 Jingjie Luo
Jingjie Luo Pengcheng Liu
Pengcheng Liu Wenhao Yang
Wenhao Yang Changhai Liang
Changhai Liang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem., 22 July 2022
Sec. Catalytic Reactions and Chemistry
Volume 10 - 2022 | https://doi.org/10.3389/fchem.2022.945028
This article is part of the Research TopicNew Innovations in Non-precious Metal Catalysts as Platinum ReplacementsView all 4 articles
The carbonylative transformation of ethylene oxide (EO) into methyl 3-hydroxypropionate (3-HPM) is a key process for the production of 1,3-propanediol (1,3-PDO), which is currently viewed as one of the most promising monomers and intermediates in polyester and pharmaceuticals industry. In this work, a homogeneous reaction system using commercial Co2(CO)8 was first studied for the carbonylation of EO to 3-HPM. The catalytic behavior was related to the electronic environment of N on aromatic rings of ligands, where N with rich electron density induced a stronger coordination with Co center and higher EO transformation. A reaction order of 2.1 with respect to EO and 0.3 with respect to CO was unraveled based on the kinetics study. The 3-HPM yield reached 91.2% at only 40°C by Co2(CO)8 coordinated with 3-hydroxypyridine. However, Co-containing colloid was formed during the reaction, causing the tough separation and impossible recycling of samples. Concerning the sustainable utilization, Co particles immobilized on pre-treated carbon nanotubes (Co/CNT-C) were designed via an in situ reduced colloid method. It is remarkable that unlike conventional Co/CNT, Co/CNT-C was highly selective toward the transformation of EO to 3-HPM with a specific rate of 52.2
Since the ring-opening carbonylation of epoxide in alcohol solvent has high atomic economy, it is very attractive to prepare downstream high value-added products using epoxide as the reactant via catalytic technique (Ocampo and Dolbier, 2004). Among the most commonly used epoxides, ethylene oxide (EO) could be obtained as a petrochemical product in excess capacity by industrial production. It also becomes a promising raw material to produce methyl 3-hydroxypropionate (3-HPM) through carbonylative conversion, with the subsequent hydrogenation product 1,3-propanediol (1,3-PDO) as one of the most expected industrial products for new polyester (PTT) and medical intermediates (Lv et al., 2010; Rajendiran et al., 2018; Wen et al., 2018; Xu and Wu, 2019). No matter how, the industrial path for perspective 3-HPM and 1,3-PDO required the key carbonylative transformation of EO with high efficiency and selectivity, which was, however, greatly challenging until current days.
In the past decades, carbonyl metallic salts have been used as operatable homogeneous catalysts for carbonylation of epoxides. In fact, researchers have reported in detail on the ring expansion and ring-opening carbonylation of epoxides (Suresh et al., 2021). However, complicated homogeneous reaction systems were dominantly used with metastable metal carbonyls with unsatisfied product yields. The early work of Lee and Alper (2004) designed a new Co2(CO)8/A(6,7-dihydro-5,8-dimethyldibenzo [b,j]-1,10-phenanthroline)/PhCH2Br catalyst to synthesize aliphatic polyesters via alternating copolymerization of propylene oxide and CO, with 55% yield of atactic polyhydroxybutrate (PHB). While homogeneous reactions were carried out using phosphorus ligands in the carcinogenic benzene under 900 psi, Rowley et al. (2007) reported a special bimetallic [(CITPP)Al(THF)2]+[Co(CO)4]− catalyst efficient in transforming epoxides to β-lactone and subsequently succinic anhydrides via one-pot double carbonylation. The rates of both epoxide and lactone carbonylation were suggested independent of CO pressure and are first-order in the catalyst concentration. They also confirmed an opposite solvent effect for the different carbonylation processes.
In fact, Lewis acids as a kind of special ligands coordinating with metal centers were also frequently applied in the epoxide-related reactions. Dunn et al. (2016) reported an improved synthesis of poly (3-hydroxypropionate) (P3HP) from ethylene oxide and CO through the intermediate β-propiolactone. The bimetallic [Lewis acid]+[Co(CO)4]− catalyst was confirmed to be suitable for an optimized carbonylation yield. Maruyama et al. (2022) also reported a selective and quantitative cyclodimerization of an epoxide with a carbonyl compound by Lewis acid–catalyzed processes, where the ring opening of EO was involved as the first step. In a recent work, Zhao et al. (2021) used a series of N,P-ligand–cooperated [LCo(CO)3]+[Co(CO)4]− to catalyze EO (hydromethoxy carbonylation) into 3-HMP and proposed a cooperative reaction mechanism by [Co(CO)4]− and its counter cation [LCo(CO)3]+ (L = Ph2PN(iPr)PPh2). The yield of 3-HPM largely depended on the type of ligands forming complexes with Co2(CO)8, and the ion-pair complexes behaved more excellently than the neutral ones.
No matter how, the research progress of Co-based catalysts has stagnated in the past 5 years, and relevant reports still focus on the carbonyl cobalt in a homogeneous reaction system. Although the selectivity for 3-HPM is better, the reaction condition is harsh and in need of thoughtful operation due to CO dissociation and the oxidation of Co0. Co2(CO)8 used as a catalyst is metastable in air and needs to be properly preserved during preparation, storage, and transportation. On the other hand, the separation process is complex, and the homogeneous metal catalyst cannot be fully recovered and reutilized in a simple way, which is not conducive to the sustainable and recycling of resources. Therefore, it is necessary to develop a more stable and durable catalyst system to replace Co2(CO)8 in advance. In addition, the reaction kinetics of carbonylation of EO to 3-HPM lacks researches in depth and requires further discussion.
Over the years, many studies have been carried out on the carbonylation reaction via heterogeneous reactions, such as hydroformylation of olefins and hydroesterification of epoxides by supported noble metals (Li et al., 2003a; Li et al., 2003b; Liu et al., 2009; Liu et al., 2013). Compared with other supports, carbon materials have attracted more attention because of their easily controlled surface specialty and chemical structure. In addition to its outstanding electronic conductivity, carbon materials also exhibit the strong adsorption of CO with manually adjusted surface acidity acting as Lewis acid and Brönsted acid (Kainulainen et al., 1997), which possibly facilitated the immobilization of Co centers as a potential substitution of coordinated Co2(CO)8.
In this work, Co2(CO)8 was first used to catalyze the carbonylative transformation of EO to 3-HPM. The reaction parameters including the dosages of reactants; the cooperated ligands and CO pressure were optimized to understand the chemical kinetics of EO carbonylation. In order to promote separation efficiency and sustainable utilization, Co particles supported by pre-treated carbon nanotubes (Co/CNT-C) were synthesized via an in situ reduced colloid method. It is necessary to mention that the Co/CNT-C sample was selective toward the transformation of EO to 3-HPM with a similar atomic efficiency to that of Co2(CO)8 cooperated with 3-hydroxypyridine. The key factors for the enhanced catalytic performances were interpreted by combining the initial properties of Co/CNT by using the conventional impregnation method.
Co2(CO)8, pyrazole, 3-hydroxypyridine, and other ligands were purchased from Macklin (A.R.). Ethylene oxide (EO) in tetrahydrofuran (THF) (3.0 mol/L) was purchased from Energy Chemical Co., Ltd. Carbon monoxide with a purity of 99.99% was commercially available and supplied by Shandong Dazhan Nano Materials Co., Ltd. All other reagents were of analytical grade and used as received.
Pre-treated carbon nanotubes (abbreviated as CNT) were used as the supporting material and were preliminarily treated by concentrated HNO3 aqueous solution at 120°C for 2 h. The treated CNT was washed with distilled water until the solution was neutral and used as supporting materials after drying overnight.
A solution of 20 wt% Co/CNT-C was synthesized by an in situ reduced colloid method, and the process was as follows. A certain amount of sodium lauryl sulfate (SDS), Co(NO3)2·6H2O, and oleic acid with a molar ratio of 10:100:1 were added in water in the presence of 1 g CNT at room temperature. The Co mass loading was kept at 20 wt%. After stirring for 30 min, fresh NaBH4 aqueous solution (1 mg/ml) was dropped into the aforementioned mixture with a molar ratio of NaBH4 to Co2+ as 1:1. The sample powder was obtained after centrifugation, washed, and dried at 90°C. The supported sample went through two-step thermal treatment: calcination at 400°C in air for 3 h and subsequently in H2/Ar (v/v = 20/50) at 350°C for 3 h and denoted as Co/CNT-C. An additional Co/CNT-I reference was prepared by incipient wetness impregnation method by impregnating the treated CNT in the aqueous solution of Co(NO3)2 and aging for 16 h. The following synthesis process (centrifugation, drying, and two-step thermal treatment) was similar to that of Co/CNT-C.
Transmission electron microscopy (TEM) was performed using a FEI Tecnai G2 F30 electron microscope at an acceleration voltage of 120 KV. Fourier transform infrared (FT-IR) spectroscopy was carried out on a Bruker Vertex 80v FT-IR spectrometer. The specific surface area and pore structure of the samples were investigated using N2 physical adsorption/desorption at −196°C using an automatic volumetric sorption analyzer (Quantachrome, Autosorb-iQ-C). X-ray diffraction (XRD) was carried out on a Shimadzu 7000S diffractometer with Cu Kα radiation (λ = 1.5418 Å), with a scanning rate of 3o/min in range of 10o–80o. X-ray photoelectron spectroscopy (XPS) was operated on the ESCALAB™ 250Xi (Thermo Fisher Scientific, United States) using the X-ray excitation source of Al Kα. The charge correction is based on C1s peak at 284.6 eV. H2 temperature–programmed reduction (H2-TPR) was carried out using Quantachrome ChemBET 3000 Chemisorber with the gas flow rate of 30 ml/min. The reduction process was carried out by increasing the temperature from 50°C to 800°C at a heating rate of 10°C/min.
The carbonylative transformation of EO was carried out in a 50 ml stainless steel autoclave equipped with a magnetic stirrer, temperature control system, and pressure sensing device. In a typical experiment, a certain amount of Co2(CO)8 and a ligand were mixed with the methanol and EO solution in the autoclave. The aforementioned operation was done in a homemade glove box filled with N2 to keep the Co2(CO)8 stable. The autoclave was then tightened under N2 and purged with carbon monoxide three times to evacuate the residual N2 in the reactor. After filling the autoclave with CO to a desired pressure, the mixture was heated to a certain temperature (30°C–100°C) under stirring at 800 rpm min−1. After maintaining the reaction for a certain period of time, the reactor was cooled down to room temperature and the CO gas was vented off in a fume hood. The reaction mixture was homogeneous and transparent in red-brown. All the catalyst was dissolved in the solution after reaction. It is impossible to separate the catalyst from the reaction mixture by centrifugation. Herein, before further analyzing the reaction mixture by GC, two independent steps were necessary for the separation of catalyst and products. First of all, the reaction mixture was treated continuously by flowing O2 at room temperature for 2 h. Purple deposition was observed after the oxidation treatment, and the supernatant liquid became light orange. In the second step, column chromatography was used. An internal standard method was used for quantitative analysis with toluene as the standard. The product mixture was analyzed by gas chromatography using a GC (Agilent 7890B) equipped with a hydrogen flame ion detector (FID) and a capillary column HP-5 (30 m × 0.32 mm × 0.25 μm). Products were also identified using a gas chromatography–mass spectrometer (Thermo Fisher Scientific Trace1300-ISQ LT, United States). The conversion rate of EO and the selectivity of methyl 3-hydroxypropionate (3-HPM) are calculated by the following equations:
where
The carbonylative transformation of ethylene oxide (EO) was catalyzed by using the cobalt catalyst in the presence of CO. Methanol was used in excess as both solvent and reactant. Compared with Co2(CO)8 as catalyst, inorganic salts such as Co(OAc)2 and Co(NO3)2 displayed poor EO conversion of only 2.3% and 2.9% (Table 1, Entry 1–2), respectively. Also, no methyl 3-hydroxypropionate (3-HPM) was detected in the reaction mixture. The invalid catalytic performances of the inorganic salts should be resulted from the failure coordination between Co and the ligand (Xu and Wu, 2019). If the Co2(CO)8 was used as the catalyst, the EO conversion easily reached 56.5% at 40°C in 6 MPa CO pressure (Table 1, Entry 3) with 95.8% selectivity toward 3-HPM, in the absence of any ligand. For better catalytic activity, the influences of ligands were studied at first, as listed in Table 1 (Entry 4 and 8–13). A variety of N-donor ligand were used as doping ligands with Co2(CO)8 for the carbonylative transformation of EO. Among all the tested ligands, 3-hydroxypyridine could be used as a most efficient one with 98.5% selectivity toward 3-HPM at 92.6% EO conversion under only 40°C.
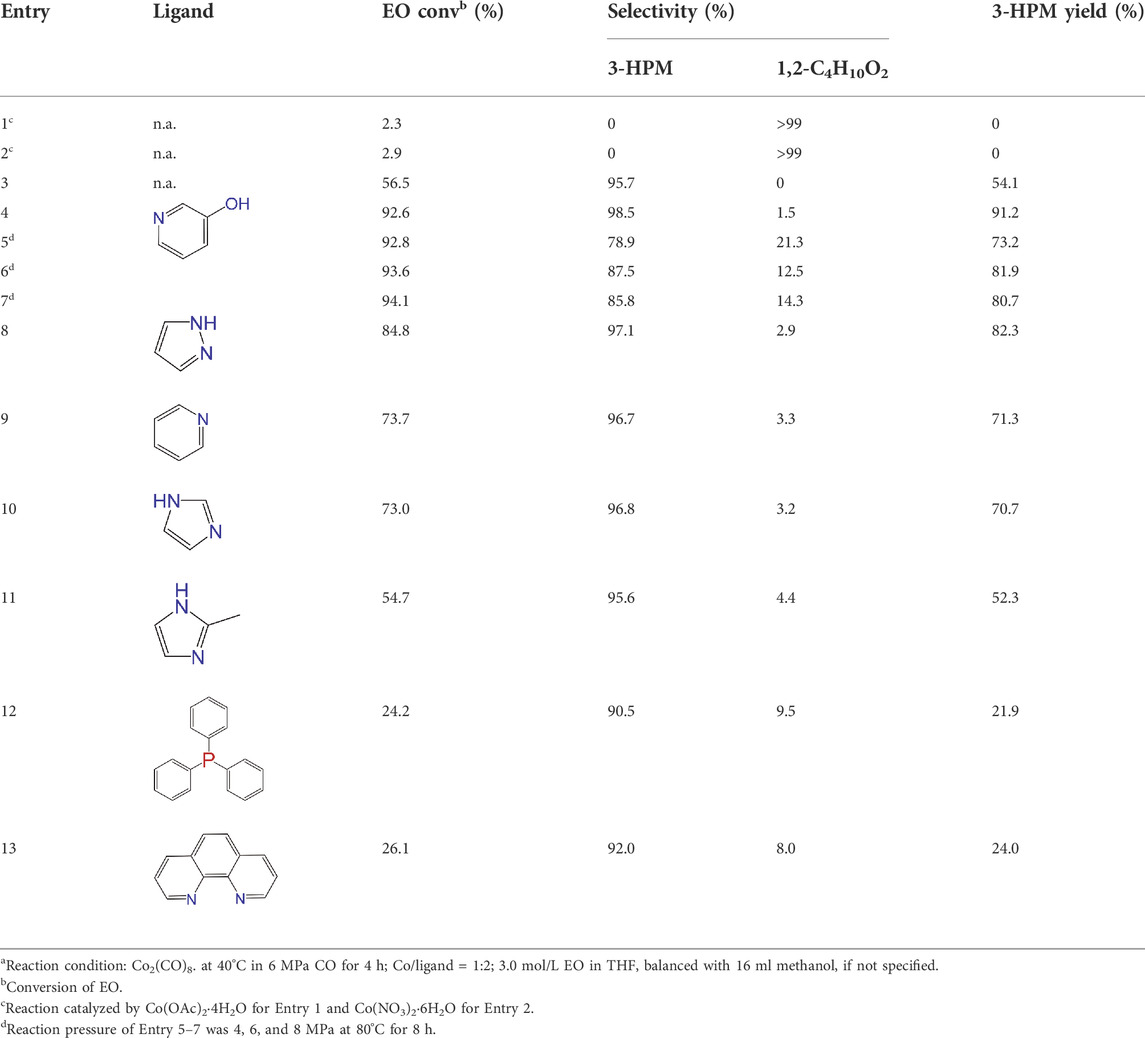
TABLE 1. Catalytic performances of Co2(CO)8 for the carbonylative transformation of EO into 3-HPM at different conditionsa.
In fact, Co2(CO)8 coordinated with the N-donor ligands to form Co-centered complexes in different paths depending on the specific electronic environment. Cobalt has an empty d-orbital and is easily complexed with the ligand by lone pair electrons on N. The nucleophilicity of a particular N atom in these ligands determines their interaction with Co as well as the subsequent catalytic performances. Compared to pyridine, the -OH group on the pyridine ring of 3-hydroxypyridine resulted in rich electron density on the aromatic ring due to p–π conjugated effect. The lone pair electrons on N may accounts for a stronger interaction between N and Co atoms in the complexes. The N-groups on pyrazole and imidazole with the
Under such circumstance, transformation of EO was further optimized by Co2(CO)8 in the presence of 3-hydroxypyridine. The experimental data suggested that the increase of CO pressure under the same temperature correlated with very slightly increases of EO conversion and 3-HPM selectivity (Table 1, Entry 5–7). The EO conversion and product selectivity change as a function of the Co/EO molar ratio was also studied, as shown in Figure 1A. The EO conversion was only 2.4% with the Co/EO ratio of 0.1/15, and most of the product was 1,2-dimethoxyethane (selectivity > 99%) as a result of nucleophilic addition without any participation of CO molecules (Kim et al., 2006). Both the EO conversion and 3-HMP selectivity grew logarithmically along with the Co/EO ratio. The EO conversion reached 92.6% with 98.5% 3-HPM selectivity at Co/EO of 1/15. Further increasing the ratio of cobalt catalyst, the selectivity of 3-HPM decreased while 1,2-dimethoxyethane (2.8%) was produced with trace amount of 1,1-dimethoxyethane (1.0% selectivity), although the EO conversion was maintained. It was suggested that CO molecules could be inserted into the skeleton of carbon chains to form 3-HPM only if the catalyst dosage kept in an appropriate Co/EO range.
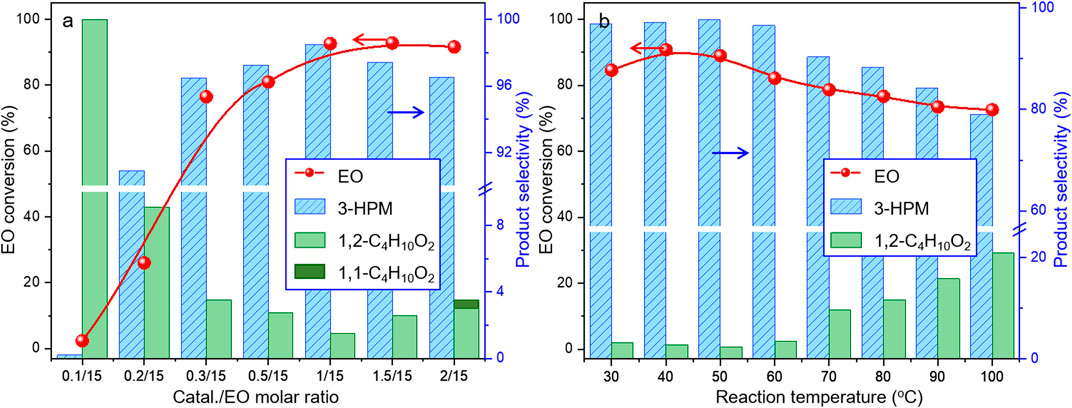
FIGURE 1. Catalytic performances of Co2(CO)8 for carbonylative transformation of EO as a function of (A) molar ratio between Co and EO at 40°C and (B) reaction temperature. Reaction condition: 6 MPa CO for 4 h; Co/3-hydroxypyridine = 1:2; and Co/EO = 1/15, balanced with methanol, 1,1-C4H10O2:1,1-Dimethoxyethane, and 1,2-C4H10O2:1,2-Dimethoxyethane.
The influence of reaction temperature was also investigated, as illustrated in Figure 1B. The EO conversion displayed a parabolic trend with the temperature, and 84.6% EO could be successfully transformed at only 30°C under 6 MPa CO with 96.8% selectivity toward 3-HPM. The catalytic performance maximized at 40°C, while the subsequent increasing of temperature induced the steadily decease of both the EO conversion and 3-HPM selectivity. The selectivity of by-product 1,2-dimethoxyethane increased to 20.9% at 100°C. At lower temperature (<60°C), the nucleophilic reaction was facilitated between EO and MtOH with the increasing temperature (Zeng et al., 2021). However, it is a common sense that the catalytic performance increases until the kinetic equilibrium, only if a stable catalyst, was in use (except for a third-order reaction). Herein, it is inferred that the as-formed complexes by Co2(CO)8 and N-donor ligand may encounter thermal decomposition under higher reaction temperature.
To the best of our knowledge, few researches were reported concerning the chemical kinetics for EO carbonylation, which is essential to suffciently understand the catalytic performances. In order to understand the key reaction factors and the kinetics of EO carbonylation, the concentration of EO changing as a function of reaction time at different temperature was studied, as shown in Figure 2A.

FIGURE 2. (A) Reaction data of the EO concentration at different reaction time and temperature, (B) linear relationship between
The conversion of EO was kept at 10%–30% by adjusting the conditions to make a precise estimation of the reaction order and other kinetic parameters. The EO was steadily consumed during the reaction and its concentration monotonously decreased with the reaction time. A higher reaction temperature in the range of 35°C–50°C resulted in the faster transformation of EO as indicated by the lower EO concentration detected under the same reaction time. The reaction data was subsequently fitted by the zero-order, first-order, and second-order reaction models in Figure 2B and Supplementary Figure S1 (supporting information). The corresponding R2 and rate constant (kA) for each linear relationship are listed in Supplementary Table S1. A much higher R2 approaching 1 based on the experimental data suggested the reaction order concerning EO was approximately 2. The reaction constant rapidly increased with the temperature, confirming the much accelerated reaction rate under higher temperature in the range of 35°C–50°C. For better interpretation, the reaction rate equation can be written in terms of EO concentration and CO pressure as Eq. 1. The influences of methanol on the reaction rate could be ignored in Eq. 1, since methanol is used as both reactant and solvent in excessive quantity.
The parameter
The reaction order (α) in terms of EO was thus obtained by the slope of a linear relationship between the logarithm of EO concentration and the logarithm of the reaction rate (Figure 2C). The mathematics fitting result suggested an apparent reaction order of 2.1 with respect to EO, further confirming the previous fitting results.
The reaction order (β) in terms of CO can be detected based on Eqs 4,5 under low EO conversion (superfluous EO concentration with nearly consistent quantity during reaction). As revealed by the slope of
According to the aforementioned investigation on kinetics, the differential rate equation could be written in the following form:
The activation energy (Ea) of the catalytic system reflected the necessary energy barrier to activate the reactant molecules and can be viewed as a parameter describing the catalytic efficiency (Joshi et al., 2018; Nazimov et al., 2021). According to the Arrhenius equation, the Ea based on Co2(CO)8 using 3-hydroxypyridine as a ligand can be estimated. The linear relationship between lnkA and the reciprocal of T in K is shown in Supplementary Figure S2. The Ea value calculated by the slope of the Arrhenius curve is 62.9 kJ/mol. It is necessary to mention that such value was occasionally similar to the reported energy barrier for disproportionation of Co2(CO)8 to [Co(MeOH)6][Co(Co)4]2 in methanol under carbonylation reactions (Mirbach and Mirbach, 1985). It is inferred that the formation of Co-centered complex may become an indispensable course for an efficient EO transformation.
For better confirmation, the solid catalysts before and after reaction were characterized by FT-IR spectra. It has to be noted that separation of the catalyst from the reaction mixture was indeed a tough task before any other analysis of the products. The cobalt complex was uniformly dissolved in the liquid and a seemingly homogeneous solution was obtained after reaction, where metal colloid was generated as confirmed by the Tyndall phenomenon (showing a characteristic light path) in Figure 3A. Colloidal Co-containing particles may exist in the reaction mixture.
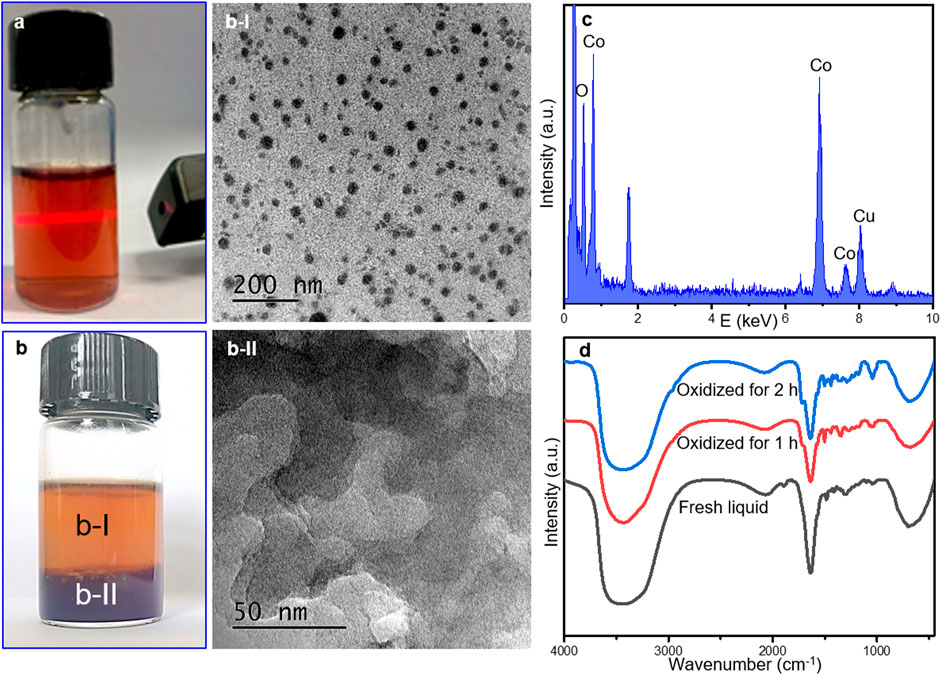
FIGURE 3. Electronic pictures of fresh mixture extracted immediately after reaction (A) and treated by oxidization with pure O2 at room temperature for 2 h (B), (B-I), and (B-II) illustrated the TEM images of supernatant liquid and under-layer deposition in b. (C) TEM/EDX analysis of the B-II sample, and (D) FT-IR spectra of fresh mixture after reaction without and with O2 flowing at room temperature.
It is impossible to separate the catalyst directly by centrifugation or column chromatography. Herein, an additional re-oxidation step was operated to recycle the cobalt-containing composition. The fresh liquid after reaction was subsequently treated by the flowing O2 for 2 h continuously to oxidize the Co colloid into Co-containing deposition (Figure 3B). The TEM images of the transparent solution in Figure 3A displayed the well dispersed particles as shown in Figure 3B-I, indicating the as-formed Co colloidal particles protected by organic compounds. The under-layer deposition was also collected and tested by TEM and EDX analysis. Amorphous CoOx was detected as a main composition (Figures 3B-II,C).
FT-IR spectra of the fresh mixture after reaction without and with oxidation treatment were displayed in Figure 3D. Infrared spectroscopy is one of most useful methods for characterizing the presence of different bands. The dominant peaks of -OH stretching and -CH deformation vibration at approximately 3,400 cm−1 and 692 cm−1 were observed in all samples (Li et al., 2012; AlOmar et al., 2016). The adsorption peaks at 1,644 cm−1 and 1,724 cm−1 originated from the aromatic skeleton vibration and the stretching vibration of the C=O bond in the coordinated cobalt catalyst (Xie and Wan, 2018), while the peak at 1,888 cm−1 suggests the presence of
No matter how, the high efficiency of Co2(CO)8 collaborated with 3-hydroxypyridine happened at the expense of pseudo-homogeneous reaction with untoward separation of the reaction liquid. It makes the separation of the metal catalyst and the recycle more difficult to be operated with considerable cost, which is also a big concern for the industrial production unit. Research works focusing on the carbonylative transformation of different chemical compounds by supported catalysts were also studied in recent years. However, tough reaction conditions including high temperature and pressure were requisite for better EO conversion and 3-HPM selectivity. The leaching of metal species was also one of the challenges during the liquid-phase heterogeneous reactions. Considering the in situ formed Co colloid within the reaction process as well as the potential advantages of the easily controlled carbon surface, a specific sample with cobalt active centers supported by CNT were herein synthesized via an in situ reduced Colloid method and denoted as Co/CNT-C. A reference synthesized by conventional Impregnation was also synthesized for comparison and denoted as Co/CNT-I.
It is remarkable that compared to the poor EO conversion by Co/CNT-I (7.2%), EO conversion of 33.7% could be reached using Co/CNT-C by the in situ reduced colloid method. The EO conversion was 77.5% in the presence of Co2(CO)8 with 3-hydroxypyridine as co-catalyst at the same condition. The selectivity toward 3-HPM was 92.5% by Co/CNT-C and slightly higher than that using Co2(CO)8. Concerning the excessive Co content used in the Co2(CO)8 system, the specific rate based on per gram Co per hour was calculated, as displayed in Figure 4A. The Co/CNT-I synthesized by conventional impregnation was only 13.3
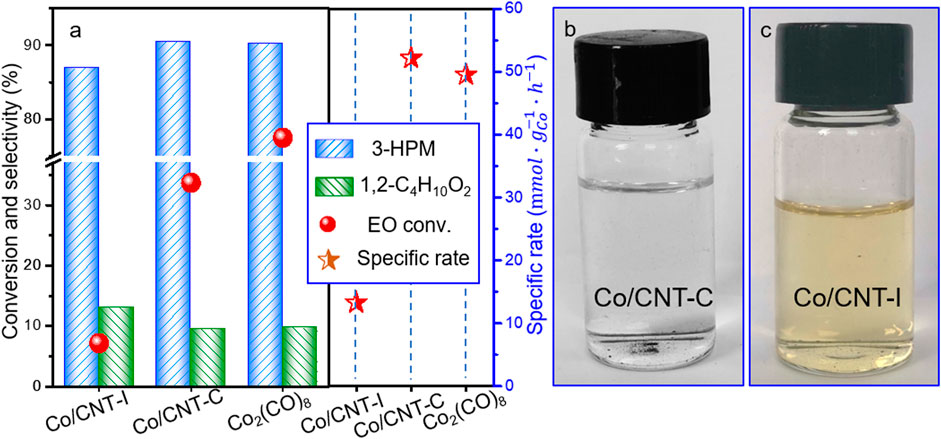
FIGURE 4. (A) Catalytic performances and specific rates and (B,C) electronic of the up-layer solution by centrifugation after reaction using Co/CNT-C and Co/CNT-I. Reaction condition: 60°C, 5 MPa CO for 5 h; Co/EO = 0.3/15 (0.1 g catalyst) for 20 wt% Co/CNT-I or Co/CNT-C; and Co/3-hydroxypyridine = 1:2, Co/EO = 1/15 for Co2(CO)8, balanced with methanol.
For better information, the FT-IR spectra of different Co/CNT-C and precursors during the synthesis are studied in Figure 5A to understanding the formation of surface functional groups on these samples. Nitric acid–treated CNT was used as the supporting material. The adsorption peak at 1,640 cm−1 was ascribed to the aromatic skeleton vibration in the main structure of CNT. After HNO3 treatment, large amount of carboxyl species was generated as revealed by the C-O stretching vibration and O-H deformation vibration at with adsorption peak at 1,400 cm−1. The deposition of Co particles caused the obvious decreased ratio of the later adsorption peak, suggesting the decreasing number of oxygenated groups during metal loading. Our previous research confirmed the possible consumption of surface oxygenated groups during tight metal loading (Luo et al., 2017; Luo et al., 2020). After the two-step thermal treatment, the adsorption peaks in the reduced Co/CNT-C were further exhausted.
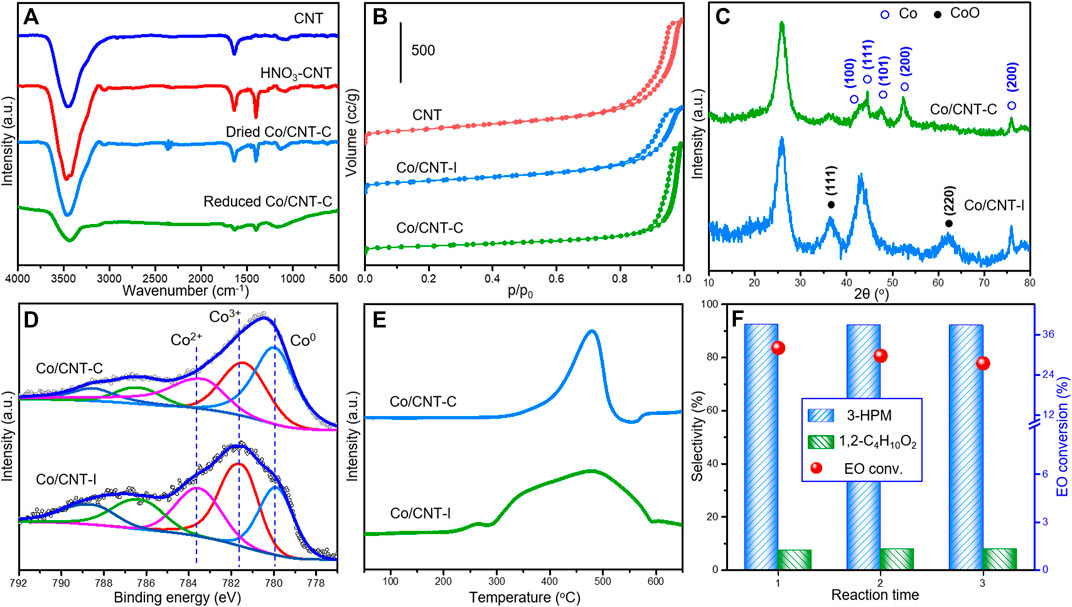
FIGURE 5. (A) FT-IR spectra of Co/CNT-C during the synthesis process, (B) N2 adsorption–desorption curves, (C) XRD patterns, (D) XPS spectra of Co 3d core level, (E) H2-TPR profiles of Co/CNT-C and Co/CNT-I by different methods, and (F) Cycle tests by Co/CNT-C. Reaction condition: 60°C, 5 MPa CO for 5 h; Co/EO = 0.3/15 (0.1 g catalyst), balanced with methanol.
The N2 adsorption–desorption curves of CNT and supported Co/CNT (Figure 5B) displayed the typical type IV isotherms, suggesting the maintained mesoporous structure after Co loading. Compared with the initial surface area of CNT (310.14 m2/g), the surface areas of Co/CNT-I and Co/CNT-C decreased to 221.8 and 204.6 m2/g, respectively. It could be attributed to the blocking of pores during the loading and growth of Co particles. Concerning the greatly differed catalytic performances of Co/CNT-I and Co/CNT-C, the XRD patterns in Figure 5C gave a clear evidence of the distinctive structures by different synthesis. Large amount of Co and CoO coexisted in Co/CNT-I, while both the diffraction peaks of Co and CoO became broadened in the Co/CNT-C, suggesting the smaller crystal sizes of Co-containing particles in such sample. The TEM image of Co/CNT-C is displayed in Supplementary Figure S3 to understand the surface dispersion of Co. Co particles existed as small black dots (Supplementary Figure S3A) on the surface of CNT. There was not any clear aggregation of Co particles. The HAADF-STEM EDX-elemental mapping of the Co/CNT-C also suggested the high dispersion of Co species on the detected area. It is also remarkable to note that Co0 was more readily enriched on the surface of Co/CNT-C and accounted for 44.3% of the total surface Co species by the Co 3d XPS core level (Figure 5D). In the Co/CNT-I reference, only 20.6% Co0 dispersed on the surface. The much lower H2 consumption by Co/CNT-C (2,500 μmol/g) than Co/CNT-I (3815 μmol/g) during H2-TPR (Figure 5E) further confirmed the accumulation of oxidative Co species in the Co/CNT-I. The combination results of XRD and XPS suggested a higher dispersion of Co particles with more Co0 sites exposed on the surface, which may correlated with the faster reaction rate via Co/CNT-C samples as reported by many carbonylation research works using supported metal catalysts (Xu et al., 2011; Zhang, 2012; Ibrahim and Denmark, 2018; Wang et al., 2021). The Co/CNT-C was further tested as a recycle catalyst for the EO carbonylation. After three reaction cycles, the conversion of EO in Figure 5F were still kept at 27.4% with the stabilized high selectivity of 3-HPM (92.1%). The discussion about Co/CNT-C suggested that it is a stable catalyst with nice specific rate based on the same Co atomic efficiency of Co2(CO)8 while avoiding the costly and complicated separation/post-treatments based on the metastable Co2(CO)8 system.
In this work, carbonylative transformation of EO was investigated using commercial Co2(CO)8 in a homogeneous system to produce 3-HPM. The reaction parameters including dosages of reactants, cooperated ligands, and CO pressure were optimized to understand the chemical kinetics of EO carbonylation. The reaction rate was interpreted as second-order in EO concentration and less sensitive to the CO pressure. However, Co-containing colloid was generated during the reaction, causing the tough separation process before any further chemical analysis. Concerning the impressive separation and sustainable utilization problems caused by the homogeneous reaction process, colloidal Co particles were directly supported on pre-oxidized CNT (Co/CNT-C) via an in situ reduced colloid method. Greatly improved EO conversion was observed via Co/CNT-C, if compared with conventional Co catalyst. The better activity was ascribed to the high dispersion of Co particles with good Co0 surface exposure and suitable amount of Co3+ as Lewis acid sites. The specific rate based on per gram Co per hour via Co/CNT-C was 52.2
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
JL and CL contributed to conception and design of the study. JL and PL organized the database and wrote the draft of the manuscript. PL, WY, HN, and SL performed the statistical analysis. All authors contributed to manuscript revision, read, and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (21978031 and 21802015), Science and Technology Innovation Fund in Dalian City (2021J12GX025), and Liaoning Revitalization Talent Program (XLYC1908033).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2022.945028/full#supplementary-material
Alomar, M. K., Hayyan, M., Alsaadi, M. A., Akib, S., Hayyan, A., Hashim, M. A., et al. 2016. Glycerol-based deep eutectic solvents: Physical properties. J. Mol. Liq., 215, 98–103. doi:10.1016/j.molliq.2015.11.032
Church, T. L., Getzler, Y. D. Y. L., and Coates, G. W. 2006. The mechanism of epoxide carbonylation by [lewis acid]+[CO(CO)4]- catalysts. J. Am. Chem. Soc., 128, 10125–10133. doi:10.1021/ja061503t
Dunn, E. W., Lamb, J. R., Lapointe, A. M., and Coates, G. W. 2016. Carbonylation of ethylene oxide to beta-propiolactone: A facile route to poly(3-hydroxypropionate) and acrylic acid. ACS Catal., 6, 8219–8223. doi:10.1021/acscatal.6b02773
Gorbunov, D. N., Nenasheva, M. V., and Kardashev, S. V. 2019. Use of nitrogenous bases as promoters of the reaction of ethylene carboalkoxylation on a cobalt catalyst. Russ. J. Appl. Chem., 92, 1069–1076. doi:10.1134/s1070427219080032
Ibrahim, M. Y. S., and Denmark, S. E. 2018. Palladium/Rhodium cooperative catalysis for the production of aryl aldehydes and their deuterated analogues using the water–gas shift reaction. Angew. Chem. Int. Ed. Engl., 57, 10519–10524. doi:10.1002/ange.201806148
Joshi, S. Y., Kumar, A., Luo, J., Kamasamudram, K., Currier, N. W., Yezerets, A., et al. 2018. New insights into the mechanism of NH3-SCR over Cu- and Fe-zeolite catalyst: Apparent negative activation energy at high temperature and catalyst unit design consequences. Appl. Catal. B Environ., 226, 565–574. doi:10.1016/j.apcatb.2017.12.076
Kainulainen, T. A., Niemela, M. K., and Krause, A. O. I. 1997. Hydroformylation of 1-hexene on Rh/C and Co/SiO2 catalysts. J. Mol. Catal. A Chem., 122, 39–49. doi:10.1016/s1381-1169(96)00461-x
Kim, H. S., Bae, J. Y., Lee, J. S., Jeong, C. I., Choi, D. K., Kang, S. O., et al. 2006. An efficient catalytic system for the carbomethoxylation of ethylene oxide. Appl. Catal. A General, 301, 75–78. doi:10.1016/j.apcata.2005.11.022
Kramer, J. W., Lobkovsky, E. B., and Coates, G. W. 2006. Practical beta-lactone synthesis: Epoxide carbonylation at 1 atm. Org. Lett., 8, 3709–3712. doi:10.1021/ol061292x
Kristjansdottir, S. S., Norton, J. R., Moroz, A., Sweany, R. L., and Whittenburg, S. L. 1991. Absence of hydrogen bonding between cobalt carbonyl (HCo(CO)4) and nitrogen or oxygen bases. IR and Raman study. Organometallics, 10, 2357–2361. doi:10.1021/om00053a044
Lee, J. T., and Alper, H. 2004. Alternating copolymerization of propylene oxide and carbon monoxide to form aliphatic polyesters. Macromolecules, 37, 2417–2421. doi:10.1021/ma030419k
Liu, J. H., Chen, J., and Xia, C. G. 2006. Methoxycarbonylation of propylene oxide: A new way to beta-hydroxybutyrate. J. Mol. Catal. A Chem., 250, 232–236. doi:10.1016/j.molcata.2006.01.047
Luo, J., Qu, E., Zhou, Y., Dong, Y., and Liang, C. 2020. Re/AC catalysts for selective hydrogenation of dimethyl 1, 4-cyclohexanedicarboxylate to 1, 4-cyclohexanedimethanol: Essential roles of metal dispersion and chemical environment. Appl. Catal. A General, 602, 117669. doi:10.1016/j.apcata.2020.117669
Luo, J., Wei, H., Liu, Y., Zhang, D., Zhang, B., Chu, W., et al. 2017. Oxygenated group and structural defect enriched carbon nanotubes for immobilizing gold nanoparticles. Chem. Commun., 53, 12750–12753. doi:10.1039/c7cc06594a
Li, B. T., Li, X. H., Asami, K., and Fujimoto, K. 2003a. Hydroformylation of 1-hexene over rhodium supported on active carbon catalyst. Chem. Lett., 32, 378–379. doi:10.1246/cl.2003.378
Li, B. T., Li, X. H., Asami, K. J., and Fujimoto, K. 2003b. Low-pressure hydroformylation of middle olefins over Co and Rh supported on active carbon catalysts. Energy fuels. 17, 810–816. doi:10.1021/ef0202440
Li, J., Wei, X., Wang, Q., Chen, J., Chang, G., Kong, L., et al. 2012. Homogeneous isolation of nanocellulose from sugarcane bagasse by high pressure homogenization. Carbohydr. Polym., 90, 1609–1613. doi:10.1016/j.carbpol.2012.07.038
Liu, J. H., Wu, H., Wu, L. W., Chen, J., and Xia, C. G. 2007. A novel and highly effective catalytic system for alkoxycarbonylation of (S)-propylene oxide. J. Mol. Catal. A Chem., 269, 97–103. doi:10.1016/j.molcata.2006.12.045
Liu, X. H., Hamasaki, A., Yamane, Y., Aikawa, S., Ishida, T., Haruta, M., et al. 2013. Gold nanoparticles assisted formation of cobalt species for intermolecular hydroaminomethylation and intramolecular cyclocarbonylation of olefins. Catal. Sci. Technol., 3, 3000-, doi:10.1039/c3cy00336a
Liu, X., Hu, B., Fujimoto, K., Haruta, M., and Tokunaga, M. 2009. Hydroformylation of olefins by Au/Co3O4 catalysts. Appl. Catal. B Environ., 92, 411–421. doi:10.1016/j.apcatb.2009.08.021
Lv, Z. G., Wang, H. S., Li, J. A., and Guo, Z. M. 2010. Hydroesterification of ethylene oxide catalyzed by 1-butyl-3-methylimidazolium cobalt tetracarbonyl ionic liquid. Res. Chem. Intermed., 36, 1027–1035. doi:10.1007/s11164-010-0215-5
Maruyama, K., Kanazawa, A., and Aoshima, S. 2022. ABC-type periodic terpolymer synthesis by a one-pot approach consisting of oxirane- and carbonyl-derived cyclic acetal generation and subsequent living cationic alternating copolymerization with a vinyl monomer. Macromolecules, 55, 799–809. doi:10.1021/acs.macromol.1c02468
Mirbach, M. F., and Mirbach, M. J. 1985. Reactions of cobalt carbonyls in methanol under high pressure of carbon monoxide: A reexamination of the hydroesterification mechanism. J. Mol. Catal., 32, 59–75. doi:10.1016/0304-5102(85)85033-1
Nazimov, D. A., Klimov, O. V., Saiko, A. V., Trukhan, S. N., Glazneva, T. S., Prosvirin, I. P., et al. 2021. Effect of the K loading on effective activation energy of isobutane dehydrogenation over chromia/alumina catalysts. Catal. Today, 375, 401–409. doi:10.1016/j.cattod.2020.03.005
Ocampo, R., and Dolbier, W. R. 2004. The Reformatsky reaction in organic synthesis. Recent advances. Tetrahedron, 60, 9325–9374. doi:10.1016/j.tet.2004.07.018
Qiu, X., Wang, S., Miao, S., Suo, H., Xu, H., Hu, Y., et al. 2021. Co-immobilization of laccase and ABTS onto amino-functionalized ionic liquid-modified magnetic chitosan nanoparticles for pollutants removal. J. Hazard. Mater., 401, 123353, doi:10.1016/j.jhazmat.2020.123353
Rajendiran, S., Gunasekar, G. H., and Yoon, S. 2018. A heterogenized cobaltate catalyst on a bis-imidazolium-based covalent triazine framework for hydroesterification of epoxides. New J. Chem., 42, 12256–12262. doi:10.1039/c8nj02292e
Rowley, J. M., Lobkovsky, E. B., and Coates, G. W. 2007. Catalytic double carbonylation of epoxides to succinic anhydrides: Catalyst discovery, reaction scope, and mechanism. J. Am. Chem. Soc., 129, 4948–4960. doi:10.1021/ja066901a
Suresh, R., Massad, I., and Marek, I. 2021. Stereoselective tandem iridium-catalyzed alkene isomerization-cope rearrangement of ω-diene epoxides: Efficient access to acyclic 1, 6-dicarbonyl compounds. Chem. Sci., 12, 9328–9332. doi:10.1039/d1sc02575a
Wang, Z., Guo, S., Wang, Z., Li, F., Xue, W., Wang, Y., et al. 2021. A highly efficient rod-like-CeO2-supported palladium catalyst for the oxidative carbonylation of glycerol to glycerol carbonate. RSC Adv., 11, 17072–17079. doi:10.1039/d1ra02187g
Wen, L. Y., Zhang, Z. J., Chen, X. P., Wang, S. Q., and Yu, W. L. 2018. Preparation of sodium cobalt tetracarbonyl and optimization of process conditions for hydroesterification of ethylene oxide. China Petroleum Process. Petrochem. Technol., 20, 78–84.
Xie, W., and Wan, F. 2018. Basic ionic liquid functionalized magnetically responsive Fe3O4@HKUST-1 composites used for biodiesel production. Fuel, 220, 248–256. doi:10.1016/j.fuel.2018.02.014
Xu, J. X., and Wu, X. F. 2019. Cobalt-catalyzed alkoxycarbonylation of epoxides to beta-hydroxyesters. J. Org. Chem., 84, 9907–9912. doi:10.1021/acs.joc.9b01007
Xu, B., Madix, R. J., and Friend, C. M. 2011. Activated metallic gold as an agent for direct methoxycarbonylation. J. Am. Chem. Soc., 133, 20378–20383. doi:10.1021/ja207389z
Zeng, B., Liu, M., He, J., Liu, J., Li, Z., Yun, D., et al. 2021. On the origin of nitrogen-containing promoters in the cobalt-catalyzed methoxycarbonylation of epoxides. Appl. Catal. A General, 623, 118291, doi:10.1016/j.apcata.2021.118291
Zhang, J., Li, Y., and Zhao, B. 2012. Non-noble metallic Fe catalyst systems for the reductive carbonylation of nitrobenzene. Acta chimi. Sin., 70, 35-, doi:10.6023/a1105163
Keywords: carbonylation, colloid immobilization, ethylene oxide, methyl 3-hydroxypropionate, kinetics
Citation: Luo J, Liu P, Yang W, Niu H, Li S and Liang C (2022) Chemical kinetics and promoted Co-immobilization for efficient catalytic carbonylation of ethylene oxide into methyl 3-hydroxypropionate. Front. Chem. 10:945028. doi: 10.3389/fchem.2022.945028
Received: 17 May 2022; Accepted: 29 June 2022;
Published: 22 July 2022.
Edited by:
Simon Freakley, University of Bath, United KingdomReviewed by:
Jianbin Chen, Qilu University of Technology, ChinaCopyright © 2022 Luo, Liu, Yang, Niu, Li and Liang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Changhai Liang, Y2hhbmdoYWlAZGx1dC5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.