 José David Quezada-Borja1
José David Quezada-Borja1 Luz María Rodríguez-Valdez1
Luz María Rodríguez-Valdez1 Juan Pedro Palomares-Báez1
Juan Pedro Palomares-Báez1 Marco Antonio Chávez-Rojo1
Marco Antonio Chávez-Rojo1 Linda-Lucila Landeros-Martinez1
Linda-Lucila Landeros-Martinez1 Mayra Cristina Martínez-Ceniceros1
Mayra Cristina Martínez-Ceniceros1 Gabriel Rojas-George2
Gabriel Rojas-George2 Isui Abril García-Montoya3
Isui Abril García-Montoya3 Nora Aydeé Sánchez-Bojorge1*
Nora Aydeé Sánchez-Bojorge1*- 1Facultad de Ciencias Químicas, Universidad Autónoma de Chihuahua, Circuito Universitario, Chihuahua, México
- 2CONACYT Research Fellow, Centro de Investigación en Materiales Avanzados (CIMAV), S.C., Miguel de Cervantes, Complejo Industrial Chihuahua, Chihuahua, México
- 3Departamento de Ciencias Químico-Biológicas, Instituto de Ciencias Biomédicas, Universidad Autónoma de Ciudad Juárez, Ciudad Juarez, Chihuahua, México
New organic molecules containing five different compounds, commonly called p-linkers, located between the triphenylamine units, were theoretically designed and analyzed in order to be proposed as new hole transport materials (HTMs) in perovskite solar cells, in total ten new molecules were analyzed. The electronic, optical and hole transport properties were determined, similarly, the relationship of these properties with their molecular structure was also investigated by Density Functional Theory (DFT) and Density Functional Tight Binding (DFTB) calculations. Eight of the ten analyzed compounds exhibited the main absorption band out of the visible region; therefore these compounds did not present an overlap with the absorption spectra of the typical methylammonium lead iodide (MAPI) hybrid-perovskite. The results showed that the Highest occupied molecular orbital (HOMO) levels of the compounds are higher than the perovskite HOMO level, and in some cases these are even higher than the Spiro-OMeTAD HOMO. The calculated electronic couplings and the reorganization energy values provided useful information in order to determine if the systems were hole or electron transport materials.
Introduction
The depletion of fossil fuels reserves, growing energy demands, and the environmental crisis around the world have led the scientific community to search for new ways and technologies to use renewable energies sources efficiently. Due to the enormous amount of energy that the earth receives daily from the Sun, solar energy is considered the best choice among all renewable energy sources (Kim et al., 2020; Kumari et al., 2020; Pham et al., 2020; Sharmoukh et al., 2020). One of the most promising strategies to take advantage of solar energy is the conversion into electricity using photovoltaic devices (PV). Over the last few years, perovskite solar cells (PSCs), based on hybrid organic-inorganic halides, have achieved remarkable success as a result of their extraordinary optoelectronic properties such as their broad absorption region, direct band gap, high extinction coefficients, high charge carrier mobility, and long diffusion length (Mao et al., 2018; Mesquita et al., 2018; Ghaithan et al., 2020). PSCs offer low processing costs along with high energy conversion efficiency. These cells have the potential to replace silicon-based technology by requiring less material to obtain similar or larger power conversion efficiencies (PCE) (Zhou et al., 2018; Roy et al., 2020). Since the first report in 2009, the energy conversion efficiency of perovskite solar cells has increased from 3.81% to 25% in only a few years (Snaith, 2019; NREL, 2021). Hole transport materials play an important role in PSCs regenerating the oxidized state of perovskite and facilitating the hole transport (Pitchaiya et al., 2020; Lai et al., 2021). The importance of HTMs is not limited to charge transport, their properties stabilize the perovskite layer affecting the long-time performance of the cell (Zhang et al., 2018). Spiro-OMeTAD represents one of the most efficient HTMs to date and large efficiencies have been constantly reported using this compound (Li et al., 2021). However, the high costs of synthesis and its difficult purification have limited its commercialization (Sheibani et al., 2020). Therefore, the development of small, easy to prepare and low-cost production molecules has been an attractive target for many researchers (Gong et al., 2021). Resulting from their high hole mobility, thermal stability, and their non-crystalline or amorphous morphology, triphenylamine derivatives (TPAs) are HTMs that have been widely applied in organic electronic devices, such as OPVs, OLEDs, and OFETs (Rezaei and Mohajeri, 2021; Naeem et al., 2021). In the last few years, many research groups have developed different TPA-π-TPA HTMs by changing the π-linker due to the easy synthesis and the relatively simple structure of the compounds. The reported π-linkers include truxen (Wang et al., 2019), furan (Zhang et al., 2021a), triphenylamine (Kim et al., 2020), carbazole and fluorene (Leijtens et al., 2012), anthracene (Liu et al., 2017) pyrene (Gómez et al., 2021), silafluorene (Zhang and He, 2019), thiophene derivatives (Li et al., 2014; Zhou et al., 2020) to name a few. However, some of these materials show low power conversion efficiency, poor stability, or relatively high price. For these reasons, researchers have focused on finding new efficient and low-cost π-linkers for high-performance triphenylamine-based HTM. Pyrrole derivatives attract attention due to their low oxidation potential values (0.8 V) (Ansari, 2006), which are even lower than other heterocyclic compounds, such as thiophene which has values of 1.5 V (Camarada et al., 2011). This property and its wide use as hole transport material in organic light-emitting diodes (OLEDs) have favored the interest of expanding their application in perovskite solar cells. On the other hand, thiadiazoles have been used as polymers in PSCs, however, in the current work these molecules were used as π-linkers in the HTMs.
In the present study, new HTMs were proposed; these systems present a triarylamine-π-triarylamine architecture using pyrrole and thiadiazole as π-linkers (See Figure 1A). The electronic, optical and hole transport properties of these molecules were evaluated by Density Functional Theory (DFT) and Time Dependent-DFT. Also, the dimer of each molecule was constructed and these conformers were analyzed by Density Functional Tight Binding (DFTB) in order to obtain the intermolecular electronic coupling (transfer integral). The obtained results were compared with the reported properties of similar architectures based on fluorene, anthracene, and carbazole as π-linkers, as well as with the properties exhibited by Spiro-OMeTAD.
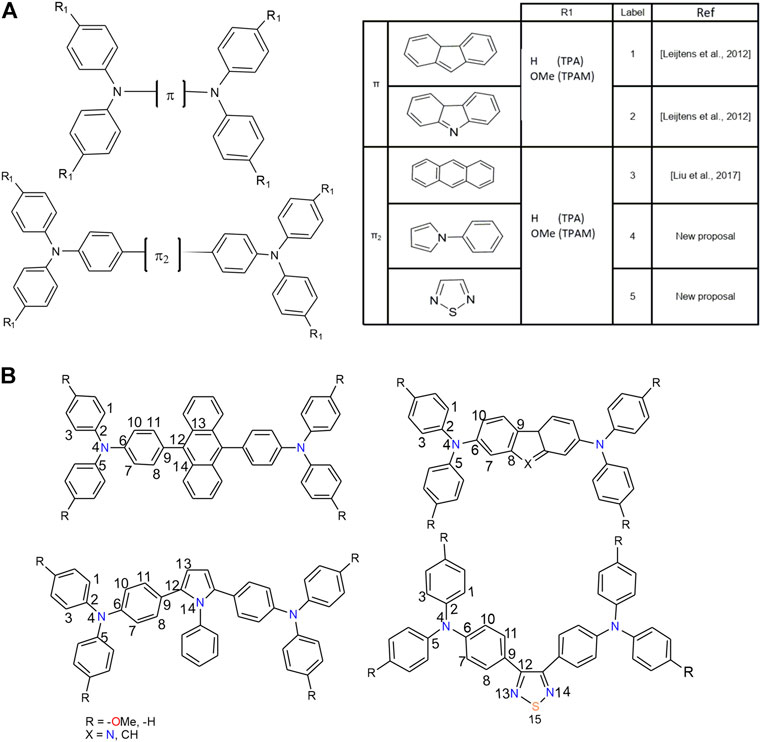
FIGURE 1. (A) Labeling scheme and general structure of the triphenylamine derivatives analyzed. (B) Numbering scheme for the atoms in the triphenylamine derivatives.
Theoretical background and computational details
In the present work, the molecular geometries of the triphenylamine derivatives were optimized by the functional M06-2X (54% HF exchange) (Zhao and Truhlar, 2008) with the 6-311++G(d,p) basis set (Hariharan and Pople, 1973; Frisch et al., 1984). The functional M06-2X was selected because this functional has shown the best agreement with experimental results of infrared spectrum in previous work with carbazole derivatives (Zhang et al., 2016). All calculated geometries are in the global minimum of their potential energy surface; this was confirmed with frequency calculations.
The analyzed triphenylamine derivatives, were denoted as nTPA and nTPAM, where n corresponds to different π-linkers (1: Fluorene, 2: Carbazole, 3: Anthracene, 4: Pyrrole and 5: Thiadiazole), TPA refers to triphenylamine units and M represents the presence of the methoxy groups in the structure (see Figure 1A).
In order to obtain the best accuracy with the experimental maximum absorption wavelength results, the already synthesized: Fluorene-TPA (1TPA), carbazole-TPA (2TPA), and anthracene-TPA (3TPAM), were analyzed using TD-DFT (Bartolotti, 1982; Runge and Gross, 1984). Several functionals: B3LYP (London, 1937), M06-L (Zhao and Truhlar, 2006), M06 (Zhao and Truhlar, 2008), M06-2X (Zhao and Truhlar, 2008) and PBE (Adamo and Barone, 1999), wB97XD (Chai and Head-Gordon, 2008), CAM-B3LYP (Yanai et al., 2004) in combination with 6-311++G(d,p) basis set, were applied for the analysis. All the calculations were carried out using chlorobenzene as solvent, with the solvation model based on density method (SMD) (Marenich et al., 2009). The TD-M06/6-311++G(d,p) was selected as the best methodology for the determination of absorption properties.
The chemical reactivity parameters, such as ionization potential (IP), electron affinity (EA), and chemical hardness (η) were calculated using DFT M06/6-311++G(d,p) The definitions of these chemical reactivity parameters can be written as:
Where
In all the analyzed compounds, the internal reorganization energy, for holes and electrons, was calculated with M06/6-31G(d) level of theory. The charge transport in each compound was calculated based on Marcus’ theory. This theory is based on the assumption that charges are located in a single molecule and charge transfer reactions take place through a jump mechanism between sites (Marcus, 1993). According to this, the carrier transport rate between a pair of molecules at a fixed temperature is expressed as:
where T is the absolute temperature, t is the charge transfer integral, kB and h are the Boltzmann and Planck constant respectively, and λ is the reorganization energy. The latter is the sum of the energies associated to the rearrangements of the molecular geometry when: i) the neutral state is ionized vertically to the cationic/anionic state and ii) the stable cationic/anionic state returns to the neutral state. Nelsen et al. (1987) have provided a convenient way to calculate the reorganization energy using a four-point model, generally called the adiabatic potential energy surface method (Nelsen et al., 1987; Touhami et al., 2015), which is illustrated in Figure 2.
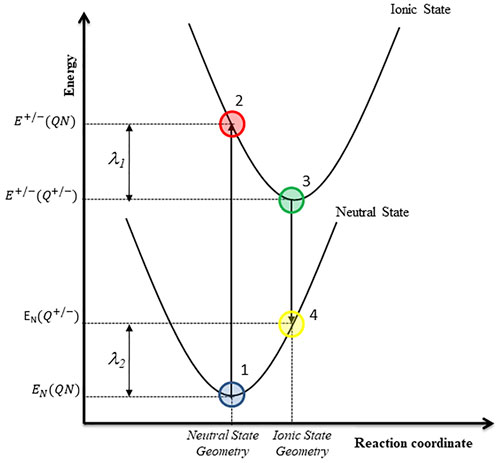
FIGURE 2. Four-point model used for the reorganization energy calculations.
The reorganization energy for the hole (λ+) and electron (λ-) transfer was calculated with the Eqs 5–7.
Where
The intermolecular electronic coupling (transfer integral) is another key parameter to calculate the charge transfer rate. The transfer integrals were calculated using the dimer projection method (Valeev et al., 2006; Baumeier et al., 2010) as implemented in the CATNIP software (Brown, 2022). In this method, the electronic coupling between donor and acceptor (molecules A and B) is given by,
where
Here,
The dimers of every molecule were built using a Montecarlo optimization algorithm at zero temperature as described below. First, the coordinates of the original (optimized) monomers were duplicated to produce a second monomer at a distance equal to twice the length of the molecule. Second, the molecule was rotated randomly in order to generate the starting configuration. A series of translations were then performed on the second monomer coordinates keeping both molecules as rigid bodies. After every translation, the total intermolecular (atom-atom) Lennard-Jones potential energy was calculated in order to accept only those displacements that minimized the energy. Once stabilized, the second molecule was allowed to perform rotations looking for the minimum energy configuration. This procedure was repeated hundreds of times, starting from different initial positions and configurations. The structure obtained by this method was further optimized using the DFTB (Density Functional based Tight Binding) method, at the DFTB-3DBJ level, as implemented in the DFTB + software (Hourahine et al., 2020).
Results and discussion
Geometrical structure
The optimized structures of triphenylamine derivatives at the M06-2X/6-311++G(d,p) level of theory are shown in Figure 3. The vibration frequencies were calculated at the same level of theory to verify that the stationary points correspond to the global minimum of the potential energy hypersurface. The compound 4TPA exhibited an almost similar structure to those compounds that incorporate carbazole and fluorene as π-linker, therefore, it can be expected that its properties were similar to the carbazol and fluorene derivatives. 4TPA presented a nearly planar structure, mainly in the region of the π-linker, this planarity gives more uniformity to the electronic density, for this reason it is expected the charge transport to be promoted.
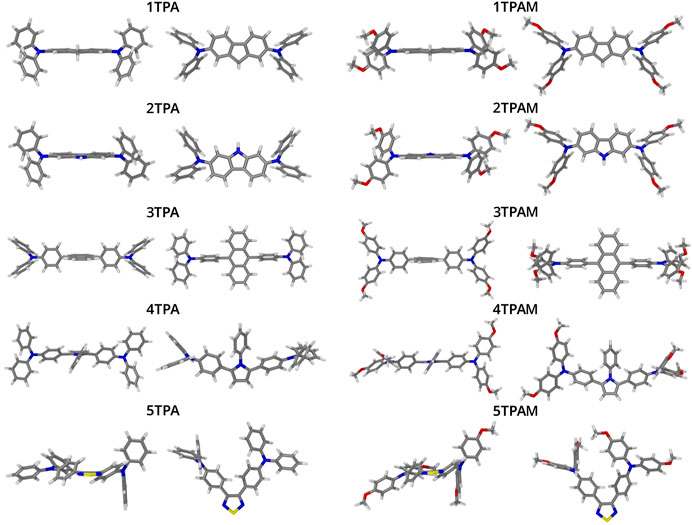
FIGURE 3. Optimized geometrical structures of the triphenylamine derivatives obtained with M06-2X/6-311++G(d,p).
On the other hand, some derivatives showed torsion angles between π-linker and triphenylamine units, which could affect the charge transport process. Table 1 lists the geometrical parameters for all the systems, such as angles and torsion angles related to π-linker, and Figure 1B shows the used labels.
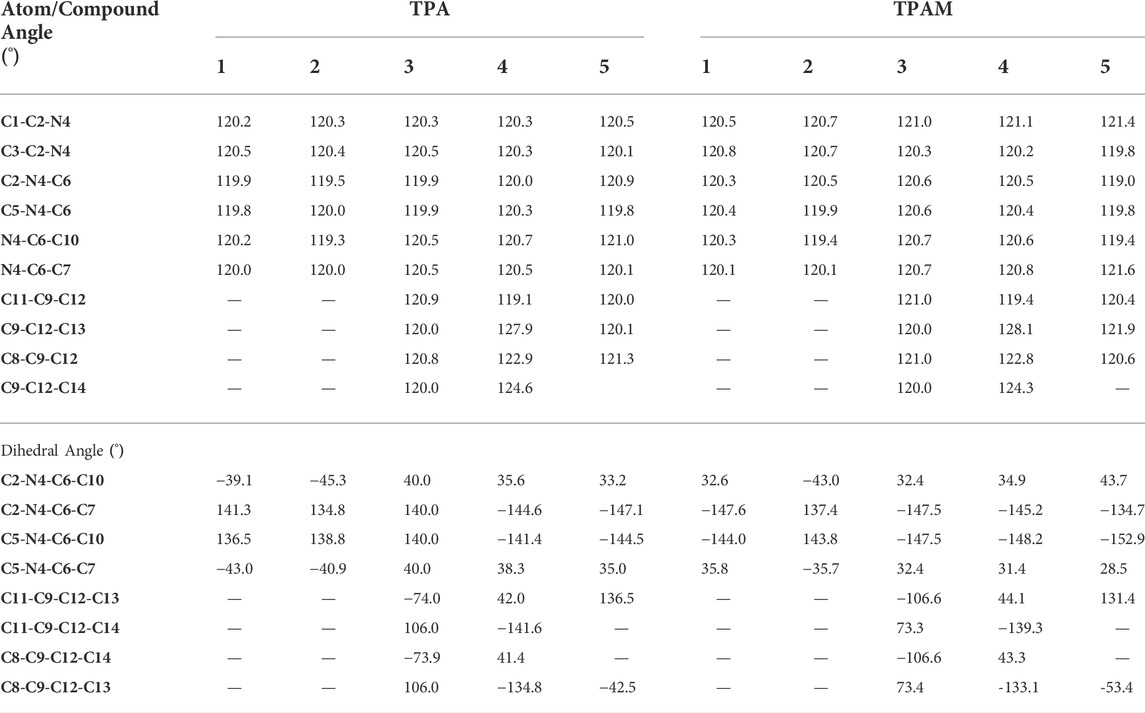
TABLE 1. Geometrical parameters for all triphenylamine derivatives calculated with M06-2X/6-311++G(d,p).
The torsion angles analysis was carried out through the comparison of the same torsion angle present in different derivatives. The 3, 4, 5TPA and 3, 4, 5TPAM derivatives showed a torsion in C11-C9-C12-C13 atoms due to the bond type C—C between the triphenylamine unit and π-linker. As can be observed, 5TPA and 5TPAM exhibited the greatest values of torsion angles. This behavior is due to the inclusion of a thiadiazole heterocycle in the structure. Further, a difference was observed in the torsion angles of 3TPA and 3TPAM molecules; this difference could be attributed to the effect of the methoxy group in the triphenylamine unit. It was observed that the addition of methoxy groups in 3TPA and 3TPAM breaks the planarity of the molecules. Nevertheless, 4TPA and 4TPAM derivatives did not show a significant difference in the torsion angle values, which denote that the molecules with a benzopyrrole unit are not affected when methoxy groups are added. This similar behavior is observed in 1TPA, 1TPAM, 2TPA, and 2TPAM.
A significant difference was observed in the torsion angle C2-N4-C6-C1O only between 5TPA and 5TPAM, which registered a difference of 10.5°, however, in the other compounds, this torsion angle did not show significant changes because it is part of the triphenylamine rings. This difference in the torsion angle (5TPA and 5TPAM) could also be ascribed to inclusion of methoxy groups in derivatives with thiadiazole as π-linker, which could affect the planarity of the molecule.
Electronic properties
One of the most significant characteristics of HTMs is to be transparent to visible light when an inverse cell is used, to be precise, these must not absorb energy in the visible region of the electromagnetic spectrum (400–700 nm). Therefore, several functionals were probed to determine the electronic properties of triphenylamine derivatives. The comparison between calculated maximum absorption wavelengths and the reported experimental values was used as a criterion to select the employed methodology.
The theoretical maximum absorption wavelengths for 1, 2, 3TPAM calculated with several functionals and their corresponding experimental values are shown in Table 2 and Figure 4. The results obtained with M06 functional agree with experimental absorption peaks, where a slight difference was observed (less than 18 nm) between the calculated and reported values for carbazole (AS37) and fluorene (AS44) derivatives (Leijtens et al., 2012). Nevertheless, only for the anthracene derivative, the obtained value with M06 functional showed a difference of 62 nm when compared to the reported value (Liu et al., 2017). On the other hand, some studies have reported that the M06 functional describes more precisely the absorption spectra for compounds with a high degree of delocalization (Soto-Rojo et al., 2015). For this reason, the M06 functional in combination with 6-311++G(d,p) basis set, was selected to calculate the absorption properties of the new compounds (pyrrole and thiadiazole derivatives).
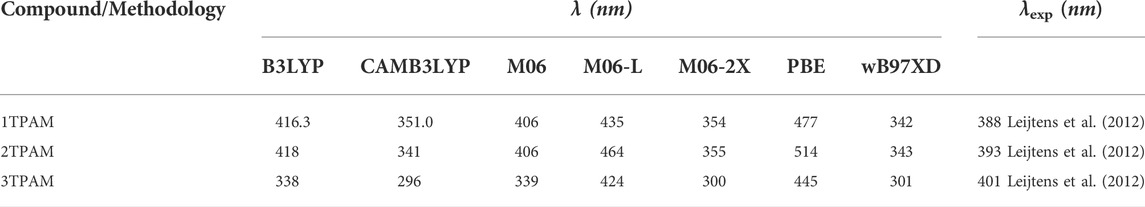
TABLE 2. Maximum absorption wavelength for 1, 2, and 3TPAM calculated with different funcionals and 6-311++G(d,p) basis set; and the comparison with the experimental value.
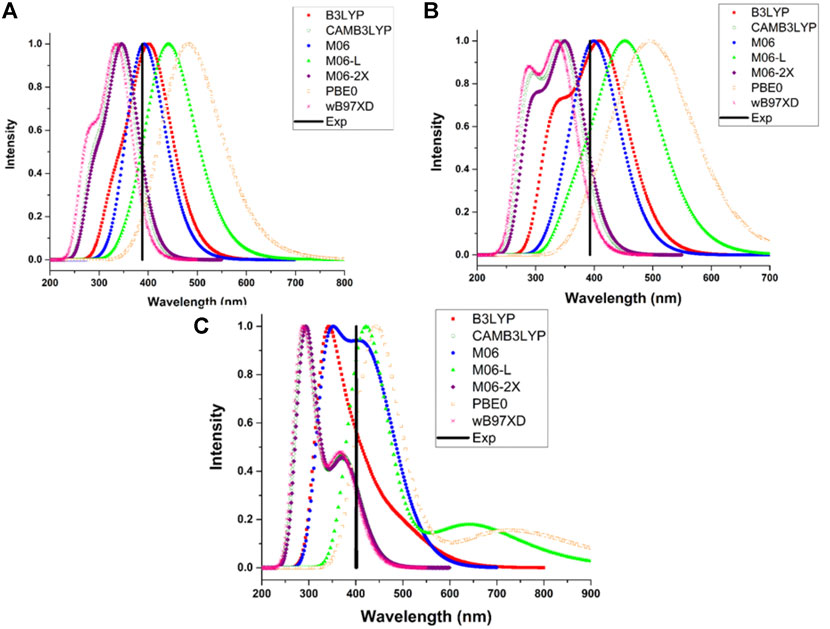
FIGURE 4. Comparison of the experimental maximum absorption wavelength with the calculated absorption spectra for the (A) 1TPAM, (B) 2TPAM and (C) 3TPAM, obtained with several functionals.
TD-DFT calculations were performed at the M06/6-311++G(d,p) level of theory using chlorobenzene as a solvent and employing SMD solvation model (Marenich et al., 2009) to simulate the absorption properties of the triphenylamine derivatives. The absorption spectra of all the analyzed compounds are shown in Figure 5. Table 3 shows the theoretical results for electronic transitions of the triphenylamine derivatives: maximum absorption wavelengths (λmax), vertical absorption energy (ΩA), oscillator strength (f), and the electronic transitions corresponding to each chemical system. As can be observed in Figure 5 all the compounds presented a minimal or null absorption in the visible region, except the anthracene derivative (3TPA and 3TPAM). Because of this, almost all the compounds could be used in a PSC, since the absorption spectra of the derivatives and the MAPI perovskite are not overlapping, which avoids a loss of efficiency due to the charge recombination (Gheno et al., 2016; Ortiz, 2019). Additionally, the absorption spectrum of pyrrole derivatives showed similar behavior to carbazole and fluorine derivatives, mainly in the TPA derivatives.
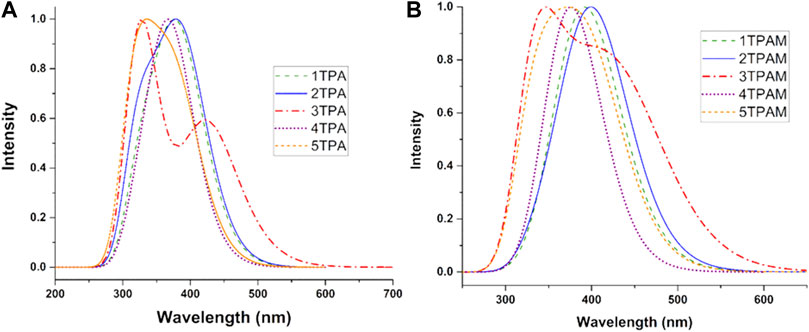
FIGURE 5. Absorption spectra of the ten analyzed triphenylamine derivatives calculated with M06/6-311++G(d,p), using chlorobenzene as solvent and employing SMD solvation model. (A) R = H and (B) R = OMe.
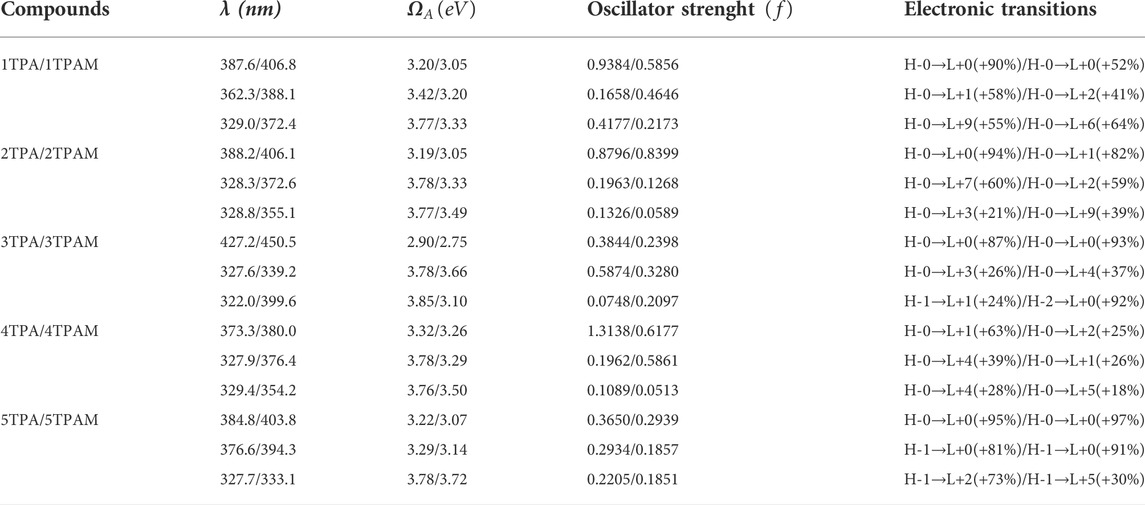
TABLE 3. Maximum absorption wavelengths (λ), vertical absorption energy (ΩA), oscillator strength (f) and the main electronic transitions of triphenylamine derivatives calculated with M06/6-311++G(d,p).
The addition of methoxy groups in the structure (TPAM) produced a bathochromic shift in the absorption spectra of all compounds. See Figures 5A,B and Table 3. The main electronic transition for almost all the derivatives was H-0→L+0 corresponding to the maximum absorption wavelength, except for the anthracene derivative, which showed an electronic transition of H-0→L+3 (3TPA in 327.6 nm) and H-0→L+4 (3TPAM in 339.2 nm) for λmax, nevertheless, the H-0→L+0 electronic transition was observed in the bands 427.2 nm (3TPA) and 450.5 nm (3TPAM) located in the visible region of the spectra. The H-0→L+0 transition corresponds to the lowest vertical absorption energy (ΩA). On the other hand, the pyrrole derivatives show the same behavior as the anthracene derivatives, since they presented electronic transitions of H-0→L+1 (4TPA in 373.3 nm) and H-0→L+2 (4TPAM in 380.0 nm), while the H-0→L+0 transition was not observed in any other band.
Molecular orbitals and energy levels
The relationship between the optical properties and the electronic structure of the compounds was analyzed by the position of the HOMOs and LUMOs orbitals (See Figure 6). In all the compounds the HOMO orbital is located over the entire molecule, which favors the holes transport (Zhang et al., 2017; Hussain et al., 2021), since the more delocalized molecular orbitals the faster charge transport, due to the reduction of the nuclear reorganization energy (Xu et al., 2018). On the other hand, the LUMO orbital is mainly concentrated in the π-linker zone, except for anthracene and pyrrole derivatives. When a methoxy group was added, only 3TPAM and 5TPAM derivatives showed a change in the HOMO distribution (See Figure 6). The methoxy groups substituted in the phenolic rings act as electron donor groups, causing the HOMO orbital extends slightly towards the periphery of the molecule.
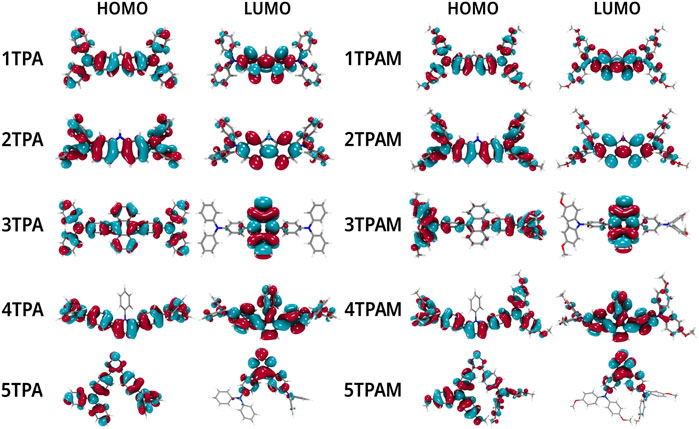
FIGURE 6. Localization of the frontier molecular orbitals calculated with M06/6-311++G(d,p) in TPA and TPAM derivatives.
The energy levels of HOMOs were compared and analyzed with the valence band of the perovskite since a good alignment improves the charge transport processes that occur in a PSC. The HOMO of the HTM should be located above the valence band of the perovskite, which provides the driven force to promote holes transport, and the LUMO should be positioned at a level higher than the conduction band of the light-sensitive material, thus preventing charge recombination. Figure 7 shows the calculated energy levels of the TPA derivatives and these are compared with the energy levels of perovskite and Spiro-OMeTAD, which is taken as HTM reference. The HOMO of almost all the derivatives is above the valence band of the perovskite, except 3TPA and 5TPA, therefore, these two derivatives could not provide the driven force to promote holes transport. The other derivatives could provide the necessary driving force to conduct electrons and holes to the perovskite at the same time. In addition, all the compounds presented HOMO energy values lower than −4.94 eV (Spiro-OMeTAD HOMO energy value); therefore, the new 4TPA could provide a better capacity to conduct holes in comparison with Spiro-OMeTAD, due to the shorter difference between the valence band of the traditional perovskite and the HOMO energy value of our proposed new HTM. Furthermore, the electron hopping from the perovskite to the metal electrode could be efficiently blocked in all derivatives, since the LUMO levels are much higher than the conduction band of the perovskite (Zhang et al., 2021b; Cheng et al., 2022). See Figure 7.
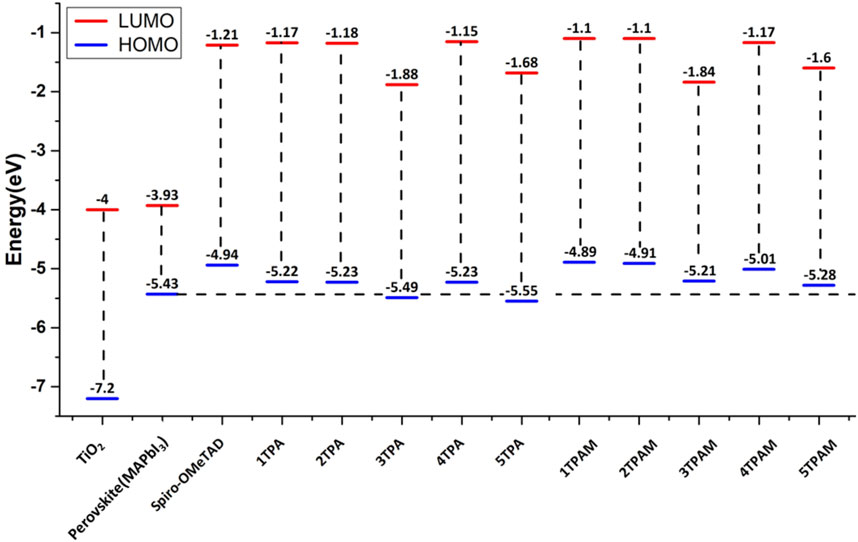
FIGURE 7. Comparison of energy levels of TPA and TPAM derivatives calculated with M06/6-311++G(d,p).
The calculated energy levels for the triphenylamine derivatives with R = OMe are shown in Figure 7 Similarly, these values were compared with the energy levels of the perovskite and the Spiro-OMeTAD. The molecules with methoxy groups also presented HOMO energy levels higher than the conduction band of the perovskite. In addition, the substitution of methoxy groups in 3TPA and 5TPA improves the HOMO energy, placing it above the perovskite valence band. Consequently, the HOMO of almost all the compounds was positioned above the HOMO of the Spiro-OMeTAD, with the exception of 3TPAM, 4TPAM, and 5TPAM, potentially converting them into hole transport materials. As can be observed, the substitution with methoxy groups produced a favorable displacement of the energy levels in comparison with the TPA derivatives, which could decrease the possible charge recombination process.
Chemical reactivity
The stability of the organic compounds is a vital factor that must be taken into account. One of the main problems of the perovskite solar cell is that it degrades rapidly even in low humidity conditions (Lu et al., 2021; Mazumdar et al., 2021). This is due to the high solubility of the perovskite. Some of the solutions proposed to solve this problem are to replace the methylammonium group of the perovskite with a more stable group in humid conditions or to process the perovskite in an inert gas environment. Also, the holes conducting solid materials have an important role in controlling the stability of the cell. Chemical reactivity parameters such as hardness (η), ionization potential (IP), and electron affinity (EA) can be used for the evaluation of the chemical stability in organic compounds. In the same way, photo-induced oxidation can be evaluated by the energy difference between the cation and the neutral molecule. For example, the higher ionization potential value, the higher stability of photo-oxidation.
Table 4 shows the calculated chemical reactivity parameters for the triphenylamine derivatives, these values were compared with the corresponding Spiro-OMeTAD parameters, which were calculated at the same level of theory. As can be observed, almost all the compounds presented IP values greater than the corresponding value of Spiro-OMeTAD (4.85 eV), except for the 1TPAM and 2TPAM, which presented IP values of 4.79 and 4.82 eV, respectively. Therefore, due to the obtained results, it is expected that these new HTMs will be more stable against oxidation caused by the incidence of light, in particular, the 5TPA thiadiazole derivative, since this compound possesses the highest IP value of the analyzed compounds. Similarly, it was also observed that the addition of the methoxy groups produced a decrease in the ionization potential.
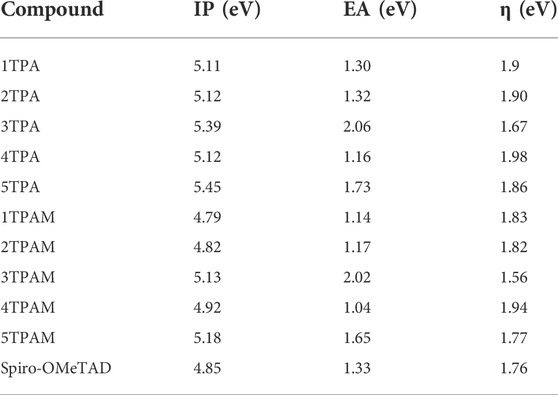
TABLE 4. Chemical reactivity parameters of the triphenylamine derivatives and Spiro-OMeTAD calculated with M06/6-311++G(d,p).
The stability of a chemical species is evaluated by absolute hardness (η). This parameter is defined as the resistance of the chemical potential to a change in the electronic configuration. The greater the value of the hardness, the more stable a chemical compound will be (Coppola et al., 2022). Nearly all compounds showed hardness values higher than the corresponding value of Spiro-OMeTAD (1.76 eV), excluding the anthracene derivatives 3TPA and 3TPAM, which presented the lower values. On other hand, the 4TPA derivative presented the highest value of chemical hardness (1.98 eV), and then this compound is expected to present greater stability.
Concerning the electron affinity (EA), high values are characteristic for n-type semiconductors materials (Chai et al., 2011, in the opposite case, low values will be characteristic of hole-conducting materials. As can be seen in Table 4, only the thiadiazole and anthracene derivatives with and without methoxy groups in the structure showed higher values than Spiro-OMeTAD electron affinity value (1.33 eV). However, this parameter does not appear to be very relevant to determine if a compound could be better holes or electrons conductor.
Reorganization energy
The hole transport reorganization energy
The reorganization energy for hole and electron transport of the triphenylamine derivatives (TPA and TPAM) is presented in Table 5. In almost all compounds, the hole transport reorganization energy showed lower values than the electron transport reorganization energy (
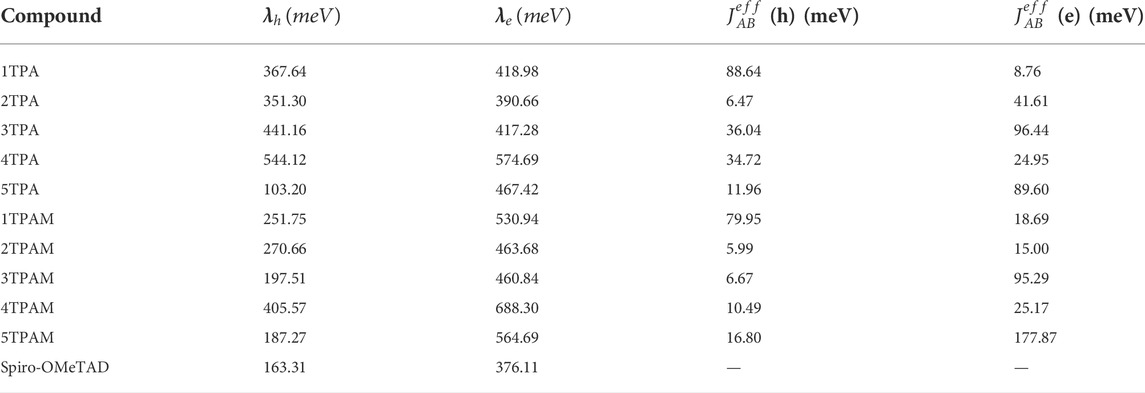
TABLE 5. Reorganization energies and electronic coupling values calculated for triphenylamine derivatives and Spiro-OMeTAD.
These data were compared with the reorganization energy of the Spiro-OMeTAD, calculated at the same level of theory. The thiadiazole derivatives presented a hole transport reorganization energy values similar to those calculated for the Spiro-OMeTAD. As shown in Table 5, the 5TPA, 3TPAM, and 5TPAM derivatives exhibited similar values of hole transport reorganization energy in comparison with the Spiro-OMeTAD, even more, the 5TPA thiadiazole derivative presented lower reorganization energy (103.20 meV) than the Spiro-OMeTAD (163.31 meV). This indicates that the 5TPA thiadiazole derivative could be a better hole conductor. On the other hand, although the pyrrole derivatives (4TPA and 4TPAM) presented the highest values for the hole transport reorganization energy, these values are still smaller than their respective electron transport reorganization energies, and thus, these derivatives are expected to behave as hole conductors.
Intermolecular electronic coupling
Another crucial parameter for the charge transport properties in organic semiconductors is the electronic coupling. The combination of low reorganization energies and high electronic couplings can maximize the carrier transport rate.
Table 5 shows the calculated electronic couplings for the dimer systems. These values are in agreement with reorganization energy predictions only for 4 molecules: 1TPA, 1TPAM, 4TPA, and 3TPA. While reorganization energy values in monomers exhibited that all the molecules, except 3TPA, are mainly hole conductors, the electronic coupling calculations presented high values for hole transport for 1TPA (88.64 meV), 1TPAM (79.95 meV), and 4TPA (34.72 meV) and, low values for the case of anthracene derivatives (3TPA and 3TPAM). This agrees with the prediction of reorganization energy values which showed higher tendency for the electron transport than for the hole transport in the anthracene derivatives.
A detailed structural analysis of the monomers and dimers provided valuable information to understand the consistency in these parameters for the above mentioned molecules. As will be shown, the dimers with less deformation and with a π-π stacking, presented the best agreement between reorganization energy and electronic coupling predictions. Figures 3, 8 allow to identify qualitatively those dimers with less deformation. In addition, values of the root mean squared displacement (RMSD) calculated between each molecule forming the dimer and the optimized monomeric structure (See Supplementary Table S1), showed that the structures of 1TPA, 1TPAM, and 3TPA were those with lowest deformations, followed by 2TPA and 4TPA. However, even though the RMSD values of 2TPA are lower than those for 4TPA, in 2TPA the interaction of the triphenylamine rings causes a rotation of one monomer around its longitudinal axis, causing the π-π interaction to decrease. The effect of a geometric change can also be noted on the site energies of monomers A and B shown in Supplementary Table S2, where the largest difference in this energy occurs in those molecules with the largest RMSD. It has recently been reported (Blaskovits et al., 2021), that flexible molecules can present a wide range of reorganization energies, strongly dependent on their geometry. And, since this geometry is affected by the restrictions imposed due to the interactions with neighbor molecules, the approach of computing λ from a lowest energy conformer in the gas phase is not appropriate for flexible structures. This could be the reason for the disagreement between the reorganization energy and electronic coupling for some of the molecules.
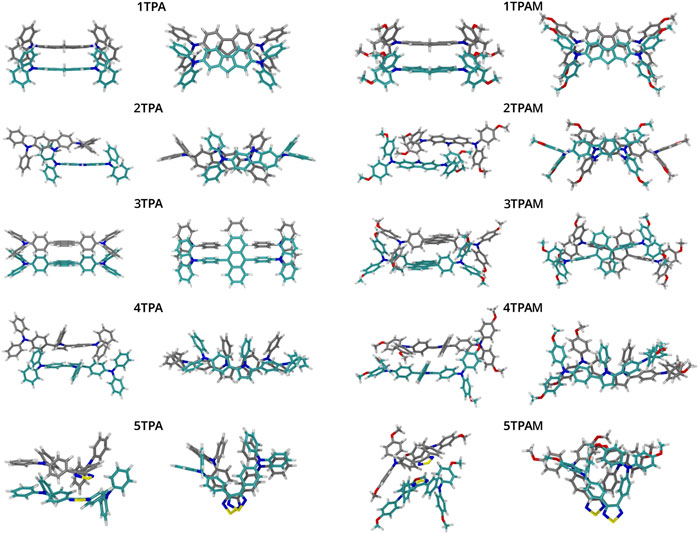
FIGURE 8. Optimized geometrical structures of the dimers obtained with B3LYP/6-311+G(d).
The HOMO and LUMO topologies of monomers and dimers are shown in Figures 6, 9, respectively. As can be observed, when the dimer is formed by 1TPA and 1TPAM, the shape of the HOMO wave function in the dimer is almost the same compared to that in the monomer. On the opposite, the LUMO wave function is mainly concentrated in one molecule of the dimer, making the LUMO overlap (
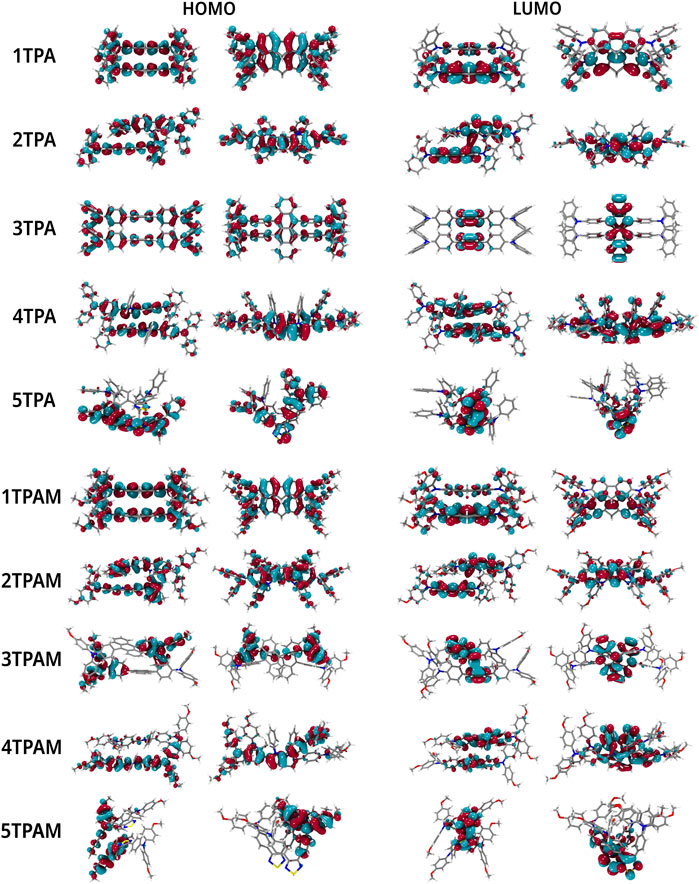
FIGURE 9. Localization of the frontier molecular orbitals in dimers of TPA and TPAM.
The dimers with the highest electronic coupling values for hole transport were 1TPA and 1TPAM, with 88.64 and 79.95 meV, respectively, while for electron transport, the highest values were obtained for 5TPAM, 3TPA, 3TPAM, and 5TPA with 177.87, 96.49, 95.29, and 89.60 meV, respectively. In fact, for the molecules studied in this work, if the dimer is a better hole or electron conductor, can be inferred from the spatial overlap values,
In the new proposed HTMs, 4TPA and 4TPAM (pyrrole derivatives), the electronic coupling values for hole transport were 34.72 and 10.49 meV, respectively; while for 5TPA and 5TPAM (thiadiazole derivatives) the calculated values were 11.96 and 16.80 meV, respectively. Based on these results, 4TPA could be a hole conductor material, whereas the 4TPAM, 5TPA, and 5TPAM seem to be electron conductor materials. After a comparison with fluorene and carbazole derivatives, it can be concluded that 4TPA presented a better hole conducting capacity than carbazole derivatives. Amongst all the dimers, 5TPA and 5TPAM showed the lowest reorganization energies, which combined with a reasonable electronic coupling value, could result in a good carrier transport rate.
In summary, the quantum-chemical calculations presented in this study are useful to understand the relationship between the chemical structure and the electronic properties in the studied molecules. In this case, the molecular structure analysis using different π-linkers revealed that these influence the planarity of the structures which could give more uniformity to the electronic density, thus favoring the charge transport process in the molecules. Additionally, the analysis of chemical reactivity parameters such as hardness (η), ionization potential (IP), and electron affinity (EA) were used for the study of the chemical stability in the analyzed organic compounds. Also, the examination of the molecular orbitals positions, energy levels, electronic transitions, and reorganization energies were extremely helpful to understand the relationship between the electronic structure and the optical properties of the studied molecules. The dimer construction for each molecule was necessary to make a detailed analysis of the charge transfer capacity; being the reorganization energies and the intermolecular electronic coupling the key parameters to define the charge transport capacity of a material.
Conclusion
New HTMs with triphenylamine units were theoretically analyzed using DFT, TD-DFT and DFTB calculations. The influence of different π-linkers on the absorption spectrum, frontier orbitals, chemical reactivity parameters, reorganization energy and intermolecular electronic coupling were studied. The obtained results suggest that all the compounds absorb energy in the ultraviolet range, which is favorable for an HTM. In addition, almost all the triphenylamine derivatives presented a HOMO energy level that is favorably aligned with the perovskite valence band, except 3TPA and 5TPA. In comparison with the Spiro-OMeTAD, practically all the derivatives showed lower values than the Spiro-OMeTAD HOMO, except 1TPAM and 2TPAM.
Regarding the new proposals, the reorganization energy values suggested that both, pyrrole and thiadiazole derivatives, could be hole transport materials. The 5TPA (thiadiazole derivative) exhibited the lowest hole transport reorganization energy (103.20 meV), being even lower than the reported value for the Spiro-OMeTAD (163.3 meV).
The analysis of the intermolecular electronic coupling carried out on dimers showed different trends compared to the reorganization energy values, which could be due to the flexibility of the molecules. However, in 4TPA derivative both values were consistent with the hole conducting capacity of this material. However, these properties would be better described considering the organic molecules in a single crystal, where there would be more conduction channels.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author contributions
All authors listed have made a substantial contribution to the work and approved its publication. JQ-B performed de DFT calculations. LR-V supervised the reorganization energy analysis, absorption spectra and contributed to the discussions. JP-B performed de DFTB calculations. L-LM contributed to the discussion of the reactivity analysis. MC-R developed the algorithm to find the dimer configurations and contributed to the theoretical background. MM-C contributed to the discussions of the reactivity analysis. GR-G contributed to the discussion in the manuscript. IG-M contributed to the discussions in the manuscript. NS-B developed the plan, supervised DFT calculations, and contributed to the discussions. All authors contributed to the writing of the manuscript.
Acknowledgments
The authors thankfully acknowledge the computer resources provided by the IPICYT’s National Supercomputing Center (CNS), under the computational time grant TKII-JPPB001.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2022.907556/full#supplementary-material
References
Adamo, C., and Barone, V. (1999). Toward reliable density functional methods without adjustable parameters: the PBE0 model. J. Chem. Phys. 110, 6158–6170. doi:10.1063/1.478522
Ansari, R. (2006). Polypyrrole conducting electroactive polymers: synthesis and stability studies. E-J. Chem. 3, 186–201. doi:10.1155/2006/860413
Bartolotti, L. J. (1982). Time-dependent Kohn-Sham density-functional theory. Phys. Rev. A 26, 2243–2244. doi:10.1103/PhysRevA.26.2243
Baumeier, B., Kirkpatrick, J., and Andrienko, D. (2010). Density-functional based determination of intermolecular charge transfer properties for large-scale morphologies. Phys. Chem. Chem. Phys. 12, 11103. doi:10.1039/C002337J
Blaskovits, J. T., Lin, K. H., Fabregat, R., Swiderska, I., Wu, H., Corminboeuf, C., et al. (2021). Is a single conformer sufficient to describe the reorganization energy of amorphous organic transport materials? J. Phys. Chem. C 125, 17355–17362. doi:10.1021/acs.jpcc.1c04067
Brown, J. S. (2022). CATNIP, version 1.9. Available at: https://github.com/JoshuaSBrown/QC_Tools (Accessed June 15, 2022).
Camarada, M. B., Jaque, P., Díaz, F. R., and Del Valle, M. A. (2011). Oxidation potential of thiophene oligomers: theoretical and experimental approach. J. Polym. Sci. B. Polym. Phys. 49, 1723–1733. doi:10.1002/polb.22360
Chai, J. D., and Head-Gordon, M. (2008). Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 10, 6615. doi:10.1039/B810189B
Chai, S., Wen, S. H., Huang, J. D., and Han, K. L. (2011). Density functional theory study on electron and hole transport properties of organic pentacene derivatives with electron-withdrawing substituent. J. Comput. Chem. 32, 3218–3225. doi:10.1002/jcc.21904
Cheng, P., Chen, Q., Liu, H., and Liu, X. (2022). Exploration of conjugated π-bridge units in N, N-bis (4-methoxyphenyl) naphthalen-2-amine derivative-based hole transporting materials for perovskite solar cell applications: a DFT and experimental investigation. RSC Adv. 12, 1011–1020. doi:10.1039/D1RA08133K
Coppola, C., Infantino, R., Dessì, A., Zani, L., Parisi, M. L., Mordini, A., et al. (2022). DFT and TDDFT investigation of four triphenylamine/phenothiazine-based molecules as potential novel organic hole transport materials for perovskite solar cells. Mat. Chem. Phys. 278, 125603. doi:10.1016/j.matchemphys.2021.125603
Delgado-Montiel, T., Baldenebro-López, J., Soto-Rojo, R., and Glossman-Mitnik, D. (2016). Quantum chemical study of the effect of π-bridge on the optical and electronic properties of sensitizers for DSSCs incorporating dioxythiophene and thiophene units. Theor. Chem. Acc. 135, 235. doi:10.1007/s00214-016-1989-3
Dev, P., Agrawal, S., and English, N. J. (2012). Determining the appropriate exchange-correlation functional for time-dependent density functional theory studies of charge-transfer excitations in organic dyes. J. Chem. Phys. 136, 224301. doi:10.1063/1.4725540
Frisch, M. J., Pople, J. A., and Binkley, J. S. (1984). Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys. 80, 3265–3269. doi:10.1063/1.447079
Ghaithan, H. M., Alahmed, Z. A., Lyras, A., Qaid, S. M., and Aldwayyan, A. S. (2020). Computational investigation of the folded and unfolded band structure and structural and optical properties of CsPb (I1− xBrx) 3 perovskites. Crystals 10, 342. doi:10.3390/cryst10050342
Gheno, A., Vedraine, S., Ratier, B., and Bouclé, J. (2016). π-Conjugated materials as the hole-transporting layer in perovskite solar cells. Metals 6, 21. doi:10.3390/met6010021
Gómez, P., Wang, J., Más-Montoya, M., Bautista, D., Weijtens, C. H., Curiel, D., et al. (2021). Pyrene-based small-molecular hole transport layers for efficient and stable narrow-bandgap perovskite solar cells. Sol. RRL 5, 2100454. doi:10.1002/solr.202100454
Gong, Z., Wang, R., Jiang, Y., Kong, X., Lin, Y., Xu, Z., et al. (2021). Novel DAD type small-molecular hole transport materials for stable inverted perovskite solar cells. Org. Electron. 92, 106102. doi:10.1016/j.orgel.2021.106102
Hariharan, P. C., and Pople, J. A. (1973). The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 28, 213–222. doi:10.1007/BF00533485
Hourahine, B., Aradi, B., Blum, V., Bonafé, F., Buccheri, A., Camacho, C., et al. (2020). DFTB+, a software package for efficient approximate density functional theory based atomistic simulations. J. Chem. Phys. 15, 124101. doi:10.1063/1.5143190
Hussain, R., Mehboob, M. Y., Khan, M. U., Khalid, M., Irshad, Z., Fatima, R., et al. (2021). Efficient designing of triphenylamine-based hole transport materials with outstanding photovoltaic characteristics for organic solar cells. J. Mat. Sci. 56, 5113–5131. doi:10.1007/s10853-020-05567-6
Kim, G. W., Choi, H., Kim, M., Lee, J., Son, S. Y., Park, T., et al. (2020). Hole transport materials in conventional structural (n–i–p) perovskite solar cells: from past to the future. Adv. Energy Mat. 10, 1903403. doi:10.1002/aenm.201903403
Kumari, K., Jana, A., Dey, A., Chakrabarti, T., and Sarkar, S. K. (2020). Lead free CH3NH3SnI3 based perovskite solar cell using ZnTe nano flowers as hole transport layer. Opt. Mat. (Amst). 111, 110574. doi:10.1016/j.optmat.2020.110574
Lai, K. W., Chang, C. C., and Chu, C. W. (2021). Benzodithiophene-based small molecules with various termini as hole transporting materials in efficient planar perovskite solar cells. Org. Electron. 89, 106010. doi:10.1016/j.orgel.2020.106010
Leijtens, T., Ding, I. K., Giovenzana, T., Bloking, J. T., McGehee, M. D., and Sellinger, A. (2012). Hole transport materials with low glass transition temperatures and high solubility for application in solid-state dye-sensitized solar cells. ACS Nano 6, 1455–1462. doi:10.1021/nn204296b
Li, H., Fu, K., Hagfeldt, A., Grätzel, M., Mhaisalkar, S. G., Grimsdale, A. C., et al. (2014). A simple 3, 4-ethylenedioxythiophene based hole-transporting material for perovskite solar cells. Angew. Chem. Int. Ed. Engl. 126, 4169–4172. doi:10.1002/ange.201310877
Li, S., Cao, Y. L., Li, W. H., and Bo, Z. S. (2021). A brief review of hole transporting materials commonly used in perovskite solar cells. Rare Met. 40, 2712–2729. doi:10.1007/s12598-020-01691-z
Liu, X., Kong, F., Ghadari, R., Jin, S., Yu, T., Chen, W., et al. (2017). Anthracene–arylamine hole transporting materials for perovskite solar cells. Chem. Commun. 53, 9558–9561. doi:10.1039/C7CC03444J
London, F. (1937). Théorie quantique des courants interatomiques dans les combinaisons aromatiques. J. Phys. Radium 8, 397–409. doi:10.1051/jphysrad:01937008010039700
Lu, Q., Yang, Z., Meng, X., Yue, Y., Ahmad, M. A., Zhang, W., et al. (2021). A review on encapsulation technology from organic light emitting diodes to organic and perovskite solar cells. Adv. Funct. Mat. 31, 2100151. doi:10.1002/adfm.202100151
Mao, X., Sun, L., Wu, T., Chu, T., Deng, W., Han, K., et al. (2018). First-principles screening of all-inorganic lead-free ABX3 perovskites. J. Phys. Chem. C 122, 7670–7675. doi:10.1021/acs.jpcc.8b02448
Marcus, R. A. (1993). Electron transfer reactions in chemistry. Theory and experiment. Rev. Mod. Phys. 65, 599–610. doi:10.1103/RevModPhys.65.599
Marenich, A. V., Cramer, C. J., and Truhlar, D. G. (2009). Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 113, 6378–6396. doi:10.1021/jp810292n
Mazumdar, S., Zhao, Y., and Zhang, X. (2021). Stability of perovskite solar cells: Degradation mechanisms and remedies. Front. Electron. 8, 712785. doi:10.3389/felec.2021.712785
Mesquita, I., Andrade, L., and Mendes, A. (2018). Perovskite solar cells: materials, configurations and stability. Renew. Sustain. Energy Rev. 82, 2471–2489. doi:10.1016/j.rser.2017.09.011
Naeem, N., Shehzad, R. A., Ans, M., Akhter, M. S., and Iqbal, J. (2021). Dopant free triphenylamine-based hole transport materials with excellent photovoltaic properties for high-performance perovskite solar cells. Energy Tech. 10, 2100838. doi:10.1002/ente.202100838
Nelsen, S. F., Blackstock, S. C., and Kim, Y. (1987). Estimation of inner shell Marcus terms for amino nitrogen compounds by molecular orbital calculations. J. Am. Chem. Soc. 109, 677–682. doi:10.1021/ja00237a007
NREL (2021). Best research-cell efficiencies. Available at: http://www.nrel.gob/ncpv/images/efficiency_chart.jpg (accessed March 19, 2021).
{kind=link}
Ortiz, A. (2019). Triarylamine-BODIPY derivatives: a promising building block as hole transporting materials for efficient perovskite solar cells. Dyes Pigm. 171, 107690. doi:10.1016/j.dyepig.2019.107690
Pham, H. D., Yang, T. C. J., Jain, S. M., Wilson, G. J., and Sonar, P. (2020). Development of dopant-free organic hole transporting materials for perovskite solar cells. Adv. Energy Mat. 10, 1903326. doi:10.1002/aenm.201903326
Pitchaiya, S., Natarajan, M., Santhanam, A., Asokan, V., Yuvapragasam, A., Ramakrishnan, V. M., et al. (2020). A review on the classification of organic/inorganic/carbonaceous hole transporting materials for perovskite solar cell application. Arab. J. Chem. 13, 2526–2557. doi:10.1016/j.arabjc.2018.06.006
Rezaei, F., and Mohajeri, A. (2021). Molecular designing of triphenylamine-based hole-transporting materials for perovskite solar cells. Sol. Energy 221, 536–544. doi:10.1016/j.solener.2021.04.055
Rostami, Z., Hosseinian, A., and Monfared, A. (2018). DFT results against experimental data for electronic properties of C60 and C70 fullerene derivatives. J. Mol. Graph. Model. 81, 60–67. doi:10.1016/j.jmgm.2018.02.009
Roy, P., Sinha, N. K., Tiwari, S., and Khare, A. (2020). A review on perovskite solar cells: evolution of architecture, fabrication techniques, commercialization issues and status. Sol. Energy 198, 665–688. doi:10.1016/j.solener.2020.01.080
Runge, E., and Gross, E. K. (1984). Density-functional theory for time-dependent systems. Phys. Rev. Lett. 52, 997–1000. doi:10.1103/PhysRevLett.52.997
Sharmoukh, W., Al Kiey, S. A., Ali, B. A., Menon, L., and Allam, N. K. (2020). Recent progress in the development of hole-transport materials to boost the power conversion efficiency of perovskite solar cells. Sustain. Mater. Technol. 26, e00210. doi:10.1016/j.susmat.2020.e00210
Sheibani, E., Yang, L., and Zhang, J. (2020). Recent advances in organic hole transporting materials for perovskite solar cells. Sol. RRL 4, 2000461. doi:10.1002/solr.202000461
Snaith, H. J. (2019). A decade of perovskite photovoltaics. Nat. Energy. 4, 1. doi:10.1038/s41560-018-0323-9
Soto-Rojo, R., Baldenebro-López, J., and Glossman-Mitnik, D. (2015). Study of chemical reactivity in relation to experimental parameters of efficiency in coumarin derivatives for dye sensitized solar cells using DFT. Phys. Chem. Chem. Phys. 17, 14122–14129. doi:10.1039/C5CP01387A
Touhami, A., Chaabane, R. B., and Allouche, A. R. (2015). Theoretical investigation on electronic, optical, and charge transport properties of new anthracene derivatives. Comput. Theor. Chem. 1073, 123–130. doi:10.1016/j.comptc.2015.09.024
Valeev, E. F., Coropceanu, V., da Silva Filho, D. A., Salman, S., and Brédas, J. L. (2006). Effect of electronic polarization on charge-transport parameters in molecular organic semiconductors. J. Am. Chem. Soc. 128, 9882–9886. doi:10.1021/ja061827h
Wang, B., Zeng, Q., Sun, Z., Xue, S., and Liang, M. (2019). Molecularly engineering of truxene-based dopant-free hole-transporting materials for efficient inverted planar perovskite solar cells. Dyes Pigm. 165, 81–89. doi:10.1016/j.dyepig.2019.01.052
Xu, Y. L., Ding, W. L., and Sun, Z. Z. (2018). How to design more efficient hole-transporting materials for perovskite solar cells? Rational tailoring of the triphenylamine-based electron donor. Nanoscale 10, 20329–20338. doi:10.1039/C8NR04730H
Yanai, T., Tew, D., and Handy, N. (2004). A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 393, 51–57. doi:10.1016/j.cplett.2004.06.011
Zhang, H., Hu, J., Zhao, J., and Zhang, Y. (2016). Spectrometric measurements and DFT studies on new complex of copper (II) with 2-((E)-9-ethyl-3-(2-(6-(4-methylpyridin-2-yl) pyridin-3-yl) vinyl)-9H-carbazole. Spectrochim. Acta Part A 168, 78–85. doi:10.1016/j.saa.2016.05.051
Zhang, X., Ghadari, R., Liu, X., Wang, W., Ding, Y., Cai, M., et al. (2021b). CN-based carbazole-arylamine hole transporting materials for perovskite solar cells: substitution position matters. J. Energy Chem. 62, 563–571. doi:10.1016/j.jechem.2021.04.021
Zhang, X., Ghadari, R., Liu, X., Wang, W., Ding, Y., Cai, M., et al. (2021a). Heteroatom effect on linear-shaped dopant-free hole transporting materials for perovskite solar cells. Sol. Energy 221, 323–331. doi:10.1016/j.solener.2021.04.034
Zhang, Y., Guo, J., Su, H., Li, J., Wu, W., and Wang, L. (2018). A rational design of hole-transport small molecules based on fluorene with different modified groups for organic lead-halide perovskite solar cells. Dyes Pigm. 154, 275–281. doi:10.1016/j.dyepig.2018.03.019
Zhang, Y., Li, Y., Chen, C., Wang, L., and Zhang, J. (2017). Design new hole transport materials for efficient perovskite solar cells by suitable combination of donor and core groups. Org. Electron. 49, 255–261. doi:10.1016/j.orgel.2017.06.064
Zhang, Z., and He, R. (2019). Effect of heterocyclic spacer on property of hole-transporting materials with silafluorene core for perovskite solar cells. Comput. Theor. Chem. 1161, 10–17. doi:10.1016/j.comptc.2019.06.002
Zhao, Y., and Truhlar, D. G. (2006). A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J. Chem. Phys. 125, 194101. doi:10.1063/1.2370993
Zhao, Y., and Truhlar, D. G. (2008). The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 120, 215–241. doi:10.1007/s00214-007-0310-x
Zhou, D., Zhou, T., Tian, Y., Zhu, X., and Tu, Y. (2018). Perovskite-based solar cells: materials, methods, and future perspectives. J. Nanomater. 2018, 8148072. doi:10.1155/2018/8148072
Zhou, Y., Zhang, Z., and Cui, J. (2020). Effect of π-linker extension on property of fluorene-based hole-transporting materials for perovskite solar cells. Comput. Theor. Chem. 1192, 113049. doi:10.1016/j.comptc.2020.113049
Keywords: triphenylamine (TPA), DFT-density functional theory, theoretical chemical reactivity, hole transport material (HTM), electronic properties
Citation: Quezada-Borja JD, Rodríguez-Valdez LM, Palomares-Báez JP, Chávez-Rojo MA, Landeros-Martinez L-L, Martínez-Ceniceros MC, Rojas-George G, García-Montoya IA and Sánchez-Bojorge NA (2022) Design of new hole transport materials based on triphenylamine derivatives using different π-linkers for the application in perovskite solar cells. A theoretical study. Front. Chem. 10:907556. doi: 10.3389/fchem.2022.907556
Received: 29 March 2022; Accepted: 12 July 2022;
Published: 05 August 2022.
Edited by:
Juan Frau, University of the Balearic Islands, SpainReviewed by:
José Carlos Germino, University of Aveiro, PortugalWeijie Chi, Hainan University, China
Lorenzo Franco, University of Padua, Italy
Copyright © 2022 Quezada-Borja, Rodríguez-Valdez, Palomares-Báez, Chávez-Rojo, Landeros-Martinez, Martínez-Ceniceros, Rojas-George, García-Montoya and Sánchez-Bojorge. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nora Aydeé Sánchez-Bojorge, bnNhbmNoZXpAdWFjaC5teA==