 Valentina Villamil1,2
Valentina Villamil1,2 Graciela Mahler
Graciela Mahler
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem., 22 August 2022
Sec. Organic Chemistry
Volume 10 - 2022 | https://doi.org/10.3389/fchem.2022.934376
This article is part of the Research TopicWomen in Chemistry 2022View all 28 articles
The reversibility of the thiol-thioester linkage has been broadly employed in many fields of biochemistry (lipid synthesis) and chemistry (dynamic combinatorial chemistry and material science). When the transthioesterification is followed by a S-to-N acyl transfer to give an amide bond, it is called Native Chemical Ligation (NCL), a high-yield chemoselective process used for peptide synthesis. Recently, we described thioglycolic acid (TGA) as a useful reagent for thioester deprotection both in solution and anchored to a solid-support under mild conditions. Inspired by NCL, in this work, we extended this approach and explored the use of 2-aminothiols for the deprotection of thiols bearing an acyl group. The best results were obtained using cysteamine or L-cysteine in an aqueous buffer pH 8 at room temperature for 30 min. The described approach was useful for S-acetyl, S-butyryl, and S-benzoyl heterocycles deprotection with yields up to 84%. Employing this methodology, we prepared six new analogs 2 of mercaptomethyl bisthiazolidine 1, a useful inhibitor of a wide-range of Metallo-β-Lactamases (MBLs). Compared with the previous methodologies (TGA polymer supported and TGA in solution), the biomimetic deprotection herein described presents better performance with higher yields, shorter reaction times, less time-consuming operations, easier setup, and lower costs.
The thiol-thioester exchange occurs smoothly in neutral aqueous media at room temperature and is a highly chemoselective process. Due to these ideal features, this exchange reaction is found in many natural processes, including energy metabolism, mitosis, and autophagy (Pietrocola et al., 2015). Moreover, the abundance of thioester-mediated processes in nature has led to speculation regarding the role of these derivatives in the origin of life (Chandru et al., 2016; Vallee et al., 2017).
Besides the importance of the thiol-thioester exchange in biochemical processes, the reversibility of this linkage has been broadly used in the field of dynamic combinatorial chemistry (Mondal and Hirsch, 2015) and, more recently, applied in material science (Worrell et al., 2018).
In particular, Native Chemical Ligation (NCL) is a methodology used for peptide synthesis. It is a high-yielding chemoselective process, successfully applied to peptide and protein synthesis, chemical modification of proteins, protein-protein ligation, and the development of probes and molecular machines (Dawson et al., 1994; Gui et al., 2016; Burke et al., 2017). The NCL process begins with a reversible transthioesterification between two peptides. The mercapto group of an N-terminal cysteine residue of a peptide B attacks the C-terminal thioester of a second unprotected peptide A in an aqueous buffer at pH 7.0 (see Figure 1, Step 1). A second step consists of an S-to-N acyl transfer, leading to the formation of an amide bond between both peptides A and B (Figure 1, Step 2) (Serra et al., 2020).
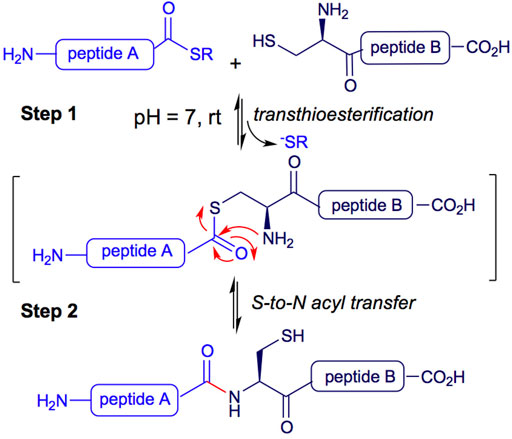
FIGURE 1. Native chemical ligation peptide synthesis.
Recently, we described another application of this reversible process applied to thiol deprotection in solution under mild conditions. This methodology is a valuable tool, especially when the substrate does not support harsh conditions like strong basic media. The deprotection is carried out using thioglycolic acid anchored to a solid support like TentaGel® resin (TG-NCO-SH, Method A) or in solution (Method B), Figure 2A (Villamil et al., 2021). Both homogeneous and heterogeneous approaches were conveniently carried out at room temperature, in aqueous buffer at pH 8, and the methods were useful for thioacetyl deprotection of several substrates, affording the free thiol after 24 h of reaction.
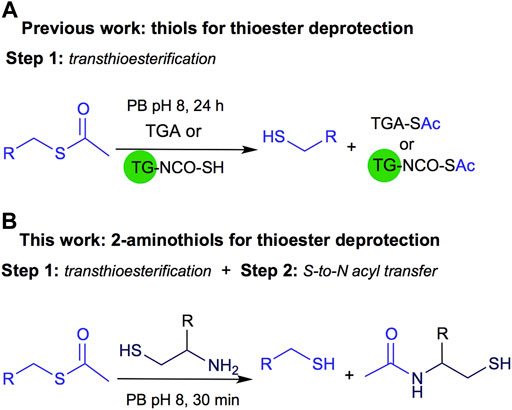
FIGURE 2. (A) Previous work on thioester deprotection using Method A polymer supported TGA (TG-NCO-SH) or Method B TGA in solution. (B) This work uses 2-aminothiols for thioacetyl deprotection.
Bisthiazolidines 1 (BTZ) are cross-class metallo-β-lactamases (MBLs) inhibitors active in vitro and against MBL-expressing bacterial pathogens, Figure 3. These compounds were designed as penicillin analogs and behaved as competitive inhibitors of MBLs from all subclasses, with Ki values in the micromolar range. BTZ were effective against NDM-1, VIM-2, L1, and IMP-1, all MBLs of clinical importance (Mojica et al., 2015; González et al., 2016; Hinchliffe et al., 2016).
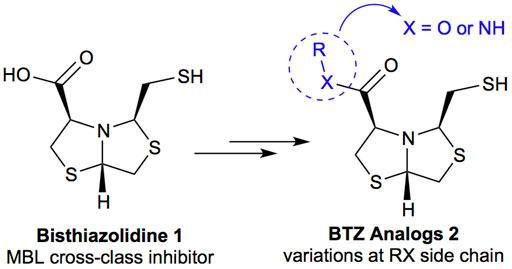
FIGURE 3. Bisthiazolidines 1 and BTZ analogs 2 with variations at RX.
Previous studies showed that the mercaptomethyl group present in 1 is essential for MBL inhibition (Hinchliffe et al., 2016). With this in mind, we aimed to prepare new bisthiazolidine analogs 2 with variations on the carboxylic acid (R) in order to improve the inhibition activity. The synthetic route proposed includes thiol deprotection as the key step, see Figure 3.
Inspired by the NCL approach, in this work we propose to use 2-aminothiols for the deprotection of the thioacetyl derivatives as shown in Figure 2B. Although NCL is widely used in several fields of organic chemistry, to the best of our knowledge, it has not been explored for thioester deprotection in solution.
In order to prepare new BTZ analogs 2, first we synthesized S-acetylated bisthiazolidine 3 as previously described (Villamil et al., 2021). Then, amides 4a-c, 4f, and esters 4d-e were obtained starting from 3 and the corresponding amine or alcohol (R-XH), using HBTU as a coupling agent in DCM with yields ranging from 38 to 79%, see Scheme 1.
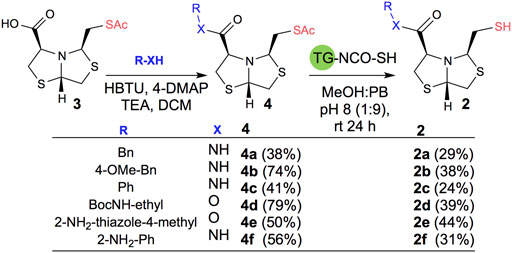
SCHEME 1. Synthesis of 2a-f using acetyl deprotection: Method A: TG-NCO-SH (2 eq), MeOH:PB pH 8 (1:9), 24 h.
Based on our previous results, we performed the deprotection of thioacetylated compounds 4a-f using TGA anchored to a TentaGel® resin (TG-NCO-SH), Method A. In this reaction, a transthioesterification process occurs between the thioester and the thiol. The mercaptomethyl derivatives 2a-f were obtained after chromatographic column purification with yields ranging from 24 to 44%, as shown in Scheme 1.
Inspired by the Native Chemical Ligation (NCL) mechanism, where a transthioesterification reaction is followed by a 1-5 S,N-acyl transfer to form an amide and shift the equilibrium toward the product (Figure 1; Agouridas et al., 2019) and the report for the use of cysteamine (Cym) in acetonitrile under reflux for the conversion of simple and unfunctionalized thioesters into thiols (Endo et al., 1974), we envisioned that 2-aminothiols 5a-c could be useful for the deprotection of acyl thioesters, as shown in Figure 2B.
In order to rapidly evaluate several sets of reaction conditions, we used reaction miniaturization, a concept developed by High Throughput Experimentation (HTE) to find the best reagents and conditions for a desired product. HTE is a tool that allows to perform a large number of experiments in parallel. The advantages of the reaction miniaturization studies is that they require a few milligrams of reagents (microscale) and shorter manipulation times compared to traditional experimentation (Wong and Cernak, 2018; Mennen et al., 2019).
Experimental and computational studies support the notion of the thiolate as the effective nucleophilic species in NCL (Wang et al., 2011; Diemer et al., 2022). In this sense, the aminothiols 5a-c to be screened were selected basically based on the pKa value of the thiol, dismissing other possible ionic species (Maguire et al., 2020).
We reasoned that L-Cysteine ethyl ester (L-CysOEt, 5a) bearing a thiol with a pKa = 6.5 could be first explored since it is close to neutral pH (Friedman et al., 1965). According to the Henderson-Hasselbach equation, the ratio of the L-CysOEt species SH/S− is 1/2.5 in H2O at pH 7.30 (Henderson, 1908). A different aspect to consider is that thiolates are susceptible to oxidation (leading to disulfide species), a process that is accelerated when the pH of the solution is close to or above the thiol pKa.
Taking these considerations into account, the deprotection reaction of 3 was screened in aqueous buffers pH range from 5 to 8, aminothiol 5a equivalents from 1 to 10 and the time from 0.5 to 6 h, see Supplementary Table S1. Aliquots were taken at different times and diluted for HPLC analysis. Overall, the best conditions for 3 deprotection were achieved using 2 eq L-CysOEt at pH 8, during 2 h to give 1 in 90% yield (see Supplementary Table S1). Since the thiol−thioester exchange is an equilibrium (Bongiardina et al., 2021), an excess of the deprotecting agent 5a is required to shift the equilibrium toward the desired product. The reactions performed at pH 5 and 6 provided the less favorable yields, even when using more equivalents of 2-aminothiol or longer reaction times.
Based on these results, we extended the screening of 2-aminothiols including L-Cysteine (L-Cys, 5b) and Cysteamine (Cym, 5c) for the deprotection of thioacetyl bisthiazolidine 3. These 2-aminothiols bear a thiol pKa value of 8.3 and 8.2 respectively (Pitman and Morris, 1979). Thus, to ensure a significant amount of thiolate, the reaction was performed at pH 7 (Table 1 entry 1–3) and pH 8 (Table 1, entry 4–7), and the reaction time was screened from 0.25 to 2 h using 2 equivalents of 5b/c.
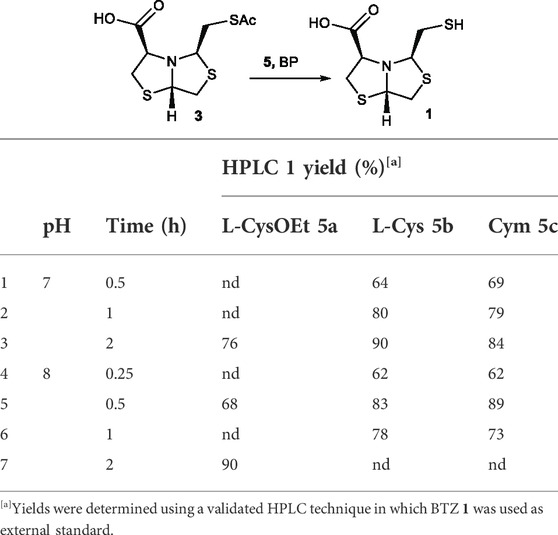
TABLE 1. Optimization of thioester deprotection using different 2-aminothiols 5a-c for the deprotection of 3.
Both 2-aminothiols, L-Cys 5b and Cym 5c, gave similar yields compared to L-Cys-OEt 5a. Overall, Cym 5c was the best deprotecting agent, using 2 equivalents at pH 8 after only 30 min, affording 1 in 89% HPLC yield, Table 1, entry 5.
The HTE allowed us to find the best reaction conditions by running several experiments simultaneously rather than carrying out single ones, reducing the cost and time of the experiments. Overall, we performed 23 reactions in aqueous media, using less than 30 mg of compound 3 to find the optimal conditions for the deprotection reaction. This example shows the potential of the reaction miniaturization tool, mainly due to waste reduction and shorter time-consuming operations.
Once the best reaction conditions were established, the reaction was scaled-up for deprotection of 3 using 2-aminothiols L-CysOEt 5a, L-Cys 5b, and Cym 5c at pH 8 for 30 min. The isolated yields of 1 were 65%, 75%, and 78%, respectively, see Table 2, entries 1, 2, and 3. These results correlate well with the yields determined by HPLC at 68%, 83%, and 89%, respectively (Table 1), validating the results obtained by HTE. The three 2-aminothiols employed afford the free thiols in better yields than previously described TGA in solution (56% yield after 30 min), Table 2. The yield values for TGA in solution was obtained in a previous work (Villamil et al., 2021).
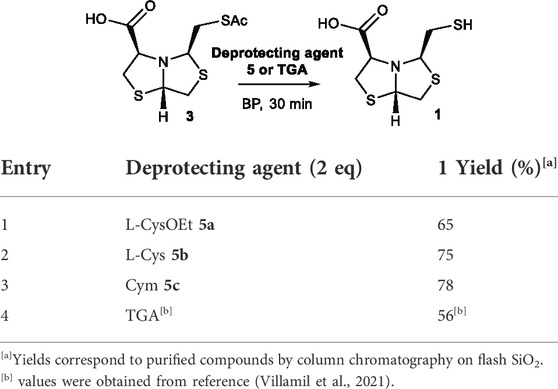
TABLE 2. Deprotection of thioester 3 under optimized conditions MeOH:BP (1:9), pH 8, rt, 30 min, aminothiol 5 (2 eq) or TGA (2 eq).
Deprotection of thioacetyl bicycles 4a-f using the optimized conditions (Method C: Cym 5c pH 8, 30 min at rt) led to analogs 2a-f in yields ranging from 50 to 78%, Table 3. The average yield was 65%, remarkably higher than those obtained using the TGA solid-supported approach (Method A, average yield of 34%). In addition, the reaction time decreased from 24 h to 30 min. This aspect is crucial since products 2a-f, containing a free thiol, are easily oxidized in aqueous buffer. 2-aminothiols 5 allowed to set up reactions in shorter reaction times compared with TGA, affording better yields, probably by avoiding product oxidation.
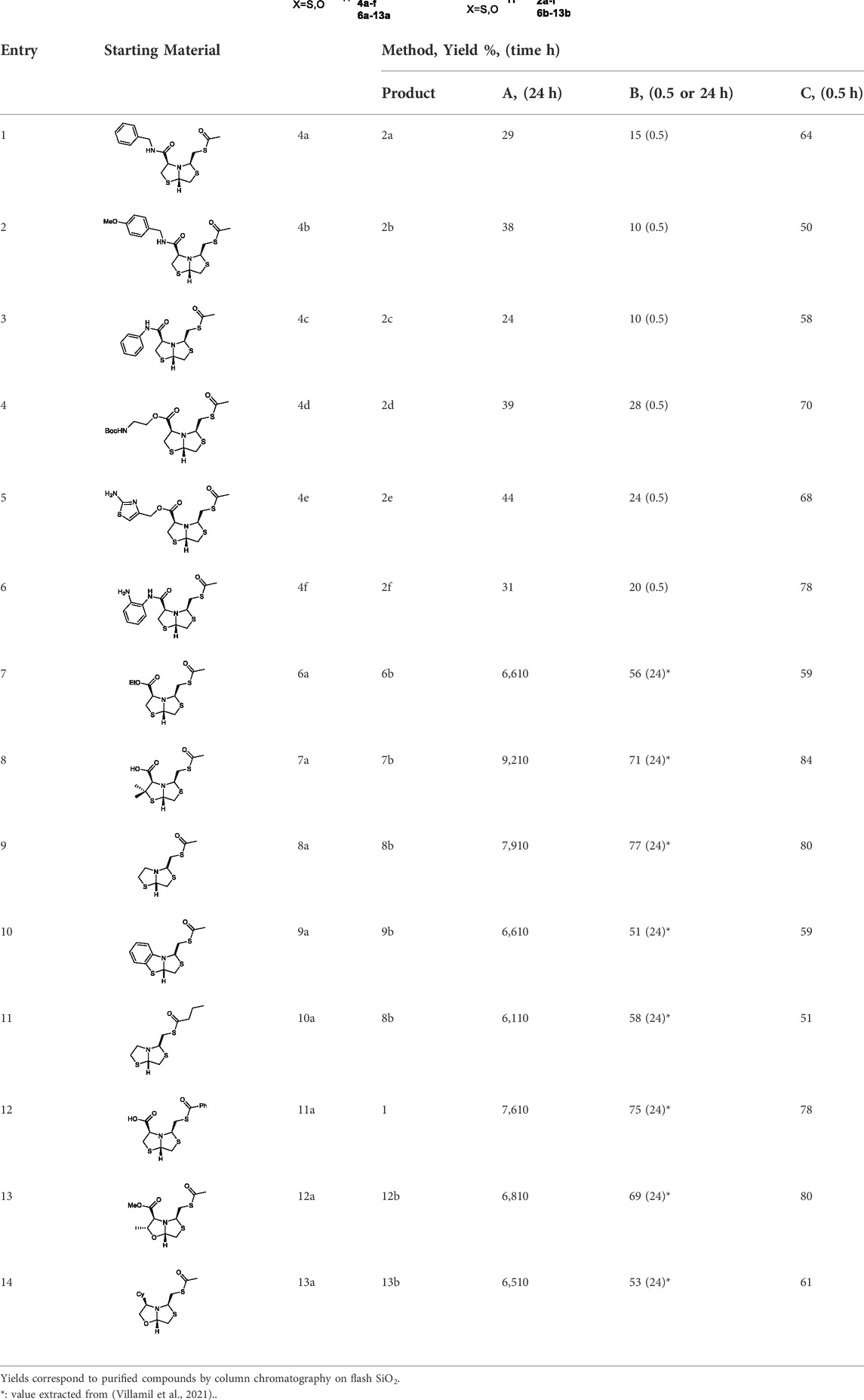
TABLE 3. Thiol deprotection yield (%) using Method A, B and C of different substituted BTZ. Method A: TG-NCO-SH (2 eq) MeOH:BP pH 8 (1:9), 24 h; Method B: TGA (2 eq), MeOH:PB pH 8 (1:9), 0.5 or 24 h; Method C: Cym (2 eq), MeOH:PB pH 8 (1:9)0.5 h.
In order to highlight the best performance of Cym regarding TGA in solution, we carried out the deprotection reaction of the new derivatives 4a-f using method B (TGA in solution) after 30 min instead of 24 h, to compare the yields with method C (Cym in solution). The average yields were methods B and C, was 18% and 65%, respectively, significantly higher when using Cym, Table 3. The yield values for methods A and B (24 h) were obtained in a previous work (Villamil et al., 2021).
To extend the methodology scope, different S-acyl bisthiazolidines 6a-11a, and S-acetyl oxazolidines 12a and 13a were assayed using Cym 5c (Method C) and compared with our previous results using TGA solid-supported and TGA in solution (Method A and B, respectively) (Villamil et al., 2021), see Table 3.
Method C smoothly deprotected S-acetyl bisthiazolidines/oxazolidines 6a-9a, 12a, and 13a with yields ranging from 59 to 84%, Table 3, entries 7–10, 13, and 14. In addition, different acyl side chains like propyl thioester 10a and benzoyl thioester 11a were deprotected in moderate to good yields, 51 and 78%, respectively, in Table 3, entries 11 and 12.
Altogether, the results obtained with our previous approaches (Methods A and B) led to slightly lower average yields compared to Method C (56, 64, and 67%, respectively) and shorter reaction times, 24 h vs 30 min. In addition, regarding method A, Cym is a common reagent in organic laboratories and is less expensive than Tentagel® resin.
Overall, Method C herein reported provides better yields in a shorter time with a lower-cost reagent and easier setup.
As described in NCL, the presence of a 2-aminothiol enables an intramolecular S-to-N-acyl migration, releasing the thiol and displacing the equilibrium toward the products (Wang et al., 2011). This fact suggests that when using Cym for thioester deprotection, an analogous process is occurring. In order to confirm the deprotection mechanism, NMR experiments were performed. Three sets of conditions were assayed in PB pH 8 using different deprotecting agents: 1) thioglycolic acid (TGA, 2 eq); 2) ethylenediamine (ETN, 2 eq); and 3) Cysteamine 5c (2 eq).
In the first experiment, when using TGA for thiol deprotection, after 40 min of reaction, 50% of the deprotection product was observed and thioacetylated TGA could be detected, Figure 4A.
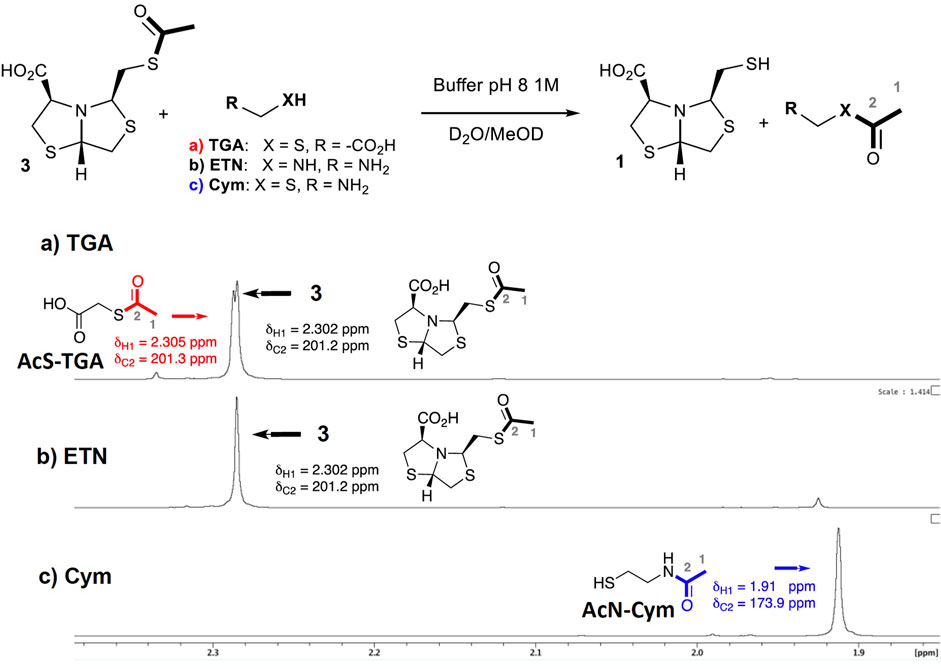
FIGURE 4. Comparison of 1H-NMR spectra for the deprotection reaction of 3 using different additives. Reaction of 3 (0.04 mmol) in D2O, PB [1 M] at pH 8, and MeOD-d4 (10%) at 27°C after 40 min: (A) TGA (0.08 mmol); (B) ethylenediamine (0.08 mmol); (C) Cym 5c (0.08 mmol). The 13C chemical shift of C2 atoms was obtained from 1H,13C-HMBC correlations to the respective acetyl protons.
Transthioesterification is the main deprotection mechanism in these conditions. A complete deprotection was observed after 9 h of incubation, Supplementary Figure S1.
On the other hand, when ethylenediamine (ETN) is used, the deprotection reaction is not observed by 1H NMR after 40 min, Figure 4B. This result indicates that only an amine group is not able to deprotect the thiol.
Finally, when Cym 5c is used as a deprotecting agent, a complete thiol deprotection is observed after 40 min of incubation. Only N-acetylated cysteamine and free-thiol 1 were found in the 1H-NMR spectra, while S-acetylated cysteamine was not detected, see Figure 4C. The 1H-NMR spectra at t = 40 min comparison of experiments 1) and c) shows that the reaction rate is faster for c), observing a complete deprotection of 3.
Based on the obtained results and the numerous studies of the NCL mechanism (Barnett and Jencks, 1969; Wang et al., 2011), we propose, as a first step, a reversible S,S-acyl transfer involving an anionic concerted SN2 displacement mechanism with the formation of a tetrahedral intermediate, Figure 5, Step I (Barnett and Jencks, 1969). This is a common intermediate for the deprotection of thioesters using 2-aminothiols or thiols lacking the amino group.
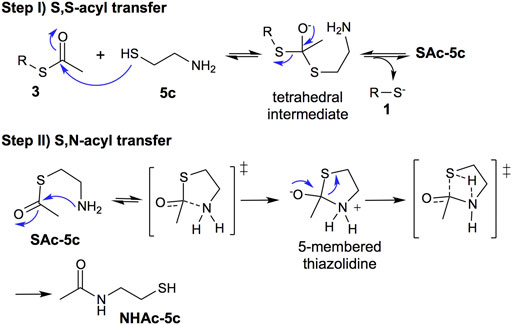
FIGURE 5. Mechanism proposal: step (I) reversible thiol−thioester exchange through a tetrahedral intermediate; step (II) irreversible thiazolidine intermediate to S,N-acyl transfer process.
The second step involves the S,N-acyl transfer, which could proceed by an 5-endo-trig cyclization to form a thiazolidine intermediate, an irreversible step faster than the thiol−thioester exchange (Figure 5, step II).
In summary, the thiol-thioester exchange step is reversible, whereas the S,N-acyl transfer is irreversible, displacing the thiol-thioester exchange equilibrium toward the product 1 and N-acetylated cysteamine (NHAc-5c), increasing the deprotection reaction rate.
In summary, we found that 2-aminothiols are suitable deprotecting agents for acyl thioester groups under aqueous mild conditions, mimicking a NCL approach. The reversibility of thiol−thioester exchange and the high efficacy of the intramolecular S,N-acyl migration process are driving forces to increase the reaction rate, affording shorter reaction times. In addition to this, the mild experimental conditions, near neutral pH, disfavor oxidation processes or bisthiazolidine-ring opening, increasing the deprotection reaction yields.
The new Cym deprotection methodology allowed us to prepare six new analogs of BTZ 1 with different substitutions at carboxylic acid (2a-f) to further be evaluated as MBL inhibitors.
The biomimetic deprotection herein described presents better performance than previous reported methodologies (TGA polymer supported and TGA in solution), with higher yields, less time-consuming operation, shorter reaction times, easier setup, and lower costs. We envision that these conditions could provide a simple and practical method for labile thioester hydrolysis.
General Methods. All reactions were carried out in dry, freshly distilled solvents under anhydrous conditions unless otherwise stated. Reactions were monitored by analytical thin layer chromatography (TLC) on 0.25 mm silica gel coated plastic sheets (SIL G/UV 254). Flash chromatography on Silica gel 60 (40 µm average particle diameter) was used to purify the crude reaction mixtures. Yields are reported for chromatographic and spectroscopically (1H and 13C NMR) pure compounds unless otherwise stated. 1H and 13C NMR spectra were recorded on a Bruker Avance 400 instrument at 400 and 100 MHz, respectively. Chemical shifts (δ) are expressed in ppm downfield from TMS as an internal standard unless otherwise stated. Multiplicities are indicated as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), b (broad). Assignments of 1H and 13C NMR peaks were made based on a combination of COSY, HSQC, and HMBC spectra. Electrospray ESI high-resolution mass spectra (HRMS) were recorded on a MicroTOF-Q spectrometer from Bruker Daltronics. Optical rotation was measured using a Jasco p-2000 polarimeter with a 2.0 ml cell, optical path length of 100 mm and sodium lamp (λ = 589 nm) at room temperature. The concentration c is given as g/100 ml.
HPLC Equipment and Method Validation. The liquid chromatography analysis was performed using Waters HPLC equipment, with binary pumps (Waters 1,525) and a photodiode array detector (Waters 2,996), with a loop injection of 20 μL (Rheodyne 1727). A reverse phase C18 separation column was used (Kinetex NUCLEOSIL® C18, 150 mm × 4 mm, 5 µm) with detection at λ = 205 nm for all compounds at 37°C. The eluent consisted of TFA 0.003 M (mobile phase A) and MeCN (mobile phase B) at a flow rate of 1.2 ml/min. The injection volume was 20 µl. Initial conditions 70/30 (mobile phase A/mobile phase B) changed in 2 min–55/45, maintained for 3.5 min, and changed in 0.1 min–5/95, maintained for 2.4 min. Data and chromatograms were collected and analyzed using the Empower System program by Waters Corporation, 2002. System linearity was verified in the concentration range: 0.01, 0.041, 0.102, 0.163, 0.203, and 0.244 mg/ml, prepared from a stock solution of BTZ 1, 1 mM in MeOH. Linearity was established from the calibration curve using least squares linear regression analysis and a correlation coefficient (R2) value of 0.9991 was found. A standard of 0.203 mg/ml was injected five times to evaluate the precision system, and a % RSD value of 1.0% was found.
Compounds 1, (González et al., 2016; Hinchliffe et al., 2016), 3, (Villam il et al., 2021), 6–13 (Saiz et al., 2014; Villamil et al., 2021) were prepared according to previous reports.
A solution of 4a (0.0167 g, 0.0454 mmol) in MeOH (0.5 ml) and degassed BP pH 8 1M (1.0 ml) was added to the polymer-supported deprotecting agent (TG-NCO-SH, 0.222 g, 0.0908 mmol) prepared as previously described (Villamil et al., 2021) and stirred for 24 h at room temperature under a nitrogen atmosphere. Then, the polymer was filtered and washed with MeOH. The organic layer was dried over Na2SO4, filtered and evaporated. The mixture was poured into a NaCl saturated solution and extracted with EtOAc (3 × 30 ml). The combined organic layers were dried, filtered, and concentrated to dryness. The crude was purified by column chromatography on flash SiO2 (CH2Cl2/nHex 3:1) to yield 2a (0.0043 g, 29%) as a colorless oil.
In a two-necked flask, 4a (0.04 g, 0.11 mmol) was dissolved in MeOH (2 ml) and BP pH 8 1M (18 ml) and cysteamine hydrochloride 5c (0.026 g, 0.22 mmol) was added. The reaction mixture was stirred for 30 min at room temperature, then poured into a NaCl saturated aqueous solution (30 ml) and extracted with EtOAc (3 × 30 ml). The combined organic layers were dried, filtered, and concentrated to dryness. The crude was purified by column chromatography on flash SiO2 (CH2Cl2/nHex 3:1) to yield 2a (0.023 g, 64%) as a colorless oil.
Compound 3 was incubated into separate 2 ml Eppendorf® tubes at different pH and L-Cys-OEt 5a concentrations, as described: 3 (100 µL of a solution of 42 mM in MeOH) was added to aqueous buffer 1M (pH 5, 6, 7 or 8, final volume 1 ml) and L-Cys-OEt 5a (10, 20, 60 or 100 µl of a solution 420 mM in MeOH, corresponding to 1, 2, 6 or 10 equivalents) was added. The reaction solution was kept at 25°C and at suitable time intervals, aliquots of 100 µl were taken and acetonitrile (400 µl) was added. Then, the solution was vortexed, filtered, and chromatographed (Supplementary Table S1).
Optimization of the deprotection conditions of 3 using L-Cys 5b and Cym 5c was carried out using the same methodology as described for L-Cys-OEt 5a (Table 1).
(Table 2) In a two-necked flask, compound 3 was dissolved (0.05 g, 0.18 mmol) in MeOH (1 ml) and added BP at pH 8 1M (19 ml) and L-Cys-OEt 5a hydrochloride (0.067 g, 0.36 mmol). The reaction mixture was stirred for 30 min at room temperature. It was poured into HCl 5% and extracted with EtOAc (3 × 30 ml). The combined organic extracts were dried, filtered, and concentrated to dryness, and the crude was purified by column chromatography on flash SiO2 (nHex/EtOAc/AcOH 7:3:0.01) to yield 1 (0.028 g, 65%).
Deprotection of 3 using thiols 5b, 5c, and TGA was carried out using the same methods as described for 5a (Table 2).
Into a two-necked flask under nitrogen atmosphere, 3 (0.1 , 0.36 mmol) was dissolved in dry CH2Cl2 (8 ml) and cooled in an ice bath. To the mixture was added HBTU (0.163 g, 0.43 mmol), DIPEA (0.139 g, 1.08 mmol), 4-DMAP (0.005 g, 0.036 mmol) and benzylamine (0.043 g, 0.4 mmol) and warmed to room temperature with stirring for 2 h. Then it was poured into a NaCl saturated aqueous solution (30 ml) and extracted with CH2Cl2 (3 × 30 ml). The combined organic layers were dried, filtered, and concentrated in vacuo to dryness. The crude product was purified by column chromatography on flash SiO2 (nHex/EtOAc 3:1) to yield 4a (0.051 g, 38%) as a white solid: MP = 61–65°C [α]D20 -54.9 (c = 0.91 in CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 7.51 (br, 1H, NH), 7.37–7.28 (m, 5H, ArH), 4.94 (dd, J = 5.8, 4.1 Hz, 1H, CH), 4.53 (dd, J = 14.9, 6.3 Hz, 1H, CH-H), 4.44 (dd, J = 14.9, 5.7 Hz, 1H, CH-H), 4.33 (dd, J = 8.1, 5.2 Hz, 1H, CH), 4.09 (dd, J = 7.1, 2.9 Hz, 1H, CH), 3.55 (dd, J = 11.9, 5.8 Hz, 1H, CH-H), 3.52 (dd, J = 11.5, 2.5 Hz, 1H, CH-H), 3.27 (dd, J = 11.1, 7.1 Hz, 1H, CH-H), 3.19 (dd, J = 14.0, 5.2 Hz, 1H, CH-H), 3.13 (dd, J = 14.0, 8.1 Hz, 1H, CH-H), 3.09 (dd, J = 11.9, 4.0 Hz, 1H, CH-H), 2.14 (s, 3H, CH3); 13C{1H} NMR (100 MHz, CDCl3) δ 195.1, 170.3, 138.2, 128.9, 127.8, 127.7, 73.3, 72.8, 72.0, 43.6, 39.7, 37.8, 33.6, 30.6, HRMS (ESI/Q-TOF) m/z: [M + Na]+ Calcd for C16H20N2O2NaS3 391.0585, found 391.0585.
Prepared in an analogous route as described for 4a, starting from 3 (0.12 g, 0.43 mmol) and 4-methoxybenzylamine (0.065 g, 0.47 mmol), purified by column chromatography on flash silica gel (nHex/EtOAc 2:1) to yield 4b (0.127 g, 74%) as a colorless oil [α]D20 −73.6 (c = 1.96 in CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 7.43 (t, J = 5.9 Hz, 1H, ArH), 7.22 (dt, J = 4.9, 2.9 Hz, 2H, ArH), 6.88 (dt, J = 5.0, 3.0 Hz, 2H, ArH), 4.92 (dd, J = 5.8, 4.1 Hz, 1H, CH), 4.46 (dd, J = 14.6, 6.3 Hz, 1H, CH-H), 4.36 (dd, J = 14.6, 5.6 Hz, 1H, CH-H), 4.31 (dd, J = 7.9, 5.4 Hz, 1H, CH), 4.07 (dd, J = 7.1, 2.9 Hz, 1H, CH), 3.80 (s, 3H, CH3), 3.54 (dd, J = 11.9, 5.8 Hz, 1H, CH-H), 3.50 (dd, J = 11.1, 2.9 Hz, 1H, CH-H), 3.26 (dd, J = 11.1, 7.1 Hz, 1H, CH-H), 3.17 (dd, J = 13.6, 5.0 Hz, 1H, CH-H), 3.12 (dd, J = 13.7, 7.5 Hz, 1H, CH-H), 3.08 (dd, J = 11.9, 4.1 Hz, 1H CH-H), 2.17 (s, 3H, CH3); 13C{1H} NMR (100 MHz, CDCl3) δ 195.1, 170.1, 159.2, 130.3, 129.2, 114.2, 73.3, 72.7, 71.9, 55.4, 43.1, 39.6, 37.8, 33.6, 30.6. HRMS (ESI/Q-TOF) m/z: [M + Na]+ Calcd for C17H22N2O3NaS3 421.0690, found 421.0687.
Prepared in an analogous route as described for 4a, starting from 3 (0.15 g, 0.54 mmol) and aniline (0.055 g, 0.59 mmol), purified by column chromatography on flash SiO2 (nHex/EtOAc 5:2) to yield 4c (0.077 g, 41%) as a colorless oil [α]D20 −177.5 (c = 1.84 in CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 9.03 (br, 1H, NH), 7.65 (d, J = 7.7 Hz, 2H, ArH), 7.35 (t, J = 7.9 Hz, 2H, ArH), 7.13 (t, J = 7.4 Hz, 1H, ArH), 5.03 (dd, J = 5.7, 4.6 Hz, 1H, CH), 4.42 (dd, J = 9.0, 4.2 Hz, 1H, CH), 4.12 (dd, J = 7.2, 3.1 Hz, 1H, CH), 3.63 (dd, J = 11.9, 5.9 Hz, 1H, CH-H), 3.54 (dd, J = 11.2, 3.0 Hz, 1H, CH-H), 3.33 (dd, J = 14.4, 3.9 Hz, 1H CH-H), 3.32 (dd, J = 11.7, 7.9 Hz, 1H, CH-H), 3.20 (dd, J = 14.1, 9.1 Hz, 1H, CH-H), 3.12 (dd, J = 11.9, 4.3 Hz, 1H, CH-H), 2.25 (s, 3H, CH3); 13C{1H} NMR (100 MHz, CDCl3) δ 195.3, 168.3, 137.6, 129.2, 124.7, 119.7, 73.2, 73.1, 71.9, 39.8, 37.8, 33.5, 30.8; HRMS (ESI/Q-TOF) m/z: [M + Na]+ Calcd for C15H18N2O2NaS3 377.0428, found 377.0428.
Prepared in an analogous route as described for 4a starting from 3 (0.1 g, 0.36 mmol) and N-boc-ethanolamine (0.069 g, 0.43 mmol), purified by column chromatography on flash SiO2 (nHex/EtOAc 7:3) to yield 4d (0.12 g, 79%) as a white powder: MP = 65–71°C [α]D20 -74.0 (c = 3.25 in CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 5.15 (dd, J = 5.3, 3.4 Hz, 1H, CH), 5.05 (br, 1H, NH), 4.31 (t, J = 6.7 Hz, 1H, CH), 4.25–4.22 (m, 3H, CH, CH2), 3.58, (dd, J = 11.9, 5.4 Hz, 1H, CH-H), 3.45–3.43 (m, 2H, CH2), 3.34–3.26 (m, 3H, CH2, CH-H), 3.10 (dd, J = 11.6, 3.7 Hz, 1H, CH-H), 3.08 (dd, J = 13.9, 7.4 Hz, 1H, CH-H), 2.36 (s, 3H, CH3), 1.45 (s, 9H, (CH3)3); 13C{1H} NMR (100 MHz, CDCl3) δ 195.8, 170.5, 155.9, 79.7, 74.1, 71.6, 70.0, 64.9, 39.6, 38.4, 37.7, 34.3, 30.7, 28,5; HRMS (ESI/Q-TOF) m/z: [M + Na]+ Calcd for C16H26N2O5NaS3 445.0902, found 445.0906.
Prepared in an analogous route as described for 4a starting from 3 (0.2 g, 0.7 mmol) and (2-aminothiazol-4-yl)methanol (0.1 g, 0.77 mmol), purified by column chromatography on flash silica gel (nHex/EtOAc 2:3) to yield 4e (0.137 g, 50%) as a colorless oil [α]D20 −33.8 (c = 2.3 in CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 6.51 (s, 1H, H (4)-thiazol), 5.64 (br, 2H, NH2), 5.18 (dd, J = 5.3, 3.3 Hz, 1H, CH), 5.06 (d, J = 12.6 Hz, 1H, CH-H), 5.02 (d, J = 12.6 Hz, 1H, CH-H), 4.29 (t, J = 6.8 Hz, 1H, CH), 4.28 (dd, J = 6.5, 5.0 Hz, 1H, CH), 3.58 (dd, J = 11.9, 5.4 Hz, 1H, CH-H), 3.34–3.24 (m, 3H, CH-H, CH2), 3.12–3.06 (m, 2H, CH2), 2.34 (s, 3H, CH3); 13C{1H} NMR (100 MHz, CDCl3) δ 195.7, 170.3, 168.7, 146.1, 107.6, 74.1, 71.5, 69.9, 62.7, 38.5, 37.7, 34.5, 30.7; HRMS (ESI/Q-TOF) m/z: [M + Na]+ Calcd for C13H17N3O3NaS4 414.0050, found 414.0041.
Prepared in an analogous route as described for 4a starting from 3 (0.15 g, 0.54 mmol) and o-phenylenediamine (0.203 g, 1.88 mmol), purified by column chromatography on flash SiO2 (CH2Cl2/EtOAc 9:1) to yield 4f (0.111 g, 56%) as a colorless oil: [α]D20 −100.7 (c = 4.5 in CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 8.87 (br, 1H, NH), 7.23 (d, J = 7.8 Hz, 1H, ArH), 7.06 (t, J = 7.6 Hz, 1H, ArH), 6.80 (t, J = 7.5 Hz, 2H, ArH), 5.07 (t, J = 5.0 Hz, 1H, CH), 4.43 (dd, J = 8.1, 5.1 Hz, 1H, CH), 4.16 (dd, J = 7.0, 2.4 Hz, 1H, CH), 3.90 (br, 2H, NH2), 3.57 (dd, J = 11.9, 5.9 Hz, 1H, CH-H), 3.51 (dd, J = 11.3, 2.3 Hz, 1H, CH-H), 3.30–3.18 (m, 3H, CH2, CH-H), 3.09 (dd, J = 11.9, 4.1 Hz, 1H, CH-H), 2.26 (s, 3H, CH3); 13C{1H} NMR (100 MHz, CDCl3) δ 195.4, 169.1, 140.8, 127.5, 125.4, 123.4, 119.4, 117.9, 73.2, 72.9, 72.0, 39.7, 37.7, 33.6, 30.7; HRMS (ESI/Q-TOF) m/z: [M + Na]+ Calcd for C15H19N3O2NaS3 392.0537, found 392.0540.
Yield: Method A (29%): Method C (64%)
1H NMR (400 MHz, CDCl3) δ 7.77 (br, 1H, NH), 7.36–7.28 (m, 5H, ArH), 4.88 (dd, J = 5.8, 5.0 Hz, 1H, CH), 4.49 (qd, J = 14.8, 6.0 Hz, 2H, CH2), 4.31 (dd, J = 8.1, 5.3 Hz, 1H, CH), 4.07 (dd, J = 7.3, 3.7 Hz, 1H, CH), 3.49 (dd, J = 11.0, 3.9 Hz, 1H, CH-H), 3.46 (dd, J = 11.6, 5.7 Hz, 1H, CH-H), 3.34 (dd, J = 11.2, 7.3 Hz, 1H, CH-H), 3.06 (dd, J = 11.8, 4.8 Hz, 1H, CH-H), 2.77 (ddd, J = 13.4, 8.1, 5.3 Hz, 1H, CH-H), 2.69 (dd, J = 13.9, 8.4 Hz, 1H, CH-H), 1.55 (t, J = 8.4 Hz, 1H, SH); 13C{1H} NMR (100 MHz, CDCl3) δ 170.3, 138.2, 128.9, 127.9, 127.7, 74.9, 72.9, 72.1, 43.6, 39.5, 33.8, 33.3; HRMS (ESI/Q-TOF) m/z [M + Na]+ Calcd for C14H18N2ONaS3 349.0479, found 349.0478; [α]D20 -80.0 (c = 0.66 in CH2Cl2).
Prepared in an analogous route as described for 2a starting from 4b (0.0181 g, 0.0454 mmol), purified by column chromatography on flash SiO2 (nHex/EtOAc 3:1) to yield 2b (Method A: 38%, Method C: 50%) as a colorless oil [α]D20 -108.5 (c = 1.0 in CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 7.69 (br, 1H, NH), 7.23–7.19 (m, 2H, ArH), 6.88–6.85 (m, 2H, ArH), 4.86 (dd, J = 5.8, 4.9 Hz, 1H, CH), 4.47–4.36 (m, 2H, CH2), 4.29 (dd, J = 8.1, 5.3 Hz, 1H, CH), 4.05 (dd, J = 7.3, 3.8 Hz, 1H, CH), 3.80 (s, 3H, CH3), 3.46 (dd, J = 11.2, 3.8 Hz, 1H, CH-H), 3.44 (dd, J = 11.8, 6.0 Hz, 1H, CH-H), 3.32 (dd, J = 11.2, 7.3 Hz, 1H, CH-H), 3.05 (dd, J = 11.8, 4.8 Hz, 1H, CH-H), 2.76 (ddd, J = 13.6, 8.1, 5.3 Hz, 1H, CH-H), 2.68 (dt, J = 14.0, 8.4 Hz, 1H, CH-H), 1.56 (t, J = 8.4 Hz, 1H, SH); 13C{1H} NMR (100 MHz, CDCl3) δ 170.2, 159.2, 130.3, 129.2, 114.3, 74.9, 72.8, 72.1, 55.4, 43.0, 39.5, 33.8, 33.3; HRMS (ESI/Q-TOF) m/z: [M + Na]+ Calcd for C15H20N2O2NaS3 379.0585, found 379.0591.
Prepared in an analogous route as described for 2a starting from 4c (0.0161 g, 0.0454 mmol), purified by column chromatography on flash SiO2 (nHex/EtOAc 3:1) to yield 2c (Method A: 24%, Method C: 58%) as a colorless oil [α]D20 −116.6 (c = 0.25 in CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 9.44 (br, 1H, NH), 7.65 (dd, J = 8.5, 1.0 Hz, 2H, ArH), 7.36–7.31 (m, 2H, ArH), 7.15–7.11 (m, 1H, ArH), 4.99 (dd, J = 5.9, 5.2 Hz, 1H, CH), 4.43 (dd, J = 8.4, 4.7 Hz, 1H, CH), 4.11 (dd, J = 7.5, 3.5 Hz, 1H, CH), 3.57 (dd, J = 11.5, 3.7 Hz, 1H, CH-H), 3.54 (dd, J = 12.0, 5.9 Hz, 1H, CH-H), 3.38 (dd, J = 11.4, 7.5 Hz, 1H, CH-H), 3.12 (dd, J = 11.8, 5.0 Hz, 1H, CH-H), 2.96 (ddd, J = 14.0, 7.8, 4.7 Hz, 1H, CH-H), 2.84 (dd, J = 14.1, 8.5 Hz, 1H, CH-H), 1.82 (t, J = 8.2 Hz, 1H, SH); 13C{1H} NMR (100 MHz, CDCl3) δ 168.6, 137.7, 129.2, 124.7, 119.8, 74.6, 72.8, 72.4, 39.5, 33.6, 33.3. HRMS (ESI/Q-TOF) m/z: [M + Na]+ Calcd for C13H16N2ONaS3 335.0322, found 335.0321.
Prepared in an analogous route as described for 2a starting from 4d (0.0192 g, 0.0454 mmol), purified by column chromatography on flash SiO2 (nHex/EtOAc 8:2) to yield 2d (Method A: 39%, Method C: 70%) as a colorless oil [α]D20 −35.9 (c = 2.13 in CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 5.08 (dd, J = 5.3, 3.8 Hz, 1H, CH), 4.83 (br, 1H, NH), 4.31 (dd, J = 7.8, 5.9 Hz, 1H, CH), 4.25 (dd, J = 6.6, 4.8 Hz, 1H, CH), 4.24 (t, J = 5.3 Hz, 2H, CH2), 3.54 (dd, J = 12.0, 5.4 Hz, 1H, CH-H), 3.43 (t, J = 4.9 Hz, 2H, CH2), 3.34 (dd, J = 11.0, 4.9 Hz, 1H, CH-H), 3.27 (dd, J = 11.0, 6.6 Hz, 1H, CH-H), 3.09 (dd, J = 11.9, 3.8 Hz, 1H, CH-H), 2.88 (dt, J = 13.6, 7.5 Hz, 1H, CH-H), 2.64 (ddd, J = 13.6, 9.6, 5.9 Hz, 1H, CH-H), 1.97 (dd, J = 9.6, 7.2 Hz, 1H, SH), 1.45 (s, 9H, (CH3)3); 13C{1H} NMR (100 MHz, CDCl3) δ 170.6, 155.9, 79.9, 75.0, 73.5, 70.6, 64.8, 39.7, 39.3, 34.3, 33.9, 28.5; HRMS (ESI/Q-TOF) m/z: [M + Na]+ Calcd for C14H24N2O4NaS3 403.0796, found 403.0796.
Prepared in an analogous route as described for 2a starting from 4e (0.0178 g, 0.0454 mmol), purified by column chromatography on flash SiO2 (nHex/EtOAc 2:3) to yield 2e (Method A: 44%, Method C: 68%) as a colorless oil [α]D20 −33.4 (c = 1.63 in CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 6.52 (s, 1H, H (4)-thiazole), 5.38 (br, 2H, NH2), 5.09 (dd, J = 5.2, 4.1 Hz, 1H, CH), 5.06 (d, J = 12.6 Hz, 1H, CH-H), 5.03 (d, J = 12.6 Hz, 1H, CH-H), 4.32 (dd, J = 7.2, 6.4 Hz, 1H, CH), 4.26 (dd, J = 6.6, 5.3 Hz, 1H, CH), 3.52 (dd, J = 11.9, 5.4 Hz, 1H, CH-H), 3.35 (dd, J = 11.0, 5.2 Hz, 1H, CH-H), 3.28 (dd, J = 11.0, 6.7 Hz, 1H, CH-H), 3.07 (dd, J = 11.9, 4.0 Hz, 1H, CH-H), 2.87 (dt, J = 13.7, 7.4 Hz, 1H, CH-H), 2.62 (ddd, J = 13.6, 9.4, 6.3 Hz, 1H, CH-H), 1.97 (dd, J = 9.4, 7.6 Hz, 1H, SH); 13C{1H} NMR (100 MHz, CDCl3) δ 170.4, 168.5, 146.2, 108.0, 75.2, 73.5, 70.4, 62.8, 39.2, 34.3, 33.8; HRMS (ESI/Q-TOF) m/z: [M + Na]+ Calcd for C11H15N3O2NaS4 371.9945, found 371.9945.
Prepared in an analogous route as described for 2a starting from 4f (0.0168 g, 0.0454 mmol), purified by column chromatography on flash SiO2 (CH2Cl2/EtOAc 9:1) to yield 2f (Method A: 31%, Method C: 78%) as a colorless oil [α]D20 -68.3 (c = 1.85 in CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 9.19 (br, 1H, NH), 7.29 (dd, J = 8.2, 1.3 Hz, 1H, ArH), 7.08 (td, J = 7.8, 1.4 Hz, 1H, ArH), 6.84–6.80 (m, 2H, ArH), 5.06 (dd, J = 5.6, 4.7 Hz, 1H, CH), 4.45 (dd, J = 8.2, 5.0 Hz, 1H, CH), 4.24 (dd, J = 7.2, 2.7 Hz, 1H, CH), 3.60 (dd, J = 11.3, 2.7 Hz, 1H, CH-H), 3.55 (dd, J = 12.0, 5.9 Hz, 1H, CH-H), 3.35 (dd, J = 11.3, 7.2 Hz, 1H, CH-H), 3.13 (dd, J = 11.9, 4.5 Hz, 1H, CH-H), 2.97–2.80 (m, 2H, CH2), 1.84 (t, J = 8.2 Hz, 1H, SH); 13C{1H} NMR (100 MHz, CDCl3) δ 169.1, 140.4, 127.5, 125.1, 123.9, 119.8, 118.3, 75.0, 73.2, 73.1, 39.6, 33.7, 33.5, HRMS (ESI/Q-TOF) m/z: [M + Na]+ Calcd for C13H17N3ONaS3 350.0431, found 350.0430.
Deprotection of 6–13 was carried out using the same approach as described for 4a, Method C).
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
Conceived and designed the experiments: VV, CS, and GM. Performed the experiments: VV and CS. Analyzed the data: VV, CS, and GM. Contributed reagents/materials/analysis tools: CS and GM. Contributed to the writing of the manuscript: VV, CS, and GM.
This work was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (NIH) to GM under Award Number R01AI100560. This study was also supported in part by funds from the Comisión Sectorial de Investigación Científica (CSIC)-Grupos from UdelaR. VV is the recipient of a fellowship from CSIC-UdelaR.
We would like to thank Horacio Pezaroglo and Gonzalo Hernández for running the NMR spectra. The Chemistry Department of the UdelaR is thankful to the CSIC for supporting acquisition of the HPLC-MS.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2022.934376/full#supplementary-material
Agouridas, V., Mahdi, O. E., Diemer, V., Cargoët, M., Monbaliu, J. C. M., Melnyk, O., et al. (2019). Native chemical ligation and extended methods: Mechanisms, catalysis, scope, and limitations. Chem. Rev. 119 (12), 7328–7443. doi:10.1021/acs.chemrev.8b00712
Barnett, R. E., and Jencks, W. P. (1969). Diffusion-controlled proton transfer in intramolecular thiol ester aminolysis and thiazoline hydrolysis. J. Am. Chem. Soc. 91 (9), 2358–2369. doi:10.1021/ja01037a029
Bongiardina, N. J., Long, K. F., Podgórski, M., and Bowman, C. N. (2021). Substituted thiols in dynamic thiol–thioester reactions. Macromolecules 54 (18), 8341–8351. doi:10.1021/acs.macromol.1c00649
Burke, H. M., McSweeney, L., and Scanlan, E. M. (2017). Exploring chemoselective S-to-N acyl transfer reactions in synthesis and chemical biology. Nat. Commun. 8, 15655. doi:10.1038/ncomms15655
Chandru, K., Gilbert, A., Butch, C., Aono, M., and Cleaves, H. J. (2016). The abiotic chemistry of thiolated acetate derivatives and the origin of life. Sci. Rep. 6, 1–11. doi:10.1038/srep29883
Dawson, P. E., Muir, T. W., Clark-Lewis, I., and Kent, S. B. (1994). Synthesis of proteins by native chemical ligation. Science 266 (5186), 776–779. doi:10.1126/science.7973629
Diemer, V., Firstova, O., Agouridas, V., and Melnyk, O. (2022). Pedal to the metal: The homogeneous catalysis of the native chemical ligation reaction. Chemistry 28 (16), e202104229. doi:10.1002/chem.202104229
Endo, T., Oda, K., and Mukaiyama, T. (1974). Facile conversion of thiolesters into thiols using cysteamine. Chem. Lett. 3, 443–444. doi:10.1246/cl.1974.443
Friedman, M., Cavins, J. F., and Wall, J. S. (1965). Relative nucleophilic reactivities of amino groups and mercaptide ions in addition reactions with α, β-unsaturated Compounds1, 2. J. Am. Chem. Soc. 87 (16), 3672–3682. doi:10.1021/ja01094a025
González, M. M., Kosmopoulou, M., Mojica, M. F., Castillo, V., Hinchliffe, P., PettinatiBrem, J., et al. (2016). Bisthiazolidines: A substrate-mimicking scaffold as an inhibitor of the NDM-1 carbapenemase. ACS Infect. Dis. 1, 544–554. doi:10.1021/acsinfecdis.5b00046
Gui, Y., Qiu, L., Li, Y., Li, H., and Dong, S. (2016). Internal activation of peptidyl prolyl thioesters in native chemical ligation. J. Am. Chem. Soc. 138 (14), 4890–4899. doi:10.1021/jacs.6b01202
Henderson, L. (1908). Concerning the relationship between the strength of acids and their capacity to preserve neutrality. Am. J. Physiology-Legacy Content 21 (4), 465. doi:10.1152/ajplegacy.1908.21.4.465-s
Hinchliffe, P., González, M. M., Mojica, M. F., Gonzalez, J. M., Castillo, V., Saiz, C., et al. (2016). Cross-class metallo-β-lactamase inhibition by bisthiazolidines reveals multiple binding modes. Proc. Natl. Acad. Sci. U. S. A. 113, E3745–E3754. doi:10.1073/pnas.1601368113
Maguire, O. R., Zhu, J., Brittain, W. D., Hudson, A. S., Cobb, S. L., O’Donoghue, A. C., et al. (2020). N-Terminal speciation for native chemical ligation. Chem. Commun. 56 (45), 6114–6117. doi:10.1039/d0cc01604g
Mennen, S. M., Alhambra, C., Allen, C. L., Barberis, M., Berritt, S., Brandt, T. A., et al. (2019). The evolution of high-throughput experimentation in pharmaceutical development and perspectives on the future. Org. Process Res. Dev. 23, 1213–1242. doi:10.1021/acs.oprd.9b00140
Mojica, M. F., Mahler, G., Bethel, C. R., Taracila, M. A., Kosmopoulou, M., Papp-Wallace, K. M., et al. (2015). Exploring the role of residue 228 in substrate and inhibitor recognition by VIM metallo-β-lactamases. Biochemistry 54, 3183–3196. doi:10.1021/acs.biochem.5b00106
Mondal, M., and Hirsch, A. K. (2015). Dynamic combinatorial chemistry: A tool to facilitate the identification of inhibitors for protein targets. Chem. Soc. Rev. 44 (8), 2455–2488. doi:10.1039/c4cs00493k
Pietrocola, F., Galluzzi, L., Bravo-San, J. M., Madeo, P. F., and Kroemer, G. (2015). Acetyl coenzyme A: A central metabolite and second messenger. Cell Metab. 21 (6), 805–821. doi:10.1016/j.cmet.2015.05.014
Pitman, I., and Morris, I. Aust. (1979). Covalent additions of glutathione, cysteine, homocysteine, cysteamine and thioglycolic acid to quinazoline cation. Aust. J. Chem. 32 (7), 1567. doi:10.1071/ch9791567
Saiz, C., Castillo, V., Fontán, P., Bonilla, M., Salinas, G., Rodríguez-Haralambides, A., et al. (2014). Discovering Echinococcus granulosus thioredoxin glutathione reductase inhibitors through site-specific dynamic combinatorial chemistry. Mol. Divers. 18 (1), 1–12. doi:10.1007/s11030-013-9485-3
Serra, G., Posada, L., and Hojo, H. (2020). On-resin synthesis of cyclic peptides via tandem N-to-S acyl migration and intramolecular thiol additive-free native chemical ligation. Chem. Commun. 56 (6), 956–959. doi:10.1039/c9cc07783a
Vallee, Y., Shalayel, I., Ly, K. D., Rao, K. R., De Paëpe, G., Märker, K., et al. (2017). At the very beginning of life on earth: The thiol-rich peptide (TRP) world hypothesis. Int. J. Dev. Biol.Dev. Biol. 61, 471–478. doi:10.1387/ijdb.170028yv
Villamil, V., Saiz, C., and Mahler, G. (2021). A new procedure for thioester deprotection using thioglycolic acid in both homogeneous and heterogeneous phase. Tetrahedron 94, 132335. doi:10.1016/j.tet.2021.132335
Wang, C., Guo, Q. X., and Fu, Y. (2011). Theoretical analysis of the detailed mechanism of native chemical ligation reactions. Chem. Asian J. 6 (5), 1241–1251. doi:10.1002/asia.201000760
Wong, H., and Cernak, T. (2018). Reaction miniaturization in eco-friendly solvents. Curr. Opin. Green Sustain. Chem. 11, 91–98. doi:10.1016/j.cogsc.2018.06.001
Keywords: thioester deprotection, transthioesterification, native chemical ligation, bisthiazolidine, thiol
Citation: Villamil V, Saiz C and Mahler G (2022) Thioester deprotection using a biomimetic NCL approach. Front. Chem. 10:934376. doi: 10.3389/fchem.2022.934376
Received: 02 May 2022; Accepted: 06 July 2022;
Published: 22 August 2022.
Edited by:
Thierry Brigaud, CY Cergy Paris Université, FranceReviewed by:
William Brittain, Durham University, United KingdomCopyright © 2022 Villamil, Saiz and Mahler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Graciela Mahler, Z21haGxlckBmcS5lZHUudXk=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.