 Liena Qin
Liena Qin Han Dai
Han Dai Junfeng Wang2,3,4*
Junfeng Wang2,3,4*
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Chem. , 01 August 2022
Sec. Chemical Biology
Volume 10 - 2022 | https://doi.org/10.3389/fchem.2022.934337
This article is part of the Research Topic Reviews in Chemistry View all 14 articles
Targeting proteins’ enzymatic functions with small molecule inhibitors, as well as functions of receptor proteins with small-molecule agonists and antagonists, were the major forms of small-molecule drug development. These small-molecule modulators are based on a conventional occupancy-driven pharmacological approach. For proteome space traditionally considered undruggable by small-molecule modulators, such as enzymes with scaffolding functions, transcription factors, and proteins that lack well-defined binding pockets for small molecules, targeted protein degraders offer the opportunity to drug the proteome with an event-driven pharmacological approach. A degrader molecule, either PROTAC or molecular glue, brings the protein of interest (POI) and E3 ubiquitin ligase in close proximity and engages the ubiquitin-proteasome system (UPS), the cellular waste disposal system for the degradation of the POI. For the development of targeted protein degraders to meet therapeutic needs, several aspects will be considered, namely, the selective degradation of disease-causing proteins, the oral bioavailability of degraders beyond Lipinski’s rule of five (bRo5) scope, demands of new E3 ubiquitin ligases and molecular glue degraders, and drug resistance of the new drug modality. This review will illustrate several under-discussed key considerations in targeted protein degradation drug discovery and development: 1) the contributing factors for the selectivity of PROTAC molecules and the design of PROTACs to selectively degrade synergistic pathological proteins; 2) assay development in combination with a multi-omics approach for the identification of new E3 ligases and their corresponding ligands, as well as molecular glue degraders; 3) a molecular design to improve the oral bioavailability of bRo5 PROTACs, and 4) drug resistance of degraders.
PROTAC (proteolysis-targeting chimera) (Sakamoto et al., 2001) is a type of small molecule capable of the engaging ubiquitin-proteasome system, the cellular waste disposal system (Salami and Crews 2017), to degrade disease-causing proteins, by recruiting E3 ubiquitin ligase and labeling the target protein with polyubiquitin for proteasomal recognition. Classical PROTAC molecules are heterobifunctional small molecules consisting of two ligands connected with a flexible or rigid linker, with one ligand binding to POI, and the other binding to E3 ubiquitin ligase (Paiva and Crews 2019). The most prevalent E3 ligases used in pharmaceutical drug development are VHL (Von Hippel-Lindau) and CRBN (Cereblon) E3 ligases (Ishida and Ciulli 2021). Molecular glue degraders represented by immunomodulatory imide drugs (IMiDs) function similarly to PROTACs by engaging PPI (protein-protein interaction) between POI and E3 ligase, and directing the target protein for proteasomal degradation. Molecular glue (Schreiber 2021) degraders lack the typical linker seen in the PROTAC molecule. They are lower in molecular weight, and the binding affinity to each individual partner is lower or undetectable, as shown in the case of sulfonamide with DCAF15 and RBM39 (Du et al., 2019). In some cases, a degrader molecule could harness features of both PROTAC and molecular glue to degrade multiple targets (I-208, Figure 1B). In this review, both PROTAC and molecular glue approaches will be treated as small molecule targeted protein degraders.
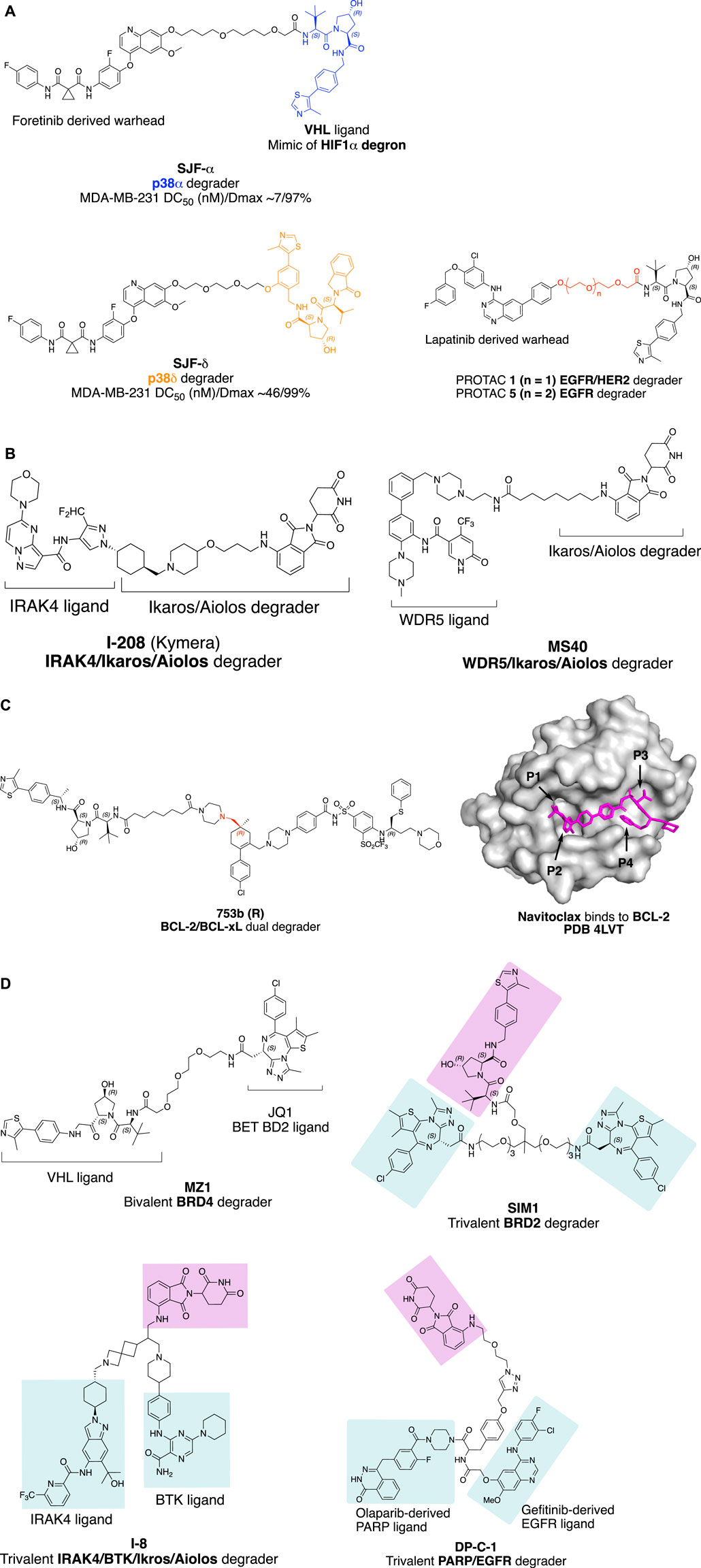
FIGURE 1. Factors influence the selectivity of degraders. (A) PROTAC with different linker attachment point to the VHL ligand selectively degrade p38α and p38β. (B) Structure of IRAK/Ikaros/Aiolos multi-targets degrader I-208 and WDR5/Ikaros/Aiolos multi-targets degrader MS40. (C) Bcl-2/Bcl-xL dual degradation by 753bR achieved with different linker attachment points to warhead; and crystal structure of Navitoclax with BCL-2 (Souers et al., 2013) with four pockets indicated with an arrow. The crystal structure was processed with PyMOL. (D) Trivalent PROTAC with multi-protein or multi-domain binding warheads. MZ1 with a preference for BRD4 degradation, SIM1 with a preference for BRD2 degradation.
Targeted protein degraders have the potential to target conventionally undruggable proteome (Samarasinghe and Crews 2021; Schneider and Chris, 2021), either as chemical biology research tools (Burslem and Crews 2020) or as new therapeutic modalities (Cromm and Crews 2017; Lai and Crews 2017; Nalawansha and Crews 2020), rapidly applied to cancer therapy (Dale et al., 2021), further applications include neurodegenerative disorders (Tomoshige and Ishikawa 2021). Pioneered by the AR (Androgen receptor) degrader ARV-110 (NCT03888612) and ER (Estrogen receptor) degrader ARV-471 (NCT04072952), developed by Arvinas Inc. for the treatment of prostate cancer and breast cancer, respectively, the field has seen at least 15 degraders in a clinical trial (Table 1) (Mullard 2021).
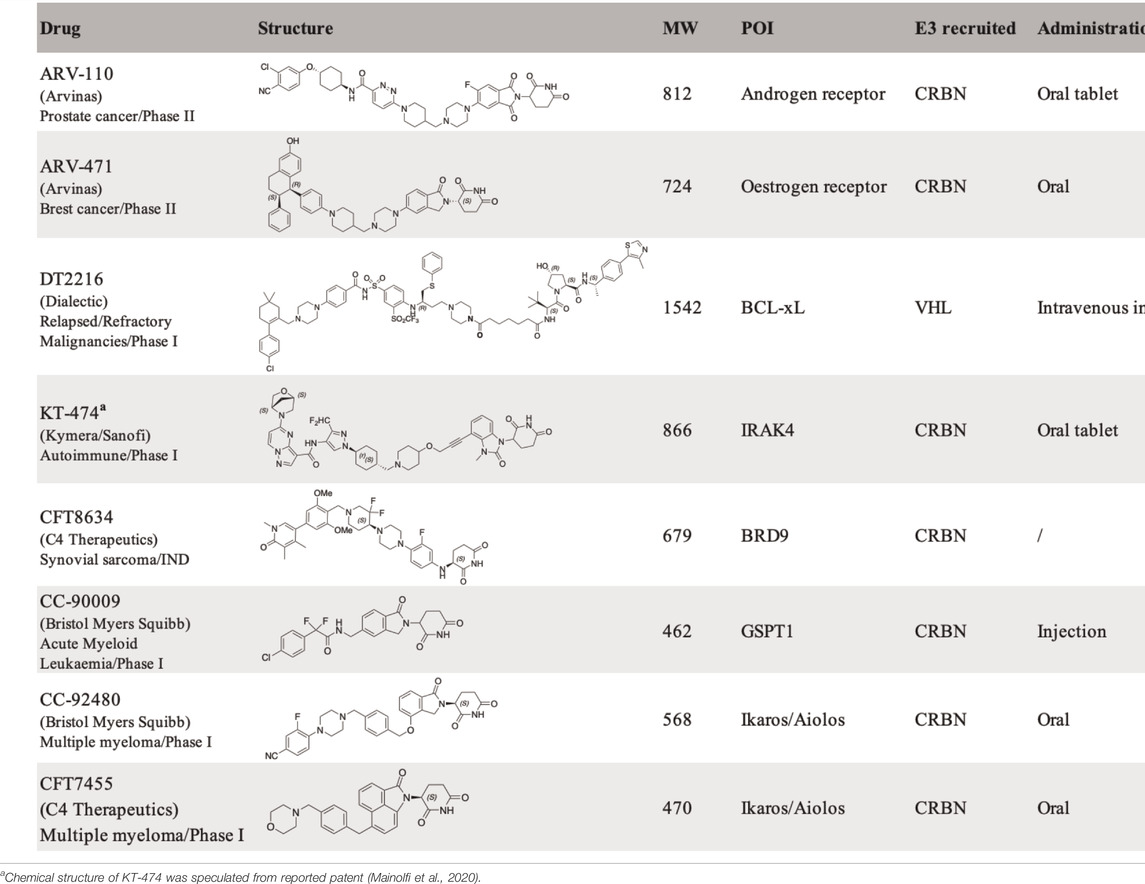
TABLE 1. Protein degraders approaching clinical trial (https://clinicaltrials.gov/) with structures disclosed.
Two aspects are key to fostering the development of targeted protein degraders in drug discovery and development, one is structure-guided design (Leissing, Luh, and Cromm 2020) of the heterobifunctional molecules, and the other is assay development driven by synthetic biology in combination with a multi-omics approach (Scholes et al., 2021) to systematically identify new E3 ubiquitin ligases and their corresponding ligands and molecular glue degraders. The design of selective PROTACs will be exemplified in the following context by the impact of linkerology on selective protein degradation, and the design of PROTACs to degrade multiple disease-causing proteins simultaneously. The impact of linkerology is also reflected in the physicochemical properties and oral bioavailability of PROTACs. Molecular design to improve oral bioavailability is important for bRo5 PROTAC drug development. The correlation of physicochemical features of PROTAC molecules with in vivo pharmacokinetics profile will be discussed. There are more than 600 E3 ubiquitin ligases encoded by the human genome, but only 2% of them have been applied in proximity-induced protein degradation. E3 ligases beyond VHL and CRBN for targeted protein degradation, for example, tissue-specific or disease-specific E3 ligases would considerably expand the application of targeted protein degradation for therapeutic purposes (Kannt and Đikić 2021; Guenette et al., 2022). Systematic searching for E3 ubiquitin ligases and their ligands is achieved using chemoproteomics methods applying cysteine reactive covalent small molecules to map the E3 ligase proteome. Assay development combined with multi-omics approaches is discussed. These benefit the targeted protein degradation field by providing the tools to systematically identify molecular glue degraders instead of being discovered by serendipity. At the end of the review, potential drug resistance mechanisms arising from targeted protein degradation will be briefly discussed. The design of degraders for therapeutic application discussed throughout the context reflects joint efforts from the chemistry and biology fronts to understand the molecular basis of disease pathways, the structure of productive ternary complex formation, as well as medicinal chemistry effort for the design of bioavailable molecules.
Turning a promiscuous small molecule inhibitor into a PROTAC, the selectivity could be rewired in the degradable proteome. A multi-kinase degrader generated by conjugating a highly promiscuous kinase inhibitor with CRBN-binding ligand was discovered to degrade a small set of kinases and CDK family proteins, using chemoproteomics method. In this study, Huang and coworkers demonstrated that target engagement is not sufficient for successful degradation (Huang et al., 2018). The selectivity of productive protein degradation is influenced by the E3 ubiquitin ligase (Lai et al., 2016), linker attachment points to the warhead, linker attachment point to E3 ligase ligand, and linker length (termed ‘linkerology’). The effect of E3 ubiquitin ligase selection and cell type on the degradation profile for PROTAC molecules will not be discussed here. This part will focus on the impact of linkerology on the selectivity of PROTAC molecules with promiscuous warheads.
The linker attachment point to a POI is usually selected at a solvent-exposed site of the warhead binding in a protein pocket. Linkers extended from a buried site may hinder the binding of a target protein and unsuccessful degradation. This can sometimes enhance the selectivity of a promiscuous warhead, as in the well-discussed case of enhancing the degradation of the cellular retinoic acid-binding protein (CRABP) over retinoic acid receptor (RAR) with the dual protein binder all-trans retinoic acid (ATRA) (Ishikawa et al., 2020).
Linker attachment point to E3 ligase ligand has the potential to influence the direction of assembly of the E3 ubiquitin complex. In a study evaluating protein degradation profile with promiscuous c-Met tyrosine kinase inhibitor Defactinib (Bondeson et al., 2018), p38α was found to be degraded (DC50 (nM)/Dmax 210/91%) with VHL recruiting E3 ligase in triple-negative breast cancer cell line MDA-MB-231, while the MAPK family homolog p38δ, which shares 61% sequence identity with p38α, was only slightly degraded (∼30%). Later, a more potent selective p38α degrader SJF-α (MDA-MB-231 DC50 (nM)/Dmax ∼7/97%) was developed (Smith et al., 2019). The linear linker and the VHL E3 ligase ligand were connected through the amide bond as depicted in Figure 1A. By changing the linker attachment point to the benzene ring of the VHL E3 ligase ligand through an ether bond, selective p38δ degradation (MDA-MB-231 DC50 (nM)/Dmax ∼46/99%) was achieved. The distinct degradation selectivity of two PROTAC molecules between two homologous MAPK family proteins was illustrated by in vitro stable ternary complex formation of VHL-SJFα-p38α and VHL-SJFδ-p38δ, respectively. Molecular dynamic simulation of the ternary complexes indicates that the VHL/p38α and VHL/p38δ interface was altered. VHL protein was recruited in a different direction approaching the p38α/p38δ protein due to the distinct linker attachment points of the PROTAC molecules.
The impact of linker length on the selectivity of PROTACs is exemplified by epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor 2 (HER2) degrader with receptor tyrosine kinase inhibitor Lapatinib derived warhead (Burslem et al., 2018). PROTAC 1 with two PEG (polyethylene glycol) linker degrades both EGFR and HER2, while PROTAC 5 with three PEG linkers selectively degrade EGFR (Figure 1A). This type of exquisite selectivity is also reported in the CDK4/6 case with a highly conserved kinase active site (Anderson et al., 2020).
Interleukin-1 receptor-associated kinase 4 (IRAK4) is a serine/threonine-protein kinase with scaffolding functions, involved in Toll-like receptor (TLR, except for TLR3) and Interleukin-1 receptor (IL-1R) signaling pathways (Hennessy et al., 2010; Picard et al., 2011). IRAK4 is a 51KD protein that consists of an N-terminal Death Domain (DD residues 1–125), a hinge domain (residues 140–150), and a C-terminal kinase domain (residues 150–460). Upon TLR activation, IRAK4 is rapidly recruited by MYD88 to the receptor-signaling complex to form the Myddosome complex, then phosphorylates initially IRAK1 via oligomerization of the N-terminal DD in each of these proteins, leading to NF-κB nuclear translocation and activation. The scaffolding function of the DD of IRAK4 is essential in IL-1 signaling, while the kinase function of IRAK4 is partially responsible (Kim et al., 2007; De Nardo et al., 2018). To target both scaffolding function and kinase activity of IRAK4, degradation is superior over of IRAK4 kinase inhibitor, similar to that reported on FAK (Law et al., 2021). On the other hand, Ikaros and Aiolos are the activators of the redundant NF-κB pathway and upstream type I INF regulator. A PROTAC molecule capable of degrading IRAK4/Ikaros/Aiolos simultaneously could meet the clinical needs in treating B cell malignancy (Yang et al., 2012).
The key question to address for an IRAK4 degrader with Ikaros/Aiolos degradation properties is the selectivity. By proteomic analysis in thalidomide-treated H9 human embryonic stem cells, C2H2 zinc finger transcription factor SALL4 was identified as a bona fide neo-substrate of the thalidomide-CRBN-DDB1-Cul4A E3 ligase complex. Loss of function of SALL4 was verified to be responsible for thalidomide-caused teratogenicity (Donovan et al., 2018). Despite the side effects of thalidomide, it was later approved by FDA for the treatment of multiple myeloma under strict restrictions. Other thalidomide analogous immunomodulatory imide drugs (IMiDs), lenalidomide, and pomalidomide, have been developed for the treatment of cancer with fewer side effects and increased potency. Other than SALL4, IMiDs degrade a set of neo-substrates, most of which are C2H2 zinc finger proteins (Krönke et al., 2014; Donovan et al., 2018; Gao et al., 2020; Kozicka and Thomä, 2021) including Ikaros (IKZF1) and Aiolos (IKZF3). More selective Ikaros and Aiolos degraders are needed to reduce the safety concern as well as gain structural insights for the selectivity of degradation. CC-92480 (Hansen et al., 2020) (NCT03989414) and CFT7455 (Henderson et al., 2020) (Table 1) (NCT04756726) have been developed for the treatment of multiple myeloma and lymphoma by Bristol Myers Squibb and C4 Therapeutics independently, and are currently in phase I clinical trials. The selectivity of the IMiD degraders is achieved by introducing substitution to the phthalimide, therefore changing the approachable interface of CRBN by neo-substrates in the presence of IMiD (Leissing et al., 2020).
Kymera Therapeutics has designed a potent multi-targets IRAK4/Ikaros/Aiolos degrader KT-413 for treating relapsed or refractory B-cell Non-Hodgkin’s lymphoma, which is in phase I clinical trial (NCT05233033). Although the structure of KT-413 has not been disclosed, the molecule I-208 (Figure 1B) (Mainolfi et al., 2020) has been disclosed to be able to induce in vivo degradation of IRAK4/Ikaros/Aiolos in OCI-LY10 tumor xenograft, correlated with tumor regression.
A similar approach of designing the PROTAC MS40 (Figure 1B) for the degradation of MDR5/Ikaros with a synergistic effect in mixed-lineage leukemia (MLL)-rearranged leukemias was reported most recently (D. Li et al., 2022). WD repeat domain 5 (WDR5) is an integral component of histone lysine methyltransferase complex and MLL complex. MLL-rearranged leukemias also exhibit high expression and dependency on Ikaros. MS40 was shown to degrade WDR5/Ikaros/Aiolos in acute lymphoblastic leukemia (ALL) RS4; 11 cells, and WDR5/Ikaros in biphnotypic B myelomonocytic leukemia MV-4–11cells at submicromolar range (MV-4-11 lack of expression of Aiolos). MS40 has also shown modular in vivo tumor suppression activity in the subcutaneous MLL-AF9+AML PDX model, dosing at 100 mpk once daily for five days per week through intraperitoneal injection.
Navitoclax is a potent Bcl-xL and Bcl-2 dual inhibitor developed by AbbVie for the treatment of relapsed or refractory lymphoid malignancies. Navitoclax failed in the phase II clinical trial, due to on-target and dose-limiting thrombocytopenia (Mohamad Anuar et al., 2020). Platelets require Bcl-xL for survival. The VHL recruiting PROTAC DT2216 (Table 1), utilizes Navitoclax as a warhead, achieved potent antitumor activity while less platelet toxicity in vivo, and is currently in Phase I clinical trial (NCT04886622). The reduced platelet toxicity was suggested to be due to the low expression level of VHL in platelet (He et al., 2019; Khan et al., 2019; He et al., 2020). The authors also validated that DT2216 selectively degrades Bcl-xL in a Lys 87-dependent manner. Single Lys 87 mutation to arginine is sufficient to induce resistance to Bcl-xL degradation; if all the other lysines of Bcl-xL except Lys 87 were mutated, the degradation of Bcl-xL was retained.
The selective degradation of Bcl-xL over Bcl-2 could be explained by the linkerology of PROTAC molecule design. The linker of DT2216 was designed by forming two amide bonds with VHL ligand and Navitoclax warhead respectively, one with the primary amine of the VHL ligand, and the other with the secondary amine of piperazine, which was converted from morpholine of Navitoclax. Linker extension from the morpholine binding site of Bcl-xL exposed the Lys 87 for ubiquitination, while Bcl-2 lacks such an accessible lysine, which results in the selective degradation of Bcl-xL over Bcl-2. The result is consistent with the finding that a productive ternary complex formation is required for targeted protein degradation (Chung et al., 2020).
Furthermore, degradation of both BCL-xL and BCL-2 with improved anti-leukemic activity was achieved by changing the linker attachment point to the methyl group, which is solvent-exposed located at pocket 1 (P1) and pocket 2 (P2) intersection of Bcl-2 or Bcl-xL as indicated in Figure 1C, to generate 753b (R enantiomer) (Figure 1C) (D. Lv et al., 2021, 2). By doing so, the Lys 17 of Bcl-2 was accessible for ubiquitination according to the computational modeling of the Bcl-2-753b-VHL E3 ligase. Meanwhile, Lys 87 and Lys 20 of Bcl-xL remain accessible for ubiquitination in the BCL-xL-753b-VHL E3 ligase composition.
Trivalent PROTAC contains a bivalent warhead, which binds to two domains of one protein or two proteins simultaneously. Degrading dual-target proteins in the complimentary pathological pathway or synthetic lethal pair could be interesting to generate a synergistic effect in drug development. However, simultaneous degrading of such protein pair by a trivalent PROTAC requires the two proteins to be present at the same time and space in a cellular context. The design of a trivalent PROTAC requires careful planning of the linkerology, which has been well illustrated by the structure-guided design of a trivalent PROTAC with a warhead binding to two domains of one protein (Imaide et al., 2021). Bromodomain-containing proteins BRD2/3/4 and BRDT are members of the bromodomain and extra terminal domain (BET) family of proteins, structurally featuring two bromodomains (BD1 and BD2), which recognize acetylated lysine during transcriptional regulation. The well-known BET BD inhibitor JQ1 (Filippakopoulos et al., 2010) was converted to MZ1 (Zengerle et al., 2015) to give a VHL E3 ligase recruiting BET degrader. A more potent bivalent BD inhibitor MT1, which binds to the BD1 and BD2 of BET family proteins, was also reported with significant in vivo efficacy (Tanaka et al., 2016). Ciulli and coworkers validated the concept of developing a trivalent PROTAC SIM1 (Figure 1D), guided by the chemical structure of MZ1, MT1, and the crystal structure of the BRD4 (BD2)-MZ1-VHL E3 ligase ternary complex (Gadd et al., 2017). The structure of BRD4 (BD2)-MZ1-VHL E3 ligase ternary complex suggests a three PEG point could be a branching point for another BD1 binding JQ1 ligand. The eight PEG linker SIM1 suggests sufficient length for linker attachment to the VHL ligand. The trivalent SIM1 binds to both BD1 and BD2 domains of the BET protein and recruits VHL E3 ligase for targeted degradation of BET proteins. SIM1 degrades BET family proteins with a preference for BRD2, which is different from MZ1’s preference for BRD4 degradation.
Trivalent PROTAC with a warhead targeting the synthetic lethal pair of EGFR and poly (ADP-ribose) polymerase (PARP) has been reported (Zheng et al., 2021). Based on the report that EGFR mutated lung cancer cells were sensitized to the treatment of PARP inhibitor Olaparib (Pfäffle et al., 2013), Zheng and coworkers designed a trivalent PROTAC with bivalent warhead derived from Olaparib and EGFR inhibitor Gefitinib (DP-C-1, Figure 1D). Both VHL and CRBN recruiting trivalent PROTACs were designed, and dose- and time-dependent degradation of EGFR and PARP was observed in non-small cell lung cancer cell line H1299 and pancreatic adenocarcinoma cell line SW1990 at μM range, respectively. Most recently, trivalent degraders targeting two synergistic protein targets, IRAK4 and BTK in B cell lymphoma, have been disclosed by Kymera Therapeutics (Weiss et al., 2022). The degraders represented by I-8 (Figure 1D) also degrade Ikaros and Aiolos. Overall, these researches set a foundation for structure-guided design of PROTAC molecules for multidomain proteins, and potentially two protein targets synergistically for therapeutic benefits.
The majority of PROTAC molecules in clinical trials recruit CRBN E3 ubiquitin ligase for targeted protein degradation (Table 1), including the AR degrader (ARV-110) (Crew et al., 2021), ER degrader (ARV-471) (Chen X. et al., 2021; Halford 2021), IRAK4 degrader (KT-474) (Mainolfi et al., 2020) (NCT04772885) and BRD9 degrader (CFT8634) (Nasveschuk et al., 2022) (NCT05355753). The Helios degrader, GSPT1 degrader (CC-90009) (Hansen et al., 2021) (NCT04336982), and Ikaros/Aiolos degraders are CRBN E3 recruiting molecular glue degraders. The only VHL E3 ligase recruiting PROTAC molecule currently in Phase I clinical trial is the Bcl-xL degrader (DT2216). Ligands of several E3 ubiquitin ligases including Nutlin-3a for MDM2 (Schneekloth et al., 2008) and Bestatin for cIAP (Itoh et al., 2010), have been reported for targeted protein degradation, and their application in drug development is still limited. The systematic approach for the identification of E3 ligases DCAF16 and RNF114 could be inspirational for the discovery of other new E3 ligases and their corresponding ligands (Spradlin et al., 2019; Zhang X. et al., 2019).
Chloroacetamide and acrylamide containing “Scout” fragments are cysteine reactive electrophiles used by the pioneer of the Cravatt research team in activity-based protein profiling (Backus et al., 2016; Bar-PeledKemper et al., 2017). The ‘Scout’ fragments KB02, KB03, and KB05 (Figure 2A) displayed broad cysteine reactivity in the human proteome, thus capable of capturing reactive cysteine residues of the E3 ubiquitin ligase pool once the scout fragments are turned into PROTAC molecules. Such PROTAC molecules are designed by linking FKBP12 binding protein-ligand SLF to the scout fragment. The molecules were tested in the HEK293 T cell line transduced with FLAG-tagged FKBP12 either with or without C-terminal NLS (nuclear localization sequence) KKKRKV. The compound KB02-SLF was found to promote the loss of nuclear FKBP12 in a Cullin E3 ligase and proteasome system-dependent manner. FLAG-mediated affinity enrichment was used to identify that the DCAF16 E3 ligase was associated with FKBP12_NLS degradation in a KB02-SLF-dependent manner (Zhang X. et al., 2019).
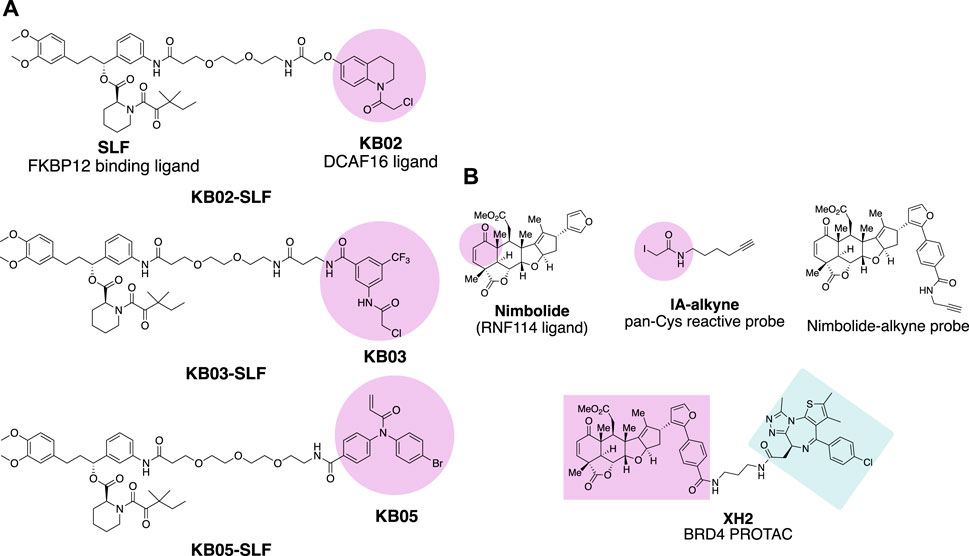
FIGURE 2. Chemical probes for the identification of E3 ligases. (A) FKBP12 degrading molecules with “scout” fragment KB02, KB03, and KB05 covalently react with Cysteine in the proteome. (B) Chemical probes for the identification of E3 ligase recruited by Nimbolide.
Nimbolide is a natural product derived from the Neem tree and possesses anticancer activity (Elumalai and Arunakaran 2014). The chemical structure of Nimbolide features an α,β-unsaturated ketone as Michael acceptor with the potential to act as an electrophile for the reactive cysteine residues of the target protein (Figure 2B). This enables isoTOP-ABPP (isotopic tandem orthogonal proteolysis-activity-based protein profiling) (Weerapana et al., 2010) to identify the direct protein targets of Nimbolide. The iodoacetamide-alkyne was used as the chemical probe in the experiment to react with those Cysteines spared by Nimbolide so that Nimbolide reactive cysteine-containing proteins will show differences in the quantitative mass spectrometry analysis. The E3 ligase RNF114 was identified to be the target of Nimbolide. The anticancer reactivity of Nimbolide arises from the inhibited ubiquitination and degradation of tumor suppressor p21 in the 231MFP breast cancer cell line by RNF114. The interaction of Nimbolide with RNF114 was further validated by pulldown of Flag-tagged RNF114 in 231MFP cells with the Nimbolide-alkyne probe (Figure 2B). The capability of E3 ligase RNF114 recruited by Nimbolide for targeted degradation was evaluated by the PROTAC molecule XH2 (Spradlin et al., 2019), to degrade BRD4 with the Bromodomain ligand JQ1 as a warhead.
An indirect chemical biology method to evaluate the function of E3 ubiquitin ligases for targeted protein degradation is induced protein proximity by using a heterobifunctional small molecule (Ottis et al., 2017). E3 ubiquitin ligase and GFP were fused with HT7 (HaloTag7) and FKBP (FK506 binding protein) respectively, a heterobifunctional small molecule was designed with one side forming a covalent bond with Asp106 of HT7 while the other side binds to FKBP in a bump-hole mode. The E3 ubiquitin ligase and POI were induced close in space to evaluate the degradation signal. In the assay, GFP was used to give a fluorescent signal for monitoring the degradation event. More recently, the HiBit technology has been developed for measuring endogenous protein dynamics (Schwinn et al., 2020); and the NanoBRET assay (Riching et al., 2018) has also been developed to measure the kinetics of cellular degradation cascades. Those assays in combination provide methods to evaluate E3 ubiquitin ligases for targeted protein degradation in a high-throughput manner.
In the past, molecular glue degraders were usually found by serendipity while searching for the mode of action of small molecule drugs (Dong et al., 2021). Examples include thalidomide (Ito et al., 2010) and RBM39 (RNA binding motif protein 39) (T. Han et al., 2017) degrading sulfonamides (Figure 3) which were discovered to be molecular glue degraders. Thalidomide was used in the late 1950s and early 1960s for the treatment of morning sickness in pregnant women, which resulted in severe tragedies in causing thousands of miscarriages and birth defects. Via affinity-based protein profiling with a thalidomide-based probe in HeLa cell extracts, CRBN was found to bind to thalidomide, a substrate recognition subunit of DDB1-Cul4A Cullin RING E3 ubiquitin ligase. Indisulam was a small molecule compound with anticancer activity. The mode of action of indisulam was not revealed until recently by using a forward genetic method. Several single amino acid mutations of Indisulam were found in common across the RBM39 resistant HCT-116 cell line. RBM39 was later found to be degraded by indisulam in a dose-dependent manner. Following co-immunoprecipitation and liquid chromatography and mass spectrometry analysis, DCAF15 E3 ligase was found to be engaged in the degradation of RBM39.

FIGURE 3. Structures of RBM39 degrading sulfonamides and CDK12 dependent Cyclin K degraders.
Small molecules targeting protein homeostasis specifically by engaging protein-E3 ligase interactions for targeted degradation might be more common than previously known. Cell biology assays in combination with multi-omics methods have been developed for systematically searching for small molecules with such capability (Mayor-Ruiz et al., 2020; Scholes et al., 2021). Phenotypic screening of 2,000 cytotoxic compounds was carried out in WT and UBE2M (E2 enzyme) mutated (hyponeddylated) myeloid leukemia cell line KBM-7 to identify correlations between the toxicity of small molecules with the neddylation level. Since the neddylation of cullin-RING E3 ligases (CRLs) is highly associated with the E3s’ activity, the small molecules identified in the screen will be E3 ligase activity-dependent cytotoxic compounds. Four compounds (dCeMM1/2/3/4) were identified in the phenotypic screening. Quantitative expression proteomics revealed dCeMM1 to be RBM39 destabilizer and dCeMM2/3/4 to be a cyclin K degrader. CRISPR-Cas9 screening against all components of known CRLs revealed that cyclin K degradation is mediated by the CUL4B complex. Affinity-based protein profiling using a dCeMM3-derived chemical probe identified drug-mediated DDB1-CDK12-cyclin K complex formation. Drug sensitivity data for 4,518 clinical and pre-clinical compounds tested in 578 cancer cell lines were compared with the mRNA expression level of 499 E3 ligase components, and the cytotoxicity of the CDK inhibitor R-CR8 (Figure 3) was found to correlate with the expression levels of CUL4 adaptor DDB1 (Słabicki et al., 2020). In the counter-confirmation experiment, sgRNA targeting DDB1 conferred resistance to R-CR8. In the proteome-wide analysis of protein abundance following R-CR8 treatment, cyclin K was the only protein shown to be consistently decreased. In the in vitro pulldown experiment of CDK12 (AA713-1052 kinase domain) bound cyclin K in the presence of R-CR8, DDB1 was significantly enriched versus in the absence of R-CR8. The crystal structure of CDK12713−1052-cycK1−267 bound to R-CR8 and DDB1ΔBPB was determined to illustrate the structural mechanism of R-CR8 acting as a molecular glue degrader for cyclin K by strengthening the CDK12-DDB1 interaction.
At the same time, serendipitously, a screening effort for NRF2 inhibitors using NRF2 activity-based luciferase reporter assay, HQ461 (Figure 3) was found to down-regulate NRF2 mRNA and expression levels (L. Lv et al., 2020). However, HQ461’s potent cytotoxicity in the A549 cell line was independent of NRF2 function. To explore the mechanism of function of the molecule, HQ461 sensitive colorectal cancer cell line HCT-116 was treated with the compound to select resistant clones. Whole-exome sequencing against the HQ461 resistant clones was performed to find the top-ranking variant was CDK12 G731E mutation. Both CDK12 and its interacting protein Cyclin K protein level were quantified, showing more than the 8-fold reduction of Cyclin K was observed after treatment with HQ461 in the CDK12 wild-type cell line. The downregulation of Cyclin K was UPS-dependent. Pulldown using Flag-tagged CDK12 in the cell lysate treated with HQ461 identified the interaction between CDK12 and DDB1. The HQ461-induced CDK12 (kinase domain)/CCNKΔC/DDB1 complex was further evaluated by AlphaScreen assay and chemical cross-linking mass spectrometry. The assay results give evidence that HQ461 function as a molecular glue between CDK12 and DDB1, which triggers UPS-dependent depletion of Cyclin K.
PROTAC molecules are usually beyond Lipinski’s rule of five (Ro5) (Caron et al., 2021) for orally administered drugs, conventionally considered to indicate poor permeability and oral bioavailability. A classical PROTAC molecule harnesses a warhead which is the small molecule ligand of the target protein, an E3 ligase ligand to recruit the VHL or CRBN subunit of Cullin ring E3 ligase, and a linker that brings the E3 ubiquitin ligase complex in close proximity to the target protein. Linkers of PROTAC molecules not only have a great influence on the degradation efficiency and selectivity of the target protein as previously reviewed (Cyrus et al., 2011; Zagidullin et al., 2020), but also had a profound impact on the in vivo PK profile of PROTACs, as shown in the cases of Androgen Receptor degraders and SMARCA2/4 degraders (Xiang et al., 2021; Kofink et al., 2022).
In castration-resistant prostate cancer (CRPC), the progression of the disease is uncontrolled despite the low testosterone level, due to AR (androgen receptor) amplification and hypersensitivity, AR mutations, and intra-tumoral androgen production (Chandrasekar et al., 2015). In resistance development, AR antagonists could also be switched to agonists after treatment with inhibitors (Culig et al., 1999). Degradation offers new opportunities to tackle these problems with the event-driven pharmacological mechanism (Salami and Crews 2017). ARV-110, an orally available AR PROTAC developed by Arvinas Inc., is currently a Phase II clinical trial for the treatment of metastatic castration-resistant prostate cancer. With the success of ARV-110, Wang and coworkers achieved an orally available AR PROTAC by linker rigidification with a CRBN E3 recruiting ligand. ARD-69 (Figure 4A) is a potent AR PROTAC with an enzalutamide analog as an AR binder and a rigid linker connected with the optimized VHL ligand (X. Han et al., 2019). ARD-69, with molecular weight >1000, calculated TPSA (topological polar surface area) = 197 and ClogP = 8 respectively, was administered intraperitoneal in the in vivo PD study. By changing the E3 recruiting element to thalidomide, the molecule ARD-2542 induced efficient in vitro degradation of AR, since both VHL and CRBN are expressed ubiquitously and could induce efficient degradation of AR. Although with significantly reduced molecular weight, calculated TPSA and ClogP, ARD-2542 exhibited a low plasma concentration of about 17 ng/ml after 1 h of oral administration at 10 mpk in a mouse pharmacokinetics study. Changing the linear linker to rigid piperazine and azetidines, ARD-2582 plasma concentration in mice was increased with Cmax at 1140 ng/ml after oral administration at 5 mpk. The oral bioavailability increased to 51% in mice (Xiang et al., 2021).
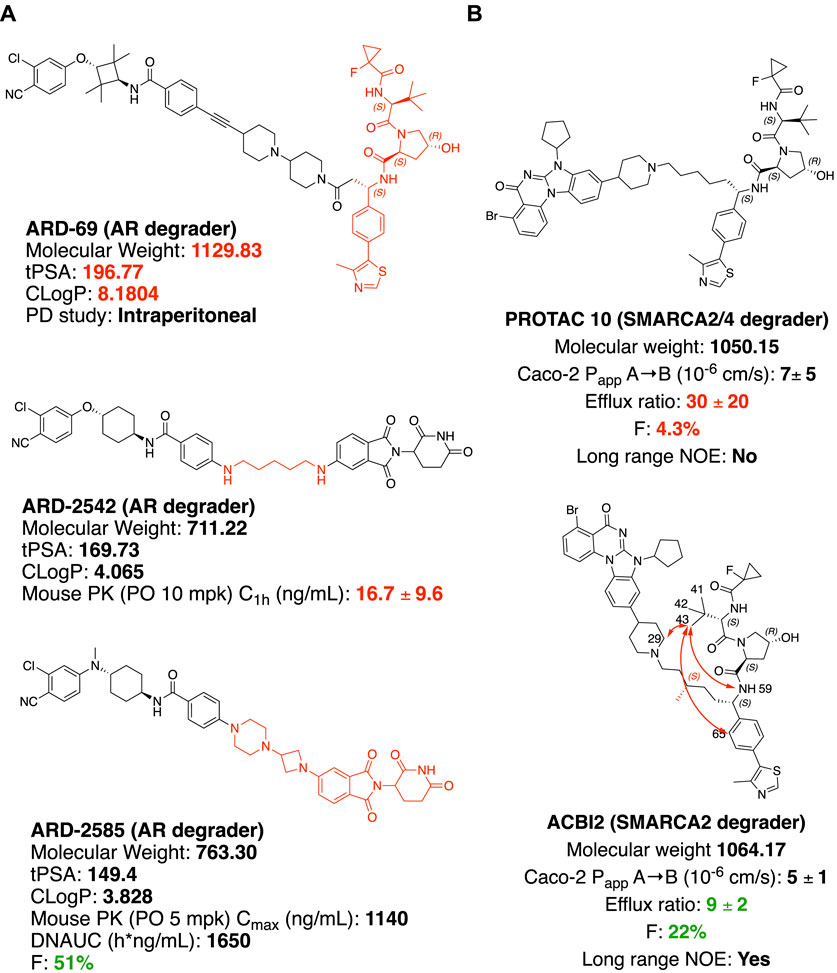
FIGURE 4. Structure modification to improve oral bioavailability. (A) Structures of AR degraders, ARD-69 with VHL ligand, ARD-2542 with a flexible linear linker, ARD-2585 with the rigid linker. (B) Structures of SMARCA2/4 degraders. Red arrows indicate atoms with long rang NOE.
SMARCA2 and SMARCA4 are close homologs as a component of the SWI/SNF complex, involved in chromatin remodeling and repair (Mashtalir et al., 2018; Chetty and Serra 2020). In SMARCA4-deficient cancer, selectively targeting SMARCA2 would be a potential synthetic lethal therapeutic method to treat cancer (Hoffman et al., 2014). SMARCA2 and SMARCA4 share 73.6% of protein sequence identity by EMBOSS Needle pairwise sequence alignment (Madeira et al., 2019), containing both ATP-dependent helicase domain and bromodomain (BD) domain. Small molecule inhibitors of the bromodomain developed so far inhibit the bromodomain of both SMARCA2 and SMARCA4 (Theodoulou et al., 2016). Additionally, the ATP-dependent helicase function of SMARCA2 is not targeted by the bromodomain inhibitor. An orally available SMARCA2 selective degrader would have potential therapeutic value over small molecule inhibitors. ACBI2 (Figure 4B) was reported by Kofinik and coworkers to be SMARCA2 selective PROTAC with improved oral bioavailability over PROTAC 10 (Figure 4B) via a minor change of the linker (Kofink et al., 2022). The PROTAC 10, with a five-carbon chain to link a SMARCA2/4 BD inhibitor and a VHL E3 ligase ligand, turned out to be a SMARCA2/4 degrader with only 4.3% oral bioavailability in mouse pharmacokinetic studies. The poor oral bioavailability was attributed to its poor permeability as indicated by the high efflux ratio from an in vitro Caco-2 permeability test. Introducing a methyl group to the full carbon chain to generate ACBI2, dramatically reduced the efflux ratio and thus increased the oral bioavailability to 22%. MD (Molecular Dynamics) simulation and NOE (Nuclear Overhauser effect) NMR spectroscopy were carried out to elucidate the link between conformational restraint and reduced efflux ratio. ACBI2 was found to have reduced TPSA by MD simulation compared to PROTAC 10. NOE is usually observed in NMR experiments between protons close in space. Long-range NOE observed in ACBI2 but not PROTAC 10 indicates that ACBI2 adopts a more constrained solution structure, which explains the reduced efflux ratio. For PROTAC with a constrained structure, macrocyclization could be a design strategy. In a case reported by Testa and coworkers, macrocyclization of MZ1 leads to a 12-fold loss of binding to BRD4, however, the comparable cellular degradation activity to MZ1 may indicate increased cell permeability (Testa et al., 2020). Because of the unique properties of bRo5 molecules, new descriptors such as EPSA and ChameLogD (Ermondi et al., 2020; Caron et al., 2021) have been introduced to take dynamic intramolecular hydrogen bond (dIMHB) into consideration for better correlation of the physicochemical properties of PROTACs with Caco-2 cell permeability profiles.
One of the advantages of PROTAC is in overcoming some of the resistance mechanisms to traditional targeted therapies (Burke et al., 2022), represented by AR PROTAC ARV-110 to address metastatic castration-resistant prostate cancer (mCRPC) (Salami and Crews 2017; Halford, 2021; Mullard 2021). Acquired drug resistance quite often occurs during the use of clinical small molecule inhibitors or antagonists, such as T790M and C797S mutation of EGFR conferred drug resistance induced by EGFR inhibitors (Thress et al., 2015). Although the resistance could be addressed by developing third- or fourth generations of EGFR inhibitors, new drug resistance will emerge. PROTAC technology has shown certain advantages in overcoming drug resistance against cancer drug targets due to the degradation of target proteins with reduced evolutionary pressure of target mutations (Shibata et al., 2017; Burslem et al., 2018; Burslem et al., 2019; Flanagan et al., 2019; Cheng et al., 2020; Robbins et al., 2020; Liu et al., 2021; Robbins et al., 2021). However, new mechanisms of drug resistance may occur (Zhang L. et al., 2019). Several research teams have revealed the vulnerabilities of UPS using siRNA-based loss-of-function screening (Ottis et al., 2019), resistance mutations by CRISPR-suppressor scanning (Gosavi et al., 2022), and potential acquired resistance mechanism against degraders by whole-exome sequencing in degrader selected cells lines (Zhang L. et al., 2019). The study of acquired resistance was carried out in SKM1, MV4; 11, LNCaP, and OVCAR8 cell lines. Resistance cell lines were selected after 4 months of treatment with BET PROTACs. The two AML cell lines (SKM-1 and MV4; 11) and the prostate cancer cell line (LNCaP) were much more sensitive to the compounds’ treatment compared with the ovarian cancer cell line OVCAR8. No stable resistant clones were obtained from SKM-1, MV4; 11, and LNCaP cell lines. The resistant clones from the BET PROTAC insensitive OVCAR8 cell line were further validated. The genomic and transcriptional analysis indicated that resistance to VHL-based BET PROTAC was caused by CUL2 loss due to multiple genetic alterations at the CUL2 locus; the resistance to CRBN-based BET PROTAC was due to chromosomal CRBN gene deletion. PROTAC is usually applied to the cancer cell lines highly dependent on the UPS system for therapeutic purposes, therefore the probability of acquired resistance due to loss of function of E3 ubiquitin ligase is small. Although there is no reported PROTAC resistance in the clinic yet, with more degraders approaching clinical trial, it is important to look for new cancer cell line essential E3 ligases for precision medicine.
In cells, DNA, RNA, and proteins are the key elements at the foundation of biological complexity, forming the backbone of what Francis Crick in 1957 termed the “Central Dogma” of molecular biology. However, according to Stuart Schreiber, there is a missing link in the network of Central Dogma: small molecules. Small molecules have critical roles at all levels of biological complexity and yet remain orphans of the Central Dogma (Schreiber 2005). Small molecule perturbation of protein functions has contributed a profound part to modern small molecule drug discovery (Beck et al., 2022). In addition to individual protein targeting by small molecules, chemically-induced proximity by heterobifunctional small molecules could redirect the biological processes of protein homeostasis. Targeted protein degrader is the type of induced-proximity molecule which targets POI for the posttranslational modification (PTM) of ubiquitination and subsequent proteasomal degradation (Figure 5). Protein homeostasis is regulated by many other types of PTMs (Uversky 2013), including but not limited to phosphorylation, acetylation, SUMOlyation, hydroxylation, farnesylation, glycosylation, and ADP-ribosylation. These PTMs of proteins regulate protein life span, protein cellular location, and protein function. Small molecules targeting the protein homeostasis by inducing PTM beyond ubiquitination may impact small molecule drug development in the pharmaceutical industry. PROTAC will offer the opportunity to target traditionally undruggable targets by an event-driven pharmacological approach, opening new therapeutic modalities (Békés et al., 2022) to expand the druggable space. Inspired by PROTAC, induced-proximity drug modalities including LYTAC (Banik et al., 2020), AUTAC (Takahashi et al., 2019), ATTEC (Z. Li et al., 2020), PhosTAC (Yamazoe et al., 2020; Chen P.-H. et al., 2021), DUBTAC (Kanner et al., 2020; Henning et al., 2021) and RIBOTAC (Haniff et al., 2020; P.; Zhang et al., 2021), are of interest to the pharmaceutical industry, allowing targeting of disease-causing proteins and even RNAs. This has resulted in the emergence of new start-up companies in the targeted protein degradation area (https://www.ventureradar.com/startup/Protein%20Degradation). The developments within Cryo-EM and X-ray crystallography technology, CRISPR-Cas technology-based assay development, and increasing sequencing capability will additionally strengthen the structure-guided design and multi-omics approach to designing small molecule therapeutics with induced-proximity mechanisms.
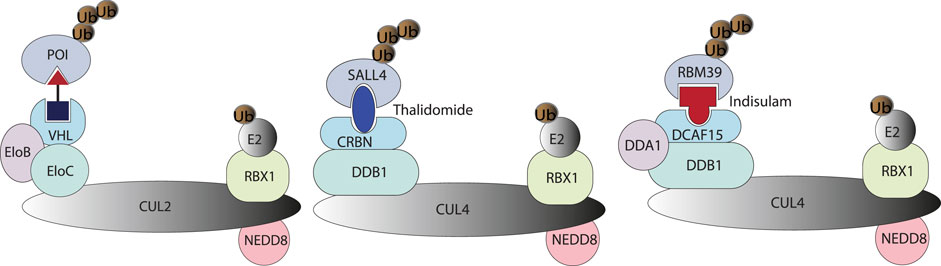
FIGURE 5. A PROTAC molecule is represented by a heterobifunctional molecule with a linker, which induces proximity between VHL and POI. Molecular glue degraders are represented by Thanlidomide and Indisulam-induced protein-protein interaction between neosubstrate and substrate-binding subunit of E3 ubiquitin ligase (Bulatov and Ciulli 2015).
LQ, HD, and JW conceived the content, drafted the manuscript, and contributed to the final version of the manuscript.
This work was primarily supported by the National Natural Science Foundation of China (Grant No. U1532269 to JW) and the Ministry of Science and Technology of China (Grant No. 2016YFA0400900 to JW). This work was also supported by the High Magnetic Field Laboratory of Anhui Province.
LQ is employed by Insilico Medicine Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors thank Feng Ren for critically reading and editing the manuscript.
Anderson, N. A., Cryan, J., Ahmed, A., Dai, H., McGonagle, G. A., Rozier, C., et al. (2020). Selective CDK6 Degradation Mediated by Cereblon, VHL, and Novel IAP-Recruiting PROTACs. Bioorg. Med. Chem. Lett. 30 (9), 127106. doi:10.1016/j.bmcl.2020.127106
Backus, K. M., Correia, B. E., Lum, K. M., Forli, S., Horning, B. D., González-Páez, G. E., et al. (2016). Proteome-Wide Covalent Ligand Discovery in Native Biological Systems. Nature 534 (7608), 570–574. doi:10.1038/nature18002
Banik, S. M., Pedram, K., Wisnovsky, S., Ahn, G., Riley, N. M., and Bertozzi, C. R. (2020). Lysosome-targeting Chimaeras for Degradation of Extracellular Proteins. Nature 584 (7820), 291–297. doi:10.1038/s41586-020-2545-9
Bar-Peled, L., Kemper, E. K., Suciu, R. M., Vinogradova, E. V., Backus, K. M., Horning, B. D., et al. (2017). Chemical Proteomics Identifies Druggable Vulnerabilities in a Genetically Defined Cancer. Cell. 171 (3), 696–709.e23. doi:10.1016/j.cell.2017.08.051
Beck, H., Härter, M., Haß, B., Schmeck, C., and Baerfacker, L. (2022). Small Molecules and Their Impact in Drug Discovery: A Perspective on the Occasion of the 125th Anniversary of the Bayer Chemical Research Laboratory. Drug Discov. Today 27 (6), 1560–1574. doi:10.1016/j.drudis.2022.02.015
Békés, M., Langley, D. R., and Crews, C. M. (2022). PROTAC Targeted Protein Degraders: The Past Is Prologue. Nat. Rev. Drug Discov. 21 (3), 181–200. doi:10.1038/s41573-021-00371-6
Bondeson, D. P., Smith, B. E., Burslem, G. M., Buhimschi, A. D., Hines, J., Jaime-Figueroa, S., et al. (2018). Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead. Cell. Chem. Biol. 25 (1), 78–87. e5. doi:10.1016/j.chembiol.2017.09.010
Bulatov, E., and Ciulli, A. (2015). Targeting Cullin-RING E3 Ubiquitin Ligases for Drug Discovery: Structure, Assembly and Small-Molecule Modulation. Biochem. J. 467 (3), 365–386. doi:10.1042/BJ20141450
Burke, M. R., Smith, A. R., and Zheng., G. (2022). Overcoming Cancer Drug Resistance Utilizing PROTAC Technology. Front. Cell. Dev. Biol. 10 (April), 872729. doi:10.3389/fcell.2022.872729
Burslem, G. M., and Crews, C. M. (2020). Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell. 181 (1), 102–114. doi:10.1016/j.cell.2019.11.031
Burslem, G. M., Schultz, A. R., Bondeson, D. P., Eide, C. A., Savage Stevens, S. L., Druker, B. J., et al. (2019). Targeting BCR-ABL1 in Chronic Myeloid Leukemia by PROTAC-Mediated Targeted Protein Degradation. Cancer Res. 79 (18), 4744–4753. doi:10.1158/0008-5472.CAN-19-1236
Burslem, G. M., Smith, B. E., Lai, A. C., Jaime-Figueroa, S., McQuaid, D. C., Bondeson, D. P., et al. (2018). The Advantages of Targeted Protein Degradation over Inhibition: An RTK Case Study. Cell. Chem. Biol. 25 (1), 67–77.e3. doi:10.1016/j.chembiol.2017.09.009
Caron, G., Kihlberg, J., Goetz, G., Ratkova, E., Poongavanam, V., and Ermondi, G. (2021). Steering New Drug Discovery Campaigns: Permeability, Solubility, and Physicochemical Properties in the BRo5 Chemical Space. ACS Med. Chem. Lett. 12 (1), 13–23. doi:10.1021/acsmedchemlett.0c00581
Chandrasekar, T., Yang, J. C., Gao, A. C., and Evans, C. P. (2015). Mechanisms of Resistance in Castration-Resistant Prostate Cancer (CRPC). Transl. Androl. Urol. 4 (3), 365–380. doi:10.3978/j.issn.2223-4683.2015.05.02
Chen, P.-H., Hu, Z., An, E., Okeke, I., Zheng, S., Luo, X., et al. (2021). Modulation of Phosphoprotein Activity by Phosphorylation Targeting Chimeras (PhosTACs). ACS Chem. Biol. 16 (12), 2808–2815. doi:10.1021/acschembio.1c00693
Chen, X., Crew, A., Flanagan, J., Sheryl, G., Royal, H., III, Marcia, M., et al. (2021). Methods of Treating Breast Cancer with Tetrahydronaphthalene Derivatives as Estrogen Receptor Degraders. United States Patent Application Publication US 2021/0060008 A1.
Cheng, M., Yu, X., Lu, K., Xie, L., Wang, L., Meng, F., et al. (2020). Discovery of Potent and Selective Epidermal Growth Factor Receptor (EGFR) Bifunctional Small-Molecule Degraders. J. Med. Chem. 63 (3), 1216–1232. doi:10.1021/acs.jmedchem.9b01566
Chetty, R., and Serra, S. (2020). SMARCA Family of Genes. J. Clin. Pathol. 73 (5), 257–260. doi:10.1136/jclinpath-2020-206451
Chung, C.-w., Dai, H., Fernandez, E., Tinworth, C. P., Churcher, I., Cryan, J., et al. (2020). Structural Insights into PROTAC-Mediated Degradation of Bcl-XL. ACS Chem. Biol. 15 (9), 2316–2323. doi:10.1021/acschembio.0c00266
Crew, A., Snyder, L., Wang, J., Royal, H., III, and Marcia, M. (2021). Methods of Treating Prostate Cancer. United States Patent Application Publication US 2021/0113557 A1.
Cromm, P. M., and Crews, C. M. (2017). Targeted Protein Degradation: From Chemical Biology to Drug Discovery. Cell. Chem. Biol. 24 (9), 1181–1190. doi:10.1016/j.chembiol.2017.05.024
Culig, Z., Hoffmann, J. M., Erdel, M., Eder, I. E., Hobisch, A., Hittmair, A., et al. (1999). Switch from Antagonist to Agonist of the Androgen Receptor Blocker Bicalutamide Is Associated with Prostate Tumour Progression in a New Model System. Br. J. Cancer 81 (2), 242–251. doi:10.1038/sj.bjc.6690684
Cyrus, K., Wehenkel, M., Choi, E.-Y., Han, H.-J., Lee, H., Swanson, H., et al. (2011). Impact of Linker Length on the Activity of PROTACs. Mol. Biosyst. 7 (2), 359–364. doi:10.1039/C0MB00074D
Dale, B., Cheng, M., Park, K.-S., Kaniskan, H. Ü., Xiong, Y., and Jin, J. (2021). Advancing Targeted Protein Degradation for Cancer Therapy. Nat. Rev. Cancer 21 (10), 638–654. doi:10.1038/s41568-021-00365-x
De Nardo, D., Balka, K. R., Cardona Gloria, Y., Rao, V. R., Latz, E., and Masters, S. L. (2018). Interleukin-1 Receptor-Associated Kinase 4 (IRAK4) Plays a Dual Role in Myddosome Formation and Toll-like Receptor Signaling. J. Biol. Chem. 293 (39), 15195–15207. doi:10.1074/jbc.RA118.003314
Dong, G., Ding, Y., He, S., and Sheng, C. (2021). Molecular Glues for Targeted Protein Degradation: From Serendipity to Rational Discovery. J. Med. Chem. 64 (15), 10606–10620. doi:10.1021/acs.jmedchem.1c00895
Donovan, K. A., An, J., Nowak, R. P., Yuan, J. C., Fink, E. C., Berry, B. C., et al. (2018). Thalidomide Promotes Degradation of SALL4, a Transcription Factor Implicated in Duane Radial Ray Syndrome. ELife 7 (August), e38430. doi:10.7554/eLife.38430
Du, X., Volkov, O. A., Czerwinski, R. M., Tan, H., Huerta, C., Morton, E. R., et al. (2019). Structural Basis and Kinetic Pathway of RBM39 Recruitment to DCAF15 by a Sulfonamide Molecular Glue E7820. Structure 27 (11), 1625–1633.e3. doi:10.1016/j.str.2019.10.005
Elumalai, P., and Arunakaran, J. (2014). Review on Molecular and Chemopreventive Potential of Nimbolide in Cancer. Genomics Inf. 12 (4), 156. doi:10.5808/GI.2014.12.4.156
Ermondi, G., Vallaro, M., Goetz, G., Shalaeva, M., and Caron, G. (2020). Updating the Portfolio of Physicochemical Descriptors Related to Permeability in the beyond the Rule of 5 Chemical Space. Eur. J. Pharm. Sci. 146 (April), 105274. doi:10.1016/j.ejps.2020.105274
Filippakopoulos, P., Qi, J., Picaud, S., Shen, Y., Smith, W. B., Fedorov, O., et al. (2010). Selective Inhibition of BET Bromodomains. Nature 468 (7327), 1067–1073. doi:10.1038/nature09504
Flanagan, J., Qian, Y., Gough, S., Andreoli, M., Bookbinder, M., Cadelina, G., et al. (2019). “Abstract P5-04-18: ARV-471, an Oral Estrogen Receptor PROTAC Degrader for Breast Cancer,” in Poster Session Abstracts, P5-04-18-P5-04–18 (American Association for Cancer Research). doi:10.1158/1538-7445.SABCS18-P5-04-18
Gadd, M. S., Testa, A., Lucas, X., Chan, K.-H., Chen, W., Lamont, D. J., et al. (2017). Structural Basis of PROTAC Cooperative Recognition for Selective Protein Degradation. Nat. Chem. Biol. 13 (5), 514–521. doi:10.1038/nchembio.2329
Gao, S., Wang, S., and Song, Y. (2020). Novel Immunomodulatory Drugs and Neo-Substrates. Biomark. Res. 8 (1), 2. doi:10.1186/s40364-020-0182-y
Gosavi, P. M., Ngan, K. C., Yeo, M. J. R., Su, C., Li, J., Lue, N. Z., et al. (2022). Profiling the Landscape of Drug Resistance Mutations in Neosubstrates to Molecular Glue Degraders. ACS Cent. Sci. 8, 417–429. doi:10.1021/acscentsci.1c01603
Guenette, R. G., Yang, S. W., Min, J., Pei, B., and Potts, P. R. (2022). Target and Tissue Selectivity of PROTAC Degraders. Chem. Soc. Rev. 101039, D2CS00200K. doi:10.1039/D2CS00200K
Han, T., Goralski, M., Gaskill, N., Capota, E., Kim, J., Ting, T. C., et al. (2017). Anticancer Sulfonamides Target Splicing by Inducing RBM39 Degradation via Recruitment to DCAF15. Science 356 (6336), eaal3755. doi:10.1126/science.aal3755
Han, X., Wang, C., Qin, C., Xiang, W., Fernandez-Salas, E., Yang, C.-Y., et al. (2019). Discovery of ARD-69 as a Highly Potent Proteolysis Targeting Chimera (PROTAC) Degrader of Androgen Receptor (AR) for the Treatment of Prostate Cancer. J. Med. Chem. 62 (2), 941–964. doi:10.1021/acs.jmedchem.8b01631
Haniff, H. S., Tong, Y., Liu, X., Chen, J. L., Suresh, B. M., Andrews, R. J., et al. (2020). Targeting the SARS-CoV-2 RNA Genome with Small Molecule Binders and Ribonuclease Targeting Chimera (RIBOTAC) Degraders. ACS Cent. Sci. 6 (10), 1713–1721. doi:10.1021/acscentsci.0c00984
Hansen, J. D., Correa, M., Alexander, M., Nagy, M., Huang, D., Sapienza, J., et al. (2021). CC-90009: A Cereblon E3 Ligase Modulating Drug that Promotes Selective Degradation of GSPT1 for the Treatment of Acute Myeloid Leukemia. J. Med. Chem. 64 (4), 1835–1843. doi:10.1021/acs.jmedchem.0c01489
Hansen, J. D., Correa, M., Nagy, M. A., Alexander, M., Plantevin, V., Grant, V., et al. (2020). Discovery of CRBN E3 Ligase Modulator CC-92480 for the Treatment of Relapsed and Refractory Multiple Myeloma. J. Med. Chem. 63 (13), 6648–6676. doi:10.1021/acs.jmedchem.9b01928
He, Y., Koch, R., Budamagunta, V., Lv, D., Khan, S., Zhang, X., et al. (2019). DT2216, a BCL-XL Proteolysis Targeting Chimera (PROTAC), Is a Potent Anti T-Cell Lymphoma Agent that Does Not Induce Significant Thrombocytopenia. Blood 134 (Suppl. ment_1), 303. doi:10.1182/blood-2019-125820
He, Y., Koch, R., Budamagunta, V., Zhang, P., Zhang, X., Khan, S., et al. (2020). DT2216-a Bcl-xL-specific Degrader Is Highly Active against Bcl-xL-dependent T Cell Lymphomas. J. Hematol. Oncol. 13 (1), 95. doi:10.1186/s13045-020-00928-9
Henderson, J., He, M., Good, A., and Andrew, P. (2020). Tricyclic Degraders of Ikaros and Aiolos. WO 2020/210630 A1.
Hennessy, E. J., Parker, A. E., and O'Neill, L. A. J. (2010). Targeting Toll-like Receptors: Emerging Therapeutics? Nat. Rev. Drug Discov. 9 (4), 293–307. doi:10.1038/nrd3203
Henning, N. J., Boike, L., Spradlin, J. N., Ward, C. C., Belcher, B., Brittain, S. M., et al. (2021). Deubiquitinase-Targeting Chimeras for Targeted Protein Stabilization. Prepr. Biochem. doi:10.1101/2021.04.30.441959
Hoffman, G. R., Rahal, R., Buxton, F., Xiang, K., McAllister, G., Frias, E., et al. (2014). Functional Epigenetics Approach Identifies BRM/SMARCA2 as a Critical Synthetic Lethal Target in BRG1-Deficient Cancers. Proc. Natl. Acad. Sci. U.S.A. 111 (8), 3128–3133. doi:10.1073/pnas.1316793111
Huang, H.-T., Dobrovolsky, D., Paulk, J., Yang, G., Weisberg, E. L., Doctor, Z. M., et al. (2018). A Chemoproteomic Approach to Query the Degradable Kinome Using a Multi-Kinase Degrader. Cell. Chem. Biol. 25 (1), 88–99.e6. doi:10.1016/j.chembiol.2017.10.005
Imaide, S., Riching, K. M., Makukhin, N., Vetma, V., Whitworth, C., Hughes, S. J., et al. (2021). Trivalent PROTACs Enhance Protein Degradation via Combined Avidity and Cooperativity. Nat. Chem. Biol. 17 (11), 1157–1167. doi:10.1038/s41589-021-00878-4
Ishida, T., and Ciulli, A. (2021). E3 Ligase Ligands for PROTACs: How They Were Found and How to Discover New Ones. SLAS Discov. 26 (4), 484–502. doi:10.1177/2472555220965528
Ishikawa, M., Tomoshige, S., Demizu, Y., and Naito, M. (2020). Selective Degradation of Target Proteins by Chimeric Small-Molecular Drugs, PROTACs and SNIPERs. Pharmaceuticals 13 (4), 74. doi:10.3390/ph13040074
Ito, T., Ando, H., Suzuki, T., Ogura, T., Hotta, K., Imamura, Y., et al. (2010). Identification of a Primary Target of Thalidomide Teratogenicity. Science 327 (5971), 1345–1350. doi:10.1126/science.1177319
Itoh, Y., Ishikawa, M., Naito, M., and Hashimoto, Y. (2010). Protein Knockdown Using Methyl Bestatin−Ligand Hybrid Molecules: Design and Synthesis of Inducers of Ubiquitination-Mediated Degradation of Cellular Retinoic Acid-Binding Proteins. J. Am. Chem. Soc. 132 (16), 5820–5826. doi:10.1021/ja100691p
Kanner, S. A., Shuja, Z., Choudhury, P., Jain, A., and Colecraft, H. M. (2020). Targeted Deubiquitination Rescues Distinct Trafficking-Deficient Ion Channelopathies. Nat. Methods 17 (12), 1245–1253. doi:10.1038/s41592-020-00992-6
Kannt, A., and Đikić, I. (2021). Expanding the Arsenal of E3 Ubiquitin Ligases for Proximity-Induced Protein Degradation. Cell. Chem. Biol. 28 (7), 1014–1031. doi:10.1016/j.chembiol.2021.04.007
Khan, S., Zhang, X., Lv, D., Zhang, Q., He, Y., Zhang, P., et al. (2019). A Selective BCL-XL PROTAC Degrader Achieves Safe and Potent Antitumor Activity. Nat. Med. 25 (12), 1938–1947. doi:10.1038/s41591-019-0668-z
Kim, T. W., Staschke, K., Bulek, K., Yao, J., Peters, K., Oh, K.-H., et al. (2007). A Critical Role for IRAK4 Kinase Activity in Toll-like Receptor-Mediated Innate Immunity. J. Exp. Med. 204 (5), 1025–1036. doi:10.1084/jem.20061825
Kofink, C., Trainor, N., Mair, B., Wöhrle, S., Wurm, M., Mischerikow, N., et al. (2022). A Selective and Orally Bioavailable VHL-Recruiting PROTAC Achieves SMARCA2 Degradation In Vivo. Prepr. Chem. doi:10.26434/chemrxiv-2022-q63s3
Kozicka, Z., and Thomä, N. H. (2021). Haven't Got a Glue: Protein Surface Variation for the Design of Molecular Glue Degraders. Cell. Chem. Biol. 28 (7), 1032–1047. doi:10.1016/j.chembiol.2021.04.009
Krönke, J., Udeshi, N. D., Narla, A., Grauman, P., Hurst, S. N., McConkey, M., et al. (2014). Lenalidomide Causes Selective Degradation of IKZF1 and IKZF3 in Multiple Myeloma Cells. Science 343 (6168), 301–305. doi:10.1126/science.1244851
Lai, A. C., and Crews, C. M. (2017). Induced Protein Degradation: An Emerging Drug Discovery Paradigm. Nat. Rev. Drug Discov. 16 (2), 101–114. doi:10.1038/nrd.2016.211
Lai, A. C., Toure, M., Hellerschmied, D., Salami, J., Jaime-Figueroa, S., Ko, E., et al. (2016). Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew. Chem. Int. Ed. 55 (2), 807–810. doi:10.1002/anie.201507634
Law, R. P., Nunes, J., Chung, C. w., Bantscheff, M., Buda, K., Dai, H., et al. (2021). Discovery and Characterisation of Highly Cooperative FAK‐Degrading PROTACs. Angew. Chem. Int. Ed. 60 (43), 23327–23334. doi:10.1002/anie.202109237
Leissing, T. M., Luh, L. M., and Cromm, P. M. (2020). Structure Driven Compound Optimization in Targeted Protein Degradation. Drug Discov. Today Technol. 37 (December), 73–82. doi:10.1016/j.ddtec.2020.11.005
Li, D., Yu, X., Kottur, J., Gong, W., Zhang, Z., Storey, A. J., et al. (2022). Discovery of a Dual WDR5 and Ikaros PROTAC Degrader as an Anti-cancer Therapeutic. Oncogene. doi:10.1038/s41388-022-02340-8
Li, Z., Zhu, C., Ding, Y., Fei, Y., and Lu, B. (2020). ATTEC: A Potential New Approach to Target Proteinopathies. Autophagy 16 (1), 185–187. doi:10.1080/15548627.2019.1688556
Liu, H., Ding, X., Liu, L., Mi, Q., Zhao, Q., Shao, Y., et al. (2021). Discovery of Novel BCR-ABL PROTACs Based on the Cereblon E3 Ligase Design, Synthesis, and Biological Evaluation. Eur. J. Med. Chem. 223 (November), 113645. doi:10.1016/j.ejmech.2021.113645
Lv, D., Pal, P., Liu, X., Jia, Y., Thummuri, D., Zhang, P., et al. (2021). Development of a BCL-xL and BCL-2 Dual Degrader with Improved Anti-leukemic Activity,. Nat. Commun. 12 (1), 6896. doi:10.1038/s41467-021-27210-x
Lv, L., Chen, P., Cao, L., Li, Y., Zeng, Z., Cui, Y., et al. (2020). Discovery of a Molecular Glue Promoting CDK12-DDB1 Interaction to Trigger Cyclin K Degradation. ELife 9 (August), e59994. doi:10.7554/eLife.59994
Madeira, F., Park, Y. M., Lee, J., Buso, N., Gur, T., Madhusoodanan, N., et al. (2019). The EMBL-EBI Search and Sequence Analysis Tools APIs in 2019. Nucleic Acids Res. 47 (W1), W636–W641. doi:10.1093/nar/gkz268
Mainolfi, N., Nan, J., Arthur, K., Weiss, J., Zhang, Y., and Zheng, X. (2020). IRAK Degraders and Uses Thereof. World Intellectural Property Organization International Bureau WO 2020/113233 A1.
Mashtalir, N., D’Avino, A. R., Michel, B. C., Luo, J., Pan, J., Otto, J. E., et al. (2018). Modular Organization and Assembly of SWI/SNF Family Chromatin Remodeling Complexes. Cell. 175 (5), 1272–1288.e20. doi:10.1016/j.cell.2018.09.032
Mayor-Ruiz, C., Bauer, S., Brand, M., Kozicka, Z., Siklos, M., Imrichova, H., et al. (2020). Rational Discovery of Molecular Glue Degraders via Scalable Chemical Profiling. Nat. Chem. Biol. 16 (11), 1199–1207. doi:10.1038/s41589-020-0594-x
Mohamad Anuar, N. N., Nor Hisam, N. S., Liew, S. L., and Ugusman, A. (2020). Clinical Review: Navitoclax as a Pro-apoptotic and Anti-fibrotic Agent. Front. Pharmacol. 11 (November), 564108. doi:10.3389/fphar.2020.564108
Mullard, A. (2021). Targeted Protein Degraders Crowd into the Clinic. Nat. Rev. Drug Discov. 20 (4), 247–250. doi:10.1038/d41573-021-00052-4
Nalawansha, D. A., and Crews, C. M. (2020). PROTACs: An Emerging Therapeutic Modality in Precision Medicine. Cell. Chem. Biol. 27 (8), 998–1014. doi:10.1016/j.chembiol.2020.07.020
Nasveschuk, C., Zeid, R., Yin, N., Jackson, K., Veits, G., and Moustakin, M. (2022). Compounds for Targeted Degradation of Brd9. United States Patent Application Publication US 2022/0098194 A1.
Ottis, P., Palladino, C., Thienger, P., Britschgi, A., Heichinger, C., Berrera, M., et al. (2019). Cellular Resistance Mechanisms to Targeted Protein Degradation Converge toward Impairment of the Engaged Ubiquitin Transfer Pathway. ACS Chem. Biol., 9b00525. October, acschembio. doi:10.1021/acschembio.9b00525
Ottis, P., Toure, M., Cromm, P. M., Ko, E., Gustafson, J. L., and Crews, C. M. (2017). Assessing Different E3 Ligases for Small Molecule Induced Protein Ubiquitination and Degradation. ACS Chem. Biol. 12 (10), 2570–2578. doi:10.1021/acschembio.7b00485
Paiva, S.-L., and Crews, C. M. (2019). Targeted Protein Degradation: Elements of PROTAC Design. Curr. Opin. Chem. Biol. 50 (June), 111–119. doi:10.1016/j.cbpa.2019.02.022
Pfäffle, H. N., Wang, M., Gheorghiu, L., Ferraiolo, N., Greninger, P., Borgmann, K., et al. (2013). EGFR-activating Mutations Correlate with a Fanconi Anemia-like Cellular Phenotype that Includes PARP Inhibitor Sensitivity. Cancer Res. 73 (20), 6254–6263. doi:10.1158/0008-5472.CAN-13-0044
Picard, C., Casanova, J.-L., and Puel, A. (2011). Infectious Diseases in Patients with IRAK-4, MyD88, NEMO, or IκBα Deficiency. Clin. Microbiol. Rev. 24 (3), 490–497. doi:10.1128/CMR.00001-11
Riching, K. M., Mahan, S., Corona, C. R., McDougall, M., Vasta, J. D., Robers, M. B., et al. (2018). Quantitative Live-Cell Kinetic Degradation and Mechanistic Profiling of PROTAC Mode of Action. ACS Chem. Biol. 13 (9), 2758–2770. doi:10.1021/acschembio.8b00692
Robbins, D. W., Kelly, A., Tan, M., McIntosh, J., Wu, J., Konst, Z., et al. (2020). Nx-2127, a Degrader of BTK and IMiD Neosubstrates, for the Treatment of B-Cell Malignancies. Blood 136 (Suppl. 1), 34. doi:10.1182/blood-2020-141461
Robbins, D. W., Noviski, M., Rountree, R., Tan, M., Brathaban, N., Ingallinera, T., et al. (2021). Nx-5948, a Selective Degrader of BTK with Activity in Preclinical Models of Hematologic and Brain Malignancies. Blood 138 (Suppl. 1), 2251. doi:10.1182/blood-2021-147473
Sakamoto, K. M., Kim, K. B., Kumagai, A., Mercurio, F., Crews, C. M., and Deshaies, R. J. (2001). Protacs: Chimeric Molecules that Target Proteins to the Skp1-Cullin-F Box Complex for Ubiquitination and Degradation. Proc. Natl. Acad. Sci. U.S.A. 98 (15), 8554–8559. doi:10.1073/pnas.141230798
Salami, J., and Crews, C. M. (2017). Waste Disposal-An Attractive Strategy for Cancer Therapy. Science 355 (6330), 1163–1167. doi:10.1126/science.aam7340
Samarasinghe, K. T. G., and Crews, C. M. (2021). Targeted Protein Degradation: A Promise for Undruggable Proteins. Cell. Chem. Biol. 28 (7), 934–951. doi:10.1016/j.chembiol.2021.04.011
Schneekloth, A. R., Pucheault, M., Seop, T., Tae, H. S., and Crews, C. M. (2008). Targeted Intracellular Protein Degradation Induced by a Small Molecule: En Route to Chemical Proteomics. Bioorg. Med. Chem. Lett. 18 (22), 5904–5908. doi:10.1016/j.bmcl.2008.07.114
Schneider, M., Radoux, C. J., Hercules, A., Ochoa, D., Dunham, I., Zalmas, L.-P., et al. (2021). The PROTACtable Genome. Nat. Rev. Drug Discov. 20 (10), 789–797. doi:10.1038/s41573-021-00245-x
Scholes, N. S., Mayor-Ruiz, C., and Winter, G. E. (2021). Identification and Selectivity Profiling of Small-Molecule Degraders via Multi-Omics Approaches. Cell. Chem. Biol. 28 (7), 1048–1060. doi:10.1016/j.chembiol.2021.03.007
Schreiber, S. L. (2005). Small Molecules: The Missing Link in the Central Dogma. Nat. Chem. Biol. 1 (2), 64–66. doi:10.1038/nchembio0705-64
Schreiber, S. L. (2021). The Rise of Molecular Glues. Cell. 184 (1), 3–9. doi:10.1016/j.cell.2020.12.020
Schwinn, M. K., Steffen, L. S., Zimmerman, K., Wood, K. V., and Machleidt., T. (2020). A Simple and Scalable Strategy for Analysis of Endogenous Protein Dynamics. Sci. Rep. 10 (1), 8953. doi:10.1038/s41598-020-65832-1
Shibata, N., Miyamoto, N., Nagai, K., Shimokawa, K., Sameshima, T., Ohoka, N., et al. (2017). Development of Protein Degradation Inducers of Oncogenic BCR ‐ ABL Protein by Conjugation of ABL Kinase Inhibitors and IAP Ligands. Cancer Sci. 108 (8), 1657–1666. doi:10.1111/cas.13284
Słabicki, M., Kozicka, Z., Petzold, G., Li, Y.-D., Manojkumar, M., Bunker, R. D., et al. (2020). The CDK Inhibitor CR8 Acts as a Molecular Glue Degrader that Depletes Cyclin K. Nature 585 (7824), 293–297. doi:10.1038/s41586-020-2374-x
Smith, B. E., Wang, S. L., Jaime-Figueroa, S., Harbin, A., Wang, J., Hamman, B. D., et al. (2019). Differential PROTAC Substrate Specificity Dictated by Orientation of Recruited E3 Ligase. Nat. Commun. 10 (1), 131. doi:10.1038/s41467-018-08027-7
Souers, A. J., Leverson, J. D., Boghaert, E. R., Ackler, S. L., Catron, N. D., Chen, J., et al. (2013). ABT-199, a Potent and Selective BCL-2 Inhibitor, Achieves Antitumor Activity while Sparing Platelets. Nat. Med. 19 (2), 202–208. doi:10.1038/nm.3048
Spradlin, J. N., Hu, X., Ward, C. C., Brittain, S. M., Jones, M. D., Ou, L., et al. (2019). Harnessing the Anti-cancer Natural Product Nimbolide for Targeted Protein Degradation. Nat. Chem. Biol. 15 (7), 747–755. doi:10.1038/s41589-019-0304-8
Takahashi, D., Moriyama, J., Nakamura, T., Miki, E., Takahashi, E., Sato, A., et al. (2019). AUTACs: Cargo-specific Degraders Using Selective Autophagy. Mol. Cell. 76 (5), 797–810.e10. doi:10.1016/j.molcel.2019.09.009
Tanaka, M., Roberts, J. M., Seo, H.-S., Souza, A., Paulk, J., Scott, T. G., et al. (2016). Design and Characterization of Bivalent BET Inhibitors. Nat. Chem. Biol. 12 (12), 1089–1096. doi:10.1038/nchembio.2209
Testa, A., Hughes, S. J., Lucas, X., Wright, J. E., and Ciulli, A. (2020). Structure‐Based Design of a Macrocyclic PROTAC. Angew. Chem. Int. Ed. 59 (4), 1727–1734. doi:10.1002/anie.201914396
Theodoulou, N. H., Tomkinson, N. C., Prinjha, R. K., and Humphreys, P. G. (2016). Clinical Progress and Pharmacology of Small Molecule Bromodomain Inhibitors. Curr. Opin. Chem. Biol. 33 (August), 58–66. doi:10.1016/j.cbpa.2016.05.028
Thress, K. S., Paweletz, C. P., Felip, E., Cho, B. C., Stetson, D., Dougherty, B., et al. (2015). Acquired EGFR C797S Mutation Mediates Resistance to AZD9291 in Non-small Cell Lung Cancer Harboring EGFR T790M. Nat. Med. 21 (6), 560–562. doi:10.1038/nm.3854
Tomoshige, S., and Ishikawa, M. (2021). PROTACs and Other Chemical Protein Degradation Technologies for the Treatment of Neurodegenerative Disorders. Angew. Chem. Int. Ed. 60 (7), 3346–3354. doi:10.1002/anie.202004746
Uversky, V. N. (2013). “Posttranslational Modification,” in Brenner’s Encyclopedia of Genetics. Editors K. Hughes, and S. Maloy (Elsevier), 425–30, 425–430. doi:10.1016/B978-0-12-374984-0.01203-1
Weiss, M., Zheng, X., and Xiao, Z. (2022)n.d. Double Degraders and Uses There of. World Intellectural Property Organization International Bureau WO/2022/087216.
Weerapana, E., Wang, C., Simon, G. M., Richter, F., Khare, S., Dillon, M. B. D., et al. (2010). Quantitative Reactivity Profiling Predicts Functional Cysteines in Proteomes. Nature 468 (7325), 790–795. doi:10.1038/nature09472
Xiang, W., Zhao, L., Han, X., Qin, C., Miao, B., McEachern, D., et al. (2021). Discovery of ARD-2585 as an Exceptionally Potent and Orally Active PROTAC Degrader of Androgen Receptor for the Treatment of Advanced Prostate Cancer. J. Med. Chem. 64 (18), 13487–13509. doi:10.1021/acs.jmedchem.1c00900
Yamazoe, S., Tom, J., Fu, Y., Wu, W., Zeng, L., Sun, C., et al. (2020). Heterobifunctional Molecules Induce Dephosphorylation of Kinases-A Proof of Concept Study. J. Med. Chem. 63 (6), 2807–2813. doi:10.1021/acs.jmedchem.9b01167
Yang, Y., Shaffer, A. L., Emre, N. C. T., Ceribelli, M., Zhang, M., Wright, G., et al. (2012). Exploiting Synthetic Lethality for the Therapy of ABC Diffuse Large B Cell Lymphoma. Cancer Cell. 21 (6), 723–737. doi:10.1016/j.ccr.2012.05.024
Zagidullin, A., Milyukov, V., Rizvanov, A., and Bulatov, E. (2020). Novel Approaches for the Rational Design of PROTAC Linkers. Explor. Target. Anti-Tumor Ther. 1 (5), 381–390. doi:10.37349/etat.2020.00023
Zengerle, M., Chan, K.-H., and Ciulli, A. (2015). Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol. 10 (8), 1770–1777. doi:10.1021/acschembio.5b00216
Zhang, L., Riley-Gillis, B., Vijay, P., and Shen, Y. (2019). Acquired Resistance to BET-PROTACs (Proteolysis-Targeting Chimeras) Caused by Genomic Alterations in Core Components of E3 Ligase Complexes. Mol. Cancer Ther. 18 (7), 1302–1311. doi:10.1158/1535-7163.MCT-18-1129
Zhang, P., Liu, X., Abegg, D., Tanaka, T., Tong, Y., Benhamou, R. I., et al. (2021). Reprogramming of Protein-Targeted Small-Molecule Medicines to RNA by Ribonuclease Recruitment. J. Am. Chem. Soc. 143 (33), 13044–13055. doi:10.1021/jacs.1c02248
Zhang, X., Crowley, V. M., Wucherpfennig, T. G., Dix, M. M., and Cravatt, B. F. (2019). Electrophilic PROTACs that Degrade Nuclear Proteins by Engaging DCAF16. Nat. Chem. Biol. 15 (7), 737–746. doi:10.1038/s41589-019-0279-5
Keywords: targeted protein degradation, PROTAC, molecular glue, chemically induced proximity, drug discovery and development
Citation: Qin L, Dai H and Wang J (2022) Key Considerations in Targeted Protein Degradation Drug Discovery and Development. Front. Chem. 10:934337. doi: 10.3389/fchem.2022.934337
Received: 02 May 2022; Accepted: 08 June 2022;
Published: 01 August 2022.
Edited by:
Hany S. Ibrahim, Martin Luther University of Halle-Wittenberg, GermanyReviewed by:
Tamer Elwaie, Cairo University, EgyptCopyright © 2022 Qin, Dai and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liena Qin, bGllbmEucWluQGluc2lsaWNvLmFp; Han Dai, ZGFpaGFuQGhtZmwuYWMuY24=; Junfeng Wang, anVuZmVuZ0BobWZsLmFjLmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.