 Yuqing Hua1
Yuqing Hua1 Bo Liu
Bo Liu Xiao Li
Xiao Li- 1Department of Clinical Pharmacy, The First Affiliated Hospital of Shandong First Medical University and Shandong Provincial Qianfoshan Hospital, Shandong Engineering and Technology Research Center for Pediatric Drug Development, Shandong Medicine and Health Key Laboratory of Clinical Pharmacy, Jinan, China
- 2Institute of Materia Medica, Shandong First Medical University & Shandong Academy of Medical Sciences, Jinan, China
- 3Department of Clinical Pharmacy, Shandong Provincial Qianfoshan Hospital, Shandong University, Jinan, China
The rapid and accurate evaluation of chemical toxicity is of great significance for estimation of chemical safety. In the past decades, a great number of excellent computational models have been developed for chemical toxicity prediction. But most machine learning models tend to be “black box”, which bring about poor interpretability. In the present study, we focused on the identification and collection of structural alerts (SAs) responsible for a series of important toxicity endpoints. Then, we carried out effective storage of these structural alerts and developed a web-server named SApredictor (www.sapredictor.cn) for screening chemicals against structural alerts. People can quickly estimate the toxicity of chemicals with SApredictor, and the specific key substructures which cause the chemical toxicity will be intuitively displayed to provide valuable information for the structural optimization by medicinal chemists.
Introduction
Nowadays, the development of chemical toxicology studies has provided us with extensive compound toxicity data. By analyzing and mining the existing toxicological experimental data, computational models can be established to predict the toxicity of chemical compounds around our lives. Compared with biological experimental methods, the computational methods were always green, fast, cheap, and accurate (Yang et al., 2018c). More importantly, toxicity can be predicted with computational models even before a chemical is synthesized or isolated. In the past decades, several expert systems, for example, Toxtree (Patlewicz et al., 2008) and OECD QSAR Toolbox (https://qsartoolbox.org/etc), and web-servers, for example, admetSAR (Yang et al., 2018a), ToxAlerts (Sushko et al., 2012), ADMETlab (Xiong et al., 2021), pkCSM (Pires et al., 2015), and vNN (Schyman et al., 2017) have also been proposed for in silico toxicity estimation.
The quantitative structure activity relationships (QSAR) method is one of the most widely used computational approaches for toxicity prediction, and many QSAR models are reported every year. However, these QSAR models based on machine learning methods tend to be “black box” models, which have limited the application in the prediction of various properties for regulatory agencies (Alves et al., 2016). In addition, the machine learning models need to face the problem of applicability domains (ADs) definition. Defining ADs are essential for regulatory acceptance of QSAR models, but there is less standard definition of AD for the global QSAR model nowadays, and many published QSAR models do not provide ADs (Wang et al., 2021).
Structural alert (SA) is another widely accepted tool for toxicity prediction in recent years, which can be defined as the key substructure which can cause specific toxicity. SA has been commonly used for assessment of many toxicity endpoints (Benigni et al., 2013; Li et al., 2017a; Limban et al., 2018; Kalgutkar, 2020; Cui et al., 2021; Huang et al., 2021; Shi et al., 2022) since Ashby and Tennant (1988) proposed the concept in 1985. The SAs can visually alert the toxicity of chemicals by displaying the key fragments responsible for drug toxicity because of the direct derivation from mechanistic knowledge. Therefore, SAs can provide valuable guidance and reference for structural optimization by medicinal chemists to reduce the risk (Yang et al., 2018c).
In the present study, we focused on screening chemicals against structural alerts, including 1) the identification of specific SAs responsible for the toxicity endpoints most concerned in drug studies based on a database with high quality toxicity data and the collection of reported SAs from research publications; and 2) the development of web-server for screening chemicals against structural alerts.
Materials and Methods
Data Collection and Preparation
The data for identification of structural alerts were collected from 1) the databases such as ChEMBL (Gaulton et al., 2011), ChemIDplus (Tomasulo, 2002), Comparative Toxicogenomics Database (CTD) (Davis et al., 2018), Carcinogenic Potency Database (CPDB) (Gold et al., 1984) and DrugBank (Wishart et al., 2017) and 2) peer-reviewed publications through manually filtering and processing. We focused on 22 toxicity endpoints which are of most concern in environmental toxicology and drug discovery, including acute oral toxicity (Li et al., 2014), chemical aquatic toxicity [Tetrahymena pyriformis (Cheng et al., 2011), Daphnia magna (Gajewicz-Skretna et al., 2021), and fathead minnow (Sun et al., 2015)], chemical-induced hematotoxicity (Hua et al., 2021), drug-induced neurotoxicity (Jiang et al., 2020), drug-induced autoimmune diseases (Wu et al., 2021), drug-induced ototoxicity (Huang et al., 2021), drug-induced rhabdomyolysis (Cui et al., 2019), endocrine disruption (Chen et al., 2014), eye irritation (Wang et al., 2017), hepatotoxicity (Li et al., 2018), hERG inhibition (Li et al., 2017c), honey bee toxicity (Li et al., 2017b), inhalation toxicity (Cui et al., 2021), mitochondrial toxicity (Nelms et al., 2015), mutagenicity (Yang et al., 2017), nephrotoxicity (Shi et al., 2022), non-genotoxic carcinogenicity (Benigni et al., 2013), reproductive and development toxicity (Fan et al., 2018; Jiang et al., 2019), skin sensitization (Di et al., 2019), and toxicity on avian species (Zhang et al., 2015). For each toxicity endpoint, we searched the literature separately and included the publications with the same definition of the toxicity endpoint and consistent toxic/non-toxic classification criteria.
The datasets were prepared in following steps to guarantee the quality and reliability of the data: 1) removing mixtures, inorganic, salts, and organic metallic compounds; 2) removing compounds without explicit description for toxicity properties or have inconsistent results in different experimental groups; 3) removing the fuzzy, uncertain, and obviously uncorrected data points; and 4) standardization and representing as canonical SMILES (O’Boyle, 2012).
Identification of Structural Alerts
The structural alerts were identified with two different methods, including SARpy (Ferrari et al., 2013) and fingerprints filter (Yang et al., 2020). Both the methods were based on frequency analysis, the general idea of which was to find some substructures presented more frequently in toxic compounds than in non-toxic ones (Yang et al., 2020). If a substructure presented far more frequently in toxic compounds than non-toxic compounds, the presence of such a substructure could alert to toxicity. Thus, this substructure should be regarded as a structural alert responsible for the specific toxicity. The flow of these two methods for identifying structural alerts was shown in Figure 1. SARpy is a python-based standalone software program for automated QSAR modeling. This program has been well-described in detail by Ferrari et al. (2013). Using SMILES-based algorithms, SARpy can cleave the compounds to obtain all possible fragments, and the potential structural alerts can be obtained by frequency analysis. In this study, rule sets were generated using standard settings; the substructures are composed of minimum two and maximum 18 atoms and occurring in a minimum of three substances. For the fingerprints filter method, the well-defined fingerprints of various lengths were utilized as the source of substructures. In the present study, the structural alerts were identified with a f-score and positive rate of each substructure from Klekota-Roth fingerprint (KRFP) calculated with a PaDEL-Descriptor (Yap, 2011), which contained 4,860 predefined structural fragments. The positive rate (PR) of a substructure is defined as Eq. 1:
where Nfragment_positive is the number of toxic compounds containing the fragment, and Nfragment is the total number of compounds containing the fragment. For each specific endpoint, only the fragments presented in six or more compounds were maintained. The fragments with f-score ≥0.005 and positive rate ≥0.65 were identified as structural alerts.

FIGURE 1. SARpy and fingerprints filter approaches implemented for structural alerts identification. (A) SARpy method is a fragment-based method, which can cut all possible bonds to obtain substructures. (B) Fingerprints filter approach would regard the predefined substructures as potential structural alerts.
The structural alerts were converted into SMARTS patterns (Hanson, 2016) and stored in the MySQL database. The SMARTS pattern is a language that allows users to specify substructures using the rules, which are straightforward extensions of SMILES. With SMARTS, flexible, and efficient substructure-search specifications can be made in terms that are meaningful to chemists, a compound can be matched against the alert in an automatic manner using one of the available chemical libraries (Sushko et al., 2012).
In addition, we also collected the structural alerts reported in the peer-reviewed publications. The collected structural alerts were also converted into SMARTS patterns, and the duplicates were removed.
Applicability Domain Definition
As emphasized by OECD principles, a well-defined applicability domain (AD) was preferred to make models more precise and robust (Yang et al., 2020). From the viewpoint of predictive performance, AD can also be helpful for improving the accuracy of SAs. Numerical relationships between chemical descriptors and toxicity values from training set are the basis of many applicability domain definition techniques, especially for QSAR models. However, AD for SAs have not been defined using these methods since the alerts are always a combination of structural information, toxic or non-toxic testing outcomes, and expert knowledge, which are used to directly link substructures with potential activity (Ellison et al., 2011). To date, there has been no single generally accepted algorithm for determining the AD on SAs. Since no chemical descriptors were used for SA model building, structural similarity could be the most appropriate measure to define the AD. Structural similarity is a popular AD definition method based on the concept that if a query chemical can be defined as similar to the chemicals in the training data, then it can be considered within the AD (Kühne et al., 2006; Ellison et al., 2011). In the present study, the similarity matrix was calculated employing the Tanimoto coefficient (Tc) (Godden et al., 2000; Bajusz et al., 2015) based on the KRFP fingerprint. The Tc is defined as Tc = Nab/(Na+ Nb− Nab), with Na being the number of bits set on in molecule a, Nb is the number of bits set on in molecule b, and Nab is the number of bits set on common to both molecules (Godden et al., 2000). The cutoff similarity value was defined as 0.5, thus if a query compound had a similarity value of ≥0.5 to at least one compound in the training set, it would be considered to be within the AD.
Toxicity Prediction With Structural Alerts
The structural alerts were assessed with the specific dataset of each endpoint. The compounds were input as SMILES and queried for matching the specific structural alerts with RDKit (Lovrić et al., 2019). If a compound contains one or more structural alerts, it would be predicted to have the specific toxicity. The evaluation was based on the counts of true positives (TP), false positives (FP), true negatives (TN), and false negatives (FN). Several statistical parameters were also calculated, including the total accuracy (Q), sensitivity (SE), specificity (SP), and positive predictive value (PPV). These parameters are calculated with Eqs. 2–5:
Web-Server Implementation
The prediction system was developed employing the Python web framework of Django. The system was deployed on an elastic compute service from Huawei Cloud running an Ubuntu Linux system. The web access was enabled via the Nginx web-server and the interactions between Django and proxy server were supported by mod_wsgi v3.3. A user-friendly web interface was provided for computational prediction using a cascading style sheet (CSS) and Python script.
Results and Discussion
Compound Libraries and Sets of Structural Alerts
In total, more than 35,716 annotated measurements of about 27,500 unique compounds were collected, including thousands of FDA-approved and experimental drugs, pesticides, environmental agents, and industrial chemicals. As shown in Table 1, these chemicals were checked and divided into 22 subsets, according to different toxicity endpoints.
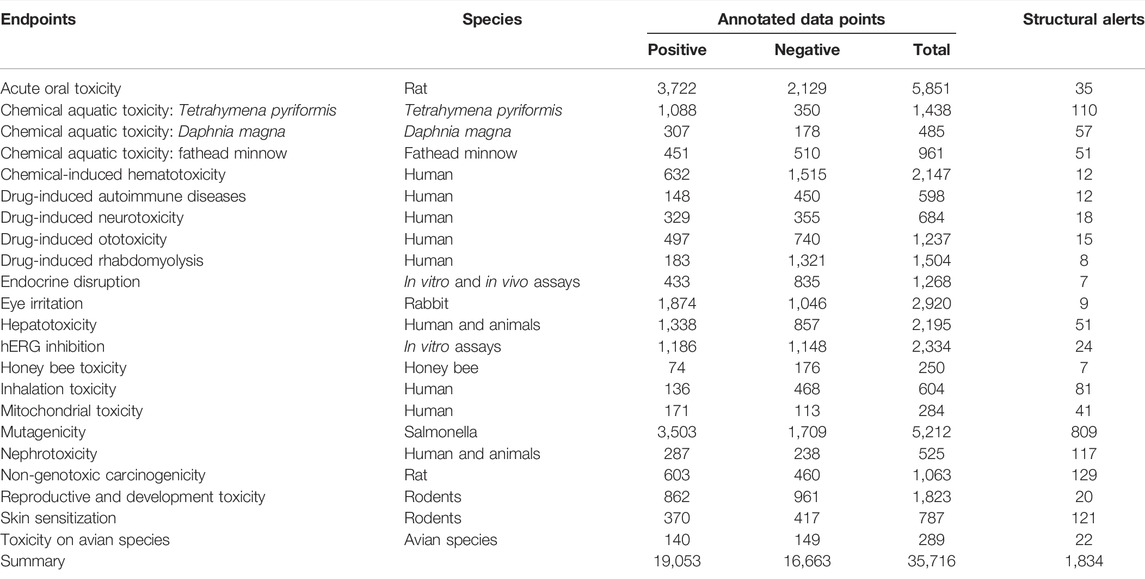
TABLE 1. Number of data points and structural alerts in the data set.
Through the identification and literature retrieval, a total of 1,834 structural alerts were identified and collected for the aforementioned 22 toxicity endpoints, as shown in Table 1. ToxAlerts and Toxtree are two popular tools for the estimation of potential adverse reactions of chemicals. ToxAlerts is a web-server of structural alerts, which collected SAs defined by experts or detected by computational tools. The latest ToxAlerts (accessed on 8 April 2022) contains 814 structural alerts for 13 toxicity endpoints, as shown in Supplementary Table S1. Toxtree is another user-friendly open-source application, which is able to estimate toxic hazard by applying a decision tree approach. In the latest version (Toxtree 3.1.0), in addition to the three Cramer Decision Trees (Cramer Rules, Revised Cramer Decision Tree, and Cramer Rules, with Extensions), it contains 499 structural alerts for 13 toxicity endpoints, as shown in Supplementary Table S2. To our knowledge, this may be the largest structural alert database with specific toxicity endpoints until now. In addition, several toxicity endpoints which get a lot of concerns (hepatotoxicity, nephrotoxicity, reproductive and development toxicity, hERG inhibition, hematotoxicity, mitochondrial toxicity, etc.) were included in SApredictor while not in ToxAlerts or Toxtree.
Performance of Toxicity Prediction With Structural Alerts
The performances of the structural alerts on toxicity prediction are shown in Table 2. The results suggested that for most endpoints, the structural alerts can well distinguish toxic compounds from non-toxic ones. For different toxicity endpoints, the disparity was observed in the performance. This can be attributed to that the complexity of the mechanisms of action (MOAs) of different toxicity endpoints vary greatly, and the sizes of the data are also different, which lead to differences in the representativeness of SAs and the ability to distinguish between toxic and non-toxic compounds.
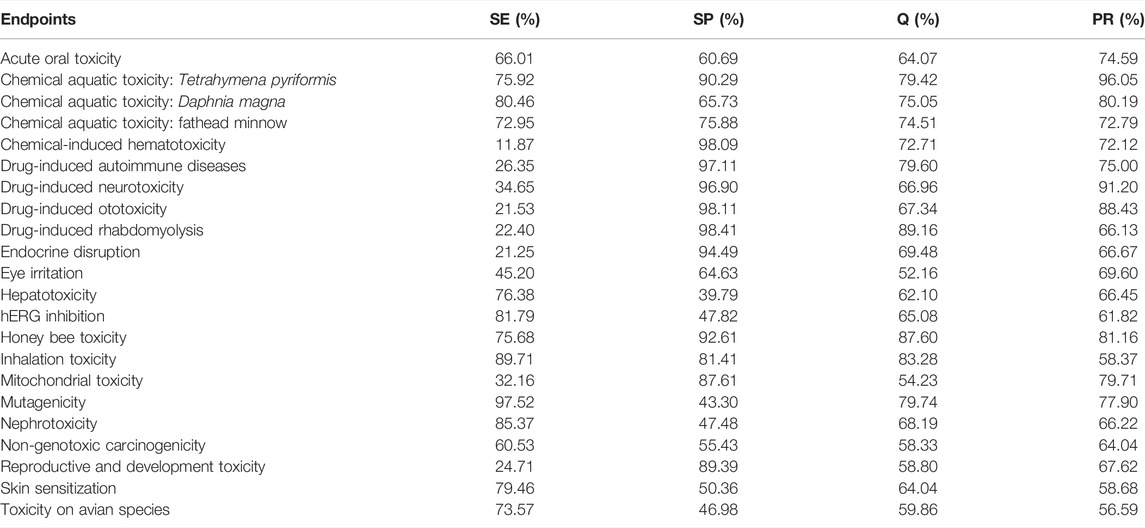
TABLE 2. Performance of toxicity prediction with structural alerts.
It was worth pointing out that compared with QSAR models, the prediction accuracy of structural alerts did not have any advantage in most cases. However, different from the QSAR’s black box model, the structural alerts can visually display the fragments that lead to specific toxicity of compounds, which is conducive to the targeted optimization of toxic structures and the study of toxic mechanisms (Yang et al., 2018c; Shi et al., 2022).
To ensure usefulness of the prediction system, it will be updated regularly with additional structural alerts based on available data, whether identified by ourselves or reported by peer-reviewed publications. If high quality datasets with new endpoints are reported, new structural alerts will be identified and implemented in our database.
Web Interface and Usage of the Structural Alerts
Based on distributed storage architectures, a piece of software for the estimation of chemical toxicity with structural alert was developed. The software provides a user-friendly interface via www.sapredictor.cn. A screenshot of the web-server is shown in Figure 2. Users can submit compound structures in two different ways: 1) enter the SMILES of small compounds in the dialog box; 2) click the “Select File” button to upload the structure file of compounds with SMILES format. After entering the verification code, users can click the “Predict” button to complete the task submission.

FIGURE 2. SApredictor main page. From this page, users can submit the query structure.
After the matching of the structural alerts, it will be redirected to the results page, as shown in Figure 3. On the left is the 2D structure of the query compound and on the right is the toxicity endpoints and corresponding predicted result. Where “Yes” indicates that the query structure contains one or more structural alerts of the specific toxic property, that is, the compound has the potential of the specific toxicity, while “No” indicates the query compound does not have the potential of the specific toxicity. For the toxicity endpoint with the result of “Yes,” click the name of the toxicity endpoint and a drop-down list will appear listing the ID of structural alerts. When clicking the ID, the fragments of the compound will be highlighted in red, and the SMARTS of the alert will also be available on the page. The researchers can view the specific substructure that causes the toxicity of the compound.
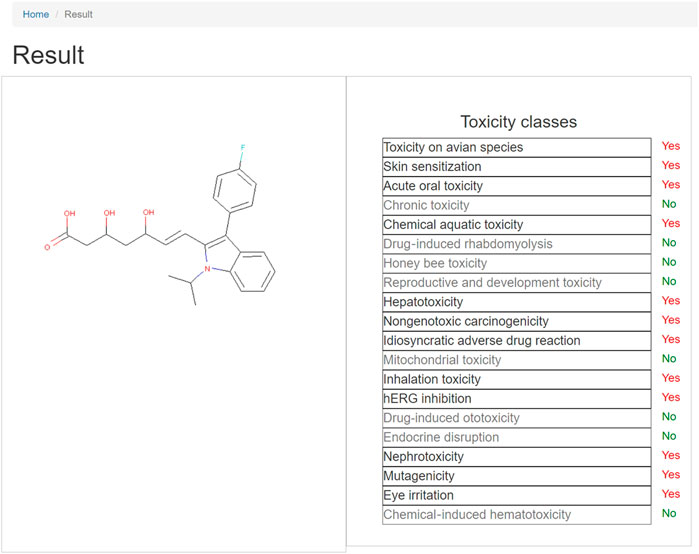
FIGURE 3. Structural alert-based toxicity predictions result page. When clicking the name of the toxicity endpoint withe positive result, a drop-down list will appear with the ID of structural alerts. Users can click the ID, and then the fragments of the compound will be highlighted in red, so that researchers can view the specific substructure that causes the toxicity of the compound.
Conclusion and Perspectives
In summary, we have described here a web-server, named SApredictor, for screening chemicals against structural alerts via www.sapredictor.cn. In SApredictor, 1,834 structural alerts for 22 different toxicity endpoints were extracted from more than 35,716 toxicity annotated data points or collected from peer-reviewed publications. Users can quickly estimate the toxicity of compounds and visually display the fragments, which contribute to their toxicity. The web-server will never retain any information submitted to it because of the confidentiality of users’ projects. We hope that the software should facilitate the process of drug discovery and development by enabling the rapid and rational screening, design, evaluation, and prioritization of drug candidates.
It is worth pointing out that use of structural alerts alone may suffer from false positives, such as skin sensitization and toxicity on avian species in the present study. The structural alerts were always identified by statistics-based methods or knowledge of toxic mechanisms, which would make them be overtly common and lead to many non-toxic structures being estimated as toxic. On the other hand, it is debatable whether compounds which do not contain any SA can be classified as non-toxic. Toxicity prediction based on SA is based on the existing knowledge. The compounds with SAs are always toxic, but whether those without SA are non-toxic needs more toxicity data support. Yang et al. (2018b) proposed a concept of non-toxic substructures, whose appearance will reduce the probability of a compound becoming toxic (Yang et al., 2018b). In Wang et al. (2012) work, modulating factors that suppressed the toxic effects of SAs were extracted and practice on carcinogens (Wang et al., 2012). Non-toxic substructures and modulating factors could be beneficial supplements to SAs. In addition to optimizing the identification method of structural alerts, defining the applicability domain for the structural alerts in a reasonable strategy may be helpful to improve the predictive performance and eliminate the worries. We will continue to work in both directions to improve the predictive ability of structural alerts and make them more useful.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding authors.
Author Contributions
XL contributed to conception and design of the study. YH, XC, and BL collected the datasets and carried out the experiments. YH, BL, HG, and RZ performed the analysis. YH, XC, BL, and XL interpreted the results and wrote the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (grant 81803433) and the Special Research project of Clinical Toxicology of Chinese Society of Toxicology (CST2020CT104).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors gratefully acknowledge the encouragement and support from Miss Chaoyue Yang and Mr. Yibo Wang.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2022.916614/full#supplementary-material
References
Alves, V. M., Muratov, E. N., Capuzzi, S. J., Politi, R., Low, Y., Braga, R. C., et al. (2016). Alarms about Structural Alerts. Green Chem. 18 (16), 4348–4360. doi:10.1039/C6GC01492E
Ashby, J., and Tennant, R. W. (1988). Chemical Structure, Salmonella Mutagenicity and Extent of Carcinogenicity as Indicators of Genotoxic Carcinogenesis Among 222 Chemicals Tested in Rodents by the U.S. NCI/NTP. Mutat. Research/Genetic Toxicol. 204 (1), 17–115. doi:10.1016/0165-1218(88)90114-0
Bajusz, D., Rácz, A., and Héberger, K. (2015). Why Is Tanimoto Index an Appropriate Choice for Fingerprint-Based Similarity Calculations? J. Cheminform 7 (1), 20. doi:10.1186/s13321-015-0069-3
Benigni, R., Bossa, C., and Tcheremenskaia, O. (2013). Nongenotoxic Carcinogenicity of Chemicals: Mechanisms of Action and Early Recognition through a New Set of Structural Alerts. Chem. Rev. 113 (5), 2940–2957. doi:10.1021/cr300206t
Chen, Y., Cheng, F., Sun, L., Li, W., Liu, G., and Tang, Y. (2014). Computational Models to Predict Endocrine-Disrupting Chemical Binding with Androgen or Oestrogen Receptors. Ecotoxicol. Environ. Saf. 110, 280–287. doi:10.1016/j.ecoenv.2014.08.026
Cheng, F., Shen, J., Yu, Y., Li, W., Liu, G., Lee, P. W., et al. (2011). In Silico prediction of Tetrahymena Pyriformis Toxicity for Diverse Industrial Chemicals with Substructure Pattern Recognition and Machine Learning Methods. Chemosphere 82 (11), 1636–1643. doi:10.1016/j.chemosphere.2010.11.043
Cui, X., Liu, J., Zhang, J., Wu, Q., and Li, X. (2019). In Silico prediction of Drug‐induced Rhabdomyolysis with Machine‐learning Models and Structural Alerts. J. Appl. Toxicol. 39 (8), 1224–1232. doi:10.1002/jat.3808
Cui, X., Yang, R., Li, S., Liu, J., Wu, Q., and Li, X. (2021). Modeling and Insights into Molecular Basis of Low Molecular Weight Respiratory Sensitizers. Mol. Divers 25 (2), 847–859. doi:10.1007/s11030-020-10069-3
Davis, A. P., Grondin, C. J., Johnson, R. J., Sciaky, D., McMorran, R., Wiegers, J., et al. (2018). The Comparative Toxicogenomics Database: Update 2019. Nucleic Acids Res. 47 (D1), D948–D954. doi:10.1093/nar/gky868
Di, P., Yin, Y., Jiang, C., Cai, Y., Li, W., Tang, Y., et al. (2019). Prediction of the Skin Sensitising Potential and Potency of Compounds via Mechanism-Based Binary and Ternary Classification Models. Toxicol. Vitro 59, 204–214. doi:10.1016/j.tiv.2019.01.004
Ellison, C. M., Sherhod, R., Cronin, M. T. D., Enoch, S. J., Madden, J. C., and Judson, P. N. (2011). Assessment of Methods to Define the Applicability Domain of Structural Alert Models. J. Chem. Inf. Model. 51 (5), 975–985. doi:10.1021/ci1000967
Fan, D., Yang, H., Li, F., Sun, L., Di, P., Li, W., et al. (2018). In Silico prediction of Chemical Genotoxicity Using Machine Learning Methods and Structural Alerts. Toxicol. Res. 7 (2), 211–220. doi:10.1039/C7TX00259A
Ferrari, T., Cattaneo, D., Gini, G., Golbamaki Bakhtyari, N., Manganaro, A., and Benfenati, E. (2013). Automatic Knowledge Extraction from Chemical Structures: the Case of Mutagenicity Prediction. SAR QSAR Environ. Res. 24 (5), 365–383. doi:10.1080/1062936X.2013.773376
Gajewicz-Skretna, A., Furuhama, A., Yamamoto, H., and Suzuki, N. (2021). Generating Accurate In Silico Predictions of Acute Aquatic Toxicity for a Range of Organic Chemicals: Towards Similarity-Based Machine Learning Methods. Chemosphere 280, 130681. doi:10.1016/j.chemosphere.2021.130681
Gaulton, A., Bellis, L. J., Bento, A. P., Chambers, J., Davies, M., Hersey, A., et al. (2011). ChEMBL: a Large-Scale Bioactivity Database for Drug Discovery. Nucleic Acids Res. 40 (D1), D1100–D1107. doi:10.1093/nar/gkr777
Godden, J. W., Xue, L., and Bajorath, J. (2000). Combinatorial Preferences Affect Molecular Similarity/diversity Calculations Using Binary Fingerprints and Tanimoto Coefficients. J. Chem. Inf. Comput. Sci. 40 (1), 163–166. doi:10.1021/ci990316u
Gold, L. S., Sawyer, C. B., Magaw, R., Backman, G. M., De Veciana, M., Levinson, R., et al. (1984). A Carcinogenic Potency Database of the Standardized Results of Animal Bioassays. Environ. Health Perspect. 58, 9–319. doi:10.1289/ehp.84589
Hanson, R. M. (2016). Jmol SMILES and Jmol SMARTS: Specifications and Applications. J. Cheminform 8 (1), 50. doi:10.1186/s13321-016-0160-4
Hua, Y., Shi, Y., Cui, X., and Li, X. (2021). In Silico prediction of Chemical-Induced Hematotoxicity with Machine Learning and Deep Learning Methods. Mol. Divers 25 (3), 1585–1596. doi:10.1007/s11030-021-10255-x
Huang, X., Tang, F., Hua, Y., and Li, X. (2021). In Silico prediction of Drug‐induced Ototoxicity Using Machine Learning and Deep Learning Methods. Chem. Biol. Drug Des. 98 (2), 248–257. doi:10.1111/cbdd.13894
Jiang, C., Yang, H., Di, P., Li, W., Tang, Y., and Liu, G. (2019). In Silico prediction of Chemical Reproductive Toxicity Using Machine Learning. J. Appl. Toxicol. 39 (6), 844–854. doi:10.1002/jat.3772
Jiang, C., Zhao, P., Li, W., Tang, Y., and Liu, G. (2020). In Silico prediction of Chemical Neurotoxicity Using Machine Learning. Toxicol. Res. 9 (3), 164–172. doi:10.1093/toxres/tfaa016
Kalgutkar, A. S. (2020). Designing Around Structural Alerts in Drug Discovery. J. Med. Chem. 63 (12), 6276–6302. doi:10.1021/acs.jmedchem.9b00917
Kühne, R., Ebert, R.-U., and Schüürmann, G. (2006). Model Selection Based on Structural Similarity−Method Description and Application to Water Solubility Prediction. J. Chem. Inf. Model. 46 (2), 636–641. doi:10.1021/ci0503762
Li, X., Chen, L., Cheng, F., Wu, Z., Bian, H., Xu, C., et al. (2014). In Silico prediction of Chemical Acute Oral Toxicity Using Multi-Classification Methods. J. Chem. Inf. Model. 54 (4), 1061–1069. doi:10.1021/ci5000467
Li, X., Chen, Y., Song, X., Zhang, Y., Li, H., and Zhao, Y. (2018). The Development and Application of In Silico Models for Drug Induced Liver Injury. RSC Adv. 8 (15), 8101–8111. doi:10.1039/C7RA12957B
Li, X., Zhang, Y., Chen, H., Li, H., and Zhao, Y. (2017a). In Silico prediction of Chronic Toxicity with Chemical Category Approaches. RSC Adv. 7 (66), 41330–41338. doi:10.1039/C7RA08415C
Li, X., Zhang, Y., Chen, H., Li, H., and Zhao, Y. (2017b). Insights into the Molecular Basis of the Acute Contact Toxicity of Diverse Organic Chemicals in the Honey Bee. J. Chem. Inf. Model. 57 (12), 2948–2957. doi:10.1021/acs.jcim.7b00476
Li, X., Zhang, Y., Li, H., and Zhao, Y. (2017c). Modeling of the hERG K+ Channel Blockage Using Online Chemical Database and Modeling Environment (OCHEM). Mol. Inf. 36 (12), 1700074. doi:10.1002/minf.201700074
Limban, C., Nuţă, D. C., Chiriţă, C., Negreș, S., Arsene, A. L., Goumenou, M., et al. (2018). The Use of Structural Alerts to Avoid the Toxicity of Pharmaceuticals. Toxicol. Rep. 5, 943–953. doi:10.1016/j.toxrep.2018.08.017
Lovrić, M., Molero, J. M., and Kern, R. (2019). PySpark and RDKit: Moving towards Big Data in Cheminformatics. Mol. Inf. 38 (6), 1800082. doi:10.1002/minf.201800082
Nelms, M. D., Mellor, C. L., Cronin, M. T. D., Madden, J. C., and Enoch, S. J. (2015). Development of an In Silico Profiler for Mitochondrial Toxicity. Chem. Res. Toxicol. 28 (10), 1891–1902. doi:10.1021/acs.chemrestox.5b00275
O’Boyle, N. M. (2012). Towards a Universal Smiles Representation - a Standard Method to Generate Canonical Smiles Based on the InChI. J. Cheminform 4 (1), 22. doi:10.1186/1758-2946-4-22
Patlewicz, G., Jeliazkova, N., Safford, R. J., Worth, A. P., and Aleksiev, B. (2008). An Evaluation of the Implementation of the Cramer Classification Scheme in the Toxtree Software. SAR QSAR Environ. Res. 19 (5-6), 495–524. doi:10.1080/10629360802083871
Pires, D. E. V., Blundell, T. L., and Ascher, D. B. (2015). pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 58 (9), 4066–4072. doi:10.1021/acs.jmedchem.5b00104
Schyman, P., Liu, R., Desai, V., and Wallqvist, A. (2017). vNN Web Server for ADMET Predictions. Front. Pharmacol. 8, 889. doi:10.3389/fphar.2017.00889
Shi, Y., Hua, Y., Wang, B., Zhang, R., and Li, X. (2022). In Silico Prediction and Insights into the Structural Basis of Drug Induced Nephrotoxicity. Front. Pharmacol. 12, 793332. doi:10.3389/fphar.2021.793332
Sun, L., Zhang, C., Chen, Y., Li, X., Zhuang, S., Li, W., et al. (2015). In Silico prediction of Chemical Aquatic Toxicity with Chemical Category Approaches and Substructural Alerts. Toxicol. Res. 4 (2), 452–463. doi:10.1039/C4TX00174E
Sushko, I., Salmina, E., Potemkin, V. A., Poda, G., and Tetko, I. V. (2012). ToxAlerts: a Web Server of Structural Alerts for Toxic Chemicals and Compounds with Potential Adverse Reactions. J. Chem. Inf. Model. 52 (8), 2310–2316. doi:10.1021/ci300245q
Tomasulo, P. (2002). ChemIDplus-super Source for Chemical and Drug Information. Med. Ref. Serv. Q. 21 (1), 53–59. doi:10.1300/J115v21n01_04
Wang, Q., Li, X., Yang, H., Cai, Y., Wang, Y., Wang, Z., et al. (2017). In Silico prediction of Serious Eye Irritation or Corrosion Potential of Chemicals. RSC Adv. 7 (11), 6697–6703. doi:10.1039/C6RA25267B
Wang, Y., Lu, J., Wang, F., Shen, Q., Zheng, M., Luo, X., et al. (2012). Estimation of Carcinogenicity Using Molecular Fragments Tree. J. Chem. Inf. Model. 52 (8), 1994–2003. doi:10.1021/ci300266p
Wang, Z., Chen, J., and Hong, H. (2021). Developing QSAR Models with Defined Applicability Domains on PPARγ Binding Affinity Using Large Data Sets and Machine Learning Algorithms. Environ. Sci. Technol. 55 (10), 6857–6866. doi:10.1021/acs.est.0c07040
Wishart, D. S., Feunang, Y. D., Guo, A. C., Lo, E. J., Marcu, A., Grant, J. R., et al. (2017). DrugBank 5.0: a Major Update to the DrugBank Database for 2018. Nucleic Acids Res. 46 (D1), D1074–D1082. doi:10.1093/nar/gkx1037
Wu, Y., Zhu, J., Fu, P., Tong, W., Hong, H., and Chen, M. (2021). Machine Learning for Predicting Risk of Drug-Induced Autoimmune Diseases by Structural Alerts and Daily Dose. Ijerph 18 (13), 7139. doi:10.3390/ijerph18137139
Xiong, G., Wu, Z., Yi, J., Fu, L., Yang, Z., Hsieh, C., et al. (2021). ADMETlab 2.0: an Integrated Online Platform for Accurate and Comprehensive Predictions of ADMET Properties. Nucleic Acids Res. 49 (W1), W5–W14. doi:10.1093/nar/gkab255
Yang, H., Li, J., Wu, Z., Li, W., Liu, G., and Tang, Y. (2017). Evaluation of Different Methods for Identification of Structural Alerts Using Chemical Ames Mutagenicity Data Set as a Benchmark. Chem. Res. Toxicol. 30 (6), 1355–1364. doi:10.1021/acs.chemrestox.7b00083
Yang, H., Lou, C., Li, W., Liu, G., and Tang, Y. (2020). Computational Approaches to Identify Structural Alerts and Their Applications in Environmental Toxicology and Drug Discovery. Chem. Res. Toxicol. 33 (6), 1312–1322. doi:10.1021/acs.chemrestox.0c00006
Yang, H., Lou, C., Sun, L., Li, J., Cai, Y., Wang, Z., et al. (2018a). admetSAR 2.0: Web-Service for Prediction and Optimization of Chemical ADMET Properties. Bioinformatics 35 (6), 1067–1069. doi:10.1093/bioinformatics/bty707
Yang, H., Sun, L., Li, W., Liu, G., and Tang, Y. (2018b). Identification of Nontoxic Substructures: a New Strategy to Avoid Potential Toxicity Risk. Toxicol. Sci. 165 (2), 396–407. doi:10.1093/toxsci/kfy146
Yang, H., Sun, L., Li, W., Liu, G., and Tang, Y. (2018c). In Silico prediction of Chemical Toxicity for Drug Design Using Machine Learning Methods and Structural Alerts. Front. Chem. 6, 30. doi:10.3389/fchem.2018.00030
Yap, C. W. (2011). PaDEL-descriptor: An Open Source Software to Calculate Molecular Descriptors and Fingerprints. J. Comput. Chem. 32 (7), 1466–1474. doi:10.1002/jcc.21707
Keywords: SApredictor, structural alerts, web-server, toxicity prediction, expert system
Citation: Hua Y, Cui X, Liu B, Shi Y, Guo H, Zhang R and Li X (2022) SApredictor: An Expert System for Screening Chemicals Against Structural Alerts. Front. Chem. 10:916614. doi: 10.3389/fchem.2022.916614
Received: 09 April 2022; Accepted: 20 June 2022;
Published: 13 July 2022.
Edited by:
Xiaoming Zhang, Hebei University of Technology, ChinaReviewed by:
Ramakrishnan Parthasarathi, Indian Institute of Toxicology Research (CSIR), IndiaCrtomir Podlipnik, University of Ljubljana, Slovenia
Copyright © 2022 Hua, Cui, Liu, Shi, Guo, Zhang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiao Li, bGl4aWFvMTY4OEAxNjMuY29t, eC5saUBzZHUuZWR1LmNu, b3JjaWQub3JnLzAwMDAtMDAwMi0xMTQ4LTk4OTg=