 Yunze Wang
Yunze Wang Qingyu Lin1,2,3
Qingyu Lin1,2,3 Dengfeng Cheng
Dengfeng Cheng- 1Department of Nuclear Medicine, Zhongshan Hospital, Fudan University, Shanghai, China
- 2Institute of Nuclear Medicine, Fudan University, Shanghai, China
- 3Shanghai Institute of Medical Imaging, Shanghai, China
The positron emission tomography (PET) molecular imaging technology has gained universal value as a critical tool for assessing biological and biochemical processes in living subjects. The favorable chemical, physical, and nuclear characteristics of fluorine-18 (97% β+ decay, 109.8 min half-life, 635 keV positron energy) make it an attractive nuclide for labeling and molecular imaging. It stands that 2-[18F]fluoro-2-deoxy-D-glucose ([18F]FDG) is the most popular PET tracer. Besides that, a significantly abundant proportion of PET probes in clinical use or under development contain a fluorine or fluoroalkyl substituent group. For the reasons given above, 18F-labeled radiotracer design has become a hot topic in radiochemistry and radiopharmaceutics. Over the past decades, we have witnessed a rapid growth in 18F-labeling methods owing to the development of new reagents and catalysts. This review aims to provide an overview of strategies in radiosynthesis of [18F]fluorine-containing moieties with nucleophilic [18F]fluorides since 2015.
Introduction
Positron emission tomography (PET) is a non-invasive and quantitative imaging technology for assessing biological processes in vivo (Preshlock et al., 2016). Due to the high sensitivity of PET, the concentration of radiolabeled probes required was as few as the picomolar scale (10−6–10−8 g). Therefore, the mass effect is not to be highly considered in probe design and labeling experiments. Compared with alternative positron-emitting radioisotopes (e.g., 11C, 13N, 15O, 68Ga, 89Zr), 18F has distinct physical advantages, including 1) simple decay profile (97% positron emission and 3% electron capture), 2) lower positron energy (maximal positron energy of 0.635 MeV) resulting in short positron range and high resolution, 3) favorable physical half-life (109.8 min half-life) suitable for labeling and in vivo evaluation, and 4) flexible for labeling viable molecules by different labeling strategies. Based on these unique and advantageous characteristics of fluorine-18, 18F-labeled radiotracers have become a hot topic in molecular probe design. Whereas challenges still exist, considering fast labeling and favorable radiochemical yields have to be given higher priority in clinical practice. In recent years, many efforts have been made to develop new methods and new reagents for radiosynthesis of [18F]fluorine-containing moieties. Radiofluorination and radiofluoroalkylation reactions have been excellently reviewed by Gouverneur and co-workers (Preshlock et al., 2016), and Vugts and co-workers (Born et al., 2017) from 2010. Herein, this review focused on summarizing the recent developments in 18F-labeling methods and application in PET tracer design since 2015, according to the structures of desired radiolabeled complexes in each case, the following characteristics will be discussed: 1) radiosynthesis of fluoroalkanes with [18F]fluorides; 2) radiosynthesis of fluoroarenes with [18F]fluorides; 3) radiosynthesis of fluoroalkenes with [18F]fluorides; 4) heteroatom-18F bonds formation (Figure 1). Unless otherwise mentioned, radiochemical yield (RCY) and radiochemical conversion (RCC) are calculated without time-decay; R means both electron-donating groups and electron-withdrawing groups are capable of this reaction.
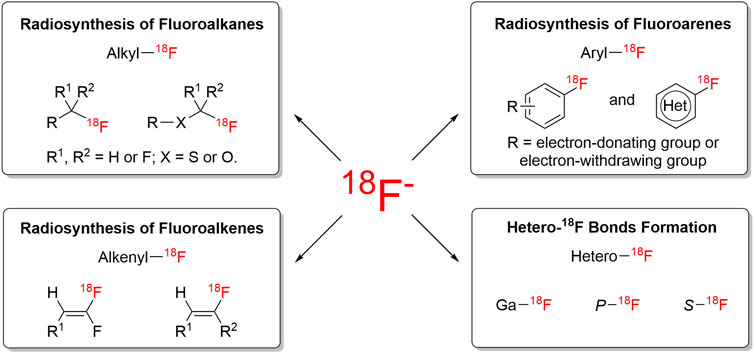
FIGURE 1. Overview of 18F-chemistry.
Radiosynthesis of Fluoroalkanes With [18F]Fluorides
Nucleophilic [18F]fluorination has been commonly used to generate aliphatic C-18F bonds (Deng et al., 2019). However, labeling precursors with alcohol-derived leaving groups or halides are easily decomposed or eliminated to the corresponded alkenes under harsh conditions (high temperatures and basicity) (D'Angelo and Taylor, 2016; Cai et al., 2008). To resolve this issue, nucleophilic [18F]fluorination of aliphatic alcohol and halides under mild conditions remained to be discussed. Other novel labeling precursors, such as carboxylic acid, and carbene precursors will be also discussed (Figure 2).
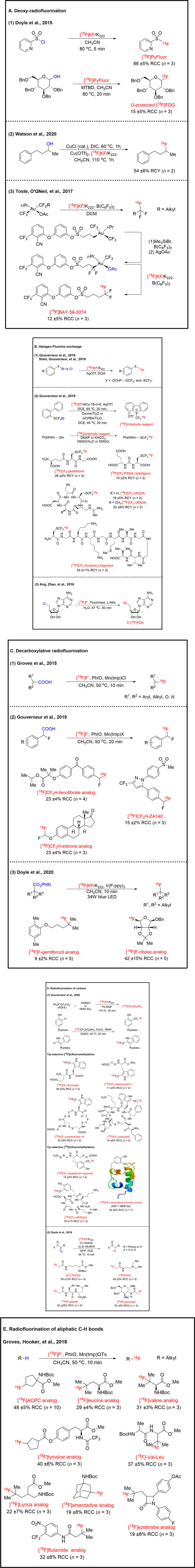
FIGURE 2. (Continued).
Aliphatic C−18F bonds are constructed by nucleophilic substitution of alcohol-derived leaving groups with [18F]fluorides in the presence of phase transfer reagents. The transformation of alcohols into corresponding 18F-labeled alkyl compounds typically involves two steps, the formation of alcohol-derived leaving groups was firstly required, including mesylate, triflate, tosylate, and followed by nucleophilic [18F]fluorination (Deng et al., 2019). Alcohols are frequently-used moiety in natural products and pharmaceutical molecules. Herein, the research on direct deoxy-radiofluorination benefits PET tracer designing. Previously, deoxy-radiofluorination required a19F carrier to generate adequate electrophile in situ to react with the tiny amount of 18F anion (Jelinski, et al., 2001). Doyle and co-workers reported the first example of a no-carrier-added deoxy-radiofluorination which applied direct conversion of alcohols into alkyl fluorides in one pot (Nielsen et al., 2015) (Figure 2A1). As proof of concept, the [18F]PyFluor was achieved in 88% of radiochemical conversion (RCC) by reacting 2-pyridinesulfonyl chloride with [18F]KF/K222 at 80°C for 5 min. Using this reagent, hydroxy protected [18F]FDG was achieved by deoxy-radiofluorination in 15% of RCC at 80°C for 20 min. This radiolabeling protocol shows particularly useful with substrates for which the sulfonate ester cannot be isolated. After that, Watson and co-workers developed an efficient method for deoxy-radiofluorination of alcohols using [18F]CuF2 (generated by Cu(OTf)2 and [18F]KF in situ) in 54% of radiochemical yield (RCY) (Sood et al., 2020) (Figure 2A2). In addition, alcohols were active by N,N′-diisopropylcarbodiimide (DIC), and CuF2. Elimination byproducts are often generated when radiofluorination of sulfonate-activated secondary alcohols (Cai et al., 2008). They obtained the pure 18F-labeled product directly from the alcohol substrate without the elimination of byproducts. Toste, O'QNeil, and co-workers developed a formal deoxy-radiofluorination for radiosynthesis of [18F]trifluoromethyl moiety (Levin et al., 2017) (Figure 2A3). The unique biological properties of the trifluoromethyl group have led to their ubiquity in pharmaceutical complexes (Tsui and Hu, 2019). They showed the borane catalyzed formal C (sp3)-CF3 reductive elimination from an Au(III) complex resulted in the formation of [18F]alkyl-CF3 compounds. Radiofluorination of the Au(III)-OAc complexes, fluorine-18 substitutes acetate group, are formal deoxy-radiofluorination reactions. The Au(III) complexes (labeling precursors) were prepared by migration-insertion of the alkyl fragment in presence of borane and bromotrimethylsilane (TMSBr) and then via anion change with AgOAc. They considered the Au(III)-OAc complexes to exhibit an appropriate blend of stability and reactivity to enable nucleophilic reductive elimination. 18F-labeled Bayer lead compound BAY 59–3,074, a cannabinoid agonist (Vry et al., 2004), was radio-synthesized in 12% of RCC with the molar activity of 0.3 GBq/μmol. This protocol provided an important proof of concept in the radiosynthesis of [18F]trifluoromethyl groups by a retrosynthetic paradigm involving C-F reductive elimination.
Except for the above-mentioned alcohol-derived leaving groups, aliphatic halides are also capable of nucleophilic [18F]fluorination, while showing lower reactivity (Deng et al., 2019). The methods to introduce [18F]fluorine anion into organic compounds via halogen-fluorine exchange rely on thermal activation only and limit to aryl-CF2X substrates (Suehiro et al., 2011; Martinez et al., 2012). Gouverneur and co-workers developed a novel strategy which for the first-time gave access to a range of respective transformations of aryl-OCHFCl, -OCF2Br and -SCF2Br to aryl-[18F]OCHF2, -[18F]OCF3 and -[18F]SCF3 derivatives with silver(I) [18F]fluoride (generated by halogenophilic silver(I) triflate and [18F]fluoride) (Khotavivattana et al., 2015) (Figure 2B1). The molar activities range from 0.04 GB GBq/μmol to 0.17 GBq/μmol. Among this research, they indicated the order of reactivity towards silver [18F]fluoride: aryl-OCHFCl > aryl-CF2Br
[18F]5-(trifluoromethyl)dibenzothiophenium trifluoromethanesulfonate ([18F]Umemoto reagent) has been prepared from [18F]fluoride by Gouverneur and co-workers via bromine-fluorine exchange and cyclization with the molar activity of 0.08 GBq/μmol (Verhoog et al., 2018) (Figure 2B2). Compared with previous [18F]trifluoromethylation methods, this method provided a direct way for [18F]trifluoromethylation of unmodified peptides at the thiol cysteine residue with high chemoselectivity. Function groups such as asparagine, glutamine, methionine, glutamic acid, proline, threonine, serine, tyrosine, lysine, or arginine were compatible for this [18F]trifluoromethylation with RCCs superior to 55%. Glutathione and ((1-carboxy-2-mercaptoethyl)-carbamoyl)-glutamic acid, a core structure found in PET radioligands targeting prostate-specific membrane antigen (PSMA) (Schwarzenboeck et al., 2017), underwent successful thiol [18F]trifluoromethylation respectively in 26 % and 10% of RCY. Radiolabeled Arg-Gly-Asp (RGD) peptides have been a focus for noninvasive assessment of angiogenesis because of their high affinity and selectivity for integrin αvβ3 (Hatley et al., 2018). The [18F]trifluoromethylation was performed with a cyclic peptide containing the RGD sequence and the radiolabeled cRGDfC and cRADfC were purified and isolated by prep-HPLC in 19% and 33% of RCY. Biodistribution studies by imaging and dissection show that [18F]CF3-cRGDfC is predominantly excreted by the hepatobiliary route and to a lesser extent by the kidneys. The absence of uptake in the bones indicated that [18F]CF3-cRGDfC is metabolically stable toward [18F]SCF3 elimination and that no [18F]F−was released (Gadais et al., 2017). Beta-amyloid peptide fragments also underwent successful [18F]trifluoromethylation in 30% of RCY.
The discovery and characterization of naturally occurring fluorinase enzymes provide an opportunity to synthesize organofluorine compounds by utilizing biocatalysts (O'Hagan and Deng, 2015). O’Hagan et al. (2002) demonstrated the application of the fluorinase enzymes in the fluorination for the first time, then in a following work, O’Hagan and co-workers radio-synthesized 5'-[18F]FDA in presence of fluorinase FlA (Martarello et al., 2003). Ang, Zhao, and co-workers employed the evolved variants, fah 2081 (A279Y) and fah2114 (F213Y, A279L), to smoothly radiosynthesize 5'-[18F]FDA, with overall radiochemical conversion more than 3-fold higher than FlA1 (Sun et al., 2016) (Figure 2B3).
During the past decades, remarkable advances have been made in the area of decarboxylative fluorination. However, these recently developed methods are mainly based on electrophilic fluorination reagents, the research on decarboxylative fluorination with nucleophilic fluorination reagents is rare (Liang et al., 2013; Brooks et al., 2014). Groves and co-workers reported manganese catalyzed decarboxylative radiofluorination and the RCCs ranged from 20 % to 50% (Huang et al., 2015) (Figure 2C1). Compared to present decarboxylative fluorination methods which are based on electrophilic fluorination reagents, the major advantage of this fluoride-based decarboxylative fluorination reaction is its applicability to 18F-labeling with [18F]fluoride. Based on their previous work, they considered the tedious azeotropic [18F]KF drying step would be eliminated by directly eluting [18F]fluoride from the ion exchange cartridge with a solution of the [Mn (salen)]OTs catalyst. Gouverneur and co-workers reported radiosynthesis of [18F]ArCF2H via Mn mediated decarboxyl-radiofluorination with [18F]fluoride (Sap et al., 2019) (Figure 2C2). This reaction is tolerated with various functional groups, such as ether, alkyl, aldehyde, ketone, pyridine, triazole, pyrazole, and dibenzofuran motifs. The higher RCCs were obtained for electron-rich arenes. Fenofibrate analog, COX-II inhibitor ZA140, and estrone analog were also successfully radiolabeled. Doyle and co-workers developed another redox-neutral decarboxylative radiofluorination of N-hydroxyphthalimide esters with [18F]KF under photoredox catalytic conditions with the typical molar activity of 36.6 GBq/μmol (Webb et al., 2020) (Figure 2C3). To stable the carbocation intermediate, the reacting carbon atom bearing bi-alkyl or aryl is necessary. A common limitation in nucleophilic fluorination methods in delivering secondary benzylic fluorides elimination to styrene byproducts (Sladojevich et al., 2013). However, less than 5% elimination products were observed in the fluorination of the secondary substrates. Gemfibrozil analog and ribose analog could be prepared in 9% and 42% of RCC. Significantly, radiosynthesis of radiolabeled ribose analogs by conventional substitution reactions was limited due to the sulfonate precursor readily decomposed at room temperature (D’Angelo and Taylor, 2016).
Difluorocarbene intermediate is a powerful tool in organic synthesis (Ni and Hu, 2014). It can readily recombine fluorine anion to prepare a trifluoromethyl group in situ (Zheng et al., 2015a; Zheng et al., 2015b; Zheng et al., 2017). Gouverneur and co-workers reported a one-step route to [18F]CF3SO2NH4 from [18F]fluoride, (triphenylphosphonio)acetate (PDFA, difluorocarbene precursor) with 1,4-diazabicyclo [2.2.2]octane bis(SO2) adduct (DABSO) or N-methylmorpholine·SO2 (NMMSO2) (SO2 source) and it applied to selective C-H bonds [18F]trifluoromethylation of peptides (Kee et al., 2020) (Figure 2D1). [18F]Trifluoromethyl radical could smoothly generate form [18F]CF3SO2NH4 in presence of Fe(III) salts and TBHP. For unmodified peptides, direct C−H bonds [18F]Trifluoromethylation selective occurred at L-tryptophan or L-tyrosine. Biologically relevant peptides, including immunomodulator thymogen (oglufanide) (Deigin et al., 2007), endomorphin-1 (a tetrapeptide related to Alzheimer’s disease) (Frydman-Marom et al., 2011), somatostatin-14 (a cyclic tetradecapeptidic hormone with a broad inhibitory effect on endocrine secretion) (Vale et al., 1972), melittin (a 26-residue venom peptide) (Raghuraman and Chattopadhyay, 2007), octreotide (an octapeptide that mimics natural somatostatin) (Pauwels et al., 2019), underwent Trp-selective [18F]trifluoromethylation; Angiotensin fragment 1–7 (a peptide with anti-inflammatory properties) (El-Hashim et al., 2012), c (RGDyK) (a peptide ligand of integrin αvβ3 receptors) (Cai et al., 2006), underwent Tyr-selective [18F]trifluoromethylation. It was worth noting that the C-H bonds [18F]trifluoromethylation of recombinant human insulin (MW = 5,808 Da) were also successful. [18F]CF3-octreotide was automated radio-synthesized with the molar activity of 0.28 GBq/μmol.
Diazo compounds are known as carbene precursors to react rapidly with transition metals to form electrophilic metal carbenoids under mild conditions (Ford et al., 2015). Doyle and co-workers developed copper-catalyzed radiofluorination of α-diazocarbonyl compounds in mild conditions with [18F]KF as a result of synthesizing α-[18F]fluorocarbonyl products with the typical molar activity of 48.1 GBq/μmol (Gray et al., 2016) (Figure 2D2). The traditional nucleophilic [18F]fluorination reaction conditions are not befitting for the radiosynthesis of most α-[18F]fluorocarbonyl targets (Liu et al., 2011). They showed the RCY of this radiofluorination protocol is significantly higher than in the previous literature. Some known radiotracers for positron emission tomography were readily accessible using this practical approach. N5-[18F]fluoroacetylornithine (N5-[18F]FAO) (Turkman et al., 2011) was labeled in 39% of RCC (only 8% of RCY by the previous SN2). N-(2-(diethylamino)ethyl)-2-[18F]fluoropropanamide ([18F]FPDA) (Liu et al., 2013) was labeled in 45% of RCC in one step (merely 3% of overall RCY with many radiochemical steps in prior method). Peptides and glycosides were also compatible with this radiofluorination.
The strategy of directly and selectively transforming C-H bonds to C-18F bonds is helpful due to the needlessness for the pre-functionalization of labeling precursors (Szpera et al., 2019). Groves, Hooker, and co-workers presented manganese porphyrin mediated direct radiofluorination of unactivated aliphatic C-H bonds with [18F]fluoride (Liu et al., 2018) (Figure 2E). Similar to the earlier mentioned protocol (Huang et al., 2015), the anion exchange cartridge was eluted by acetone/acetonitrile solution of MnIII(TPFPP)OTs. Amino acid transporter, such as ACPC, leucine, valine, tyrosine analogs and leucine containing dipeptide, Lyrica analogs (an anticonvulsant drug) (Dworkin and Kirkpatrick, 2005), amantadine analogs (an antiparkinson disease drug), ezetimibe (a cholesterol-lowering drug), flutamide (a prostate cancer drug) (Baker et al., 1967), were radiolabeled efficiently at the aliphatic C-H bonds. They hypothesized that 18F-labeled leucine and valine analogs have never been reported due to the inaccessibility of their corresponding precursors. However, C-H bonds radiofluorination occurred at the tertiary carbon atom due to the ortho-position alkyl group can stabilize the reaction intermediate. Herein, the labeling precursors with gem-dialkyl groups are required for this C-H bonds radiofluorination reaction.
Radiosynthesis of Fluoroarenes With [18F]Fluorides
Aromatic nucleophilic substitution (SNAr) reaction is a widely practiced method for the construction of [18F]fluoroarenes with [18F]fluoride (Preshlock et al., 2016). An activating group and a leaving group on the arene to stabilize the Meisenheimer complex are necessary for the highly efficient introduction of fluoride into fluoroarenes by SNAr (Bunnett and Zahler, 1951). Despite significant advances in the 18F-labeling of electron-deficient arenes, there is still a huge amount of need for efficient methods for the [18F]fluorination of electron-neutral and electron-rich arenes (Born et al., 2017). Herein, a new mechanism avoiding Meisenheimer intermediate or novel [18F]fluoroarene precursors carrying new activating and leaving groups remains to be discussed. Furthermore, novel halogen-[18F]fluorine exchange reactions and C-H bond radiofluorination will also be discussed (Figure 3).
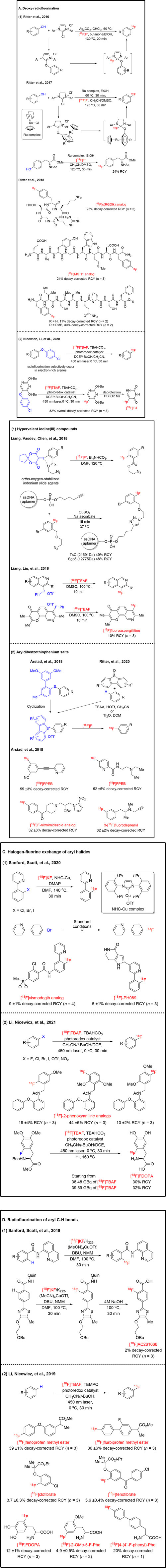
FIGURE 3. (Continued).
Phenols are frequently-used moieties in organic compounds (Qiu and Li, 2020), which makes deoxy-radiofluorination of phenols becoming an attractive strategy to achieve fluoroarenes (Tang et al., 2011). Ritter and co-workers presented a distinctive deoxy-radiofluorination method of phenols based on a concerted nucleophilic aromatic substitution (CSNAr) reaction (Neumann et al., 2016) (Figure 3A1). Compared with the traditional aromatic nucleophilic substitution (SNAr) mechanism (Bunnett and Zahler, 1951), CSNAr does not proceed via a Meisenheimer intermediate. Herein, a wide variety of functional groups including amines, amides, thioethers, and heteroarenes were tolerated for this deoxy-radiofluorination. One year later, Ritter and co-workers further utilized a ruthenium-mediated deoxy-radiofluorination of phenols (Beyzavi et al., 2017). Compared with previous work, this ruthenium-mediated deoxy-radiofluorination reaction expanded the substrate scope to even the most electron-rich phenols. Ruthenium reduced the electron density of phenols and accelerated nucleophilic aromatic substitution of phenols. They were able to perform the reaction in a fully automated mode and get the isolated protected [18F]fluorophenylalanine derivative in 24% of activity yield with the molar activity of 93 GBq/μmol. Site-specific deoxy-radiofluorination of small peptides with [18F]fluoride also had been reported by Ritter and co-workers (Rickmeier and Ritter, 2018). Small peptides that could potentially be used as PET tracers, such as the c (RGDfk) analog (an angiogenesis monitoring PET tracer) (Cai and Conti, 2013), MG 11 analog (a gastrin-releasing peptide receptor tracer) (Good et al., 2008), were successfully labeled by this protocol. In their previous work, the substrates with C-terminal free carboxylic acid suffered from low yields during radiolabeling (Neumann et al., 2016). In this work, they showed the protection of C-terminal free carboxylic acid with p-methoxybenyzl (PMB) ester effectively increased the RCY. The typical peptide was automated radio-synthesized with the molar activity of 99 GBq/μmol.
Nicewicz, Li, and co-workers demonstrated another deoxy-radiofluorination mechanism called cation-radical-accelerated SNAr (CRA- SNAr) (Tay et al., 2020) (Figure 3A2). A novel strategy for polarity-reversed photoredox catalyzed deoxy-radiofluorination of electron-rich phenol derivatives with [18F]TBAF was presented. Photoredox catalyzed deoxy-radiofluorination selectively occurred in the electron-rich arenes under mild conditions with moderate-to-excellent RCYs. Highly efficient radio-synthesized 5-[18F]fluorouracil ([18F]FU), which is an antimetabolite used to treat certain cancers (Saleem et al., 2000), was produced in two steps, including deoxy-radiofluorination and deprotection with an overall 82% of decay-corrected RCY with the molar activity of 74.7 GBq/μmol. This method was supplementary to existing ways that involve hypervalent iodoniums (Rotstein et al., 2014) and aryl nickel complexes (Hoover et al., 2016).
Hypervalent iodine (III) compounds as novel activating and leaving groups play a pivotal role in nucleophilic [18F]fluorination of non-activated arenes (Deng et al., 2019). Pike and co-workers demonstrated the first example of radiofluorination with diaryliodonium salts, whereby both electron-deficient and electron-rich arenes showed a high 18F-labeling efficiency (Pike and Aighirhio, 1995). During modifying the structure of hypervalent iodine (III) compounds, Liang, Vasdev, Chen, and co-workers utilized the ortho-effect and developed an ortho-oxygen-stabilized iodonium ylide agents (Wang et al., 2015) (Figure 3B1). Compared with Pike’s work (Pike and Aighirhio, 1995), they speculated that a secondary bonding interaction between ortho-oxygen and hypervalent iodine would provide stabilization for iodine (III) to yield thermally stable and highly reactive. The azide moiety of 18F-labeled products, the molar activity greater than 74 GBq/μmol, easily underwent [3 + 2] cycloaddition or coupling with alkyne-containing small or biological molecules, such as ssDNA aptamer TsC (21591Da) and Sgc8 (12775Da) (Jacobson et al., 2015). In previous work, TsC aptamer radiolabeled in only 1.5% of RCY. The RCY raised to 49% of RCY by using this novel method. Recently, Liang, Liu, and co-workers reported a general protocol for the preparation of [18F]fluoroisoquinolines with radiochemical conversion up to 92% with the molar activity of 56.6 GBq/μmol (Yuan et al., 2016). As proof of concept [18F]fluoroaspergillitine, a fluorinated marine natural product, was prepared in 10% of isolated radiochemical yield.
Beside the diaryliodonium salts, aryldibenzothiophenium salts can be used as another catalyzer for synthesizing non-activated arene [18F]fluorides by nucleophilic fluorination. Årstad and co-workers demonstrated regioselectively radiofluorination of dibenzothiophene sulfonium salts (prepared by biaryl thioethers via intramolecular ring-closing reaction) with [18F]fluoride afford [18F]fluoroarenes (Gendron et al., 2018) (Figure 3B2). 3-[18F]fluoro-5-(pyridine-3-ylethynyl)benzonitrile ([18F]FPEB, a metabotropic glutamate 5 receptors tracer) (Wang et al., 2007), 18F-labeled nitroimidazole analog (a hypoxia tracer), N-[2-(diethylamino)-ethyl]-5-[18F]fluoropicolinamide ([18F]P3BZA, a melanoma tracer) (Ma et al., 2019), 3-[18F]fluorodeprenyl (a monoamine oxidase B tracer) (Fowler et al., 2015), were successfully radio-synthesized by this [18F]fluorination method. 3-[18F]fluorodeprenyl was radio-synthesized with the molar activity of 17–30 GBq/μmol. To achieve radiosynthesis of [18F]FPEB, they attempted to prepare the corresponding triarylsulfonium salt but failed. For [18F]P3BZA, the reported radiosynthesis is low yielding. Followed by the new [18F]fluorination method, the RCY raised from 12% to 52%. Ritter and co-workers reported a novel site-selective late-stage aromatic [18F]fluorination method via aryl sulfonium salts (Xu et al., 2020). Significantly, they showed how electronically different dibenzothiophenes appropriately matched the electronic requirements of the arene. Heterocycles, halides, amides, and sulfonamides were tolerated for this [18F]fluorination reaction and a range of small-molecule drugs were successfully labeled.
Aryl halides are the most wildly used radiofluorination precursors due to their characteristics of being stable and synthetically accessible (Preshlock et al., 2016). Halogen-fluorine exchange reactions are limited for radiofluorination of electron-deficient arenes via SNAr (Preshlock et al., 2016). Nevertheless, it is still a huge challenge for radiofluorination of unactivated arenes via halogen-fluorine exchange. Sanford, Scott, and co-workers described the ligand-directed N-heterocyclic carbene (NHC) Cu complexes mediated radiofluorination of aryl halides with the typical molar activity of 1.6 GBq/μmol (Sharninghausen et al., 2020) (Figure 3C1). They showed that directing groups pattern on the ortho-position of halogen substituents was necessary for this reaction. These substrates with bromo-substituent on the para-position of directing groups did not afford desired products under standard conditions. Vismodegib analog, a basal cell carcinoma treatment drug (Dlugosz et al., 2012), and PH089, an MK-2 inhibitor (Anderson et al., 2007), smoothly underwent radiofluorination. Li, Nicewicz, and co-workers demonstrated a method for constructing aryl C-18F bonds through direct halogen-fluorine exchange on electron-rich arene halides under mild photoredox conditions (Chen et al., 2021) (Figure 3C2). 18F-labeled 2-phenoxyaniline analogs as translocator protein (TSPO)-specific PET tracers for neuroinflammation imaging have been investigated (Werry et al., 2019). Using their halogen-fluorine exchange method, the 18F atom was successfully introduced into potential new imaging agents targeting TSPO. This novel protocol offered an opportunity to radiosynthesis and explore a series of 18F-labeled O-methyl tyrosines as PET tracers in an MCF-7 tumor model. For clinically relevant scaling, FDA approved PET tracer [18F]FDOPA was obtained with >30% of RCY and molar activity of 1.5 GBq/μmol in 100 min by using this method. For this radiofluorination reaction, substrates with O-atom, N-atom, or S-atom at ortho- or para-position of halides were necessary.
Compare to traditional methods of radiofluorination, such as the Balz-Schiemann reaction, deoxy-fluorination, and SNAr reaction, C-H bonds radiofluorination methods are favorable due to needless pre-functionalization of the substrate (Preshlock et al., 2016; Szpera et al., 2019). Sanford, Scott, and co-workers disclosed that 8-aminoquinoline directing groups enable Cu-mediated aromatic C-H bonds activation and nucleophilic radiofluorination with [18F]KF (Lee et al., 2019) (Figure 3D1). This aromatic C-H bonds radiofluorination method was applied to a series of biologically relevant molecules [18F]AC261066, a RARβ2 agonist (Lund et al., 2005), was automated radio-synthesized in two steps, radiofluorination, and hydrolysis of the directing group, with 2% of decay-corrected RCY with the molar activity of 29.6 GBq/μmol. Li, Nicewicz, and co-workers disclosed a mild condition for direct radiofluorination of aromatic C-H bonds under organic photoredox catalyzed conditions with the typical molar activity of 51.8 GBq/μmol with 2,2,6,6-tetramethylpiperidinooxy (TEMPO) as oxidant and tetrabutylammonium [18F]fluoride ([18F]TBAF) as [18F]fluoride source (Chen et al., 2019) (Figure 3D2). Radiofluorination mainly occurred at the para-position of electron-donating groups; when the para-position was substituted, radiofluorination occurred at the ortho-position of electron-donating groups. Nonsteroidal anti-inflammatory drugs (NSAIDs) are an important class of pharmaceuticals that alleviate pain and inflammation. The NSAID derivatives (Cryer and Feldman, 1998), fenoprofen methyl ester, and flurbiprofen methyl ester were radiofluorinated in 39% and 36% of decay-corrected RCYs. Restricted by the radiolabeled method, well-studied 11C-labeled NSAID derivatives had the disadvantage of a shorter half-life than fluorine-18. The hypolipidemic agents, clofibrate and fenofibrate, were selectively fluorinated in moderate decay-corrected RCYs. [18F]FDOPA, a PD and neuroendocrine tumors PET tracer (Pretze et al., 2017), was radio-synthesized in two steps with 12% of decay-corrected RCY. Extensive and sensitive [18F]FDOPA precursors were required in published routes. The protected O-Me-ortho-tyrosine and 4-phenyl-phenylalanine were also successfully radiofluorinated, and their deprotected forms were accessed with relative ease.
Radiosynthesis of Fluoroalkenes With [18F]Fluorides
Gem-difluoroalkene moiety presents in several drug molecules, such as numerous enzyme inhibitors, due to the similar bioisosteric to a carbonyl group (Shen et al., 2014). The [18F]gem-difluoroalkenes were obtained as byproducts in radiofluorination of corresponding difluoroalkenes via an addition-elimination mechanism (Fawaz et al., 2014). Tredwell and co-workers reported the synthesis of [18F]gem-difluoroalkenes with the typical molar activity of 1.0 GBq/μmol from [18F]fluoride and fluoroalkenyl (4-methoxyphenyl)iodonium triflates (Frost et al., 2019) (Figure 4A). The [18F]gem-difluoroalkenes can be easily translated into 1,1-[18F] difluoromethylene-containing groups. This transformation supplied another method of radiosynthesis of non-benzylic geminal [18F]CF2 groups. Monofluoroalkene moiety can be used as a peptidomimetic unit in the design of protease inhibitors as well as positron emission tomography probes based on the similar charge distribution and dipole moment between amide bond and fluoroalkene moiety (Zhang et al., 2009). Xu, Hammond, and co-workers offered a reliable protocol for the synthesis of 18F-labeled monofluoroalkene via hydrogen-bonding enabled radiofluorination of ynamides (Zeng et al., 2018) (Figure 4B). They demonstrated the hydrogen-bonding network generated from hydrogen-bond-donor solvents accelerates the rate-determining proton-transfer step. To demonstrate the applicability, an 18F-labeled biologically active estrone derivative was prepared with great efficiency (Figure 4).

FIGURE 4. Radiosynthesis of fluoroalkenes.
Heteroatom-18F Bonds Formation
Expect the traditional 18F-labeling strategies of C-18F bond formation, the noncanonical strategies of hetero-18F bond formations, such as B-18F, Al-18F, Si-18F, Ga-18F, P-18F, and S-18F bonds, which show the unique properties in positron emission tomography probes design. B-18F, Al-18F, and Si-18F derivatives as PET tracers have been excellently reviewed by Gabbai and co-workers (Chansaenpak et al., 2016), and Schirrmacher, and co-workers (Wängler et al., 2012). Herein, B-18F, Al-18F, and Si-18F bond formation warrants a brief discussion. Within the Group 13 elements, B-18F derivatives are the most studied PET applications (Monzittu et al., 2018). The research on Al-18F provided the first example of a metal chelate system for [18F]fluoride capture in water (Laverman et al., 2010). However, B-18F, Al-18F, and Si-18F derivatives have obvious weaknesses (Hong et al., 2019), such as specific pH requirement (B-18F derivatives), the steric effect of bulky chelate synthons (Al-18F derivatives), limited stability and high lipophilicity (Si-18F derivatives) and potential biosafety issue due to possible metal contamination. Reid and co-workers synthesized and demonstrated 1-benzyl-4,7-dimethyl-1,4,7-triazacyclononane (BnMe2-tacn) liganded GaF3 complex is extremely stable in water (Bhalla et al., 2014). Then, they presented a simple and rapid method for 18F-labeling of [18F]GaF3(BnMe2-tacn) complex by isotopic exchange with the molar activity of 675 MBq/μmol (Monzittu et al., 2018) (Figure 5A). This 18F–19F exchange method significantly decreased the concentration of the GaF3(BnMe2-tacn) compare to the previous 18F-Cl exchange reaction (Bhalla et al., 2014). Organophosphine [18F]fluorides also had been prepared by Li, Nie, and co-workers by isotopic exchange (Hong et al., 2019). They illustrated that steric hindrance is critical for the stability of organophosphine [18F]fluorides (Figure 5B). Human serum albumin (HSA), a heat-sensitive globular protein, was radiolabeled at room temperature and applied to blood pool imaging with the molar activity of 1.1 GBq/μmol. Wu, Yang, Sharpless, and co-workers reported an ultrafast (within 30 s) isotopic exchange method for the radiosynthesis of aryl [18F]fluorosulfates (Ar-OSO2F) which was the first PET imaging application of S−18F based probes (Zheng et al., 2021) (Figure 5C). 18F-labeled olaparib analog was successfully radio-synthesized in 32% of RCY with the molar activity of 280 GBq/μmol. Aryl [18F]fluorosulfates were also successfully radio-synthesized in two modes by Chun, Hong, and co-workers (Kwon et al., 2020). The radiofluorination modes include the direct one-pot radiofluorosulfurylation of phenolic precursors (Mode 1) and the radiofluorination of isolated imidazylates (Mode 2). The radiofluorination of isolated imidazylates (Mode 2) showed higher RCYs. Both Mode 1 and 2 afforded similar molar activity. 18F-labeled acetaminophen analog was automated radio-synthesized in 9% of decay-corrected RCY with the molar activity of 42 GBq/μmol (Mode 1) and 22% of decay-corrected RCY with the molar activity of 55 GBq/μmol (Mode 2). Based on previous works, Chun, Hong, and co-workers radio-synthesized sulfamoyl [18F]fluorides by the 18F–19F exchange method (Jeon et al., 2021). 18F-labeled amoxapine derivative of sulfamoyl fluoride was automated radio-synthesized in 53% of RCY with 56 GBq/μmol (Figure 5).
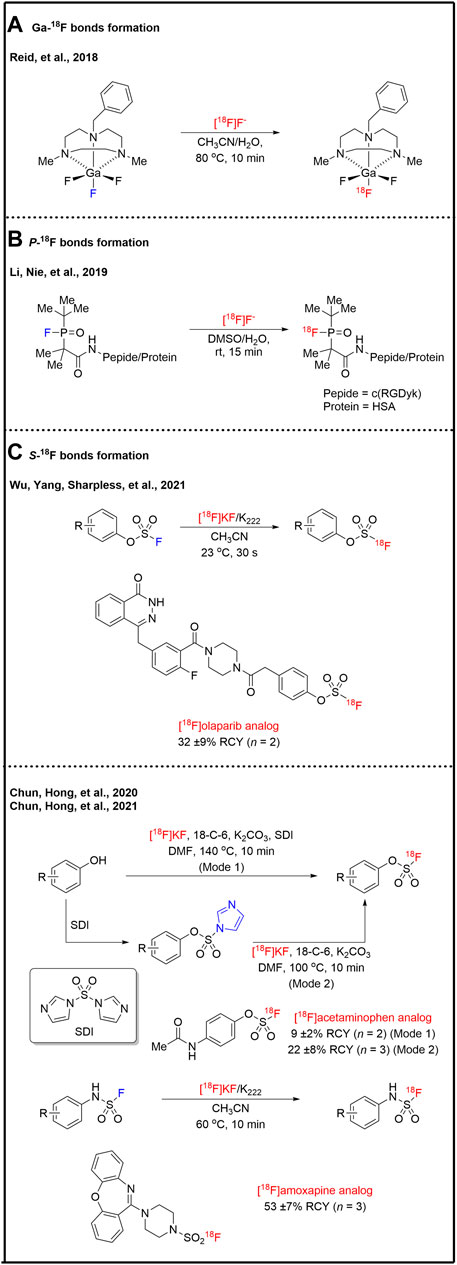
FIGURE 5. Heteroatom-18F bonds formation.
Summary
Recently, the interest in the development of novel 18F-labeled PET tracers increased rapidly. New methods, new reagents, and new structures have been investigated for synthesizing [18F]fluoroalkanes [18F]fluoroarenes [18F]fluoroalkenes, and [18F]fluorine-heteroatom containing compounds. Commercially available and inexpensive labeling precursors are beneficial for PET tracer design. Alcohols, phenols, and carboxylic acid are frequently-used moiety in natural products and pharmaceutical molecules. For this reason, deoxy-radiofluorination, decarboxylative radiofluorination, and C-H bonds radiofluorination have the advantage. Among them, C-H bonds radiofluorination has the greatest advantage, but it also has the disadvantage of poor regioselectivity. Halogen-[18F]fluorine exchange and [18F]fluorination of difluorocarbene provide reliable methods of access to [18F]fluoroalkyl groups, such as -[18F]SCF3, -[18F]CF3, et al. Innovation in reagents and structures is led by the new radiosynthesis methods. The new reagents [18F]Umemoto reagent and [18F]CF3SO2NH4, allow the introduction of [18F]trifluoromethyl into bioactive molecules and biologically relevant peptides. The new structures, [18F]fluoroalkenes, and novel [18F] fluorine-heteroatom-containing compounds have been successfully synthesized. It should also be noted that the research of PET tracer based on [18F]fluoroalkenes is rare. The application of these novel protocols accelerates the progress of PET tracer design and allows PET tracers to synthesize on a clinically relevant scale.
Author Contributions
This review was written by YW, corrected by QL, HS, and DC.
Funding
This study was funded by the National Nature Science Foundation of China (No. 82172002, 81471706, 11875114, 81871407, China), Shanghai Municipal Science and Technology Committee of Shanghai outstanding academic leaders plan (No.21XD1423500, China), Shanghai Municipal Key Clinical Specialty Project (No.SHSLCZDZK 03401) and Clinical Research Project of Zhongshan Hospital (No.2020ZSLC20, China).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Anderson, D. R., Meyers, M. J., Vernier, W. F., Mahoney, M. W., Kurumbail, R. G., Caspers, N., et al. (2007). Pyrrolopyridine Inhibitors of Mitogen-Activated Protein Kinase-Activated Protein Kinase 2 (MK-2). J. Med. Chem. 50, 2647–2654. doi:10.1021/jm0611004
Baker, J. W., Bachman, G. L., Schumacher, I., Roman, D. P., and Tharp, A. L. (1967). Synthesis and Bacteriostatic Activity of Some Nitrotrifluoro Methylanilides. J. Med. Chem. 10, 93–95. doi:10.1021/jm00313a020
Beyzavi, H., Mandal, D., Strebl, M. G., Neumann, C. N., D’Amato, E. M., Chen, J., et al. (2017). 18F-Deoxyfluorination of Phenols via Ru π-Complexes. ACS Cent. Sci. 3, 944–948. doi:10.1021/acscentsci.7b00195
Bhalla, R., Darby, C., Levason, W., Luthra, S. K., McRobbie, G., Reid, G., et al. (2014). Triaza-macrocyclic Complexes of Aluminium, Gallium and Indium Halides: fast18F and19F Incorporation via Halide Exchange under Mild Conditions in Aqueous Solution. Chem. Sci. 5, 381–391. doi:10.1039/C3SC52104D
Brooks, A. F., Topczewski, J. J., Ichiishi, N., Sanford, M. S., and Scott, P. J. H. (2014). Late-stage [18F]fluorination: New Solutions to Old Problems. Chem. Sci. 5, 4545–4553. doi:10.1039/C4SC02099E
Bunnett, J. F., and Zahler, R. E. (1951). Aromatic Nucleophilic Substitution Reactions. Chem. Rev. 49, 273–412. doi:10.1021/cr60153a002
Cai, H., and Conti, P. S. (2013). RGD-based PET Tracers for Imaging Receptor Integrin αvβ3expression. J. Label. Compd. Radiopharm. 56, 264–279. doi:10.1002/jlcr.2999
Cai, L., Lu, S., and Pike, V. W. (2008). Chemistry with [ 18 F]Fluoride Ion. Eur. J. Org. Chem. 2008, 2853–2873. doi:10.1002/ejoc.200800114
Cai, W., Zhang, X., Wu, Y., and Chen, X. (2006). A Thiol-Reactive 18F-Labeling Agent, N-[2-(4-18F-fluorobenzamido)ethyl]maleimide, and Synthesis of RDG Peptide-Based Tracer for PET Imaging of αvβ3 Integrin Expression. J. Nucl. Med. 47, 1172.
Chansaenpak, K., Vabre, B., and Gabbaï, F. P. (2016). [18F]-Group 13 Fluoride Derivatives as Radiotracers for Positron Emission Tomography. Chem. Soc. Rev. 45, 954–971. doi:10.1039/C5CS00687B
Chen, W., Huang, Z., Tay, N. E. S., Giglio, B., Wang, M., Wang, H., et al. (2019). Direct Arene C-H Fluorination with 18 F − via Organic Photoredox Catalysis. Science 364, 1170–1174. doi:10.1126/science.aav7019
Chen, W., Wang, H., Tay, N. E. S., Pistritto, V. A., Li, K.-P., Zhang, T., et al. (2021). Arene Radiofluorination Enabled by Photoredox-Mediated Halide Interconversion. Nat. Chem. 14, 216–223. doi:10.1038/s41557-021-00835-7
Cryer, B., and Feldman, M. (1998). Cyclooxygenase-1 and Cyclooxygenase-2 Selectivity of Widely Used Nonsteroidal Anti-inflammatory Drugs. Am. J. Med. 104, 413–421. doi:10.1016/S0002-9343(98)00091-6
D’Angelo, K. A., and Taylor, M. S. (2016). Borinic Acid Catalyzed Stereo- and Regioselective Couplings of Glycosyl Methanesulfonates. J. Am. Chem. Soc. 138, 11058–11066. doi:10.1021/jacs.6b06943
De Vry, J., Denzer, D., Reissmueller, E., Eijckenboom, M., Heil, M., Meier, H., et al. (2004). 3-[2-Cyano-3-(trifluoromethyl)phenoxy]phenyl-4,4,4-trifluoro-1-butanesulfonate (BAY 59-3074): A Novel Cannabinoid CB1/CB2 Receptor Partial Agonist with Antihyperalgesic and Antiallodynic Effects. J. Pharmacol. Exp. Ther. 310, 620–632. doi:10.1124/jpet.103.062836
Deigin, V. I., Semenets, T. N., Zamulaeva, I. A., Maliutina, Y. V., Selivanova, E. I., Saenko, A. S., et al. (2007). The Effects of the EW Dipeptide Optical and Chemical Isomers on the CFU-S Population in Intact and Irradiated Mice. Int. Immunopharmacol. 7, 375–382. doi:10.1016/j.intimp.2006.11.010
Deng, X., Rong, J., Wang, L., Vasdev, N., Zhang, L., Josephson, L., et al. (2019). Chemistry for Positron Emission Tomography: Recent Advances in 11 C‐, 18 F‐, 13 N‐, and 15 O‐Labeling Reactions. Angew. Chem. Int. Ed. 58, 2580–2605. doi:10.1002/anie.201805501
Dlugosz, A., Agrawal, S., and Kirkpatrick, P. (2012). Vismodegib. Nat. Rev. Drug Discov. 11, 437–438. doi:10.1038/nrd3753
Dworkin, R. H., and Kirkpatrick, P. (2005). Pregabalin. Nat. Rev. Drug Discov. 4, 455–456. doi:10.1038/nrd1756
El-Hashim, A. Z., Renno, W. M., Raghupathy, R., Abduo, H. T., Akhtar, S., and Benter, I. F. (2012). Angiotensin-(1-7) Inhibits Allergic Inflammation, via the MAS1 Receptor, through Suppression of ERK1/2- and NF-κb-dependent Pathways. Br. J. Pharmacol. 166, 1964–1976. doi:10.1111/j.1476-5381.2012.01905.x
Fawaz, M. V., Brooks, A. F., Rodnick, M. E., Carpenter, G. M., Shao, X., Desmond, T. J., et al. (2014). High Affinity Radiopharmaceuticals Based upon Lansoprazole for PET Imaging of Aggregated Tau in Alzheimer's Disease and Progressive Supranuclear Palsy: Synthesis, Preclinical Evaluation, and Lead Selection. ACS Chem. Neurosci. 5, 718–730. doi:10.1021/cn500103u
Ford, A., Miel, H., Ring, A., Slattery, C. N., Maguire, A. R., and McKervey, M. A. (2015). Modern Organic Synthesis with α-Diazocarbonyl Compounds. Chem. Rev. 115, 9981–10080. doi:10.1021/acs.chemrev.5b00121
Fowler, J. S., Logan, J., Shumay, E., Alia-Klein, N., Wang, G.-J., and Volkow, N. D. (2015). Monoamine Oxidase: Radiotracer Chemistry and Human Studies. J. Label. Compd. Radiopharm. 58, 51–64. doi:10.1002/jlcr.3247
Frost, A. B., Brambilla, M., Exner, R. M., and Tredwell, M. (2019). Synthesis and Derivatization of 1,1-[18 F]Difluorinated Alkenes. Angew. Chem. Int. Ed. 58, 472–476. doi:10.1002/anie.201810413
Frydman-Marom, A., Convertino, M., Pellarin, R., Lampel, A., Shaltiel-Karyo, R., Segal, D., et al. (2011). Structural Basis for Inhibiting β-Amyloid Oligomerization by a Non-coded β-Breaker-Substituted Endomorphin Analogue. ACS Chem. Biol. 6, 1265–1276. doi:10.1021/cb200103h
Gadais, C., Saraiva-Rosa, N., Chelain, E., Pytkowicz, J., and Brigaud, T. (2017). Tailored Approaches towards the Synthesis Ofl-S-(Trifluoromethyl)cysteine- Andl-Trifluoromethionine-Containing Peptides. Eur. J. Org. Chem. 2017, 246–251. doi:10.1002/ejoc.201601318
Gendron, T., Sander, K., Cybulska, K., Benhamou, L., Sin, P. K. B., Khan, A., et al. (2018). Ring-Closing Synthesis of Dibenzothiophene Sulfonium Salts and Their Use as Leaving Groups for Aromatic 18F-Fluorination. J. Am. Chem. Soc. 140, 11125–11132. doi:10.1021/jacs.8b06730
Good, S., Walter, M. A., Waser, B., Wang, X., Müller-Brand, J., Béhé, M. P., et al. (2008). Macrocyclic Chelator-Coupled Gastrin-Based Radiopharmaceuticals for Targeting of Gastrin Receptor-Expressing Tumours. Eur. J. Nucl. Med. Mol. Imaging 35, 1868–1877. doi:10.1007/s00259-008-0803-4
Gray, E. E., Nielsen, M. K., Choquette, K. A., Kalow, J. A., Graham, T. J. A., and Doyle, A. G. (2016). Nucleophilic (Radio)Fluorination of α-Diazocarbonyl Compounds Enabled by Copper-Catalyzed H-F Insertion. J. Am. Chem. Soc. 138, 10802. doi:10.1021/jacs.6b06770
Hatley, R. J. D., Macdonald, S. J. F., Slack, R. J., Le, J., Ludbrook, S. B., and Lukey, P. T. (2018). An αv-RGD Integrin Inhibitor Toolbox: Drug Discovery Insight, Challenges and Opportunities. Angew. Chem. Int. Ed. 57, 3298–3321. doi:10.1002/anie.201707948
Hong, H., Zhang, L., Xie, F., Zhuang, R., Jiang, D., Liu, H., et al. (2019). Rapid One-step 18F-Radiolabeling of Biomolecules in Aqueous Media by Organophosphine Fluoride Acceptors. Nat. Commun. 10, 989. doi:10.1038/s41467-019-08953-0
Hoover, A. J., Lazari, M., Ren, H., Narayanam, M. K., Murphy, J. M., van Dam, R. M., et al. (2016). A Transmetalation Reaction Enables the Synthesis of [18F]5-Fluorouracil from [18F]Fluoride for Human PET Imaging. Organometallics 35, 1008–1014. doi:10.1021/acs.organomet.6b00059
Hu, J., and Ni, C. (2014). Recent Advances in the Synthetic Application of Difluorocarbene. Synthesis 46, 842–863. doi:10.1055/s-0033-1340856
Huang, X., Liu, W., Hooker, J. M., and Groves, J. T. (2015). Targeted Fluorination with the Fluoride Ion by Manganese-Catalyzed Decarboxylation. Angew. Chem. Int. Ed. 54, 5241–5245. doi:10.1002/anie.201500399
Jacobson, O., Yan, X., Niu, G., Weiss, I. D., Ma, Y., Szajek, L. P., et al. (2015). PET Imaging of Tenascin-C with a Radiolabeled Single-Stranded DNA Aptamer. J. Nucl. Med. 56, 616–621. doi:10.2967/jnumed.114.149484
Jelinski, M., Hamacher, K., and Coenen, H. H. (2001). Direct 18F-Substitution of Hydroxy Groups in Peptides Using Nonafluorobutane-I-sulfonyl[18F]fluoride. J. Label. Compd. Radiopharm. 44, S151–S153. doi:10.1002/jlcr.2580440153
Jeon, M. H., Kwon, Y.-D., Kim, M. P., Torres, G. B., Seo, J. K., Son, J., et al. (2021). Late-Stage 18F/19F Isotopic Exchange for the Synthesis of 18F-Labeled Sulfamoyl Fluorides. Org. Lett. 23, 2766–2771. doi:10.1021/acs.orglett.1c00671
Kee, C. W., Tack, O., Guibbal, F., Wilson, T. C., Isenegger, P. G., Imiołek, M., et al. (2020). 18F-Trifluoromethanesulfinate Enables Direct C-H 18F-Trifluoromethylation of Native Aromatic Residues in Peptides. J. Am. Chem. Soc. 142, 1180–1185. doi:10.1021/jacs.9b11709
Khotavivattana, T., Verhoog, S., Tredwell, M., Pfeifer, L., Calderwood, S., Wheelhouse, K., et al. (2015). 18 F‐Labeling of Aryl‐SCF 3 , ‐OCF 3 and ‐OCHF 2 with [ 18 F]Fluoride. Angew. Chem. Int. Ed. 54, 9991–9995. doi:10.1002/anie.201504665
Kwon, Y.-D., Jeon, M. H., Park, N. K., Seo, J. K., Son, J., Ryu, Y. H., et al. (2020). Synthesis of 18F-Labeled Aryl Fluorosulfates via Nucleophilic Radiofluorination. Org. Lett. 22, 5511–5516. doi:10.1021/acs.orglett.0c01868−5516
Laverman, P., McBride, W. J., Sharkey, R. M., Eek, A., Joosten, L., Oyen, W. J. G., et al. (2010). A Novel Facile Method of Labeling Octreotide with 18F-Fluorine. J. Nucl. Med. 51, 454–461. doi:10.2967/jnumed.109.066902
Lee, S. J., Makaravage, K. J., Brooks, A. F., Scott, P. J. H., and Sanford, M. S. (2019). Copper‐Mediated Aminoquinoline‐Directed Radiofluorination of Aromatic C−H Bonds with K 18 F. Angew. Chem. Int. Ed. 58, 3119–3122. doi:10.1002/anie.201812701
Levin, M. D., Chen, T. Q., Neubig, M. E., Hong, C. M., Theulier, C. A., Kobylianskii, I. J., et al. (2017). A Catalytic Fluoride-Rebound Mechanism for C(sp 3 )-CF 3 Bond Formation. Science 356, 1272–1276. doi:10.1126/science.aan1411
Liang, T., Neumann, C. N., and Ritter, T. (2013). Introduction of Fluorine and Fluorine-Containing Functional Groups. Angew. Chem. Int. Ed. 52, 8214–8264. doi:10.1002/anie.201206566
Liu, H., Liu, S., Miao, Z., Jiang, H., Deng, Z., Hong, X., et al. (2013). A Novel Aliphatic 18F-Labeled Probe for PET Imaging of Melanoma. Mol. Pharm. 10, 3384–3391. doi:10.1021/mp400225s
Liu, S., Shen, B., T. Chin, F., and Cheng, Z. (2011). Recent Progress in Radiofluorination of Peptides for PET Molecular Imaging. Cos 8, 584–592. doi:10.2174/157017911796117197
Liu, W., Huang, X., Placzek, M. S., Krska, S. W., McQuade, P., Hooker, J. M., et al. (2018). Site-selective 18F Fluorination of Unactivated C-H Bonds Mediated by a Manganese Porphyrin. Chem. Sci. 9, 1168–1172. doi:10.1039/c7sc04545j
Lund, B. W., Piu, F., Gauthier, N. K., Eeg, A., Currier, E., Sherbukhin, V., et al. (2005). Discovery of a Potent, Orally Available, and Isoform-Selective Retinoic Acid β2 Receptor Agonist. J. Med. Chem. 48, 7517–7519. doi:10.1021/jm050891r
Ma, X., Wang, S., Wang, S., Liu, D., Zhao, X., Chen, H., et al. (2019). Biodistribution, Radiation Dosimetry, and Clinical Application of a Melanin-Targeted PET Probe, 18F-P3bza, in Patients. J. Nucl. Med. 60, 16–22. doi:10.2967/jnumed.118.209643
Martarello, L., Schaffrath, C., Deng, H., Gee, A. D., Lockhart, A., and O'Hagan, D. (2003). The First Enzymatic Method for C-18F Bond Formation: the Synthesis of 5-[18f]-Fluoro-5-Deoxyadenosine for Imaging with PET. J. Label. Cpd. Radiopharm. 46, 1181–1189. doi:10.1002/jlcr.779
Martinez, H., Rebeyrol, A., Nelms, T. B., and Dolbier, W. R. (2012). Impact of Fluorine Substituents on the Rates of Nucleophilic Aliphatic Substitution and β-elimination. J. Fluor. Chem. 135, 167–175. doi:10.1016/j.jfluchem.2011.10.008
Monzittu, F. M., Khan, I., Levason, W., Luthra, S. K., McRobbie, G., and Reid, G. (2018). Rapid Aqueous Late‐Stage Radiolabelling of [GaF 3 (BnMe 2 ‐tacn)] by 18 F/19 F Isotopic Exchange: Towards New PET Imaging Probes. Angew. Chem. Int. Ed. 57, 6658–6661. doi:10.1002/anie.201802446
Neumann, C. N., Hooker, J. M., and Ritter, T. (2016). Concerted Nucleophilic Aromatic Substitution with 19F− and 18F−. Nature 534, 369–373. doi:10.1038/nature17667
Nielsen, M. K., Ugaz, C. R., Li, W., and Doyle, A. G. (2015). PyFluor: A Low-Cost, Stable, and Selective Deoxyfluorination Reagent. J. Am. Chem. Soc. 137, 9571–9574. doi:10.1021/jacs.5b06307
O'Hagan, D., Schaffrath, C., Cobb, S. L., Hamilton, J. T. G., and Murphy, C. D. (2002). Biosynthesis of an Organofluorine Molecule. Nature 416, 279. doi:10.1038/416279a
O’Hagan, D., and Deng, H. (2015). Enzymatic Fluorination and Biotechnological Developments of the Fluorinase. Chem. Rev. 115, 634–649. doi:10.1021/cr500209t
Pauwels, E., Cleeren, F., Tshibangu, T., Koole, M., Serdons, K., Dekervel, J., et al. (2019). Al18F-NOTA-octreotide: First Comparison with 68Ga-DOTATATE in a Neuroendocrine Tumour Patient. Eur. J. Nucl. Med. Mol. Imaging 46, 2398–2399. doi:10.1007/s00259-019-04425-1
Pike, V. W., and Aigbirhio, F. I. (1995). Reactions of Cyclotron-Produced [18F]fluoride with Diaryliodonium Salts-A Novel Single-step Route to No-Carrier-Added [18]fluoroarenes. J. Chem. Soc. Chem. Commun. 2215, 2215–2216. doi:10.1039/C39950002215
Preshlock, S., Tredwell, M., and Gouverneur, V. (2016). 18F-Labeling of Arenes and Heteroarenes for Applications in Positron Emission Tomography. Chem. Rev. 116, 719–766. doi:10.1021/acs.chemrev.5b00493
Pretze, M., Franck, D., Kunkel, F., Foßhag, E., Wängler, C., and Wängler, B. (2017). Evaluation of Two Nucleophilic Syntheses Routes for the Automated Synthesis of 6-[18F]fluoro-L-DOPA. Nucl. Med. Biol. 45, 35–42. doi:10.1016/j.nucmedbio.2016.10.007
Qiu, Z., and Li, C.-J. (2020). Transformations of Less-Activated Phenols and Phenol Derivatives via C-O Cleavage. Chem. Rev. 120, 10454–10515. doi:10.1021/acs.chemrev.0c00088
Raghuraman, H., and Chattopadhyay, A. (2007). Melittin: a Membrane-Active Peptide with Diverse Functions. Biosci. Rep. 27, 189–223. doi:10.1007/s10540-006-9030-z
Rickmeier, J., and Ritter, T. (2018). Site-Specific Deoxyfluorination of Small Peptides with [18 F]Fluoride. Angew. Chem. Int. Ed. 57, 14207–14211. doi:10.1002/anie.201807983
Rotstein, B. H., Stephenson, N. A., Vasdev, N., and Liang, S. H. (2014). Spirocyclic Hypervalent Iodine(iii)-Mediated Radiofluorination of Non-activated and Hindered Aromatics. Nat. Commun. 5, 4365. doi:10.1038/ncomms5365
Saleem, A., Yap, J., Osman, S., Brady, F., Lucas, S. V., Jones, T., et al. (2000). Modulation of Fluorouracil Tissue Pharmacokinetics by Eniluracil: In-Vivo Imaging of Drug Action. Lancet 355, 2125–2131. doi:10.1016/S0140-6736(00)02380-1
Sap, J. B. I., Wilson, T. C., Kee, C. W., Straathof, N. J. W., Ende, C. W. a., Mukherjee, P., et al. (2019). Synthesis of 18F-Difluoromethylarenes Using Aryl Boronic Acids, Ethyl Bromofluoroacetate and [18F]fluoride. Chem. Sci. 10, 3237–3241. doi:10.1039/c8sc05096a
Schwarzenboeck, S. M., Rauscher, I., Bluemel, C., Fendler, W. P., Rowe, S. P., Pomper, M. G., et al. (2017). PSMA Ligands for PET Imaging of Prostate Cancer. J. Nucl. Med. 58, 1545–1552. doi:10.2967/jnumed.117.191031
Sharninghausen, L. S., Brooks, A. F., Winton, W. P., Makaravage, K. J., Scott, P. J. H., and Sanford, M. S. (2020). NHC-copper Mediated Ligand-Directed Radiofluorination of Aryl Halides. J. Am. Chem. Soc. 142, 7362–7367. doi:10.1021/jacs.0c02637
Shen, X., Liu, Q., Ni, C., and Hu, J. (2014). Nucleophilic Difluoro(phenylsulfonimidoyl)methylation of Unactivated Alkyl Bromides with PhSO(NTBS)CF2H: Facile Entry Intogem-Difluoroalkenes. Chin. J. Chem. 32, 703–708. doi:10.1002/cjoc.201400403
Sladojevich, F., Arlow, S. I., Tang, P., and Ritter, T. (2013). Late-Stage Deoxyfluorination of Alcohols with Phenofluor. J. Am. Chem. Soc. 135, 2470–2473. doi:10.1021/ja3125405
Sood, D. E., Champion, S., Dawson, D. M., Chabbra, S., Bode, B. E., Sutherland, A., et al. (2020). Deoxyfluorination with CuF 2 : Enabled by Using a Lewis Base Activating Group. Angew. Chem. Int. Ed. 59, 8460–8463. doi:10.1002/anie.202001015
Suehiro, M., Yang, G., Torchon, G., Ackerstaff, E., Humm, J., Koutcher, J., et al. (2011). Radiosynthesis of the Tumor Hypoxia Marker [18F]TFMISO via O-[18F]trifluoroethylation Reveals a Striking Difference between Trifluoroethyl Tosylate and Iodide in Regiochemical Reactivity toward Oxygen Nucleophiles. Bioorg. Med. Chem. 19, 2287–2297. doi:10.1016/j.bmc.2011.02.026
Sun, H., Yeo, W. L., Lim, Y. H., Chew, X., Smith, D. J., Xue, B., et al. (2016). Directed Evolution of a Fluorinase for Improved Fluorination Efficiency with a Non-native Substrate. Angew. Chem. Int. Ed. 55, 14277–14280. doi:10.1002/anie.201606722
Szpera, R., Moseley, D. F. J., Smith, L. B., Sterling, A. J., and Gouverneur, V. (2019). The Fluorination of C−H Bonds: Developments and Perspectives. Angew. Chem. Int. Ed. 58, 14824–14848. doi:10.1002/anie.201814457
Tang, P., Wang, W., and Ritter, T. (2011). Deoxyfluorination of Phenols. J. Am. Chem. Soc. 133, 11482–11484. doi:10.1021/ja2048072
Tay, N. E. S., Chen, W., Levens, A., Pistritto, V. A., Huang, Z., Wu, Z., et al. (2020). 19F- and 18F-Arene Deoxyfluorination via Organic Photoredox-Catalysed Polarity-Reversed Nucleophilic Aromatic Substitution. Nat. Catal. 3, 734–742. doi:10.1038/s41929-020-0495-0
Tsui, G. C., and Hu, J. (2019). Organofluorine Chemistry. Asian J. Org. Chem. 8, 566–567. doi:10.1002/ajoc.201900271
Turkman, N., Gelovani, J. G., and Alauddin, M. M. (2011). Radiosynthesis of N5-[18F]fluoroacetylornithine (N5-[18F]FAO) for PET Imaging of Ornithine Decarboxylase (ODC) in Malignant Tumors. J. Label. Compd. Radiopharm. 54, 33–37. doi:10.1002/jlcr.1799
Vale, W., Grant, G., Rivier, J., Monahan, M., Amoss, M., Blackwell, R., et al. (1972). Synthetic Polypeptide Antagonists of the Hypothalamic Luteinizing Hormone Releasing Factor. Science 176, 933–934. doi:10.1126/science.176.4037.933
van der Born, D., Pees, A., Poot, A. J., Orru, R. V. A., Windhorst, A. D., and Vugts, D. J. (2017). Fluorine-18 Labelled Building Blocks for PET Tracer Synthesis. Chem. Soc. Rev. 46, 4709–4773. doi:10.1039/c6cs00492j
Verhoog, S., Kee, C. W., Wang, Y., Khotavivattana, T., Wilson, T. C., Kersemans, V., et al. (2018). 18F-Trifluoromethylation of Unmodified Peptides with 5-18F-(Trifluoromethyl)dibenzothiophenium Trifluoromethanesulfonate. J. Am. Chem. Soc. 140, 1572–1575. doi:10.1021/jacs.7b10227
Wang, J.-Q., Tueckmantel, W., Zhu, A., Pellegrino, D., and Brownell, A.-L. (2007). Synthesis and Preliminary Biological Evaluation of 3-[18F]fluoro-5-(2-Pyridinylethynyl)benzonitrile as a PET Radiotracer for Imaging Metabotropic Glutamate Receptor Subtype 5. Synapse 61, 951–961. doi:10.1002/syn.20445
Wang, L., Jacobson, O., Avdic, D., Rotstein, B. H., Weiss, I. D., Collier, L., et al. (2015). Ortho-Stabilized18F-Azido Click Agents and Their Application in PET Imaging with Single-Stranded DNA Aptamers. Angew. Chem. Int. Ed. 54, 12777–12781. doi:10.1002/anie.201505927
Wängler, C., Kostikov, A., Zhu, J., Chin, J., Wängler, B., and Schirrmacher, R. (2012). Silicon-[18F]Fluorine Radiochemistry: Basics, Applications and Challenges. Appl. Sci. 2, 277–302. doi:10.3390/app2020277
Webb, E. W., Park, J. B., Cole, E. L., Donnelly, D. J., Bonacorsi, S. J., Ewing, W. R., et al. (2020). Nucleophilic (Radio)Fluorination of Redox-Active Esters via Radical-Polar Crossover Enabled by Photoredox Catalysis. J. Am. Chem. Soc. 142, 9493–9500. doi:10.1021/jacs.0c03125
Werry, E. L., Bright, F. M., Piguet, O., Ittner, L. M., Halliday, G. M., Hodges, J. R., et al. (2019). Recent Developments in TSPO PET Imaging as A Biomarker of Neuroinflammation in Neurodegenerative Disorders. Ijms 20, 3161. doi:10.3390/ijms20133161
Wu, J., Zhao, Q., Wilson, T. C., Verhoog, S., Lu, L., Gouverneur, V., et al. (2019). Synthesis and Reactivity of α‐Cumyl Bromodifluoromethanesulfenate: Application to the Radiosynthesis of [ 18 F]ArylSCF 3. Angew. Chem. Int. Ed. 58, 2413–2417. doi:10.1002/anie.201813708
Xu, P., Zhao, D., Berger, F., Hamad, A., Rickmeier, J., Petzold, R., et al. (2020). Site‐Selective Late‐Stage Aromatic [ 18 F]Fluorination via Aryl Sulfonium Salts. Angew. Chem. Int. Ed. 59, 1956–1960. doi:10.1002/anie.201912567
Yuan, Z., Cheng, R., Chen, P., Liu, G., and Liang, S. H. (2016). Efficient Pathway for the Preparation of Aryl(isoquinoline)Iodonium(III) Salts and Synthesis of Radiofluorinated Isoquinolines. Angew. Chem. Int. Ed. 55, 11882–11886. doi:10.1002/anie.201606381
Zeng, X., Li, J., Ng, C. K., Hammond, G. B., and Xu, B. (2018). (Radio)fluoroclick Reaction Enabled by a Hydrogen-Bonding Cluster. Angew. Chem. Int. Ed. 57, 2924–2928. doi:10.1002/anie.201711341
Zhang, W., Huang, W., and Hu, J. (2009). Highly Stereoselective Synthesis of Monofluoroalkenes from α-Fluorosulfoximines and Nitrones. Angew. Chem. Int. Ed. 48, 9858–9861. doi:10.1002/anie.200905077
Zheng, J., Cheng, R., Lin, J.-H., Yu, D.-H., Ma, L., Jia, L., et al. (2017). An Unconventional Mechanistic Insight into SCF3Formation from Difluorocarbene: Preparation of18F-Labeled α-SCF3Carbonyl Compounds. Angew. Chem. Int. Ed. 56, 3196–3200. doi:10.1002/anie.201611761
Zheng, J., Lin, J.-H., Deng, X.-Y., and Xiao, J.-C. (2015b). 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU)-Promoted Decomposition of Difluorocarbene and the Subsequent Trifluoromethylation. Org. Lett. 17, 532–535. doi:10.1021/ol503548s
Zheng, J., Wang, L., Lin, J. H., Xiao, J. C., and Liang, S. H. (2015a). Difluorocarbene‐Derived Trifluoromethylthiolation and [ 18 F]Trifluoromethylthiolation of Aliphatic Electrophiles. Angew. Chem. Int. Ed. 54, 13236–13240. doi:10.1002/anie.201505446
Keywords: fluorination, fluoroalkylation, 18 F-radiolabeling, PET radiotracers, PET imaging
Citation: Wang Y, Lin Q, Shi H and Cheng D (2022) Fluorine-18: Radiochemistry and Target-Specific PET Molecular Probes Design. Front. Chem. 10:884517. doi: 10.3389/fchem.2022.884517
Received: 26 February 2022; Accepted: 19 May 2022;
Published: 29 June 2022.
Edited by:
Zonghua Luo, ShanghaiTech University, ChinaReviewed by:
Jie Tong, Yale University, United StatesLin Qiu, Washington University in St. Louis, United States
Tianyu Huang, Washington University in St. Louis, United States
Copyright © 2022 Wang, Lin, Shi and Cheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongcheng Shi, c2hpLmhvbmdjaGVuZ0B6cy1ob3NwaXRhbC5zaC5jbg==; Dengfeng Cheng, Y2hlbmcuZGVuZ2ZlbmdAenMtaG9zcGl0YWwuc2guY24=