 Marina Porras
Marina Porras Concepción C. González
Concepción C. González Alicia Boto
Alicia Boto
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Chem. , 18 May 2022
Sec. Organic Chemistry
Volume 10 - 2022 | https://doi.org/10.3389/fchem.2022.884124
This article is part of the Research Topic New Hypervalent Iodine Reagents for Oxidative Coupling - Volume II View all 8 articles
Hypervalent iodine reagents have been applied in many metal-free, efficient synthesis of natural products and other bioactive compounds. In particular, treatment of alcohols, acetals and acids with hypervalent iodine reagents and iodine results in O-radicals that can undergo a β-scission reaction. Under these oxidative conditions, derivatives of amino acids, peptides or carbohydrates are converted into cationic intermediates, which can subsequently undergo inter- or intramolecular addition of nucleophiles. Most reported papers describe the addition of oxygen nucleophiles, but this review is focused on the addition of carbon, nitrogen and phosphorous nucleophiles. The resulting products (nucleoside and alkaloid analogs, unnatural amino acids, site-selectively modified peptides) are valuable intermediates or analogs of bioactive compounds.
Hypervalent iodine reagents have proven very useful for the synthesis of natural products and other bioactive compounds (Dohi and Kita, 2016; Wang, 2021). A variety of methodologies have been developed, in many cases combining the hypervalent iodine reagents with other compounds, such as iodine, organic peroxides, TEMPO, and organic photosensitizers or metal catalysts in catalytic photoredox processes (Singh and Wirth, 2021; Le Du et al., 2021; Zhdankin, 2020; Chen et al., 2020; Wang and Studer, 2017; Yoshimura and Zhdankin, 2016; Wang and Liu, 2016; Zhdankin and Stang, 2008; 2002).
The use of metal-free procedures is particularly important for the synthesis of bioactive products, to avoid undesired contamination of the product, especially when large-scale synthesis is needed for bioactivity assays or industrial production (Dohi and Kita, 2016). Among these metal-free methodologies, the combination of hypervalent iodine reagents and iodine (Suárez reaction) is quite interesting for its operational simplicity, low reagent toxicity, reagent degradation during aqueous work-up, and easy product purification. This method has been used to generate O- and N-radicals that can undergo different reactions depending on the substrate and reaction conditions, mainly hydrogen abstraction and scission of the Cα,Cβ-bond (β-fragmentation) (Stella, 2001; Suárez, 2001; Li et al., 2010; Wang, 2021).
Our group and others have described different applications of the β-scission of O-radicals, generated from alcohols, acetals or acids under Suárez conditions, to the preparation of bioactive products or their analogs (Saavedra et al., 2020; Cuevas et al., 2021 and references cited therein). However, in these protocols a further development was introduced: coupling these oxidative scission processes to the addition of carbon, nitrogen, phosphorous, oxygen, hydrogen, sulfur and other nucleophiles (Boto, Gallardo et al, 2006; 2007a; Boto, Hernández et al, 2008a,b; 2009; Batchu et al, 2014; Carro et al, 2017; Saavedra et al., 2018; Santana and González, 2020). In effect, the scission generates a C-radical that can be readily oxidized to a cationic intermediate when the latter is stabilized by adjacent groups. This is the case for tertiary C-radicals, or those adjacent to nitrogen or electron-rich oxygen functionalities (Boto et al., 1999–2004; 2005a; 2007a; 2007b; 2007c; 2007d; Francisco et al., 2001; Romero-Estudillo, 2013, 2015a, 2015b; Kiyokawa et al., 2017; André-Joyaux et al., 2019). When the substrates are aminoacids or β-hydroxyamines, an intermediate iminium ion is formed, while carbohydrate substrates afford oxycarbenium ion intermediates. These cationic species may then undergo nucleophilic addition; most reports describe the addition of oxygen nucleophiles (Chai et al., 1998a; 2005; Francisco et al., 2001; Boto et al., 2005a; 2007a; 2008a; Miguélez et al., 2012; Kiyokawa et al., 2018), but this minireview will focus on carbon, nitrogen and phosphorous nucleophiles. Since several transformations are carried out consecutively, with no need to purify the intermediates, these one-pot radical-polar crossover reactions save time, materials and energy with respect to the original conditions.
A selection of these one-pot scission-oxidation-addition of C, N and P nucleophiles methodologies is presented in this review, as well as the known or potential bioactivities of the products thus obtained.
One of the first applications of this process was the generation of aryl glycines, some of which displayed cytotoxic properties (Boto, Gallardo et al, 2007a; 2006). As shown in conversion 1→ 6 (Figure 1), treatment of serine derivatives 1 with hypervalent iodine reagents (such as diacetoxyiodobenzene, DIB) and iodine, under irradiation with visible light, generated an O-radical 2 that underwent β-scission to give a C-radical 3. The latter reacted with iodine or with the hypervalent iodine reagent to give an unstable intermediate 4 that was transformed into an α-acetoxyglycine 5. On treatment with a Lewis acid an acyliminium ion 6 was generated, that reacted with electron-rich arenes, to afford arylglycines such as 7 in good to excellent yields. Arylglycines are components of antibiotics such as nocardicins and vancomycin, anti-neurodegenerative agents and alkaloids (Williams and Hendrix, 1992; Boto, Gallardo et al, 2007a).
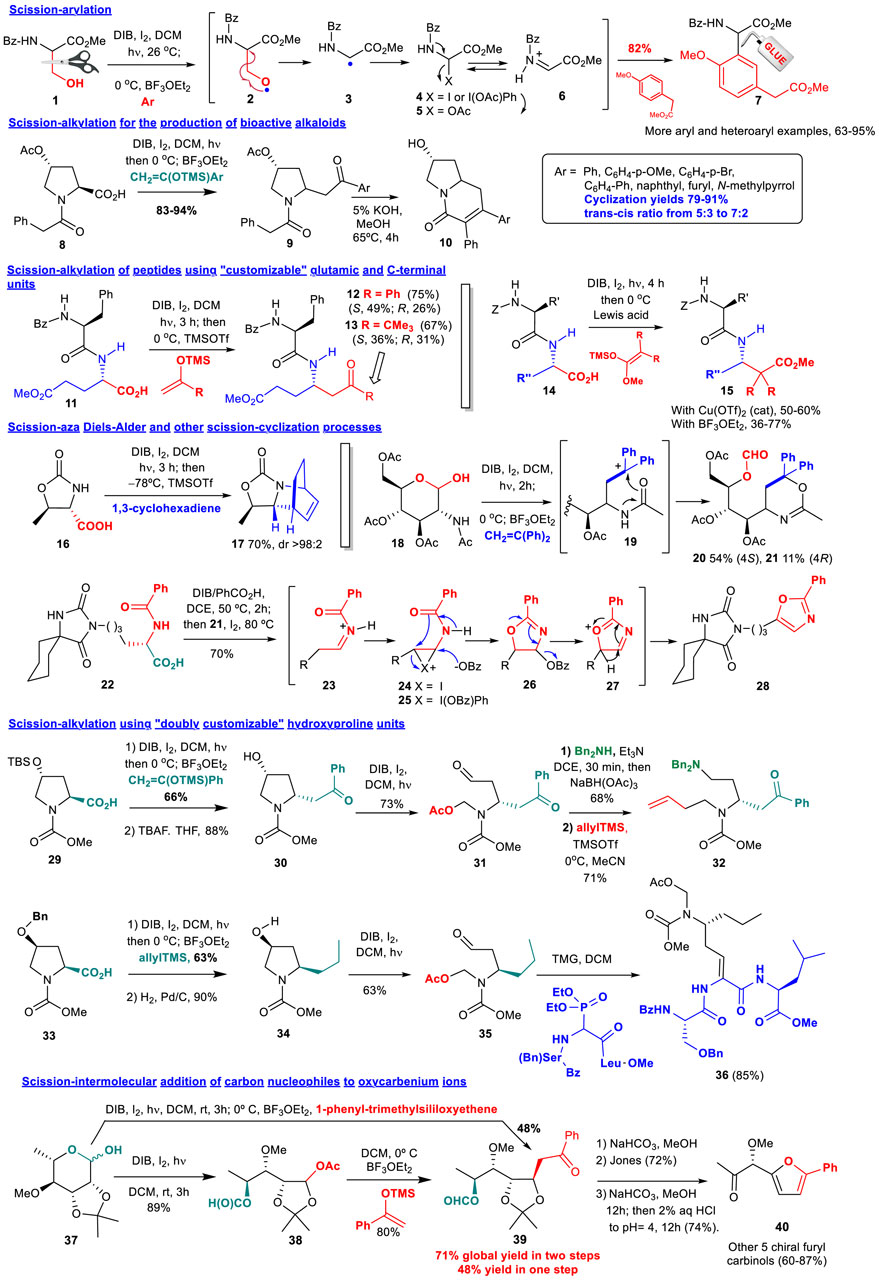
FIGURE 1. Scission-addition of C-nucleophiles.
A related scission-alkylation reaction was used to prepare analogues of cytotoxic indolizidine alkaloids (Miguélez et al., 2013a). Thus, proline amides 8 underwent decarboxylation and the addition of silyl enol ethers derived from aryl methyl ketones, to give the substituted pyrrolidines 9. Cyclization under basic conditions afforded the two isomers of the desired bicyclic systems 10, which were separated and tested. Some of these alkaloid analogues displayed a promising activity, but interestingly, some monocyclic derivatives 9 also did, which could be useful to determine SAR relationships.
A variation of the previous scission-alkylation methodology was applied to the site-selective modification of peptides using “customizable” glutamic acid (Saavedra et al., 2012c) or C-terminal residues (Saavedra et al., 2009). In both cases, an oxidative radical decarboxylation took place, followed in the first case (conversion 11→ 12/13) by the addition of silyl enol ethers to give derivatives 12 or 13 in good global yields. In that way, an ordinary α,α-unit was converted into α,γ-peptide hybrids, which have elicited interest for their antimicrobial, antitumour, antihypertensive, and anti-Alzheimer properties, as well as their superior resistance to protease degradation (Ordóñez and Cativiela, 2007; Hernández et al., 2017).
In the second example, the substrate 14 was decarboxylated and subjected to the addition of silyl ketenes, to give substituted α,ß-peptide hybrids such as compound 15. These hybrids had unusual conformations, which could be used for drug or catalyst design (Saavedra et al., 2009; 2012a; 2012b). In addition, many α,β-hybrids have displayed promising activities such as the antitumour dipeptide bestatin (Ubenimex). Moreover, they are more resistant to degradation by peptidases, as evidenced with a series of α,β-peptide bradykinin cleavage inhibitors, whose half-life was greatly increased with respect to the α,α-analogues (Bauvois and Dauzonne, 2006; Aguilar et al., 2007).
The scission reaction can also be followed by a cycloaddition reaction. Thus, when substrate 16 was decarboxylated and treated with different dienes, it provided cycloaddition products such as 17 in good yields and excellent stereoselectivities (Boto and Romero-Estudillo, 2011). Other addition-cyclization reactions have been also described, such as the transformation of the α,ß-amino sugar 18 into the oxazines 20 and 21, through intramolecular cyclization of cationic intermediate 19 (Boto, Gallardo and Alvarez, 2012). In another example, the one-pot conversion of an amino acid into an oxazol (transformation 22→ 28), the reagent (diacetoxyiodo)benzene was transformed in situ into other (diacyliodo)benzenes; then, the substrate 22 was added and underwent an oxidative radical scission to give an acyliminium intermediate 23. This ion isomerized to an enamide, which reacted with iodine (or the hypervalent iodine reagent) to afford an intermediate 24, which experienced an intramolecular cyclization with opening of the halonium ring. The halogen group was then replaced by the acyloxy moiety (benzoate in the example) to give the oxazolidine 26. However, the reaction went on, with extrusion of the benzoate, formation of a cationic intermediate 27 and aromatization, providing the oxazole 28 in 70% global yield (Romero-Estudillo et al., 2014). These heterocycles can be found in many bioactive peptides, and are considered privileged structures (Boto, González et al, 2021).
Two scission reactions can be carried out in “doubly customizable” hydroxyproline units (Hernández et al., 2021). Thus, using the hydroxyproline substrate 29, a decarboxylation-alkylation was carried out, to give a 2-substituted pyrrolidine in good yield and excellent 2,4-cis stereoselectivity. After deprotection of the 4-hydroxy group, the resultant pryrrolidine 30 underwent a second O-radical scission, to afford compound 31, which presented two new chains which could be manipulated independently (conversion 31→ 32). Thus, the α-chain was subjected to a reductive amination, while the addition of allylTMS to the N,O-acetal gave an olefinic chain, that could be further diversified using olefin metathesis (Saavedra et al., 2019; Hernández et al., 2021).
In a second example (conversion 33→ 23), the decarboxylation-allylation of compound 33, followed by hydrogenation, afforded a 2-substituted pyrrolidine 34. The stereogenic center at C-4 determined, as before, the configuration at C-2, and thus, compounds 30 and 34 had opposite C2-stereochemistry. This result was translated to the second scission product 35, which was used in a Horner-Wadsworth-Emmons reaction to afford the peptide 36. This peptide has a dehydroaminoacid unit, which is often used in peptides to provide rigidity and a better interaction with biological targets. Dehydropeptides have displayed antimicrobial, antitumour and phytotoxic activities (Jiang et al., 2015; Siodlak, 2015).
The addition of C-nucleophiles to oxycarbenium ions derived from the scission of carbohydrates has also been studied, as shown by the conversion 37→ 39 (Boto, Hernández et al, 2007c). In this case, however, the one-pot procedure was less efficient than the two-step process, where the intermediate acetate 38 was isolated and then treated with a Lewis acid and the C-nucleophile. Other examples were also studied, with similar results. The resultant products were converted in a few steps and with high optical purity into chiral furyl carbinols, which are useful precursors of bioactive products, such as the selective antifungal populacandin D (Balachari and O’Doherty, 2000), or KDO, a vital component of the Gram-negative bacteria cell wall (Martin and Zinke, 1991).
The scission-addition of nitrogen nucleophiles provides other families of potentially bioactive products, such as iminosugars and nucleoside analogues (Figure 2). Many iminosugars have displayed a potent activity as glycosidase inhibitors, while many nucleoside analogues have been used as antimicrobial or antitumour agents (Horne et al., 2011; Drug Bank, 2021). Given the potential in this field, the main goal of our research has been focused on synthesizing new classes of iminosugar derivatives starting from carbohydrates.

FIGURE 2. Scission-addition of N- and P-nucleophiles.
As shown in the conversion 41/42→ 47/48 (Figure 2), the cleavage of the carbohydrate C1-C2 bond forms a C-radical 43 that reacts with iodine or the hypervalent iodine reagent to afford a halogenated intermediate 44 (Francisco et al., 2001). Indeed, some iododerivatives (X = I) are stable enough to be isolated and characterized (Francisco et al., 1998b). But in many cases, the intermediate is rapidly converted into an acetoxy compound (eg product 45). This acetoxyderivative 45 is in equilibrium with its oxycarbenium ion 46, which can undergo the intramolecular addition of nitrogen nucleophiles, to give nitrogen heterocycles such as 47 and 48. A related conversion of aldoses to ketoses 49→ 50 also took place in satisfactory yield (Santana et al., 2013). This methodology, which represents an efficient alternative to the Lobry de Bruyn-Alberda van Ekestein alkaline isomerization, used readily available aldoses as starting material. Despite the steric hindrance to nucleophile approach, the reaction proceeded smoothly, affording ketoses such as 50, where a new quaternary center with a desired configuration has been incorporated.
Following this strategy, a variety of novel polyhydroxylated heterocyclic compounds have been recently prepared. For example, cyclic guanidines derived from carbohydrates were synthesized as shown in transformation 51→ 52/53 (Santana and González, 2020). The bidentate nucleophilic character of the guanidinium group opened the possibility to differentiate between the two non-equivalent nitrogen atoms, which made the proposed methodology more versatile, as endocyclic or exocyclic guanidines of different sizes (5-, 6-, 7- or 8-membered rings) could be generated in good yields (85% in the example shown). The guanidinium moiety appears in natural products with potent biological activities, such as saxitoxin, tetrodotoxin or crambescin, and also drugs such as antiplasmodium compounds (Alonso-Moreno et al., 2014; Perry et al., 2019).
This versatile strategy has also provided a battery of sugar tetrazoles and benzimidazoles (Paz et al., 2012; André-Joyaux et al., 2019). Remarkably, in the key cyclization step these aromatic nitrogen heterocycles were used for the first time as nucleophiles. The method efficiently afforded polycyclic systems such as 55 and 57 in good yields. Tetrazole and benzimidazole are privileged structures, and tetrazoles are found in many fungicides. These heterocycles have been used to protect crops since the 1970s, due to their low or moderate toxicity, broad fungicide spectrum and potent systemic action (Pernak et al., 2015). Benzimidazoles have displayed potent antimicrobial properties, and carbohydrate-bound benzoimidazoles (pseudonucleosides) are antiviral agents (Verma et al., 2016).
Nitrogen bases can also add to oxycarbenium ions, as shown in the conversions 58→ 59 and 60→ 61 (Boto, Hernández et al, 2007b). Both purine and pyrimidine bases reacted in good to excellent yields. The acyclic nucleosides have elicited much interest as antivirals (de Clercq, 2005). In addition, the reaction of nitrogen bases with acyliminium ions, as in transformation 62→ 63 (Boto, Hernández and Hernández, 2010a), afforded azanucleosides, a class of compounds with antimicrobial, anticancer and enzyme inhibitor properties (Hernández and Boto, 2014). Interestingly, when the scission reaction was carried out in acetonitrile instead of the more usual, less polar solvent dichloromethane (conversions 64→ 66), the initial iminium ion isomerized to an enamine derivative such as 65, which reacted with iodine and then with the nucleophile to give the final iodoazanucleosides (eg compound 66; Boto, Hernández and Hernández, 2010b).
The decarboxylation of acids to give tertiary cationic species which reacted with the solvent (acetonitrile) to afford Ritter-type products (conversion 67→ 70), was studied by Kiyokawa et al. (2017) giving hindered amines such as 70a and 70b in good yields. A decarboxylative amination where boron Lewis catalysts were used instead of iodine was reported by Narobe, König et al. (2022).
Finally, the one-pot scission-addition of phosphorous nucleophiles was studied for the site-selective modification of peptides (Boto, Gallardo et al, 2005b; Saavedra et al, 2012d; 2018). After the oxidative radical scission a phosphite was added, and an aminophosphonate such as compound 72 was formed. A Horner-Wadsworth-Emmons reaction with different aldehydes was carried out to give a peptide with a dehydroaminoacid unit, which increased the system rigidity. The reaction took place in good yields and a high Z stereoselectivity, even when interior positions were functionalized. The introduction of dehydroaminoacids into peptides can improve their interaction with their biological targets and their resistance to proteases. Therefore, dehydroaminoacids are components in a variety of bioactive natural peptides and drugs (Jiang et al., 2015; Siodlak, 2015).
The tandem decarboxylation-phosphorylation process was recently adapted by Viveros-Ceballos et al. (2021) to prepare tetrahydroisoquinoline-3-phosphonic acids, which are key components of enzyme inhibitors and other bioactive products. In another example, nucleotide analogs were formed by a decarboxylation-phosphorylation reaction (Miguélez et al., 2013b). Although the authors do not report the bioactivity for this set of compounds, clearly this methodology would be valuable for the preparation of chemical libraries for structure-activity relationships.
The use of metal-free methodologies for the synthesis of bioactive products is a hot area in pharmaceutical chemistry, and hypervalent iodine reagents have proven very useful to achieve this goal. Moreover, among metal-free synthetic methodologies, particularly interesting are those which carry out the transformation of readily available natural products into added-value bioactive compounds using one-pot ‘cut and paste’ processes.
This mireview focuses on the methods which use the generation and scission of O-radicals in their key step, followed by the addition of carbon, nitrogen and phosphorous nucleophiles. These methodologies include scission-alkylation, scission-arylation, fragmentation-Diels Alder and other inter- and intramolecular cyclization processes, scission-Ritter, fragmentation-addition of nucleobases, and scission-phosphorylation. A range of products can be obtained from simple substrates such as organic acid and alcohols, amino acids, carbohydrates and peptides. Among those interesting for their potential bioactivity are alkaloid and nucleoside analogues, heterocycles, aminophosphonates and other amino acid analogues, and site-selective modified peptides and peptide hybrids.
In most cases, these strategies afforded high added-value products in good to excellent yields, operational simplicity and easy work-up and product purification. These processes offer a quick route to families of many bioactive products (such as glycosidase inhibitors and other antimicrobial or cytotoxic compounds) and also to some new compounds containing privileged structures, whose biological properties deserve to be further studied. This could be a goal for the next future in this area.
The work carried out up to now highlights the opportunities offered by these sustainable metal-free, one-pot methodologies, where many other substrates and nucleophiles remain to be explored. Since structural diversity often translates into biological diversity, future efforts in the topic could provide new promising bioactive compounds and drug candidates.
MP collected the references and wrote a draft of the manuscript. AB, DH and CG reviewed the manuscript and the references and polished the article. AB modified part of Figures 1, 2, and DH and CG modified part of Figure 2.
This work was partly financed by RETOS project SELECTFIGHT (PID2020-116688RB-C21) from the Spanish Ministry of Science and Innovation (Plan Nacional de I+D) with European Funds for Regional Development-FEDER, and well as by project APOGEO (MAC/1.1.b/226, Cooperation Program INTERREG-MAC 2014‐2020, with European FEDER funds. DH also acknowledges her current contract (TRANSALUDAGRO) financed by Cabildo de Tenerife, Program TF INNOVA 2016-21 (with MEDI & FDCAN Funds). MP thanks predoctoral grants from Gobierno de Canarias (Convocatoria Tesis 2019, REF. TESIS2020010041) and Ministerio de Ciencia, Innovación y Universidades, Spain (FPU grants, REF. FPU19/02589).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We also acknowledge support of the publication fee by the University of La Laguna and CSIC Open Access Publication Support Initiative-through its Unit of Information Resources for Research (URICI). MP is currently a student of the PhD Program “Ciencias Médicas y Farmacéuticas, Desarrollo y Calidad de Vida” of Universidad de La Laguna (ULL).
Aguilar, M.-I., Purcell, A. W., Devi, R., Lew, R., Rossjohn, J., Smith, A. I., et al. (2007). β-Amino Acid-Containing Hybrid Peptides-New Opportunities in Peptidomimetics. Org. Biomol. Chem. 5, 2884–2890. doi:10.1039/B708507A
Alonso-Moreno, C., Antiñolo, A., Carrillo-Hermosilla, F., and Otero, A. (2014). Guanidines: From Classical Approaches to Efficient Catalytic Syntheses. Chem. Soc. Rev. 43, 3406–3425. doi:10.1039/C4CS00013G
André-Joyaux, E., Santana, A. G., and González, C. C. (2019). Synthesis of Chiral Polyhydroxylated Benzimidazoles by a Tandem Radical Fragmentation/Cyclization Reaction: A Straight Avenue to Fused Aromatic-Carbohydrate Hybrids. J. Org. Chem. 84, 506–515. doi:10.1021/acs.joc.8b01988
Balachari, D., and O'Doherty, G. A. (2000). Sharpless Asymmetric Dihydroxylation of 5-Aryl-2-Vinylfurans: Application to the Synthesis of the Spiroketal Moiety of Papulacandin D. Org. Lett. 2, 863–866. doi:10.1021/ol0000253
Batchu, V. R., Romero-Estudillo, I., Boto, A., and Miguélez, J. (2014). Metal-free, One-Pot Conversion of Proline Derivatives into 2-Aryl-3-Iodo Pyrrolidines by a Sequential Scission-Iodination-Arylation Process. Org. Biomol. Chem. 12, 9547–9556. doi:10.1039/c4ob01372g
Bauvois, B., and Dauzonne, D. (2006). Aminopeptidase-N/CD13 (EC 3.4.11.2) Inhibitors: Chemistry, Biological Evaluations, and Therapeutic Prospects. Med. Res. Rev. 26, 88–130. doi:10.1002/med.20044
Boto, A., Gallardo, J. A., and Álvarez, E. (2012). One-Pot Conversion of Serine Derivatives and Amino Sugars into Oxazine Derivatives of γ-Aryl-γ-(hydroxy)amines. Eur. J. Org. Chem. 2012, 391–397. doi:10.1002/ejoc.201101274
Boto, A., Gallardo, J. A., Hernández, D., and Hernández, R. (2007a). Synthesis of Unnatural Amino Acids from Serine Derivatives by β-Fragmentation of Primary Alkoxyl Radicals. J. Org. Chem. 72, 7260–7269. doi:10.1021/jo071155t
Boto, A., Gallardo, J. A., Hernández, R., Ledo, F., Muñoz, A., Murguía, J. R., et al. (2006). Genotoxic Activity of Halogenated Phenylglycine Derivatives. Bioorg. Med. Chem. Lett. 16, 6073–6077. doi:10.1016/j.bmcl.2006.08.111
Boto, A., Gallardo, J. A., Hernández, R., and Saavedra, C. J. (2005b). One-pot Synthesis of α-amino Phosphonates from α-amino Acids and β-amino Alcohols. Tetrahedron Lett. 46, 7807–7811. doi:10.1016/j.tetlet.2005.09.019
Boto, A., González, C. C., Hernández, D., Romero-Estudillo, I., and Saavedra, C. J. (2021). Site-Selective Modification of Peptide Backbones. Org. Chem. Front. 8, 6720–6759. doi:10.1039/d1qo00892g
Boto, A., Hernández, D., Hernández, R., and Álvarez, E. (2007b). One-Pot Synthesis of Acyclic Nucleosides from Carbohydrate Derivatives, by Combination of Tandem and Sequential Reactions. J. Org. Chem. 72, 9523–9532. doi:10.1021/jo701608p
Boto, A., Hernández, D., and Hernández, R. (2008a). Efficient Conversion of Carbohydrates into 1-C-Alditols: Application to the Synthesis of Chiral γ-Substituted Butenolides and Bicyclic Alkaloid Analogues. J. Org. Chem. 73, 5287–5297. doi:10.1021/jo800478a
Boto, A., Hernández, D., and Hernández, R. (2008b). One-pot Synthesis of Azanucleosides from Proline Derivatives. Tetrahedron Lett. 49, 455–458. doi:10.1016/j.tetlet.2007.11.113
Boto, A., Hernández, D., and Hernández, R. (2009). Enantiopure Alkaloid Analogues and Iminosugars from Proline Derivatives: Stereocontrol in Sequential Processes. Tetrahedron Lett. 50, 3974–3977. doi:10.1016/j.tetlet.2009.04.082
Boto, A., Hernández, D., Hernández, R., Montoya, A., and Suárez, E. (2007d). Synthesis of Alkaloid Analogues from β-Amino Alcohols by β-Fragmentation of Primary Alkoxyl Radicals. Eur. J. Org. Chem. 2007, 325–334. doi:10.1002/ejoc.200600720
Boto, A., Hernández, D., and Hernández, R. (2010b). One-Pot Conversion of Proline Derivatives into Iodinated Iminosugar-Based Nucleosides, Useful Precursors of Highly Functionalized Nucleoside Analogues. Eur. J. Org. Chem. 2010, 6633–6642. doi:10.1002/ejoc.201000997
Boto, A., Hernández, D., and Hernández, R. (2010a). One-Pot Synthesis of Azanucleosides from Proline Derivatives - Stereoselectivity in Sequential Processes. Eur. J. Org. Chem. 2010, 3847–3857. doi:10.1002/ejoc.201000360
Boto, A., Hernández, D., and Hernández, R. (2007c). Short and Efficient Synthesis of Chiral Furyl Carbinols from Carbohydrates. Org. Lett. 9, 1721–1724. doi:10.1021/ol070412d
Boto, A., Hernández, D., Hernández, R., and Suárez, E. (2003). β-Fragmentation of Primary Alkoxyl Radicals versus Hydrogen Abstraction: Synthesis of Polyols and α,ω-Differently Substituted Cyclic Ethers from Carbohydrates. J. Org. Chem. 68, 5310–5319. doi:10.1021/jo034442f
Boto, A., Hernández, R., de León, Y., Murguía, J. R., and Rodríguez-Afonso, A. (2005a). Synthesis of Functionalized Nitrogen Heterocycles by Radical Decarboxylation of β- and γ-Amino Acids. Eur. J. Org. Chem. 2005, 673–682. doi:10.1002/ejoc.200400698
Boto, A., Hernández, R., de León, Y., and Suárez, E. (2001a). Synthesis of 2,3-Disubstituted Pyrrolidines and Piperidines via One-Pot Oxidative Decarboxylation−β-Iodination of Amino Acids. J. Org. Chem. 66, 7796–7803. doi:10.1021/jo015877a
Boto, A., Hernández, R., Montoya, A., and Suárez, E. (2002). One-pot Synthesis of Aryl Glycines and Other Unnatural Amino Acids from Serine Derivatives. Tetrahedron Lett. 43, 8269–8272. doi:10.1016/S0040-4039(02)02023-3
Boto, A., Hernández, R., Montoya, A., and Suárez, E. (2004). Synthesis of Alkaloids from Aminol Derivatives by β-fragmentation of Primary Alkoxyl Radicals. Tetrahedron Lett. 45, 1559–1563. doi:10.1016/j.tetlet.2003.12.003
Boto, A., Hernández, R., and Suárez, E. (2001b). Synthesis of Acyclic Nucleosides and Other C-1 Substituted Alditols from Carbohydrates Using a Tandem Alkoxyl Radical β-fragmentation-nucleophilic Addition. Tetrahedron Lett. 42, 9167–9170. doi:10.1016/S0040-4039(01)02016-0
Boto, A., Hernández, R., and Suárez, E. (2000c). Synthesis of Alkaloids from Amino Acids via N-Acyliminium Ions Generated by One-Pot Radical Decarboxylation-Oxidation. Tetrahedron Lett. 41, 2899–2902. doi:10.1016/S0040-4039(00)00306-3
Boto, A., Hernández, R., and Suárez, E. (2000b). Tandem Oxidative Radical Decarboxylation-β-Iodination of Amino Acids. Application to the Synthesis of Chiral 2,3-disubstituted Pyrrolidines. Tetrahedron Lett. 41, 2495–2498. doi:10.1016/S0040-4039(00)00188-X
Boto, A., Hernández, R., and Suárez, E. (2000a). Tandem Radical Decarboxylation‑Oxidation of Amino Acids: A Mild and Efficient Method for the Generation of N-Acyliminium Ions and Their Nucleophilic Trapping. J. Org. Chem. 65, 4930–4937. doi:10.1021/jo000356t
Boto, A., Hernaández, R., and Suaárez, E. (1999). Oxidative Decarboxylation of α-amino Acids: A Mild and Efficient Method for the Generation of N-Acyliminium Ions. Tetrahedron Lett. 40, 5945–5948. doi:10.1016/S0040-4039(99)01180-6
Boto, A., and Romero-Estudillo, I. (2011). One-Pot Stereoselective Synthesis of 1,2-Amino Alcohol Derivatives. Org. Lett. 13, 3426–3429. doi:10.1021/ol201173a
Carro, C., Romero, I., and Boto, A. (2017). Microwave versus Conventional Light Activation of O-Radical Scission Processes. Eur. J. Org. Chem. 2017, 373–380. doi:10.1002/ejoc.201601034
Chai, C. L. L., Elix, J. A., and Huleatt, P. B. (2005). The Synthetic Versatility of Alkoxycarbonyl- and Hydroxymethyl-Piperazine-2,5-Diones. Tetrahedron 61, 8722–8739. doi:10.1016/j.tet.2005.06.084
Chen, C., Wang, X., and Yang, T. (2020). Recent Synthetic Applications of the Hypervalent Iodine(III) Reagents in Visible-Light-Induced Photoredox Catalysis. Front. Chem. 8, 551159. doi:10.3389/fchem.2020.551159
Cuevas, F., Saavedra, C. J., Romero‐Estudillo, I., Boto, A., Ordóñez, M., and Vergara, I. (2021). Structural Diversity Using Hyp "Customizable Units" : Proof‐of‐Concept Synthesis of Sansalvamide‐Related Antitumoral Peptides. Eur. J. Org. Chem. 2021, 933–943. doi:10.1002/ejoc.202001427
de Clercq, E. (2005). Antiviral Drug Discovery and Development: Where Chemistry Meets with Biomedicine. Antiviral Res. 67, 56–75. doi:10.1016/j.antiviral.2005.05.001
Dohi, T., and Kita, Y. (2016). Hypervalent Iodine-Induced Oxidative Couplings (New Metal-free Coupling Advances and Their Applications in Natural Product Syntheses). Top. Curr. Chem. 373, 1–23. doi:10.1007/128_2016_667
Drug Bank (2021). Iminosugar Lists in. Alberta,Canada: Drug Bank. Available at: https://go.drugbank.com/categories/DBCAT001529.
Francisco, C. G., Freire, R., González-Martín, C. C., León, E. I., Riesco-Fagundo, C., and Suárez, E. (2001). Fragmentation of Carbohydrate Anomeric Alkoxy Radicals. Synthesis of Polyhydroxy Piperidines and Pyrrolidines Related to Carbohydrates. J. Org. Chem. 66, 1861–1866. doi:10.1021/jo0057452
Francisco, C. G., González-Martín, C. C., and Suárez, E. (1998a). Fragmentation of Carbohydrate Anomeric Alkoxy Radicals. A New General Method for the Synthesis of Alduronic Acid Lactones. J. Org. Chem. 63, 2099–2109. doi:10.1021/jo971323p
Francisco, C. G., González-Martín, C. C., and Suárez, E. (1998b). Synthesis of α-Iodoalkyl Esters and α-Iodoalkyl Carbonates from Carbohydrates. Formation of Convenient Chiral Synthetic Intermediates. J. Org. Chem. 63, 8092–8093. doi:10.1021/jo981587r
Hernández, D., Boto, A., Guzmán, D., and Alvarez, E. (2017). Metal-free, Direct Conversion of α-amino Acids into α-keto γ-amino Esters for the Synthesis of α,γ-peptides. Org. Biomol. Chem. 15, 7736–7742. doi:10.1039/c7ob02033c
Hernández, D., and Boto, A. (2014). Nucleoside Analogues: Synthesis and Biological Properties of Azanucleoside Derivatives. Eur. J. Org. Chem. 2014, 2201–2220. doi:10.1002/ejoc.201301731
Hernández, D., Carro, C., and Boto, A. (2021). “Doubly Customizable” Unit for the Generation of Structural Diversity: From Pure Enantiomeric Amines to Peptide Derivatives. J. Org. Chem. 86, 2796–2809. doi:10.1021/acs.joc.0c02751
Horne, G., Wilson, F. X., Tinsley, J., Williams, D. H., and Storer, R. (2011). Iminosugars Past, Present and Future: Medicines for Tomorrow. Drug Discov. Today 16, 107–118. doi:10.1016/j.drudis.2010.08.017
Jiang, J., Ma, Z., and Castle, S. L. (2015). Bulky α,β-dehydroamino Acids: Their Occurrence in Nature, Synthesis, and Applications. Tetrahedron 71, 5431–5451. doi:10.1016/j.tet.2015.06.001
Kiyokawa, K., Okumatsu, D., and Minakata, S. (2018). Hypervalent Iodine(III)-Mediated Decarboxylative Acetoxylation at Tertiary and Benzylic Carbon Centers. Beilstein J. Org. Chem. 14, 1046–1050. doi:10.3762/bjoc.14.92
Kiyokawa, K., Watanabe, T., Fra, L., Kojima, T., and Minakata, S. (2017). Hypervalent Iodine(III)-Mediated Decarboxylative Ritter-type Amination Leading to the Production of α-Tertiary Amine Derivatives. J. Org. Chem. 82, 11711–11720. doi:10.1021/acs.joc.7b01202
Le Du, E., Garreau, M., and Waser, J. (2021). Small Peptide Diversification through Photoredox-Catalyzed Oxidative C-Terminal Modification. Chem. Sci. 12, 2467–2473. doi:10.1039/d0sc06180h
Li, W., Gan, J., and Fan, R. (2010). Base-promoted Selective β-fragmentation of Homoallylamines. Tetrahedron Lett. 51, 4275–4277. doi:10.1016/j.tetlet.2010.06.031
Martin, S. F., and Zinke, P. W. (1991). The Furan Approach to Oxygenated Natural Products. Total Synthesis of (+)-KDO. J. Org. Chem. 56, 6600–6606. doi:10.1021/jo00023a028
Miguélez, J., Batchu, V. R., and Boto, A. (2012). Stereoselective Conversion of Sugar Derivatives into C-Nucleosides. J. Org. Chem. 77, 7652–7658. doi:10.1021/jo301031t
Miguélez, J., Boto, A., Marín, R., and Díaz, M. (2013a). Simplification of Antitumoral Phenanthroindolizidine Alkaloids: Short Synthesis of Cytotoxic Indolizidinone and Pyrrolidine Analogs. Eur. J. Med. Chem. 66, 540–554. doi:10.1016/j.ejmech.2013.06.009
Miguélez-Ramos, J., Batchu, V. R., and Boto, A. (2013b). Tuning the Stereoselectivity in One-Pot Scission/Addition Processes: Synthesis of Azanucleotide Analogues from Proline Derivatives. Eur. J. Org. Chem. 2013, 846–852. doi:10.1002/ejoc.201201443
Narobe, R., Murugesan, K., Schmid, S., and König, B. (2022). Decarboxylative Ritter-type Amination by Cooperative Iodine (I/III)─Boron Lewis Acid Catalysis. ACS Catal. 12, 809–817. doi:10.1021/acscatal.1c05077
Ordóñez, M., and Cativiela, C. (2007). Stereoselective Synthesis of γ-Amino Acids. Tetrahedron: Asymmetry 18, 3–99. doi:10.1016/j.tetasy.2006.12.001
Paz, N. R., Santana, A. G., Francisco, C. G., Suárez, E., and González, C. C. (2012). Synthesis of Tetrazole-Fused Glycosides by a Tandem Fragmentation-Cyclization Reaction. Org. Lett. 14, 3388–3391. doi:10.1021/ol3013638
Pernak, J., Markiewicz, B., Łęgosz, B., Walkiewicz, F., Gwiazdowski, R., and Praczyk, T. (2015). Known Triazole Fungicides - a New Trick. RSC Adv. 5, 9695–9702. doi:10.1039/C4RA12160K
Perry, D. L., Roberts, B. F., Debevec, G., Michaels, H. A., Chakrabarti, D., and Nefzi, A. (2019). Identification of Bis-Cyclic Guanidines as Antiplasmodial Compounds from Positional Scanning Mixture-Based Libraries. Molecules 24, 1100. doi:10.3390/molecules24061100
Romero-Estudillo, I., Batchu, V. R., and Boto, A. (2014). One-Pot Conversion of Amino Acids into 2,5-Disubstituted Oxazoles: No Metals Needed. Adv. Synth. Catal. 356, 3742–3748. doi:10.1002/adsc.201400496
Romero-Estudillo, I., and Boto, A. (2013). Creating Diversity by Site-Selective Peptide Modification: A Customizable Unit Affords Amino Acids with High Optical Purity. Org. Lett. 15, 5778–5781. doi:10.1021/ol402800a
Romero-Estudillo, I., and Boto, A. (2015a). Domino Process Achieves Site-Selective Peptide Modification with High Optical Purity. Applications to Chain Diversification and Peptide Ligation. J. Org. Chem. 80, 9379–9391. doi:10.1021/acs.joc.5b00932
Romero-Estudillo, I., Saavedra, C., Boto, A., and Álvarez, E. (2015b). Site-selective Modification of Peptides: From “Customizable Units” to Novelα-Aryl Andα-Alkyl glycine Derivatives, and Components of Branched Peptides. Biopolymers 104, 650–662. doi:10.1002/bip.22642
Saavedra, C. J., Hernández, R., Boto, A., and Álvarez, E. (2009). Catalytic, One-Pot Synthesis of β-Amino Acids from α-Amino Acids. Preparation of α,β-Peptide Derivatives. J. Org. Chem. 74, 4655–4665. doi:10.1021/jo9004487
Saavedra, C. J., Boto, A., and Hernández, R. (2012d). "Customizable" Units in Di- and Tripeptides: Selective Conversion into Substituted Dehydroamino Acids. Org. Lett. 14, 3788–3791. doi:10.1021/ol301676z
Saavedra, C. J., Boto, A., Hernández, R., Miranda, J. I., and Aizpurua, J. M. (2012a). Conformation and Chiral Effects in α,β,α-Tripeptides. J. Org. Chem. 77, 5907–5913. doi:10.1021/jo300892u
Saavedra, C. J., Boto, A., and Hernández, R. (2012b). Preparation of Modified Peptides: Direct Conversion of α-amino Acids into β-amino Aldehydes. Org. Biomol. Chem. 10, 4448–4461. doi:10.1039/c2ob25433f
Saavedra, C. J., Boto, A., and Hernández, R. (2012c). Synthesis of α,γ-Peptide Hybrids by Selective Conversion of Glutamic Acid Units. Org. Lett. 14, 3542–3545. doi:10.1021/ol301552c
Saavedra, C. J., Carro, C., Hernández, D., and Boto, A. (2019). Conversion of "Customizable Units" into N-Alkyl Amino Acids and Generation of N-Alkyl Peptides. J. Org. Chem. 84, 8392–8410. doi:10.1021/acs.joc.9b00114
Saavedra, C. J., Cuevas, F., Romero‐Estudillo, I., and Boto, A. (2020). Synthesis of Diketopiperazine Scaffolds with Tailored N‐ and α‐Chains by Selective Modification of Customizable Units. Adv. Synth. Catal. 362, 3158–3169. doi:10.1002/adsc.202000470
Saavedra, C. J., Hernández, D., and Boto, A. (2018). Metal-Free, Site-Selective Peptide Modification by Conversion of "Customizable" Units into β-Substituted Dehydroamino Acids. Chem. Eur. J. 24, 599–607. doi:10.1002/chem.201703758
Santana, A. G., and González, C. C. (2020). Tandem Radical Fragmentation/Cyclization of Guanidinylated Monosaccharides Grants Access to Medium-Sized Polyhydroxylated Heterocycles. Org. Lett. 22, 8492–8495. doi:10.1021/acs.orglett.0c03091
Santana, A. G., Paz, N. R., Francisco, C. G., Suárez, E., and González, C. C. (2013). Synthesis of Branched Iminosugars through a Hypervalent Iodine(III)-Mediated Radical-Polar Crossover Reaction. J. Org. Chem. 78, 7527–7543. doi:10.1021/jo401041s
Singh, F. V., and Wirth, T. (2021). Hypervalent Iodine Chemistry and Light: Photochemical Reactions Involving Hypervalent Iodine Chemistry. Arkivoc 2021, 12–47. doi:10.24820/ark.5550190.p011.483
Siodłak, D. (2015). α,β-Dehydroamino Acids in Naturally Occurring Peptides. Amino Acids 47, 1–17. doi:10.1007/s00726-014-1846-4
Stella, L. (2001). “Nitrogen-Centered Radicals,” in Radicals in Organic Synthesis. Editors P. Renaud, and M. Sibi (Weinheim: Wiley VCH), Vol. 2, 407–426. 3-527-301 60-7. Chapter 5.1.
Suárez, E. (2001). “β-Fragmentation of Alkoxyl Radicals: Synthetic Applications,” in Radicals in Organic Synthesis. Editors P. Renaud, and M. Sibi (Weinheim: Wiley VCH), Vol. 2, 440–454. 3-527-301 60-7. Chapter 5.3.
Verma, N., Singh, R. B., Srivastava, S., and Dubey, P. (2016). Benzimidazole: A Plethro of Biological Load. J. Chem. Pharm. Res. 8 (3), 365–374.
Viveros-Ceballos, J. L., Matías-Valdez, L. A., Sayago, F. J., Cativiela, C., and Ordóñez, M. (2021). New Approaches towards the Synthesis of 1,2,3,4-tetrahydroisoquinoline-3-Phosphonic Acid (TicP). Amino Acids 53, 451–459. doi:10.1007/s00726-021-02962-4
Wang, L., and Liu, J. (2016). Synthetic Applications of Hypervalent Iodine(III) Reagents Enabled by Visible Light Photoredox Catalysis. Eur. J. Org. Chem. 2016, 1813–1824. doi:10.1002/ejoc.201501490
Wang, X., and Studer, A. (2017). Iodine(III) Reagents in Radical Chemistry. Acc. Chem. Res. 50, 1712–1724. doi:10.1021/acs.accounts.7b00148
Wang, Z. (2021). Innovation of Hypervalent(III) Iodine in the Synthesis of Natural Products. New J. Chem. 45, 509–516. doi:10.1039/d0nj05078d
Williams, R. M., and Hendrix, J. A. (1992). Asymmetric Synthesis of Arylglycines. Chem. Rev. 92, 889–917. doi:10.1021/cr00013a007
Yoshimura, A., and Zhdankin, V. V. (2016). Advances in Synthetic Applications of Hypervalent Iodine Compounds. Chem. Rev. 116, 3328–3435. doi:10.1021/acs.chemrev.5b00547
Zhdankin, V. V. (2020). Hypervalent Iodine Compounds: Reagents of the Future. Arkivoc 2020, 1–11. doi:10.24820/ark.5550190.p011.145
Zhdankin, V. V., and Stang, P. J. (2008). Chemistry of Polyvalent Iodine. Chem. Rev. 108, 5299–5358. doi:10.1021/cr800332c
Keywords: hypervalent iodine, O-radicals, one-pot processes, metal-free, radical-ion crossover, peptides, nucleosides, alkaloids
Citation: Porras M, Hernández D, González CC and Boto A (2022) “Cut and Paste” Processes in the Search of Bioactive Products: One-Pot, Metal-free O-Radical Scission-Oxidation-Addition of C, N or P-Nucleophiles. Front. Chem. 10:884124. doi: 10.3389/fchem.2022.884124
Received: 25 February 2022; Accepted: 05 April 2022;
Published: 18 May 2022.
Edited by:
Toshifumi Dohi, Ritsumeikan University, JapanCopyright © 2022 Porras, Hernández, González and Boto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alicia Boto, YWxpY2lhQGlwbmEuY3NpYy5lcw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.