
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. , 17 June 2022
Sec. Organic Chemistry
Volume 10 - 2022 | https://doi.org/10.3389/fchem.2022.914944
This article is part of the Research Topic Bioactive Natural Products-Oriented Synthetic and Functional Studies View all 24 articles
 Ya-Guang Hu1†Zhu-Peng Gao1†Ying-Ying Zheng2Chun-Mei Hu2
Ya-Guang Hu1†Zhu-Peng Gao1†Ying-Ying Zheng2Chun-Mei Hu2 Jing Lin2Xiao-Zheng Wu2Xin Zhang2Yong-Sheng Zhou3
Jing Lin2Xiao-Zheng Wu2Xin Zhang2Yong-Sheng Zhou3 Zhuang Xiong2*
Zhuang Xiong2* Dao-Yong Zhu1*
Dao-Yong Zhu1*In order to find potential inhibitors of tyrosinase, two series of pyrrole derivatives A (1–17) and B (1–8) were synthesized and screened for their inhibitory activities on tyrosinase. Most of the 2-cyanopyrrole derivatives exhibited effective inhibitory activities. In particular, A12 exhibited the strongest inhibitory activities, with the IC50 values of 0.97 μM, which is ∼30 times stronger than the reference inhibitor kojic acid (IC50: 28.72 μM). The inhibitory mechanism analysis results revealed that A12 was a reversible and mixed-type inhibitor. Molecular docking experiments clarified the interaction between A12 with tyrosinase. Furthermore, A12 (100 μM) presented effective inhibitory effect on tyrosinase in B16 melanoma cells with inhibition of 33.48%, which was equivalent to that of Kojic acid (39.81%). Accordingly, compound A12 may serve as the lead structure for the further design of potent tyrosinase inhibitors. Molecular docking studies confirmed the interaction between the compound and tyrosinase.
The browning of food including fruits, vegetables, and beverages seriously threats the development of agriculture and the food industry (Chai et al., 2018). Recently, quality loss of food during postharvest handling and processing occur frequently in the food industry (Zhu et al., 2011). The recognized mechanism of the corresponding browning includes two types of procedures, namely, enzymatic and non-enzymatic browning (Todaro et al., 2011). Tyrosinase, a metalloenzyme containing dinuclear copper ions, is one of the key catalytic proteins in the process of enzymatic browning (Liu et al., 2012; Asthana et al., 2015; Chai et al., 2015; Xie et al., 2016). It is widely distributed in animals, insects, plants, and microorganisms and is a rate-limiting enzyme involved in two-step oxidation reactions including mono phenols into diphenols and o-diphenols into o-quinones, followed by further transformations into dark brown pigments (Ullah et al., 2016; Cai et al., 2019; Ielo et al., 2019; Zolghadri et al., 2019). For plant-derived foods, enzymatic browning would result in abnormal variations of color, flavor, and nutritional quality (Larik et al., 2017). In addition, it has been believed that excessively active tyrosinase leads to the accumulation of melanin in the human skin, which results in various common skin diseases, such as freckles, melasma, and melanosis (Ullah et al., 2019a; Ullah et al., 2019b; Butt et al., 2019; Haldys and Latajka, 2019; Huang et al., 2019; Lee et al., 2019). In the biosynthesis pathway of melanin, tyrosinase oxidizes l-tyrosine into L-3,4-dihydroxyphenylalanine (l-DOPA), and l-DOPA into o-quinones (Akın et al., 2019; Ashooriha et al., 2019; Santi et al., 2019). Tyrosinase is also proven to be related to Parkinson’s and other neurodegenerative diseases (Ali et al., 2019). Moreover, tyrosinase plays a key role in the processes of cuticle formation in insects (Yu, 2003). Hence, developing tyrosinase inhibitors is very important in the agriculture and food industry, medicine, and cosmetic products.
Although hundreds of tyrosinase inhibitors have been obtained in the laboratory, only a few are sufficiently potent for practical use, such as arbutin, kojic acid, and ascorbic acid (Demirkiran et al., 2013; Crespo et al., 2019; Rodriguez et al., 2020; Wang et al., 2020). However, they still exhibit the defect of undesirable side effects, including cytotoxicity, dermatitis, and neurodegenerative disorders. Therefore, it is extremely urgent to develop alternatively safe and effective tyrosinase inhibitors.
Pyrrole, aromatic five-membered heterocyclic compounds containing nitrogen, have gotten increased attention due to their extensive biological, agrochemical, and pharmaceutical activities, as well as reactivity and stability (Deibl et al., 2015; Liao et al., 2015; Qin et al., 2015). Moreover, heterocyclic compounds containing nitrogen exhibit good water solubility and salt formation ability. Until now, more and more biologically active molecules were designed based on the pyrrole skeleton (Insuasty et al., 2010; Jamwal et al., 2013; Kumar et al., 2013; Ansari et al., 2017). Also, for searching tyrosinase inhibitors, N-heterocycle derivatives have received much attention.
Recently, we have reported some efficient synthetic methodologies of N-heterocycles, which are the key skeleton of many bioactive molecules (Mou et al., 2016; Zhang et al., 2017). As the systematic continuation of aforementioned research results, we synthesized two series of pyrrole derivatives and screened their tyrosinase inhibitory activity.
Mushroom tyrosinase (EC 1.14.18.1), L-3,4-dihydroxy phenylalanine (l-DOPA), and kojic acid were obtained from Sigma (St. Louis, MO, United States). Dimethyl sulfoxide (DMSO) of molecular biological grade was purchased from J&K Chemical Co., Ltd. (Shanghai, China). Fetal bovine serum (FBS) and Dulbecco’s Modified Eagle’s Medium (DMEM) were obtained from Gibco (Grand Island, NY, United States). The 3-(4,5-Dimethythiazol-2-yl)-2,5-diphenyl-tetra-zoliumbromide (MTT) dye was supplied by Sigma-Aldrich (St. Louis, MO, United States). All other reagents were of analytical grade.
1H and 13C NMR spectra were recorded in CDCl3 on 400/300 MHz instruments, and spectral data were reported in ppm. High-resolution mass spectral analysis (HRMS) data were measured on Apex II by means of the ESI technique.
2-Cyanopyrrole derivatives A (1–17) were synthesized according to our previous article (Mou et al., 2016).
The general synthetic procedure for derivative A (1–12) is as follows. To a stirred mixture of aromatic olefin or alkyne (0.5 mmol, 1 equiv), DMF (15 equiv), TMSCN (trimethylsilanecarbonitrile, 5 equiv), and Cu(OTf)2 (0.2 equiv), was added DDQ (2 equiv) through a batch-wise mode (the first equiv was added 0.2 equiv/2 h for 5 times, and the second equiv of DDQ was then introduced at once) at 80°C under an argon atmosphere. Then, the reaction mixture was stirred for an additional 14 h. Upon the completion of the reaction (monitored by TLC), the mixture was quenched by the addition of water. The aqueous layer was extracted three times with EtOAc, and the combined organic layers were washed with brine, dried over sodium sulfate, evaporated to dryness, and purified by column chromatography to afford desired products A (1–12).
The general synthetic procedure for derivative A (13–17) is as follows. To a mixed solvent of n-heptane and N,N-diethylacetamide (v/v = 6:1, 1 ml), aromatic olefin (0.5 mmol, 1 equiv), N,N-disubstituted formamide (3 equiv), TMSCN (6 equiv), and Cu(OTf)2 (0.2 equiv) were successively added at 80°C under an argon atmosphere. Next, DDQ was added through a batch-wise mode (the first equiv was added 0.2 equiv/2 h five times, and the second equiv of DDQ was then introduced at once) at the same temperature. The reaction mixture was refluxed for an additional 14 h. Upon completion of the reaction (monitored by TLC), the reaction mixture was quenched by the addition of water. The aqueous layer was extracted three times with EtOAc, and the combined organic layers were washed with brine, dried over sodium sulfate, evaporated to dryness, and purified by column chromatography to afford desired products A (13–17).
The general synthetic procedure for derivative B (1–2) is as follows. To a stirred mixture of A5 or A6 (1 mmol) in DMSO (10 ml), K2CO3 (2 mmol) and hydrogen peroxide (30% aqueous, 30 ml) were added. The mixture was stirred at room temperature for 8 h. When the substrate was completely disappeared (monitored by TLC), the reaction mixture was extracted with ether and dichloromethane (2: 1) three times. The combined organic layers were washed with saturated brine (50 ml), dried over sodium sulfate, filtered, and concentrated in vacuo. The residue was purified by column chromatography to give B (1–2).
The general synthetic procedure for derivative B (3–8) is as follows. To a stirred mixture of A6, A3, A5, A1, A9, or A2 (1 mmol) in ether (10 ml), lithium aluminum tetrahydride (1.2 mmol) was added slowly at 0°C. Then, the mixture was stirred at reflux for 3 h and quenched with water (10 ml). The mixture was extracted with ether three times. The combined organic layers were washed with brine (50 ml), dried over magnesium sulfate, and concentrated in vacuo to give crude primary amine 2. Then, the crude primary amine 2 (1 mmol), (Boc)2O (1.3 mmol), and Et3N (5 mmol) were added into methanol (10 ml) and refluxed for 20 min. The mixture was quenched with saturated aqueous ammonium chloride solution and extracted with diethyl ether three times. The combined organic layers were washed with brine (50 ml), dried over magnesium sulfate, and concentrated in vacuo. The crude residue was purified by column chromatography to afford B (3–8).
3-(3-Bromophenyl)-1-methyl-1H-pyrrole-2-carboxamide (B1). Amorphous solid; 39% yield; 1H NMR (400 MHz, CDCl3) δ 7.53–7.52 (m, 1H), 7.52–7.49 (m, 1H), 7.31–7.30 (m, 1H), 7.29–7.28 (m, 1H), 6.74 (d, J = 2.4 Hz, 1H), 6.09 (d, J = 2.4 Hz, 1H), 5.43–5.29 (m, 2H), 3.93 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 163.45, 134.99, 131.81, 131.07, 127.98, 127.14, 121.46, 109.32, 109.28, 98.48, 37.31, and 37.27; HRMS (ESI) calcd for C12H11BrN2O [M + H]+: 279.0128, found 279.0124.
3-(4-Bromophenyl)-1-methyl-1H-pyrrole-2-carboxamide (B2). White solid; mp 172–174°C; 33% yield; 1H NMR (400 MHz, CDCl3) δ 7.52 (d, J = 8.4 Hz, 2H), 7.30 (d, J = 8.4 Hz, 2H), 6.73 (d, J = 2.4 Hz, 1H), 6.09 (d, J = 2.4 Hz, 2H), 5.48 (d, J = 9.6 Hz, 2H), 3.92 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 163.52, 134.94, 131.79, 131.06, 127.95, 127.13, 121.44, 109.28, and 37.31; HRMS (ESI) calcd for C12H11BrN2O [M + H]+:279.0128, found 279.0123.
tert-Butyl ((3-(4-bromophenyl)-1-methyl-1H-pyrrol-2-yl)methyl)carbamate (B3). White solid; mp 109–113°C; 29% yield; 1H NMR (400 MHz, CDCl3) δ 7.48 (d, J = 8.4 Hz, 2H), 7.19 (d, J = 8.4 Hz, 2H), 6.64 (d, J = 2.4 Hz, 1H), 6.19 (d, J = 2.8 Hz, 1H), 4.59 (s, 1H), 4.39 (d, J = 5.2 Hz, 2H), 3.65 (s, 3H), and 1.46 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 155.25, 135.28, 131.54, 129.55, 125.03, 123.82, 122.73, 119.67, 107.40, 79.65, 34.57, 34.10, and 28.33; HRMS (ESI) calcd for C17H21BrN2O2 [M + H]+: 365.0859, found 365.0853.
tert-Butyl ((3-(3-fluorophenyl)-1-methyl-1H-pyrrol-2-yl)methyl)carbamate (B4). White solid; mp 103–104°C; 44% yield; 1H NMR (400 MHz, CDCl3) δ 7.35–7.29 (m, 1H), 7.10–7.08 (m, 1H), 6.95–6.91 (m, 1H), 6.64 (d, J = 2.4 Hz, 1H), 6.21 (d, J = 2.4 Hz, 1H), 4.63 (s, 1H), 4.42–4.41 (d, J = 4.8 Hz, 2H), 3.66 (s, 3H), and 1.47 (s, 9H); 13C NMR (100 MHz,CDCl3) δ 162.93 (d, J = 243 Hz), 155.28, 138.62 (d, J = 7.9 Hz), 129.85 (d, J = 8.6 Hz), 125.19, 123.89, 123.53, 122.71, 114.68 (d, J = 21.4 Hz), 112.54 (d, J = 21 Hz), 107.50, 79.64, 34.55, 34.07, and 28.32; HRMS (ESI) calcd for C17H21FN2O2 [M + Na]+: 327.1479, found 324.1472.
tert-Butyl ((3-(3-bromophenyl)-1-methyl-1H-pyrrol-2-yl)methyl)carbamate (B5). White solid; mp 124–127°C; 40% yield; 1H NMR (400 MHz, CDCl3) δ 7.46 (s, 1H), 7.35–7.32 (m, 1H), 7.23–7.20 (m, 2H), 6.61 (d, J = 2.8 Hz, 1H), 6.17 (d, J = 2.8 Hz, 1H), 4.59 (s, 1H), 4.37 (d, J = 5.2 Hz, 2H), 3.62 (s, 3H), and 1.44 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 155.25, 138.53, 130.89, 129.94, 128.70, 126.49, 125.23, 123.57, 122.75, 122.54, 107.50, 79.65, 34.52, 34.09, and 28.33; HRMS (ESI) calcd for C17H21BrN2O2 [M + H]+: 365.0859, found 365.0857.
tert-Butyl ((1-methyl-3-phenyl-1H-pyrrol-2-yl)methyl)carbamate (B6). White solid; mp 87–90°C; 46% yield; 1H NMR (400 MHz, CDCl3) δ 7.37–7.30 (m, 4H), 7.24–7.20 (m, 1H), 6.61 (d, J = 2.4 Hz, 1H), 6.20 (d, J = 2.4 Hz, 1H), 4.67 (s, 1H), 4.40 (d, J = 4.8 Hz, 2H), 3.62 (s, 3H), and 1.45 (s, 9H); 13C NMR (100 MHz,CDCl3) δ 155.26, 136.30, 128.41, 127.98, 125.73, 124.92, 122.43, 107.46, 79.40, 34.56, 33.91, and 28.24; HRMS (ESI) calcd for C17H22N2O2 [M + H]+: 287.1754, found 287.1750.
tert-Butyl ((1-methyl-3-(4-(trifluoromethyl)phenyl)-1H-pyrrol-2-yl)methyl)carbamate (B7). White solid; mp 120–122°C; 37% yield; 1H NMR (400 MHz, CDCl3) δ 7.62 (d, J = 8 Hz, 2H), 7.43 (d, J = 8.0 Hz, 2H), 6.67 (d, J = 2.8 Hz, 1H), 6.25 (d, J = 2.8 Hz, 1H), 4.66 (s, 1H), 4.42 (d, J = 4.8 Hz, 2H), 3.68 (s, 3H), and 1.47 (s, 9H); 13C NMR (100 MHz,CDCl3) δ 155.27, 140.06, 127.99, 127.74 (q, J = 32.3 Hz), 125.56, 125.43 (q, J = 3.8 Hz), 124.41 (q, J = 270 Hz), 123.70, 122.98, 107.63, 79.75, 34.64, 34.12, and 28.34; HRMS (ESI) calcd for C18H21F3N2O2 [M + H]+: 355.1628, found 355.1623.
tert-Butyl ((3-(2-fluorophenyl)-1-methyl-1H-pyrrol-2-yl)methyl)carbamate (B8). White solid; mp 81–84°C; 39% yield; 1H NMR (400 MHz, CDCl3) δ 7.31–7.27 (m, 1H), 7.25–7.21 (m, 1H), 7.17–7.08 (m, 2H), 6.68 (d, J = 2.4 Hz, 1H), 6.17 (d, J = 2.8 Hz, 1H), 4.81 (s, 1H), 4.29 (d, J = 5.6 Hz, 2H), 3.68 (s, 3H), and 1.45 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 159.50 (d, J = 241.7 Hz), 155.45, 131.63 (d, J = 3.7 Hz), 127.78 (d, J = 8.3 Hz), 126.72, 124.19 (d, J = 3.5 Hz), 124.05 (d, J = 15.3 Hz), 122.71, 117.68, 115.60 (d, J = 23.3 Hz), 108.39, 79.36, 34.81, 34.21, and 28.35; HRMS (ESI) calcd for C17H21FN2O2 [M + H]+: 305.1660, found 305.1657.
The mushroom tyrosinase activity assays of synthetic compounds were carried out according to the previously reported modification (Qin et al., 2015). l-DOPA was used as the substrate. Briefly, 800 μl of phosphate buffer (50 mM, pH 6.8), 50 μl of mushroom tyrosinase (666.67 U/ml, dissolved in PBS), and 50 μl of inhibitor (dissolved in DMSO) were placed in the plastic centrifuge tube. Then, 100 μl of l-DOPA (5 mM, dissolved in PBS) was added. Subsequently, its change in absorbance at 475 nm was measured in a time-dependent manner using a Beckman UV-650 spectrophotometer. The percentage inhibition was calculated by using [(OD0 - OD1)/OD0] × 100%, where OD0 was the absorbance without inhibitor, and OD1 was the absorbance with the inhibitor. The IC50 value of the potential compound was calculated from dose-response curves of percentage inhibition. Kojic acid was used as a standard control for comparison. All experiments were performed in duplicate.
Experiments were performed to analyze the inhibitory mechanism by the following method. For the text, the mushroom tyrosinase at a final concentration of 16.67–100.00 U/ml and l-DOPA at a final concentration of 0.5 mM were chosen. Then, the mushroom tyrosinase activity was measured in the presence of the inhibitor.
The inhibition type of the inhibitor on mushroom tyrosinase was evaluated by the Lineweaver–Burk plot, and the inhibition constants were obtained from the second plots of the apparent 1/Vmax and Km/Vmax against the inhibitor concentration. In this assay, mushroom tyrosinase at a final concentration of 33.33 U/ml and l-DOPA at a final concentration of 0.25–2 mM were used. The equation for the Lineweaver–Burk plot can be written as follows:
where v is the reaction velocity, Km is the Michaelis constant, Vm is the maximal velocity, [I] is the concentration of the inhibitor, [S] is the concentration of the substrate, KI is the constant for the inhibitor binding with the free enzyme, and KIS is the constant for the inhibitor binding with the enzyme-substrate complex. KI and KIS were obtained from the slope or the vertical intercept vs. the inhibitor concentration, respectively.
Sybyl 2.1.1 (Tripos, United States) was used for the docking simulations between mushroom tyrosinase and the inhibitor. First, compound A12 was prepared by the energy minimization using the MM2 program, with energy termination of 0.01 kcal/mol, iteration of 1,000 times, and charges of Gasteiger-Hückle. Second, the crystal structure of mushroom tyrosinase (PDB ID: 2Y9X) was prepared by extracting ligands, removing H2O, termini treatment, and adding hydrogens. The active pocket of tyrosinase was generated using the ligand mode. Finally, the docking simulation between tyrosinase and compound A12 was carried out in the default format. PyMOL software was used to draw the view based on the Sybyl results.
B16 mouse melanoma cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum and 1% penicillin/streptomycin at 37°C in a 5% CO2 incubator.
B16 cells (100 µl) were plated in a 96-well plate (5 × 103 cells/well) for 24 h and then treated with a 100 µl complete medium containing A12 or kojic acid for 24 h. MTT solution (0.5 mg/ml, final concentration) was added to each well, and plates were incubated at 37°C for another 3 h. The absorbance was measured at 490 nm using a multi-detection microplate reader.
The inhibitory activity of A12 on tyrosinase in B16 cells was evaluated. B16 cells (5 × 104 cells/well) were plated in 96-well plates and incubated for 24 h. A measure of 100 µl of complete medium including A12 or kojic acid was added for further incubation of 48 h. Then, cells were added to 50 µl Triton X-100 (1%) and allowed for freezing for 1 h at −80°C. After thawing at room temperature, 10 μl l-DOPA solutions (0.1%, w/v) were added and co-incubated for 1 h at 37°C. The absorbance at 405 nm was measured.
Pyrrole derivatives A (1–17) and B (1–8) were synthesized according to the synthetic route shown in Scheme 1. A1–A12 were obtained using aromatic olefin or alkyne, DMF, and TMSCN as starting materials under the catalysis of Cu(OTf)2 and DDQ. A (13–17) were synthesized from aromatic olefin, N,N-disubstituted formamide, and TMSCN with Cu(OTf)2 as the catalyst and DDQ as additive. B 1–2 could be synthesized from compound A5 or A6 through a hydrolysis reaction. Meanwhile, the reduction of compounds A6, A3, A5, A1, A9, or A2 by LAH with subsequent amine derivatization by (Boc)2O would produce B (3–8). 1H NMR, 13C NMR, and HRMS were applied to confirm their structures.
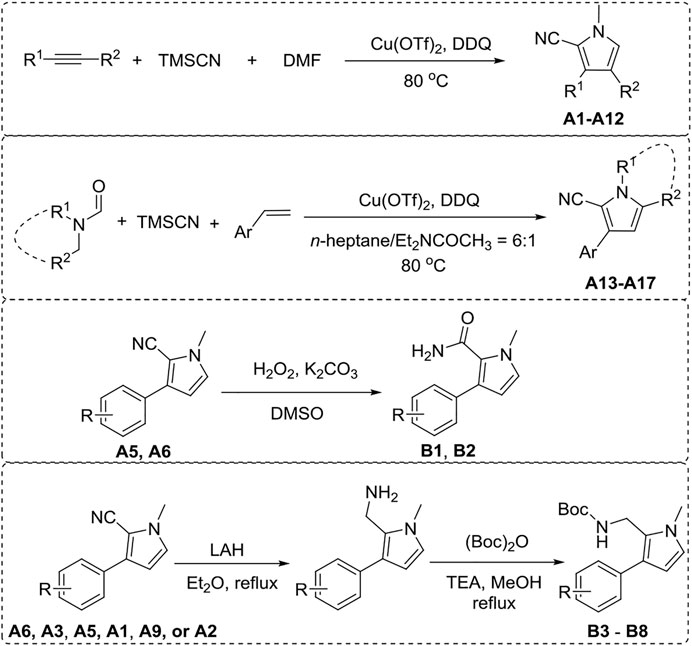
Scheme 1. General synthesis of A (1–17) and B (1–8).
Tyrosinase inhibitory assay of all synthesized pyrrole derivatives A (1–17) and B (1–8) was performed using l-DOPA as the substrate. As shown in Table 1, compounds A (1–17) exhibited moderate to excellent inhibitory activities against tyrosinase, with IC50 values ranging from 0.97 to 89.15 μM. In particular, A12 (IC50 = 0.97 μM) showed the strongest inhibitory activities, which were ∼30 times stronger than the reference inhibitor kojic acid (IC50 = 28.72 μM).
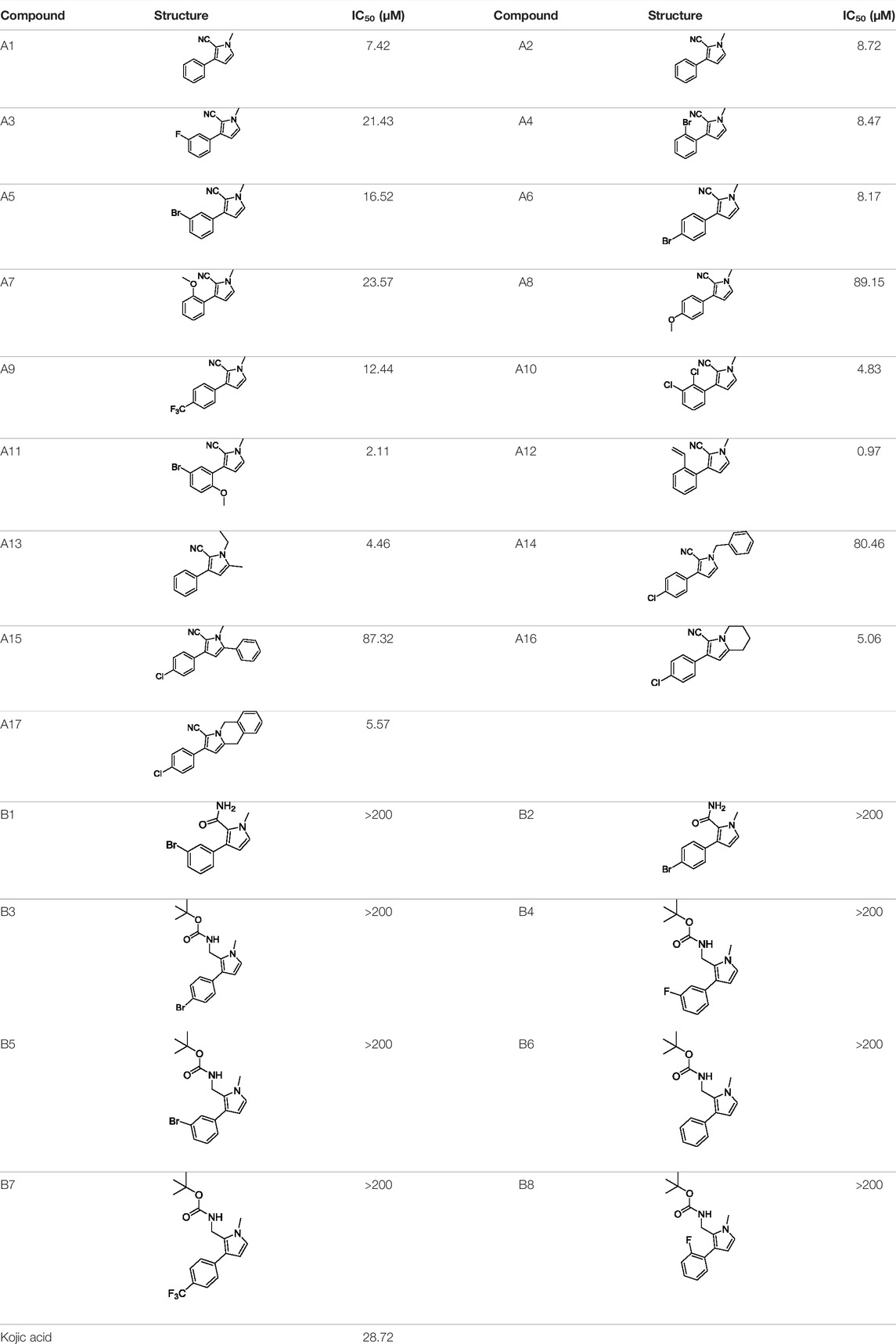
TABLE 1. Inhibitory activity of N-heterocycle derivatives on tyrosinase.
Based on the data displayed in Table 1, the SAR of the tested compounds against tyrosinase was analyzed. For compounds A (1–17), compound A1 (IC50 = 7.24 μM) with no substitute group was selected as the template compound. It was clear that the introduction of a substituent on the phenyl ring at the 3-position of the pyrrole skeleton did show the clear influence of the corresponding inhibition potency. Among them, compound A2 with the 2-fluorine group on the benzene ring (IC50 = 8.72 μM), A3 with the 3-fluorine group (IC50 = 21.43 μM), A4 with the 2-bromine group (IC50 = 8.47 μM), A5 with the 3-bromine group (IC50 = 16.52 μM), A6 with the 4-bromine group (IC50 = 8.17 μM), A7 with the 2-methoxy group (IC50 = 23.57 μM), A8 with the 4-methoxy group (IC50 = 89.15 μM), and A9 with the 4-trifluoromethyl group (IC50 = 12.44 μM) all presented lower inhibitory activities than compound A1. These results indicated that the introduction of fluorine, bromine, methoxy and trifluoromethyl alone might cause a decrease in tyrosinase inhibitory activity. In contrast, two compounds A10 and A11 with dual substituents on the benzene ring provided better inhibitory results than compound A1 and showed an IC50 value of 4.83 and 2.11 μM, respectively. Moreover, A12 with a 2-vinyl group on the benzene ring (IC50 = 0.97 μM) also appeared to have much higher inhibitory activities than compound A1. This result revealed the special property of the 2-vinyl group during the interaction between A12 and tyrosinase. As for the substituent on the 1- and 5-positions of the pyrrole ring, additional five compounds A (13–17) were further tested. Based on the results, it is obvious that the presence of an untethered benzyl at 1-position (A14) or the phenyl group at 5-position (A15) would dramatically affect the inhibitory activities. On the contrary, A13 with the ethyl group on nitrogen of the pyrrole ring (IC50 = 4.46 μM), A16, and A17 with the pyrrole as a part of a fused ring system showed higher inhibitory activities (IC50 = 5.06 and 5.57 μM, respectively) than compound A1, which revealed that the alkyl group at 1-position or a planar type of the molecule might increase the inhibitory activity against tyrosinase. Therefore, based on the previous SAR results of all compounds, further derivatization of A1 with other ring systems at 3-position of the pyrrole ring or other fused ring systems will be designed and screened.
For investigating the effect of the cyano group on the inhibitory activities, it was transformed into an amide group (B1, B2) or carbamate group (B3 ∼ 8), and the inhibitory activities of corresponding derivatives were obviously reduced. These results indicated that the cyano group of compounds A1-A17 was crucial for tyrosinase inhibition.
Compound A12 with the best inhibition activity was selected as the lead compound to ascertain the inhibition mechanism of mushroom tyrosinase. Figure 1A shows the plots of initial velocity vs. tyrosinase concentrations at different concentrations of compound A12. As can be seen, the plots gave a family of straight lines, respectively, which passed through the origin point. Moreover, the slopes of the lines extended a descent with the increase of concentrations of compound A12. These results revealed that the presence of compound A12 did not bring down the amount of active enzyme but just resulted in the reduction of the enzyme activity. Hence, the tyrosinase inhibition of compound A12 was reversible.
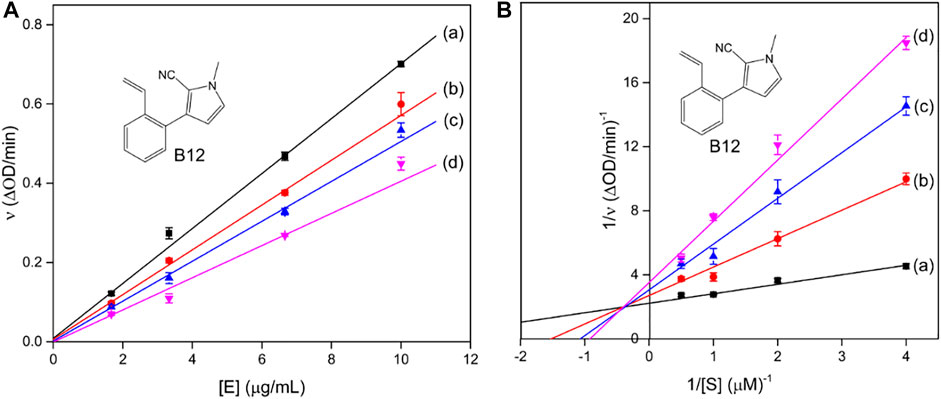
FIGURE 1. Plots of ν vs. [E] and the Lineweavere–Burk plot of compound A12 (concentrations: (a) 0, (b) 0.75, (c) 1 μM, and (d) 1.5 μM).
To further explore the inhibition kinetic behavior of the most promising compound A12 on mushroom tyrosinase, the tyrosinase activity was determined under different concentrations of l-DOPA in the presence or absence of an inhibitor. The results were analyzed using Lineweaver–Burk double reciprocal plots. For compound A12 (Figure 2B), the results presented that the plots of 1/ν vs. 1/[S] gave straight lines with different slopes intersecting one another in the second quadrant. The values of Vm and Km all descended with the increase in concentration of A12, which suggested that compound A12 induced mixed-type inhibition of mushroom tyrosinase. In other words, compound A12 could bind with both free enzyme and the enzyme-substrate complex, which was similar to the inhibition type of kojic acid. Hence, we determined the equilibrium constant for inhibitor binding with free enzyme (KI) and the enzyme-substrate complex (KIS) through the second plots of the apparent Km/Vmax and 1/Vmax vs. inhibitor concentrations. The KI and KIS values of the compound A12 were calculated as 1.11 and 1.00 μM.
FIGURE 2. Representative tyrosinase inhibitors.
To better understand the inhibitory activity of compound A12, Sybyl and PyMOL software were conducted to understand the interaction pattern between the A12 and the active site of tyrosinase. Mushroom tyrosinase (PDB: 2Y9X) from the RCSB Protein Data Bank was selected as the target protein for simulation. The docking simulation results showed that A12 was well accommodated and bound in the active pocket of tyrosinase (Figure 3A) and had in-depth interactions with the active site residues (Figure 3B). The cyano group of A12 resided adjacent to the dicopper nucleus, indicating that there was a potential metal-ligand interaction, which was considered to be one of the key interactions between tyrosinase and the ligand compound. The pyrrole ring formed a π-π interaction with His263. Furthermore, A12 formed hydrophobic interactions with Ala286, Asn260, Gly281, His244, His259, Met280, Phe264, Ser282, Val248, and Val283.

FIGURE 3. Molecular docking of compound A12 with tyrosinase: (A) in the active pocket; (B) interactions with amino acid residues.
For evaluating the effect of compound A12 on cellular tyrosinase activity, its cell cytotoxicity was first tested. The results showed that compound A12 and kojic acid showed no cytotoxicity on B16 melanoma cells under a concentration of 100 μM (Figure 4A). Thence, the concentration of A12 (under 100 μM) was selected for the assay of the cellular tyrosinase activity. A12 could effectively inhibit the tyrosinase activity in B16 melanoma cells in a dose-dependent manner (Figure 4B) and present an effective inhibitory of 33.48% at a concentration of 100 μM, which was equivalent to that of Kojic acid (39.81%).

FIGURE 4. (A) Cell cytotoxicityof A12 and kojic acid (B) and inhibition of cell tyrosinase activity of A12.
To conclude, the inhibitory activities of two series of pyrrole derivatives A (1–17) and B (1–8) were screened against tyrosinase. Compound A12 showed the highest inhibition activity with reversible and mixed-type inhibition types. Compound A12 (IC50: 0.97 μM) showed ∼30 times stronger inhibition activity than the reference inhibitor kojic acid (IC50: 28.72 μM). Molecular docking studies showed that the metal–ligand interaction, π-π interaction, and hydrophobic interactions played important roles in the interaction between tyrosinase and A12. Furthermore, A12 (100 μM) presented effective inhibitory on tyrosinase in B16 melanoma cells with an inhibition rate of 33.48%, which was equivalent to that of kojic acid (39.81%). Our studies have indicated that these pyrrole derivatives have the potential to be developed as anti-tyrosinase agents for use in medicine, agriculture and food industry, and cosmetic products.
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding authors.
Y-GH and Z-PG carried out the synthetic work. ZX and D-YZ were responsible for the initial study design and overall supervision. All the authors participated in the writing of the manuscript.
This work was supported by the Foundation from the National Natural Science Foundation of China (Nos. 21472077 and 21772071), the Department of Education of Guangdong Province (Nos. 2017KSYS010, 2019KZDXM035, 2021KCXTD044, and 2021KTSCX135), the Science and Technology Program of Gansu Province (20JR10RA608), the Fundamental Research Funds for the Central Universities (lzujbky-2021-kb40), the Special Funds for the Cultivation of Guangdong College Students’ Scientific and Technological Innovation (“Climbing Program” Special Funds, pdjh 2021a0504), and the Jiangmen City Basic and Application Basic Research key Project (No. 2021030103150006664).
Author Y-SZ is employed by Guangzhou Yuming Biological Technology Co, LTD.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2022.914944/full#supplementary-material
Akın, Ş., Demir, E. A., Colak, A., Kolcuoglu, Y., Yildirim, N., and Bekircan, O. (2019). Synthesis, Biological Activities and Molecular Docking Studies of Some Novel 2,4,5-Trisubstituted-1,2,4-Triazole-3-One Derivatives as Potent Tyrosinase Inhibitors. J. Mol. Struct. 1175, 280–286.
Ali, A., Ashraf, Z., Rafiq, M., Kumar, A., Jabeen, F., Lee, G. J., et al. (2019). Novel Amide Derivatives as Potent Tyrosinase Inhibitors; In-Vitro, In-Vivo Antimelanogenic Activity and Computational Studies. Mc 15, 715–728. doi:10.2174/1573406415666190319101329
Ansari, A., Ali, A., Asif, M., and Shamsuzzaman, S. (2017). Review: Biologically Active Pyrazole Derivatives. New J. Chem. 41 (1), 16–41. doi:10.1039/c6nj03181a
Ashooriha, M., Khoshneviszadeh, M., Khoshneviszadeh, M., Moradi, S. E., Rafiei, A., Kardan, M., et al. (2019). 1,2,3-Triazole-based Kojic Acid Analogs as Potent Tyrosinase Inhibitors: Design, Synthesis and Biological Evaluation. Bioorg. Chem. 82, 414–422. doi:10.1016/j.bioorg.2018.10.069
Asthana, S., Zucca, P., Vargiu, A. V., Sanjust, E., Ruggerone, P., and Rescigno, A. (2015). Structure-Activity Relationship Study of Hydroxycoumarins and Mushroom Tyrosinase. J. Agric. Food Chem. 63 (32), 7236–7244. doi:10.1021/acs.jafc.5b02636
Butt, A. R. S., Abbasi, M. A., Aziz-ur-Rehman, R., Siddiqui, S. Z., Raza, H., Hassan, M., et al. (2019). Synthesis and Structure-Activity Relationship of Tyrosinase Inhibiting Novel Bi-heterocyclic Acetamides: Mechanistic Insights through Enzyme Inhibition, Kinetics and Computational Studies. Bioorg. Chem. 86, 459–472. doi:10.1016/j.bioorg.2019.01.036
Cai, P., Xiong, Y., Yao, Y., Chen, W., and Dong, X. (2019). Synthesis, Screening and Biological Activity of Potent Thiosemicarbazone Compounds as a Tyrosinase Inhibitor. New J. Chem. 43 (35), 14102–14111. doi:10.1039/c9nj02360g
Chai, W.-M., Huang, Q., Lin, M.-Z., Ou-Yang, C., Huang, W.-Y., Wang, Y.-X., et al. (2018). Condensed Tannins from Longan Bark as Inhibitor of Tyrosinase: Structure, Activity, and Mechanism. J. Agric. Food Chem. 66 (4), 908–917. doi:10.1021/acs.jafc.7b05481
Chai, W.-M., Wei, M.-K., Wang, R., Deng, R.-G., Zou, Z.-R., and Peng, Y.-Y. (2015). Avocado Proanthocyanidins as a Source of Tyrosinase Inhibitors: Structure Characterization, Inhibitory Activity, and Mechanism. J. Agric. Food Chem. 63 (33), 7381–7387. doi:10.1021/acs.jafc.5b03099
Crespo, M. I., Chabán, M. F., Lanza, P. A., Joray, M. B., Palacios, S. M., Vera, D. M. A., et al. (2019). Inhibitory Effects of Compounds Isolated from Lepechinia Meyenii on Tyrosinase. Food Chem. Toxicol. 125, 383–391. doi:10.1016/j.fct.2019.01.019
Deibl, N., Ament, K., and Kempe, R. (2015). A Sustainable Multicomponent Pyrimidine Synthesis. J. Am. Chem. Soc. 137 (40), 12804–12807. doi:10.1021/jacs.5b09510
Demirkiran, O., Sabudak, T., Ozturk, M., and Topcu, G. (2013). Antioxidant and Tyrosinase Inhibitory Activities of Flavonoids fromTrifolium nigrescensSubsp.Petrisavi. J. Agric. Food Chem. 61 (51), 12598–12603. doi:10.1021/jf403669k
Haldys, K., and Latajka, R. (2019). Thiosemicarbazones with Tyrosinase Inhibitory Activity. Medchemcomm 10 (3), 378–389.
Huang, H.-W., Cheng, M.-C., Chen, B.-Y., and Wang, C.-Y. (2019). Effects of High Pressure Extraction on the Extraction Yield, Phenolic Compounds, Antioxidant and Anti-tyrosinase Activity of Djulis Hull. J. Food Sci. Technol. 56 (9), 4016–4024. doi:10.1007/s13197-019-03870-y
Ielo, L., Deri, B., Germanò, M. P., Vittorio, S., Mirabile, S., Gitto, R., et al. (2019). Exploiting the 1-(4-fluorobenzyl)piperazine Fragment for the Development of Novel Tyrosinase Inhibitors as Anti-melanogenic Agents: Design, Synthesis, Structural Insights and Biological Profile. Eur. J. Med. Chem. 178, 380–389. doi:10.1016/j.ejmech.2019.06.019
Insuasty, B., Tigreros, A., Orozco, F., Quiroga, J., Abonía, R., Nogueras, M., et al. (2010). Synthesis of Novel Pyrazolic Analogues of Chalcones and Their 3-Aryl-4-(3-Aryl-4,5-Dihydro-1h-Pyrazol-5-Yl)-1-Phenyl-1h-Pyrazole Derivatives as Potential Antitumor Agents. Bioorg. Med. Chem. 18 (14), 4965–4974. doi:10.1016/j.bmc.2010.06.013
Jamwal, A., Javed, A., and Bhardwaj, V. A. (2013). Review on Pyrazole Derivatives of Pharmacological Potential. J. Pharm. Biosci. 3, 114–123.
Kumar, V., Kaur, K., Gupta, G. K., and Sharma, A. K. (2013). Pyrazole Containing Natural Products: Synthetic Preview and Biological Significance. Eur. J. Med. Chem. 69, 735–753. doi:10.1016/j.ejmech.2013.08.053
Larik, F. A., Saeed, A., Channar, P. A., Muqadar, U., Abbas, Q., Hassan, M., et al. (2017). Design, Synthesis, Kinetic Mechanism and Molecular Docking Studies of Novel 1-Pentanoyl-3-Arylthioureas as Inhibitors of Mushroom Tyrosinase and Free Radical Scavengers. Eur. J. Med. Chem. 141, 273–281. doi:10.1016/j.ejmech.2017.09.059
Lee, S., Ullah, S., Park, C., Won Lee, H., Kang, D., Yang, J., et al. (2019). Inhibitory Effects of N-(acryloyl)benzamide Derivatives on Tyrosinase and Melanogenesis. Bioorg. Med. Chem. 27 (17), 3929–3937. doi:10.1016/j.bmc.2019.07.034
Liao, J.-Y., Shao, P.-L., and Zhao, Y. (2015). Catalytic Divergent Synthesis of 3H or 1H Pyrroles by [3 + 2] Cyclization of Allenoates with Activated Isocyanides. J. Am. Chem. Soc. 137 (2), 628–631. doi:10.1021/ja511895q
Liu, J., Wu, F., Chen, L., Zhao, L., Zhao, Z., Wang, M., et al. (2012). Biological Evaluation of Coumarin Derivatives as Mushroom Tyrosinase Inhibitors. Food Chem. 135 (4), 2872–2878. doi:10.1016/j.foodchem.2012.07.055
Mou, X.-Q., Xu, Z.-L., Xu, L., Wang, S.-H., Zhang, B.-H., Zhang, D., et al. (2016). The Synthesis of Multisubstituted Pyrroles via a Copper-Catalyzed Tandem Three-Component Reaction. Org. Lett. 18 (16), 4032–4035. doi:10.1021/acs.orglett.6b01883
Qin, H.-L., Shang, Z.-P., Jantan, I., Tan, O. U., Hussain, M. A., Sher, M., et al. (2015). Molecular Docking Studies and Biological Evaluation of Chalcone Based Pyrazolines as Tyrosinase Inhibitors and Potential Anticancer Agents. RSC Adv. 5 (57), 46330–46338. doi:10.1039/c5ra02995c
Rodriguez, R., Escobedo, B., Lee, A. Y., Thorwald, M., Godoy‐Lugo, J. A., Nakano, D., et al. (2020). Simultaneous Angiotensin Receptor Blockade and Glucagon‐like Peptide‐1 Receptor Activation Ameliorate Albuminuria in Obese Insulin‐resistant Rats. Clin. Exp. Pharmacol. Physiol. 47 (3), 422–431. doi:10.1111/1440-1681.13206
Santi, M. D., Bouzidi, C., Gorod, N. S., Puiatti, M., Michel, S., Grougnet, R., et al. (2019). In Vitro biological Evaluation and Molecular Docking Studies of Natural and Semisynthetic Flavones from Gardenia Oudiepe (Rubiaceae) as Tyrosinase Inhibitors. Bioorg. Chem. 82, 241–245. doi:10.1016/j.bioorg.2018.10.034
Todaro, A., Cavallaro, R., Argento, S., Branca, F., and Spagna, G. (2011). Study and Characterization of Polyphenol Oxidase from Eggplant (Solanum Melongena L.). J. Agric. Food Chem. 59 (20), 11244–11248. doi:10.1021/jf201862q
Ullah, S., Park, C., Ikram, M., Kang, D., Lee, S., Yang, J., et al. (2019). Tyrosinase Inhibition and Anti-melanin Generation Effect of Cinnamamide Analogues. Bioorg. Chem. 87, 43–55. doi:10.1016/j.bioorg.2019.03.001
Ullah, S., Park, Y., Park, C., Lee, S., Kang, D., Yang, J., et al. (2019). Antioxidant, Anti-tyrosinase and Anti-melanogenic Effects of (E)-2,3-diphenylacrylic Acid Derivatives. Bioorg. Med. Chem. 27 (11), 2192–2200. doi:10.1016/j.bmc.2019.04.020
Ullah, S., Son, S., Yun, H. Y., Kim, D. H., Chun, P., and Moon, H. R. (2016). Tyrosinase Inhibitors: a Patent Review (2011-2015). Expert Opin. Ther. Pat. 26 (3), 347–362. doi:10.1517/13543776.2016.1146253
Wang, Y., Hao, X., Wang, Z., Dong, M., and Cui, L. (2020). Facile Fabrication of Mn 2+ -doped ZnO Photocatalysts by Electrospinning. R. Soc. open Sci. 7 (4), 191050. doi:10.1098/rsos.191050
Xie, J., Dong, H., Yu, Y., and Cao, S. (2016). Inhibitory Effect of Synthetic Aromatic Heterocycle Thiosemicarbazone Derivatives on Mushroom Tyrosinase: Insights from Fluorescence, 1 H NMR Titration and Molecular Docking Studies. Food Chem. 190, 709–716. doi:10.1016/j.foodchem.2015.05.124
Yu, L. (2003). Inhibitory Effects of (S)- and (R)-6-Hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic Acids on Tyrosinase Activity. J. Agric. Food Chem. 51, 2344–2347. doi:10.1021/jf0208379
Zhang, B.-H., Lei, L.-S., Liu, S.-Z., Mou, X.-Q., Liu, W.-T., Wang, S.-H., et al. (2017). Zinc-promoted Cyclization of Tosylhydrazones and 2-(dimethylamino)malononitrile: an Efficient Strategy for the Synthesis of Substituted 1-Tosyl-1h-Pyrazoles. Chem. Commun. 53 (61), 8545–8548. doi:10.1039/c7cc04610c
Zhu, Y.-J., Zhou, H.-T., Hu, Y.-H., Tang, J.-Y., Su, M.-X., Guo, Y.-J., et al. (2011). Antityrosinase and Antimicrobial Activities of 2-phenylethanol, 2-phenylacetaldehyde and 2-phenylacetic Acid. Food Chem. 124 (1), 298–302. doi:10.1016/j.foodchem.2010.06.036
Keywords: tyrosinase, reversible inhibitor, mixed-type inhibitor, 2-cyanopyrrole, molecular docking
Citation: Hu Y-G, Gao Z-P, Zheng Y-Y, Hu C-M, Lin J, Wu X-Z, Zhang X, Zhou Y-S, Xiong Z and Zhu D-Y (2022) Synthesis and Biological Activity Evaluation of 2-Cyanopyrrole Derivatives as Potential Tyrosinase Inhibitors. Front. Chem. 10:914944. doi: 10.3389/fchem.2022.914944
Received: 07 April 2022; Accepted: 25 April 2022;
Published: 17 June 2022.
Edited by:
Xi Zheng, The State University of New Jersey, United StatesCopyright © 2022 Hu, Gao, Zheng, Hu, Lin, Wu, Zhang, Zhou, Xiong and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhuang Xiong, d3l1Y2hlbXh6QDEyNi5jb20=; Dao-Yong Zhu, emh1ZHlAbHp1LmVkdS5jbg==
†These authors have contributed equally to this work and share the first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.