 Shiwei Guo
Shiwei Guo Yinghua Li
Yinghua Li Weibin Fan
Weibin Fan Zhiqi Liu
Zhiqi Liu Deguang Huang
Deguang Huang- 1State Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, China
- 2College of Materials Science and Opto Electronic Technology, University of Chinese Academy of Sciences, Beijing, China
A simple and efficient method for the synthesis of dialdehyde aniline in good yields (up to 83%) is explored using Cu(OTf)2 as the catalyst, Selectfluor as the radical initiator, and DMSO as both the carbon and oxygen sources. Experimental studies indicate that the reaction is achieved by the formylation of two CAr-H bonds, first at the para-position and then at the ortho-position. A possible mechanism is proposed, including the thermal homolysis of Selectfluor, the Cu(II)-facilitated formylation of the CAr-H bonds, and the hydrolysis of the amide under alkaline conditions in air atmosphere.
Introduction
Aromatic aldehydes are valuable synthetic intermediates in the fields of pharmaceuticals, agrochemicals, pesticides, and chemical sciences. The formyl groups display good stability and exhibit high activity toward coupling reactions with the formation of C–X (X = C, N, S, etc.) bonds (Dirksen and Dawson., 2008; Waddell and Mack., 2009; Ambreen and Wirth., 2014; Liang et al., 2017; Tamang and Findlater., 2017). A couple of reactions have been developed for the synthesis of pharmaceuticals and functional materials. For example, phenanthrene-9-carbonitrile and its derivatives, a kind of anticancer drugs, were synthesized by the reaction of 4-substituted benzaldehydes with phenylacetonitrile in MeOH under alkaline conditions, followed by the coupling reaction for forming the CAr-CAr bond (Scheme 1A) (Perin et al., 2020). (2Z, 2′Z)-3,3'-[4-(dimethylamino)-1,3-phenylene]bis[2-(4-aminophenyl)acrylonitrile] (DPAA), a stimuli-responsive organic fluorescent material, was obtained by the condensation of 4-(dimethylamino)isophthalaldehyde with p-nitrophenylacetonitrile in EtOH with the reduction of the nitro groups to the amine groups (Scheme 1B) (Fang et al., 2018). Trans-4-[4-(dimethylamino)styryl]-1-methylpyridinium iodide (DSMI), a cationic hemicyanine dye, commonly used as a fluorescent probe for DNA analysis, was also synthesized by the reaction of 4-(dimethylamino)isophthalaldehyde with 1,4-dimethylpyrinium iodide under reflux conditions (Scheme 1C) (Sun et al., 2014).
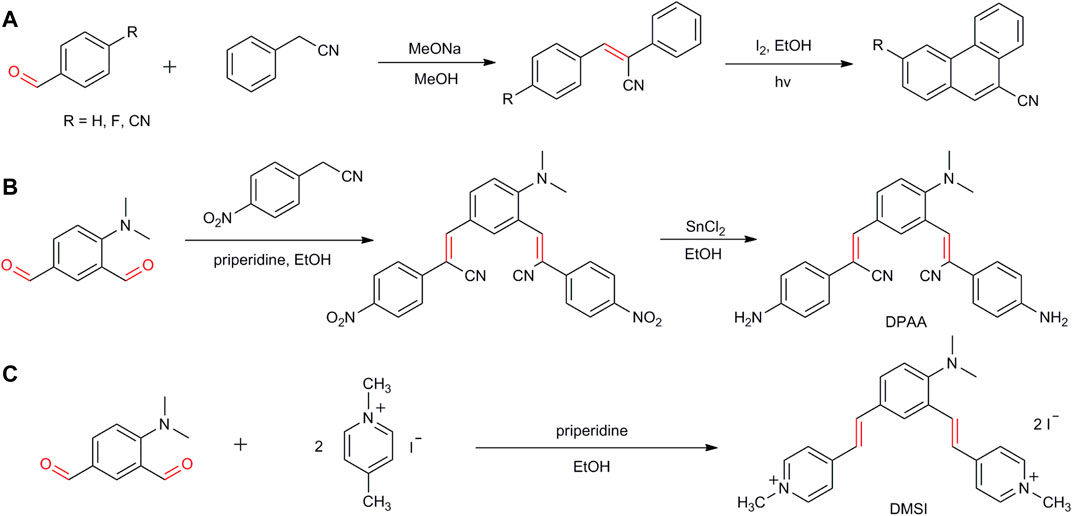
SCHEME 1. Pharmaceuticals and functional materials synthesized by aromatic aldehydes.
To date, a number of methodologies have been developed for the synthesis of aromatic aldehydes. One of the most efficient methods is the direct installation of formyl groups on the aromatic rings. For example, the reaction of N,N-dimethylaniline with phosphorus oxychloride using N, N-dimethylformamide (DMF) as the formyl source yielded the dialdehyde compound (dimethylamino)isophthalaldehyde, in which the amino group was protected by two methyl groups (Scheme 2A) (Fang et al., 2018; Sun et al., 2014). It will be more difficult to synthesize aminobenzaldehydes in one step. A common practice is to generate the amino group and formyl group in different reactions, including the oxidation of the methyl group to the formyl group first and then the reduction of the nitro group to the amino group (Scheme 2B) (Wheeler., 1958; Kijima et al., 1984; Dai et al., 2011), the reduction of the nitro group to the amino group first and then the oxidation of the alcohol group to the formyl group (Scheme 2C) (Sorkin and Hinden., 1949; Liu et al., 2015), and the addition of the formyl group to the BOC-protected aniline first and then the deprotection of the protected amino group (Scheme 2D) (Ma et al., 2002). In addition, a couple of small molecules have been studied as the formyl source for the construction of heterocyclic aromatic aldehydes, such as dimethyl sulfoxide (Scheme 2E) (Zhang et al., 2014; Suvam et al., 2021), N, N-dimethylformamide (Koeller and Lellouche., 1999; Tang and Shi., 2008; Khadka et al., 2012; Badalyan et al., 2014; Popov et al., 2019), and carbon monoxide (Schoenbe et al., 1974; Klaus et al., 2005; Sergeev et al., 2008). All these methods have their limitations for the production of aminobenzaldehydes, such as the rigorous reaction conditions, strong reducing agents, multistep processes, and/or low yields. A simple and efficient method is required for the synthesis of aminobenzaldehydes in one-step, consistent with the development of aldehyde–amine chemistry and its related pharmacological chemistry.
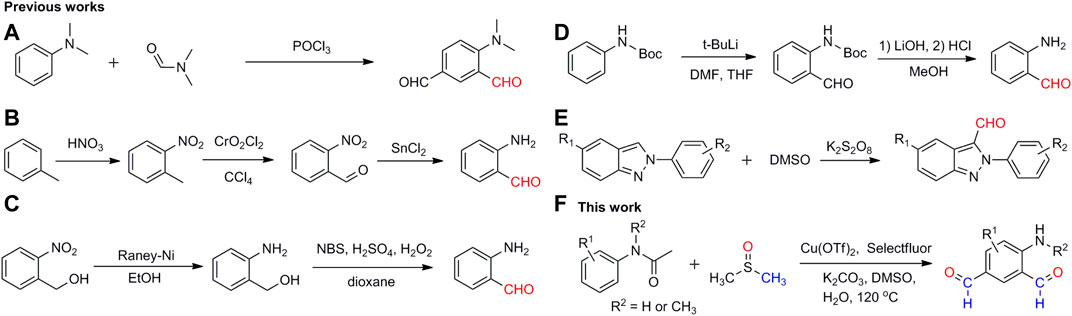
SCHEME 2. Different strategies to synthesize aromatic aldehydes.
Selectfluor is an efficient oxidant and/or radical initiator in organic synthesis (Galloway et al., 2017; Gao et al., 2018; Hu et al., 2019; Niu et al., 2019). We have used it as an oxidant for the generation of methylene groups via C–N/C–O bond break (Jin et al., 2021; Zhang et al., 2022). Herein, we report a facile and efficient method for the preparation of 4-aminoisophthalaldehyde, in which DMSO, Cu(OTf)2, and Selectfluor were used as the formyl source, catalyst and radical initiator, respectively Scheme 2F.
Materials and Methods
General Information
The single-crystal data of compounds were collected by a Cu–/Mo–Kα rotating anode source, using a Supernova diffractometer with the ω-scan method. ESI-MS was performed using a Bruker Impact II quardrupole time-of-flight mass spectrometer. 1H NMR and 13C NMR spectra were recorded on a Bruker Avance III (400 MHz) and JNM ECS400S (400 MHz), respectively. Chemical shifts are expressed in δ ppm values with reference to tetramethylsilane (TMS) as the internal standard. NMR multiplicities are abbreviated as follows: s = singlet; d = doublet; m = multiplet. Coupling constants (J) are expressed in Hz. Volume reduction and drying steps were performed in vacuo. All the reagents were purchased from commercial sources and used as received. Dimethyl sulfoxide and dichloromethane were freshly distilled over CaH2. Tetrahydrofuran, diethyl ether, toluene, and n-hexane were distilled over sodium under N2. The starting material acetanilides were synthesized according to the procedure given by Ofori et al., (2021).
General Procedure for the Synthesis of Starting Materials
A mixture of aniline derivatives (4.0 mmol) and triethylamine (4.4 mmol) in dry CH2Cl2 (50 mL) was added to acetyl chloride (4.4 mmol) dropwise while stirring. After addition, the mixture was allowed to stir for 4 h at room temperature. The reaction was quenched by adding water (20 mL). The organic layer was washed with water (2 × 20 mL), dried over Na2SO4, and concentrated in vacuo to yield products as solid.
General Procedure for Compounds 3a–3x
A mixture of acetanilides 1 (0.2 mmol), Cu(OTf)2 (0.04 mmol), Selectfluor (0.5 mmol), K2CO3 (0.4 mmol), and H2O (50 μL) in DMSO (1 mL) was stirred at 120°C for 12 h in air. After cooling to room temperature, the reaction mixture was diluted with CH2Cl2 (20 mL) and filtered through a pad of silica gel. The organic solution was washed with brine (2 × 30 mL). The aqueous solution was extracted with CH2Cl2 (3 × 30 mL). The organic layers were combined and dried over anhydrous Na2SO4 overnight. The solvent was removed under reduced pressure. The crude product was purified on a silica gel column eluted with petroleum ether/ethyl acetate (3:1 to 2:1 v/v) to yield the products 3a–3x.
General Procedure for Mechanistic Exploration
1) A mixture of N-(4-formylphenyl)acetamide 1y (0.2 mmol), Cu(OTf)2 (0.04 mmol), Selectfluor (0.5 mmol), K2CO3 (0.4 mmol), and H2O (50 μL) in DMSO (1 mL) was stirred at 120°C for 12 h in air. After cooling to room temperature, the reaction mixture was diluted with CH2Cl2 (20 mL) and filtered through a pad of silica gel. The organic solution was washed with brine (2 × 30 mL), and the aqueous solution was extracted with CH2Cl2 (3 × 30 mL). The organic layers were combined and dried over anhydrous Na2SO4 overnight. The solvent was removed under reduced pressure. The crude product was purified on a silica gel column eluted with petroleum ether/ethyl acetate (2:1 v/v) to afford the product 3a in 81% yield (23.8 mg).
2) A mixture of N-(2-formylphenyl)acetamide 1z (0.2 mmol), Cu(OTf)2 (0.04 mmol), Selectfluor (0.5 mmol), K2CO3 (0.4 mmol), and H2O (50 μL) in DMSO (1 mL) was stirred at 120°C for 12 h in air. After cooling to room temperature, the reaction mixture was diluted with CH2Cl2 (20 mL) and filtered through a pad of silica gel. No signal of the product 3a was observed on the TLC plate.
3) A mixture of acetanilide 1a (0.2 mmol), Cu(OTf)2 (0.04 mmol), Selectfluor (0.5 mmol), K2CO3 (0.4 mmol), and H2O (50 μL) in DMSO-d6 (1 mL) was stirred at 120°C for 12 h in air. After cooling to room temperature, the reaction mixture was diluted with CH2Cl2 (20 mL) and filtered through a pad of silica gel. The organic solution was washed with brine (2 × 30 mL), and the aqueous solution was extracted with CH2Cl2 (3 × 30 mL). The organic layers were combined and dried over anhydrous Na2SO4 overnight. The solvent was removed under reduced pressure. The crude product was purified on a silica gel column eluted with petroleum ether/ethyl acetate (2:1 v/v) to afford the product 3a′ (76%, 22.2 mg) with 49% and 45% deuteration of the two formyl groups, respectively.
4) A mixture of acetanilide 1a (0.2 mmol), Cu(OTf)2 (0.04 mmol), Selectfluor (0.5 mmol), K2CO3 (0.4 mmol), and H2O (50 μL) in DMSO (1 mL) was stirred at 120°C for 12 h in N2. After cooling to room temperature, the reaction mixture was diluted in dichloromethane (20 mL) and filtered through a pad of silica gel. The organic solution was washed with brine (2 × 30 mL), and the aqueous solution was extracted with CH2Cl2 (3 × 30 mL). The organic layers were combined and dried over anhydrous Na2SO4 overnight. The solvent was removed under reduced pressure. The crude product was purified on a silica gel column eluted with petroleum ether/ethyl acetate (2:1 v/v) to afford the product 3a in 79% yield (23.3 mg).
5) A mixture of acetanilide 1a (0.2 mmol), Cu(OTf)2 (0.04 mmol), Selectfluor (0.5 mmol), K2CO3 (0.4 mmol), and H218O (50 μL) in DMSO (1 mL) was stirred at 120°C for 12 h in air. After cooling to room temperature, the reaction mixture was diluted with dichloromethane (20 mL) and filtered through a pad of silica gel. The organic solution was washed with brine (2 × 30 mL), and the aqueous solution was extracted with CH2Cl2 (3 × 30 mL). The organic layers were combined and dried over anhydrous Na2SO4 overnight. The organic layers were combined, and the solvent was removed under reduced pressure. The crude product was purified on a silica gel column eluted with petroleum ether/ethyl acetate (2:1 v/v) to afford the product 3a in 79% yield (23.5 mg) rather than 3a” [(18O)-3a].
6) A mixture of acetanilide 1a (0.2 mmol), Cu(OTf)2 (0.04 mmol), Selectfluor (0.5 mmol), K2CO3 (0.4 mmol), and H2O (50 μL) in DMSO (1 mL) was stirred at 120°C for 12 h in air. After cooling to room temperature, the reaction mixture was diluted with dichloromethane (20 mL) and filtered through a pad of silica gel. The organic solution was washed with brine (2 × 30 mL), and the aqueous solution was extracted with CH2Cl2 (3 × 30 mL). The organic layers were combined and dried over anhydrous Na2SO4 overnight. The solvent was removed under reduced pressure. The crude product was purified on a silica gel column eluted with petroleum ether/ethyl acetate (1:1 v/v) to afford the product 4a in 78% yield (38.7 mg).
7) A mixture of acetanilide 1a (0.2 mmol), Cu(OTf)2 (0.04 mmol), Selectfluor (0.5 mmol), K2CO3 (0.4 mmol), 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO, 1 mmol) [or butylated hydroxytoluene (BHT, 1 mmol)], and H2O (50 μL) in DMSO (1 mL) was stirred at 120°C for 12 h in air. After cooling to room temperature, the reaction mixture was diluted with dichloromethane (20 mL) and filtered through a pad of silica gel. No signal of the product 3a was observed on the TLC plate.
NMR DATA (Note: The NH2 protons of all the compounds could not be detected in 1H NMR spectra).
4-Aminoisophthalaldehyde (3a)
Yield, 79% (23.5 mg); yellow solid; melting point, 110–112°C; 1H NMR (400 MHz, CDCl3) δ 9.96 (s, 1H), 9.81 (s, 1H), 8.05 (d, J = 1.8 Hz, 1H), 7.86 (dd, J = 8.7, 1.8 Hz, 1H), and 6.76 (d, J = 8.7 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 193.4 (s, 1C), 189.3 (s, 1C), 154.2 (s, 1C), 140.5 (s, 1C), 134.8 (s, 1C), 126.2 (s, 1C), 117.9 (s, 1C), and 116.6 (s, 1C). HRMS m/z (ESI) [M + Na]+ calculated for C8H7NO2Na, 172.0374; found, 172.0369. IR neat: 3,434, 2,968, 2,926, 1,656, and 1,616 cm−1.
4-Amino-5-Methoxyisophthalaldehyde (3b)
Yield, 83% (29.7 mg); light yellow solid; melting point, 125–127°C; 1H NMR (400 MHz, CDCl3) δ 9.95 (s, 1H), 9.78 (s, 1H), 7.66 (d, J = 1.5 Hz, 1H), 7.38 (s, 1H), and 3.96 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 193.0 (s, 1C), 189.6 (s, 1C), 147.0 (s, 1C), 146.3 (s, 1C), 134.4 (s, 1C), 125.3 (s, 1C), 116.6 (s, 1C), 109.2 (s, 1C), and 56.1 (s, 1C). HRMS m/z (ESI) [M + Na]+ calculated for C9H9NO3Na, 202.0480; found, 202.0475. IR neat: 3,406, 2,812, 2,749 1,631, and 1,593 cm−1.
4-Amino-5-(Methylthio)Isophthalaldehyde (3c)
Yield, 80%, (31.2 mg); light yellow solid melting point, 123–125°C, 1H NMR (400 MHz, CDCl3) δ 9.95 (s, 1H), 9.80 (s, 1H), 8.09 (d, J = 1.8 Hz, 1H), 7.98 (d, J = 1.8 Hz, 1H), and 2.41 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 193.2 (s, 1C), 189.0 (s, 1C), 153.7 (s, 1C), 139.8 (s, 1C), 138.3 (s, 1C), 125.9 (s, 1C), 123.1 (s, 1C), 117.5 (s, 1C), and 17.9 (s, 1C). HRMS m/z (ESI) [M + Na]+ calculated for C9H9NO2SNa 218.0252; found, 218.0247. IR neat: 3,422, 2,933, 2,926, 1,654, and 1,590 cm−1.
4-Amino-5-Phenoxyisophthalaldehyde (3d)
Yield, 82% (39.5 mg); light yellow solid; melting point, 135–137°C; 1H NMR (400 MHz, CDCl3) δ 10.00 (s, 1H), 9.74 (s, 1H), 7.82 (d, J = 1.6 Hz, 1H), 7.43–7.36 (m, 3H), 7.20 (t, J = 7.4 Hz, 1H), and 7.10–7.03 (m, 1H). 13C NMR (101 MHz, CDCl3) δ 193.0 (s, 1C), 189.1 (s, 1C), 155.3 (s, 1C), 146.9 (s, 1C), 145.2 (s, 1C), 135.3 (s, 1C), 130.3 (s, 2C), 125.3 (s, 1C), 124.8 (s, 1C), 119.2 (s, 2C), 118.3 (s, 1C), and 118.2 (s, 1C). HRMS m/z (ESI) [M + Na]+ calculated for C14H11NO3Na, 264.0637; found, 264.0632. IR neat: 3,412, 2,915, 2,853, 1,628, and 1,588 cm−1.
4-Amino-5-(Benzyloxy)isophthalaldehyde (3e)
Yield, 81% (41.3 mg); yellow solid; melting point, 142–144°C; 1H NMR (400 MHz, CDCl3) δ 9.96 (s, 1H), 9.78 (s, 1H), 7.68 (d, J = 1.4 Hz, 1H), 7.47 (d, J = 1.0 Hz, 1H), 7.45–7.41 (m, 3H), 7.40 (t, J = 2.4 Hz, 1H), and 7.39–7.35 (m, 1H), 5.16 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 193.0 (s, 1C), 189.5 (s, 1C), 146.4 (s, 1C), 146.1 (s, 1C), 135.6 (s, 1C), 134.5 (s, 1C), 128.9 (s, 2C), 128.7 (s, 1C), 128.0 (s, 2C), 125.3 (s, 1C), 116.8 (s, 1C), 110.6 (s, 1C), and 71.0 (s, 1C). HRMS m/z (ESI) [M + Na]+ calculated for C15H13NO3Na, 278.0793; found, 278.0788. IR neat 3,395, 2,979, 2,916, 1,656, and 1,593 cm−1.
4-Amino-5-Isopropylisophthalaldehyde (3f)
Yield, 81% (30.9 mg); yellow solid; melting point, 106–108°C, 1H NMR (400 MHz, CDCl3) δ 9.98 (s, 1H), 9.84 (s, 1H), 7.93 (s, 1H), 7.88 (s, 1H), 2.90 (dt, J = 13.5, 6.7 Hz, 1H), and 1.34 (d, J = 6.8 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 193.8 (s, 1C), 189.8 (s, 1C), 152.1 (s, 1C), 139.2 (s, 1C), 133.5 (s, 1C), 130.1 (s, 1C), 125.8 (s, 1C), 117.7 (s, 1C), 26.8 (s, 1C), and 21.7 (s, 2C). HRMS m/z (ESI) [M + Na]+ calculated for C11H13NO2Na, 214.0844; found, 214.0839. IR neat: 3,411, 2,965, 2,905, 1,646, and 1,599 cm−1.
4-Amino-5-Ethylisophthalaldehyde (3g)
Yield, 81% (28.3 mg); light yellow solid; melting point, 113–115°C, 1H NMR (400 MHz, CDCl3) δ 9.98 (s, 1H), 9.81 (s, 1H), 7.92 (d, J = 1.8 Hz, 1H), 7.80 (d, J = 0.8 Hz, 1H), 2.55 (q, J = 7.4 Hz, 2H), and 1.33 (t, J = 7.5 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 193.8 (s, 1C), 189.8 (s, 1C), 152.2 (s, 1C), 139.2 (s, 1C), 133.5 (s, 1C), 130.1 (s, 1C), 125.8 (s, 1C), 117.7 (s, 1C), 26.8 (s, 1C), and 21.7 (s, 1C). HRMS m/z (ESI) [M + Na]+ calculated for C10H11NO2Na, 200.0688; found, 200.0683. IR neat: 3,499, 2,988, 2,937, 1,656, and 1,593 cm−1.
4-Amino-5-Methylisophthalaldehyde (3h)
Yield, 81% (26.2 mg); light yellow solid; melting point, 100–102°C, 1H NMR (400 MHz, CDCl3) δ 9.97 (s, 1H), 9.81 (s, 1H), 7.94 (t, J = 3.3 Hz, 1H), 7.79 (d, J = 5.5 Hz, 1H), and 2.26 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 193.6 (s, 1C), 189.6 (s, 1C), 153.1 (s, 1C), 139.1 (s, 1C), 134.5 (s, 1C), 125.7 (s, 1C), 123.4 (s, 1C), 117.3 (s, 1C), and 16.5 (s, 1C). HRMS m/z (ESI) [M + Na]+ calculated for C9H9NO2Na, 186.0531; found, 186.0526. IR neat: 3,396, 2,925, 2,835, 1,664, and 1,597 cm−1.
2-Amino-[1,1′-Biphenyl]-3,5-Dicarbaldehyde (3i)
Yield, 80% (36.1 mg); yellow solid; melting point, 136–138°C, 1H NMR (400 MHz, CDCl3) δ 10.03 (s, 1H), 9.87 (s, 1H), 8.08 (d, J = 1.9 Hz, 1H), 7.83 (d, J = 1.9 Hz, 1H), 7.54 (dd, J = 7.9, 6.5 Hz, 2H), 7.50–7.46 (m, 1H), and 7.46–7.40 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 193.5 (s, 1C), 189.4 (s, 1C), 152.0 (s, 1C), 139.7 (s, 1C), 136.0 (s, 1C), 135.2 (s, 1C), 129.5 (s, 2C), 129.2 (s, 1C), 129.0 (s, 2C), 128.7 (s, 1C), 125.8 (s, 1C), and 118.0 (s, 1C). HRMS m/z (ESI) [M + Na]+ calculated for C14H11NO2Na, 248.0688; found, 248.0683. IR neat: 3,463, 2,833, 2,749, 1,664, and 1,597 cm−1.
4-Amino-5-Bromoisophthalaldehyde (3j)
Yield, 68% (31.0 mg); light yellow solid; melting point, 167–169°C, 1H NMR (400 MHz, CDCl3) δ 9.93 (s, 1H), 9.81 (s, 1H), 8.19 (d, J = 1.7 Hz, 1H), and 8.05 (d, J = 1.6 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 192.6 (s, 1C), 188.1 (s, 1C), 151.0 (s, 1C), 138.8 (s, 1C), 137.4 (s, 1C), 126.5 (s, 1C), 118.4 (s, 1C), and 110.9 (s, 1C). HRMS m/z (ESI) [M + Na]+ calculated for C8H6BrNO2Na, 249.9480; found, 249.9475. IR neat: 3,467, 2,915, 2,853 1,656, and 1,572 cm−1.
4-Amino-5-Chloroisophthalaldehyde (3k)
Yield, 63% (23.1 mg); light yellow solid; melting point, 144–146°C, 1H NMR (400 MHz, CDCl3) δ 9.98 (s, 1H), 9.82 (s, 1H), and 8.02 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 192.7 (s, 1C), 188.3 (s, 1C), 150.1 (s, 1C), 138.1 (s, 1C), 133.8 (s, 1C), 125.9 (s, 1C), 120.7 (s, 1C), and 118.4 (s, 1C). HRMS m/z (ESI) [M + Na]+ calculated for C8H6ClNO2Na, 205.9985; found, 205.9980. IR neat: 3,436, 2,843, 2,770, 1,656, and 1,603 cm−1.
4-Amino-5-Fluoroisophthalaldehyde (3l)
Yield, 54% (18.1 mg); yellow solid; melting point, 152–154°C, 1H NMR (400 MHz, CDCl3) δ 10.00 (d, J = 1.8 Hz, 1H), 9.83 (d, J = 2.6 Hz, 1H), 7.91 (d, J = 1.2 Hz, 1H), and 7.71 (dd, J = 11.1, 1.6 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 192.7 (s, 1C), 188.6 (s, 1C), 150.8 (d, 1JC-F = 245.4 Hz), 143.9 (d, 2JC-F = 14.2 Hz), 135.5 (s), 125.2(s), 119.2 (s), and 117.1 (d, 2JC-F = 18.2 Hz). HRMS m/z (ESI) [M + Na]+ calculated for C8H6FNO2Na, 190.0280; found, 190.0275. IR neat: 3,447, 2,874, 2,822, 1,635, and 1,583 cm−1.
4-Amino-6-Methoxyisophthalaldehyde (3m)
Yield, 78% (27.9 mg); yellow solid; melting point, 132–135°C, 1H NMR (400 MHz, CDCl3) δ 10.18 (s, 1H), 9.80 (s, 1H), 8.11 (s, 1H), 6.06 (s, 1H), and 3.96 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 192.6 (s, 1C), 186.9 (s, 1C), 166.3 (s, 1C), 156.2 (s, 1C), 139.7 (s, 1C), 116.7 (s, 1C), 113.5 (s, 1C), 95.8 (s, 1C), and 55.9 (s, 1C). HRMS m/z (ESI) [M + Na]+ calculated for C9H9NO3Na, 202.0480; found, 202.0475. IR neat: 3,426, 1,656, and 1,603 cm−1.
4-Amino-6-Isopropylisophthalaldehyde (3n)
Yield, 76% (29.1 mg); yellow solid; melting point, 100–102°C, 1H NMR (400 MHz, CDCl3) δ 10.05 (s, 1H), 9.92 (s, 1H), 8.03 (s, 1H), 6.63 (s, 1H), 4.01 (dt, J = 13.6, 6.8 Hz, 1H), and 1.29 (d, J = 6.8 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 193.1 (s, 1C), 189.4 (s, 1C), 158.9 (s, 1C), 153.5 (s, 1C), 143.5 (s, 1C), 123.6 (s, 1C), 116.6 (s, 1C), 112.9 (s, 1C), 28.3 (s, 1C), and 23.2 (s, 2C). HRMS m/z (ESI) [M + Na]+ calcd for C11H13NO2Na, 214.0844; found, 214.0839. IR neat: 3,426, 2,968, 2,874, 1,656, and 1,614 cm−1.
4-Amino-6-Ethylisophthalaldehyde (3o)
Yield, 78% (27.6 mg); yellow solid; melting point, 129–131°C, 1H NMR (400 MHz, CDCl3) δ 9.96 (s, 1H), 9.88 (s, 1H), 7.99 (s, 1H), 6.47 (s, 1H), 3.00 (q, J = 4.8 Hz, 2H), and 1.24 (t, J = 4.8 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 193.1 (s, 1C), 189.5 (s, 1C), 154.4 (s, 1C), 153.4 (s, 1C), 143.4 (s, 1C), 124.1 (s, 1C), 116.9 (s, 1C), 116.5 (s, 1C), 26.5 (s, 1C), and 15.2 (s, 1C). HRMS m/z (ESI) [M + Na]+ calculated for C10H11NO2Na, 200.0688; found, 200.0683. IR neat: 3,411, 2,957, 2,926, 1,646, and 1,610 cm−1.
4-Amino-6-Methylisophthalaldehyde (3p)
Yield, 80% (26.4 mg); yellow solid; melting point, 134–136°C, 1H NMR (400 MHz, CDCl3) δ 10.00 (s, 1H), 9.91 (s, 1H), 7.99 (s, 1H), 6.48 (s, 1H), and 2.64 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 193.0 (s, 1C), 189.8 (s, 1C), 153.1 (s, 1C), 148.2 (s, 1C), 143.1 (s, 1C), 124.8 (s, 1C), 118.1 (s, 1C), 116.8 (s, 1C), and 20.5 (s, 1C). HRMS m/z (ESI) [M + Na]+ calculated for C9H9NO2Na, 186.0531; found, 186.0526. IR neat: 3,426, 2,957, 2,915, 1,645, and 1,603 cm−1.
4-Amino-6-Bromoisophthalaldehyde (3q)
Yield, 52%, (23.7 mg); yellow solid; melting point, 152–154°C, 1H NMR (400 MHz, CDCl3) δ 10.08 (s, 1H), 9.89 (s, 1H), 8.18 (s, 1H), and 6.92 (s, 1H). 13C NMR (400 MHz, CDCl3) δ 192.4 (s, 1C), 188.3 (s, 1C), 137.9 (s, 1C), 133.3 (s, 1C), 123.9 (s, 1C), 122.2 (s, 1C), 118.7 (s, 1C), and 116.8 (s, 1C). HRMS m/z (ESI) [M + Na]+ calculated for C8H6BrNO2Na, 249.9480; found, 249.9475. IR neat: 3,436, 2,957, 2,915, 1,666, and 1,614 cm−1.
4-Amino-6-Chloroisophthalaldehyde (3r)
Yield, 52% (19.1 mg); yellow solid; melting point 140–142°C, 1H NMR (400 MHz, CDCl3) δ 10.20 (s, 1H), 9.89 (s, 1H), 8.17 (s, 1H), and 6.69 (s, 1H). 13C NMR (101 MHz, CDCl3) δ 1 92.2 (s, 1C), 186.2 (s, 1C), 152.5 (s, 1C), 143.1 (s, 1C), 137.9 (s, 1C), 133.5 (s, 1C), 121.3 (s, 1C), and 115.1 (s, 1C). HRMS m/z (ESI) [M + Na]+ calculated for C8H6ClNO2Na, 205.9985; found, 205.9980. IR neat: 3,436, 2,968, 2,926, 1,656, and 1,603 cm−1.
4-Amino-6-Fluoroisophthalaldehyde (3s)
Yield, 50% (16.7 mg); yellow solid; melting point, 121–123°C, 1H NMR (400 MHz, CDCl3) δ 10.08 (s), 9.88 (s), 8.15 (d, J = 7.6 Hz), and 6.34 (d, J = 12.4 Hz). 13C NMR (101 MHz, CDCl3) δ 192.8 (s, 1C), 184.6 (s, 1C), 155.8 (s, 1C), 143.3 (s, 1C), 139.8 (d, 2JC-F = 8.6 Hz, 1C), 123.0 (d, 1JC-F = 211.99 Hz, 1C), 116.2 (s, 1C), and 101.3 (d, 2JC-F = 24.1 Hz, 1H). HRMS m/z (ESI) [M + Na]+ calculated for C8H6FNO2Na, 190.0280; found, 190.0275. IR neat: 3,430, 2,979, 2,926, 1,660, and 1,609 cm−1.
4-Amino-5,6-Dimethylisophthalaldehyde (3t)
Yield, 70% (24.8 mg); yellow solid; melting point, 134–136°C, 1H NMR (400 MHz, CDCl3) δ 10.03 (s, 1H), 9.89 (s, 1H), 7.89 (s, 1H), 2.67 (s, 3H), and 2.12 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 192.4 (s, 1C), 189.5 (s, 1C), 150.9 (s, 1C), 144.4 (s, 1C), 140.4 (s, 1C), 123.7 (s, 1C), 120.5 (s, 1C), 115.0 (s, 1C), 14.9 (s, 1C), and 10.7 (s, 1C). HRMS m/z (ESI) [M + Na]+ calculated for C10H11NO2Na, 200.0688; found, 200.0683. IR neat: 3,412, 2,959, 2,911, 1,625, and 1,603 cm−1.
7-Amino-2,3-Dihydro-1H-Indene-4,6-Dicarbaldehyde (3u)
Yield, 65%, (24.5 mg); yellow solid; melting point 134–136°C; 1H NMR (400 MHz, CDCl3) δ 9.92 (s, 1H), 9.89 (s, 1H), 7.87 (s, 1H), 3.33 (t, J = 7.6 Hz, 2H), 2.71 (t, J = 7.5 Hz, 2H), and 2.27–2.19 (m, 2H).13C NMR (101 MHz, CDCl3) δ 193.2 (s, 1C), 189.9 (s, 1C), 153.2 (s, 1C), 149.9 (s, 1C), 141.6 (s, 1C), 129.9 (s, 1C), 123.1 (s, 1C), 117.2 (s, 1C), 33.2 (s, 1C), 28.0 (s, 1C), and 24.1 (s, 1C). HRMS m/z (ESI) [M + Na]+ calculated for C14H11NO2Na, 248.0687; found, 248.0682. IR neat: 3,446, 2,853, 2,824, 1,603, and 1,572 cm−1.
4-Aminonaphthalene-1,3-Dicarbaldehyde (3v)
Yield, 51% (20.3 mg); yellow solid; melting point, 192–194°C, 1H NMR (400 MHz, CDCl3) δ 10.00 (s, 1H), 9.93 (s, 1H), 9.35 (d, J = 8.6 Hz, 1H), 7.94 (s, 1H), 7.89 (d, J = 8.5 Hz, 1H), 7.76–7.71 (m, 1H), and 7.57–7.52 (m, 1H). 13C NMR (101 MHz, CDCl3) δ 192.8 (s, 1C), 191.1 (s, 1C), 153.2 (s, 1C), 148.5 (s, 1C), 145.0(s, 1C), 132.4 (s, 1C), 126.8 (s, 1C), 126.5 (s, 1C), 122.1 (s, 1C), 121.6 (s, 1C), 121.4 (s, 1C), and 111.4 (s, 1C). HRMS m/z (ESI) [M + Na]+ calculated for C12H9NO2Na, 222.0531; found, 222.0526. IR neat: 3,400, 2,843, 2,739 1,635, and 1,600 cm−1.
4-(Methylamino)Isophthalaldehyde (3w)
Yield, 79%, (25.8 mg); white solid; melting point, 107–109°C, 1H NMR (400 MHz, CDCl3) δ 9.91 (s, 1H), 9.81 (s, 1H), 8.94 (s, 1H), 8.03 (d, J = 2.0 Hz, 1H), 7.96 (dd, J = 8.9, 1.6 Hz, 1H), 6.80 (d, J = 8.9 Hz, 1H), and 3.06 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 193.6 (s, 1C), 189.3 (s, 1C), 155.3 (s, 1C), 141.1 (s, 1C), 135.7 (s, 1C), 124.8 (s, 1C), 117.8 (s, 1C), 111.1 (s, 1C), and 29.4 (s, 1C). HRMS m/z (ESI) [M + Na]+ calculated for C9H9NO2Na, 186.0531; found, 186.0526. IR neat: 3,312, 2,926, 2,853, 1,656, and 1,614 cm−1.
2-Amino-5-Benzoylbenzaldehyde (3x)
Yield, 78%, (35.1 mg); yellow solid; melting point, 122–125°C, 1H NMR (400 MHz, CDCl3) δ 9.87 (s, 1H), 8.05 (d, J = 1.9 Hz, 1H), 7.91 (dd, J = 8.7, 2.0 Hz, 1H), 7.78–7.70 (m, 2H), 7.58 (t, J = 7.4 Hz, 1H), 7.49 (t, J = 7.5 Hz, 2H), and 6.72 (d, J = 8.7 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 194.3 (s, 1C), 193.8 (s, 1C), 153.0 (s, 1C), 140.1 (s, 1C), 138.2 (s, 1C), 136.8 (s, 1C), 131.9 (s, 1C), 129.5 (s, 2C), 128.4 (s, 2C), 125.9 (s, 1C), 117.5 (s, 1C), and 115.9 (s, 1C). HRMS m/z (ESI) [M + Na]+ calculated for C11H11NO2Na 212.0687; found, 212.0682. IR neat: 3,436, 2,957, 2,926, 1,656, and 1,593 cm−1.
Methyl[(Methylsulfinyl)Methyl]Sulfane (4a)
Yield, 78%, (38.7 mg) transparent liquid; 1H NMR (400 MHz, CDCl3) δ 3.84 (s, 2H), 3.05 (s, 3H), and 2.45 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 56.2 (s), 37.8 (s), and 16.8 (s). HRMS m/z (ESI) [M + Na]+ calculated for C3H8OS2Na, 146.9909; found, 146.9913.
Results and Discussion
We commenced the study by choosing acetanilide (1a) as the model substrate and optimized the reaction conditions through catalyst, oxidants/radical initiators, and temperature (Table 1). Based on the yields obtained, it was found that the reaction of acetanilide (1a, 0.2 mmol), Selectfluor (0.5 mmol), copper (II) trifluoromethanesulfonate (0.04 mmol), water (50 μL), and potassium carbonate (0.4 mmol) in dimethyl sulfoxide (1 mL) at 120°C for 12 h provided the best result, yielding the product 4-aminobenzene-1,3-dicarbaldehyde (3a) in 78% based on 1a (entry 1). The use of other copper (II) salts such as Cu(NO3)2·6H2O, CuSO4·5H2O, and CuCl2·H2O as the catalysts would lead to the generation of 3a in lower yields 13%–45% (entries 2–4). It seems that the process of reaction was related to Cu2+ ions rather than anions. The decrease of yields was probably due to the strength of the coordination bonds between Cu2+ and their anions. This result was consistent with the finding that the use of other metal salts such as Ni(OTf)2, Zn(OTf)2, and CoSO4·H2O as the catalysts did not provide the corresponding product at all (entries 5–7). Selectfluor worked as a highly efficient radical initiator in this reaction. When Selectfluor was replaced with K2S2O8 or TBHP under the same conditions, no product could be obtained (entries 8 and 9). A temperature range of 120°C–140°C was found necessary for the formation of 3a. Lower temperature would decrease the yield dramatically down to trace (100°C), and higher temperature did not bring about additional yield (76%, 140°C).
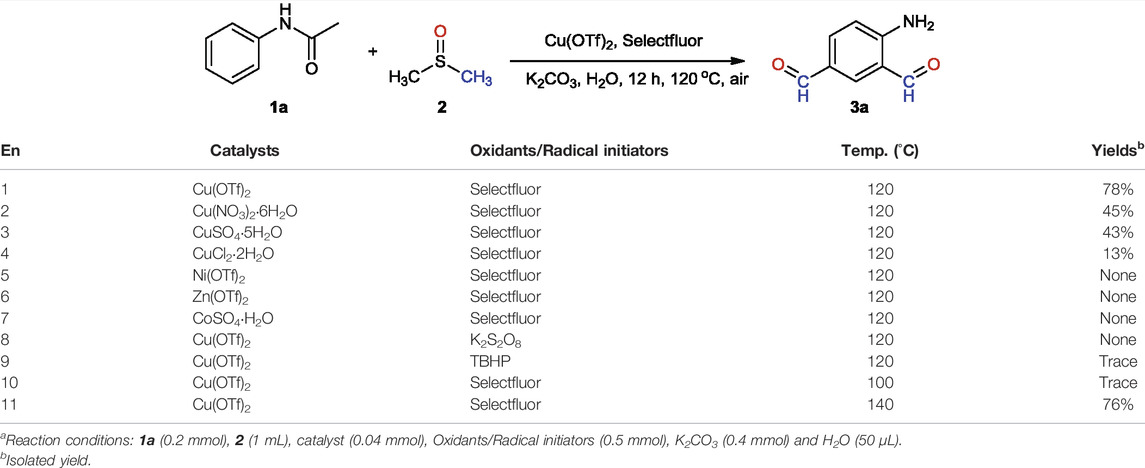
TABLE 1. Optimization of the formation of 4-aminobenzene-1,3-dicarbaldehyde (3a)a.
As the optimal reaction conditions were determined, we set out to explore the scope of 1 for the generation of compounds 3 (Scheme 3). The yields and production efficiency were discussed in terms of the electronic effect, steric effect, and synergic effect of the functional groups on the substrates. First, experimental results showed that the substitution of electron donating groups (EDGs) at the ortho-position of the phenyl rings [-OMe (3b), -SMe (3c), -OPh (3d), -OBn (3e), -iPr (3f), -Et (3g), -Me (3h), and -Ph (3i)] would be advantageous to the formation of compounds 3, providing the corresponding products at similar yields (80%–83%). On the contrary, the substitution of electron withdrawing groups (EWGs) at the same position would have a negative effect on the proceeding of reaction. The stronger the electron-withdrawing ability, the poorer the production efficiency (3j–3l). No products could be obtained when acetanilides were substituted with the strong EWGs such as –CF3, –CN and –NO2. It is thought that the strong EWGs at the ortho-position of N-acetyl made the C–H bond inactive at the para-position of N-acetyl, which would stop the formylation at the para-position (3′-position) and thereby the ortho-position (5′-position) of the N-acetyl group. Similar results were obtained with the substitution of the functional groups at the meta-position (2′-position) of N-acetyl (3m–3s).
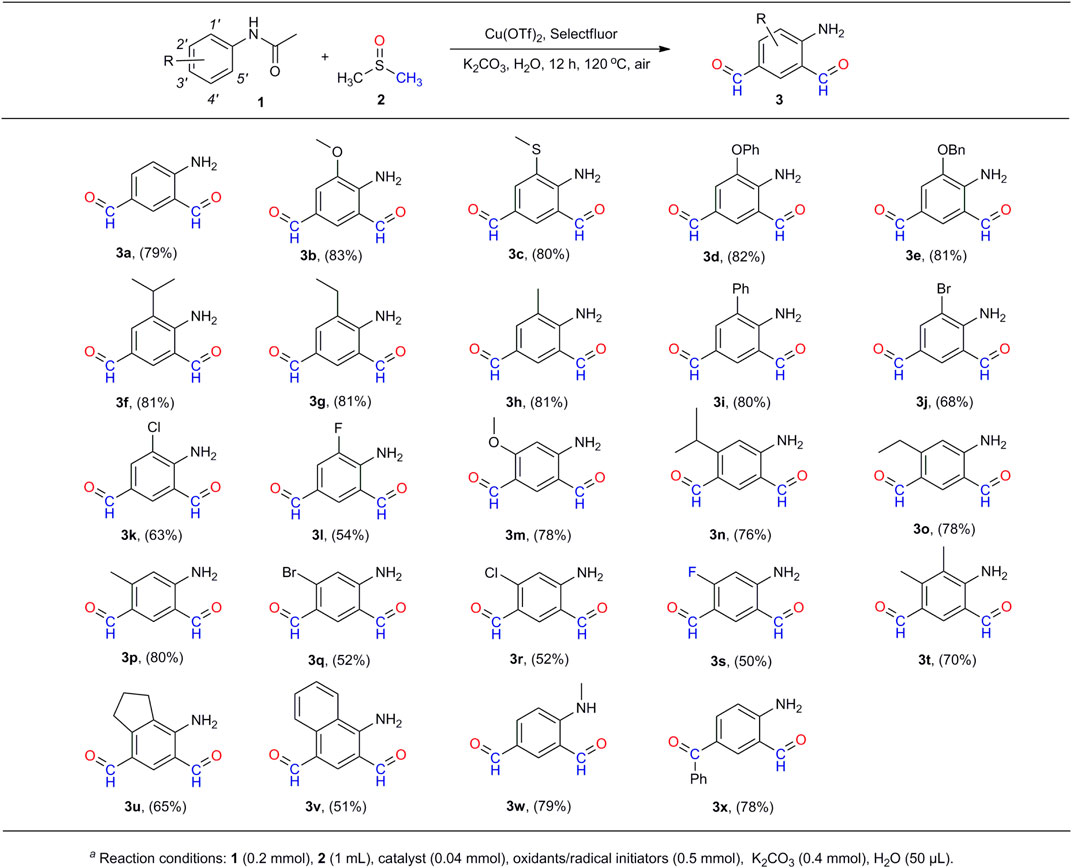
SCHEME 3. Scope of acetanilides with respect to 4-aminobenzene-1,3-dicarbaldehydes (3)a.
Second, the steric hindrance of the functional groups on the substrates to the reactions was studied by the use of methoxyl- (3b, 3m), isopropyl- (3f, 3n), ethyl- (3g, 3o), methyl- (3h 3p), bromo- (3j, 3q), chloro- (3k, 3r), and fluoro- (3l, 3s) groups at the ortho- (1′-position) and meta-positions (2′-position) of the N-acetyl group. It is found that substitution of the functional groups at either of these two positions had a relatively weak impact on the production of the target compounds. Added to this, substitution of both these positions (both 1′- and 2′-positions) have certain unfavorable effects on the formation of 3, with the decline of yields by about 15% (3t–3v). It is notable that the reaction of the disubstituted acetanilides at the 1′, 4′-positions (5′, 2′-positions) or 2′, 4′-positions did not generate the desired compounds. The presence of functional groups at the 4′-position would push back the substrates from the dinuclear copper (II) core and stop the reaction (Scheme 6). Moreover, our method is also compatible with the substrates on which the N-acetyl group is substituted with functional groups. The reaction of N-methyl–substituted acetanilide afforded the corresponding compound 3w in 79% yield. In addition, the reaction of p-benzoyl-acetanilide provided the mono-aldehyde compound 3x in a similar yield (78%). The structures of compound 3j and 3s were determined by X-ray crystallography (Scheme 4).
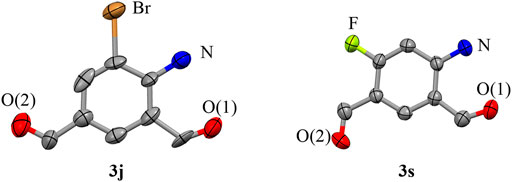
SCHEME 4. Crystal structures of compounds 3j (CCDC 2153612) and 3s (CCDC 2156314) with all non-hydrogen atoms shown as 50% probability ellipsoids.
With the progress of CAr-H bond formylation, several experiments were carried out for the investigation of the reaction mechanism (Scheme 5). The reaction of N-(4-formylphenyl)acetamide (1y) with DMSO under the standard conditions afforded the compound 3a in 81% yield (Scheme 5A). By comparison, the reaction of N-(2-formylphenyl)acetamide (1z) with DMSO at the same conditions did not generate 3a (Scheme 5B), from which the starting material was recovered. It indicates that the formylation reaction took place in two consecutive steps. First, acetanilide was formylated at the para-position to form compound 1y, and then 1y was formylated at the ortho-position to produce the target compound 3a. The reaction of acetanilide with DMSO-d6 afforded the compound 3a′ in 76% yield with 49% and 45% deuteration of the two formyl groups (Scheme 5C; Supplementary Figure S1). It suggests that the carbon atoms of the generated formyl groups likely came from DMSO. On the other hand, compound 3a was obtained in similar yield when the experiment was conducted under nitrogen atmosphere (Scheme 5D). The use of H218O as the additive to the reaction yielded the compound 3a again (Scheme 5E; Supplementary Figure S2). Methyl [(methylsulfinyl)methyl]sulfane (4a) was isolated as a by-product in 78% yield in the reaction to form compound 3a (Scheme 5F; Supplementary Figure S3). For these reasons, we proposed that the oxygen atom could come from DMSO rather than O2 and/or H2O. In addition, the formylation reaction was repeated under the standard conditions with the addition of either of the radical scavengers, 2,2,6,6-tetramethylpiperidinooxy (TEMPO) or butylated hydroxytoluene (BHT). No product was obtained with the addition of TEMPO or BHT, which indicated the generation of radicals in the process of the formylation of acetanilide (Scheme 5G). The attempt to detect the organic radical signals was unsuccessful since the presence of copper(II) compound in the solution would disturb the characteristic signals.
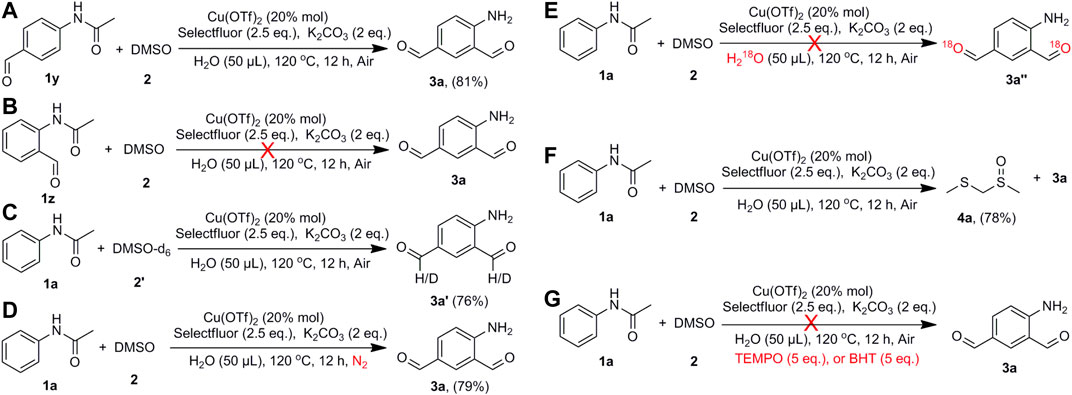
SCHEME 5. Mechanistic exploration of the formylation reaction.
The aforementioned results clearly indicated that first, a radical process was involved in this formylation; second, the carbon and oxygen atoms of the generated formyl groups come from DMSO rather than others; third, compound 1y should be an intermediate in the reaction. On the basis of the experimental results and literature research (Gao et al., 2018; Sun et al., 2018; Niu et al., 2019; Yang et al., 2020), a possible mechanism was proposed in Scheme 6. The heating of Selectfluor generated the nitrogen radical [SF-F]•. The [SF-F]• attack DMSO and acetanilide (1a) to generate [DMSO-H]• and an N-acetyl radical (a), respectively. The latter undergoes intramolecular charge transfer to form 1,4-cyclohexadiene radical (b). Intermediate (b) reacts with [DMSO-H]• to yield (c). Addition of (c) to the dinuclear copper (II) core, followed by the attack of the deprotonated DMSO anion [(DMSO-H)−] results in (f), accompanied with the release of 4a into the solution. Reduction of Cu(II) to Cu(I) leads to the decomposition of (f), by which N-(4-formylphenyl)acetamide (1y) is formed. Compound 1y was attacked by [SF-F]• and then [DMSO-H]• to produce (h). Intermediate (h) is added to the dinuclear copper (II) core, followed by attack of [DMSO-H]- and the reduction of Cu(II) to Cu(I) again to produce the final product 3a.
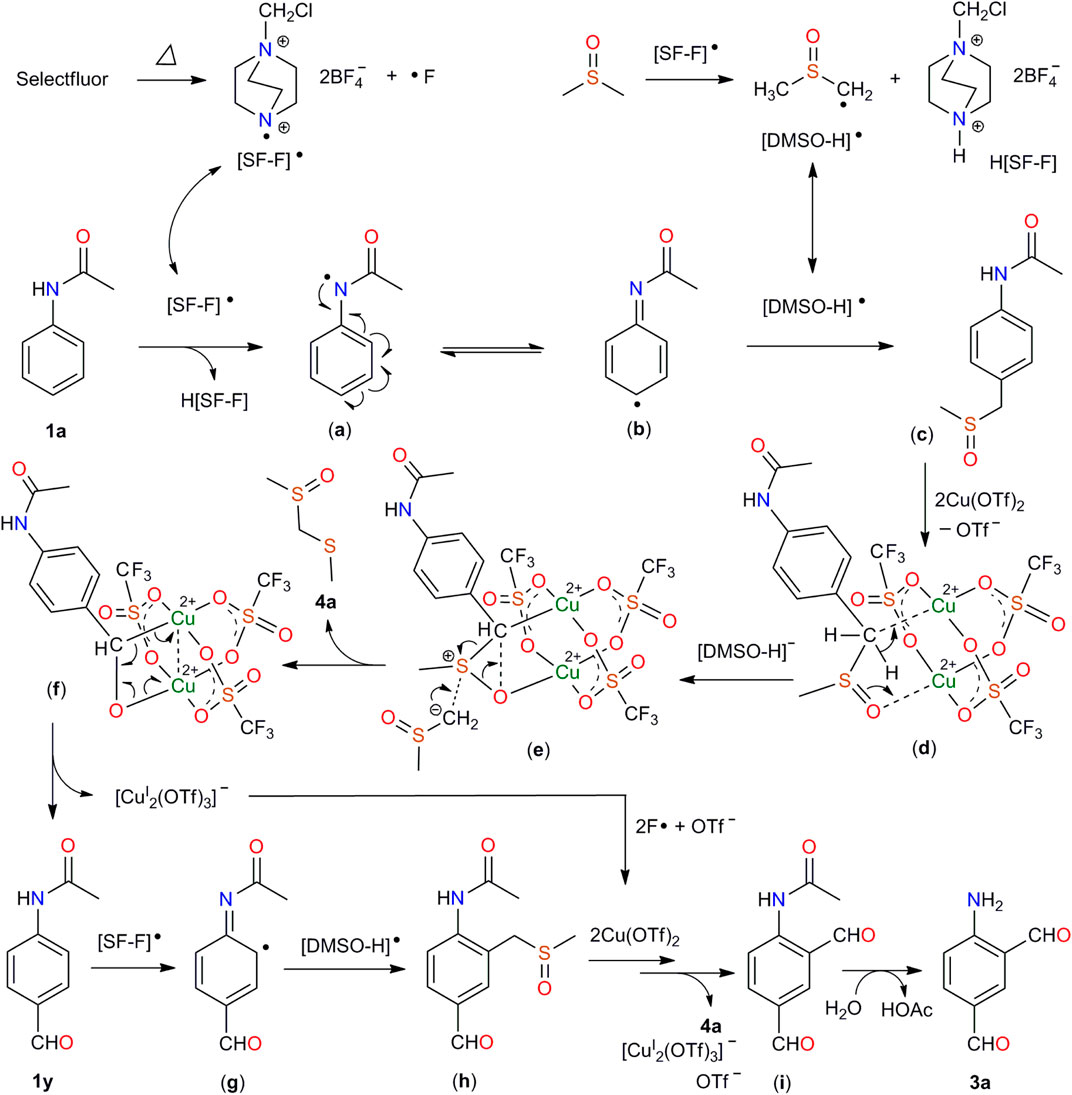
SCHEME 6. Proposed mechanism for the generation of compound 3a.
Conclusion
In summary, we have developed a simple and efficient method for the synthesis of 4-aminobenzene-1,3-dicarbaldehydes by formylation of acetanilides. The scope and versatility of the method have been successfully demonstrated with 24 examples. Experimental and mechanistic studies revealed a heat-induced radical reaction mechanism, in which copper(II) salt works as the catalyst; Selectfluor works as a radical initiator; DMSO works as a solvent and as the formyl source. In comparison with the description in the literature, our method is superior because of its simple work-up procedure, low-cost commercial available materials, air atmosphere, and one-step reaction. This work might provide a clue to synthesize valuable aminobenzaldehydes for further catalytic investigation.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author Contributions
SG: conceptualization, methodology, data curation, and original draft; YL: investigation; WF: validation and formal analysis; ZL: software; DH: project administration, funding acquisition, resources, supervision, and review and editing
Funding
This research was supported by the National Natural Science Foundation of China (Grant No. 21371171) and the Natural Science Foundation of Fujian Province (Grant No. 2020J01114).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We acknowledge the facilities of the State Key Laboratory of Structural Chemistry at the Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2022.891858/full#supplementary-material
References
Ambreen, N., and Wirth, T. (2014). High-Temperature Synthesis of Amides from Alcohols or Aldehydes by Using Flow Chemistry. Eur. J. Org. Chem. 2014 (34), 7590–7593. doi:10.1002/ejoc.201403280
Badalyan, K. S., Akopyan, A. E., Attaryan, H. S., and Asratyan, G. V. (2014). Vilsmeier-Haack Formylation of 1H-Pyrazoles. Russ. J. Gen. Chem. 84 (4), 793–795. doi:10.1134/S1070363214040331
Dai, L., Yu, J., Chen, Y., and Yu, S. (2011). Facile, Environmentally Friendly Synthesis of Benzaldehyde and Phenylacetaldehyde Analogs from Readily Available Toluene Derivatives. Synth. Commun. 41 (20), 3078–3084. doi:10.1080/00397911.2010.517366
Dirksen, A., and Dawson, P. E. (2008). Rapid Oxime and Hydrazone Ligations with Aromatic Aldehydes for Biomolecular Labeling. Bioconjug. Chem. 19 (12), 2543–2548. doi:10.1021/bc800310p
Fang, W., Zhao, W., Pei, P., Liu, R., Zhang, Y., Kong, L., et al. (2018). A Λ-shaped Cyanostilbene Derivative: Multi-Stimuli Responsive Fluorescence Sensors, Rewritable Information Storage and Colour Converter for W-LEDs. J. Mater. Chem. C 6 (34), 9269–9276. doi:10.1039/C8TC02973C
Galloway, J. D., Mai, D. N., and Baxter, R. D. (2017). Silver-Catalyzed Minisci Reactions Using Selectfluor as a Mild Oxidant. Org. Lett. 19 (21), 5772–5775. doi:10.1021/acs.orglett.7b02706
Gao, D.-W., Vinogradova, E. V., Nimmagadda, S. K., Medina, J. M., Xiao, Y., Suciu, R. M., et al. (2018). Direct Access to Versatile Electrophiles via Catalytic Oxidative Cyanation of Alkenes. J. Am. Chem. Soc. 140 (26), 8069–8073. doi:10.1021/jacs.8b03704
Hu, J., Zhou, G., Tian, Y., and Zhao, X. (2019). Selectfluor-promoted Regioselective Chlorination/Bromination of 2-Aminopyridines and 2-Aminodiazines Using LiCl/LiBr. Org. Biomol. Chem. 17 (26), 6342–6345. doi:10.1039/c9ob00972h
Jin, J., Li, Y., Xiang, S., Fan, W., Guo, S., and Huang, D. (2021). Selectfluor Facilitated Bridging of Indoles to Bis(indolyl)methanes Using Methyl Tert-Butyl Ether as a New Methylene Precursor. Org. Biomol. Chem. 19 (18), 4076–4081. doi:10.1039/d1ob00120e
Khadka, D. B., Yang, S. H., Cho, S. H., Zhao, C., and Cho, W.-J. (2012). Synthesis of 12-oxobenzo[c]phenanthridinones and 4-substituted 3-arylisoquinolones via Vilsmeier-Haack Reaction. Tetrahedron 68 (1), 250–261. doi:10.1016/j.tet.2011.10.053
Kijima, M., Nambu, Y., Endo, T., and Okawara, M. (1984). Selective Reduction of Monosubstituted Nitrobenzenes to Anilines by dihydrolipoamide-Iron(II). J. Org. Chem. 49 (8), 1434–1436. doi:10.1021/jo00182a023
Koeller, S., and Lellouche, J.-P. (1999). Preparation of Formate Esters from O-TBDMS/O-TES Protected Alcohols. A One-step Conversion Using the Vilsmeier-Haack Complex. Tetrahedron Lett. 40 (38), 7043–7046. doi:10.1016/S0040-4039(99)01453-7
Liang, G., Wang, A., Li, L., Xu, G., Yan, N., and Zhang, T. (2017). Production of Primary Amines by Reductive Amination of Biomass-Derived Aldehydes/Ketones. Angew. Chem. Int. Ed. 56 (11), 3050–3054. doi:10.1002/ange.20161096410.1002/anie.201610964
Liu, C., Fang, Z., Yang, Z., Li, Q., Guo, S., Zhang, K., et al. (2015). Highly Practical Oxidation of Benzylic Alcohol in Continuous-Flow System with Metal-free Catalyst. Tetrahedron Lett. 56 (44), 5973–5976. doi:10.1016/j.tetlet.2015.07.069
Ma, Y., Kolotuchin, S. V., and Zimmerman, S. C. (2002). Supramolecular Polymer Chemistry: Self-Assembling Dendrimers Using the DDA·AAD (GC-like) Hydrogen Bonding Motif. J. Am. Chem. Soc. 124 (46), 13757–13769. doi:10.1021/ja0202006
Niu, L., Liu, J., Liang, X.-A., Wang, S., and Lei, A. (2019). Visible Light-Induced Direct α C-H Functionalization of Alcohols. Nat. Commun. 10 (1), 467–474. doi:10.1038/s41467-019-08413-9
Ofori, E., Onyameh, E. K., Gonela, U. M., Voshavar, C., Bricker, B., Swanson, T. L., et al. (2021). New Dual 5-HT1A and 5-HT7 Receptor Ligands Derived from SYA16263. Eur. J. Med. Chem. 214, 113243–113258. doi:10.1016/j.ejmech.2021.113243
Perin, N., Rep, V., Sović, I., Juričić, Š., Selgrad, D., Klobučar, M., et al. (2020). Antiproliferative Activity and Mode of Action Analysis of Novel Amino and Amido Substituted Phenantrene and Naphtho[2,1-B]thiophene Derivatives. Eur. J. Med. Chem. 185, 111833–111866. doi:10.1016/j.ejmech.2019.111833
Popov, A. V., Kobelevskaya, V. A., Larina, L. I., and Rozentsveig, I. B. (2019). Synthesis of Poly-Functionalized Pyrazoles under Vilsmeier-Haack Reaction Conditions. Arkivoc 2019, 1–14. doi:10.24820/ark.5550190.p010.934
Sorkin, E., and Hinden, W. (1949). Untersuchungen über 4,5-Benzo-1,3-thiazine–. Hca 32, 63–68. doi:10.1002/hlca.19490320113
Sun, B., Yin, S., Zhuang, X., Jin, C., and Su, W. (2018). Selectfluor-induced C(sp2)-O Coupling Reaction of N-Substituted Anilines with Hydroxylamine Derivatives. Org. Biomol. Chem. 16 (33), 6017–6024. doi:10.1039/c8ob01348a
Sun, S., Yuan, Y., Li, Z., Zhang, S., Zhang, H., and Peng, X. (2014). Interaction of a Hemicyanine Dye and its Derivative with DNA and Cucurbit[7]uril. New J. Chem. 38 (8), 3600–3605. doi:10.1039/c4nj00068d
Suvam, B., Sudip, L., Payel, G., and Alakananda, H. (2021). Potassium Persulfate Mediated Chemodivergent C-3 Functionalization of 2H-Indazoles with DMSO as C1 Source. J. Org. Chem. 86 (15), 10866–10873. doi:10.1021/acs.joc.1c01188
Tamang, S. R., and Findlater, M. (2017). Iron Catalyzed Hydroboration of Aldehydes and Ketones. J. Org. Chem. 82 (23), 12857–12862. doi:10.1021/acs.joc.7b02020
Tang, X.-Y., and Shi, M. (2008). Vilsmeier−Haack Reaction of 1-Cyclopropyl-2-Arylethanones. J. Org. Chem. 73 (21), 8317–8320. doi:10.1021/jo801492k
Waddell, D. C., and Mack, J. (2009). An Environmentally Benign Solvent-free Tishchenko Reaction. Green. Chem. 11 (1), 79–82. doi:10.1039/b810714a
Wheeler, O. H. (1958). Étard Reaction: I its Scope and Limitation with Substituted Toluenes. Can. J. Chem. 36 (4), 667–671. doi:10.1139/v58-093
Yang, K., Song, M., Ali, A. I. M., Mudassir, S. M., and Ge, H. (2020). Recent Advances in the Application of Selectfluor as a "Fluorine‐free" Functional Reagent in Organic Synthesis. Chem. Asian J. 15 (6), 729–741. doi:10.1002/asia.202000011
Zhang, Y., Liu, Z., Zhu, T., Huang, Y., Fan, W., and Huang, D. (2022). Synthesis of Methylene-Bridged α,β-unsaturated Ketones: α-Csp3-H Methylenation of Aromatic Ketones Using Selectfluor as a Mild Oxidant. Org. Biomol. Chem. 20 (2), 415–419. doi:10.1039/d1ob02043a
Keywords: formylation, C-H bond activation, copper catalyzed, Selectfluor, radical, carbon and oxygen sources
Citation: Guo S, Li Y, Fan W, Liu Z and Huang D (2022) Copper(II)-Catalyzed Selective CAr-H Bond Formylation: Synthesis of Dialdehyde Aniline. Front. Chem. 10:891858. doi: 10.3389/fchem.2022.891858
Received: 08 March 2022; Accepted: 07 April 2022;
Published: 24 May 2022.
Edited by:
Indubhusan Deb, Indian Institute of Chemical Biology (CSIR), IndiaReviewed by:
Zhiyuan Chen, Jiangxi Normal University, ChinaChinmoy Hazra, Indian Institute of Technology Delhi, India
Raghunath Chowdhury, Bhabha Atomic Research Centre (BARC), India
Copyright © 2022 Guo, Li, Fan, Liu and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Deguang Huang, ZGh1YW5nQGZqaXJzbS5hYy5jbg==