid from glycosyl donor 10 and acceptor 12 in 84% yield (α/β = 1.0/1). Selected analytical data for α-18: Rf = 0.60 (ethyl acetate/toluene, 1/4, v/v); 1H NMR (400 MHz, CDCl3): δ 8.07 (d, 2H, J = 8.3 Hz, aromatic), 7.58–7.24 (m, 23H, aromatic), 5.59 (s, 1H, >CHPh), 5.35 (dd, 1H, J3’,4’ = 3.1 Hz, H-3′), 5.13 (s, 1H, H-1′), 4.98 (d, 1H, 2J = 11.2 Hz, CHPh), 4.68 (d, 1H, 2J = 12.0 Hz, CHPh), 4.59–4.53 (m, 4H, H-1, 3 × CHPh), 4.46 (d, 1H, 2J = 11.2 Hz, CHPh), 4.42–4.33 (m, 1H, H-5′), 4.15 (dd, 1H, J6a’,6b’ = 10.6 Hz, H-6a′), 4.06 (dd, 1H, J4’,5’ = 9.7 Hz, H-4′), 3.98 (d, 1H, J2’, 3’ = 3.2 Hz, H-2′), 3.75–3.68 (m, 5H, H-3, 5, 6a, 6b, 6 b’), 3.57–3.54 (m, 1H, H-4) 3.50 (dd, 1H, J2,3 = 9.2 Hz, H-2), and 3.31 (s, 3H, OCH3) ppm; 13C NMR (100 MHz, CDCl3): δ 165.7, 165.1, 139.3, 138.2 (×2), 138.0, 137.7, 137.5, 137.1, 137.0, 133.6, 133.3, 130.2, 129.9, 129.8 (×2), 129.7 (×2), 129.3, 129.2, 129.1 (×2), 128.8, 128.6 (×4), 128.5 (×2), 128.4 (×8), 128.2 (×5), 128.1 (×4), 127.9, 127.8, 127.7 (×2), 127.6 (×2), 127.5, 127.4 (×3), 127.2 (×2), 126.1 (×4), 102.2, 102.1, 100.2, 99.4, 98.2, 97.7, 81.4, 80.2 (×2), 79.5, 78.0, 77.9, 77.2, 75.4, 75.2, 74.1, 74.0, 73.8, 73.5, 73.2 (×2), 69.9, 69.7, 69.4, 69.0 (×2), 68.4, 68.1, 65.0, 61.4, 60.2, 59.9, 55.4, and 55.2 ppm; and HRMS [M + H]+ calcd for C48H50O11N3 844.3443; found 844.3440.
Methyl 3-O-(2-azido-4,6-O-benzylidene-2-deoxy-3-O-picoloyl-D-altropyranosyl)-2,4,6-tri-O-benzyl-α-D-glucopyranoside (19): the title compound was obtained as a pale yellow amorphous solid from glycosyl donor 9 and acceptor 13 in 63% yield (α/β = 2.2/1). Selected analytical data for α-19: Rf = 0.60 (ethyl acetate/toluene, 1/4, v/v); 1H NMR (400 MHz, CDCl3): δ 8.82 (dd, 1H, aromatic), 8.08 (dd, 1H, aromatic), 7.54–7.09 (m, 22H, aromatic), 5.58 (s, 1H, >CHPh), 5.43 (dd, 1H, J3’,4’ = 3.1 Hz, H-3′), 5.11 (s, 1H, H-1′), 4.84–4.77 (m, 2H, H-5′, CHPh), 4.55–4.46 (m, 3H, H-1, 2 x CHPh,), 4.43–4.42 (m, 3H, 3 x CHPh), 4.22 (dd, 1H, J6’a,6’b = 10.4 Hz, H-6′a), 4.07 (dd, 1H, J4,5 = 9.9 Hz, H-4′), 4.00 (dd, 1H, J3,4 = 9.4 Hz, H-3), 3.93 (d, 1H, J1’,2’ = 3.0 Hz, H-2′), 3.76–3.63 (m, 3H, H-5, 6a, 6b′), 3.60 (d, 1H, H-6b), 3.50 (dd, 1H, J4,5 = 9.6 Hz, H-4), 3.31 (dd, 1H, J2, 3 = 9.7 Hz, H-2), and 3.28 (s, 3H, CH3) ppm; 13C NMR (100 MHz, CDCl3): δ 164.0, 150.4, 149.8, 147.9, 137.7, 137.6, 137.5 (×2), 137.4 (×4), 137.1, 136.9, 129.1, 129.0, 128.6 (×10), 128.5 (×2), 128.4 (×2), 128.2 (×8), 128.1 (×2), 128.0 (×3), 127.9 (×3), 127.7, 127.6, 127.4, 127.3 (×3), 127.2 (×3) 127.0 (×2), 126.3, 126.2, 126.1, 125.2 (×2), 102.2, 102.1, 99.1 (×2), 97.7, 97.5, 79.1, 78.9, 78.7, 78.2, 78.1, 77.6, 76.0, 74.6, 74.5, 73.9, 73.7 (×2), 73.2, 72.9, 69.8, 69.5, 69.4, 69.1, 69.0, 68.3, 68.1, 67.9, 61.9, 60.0, 59.2, 58.3, and 55.1 (×2) ppm; and HRMS [M + H]+ calcd for C48H50O11N3 844.3443; found 844.3440.
Methyl 3-O-(2-azido-3-O-benzoyl-4,6-O-benzylidene-2-deoxy-D-altropyranosyl)-2,4,6-tri-O-benzyl-α-D-glucopyranoside (20): the title compound was obtained as a pale yellow amorphous solid from glycosyl donor 10 and acceptor 13 in 80% yield (α/β = 2.5/1). Selected analytical data for α-20: Rf = 0.60 (ethyl acetate/toluene, 1/4, v/v); 1H NMR (400 MHz, CDCl3): δ 8.17–8.10 (m, 2H, aromatic), 7.64–7.60 (m, 1H, aromatic), 7.48–7.11 (m, 22H, aromatic), 5.57 (s, 1H, >CHPh), 5.37 (dd, 1H, J3’,4’ = 3.0 Hz, H-3′), 5.09 (s, 1H, H-1′), 4.76 (m, 2H, H-5′, CHPh), 4.60–4.41 (m, 6H, H-1, 5 x CHPh,), 4.19 (dd, 1H, J6’a,6b = 10.3 Hz, H-6′a), 4.05 (dd, 1H, J = 10.0 Hz, H-4′), 3.99 (dd, 1H, J3,4 = 9.4 Hz, H-3), 3.91 (d, 1H, J2’,3’ = 2.9 Hz, H-2′), 3.76–3.54 (m, 4H, H-5, 6a, 6b, 6b′), 3.48 (dd, 1H, J4,5 = 9.5 Hz, H-4), 3.32 (d, 1H, J2,3 = 9.7 Hz, H-2), and 3.26 (s, 3H, CH3) ppm; 13C NMR (100 MHz, CDCl3): δ 165.9, 137.7, 137.5 (×2), 137.4, 133.4, 130.2, 129.8 (×3), 129.0, 128.6 (×4), 128.5 (×4), 128.4 (×3), 128.2 (×2), 128.1 (×3), 128.0, 127.9, 127.4, 126.3, 102.1, 99.3, 97.8, 79.4, 78.7, 78.4, 77.2, 74.6, 74.0, 73.7, 73.3, 69.6, 69.1, 68.7, 68.2, 60.2, 59.3, and 55.1 ppm; and HRMS [M + H]+ calcd for C48H50O11N3 844.3443; found 844.3440.
Methyl 2-O-(2-azido-4,6-O-benzylidene-2-deoxy-3-O-picoloyl-α-D-altropyranosyl)-3,4,6-tri-O-benzyl-α-D-glucopyranoside (21): the title compound was obtained as a pale yellow amorphous solid from glycosyl donor 9 and acceptor 1429 in 65% yield. Analytical data for 21: Rf = 0.40 (ethyl acetate/toluene, 3/7, v/v); [α]D24 +69.9 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ 8.56 (dd, 1H, aromatic), 7.92 (d, 1H, aromatic), 7.46–6.97 (m, 22H, aromatic), 5.63 (s, 1H, >CHPh), 5.49 (dd, 1H, J3’,4’ = 3.1 Hz, H-3′), 4.95 (d, 1H, CHPh) 4.93 (s, 1H, H-1′), 4.83 (d, 1H, J1,2 = 3.4 Hz, H-1), 4.75–4.65 (m, 2H, J5’,6’a = 5.4 Hz, H-5′, CHPh), 4.59 (d, 1H, CHPh), 4.52–4.38 (m, 3H, 3 × CHPh), 4.29 (dd, 1H, J6’a,6’b = 10.4 Hz, H-6′a), 4.26 (d, 1H, J2’,3’ = 2.8 Hz, H-2′), 4.11 (dd, 1H, J4’,5’ = 9.9 Hz, H-4′), 3.88 (dd, 1H, J2,3 = 9.6 Hz, H-2), 3.78 (dd, 1H, J3,4 = 9.7 Hz, H-3), 3.76 (dd, 1H, H-6′b) 3.71–3.60 (m, 4H, H-4, 5, 6a, 6b), and 3.29 (s, 3H, CH3) ppm; 13C NMR (100 MHz, CDCl3): δ 164.3, 149.5, 147.1, 137.9, 137.6, 137.4, 136.8, 136.2, 128.7, 128.0, 127.9 (×4), 127.8 (×5), 127.6 (x2), 127.4 (×2), 127.3 (×3), 127.2, 127.1, 126.4, 125.8, 124.9, 101.8, 96.0, 95.6, 80.0, 77.3, 76.3, 75.0, 74.5, 73.3, 73.1, 69.5, 68.9, 68.6, 67.9, 60.1, 58.9, and 54.5 ppm; and HRMS [M + Na]+ calcd for C47H48N4O11Na 867.3238; found 867.3212.
Methyl 2-O-(2-azido-3-O-benzoyl-4,6-O-benzylidene-2-deoxy-D-altropyranosyl)-3,4,6-tri-O-benzyl-α-D-glucopyranoside (22): the title compound was obtained as a yellowish sticky semi-solid from glycosyl donor 10 and acceptor 14 in 66% yield (α/β = 1.5/1). Selected analytical data for α-22: Rf = 0.60 (ethyl acetate/toluene, 1/4, v/v); 1H NMR (400 MHz, CDCl3): δ 8.01–7.93 (m, 2H, aromatic), 7.52–6.98 (m, 23H, aromatic), 5.61 (s, 1H, >CHPh), 5.39 (dd, 1H, J3’,4’ = 3.1 Hz, H-3′), 4.94 (s, 1H, H-1′), 4.87–4.84 (d, 1H, CHPh), 4.79 (d, 1H, J1,2 = 3.3 Hz, H-1), 4.68 (dd, 1H, J5’,6’a = 4.7, J5’,6’b = 10.4 Hz, H-5′), 4.60–4.28 (m, 5H, 5 × CHPh), 4.29 (dd, 1H, J6’a,6’b = 10.5 Hz, H-6′a), 4.25 (d, 1H, J2’,3’ = 2.7 Hz, H-2′), 4.09 (dd, 1H, J4’,5’ = 9.8 Hz, H-4′), 3.84 (dd, 1H, J2,3 = 9.6 Hz, H-2), 3.81–3.75 (m, 2H, H-3, 6′b), 3.72–3.58 (m, 4H, H-4, 5, 6a, 6 b), and 3.24 (s, 3H, CH3) ppm; 13C NMR (100 MHz, CDCl3): δ 166.5, 165.2, 138.3, 138.1, 137.9, 137.8, 137.2, 136.9, 133.7, 133.5, 133.2, 129.9, 129.8 (×4), 129.7, 129.6, 129.2 (×3), 128.7, 128.6, 128.5, 128.4 (×8), 128.3 (×6), 128.2 (×2), 128.0, 127.9 (×8), 127.8 (×3), 127.7 (x2), 127.6 (x2), 127.5, 126.3 (x2), 126.1 (x2), 126.0, 102.4, 102.2, 96.7, 96.6, 93.0, 92.5, 80.5, 77.8, 77.1, 75.5, 75.1, 75.0, 74.1, 74.0, 73.8, 73.5 (×2), 70.0, 69.5, 69.2, 69.1, 68.9 (×2), 68.4, 68.3, 65.0, 62.3, 60.5, 60.4, 60.3, 59.5, 59.3, and 54.9 ppm; and HRMS [M + H]+ calcd for C48H50O11N3 844.3443; found 844.3440.
Preparation of disaccharide 23 to understand stereoselectivity
Methyl 6-O-(2-azido-4,6-O-benzylidene-2-deoxy-α-D-altropyranosyl)-2,3,4-tri-O-benzyl-α-D-glucopyranoside (23) from compound 15: Copper acetate monohydrate (12.4 mg, 0.070 mmol) was added to a solution of compound 15 (38 mg, 0.05 mmol) in CH2Cl2/MeOH (1.2 ml, 3/1, v/v), and the resulting mixture was stirred for 1 h at room temperature. After that, the reaction mixture was diluted with CH2Cl2 (∼30 ml) and washed with water (10 ml), sat. aq. NaHCO3 (10 ml), and water (2 × 10 ml). The organic phase was separated, dried with Na2SO4, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (ethyl acetate–hexane gradient elution) to afford the title compound as a white amorphous solid. Analytical data for 23: Rf = 0.40 (ethyl acetate/toluene, 3/7, v/v); [α]D22 +8.8 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ 7.46 (dd, 2H, aromatic), 7.41–7.21 (m, 18H, aromatic), 5.61 (s, 1H, >CHPh), 5.01–4.93 (m, 2H, 2 × CHPh), 4.86 (s, 1H, H-1′), 4.82–4.60 (m, 4H, 4 × CHPh), 4.61 (d, 1H, J1,2 = 2.1 Hz, H-1), 4.25 (dd, 1H, J5’,6’a = 5.1, J5,6’b = 10.0 Hz, H-5′), 4.19–4.11 (m, 2H, H-3′, 6′a), 4.01 (dd, 1H, J3,4 = 9.2 Hz, H-3), 3.93 (d, 1H, J2’,3’ = 3.1 Hz, H-2′), 3.85 (dd, 1H, J4’,5’ = 9.7 Hz, H-4′), 3.83–3.76 (m, 3H, H-5, 6a, 6 b′), 3.64 (dd, 1H, H-6b), 3.52 (dd, 1H, J2,3 = 9.6 Hz, H-2), 3.42 (br d, 1H, J4,5 = 9.0 Hz, H-4), 3.37 (s, 3H, CH3), and 3.02 (br d, 1H, J = 7.9 Hz, OH) ppm; 13C NMR (100 MHz, CDCl3): δ 138.1, 137.6 (×2), 136.6, 128.7, 128.1 (×2), 128.0 (×2), 127.8 (×4), 127.7 (×2), 127.6, 127.5 (×2), 127.4, 127.3 (×2), 127.2, 125.8 (×2), 101.9, 98.0, 97.5, 81.5, 79.5, 77.5, 75.5, 75.3, 74.6, 72.9, 69.0, 68.5, 67.3, 66.7, 61.3, 58.0, and 54.9 ppm; and HRMS [M + Na]+ calcd for C41H45N3O10Na 867.3238; found 867.3212.
From compound 16, a 1 N solution of sodium methoxide in MeOH was added to a solution of compound 15 (38 mg, 0.05 mmol) in MeOH (1.2 ml) until pH reached ∼ 9, and the resulting mixture was kept for 1 h at room temperature. After that, the reaction mixture was neutralized with Dowex (H+); the resin was filtered off and washed successively with MeOH. The combined filtrate (∼25 ml) was concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (ethyl acetate–hexane gradient elution) to afford the title compound as a white amorphous solid.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This work was supported by an award from the NIGMS (GM GM111835).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2022.945779/full#supplementary-material
References
Ágoston, K., Streicher, H., and Fügedi, P. (2016). Orthogonal protecting group strategies in carbohydrate chemistry. Tetrahedron Asymmetry 27, 707–728. doi:10.1016/j.tetasy.2016.06.010
Alex, C., and Demchenko, A. V. (2022). Direct synthesis of glycans containing challenging ManNAcA residues. J. Org. Chem. 87, 271–280. doi:10.1021/acs.joc.1c02351
Alex, C., and Demchenko, A. V. (2021). Recent advances in stereocontrolled mannosylation: Focus on glycans comprising acidic and/or amino sugars. Chem. Rec. 21, 3278–3294. doi:10.1002/tcr.202100201
Alex, C., Visansirikul, S., and Demchenko, A. V. (2021). A versatile approach to the synthesis of glycans containing mannuronic acid residues. Org. Biomol. Chem. 19, 2731–2743. doi:10.1039/d1ob00188d
Alex, C., Visansirikul, S., and Demchenko, A. V. (2020). A versatile approach to the synthesis of mannosamine glycosides. Org. Biomol. Chem. 18, 6682–6695. doi:10.1039/d0ob01640c
Alex, C., Visansirikul, S., Zhang, Y., Yasomanee, J. P., Codee, J., and Demchenko, A. V. (2020). Synthesis of 2-azido-2-deoxy- and 2-acetamido-2-deoxy-D-manno derivatives as versatile building blocks. Carbohydr. Res. 488, 107900. doi:10.1016/j.carres.2019.107900
Araujo, N., Jenkinson, S. F., Martınez, R. F., Glawar, A. F. G., Wormald, M. R., Butters, T. D., et al. (2012). Synthesis from D-altrose of (5R, 6R, 7R, 8S)-5, 7-Dihydroxy-8-hydroxymethylconidine and 2, 4-dideoxy-2, 4-imino-D-glucitol, azetidine analogues of swainsonine and 1, 4-dideoxy-1, 4-imino-D-mannitol. Org. Lett. 14, 4174–4177. doi:10.1021/ol301844n
Baek, J. Y., Lee, B. Y., Jo, M. G., and Kim, K. S. (2009). β-Directing effect of electron-withdrawing groups at O-3, O-4, and O-6 positions and α-directing effect by remote participation of 3-O-acyl and 6-O-acetyl groups of donors in mannopyranosylations. J. Am. Chem. Soc. 131, 17705–17713. doi:10.1021/ja907252u
Behera, A., and Kulkarni, S. S. (2018). Chemical synthesis of rare, deoxy-amino sugars containing bacterial glycoconjugates as potential vaccine candidates. Molecules 23, 1997. doi:10.3390/molecules23081997
Chiu, T. M. K., Sigillo, K., Gross, P. H., and Franz, A. H. (2007). Synthesis of anomerically pure, furanose‐free α‐Benzyl‐2‐amino‐2‐deoxy‐d‐altro‐ andd‐manno‐pyranosides and some of their derivatives. Synth. Commun. 37, 2355–2381. doi:10.1080/00397910701410871
Crich, D., Cai, W., and Dai, Z. (2000). Highly diastereoselective α-mannopyranosylation in the absence of participating protecting groups. J. Org. Chem. 65, 1291–1297. doi:10.1021/jo9910482
Emmadi, M., and Kulkarni, S. S. (2014). Recent advances in synthesis of bacterial rare sugar building blocks and their applications. Nat. Prod. Rep. 31, 870–879. doi:10.1039/c4np00003j
Jager, M., and Minnaard, A. J. (2016). Regioselective modification of unprotected glycosides. Chem. Commun. 52, 656–664. doi:10.1039/c5cc08199h
Knapp, S., Kukkola, P. J., Sharma, S., Dhar, T. G. M., and Naughton, A. B. J. (1990). Amino alcohol and amino sugar synthesis by benzoylcarbamate cyclization. J. Org. Chem. 55, 5700–5710. doi:10.1021/jo00309a013
Knapp, S., Naughton, A. B. J., Jaramillo, C., and Pipik, B. (1992). Reactions of some pyranoside diol monotriflates with nucleophiles and bases. J. Org. Chem. 57, 7328–7334. doi:10.1021/jo00052a058
Komarova, B. S., Orekhova, M. V., Tsvetkov, Y. E., and Nifantiev, N. E. (2014). Is an acyl group at O-3 in glucosyl donors able to control α-stereoselectivity of glycosylation? The role of conformational mobility and the protecting group at O-6. Carbohydr. Res. 384, 70–86. doi:10.1016/j.carres.2013.11.016
Komarova, B. S., Tsvetkov, Y. E., and Nifantiev, N. E. (2016). Design of α‐selective glycopyranosyl donors relying on remote anchimeric assistance. Chem. Rec. 16, 488–506. doi:10.1002/tcr.201500245
Komarova, B. S., Ustyuzhanina, N. E., Tsvetkov, Y. E., and Nifantiev, N. E. (2013). “Stereocontrol of 1,2-cis-Glycosylation by remote O-acyl protecting groups,” in Modern SyntheticMethods in carbohydrate Chemistry: From monosaccharides to complex glycoconjugates. Editors B. Werz, D. S. Vidal, and D. Crich (Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA), 125–161.
Kulkarni, S. S., Wang, C. C., Sabbavarapu, N. M., Podilapu, A. R., Liao, P. H., and Hung, S. C. (2018). One-pot" protection, glycosylation, and protection-glycosylation strategies of carbohydrates. Chem. Rev. 118, 8025–8104. doi:10.1021/acs.chemrev.8b00036
Mannino, M. P., and Demchenko, A. V. (2021). “Hydrogen-bond-mediated Aglycone Delivery (HAD) and related methods in carbohydrate chemistry,” in Carbohydrate Chemistry: Chemical and biological approaches (specialist periodical reports). Editors A. P. Rauter, T. K. Lindhorst, and Y. Queneau (Croydon: RSC), 44, 93–116.
Nilsson, M., and Norberg, T. (2000). Synthesis of a spacer-containing disaccharide fragment of Bordetella pertussis lipopolysaccharide. Carbohydr. Res. 327, 261–267. doi:10.1016/s0008-6215(99)00318-3
Panza, M., Pistorio, S. G., Stine, K. J., and Demchenko, A. V. (2018). Automated chemical oligosaccharide synthesis: Novel approach to traditional challenges. Chem. Rev. 118, 8105–8150.
Polyakova, S. M., Nizovtsev, A. V., Kuznetskiy, R. A., and Bovin, N. V. (2015). New protecting groups in oligosaccharide synthesis. Russ. Chem. Bull., 974–989.
Ranade, S. C., Kaeothip, S., and Demchenko, A. V. (2010). Glycosyl alkoxythioimidates as complementary building blocks for chemical glycosylation. Org. Lett. 12, 5628–5631. doi:10.1021/ol1023079
Ritchmyer, N. K., Hann, R. M., and Hudson, C. S. (1939). The oxidative degradation of sedoheptulose to D-altronic acid. J. Am. Chem. Soc. 61, 343–345. doi:10.1021/ja01871a031
Sanapala, S. R., and Kulkarni, S. S. (2016). Expedient route to access rare deoxy amino L-sugar building blocks for the assembly of bacterial glycoconjugates. J. Am. Chem. Soc. 138, 4938–4947. doi:10.1021/jacs.6b01823
Sanapala, S. R., and Kulkarni, S. S. (2016). From L-rhamnose to rare 6-deoxy-L-hexoses. Org. Lett. 18, 3790–3793. doi:10.1021/acs.orglett.6b01796
Shrestha, G., Panza, M., Singh, Y., Stine, K. J., and Demchenko, A. V. (2022). N-Alkylated analogues of indolylthio glycosides as glycosyl donors with enhanced activation profile. Eur. J. Org. Chem. 2022, e202200300. doi:10.1002/ejoc.202200300
Takeo, K., Maki, K., Wada, Y., and Kitamura, S. (1993). Synthesis of the laminara-oligosaccharide methyl β-glycosides of dp 3-8. Carbohydr. Res. 245, 81–96. doi:10.1016/0008-6215(93)80061-i
Ustyuzhanina, N., Komarova, B., Zlotina, N., Krylov, V., Gerbst, A. G., Tsvetkov, Y., et al. (2006). Stereoselective α-glycosylation with 3-O-acetylated D-gluco donors. Synlett 6, 921–923. doi:10.1055/s-2006-939037
Vega-Perez, J. M., Candela, J. I., Vega, Margarita, and Iglesias-Guerra, F. (1995). A general method for Synthesis of alkyl 2-N-substituted and 2-N, N-disubstituted D-altrosamines. Carbohydr. Res. 279, C5–C8. doi:10.1016/0008-6215(96)83597-x
Volbeda, A. G., van der Marel, G. A., and Codée, J. D. C. (2019). “Protecting group strategies in carbohydrate chemistry,” in Protecting groups – strategies and applications in carbohydrate Chemistry. Editor S. Vidal (Weinheim: Wiley VCH), 1–28.
Walvoort, M. T., Moggre, G. J., Lodder, G., Overkleeft, H. S., Codee, J. D., and van der Marel, G. A. (2011). Stereoselective synthesis of 2, 3-diamino-2, 3-dideoxy-beta-D-mannopyranosyl uronates. J. Org. Chem. 76, 7301–7315. doi:10.1021/jo201179p
Wang, P., Huo, C. X., Lang, S., Caution, K., Nick, S. T., Dubey, P., et al. (2020). Chemical synthesis and immunological evaluation of a pentasaccharide bearing multiple rare sugars as a potential anti-pertussis vaccine. Angew. Chem. Int. Ed. Engl. 59, 6513–6520. doi:10.1002/ange.201915913
Wang, T., and Demchenko, A. V. (2019). Synthesis of carbohydrate building blocks via regioselective uniform protection/deprotection strategies. Org. Biomol. Chem. 17, 4934–4950. doi:10.1039/c9ob00573k
Keywords: glycosylation, synthesis, sugars, glycan, oligosaccharides
Citation: Novakova M, Das A, Alex C and Demchenko AV (2022) Synthesis and glycosidation of building blocks of D-altrosamine. Front. Chem. 10:945779. doi: 10.3389/fchem.2022.945779
Received: 16 May 2022; Accepted: 05 August 2022;
Published: 26 September 2022.
Edited by:
Clay Bennett, Tufts University, United StatesReviewed by:
George A. O’Doherty, Northeastern University, United StatesJustin Ragains, Louisiana State University, United States
Copyright © 2022 Novakova, Das, Alex and Demchenko. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexei V. Demchenko, YWxleGVpLmRlbWNoZW5rb0BzbHUuZWR1