 Robin Huber1,2
Robin Huber1,2 Alexey Koval3
Alexey Koval3 Laurence Marcourt1,2
Laurence Marcourt1,2 Margaux Héritier1,2
Margaux Héritier1,2 Sylvain Schnee4
Sylvain Schnee4 Emilie Michellod4
Emilie Michellod4 Leonardo Scapozza1,2
Leonardo Scapozza1,2 Vladimir L. Katanaev3,5*†
Vladimir L. Katanaev3,5*† Jean-Luc Wolfender1,2
Jean-Luc Wolfender1,2 Katia Gindro4*†
Katia Gindro4*† Emerson Ferreira Queiroz1,2*†
Emerson Ferreira Queiroz1,2*†- 1School of Pharmaceutical Sciences, University of Geneva, CMU, Geneva, Switzerland
- 2Institute of Pharmaceutical Sciences of Western Switzerland, University of Geneva, CMU, Geneva, Switzerland
- 3Department of Cell Physiology and Metabolism, Translational Research Centre in Oncohaematology, Faculty of Medicine, University of Geneva, CMU, Geneva, Switzerland
- 4Mycology Group, Research Department Plant Protection, Agroscope, Nyon, Switzerland
- 5Institute of Life Sciences and Biomedicine, Far Eastern Federal University, Vladivostok, Russia
The Wnt signaling pathway controls multiple events during embryonic development of multicellular animals and is carcinogenic when aberrantly activated in adults. Breast cancer and triple-negative breast cancer (TNBC) in particular depend upon Wnt pathway overactivation. Despite this importance, no Wnt pathway-targeting drugs are currently available, which necessitates novel approaches to search for therapeutically relevant compounds targeting this oncogenic pathway. Stilbene analogs represent an under-explored field of therapeutic natural products research. In the present work, a library of complex stilbene derivatives was obtained through biotransformation of a mixture of resveratrol and pterostilbene using the enzymatic secretome of Botrytis cinerea. To improve the chemodiversity, the reactions were performed using i-PrOH, n-BuOH, i-BuOH, EtOH, or MeOH as cosolvents. Using this strategy, a series of 73 unusual derivatives was generated distributed among 6 scaffolds; 55 derivatives represent novel compounds. The structure of each compound isolated was determined by nuclear magnetic resonance and high-resolution mass spectrometry. The inhibitory activity of the isolated compounds against the oncogenic Wnt pathway was comprehensively quantified and correlated with their capacity to inhibit the growth of the cancer cells, leading to insights into structure-activity relationships of the derivatives. Finally, we have dissected mechanistic details of the stilbene derivatives activity within the pathway.
Introduction
In developed countries, breast cancer is the most frequent malignancy and consequently the first cause of cancer-related death among women. The development of new targeted therapies, such as aromatase inhibitors, monoclonal antibodies (trastuzumab, pertuzumab, T-DM1), and lapatinib, has improved remarkably the outcome for endocrine-sensitive or HER2-positive breast cancer subtypes (Giordano et al., 2004). Some interesting developments resulted in further improvement of these agents, such as creation of clinically approved trastuzumab-based antibody-drug conjugates (Ado-Trastuzumab Emtansine and Fam-Trastuzumab Deruxtecan) (Mukohara et al., 2021). Additionally, HER2-positive tumors respond well to several clinically approved tyrosine kinase inhibitors (Schlam and Swain, 2021). However, the prognosis of patients with tumors negative for these factors, defined as triple-negative breast cancer subtype (TNBC), remains poor since this form of breast cancer elicits an aggressive behavior with early recurrence and a higher rate of visceral and brain metastases. Targeted therapies are virtually non-existent for TNBC with a notable exception of recent approval of sacituzumab govitecan—an anti-TROP2 antibody drug conjugate shown to be effective as a last resort against resistant and metastatic disease (Seligson et al., 2020). The treatment options are thus generally limited to cytotoxic chemotherapies, to which the disease becomes rapidly resistant (Kassam et al., 2009; Bianchini et al., 2021). There is therefore an urgent need to develop specific therapies for this subgroup of patients.
The Wnt signaling pathway controls multiple events during embryonic development of multicellular animals of almost all taxa starting from sponges, and, when aberrantly activated in the adult, is carcinogenic (Barker and Clevers, 2006; Blagodatski et al., 2014). The pathway is triggered by Wnt lipoglycoproteins binding the co-receptor pair, Frizzled (FZD) and LRP5/6; the signal is subsequently transmitted by Dishevelled (Dvl) and heterotrimeric G proteins. Their coordinate action dissociates the Axin-APC-GSK3β-casein kinase complex, which in its active state destabilizes β-catenin. Thus, when the pathway is “on,” β-catenin accumulates in the cytoplasm and consequently in the nucleus where it acts as a co-factor for transcription of the target genes through interaction with the TCF/LEF transcription factor.
Breast cancers are dependent on Wnt pathway overactivation mostly through aberrations in the expression of the pathway components rather than through mutations (Turashvili et al., 2006; Khramtsov et al., 2010; Dey et al., 2013; Koval and Katanaev, 2018). Genetic targeting of the Wnt pathway components identified the role of this pathway in TNBC proliferation and metastasis (Yang et al., 2011; Gurney et al., 2012; Wend et al., 2013; Simmons et al., 2014; Yang et al., 2014; Le et al., 2015) as well as breast cancer stem cells (CSC) proliferation and maintenance (Jang et al., 2015; Wang et al., 2016) that contribute to the development of tumor resistance to conventional chemotherapies (Abdullah and Chow, 2013; O’toole et al., 2013; O’reilly et al., 2015), with a specific role belonging to the receptor proteins in the pathway, Frizzleds (Larasati et al., 2022). Collectively, these findings identify the Wnt pathway as an excellent target to develop novel selective therapies and co-therapies, both for the primary TNBC and the prevention of tumor metastasis and relapse.
In context of these findings and inspired by our recent success on the subject, such as repositioning of the lichen-sourced compound clofazimine and its derivatives against Wnt signaling (Xu et al., 2020; Koval et al., 2021) we have embarked on a discovery of the novel anti-Wnt therapies using our platform for targeted screening of the such compounds (Shaw et al., 2019; Katanaev et al., 2021). Since there are multiple attractive targets within pathway which might mediate pharmacologically relevant anti-cancer effect, we have created a high-throughput system based on the triple-negative breast cancer cells stably transfected with TopFlash reporter, thus accurately representing the entire repertoire of pathway aberrations driving the disease (Veeman et al., 2003). In this adapted system, the activation of the pathway by Wnt3a ligand results in complete engagement of all the downstream components since it measures firefly luciferase expression levels induced by Wnt-specific transcription factors of TCF/LEF family, and thus the compound engaging any of the intermediate target within the pathway will be identified (Veeman et al., 2003). Furthermore, employment of a counter-screen using constitutively expressed Renilla luciferase allows us to immediately distinguish compounds inhibiting TopFlash unspecifically, usually by induction of toxicity, though other mechanisms are possible as well (Shaw et al., 2019). Moreover, using pharmacological approach, such as use of GSK3β inhibitor CHIR99021, we can rapidly dissect a more exact mode of action of active compounds (An et al., 2010).
Natural products have been one of the main historical sources of anticancer drugs (Newman and Cragg, 2020). Stilbenes are phenolic natural products produced by the phenylpropanoid pathway present in numerous families of plants (Shen et al., 2009). Some of them, such as resveratrol and pterostilbene, have been ascribed to a wide range of biological activities against cancer. Most notable of them are associated with effects on oxidative homeostasis, membrane structure and integrity, as well as a broad scope of intracellular pathways, ultimately activating responses resulting in tumor cell inhibition and chemosensitization—these activities are summarized in the recent excellent reviews (Obrador et al., 2021; Quarta et al., 2021; Ren et al., 2021). It has been further reported that resveratrol and pterostilbene have a potential Wnt-inhibition activity in several models and tissues (Blagodatski et al., 2020). However, this dependency on the promiscuous interactions of these molecules with multiple targets, has a backlash in being strongly context-dependent and poorly reproducible in vivo (Baell and Walters, 2014). Despite that, resveratrol and pterostilbene possess interesting properties as the starting point for the production of more complex derivatives with potentially specific and targeted activities.
Recently, we successfully used an enriched fraction of enzymes from fungi (“fungal secretome”) for the biotransformation of natural products to create complex derivative compounds with improved biological activity (Gindro et al., 2017; Righi et al., 2020; Huber et al., 2021). The enzymatic diversity produced by fungi used for the degradation and/or transformation of various substrates has the potential to catalyze different chemical reactions (Gindro et al., 2017). The use of such fungal secretomes for biotransformation thus has great potential to transform various types of substrates with less specificity than pure enzymes (Lopes et al., 2014) and is probably well suited to generate high chemodiversity by the transformation of related rather simple initial natural compound substrates. In addition, the use of a fungal secretome rather than whole-cell incubation facilitates the isolation of biotransformed compounds due to the absence of fungal metabolites in the final mixture.
It was also demonstrated that biotransformation reactions can be performed in the presence of high amounts of organic solvents (Righi et al., 2020). These solvents can improve the solubility of substrates in aqueous media and also allow a relatively easy product recovery (Klibanov, 2001). Moreover, the use of MeOH as a cosolvent was shown to generate new compounds by unexpected solvolysis reactions (Righi et al., 2020).
In this context, we extended this approach to 4 other cosolvents (i-PrOH, n-BuOH, i-BuOH and EtOH) in order to increase the chemodiversity of the stilbene dimers library obtained by the biotransformations of a mixture of resveratrol and pterostilbene using the Botrytis cinerea Pers. enzymatic secretome. This library was generated in view of a Wnt inhibition screening; the generation of close analogs allowing a study of the structure-activity relationship with the aim of determining a possible pharmacophore.
Results and Discussion
Generation of Stilbene Analogs by Biotransformation Using Organic Solvents and the Enzymatic Secretome of Botrytis cinerea
In previous studies, biotransformation reactions with the B. cinerea secretome allowed us to obtain 6 families of complex dimers with several stereochemical centers starting from simple stilbenes without asymmetric carbon (Gindro et al., 2017). To overcome solubility issues, a series of biotransformation reactions were performed with a mixture of resveratrol and pterostilbene in presence of different amounts of acetone or methanol as a cosolvent with water (Righi et al., 2020). The reactions occurred even at a high organic solvent ratio, and methoxylated derivatives with interesting biological activities were successfully obtained when the reaction was performed with methanol (Righi et al., 2020).
The study of the reactions at the origin of these unexpected compounds indicated that it was a nucleophilic attack of the solvent (solvolysis) on a sp2 carbon of an intermediate structure that generated them (Righi et al., 2020). Taking advantage of this observation, we designed a series of biotransformation reactions with a mixture of resveratrol and pterostilbene in presence of different alcohols to obtain a series of analogs. This ability to generate a variety of close analogs combined with the existence of results concerning the ability of resveratrol to inhibit the Wnt pathway in several models and tissues led us to generate a library of compounds based on this pharmacophore to evaluate their activity.
To this end, resveratrol and pterostilbene (total concentration of 1 mg/ml; 0.5 mg/ml of each compound) were diluted in the organic solvent (i-PrOH, n-BuOH, i-BuOH, EtOH, or MeOH) and H2O, before incubation with the secretome of B. cinerea over 24 h. Each reaction was monitored by UHPLC-PDA-ELSD-MS (data not shown). The biotransformation reactions were performed with 50% of organic solvents according to our previous study (Righi et al., 2020). With this amount of organic solvent, the compounds were soluble, and based on the LC-MS monitoring, a series of molecular ion species corresponding to the presence of new dimeric analogs were observed (Figure 1).
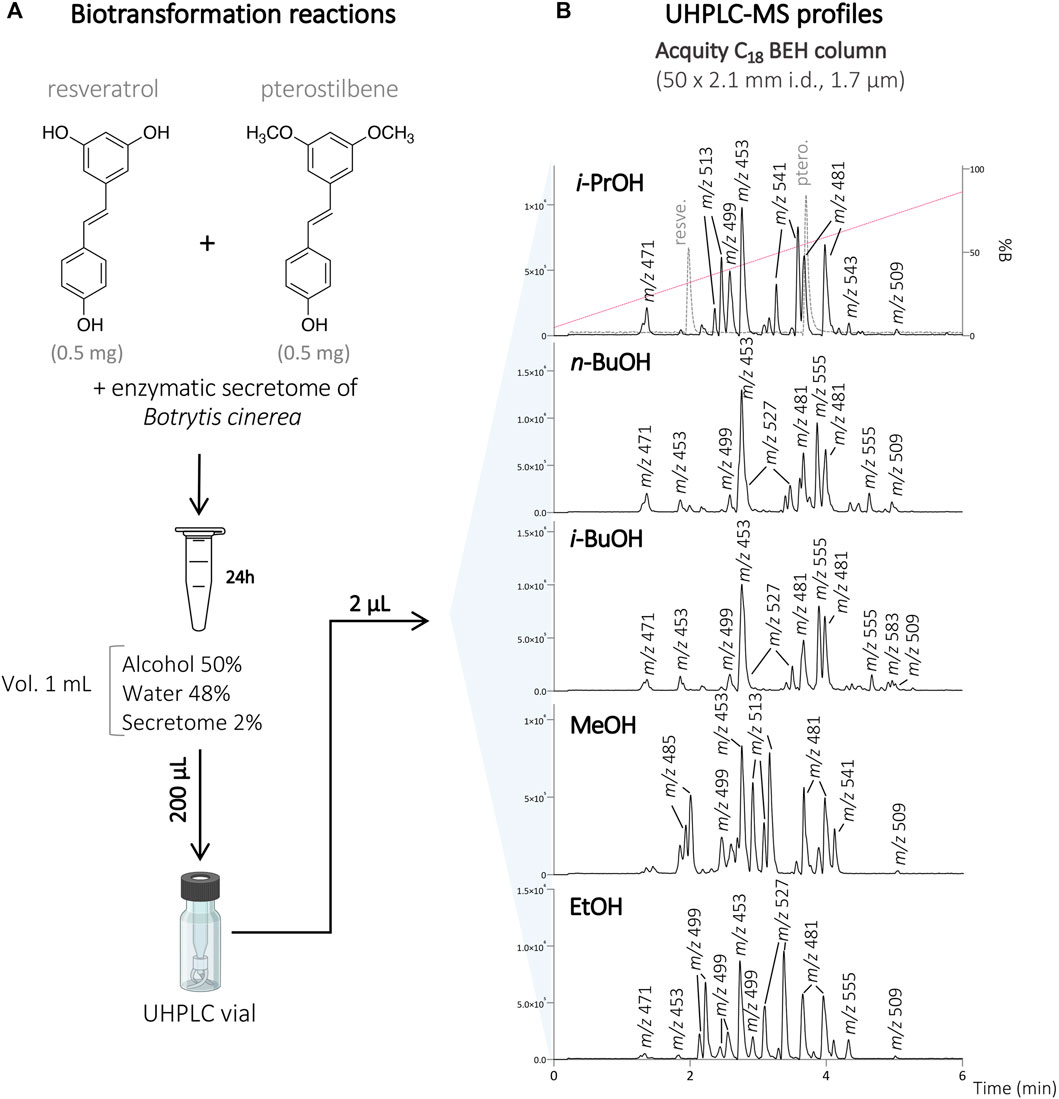
FIGURE 1. (A) Biotransformation reactions of the mixture of resveratrol and pterostilbene with the secretome of Botrytis cinerea in the presence of 50% of different organic solvents (i-PrOH, n-BuOH, i-BuOH, MeOH, EtOH). (B) UHPLC-MS profile of the different reactions at the analytical scale using an Acquity C18 BEH column 50 × 2.1 mm i.d., 1.7 µm. The retention times of the starting compounds resveratrol and pterostilbene are indicated by the dashed line on the reaction with i-PrOH. The molecular ion species for all the main compounds detected are indicated with their corresponding m/z values.
To isolate these compounds, the biotransformation reactions with each cosolvent were scaled-up keeping the same conditions, except the amount of substrate which was increased to 200–400 mg (see Experimental section). After 24 h, the reactions were monitored by UHPLC-PDA-ELSD-MS. As expected, resveratrol and pterostilbene were fully consumed, demonstrating a good enzymatic activity under such optimized conditions.
After initial reaction monitoring, separation conditions were optimized at the UHPLC scale, allowing for rapid and low solvent consumption runs. These conditions were transferred to the analytical (as a control) and semi-preparative HPLC scale using geometric gradient transfer. The same column chemistry was used on each instrument to maintain the same chromatographic selectivity (Figure 2; Supplementary Figures S1–S4) (Guillarme et al., 2007; Guillarme et al., 2008). At the semi-preparative scale, the samples were injected by dry-load instead of a conventional loop injection, following a protocol recently developed in our laboratory (Queiroz et al., 2019). This approach avoids the loss of resolution caused by the organic solvent during the sample injection. Thanks to this approach, the separations obtained at the semi-preparative HPLC scale perfectly matched those obtained at the analytical scale, allowing the isolation of a majority of compounds in one step. Using this approach, the crude mixtures of the five biotransformation reactions were efficiently purified affording a total of 73 pure compounds; compounds 1–24 from the reaction with i-PrOH, 25–40 from n-BuOH, 41–53 from i-BuOH, 54–64 from MeOH and 65–73 from EtOH (Figure 3).
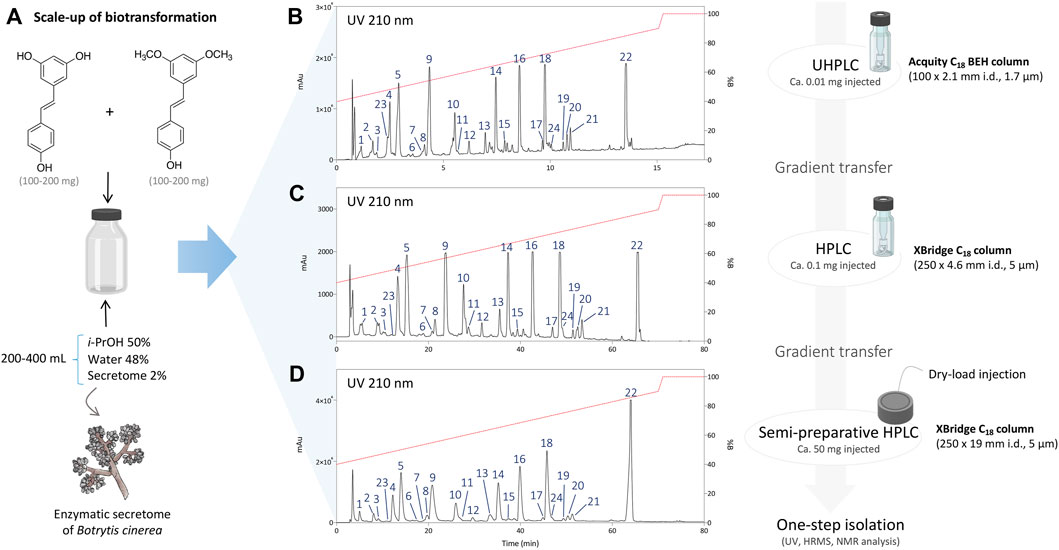
FIGURE 2. (A) Scale-up conditions of the biotransformation reaction of resveratrol and pterostilbene with the secretome of Botrytis cinerea with 50% of i-PrOH, 2% of enzymatic secretome and 48% of water (total volume 200–400 ml). (B) Metabolite profile of the reaction after 24 h using UHPLC-PDA with optimized chromatographic conditions. (C) Analytical HPLC-PDA chromatogram after gradient transfer of the UHPLC-optimized conditions. (D) Semi-preparative HPLC-PDA chromatogram after gradient transfer of the UHPLC-optimized conditions.
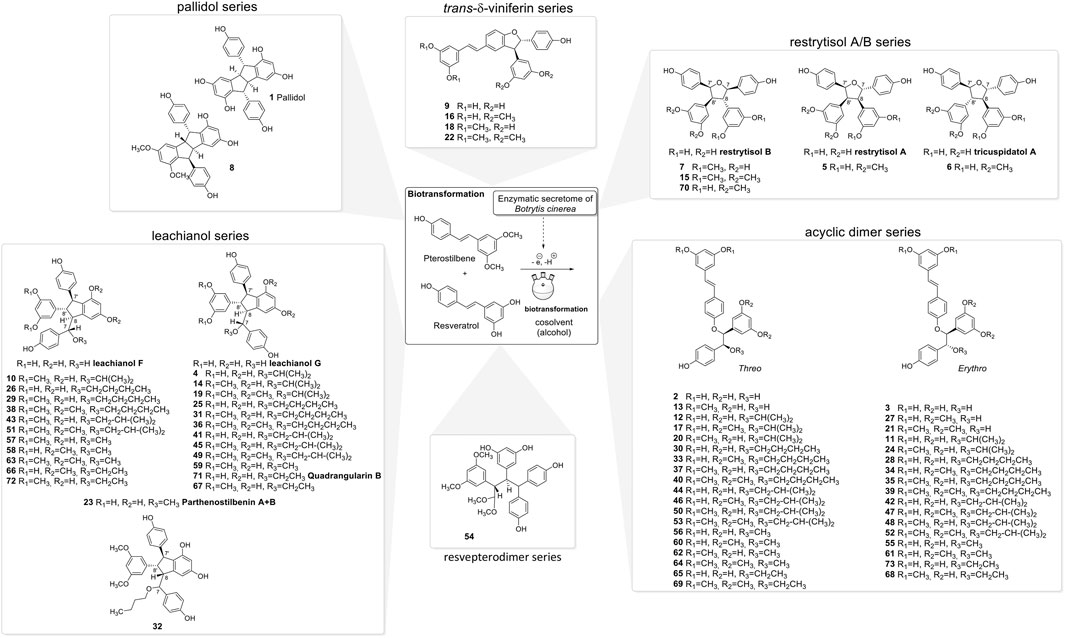
FIGURE 3. Compounds isolated from the biotransformation reactions of the mixture of resveratrol and pterostilbene with the secretome of Botrytis cinerea in the presence of 50% of different organic solvents (i-PrOH, n-BuOH, i-BuOH, EtOH, or MeOH).
The NMR analysis of the compounds generated by the biotransformation reactions of resveratrol and pterostilbene indicates that 6 main scaffolds are formed following the dimerization of the 2 starting entities (Figure 3).
The first one is the well-known trans-δ-viniferin series, resulting from a 3-8′ coupling (Figure 4). This scaffold includes only 4 compounds: trans-δ-viniferin (9), 11′,13′-di-O-methyl-trans-δ-viniferin (16), 11,13-di-O-methyl-trans-δ-viniferin (18), and pterostilbene-trans-dehydromer (22). These compounds have already been isolated from our previous biotransformations (Gindro et al., 2017). The characteristic 1H NMR signals of these compounds are 2 doublets (3JHH = 16.3–16.4 Hz) at δH 6.97–7.22 for H-7 and δH 6.82–6.96 for H-8 of the trans double bond and the H-2, H-5, and H-6 protons from the 1,3,4-trisubstituted aromatic cycle at δH 7.12–7.24 (1H, d, J = 1.9 Hz, H-2), 6.85–6.90 (1H, d, J = 8.3 Hz, H-5) and 7.38–7.44 (1H, dd, J = 8.3, 1.9 Hz, H-6) (Figure 5).
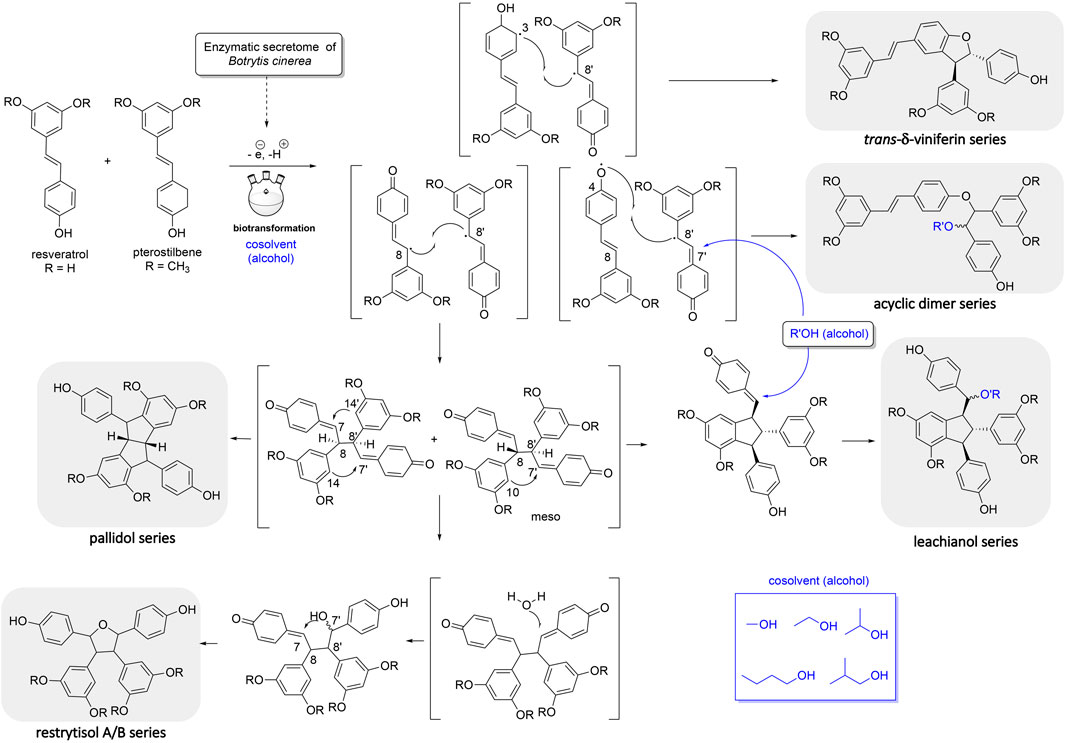
FIGURE 4. Mechanism of reaction explaining the generation of the 5 main scaffolds (trans-δ-viniferin, acyclic dimer, leachianol, pallidol, restrytisol A/B) of stilbene dimers obtained by the biotransformation reaction of resveratrol and pterostilbene using the enzymatic secretome of Botrytis cinerea in the presence of alcohol as cosolvents.
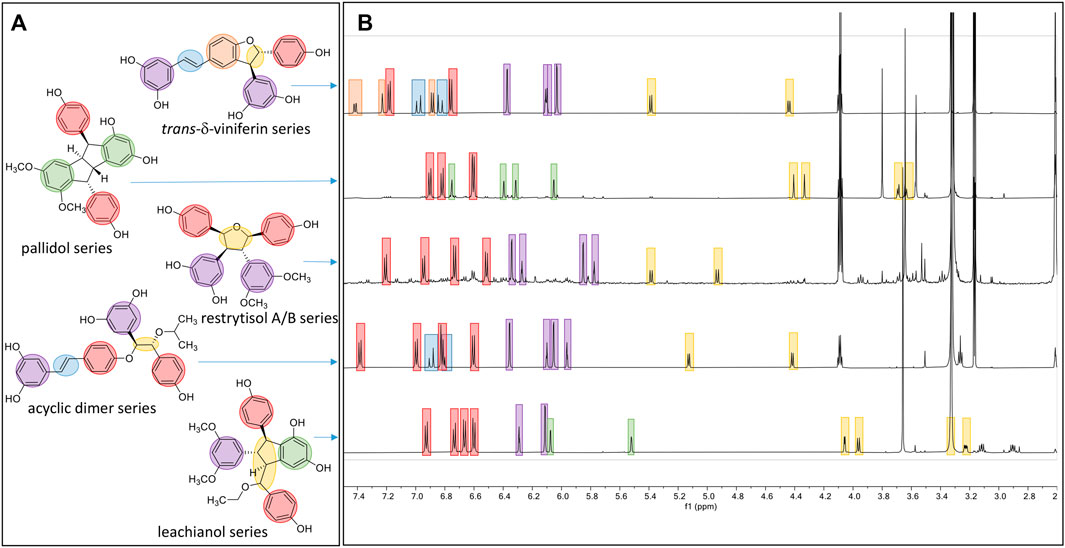
FIGURE 5. Overview of the typical 1H-NMR shifts of the different scaffolds generated. (A) Five compounds representing the five main scaffolds obtained by the biotransformations reactions. (B) The typical 1H-NMR spectra of each scaffold is highlighted in blue for the trans double-bound; orange for the 1,3,4-trisubstituted aromatic cycle; red for a para-disubstituted system; purple for the 1,3,5-trisubstituted aromatic cycle; green for the two meta coupled proton; and yellow for the methine protons.
The second one is the pallidol series, resulting from an 8-8′ coupling followed by a 7-14′ and 14-7′ cyclization (Figure 4). This scaffold is represented by pallidol (1) and 11, 13-di-O-methylisopallidol (8). The double cyclization (7-14′ and 14-7′) leads to the disappearance of the 2 systems consisting of a triplet integrating for one proton and a doublet integrating for two protons (J = 2.1–2.3 Hz) and the appearance of 2 pairs of doublets (J = 2.1–2.3 Hz) (Figure 5). Due to the symmetrical structure of pallidol (1), only the signals of one monomer are visible which is not the case for 11,13-di-O-methylisopallidol (8). These 2 compounds have already been isolated from our previous works (Gindro et al., 2017).
The third one is the restrytisol A/B series, resulting from an 8-8′ coupling followed by water addition in 7 or 7′ and O-7 or 7′ cyclization (Figure 4). 5 compounds belong to this scaffold. In this case, every aromatic signal of the resveratrol is preserved, i.e., the 2 doublets (2H each) with ortho coupling (J = 8.3–8.6 Hz) from the para-disubstituted system and the triplet (1H)/doublet (2H) with meta coupling (J = 2.1–2.3 Hz) from the trisubstituted aromatic (Figure 5). One di-O-methyl derivative (5) has the same configuration as restrytisol A as indicated by the ROE correlations from H-7 to H-10/H-14 and H-10′/H-14′, from CH3O-11′/CH3O-13′ to H-10′/H-14′ and from H-7′ to H-2/H-6 and from H-8. This compound is a new derivative named 11′,13′-O-dimethylrestrytisol A (5). A second di-O-methyl derivative (6) is also a new derivative. It has the same configuration as tricuspidatol A as confirmed by the ROE correlations from H-7 to H-8′ and H-10/H-14 and from H-7′ to H-8 and H-10′/H-14′. It was thus named 11′,13′-O-dimethyl tricuspidatol A (6). The three remaining compounds belong to the restrytisol B configuration and correspond to the two possible di-O-methyl derivatives (7 already isolated from previous biotransformations and one new compound 70 identified as 11′,13′-di-O-methylrestrytisol B) and the tetra-O-methyl derivative 15 also isolated from previous biotransformations (Gindro et al., 2017). The following ROE correlations were key to confirm the restrytisol B isomerism: from H-7 to H-8′, H-7′ and H-10/H-14 and from H-8 to H-2/H-6, H-2′/H-6′ and H-10′/H-14′. In summary for this restrytisol A/B series, 5 compounds were synthesized and 3 are described here for the first time.
The compounds belonging to these first three series were not able to incorporate solvent in their structure unlike those of the fourth one, named the acyclic dimer series and which results from an 8′-O-4 coupling (Figure 4). The compounds of this series were recognized by 1H-NMR by the presence of 2 doublets (3JHH = 16.3–16.6 Hz) at δH 6.86–7.14 for H-7 and δH 6.79–6.96 for H-8 from the trans double bound, 4 doublets (2H each) with ortho coupling (J = 8.3–8.6 Hz) from the 2 para-disubstituted ring and 2 triplets (1H each)/2 doublets (2H each) with meta coupling (J = 2.1–2.3 Hz) from the 2 trisubstituted rings, as well as 2 oxymethine between δH 4.29–4.73 for H-7′ and δH 5.00–5.34 for H-8′ (Figure 5). This series contains 36 compounds because a nucleophilic addition of the alcohol used as cosolvent happens in C-7′ after the 8′-O-4 coupling between the 2 monomer radicals (Figure 4). Moreover, 2 diastereoisomers in C-7′ and C-8′ are possible and are defined as erythro and threo. Compounds 2, 55, and 56 were already prepared by oxidative coupling reaction using AgOAc as oxidant by Zhang et al. (2014). They identified each diastereoisomer based on the larger 3JH-7′-H-8′ coupling constant in the threo form compared to the erythro and from the weak NOE effect between H-7′ and H-8′ and from H-7′ to H-2′/H-6′ and H-10′/H-14′. They also indicated that the chemical shift values of C-7′ and C-8′ were discriminant: in the threo form, these signals were shifted to a lower field than those in the erythro form. This was also described by Braga et al. (1984) in their conformational analysis of 8-O-4′ neolignans. In our series of data, both the coupling constant between H-7′ and H-8′ and the ROE correlations were the same for each diastereoisomer. Only the 13C chemical shifts values were slightly different, for example in the case of a hydroxy group in C-7′ the values were [δC: 75.6–75.7 (C-7′), 83.1–83.7 (C-8′)] in the erythro form and [δC: 76.1 (C-7′), 84.0–84.1 (C-8′)] in the threo form or in the case of an isobutanol group in C-7′ the values were [δC: 83.5–84.0 (C-7′), 81.6–81.9 (C-8′)] in the erythro form and [δC: 84.4–84.6 (C-7′), 82.6–82.9 (C-8′)] in the threo form. Due to the small difference of C-7′ and C-8′ 13C chemical shift in DMSO-d6 between the threo and erythro form and the lack of difference of the 3JH-7′-H-8′ coupling constant and ROE correlations, the 2 hydroxylated diastereoisomer 2 and 3 were analyzed again in acetone-d6. Compound 2 displayed the same chemical shift than threo-5-[1-[4-[(1E)-2-(3,5-dihydroxyphenyl)ethenyl]phenoxy]-2-hydroxy-2-(4-hydroxyphenyl)ethyl]-1,3-benzenediol described by Zhang et al. (2014) and the 3JH-7′-H-8′ coupling constant was 7.0 Hz for 2 (threo) and 5.8 Hz for 3 (erythro). These data confirmed the threo/erythro assignment made in DMSO-d6. A more probable free rotation along the C-7′-C-8′ bond in DMSO-d6 giving an average value of coupling constant compared to acetone-d6 could explain the differences observed between these two solvents. Among the 36 compounds of this acyclic series, 2 and one of the two diastereoisomers 55 or 56 were already described by Zhang et al. (2014), 21 by Ponzoni et al. (2007), 65 and 73 by Shingai et al. (2011) as well as 60 and 62 from our previous work (Righi et al., 2020). The remaining 29 derivatives are therefore compounds that are described here for the first time.
The fifth series is called leachianol, resulting from a 8-8′ coupling followed by a 10-7′ cyclization and a nucleophilic addition of the alcohol in C-7 (Figure 4). The 1H NMR spectrum of this series was recognized by the lack of doublets corresponding to the trans double bond, the presence of 4 doublets (2H each) with ortho coupling (J = 8.3–8.6 Hz) from the 2 para-disubstituted rings, and a triplet (1H)/doublet (2H) with meta coupling (J = 2.1–2.3 Hz) from the trisubstituted ring and especially the presence of four methines signals from H-7, H-8, H-7′ and H-8′ (Figure 5). Two diastereoisomers can be formed during the dimerization process and were called F and G regarding the configuration of leachianol F and G isolated first by Ohyama et al. (1995) from the roots of Sophora leachiana. The distinction between these two diastereoisomers can be made by NMR first by comparing the 1H chemical shifts of the aromatics H-14 and H-10′/H-14′ and the methine H-8′. Due to the proximity of the phenol in C-7 in the case of the F diastereoisomer, H-8′ and H-10′/H-14′ are shifted to a higher field (δH 2.59–2.76 for H-8′ and 5.59–5.73 for H-10′/H-14′) than that in the G diastereoisomer (δH 3.20–3.46 for H-8′ and 5.92–6.12 for H-10′/H-14′) (Figure 6). In the case of the G diastereoisomer, the phenol group is on the side of H-14 which is shielded compared to F diastereoisomer (δH 5.17–5.57 and 6.41–6.75 for G and F diastereoisomer, respectively). The ROE correlation from H-2/H-6 to H-10′/H14′ in the case of F diastereoisomer and to H-14 in the case of G diastereoisomer confirmed this assignment (Figure 6). Among the 26 compounds of this leachianol series, parthenostilbenin A and B (23), already isolated by Kim et al. (2005) from Parthenocissus tricuspidata were isolated as a mixture and quadrangulain B already isolated from Cissus quadrangularis by Adesanya et al. (1999) was also produced from this biotransformation reaction (71). Compound 32 could be considered apart as this was the only compound with a different configuration of the cyclopentane ring. The ROE correlations from H-7′ to H-10′/H14′ and from H8′ to H-2′/H-6′ indicated a trans configuration of H-7′ and H-8′ protons, the correlation from H-8 to H-2′/H-6′ indicated a cis configuration of H-8 and H-8′ protons and the correlation from H-2/H-6 to H-14 allowed to define the C-7 configuration (Figure 6). Finally, 23 compounds from this series are described here for the first time.
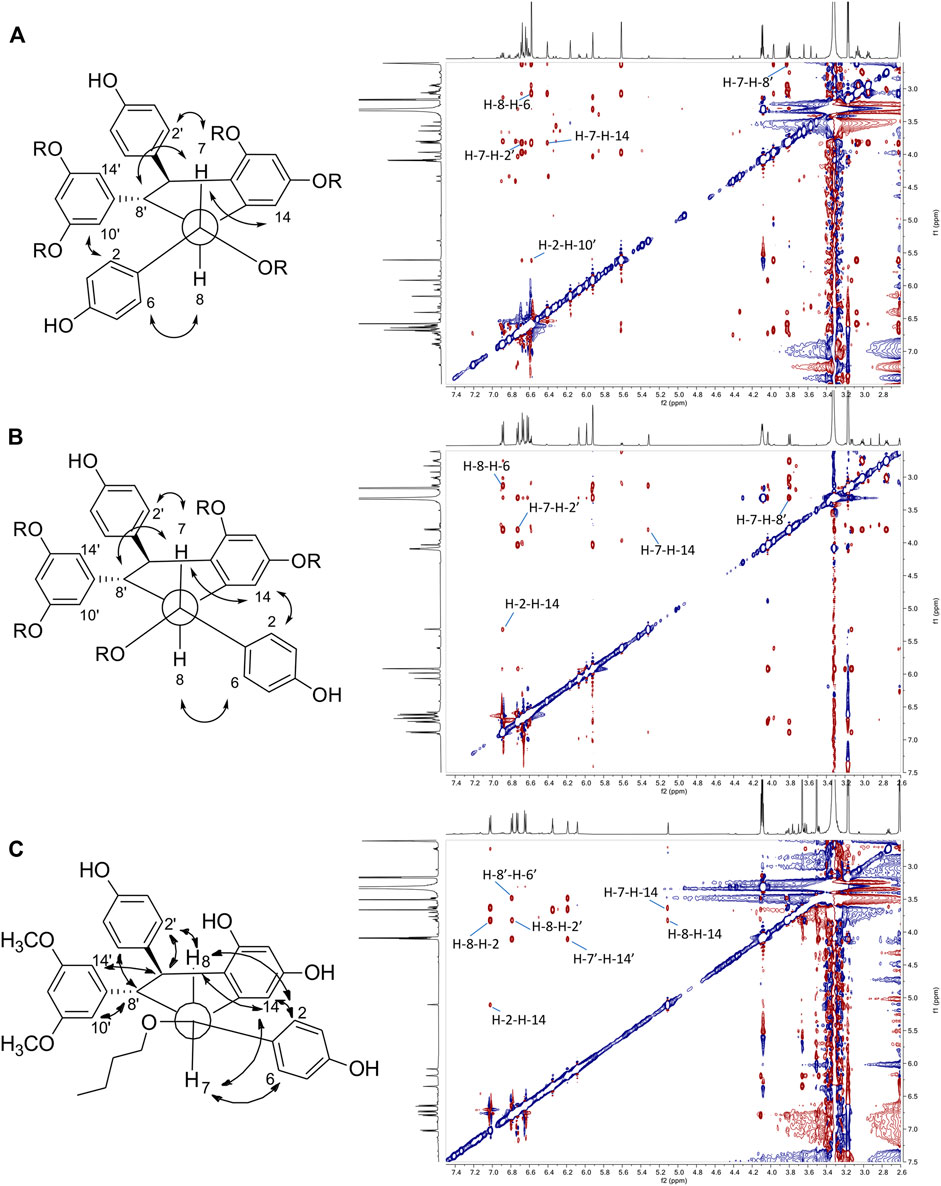
FIGURE 6. Key ROESY correlations observed for (A) leachianol F series (B) leachianol G series and (C) for the different isomer (compound 32).
Compound 54 belongs to a new scaffold (sixth series) already produced and described from previous biotransformation reactions and named resvepterodimer A (Righi et al., 2020). No other derivatives belonging to this series have been isolated, probably due to steric hindrance caused by the longer carbon chains of solvents other than methanol.
In summary, the biotransformation reactions performed with resveratrol and pterostilbene in five different alcoholic solvents allowed the isolation of 73 derivatives distributed among 6 scaffolds, 55 being described here for the first time (Figure 3).
To understand further how different or similar compounds are, we measured the Tanimoto coefficient and represented it in the form of a similarity matrix (Figure 7A). The analysis resulted in defined clusters corresponding to each series of compounds. Most clusters exhibit coefficient value superior than 0.7, showing the similarity within the clusters and the presence of a common scaffold. In contrast, when comparing the scaffolds of the different clusters to each other, the analysis reveals diversity with value lower than 0.5 indicating that the biotransformation methodology is able to deliver chemically diverse scaffolds starting from the same material.
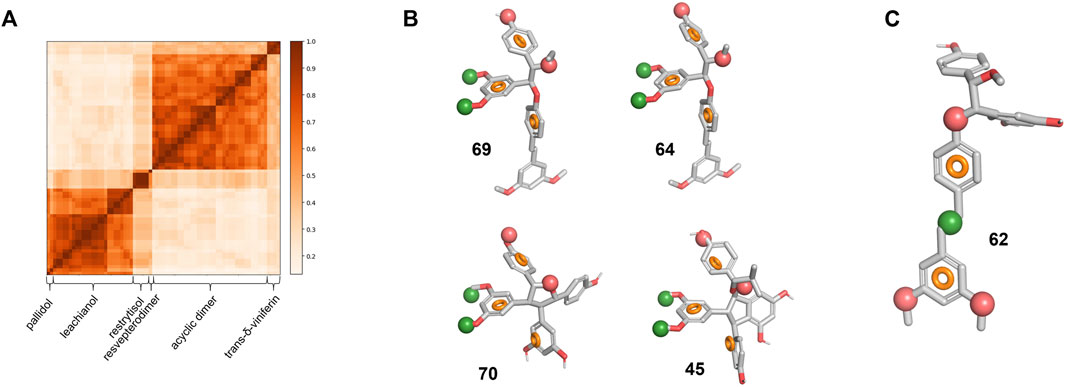
FIGURE 7. Library chemical similarity and pharmacophore models. (A) Similarity matrix for all six series. A value of 1 corresponds to the identity. (B) Pharmacophore model for specific actives overlapped on the four compounds with an SI > 10. (C) Pharmacophore model for non-specific actives overlapped on 62. The red and green spheres represent respectively an H-bond acceptor and a hydrophobic region. The orange circle represents the ideal 3D positioning of aromatic rings.
Biotransformation Leads to Novel Derivatives With Specific Activity Against the Wnt Pathway in Triple-Negative Breast Cancer
Our main building blocks resveratrol and pterostilbene have previously been reported to inhibit Wnt signaling in breast cancer (Wang et al., 2012) and inhibit or activate the pathway in many other cancers or contexts (Ashrafizadeh et al., 2020). On the other hand, both compounds are well known to be highly promiscuous and are defined as pan-assay interfering compounds (PAINS) (Baell and Walters, 2014). Thus, we first investigated if these principal building blocks might show specific anti-Wnt activity in our assay and thus confer such activity to the various derivatives generated. We analyzed activation of the TopFlash Wnt-specific reporter in the TNBC cell line BT-20 by purified Wnt3a simultaneously measuring the constitutively expressed Renilla luciferase activity as a readout of unspecific and/or cytotoxic action of compounds. Our results (Supplementary Figure S5) indicate that resveratrol possesses only certain unspecific cytotoxic effects at concentrations exceeding 100 μM, while pterostilbene did not show any activity.
We further tested all the derivatives 1–73 in the same assay, discovering that the biotransformation of resveratrol/pterostilbene results in a large family of compounds with specific anti-Wnt properties (Table 1, columns “Wnt” and “Renilla”). The most interesting derivatives are highlighted by plotting the selectivity index (SI), expressed as a ratio of Renilla and Wnt IC50s, against the compound potency for the Wnt pathway inhibition (Figure 8A). We saw a general loose trend for increasing pathway inhibition selectivity with increased potency (i.e., with the decreased IC50). The three most selective compounds (64, 69, and 70), two belonging to the threo acyclic dimers series (64 and 69) both resulting from two pterostilbene units and one being the 11′,13′-di-O-methylrestrytisol B (70), demonstrate only average potency (18–27 µM Wnt IC50). It should be noted, that this analysis also showed the absence of a well-defined dependency of the compound potency or selectivity on a particular scaffold, with most of their representatives demonstrating activity within a broad range of values. However, some trends were observed by representing the data from Figure 8A by scaffold and mapping information such as the nature of the monomers (Supplementary Figures S6A–D). The restrytisol series shows little potency (>20 µM for the best compound). The leachianol, pallidol and viniferin series have broad activity values, spanning from high (<5 µM) to no activity. A general trend for these scaffolds is a lower activity for the dimers resulting from the coupling of two resveratrol units. The presence of methoxy groups (coming from the pterostilbene unit) therefore seems to impact positively the ability to inhibit the Wnt activity. Moreover, mixed dimers resulting from the coupling of a resveratrol and pterostilbene unit generally have the highest activities in the leachianol series. For this series with a clear dependence on the nature of the monomers, the influence of the side-chain coming from the solvolysis was also investigated (Supplementary Figure S6E) but did not show any clear tendency. For the acyclic dimer series, no visible trend between structure and activity was observed, probably due to the high flexibility of this scaffold. For all the scaffolds, no clear difference in potency or selectivity was observed between the stereoisomers (leachianol F or G; acyclic dimer erythro or threo; restrytisol A or B).
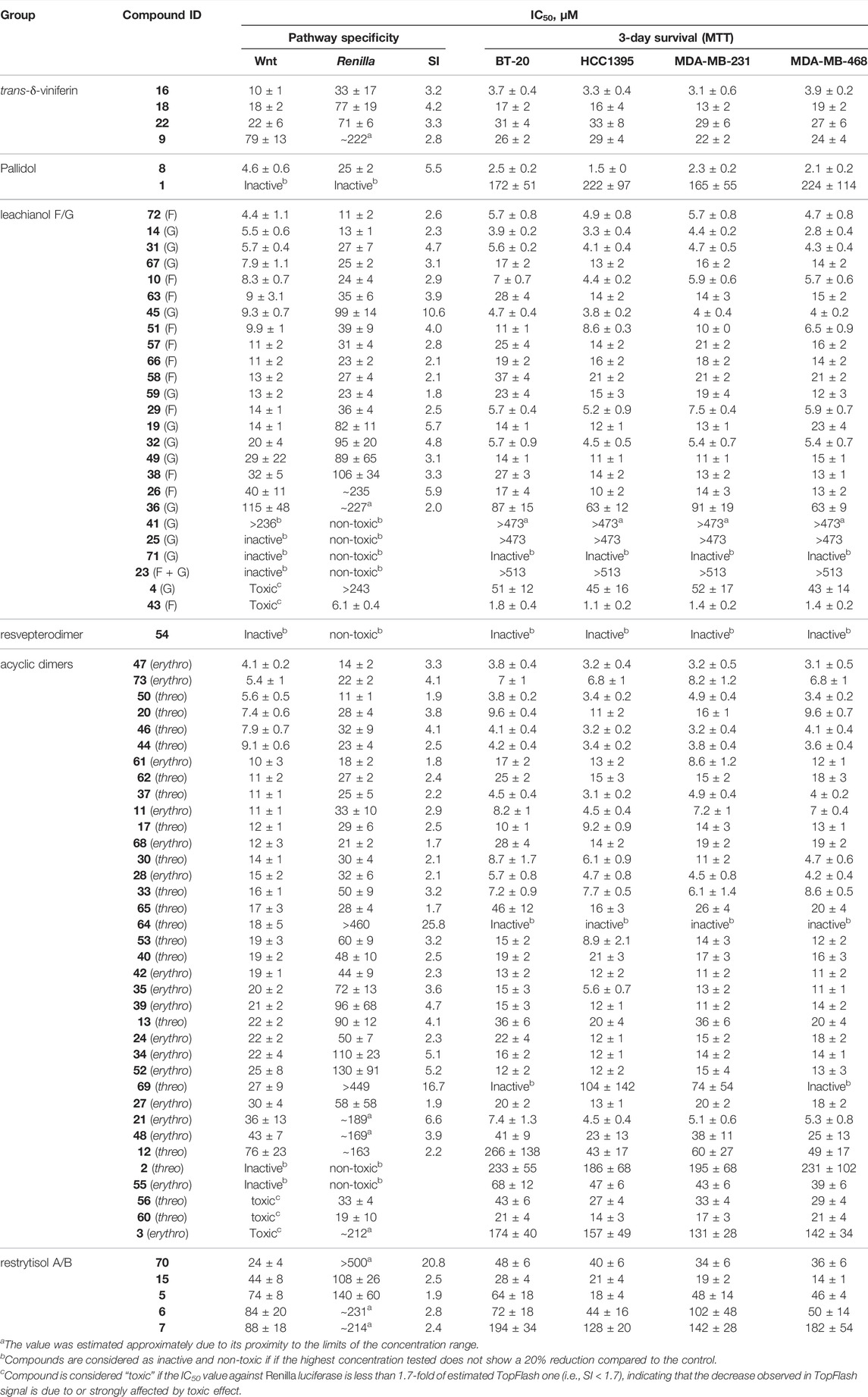
TABLE 1. IC50s of the investigated compounds against Wnt pathway and breast cancer cell survival.
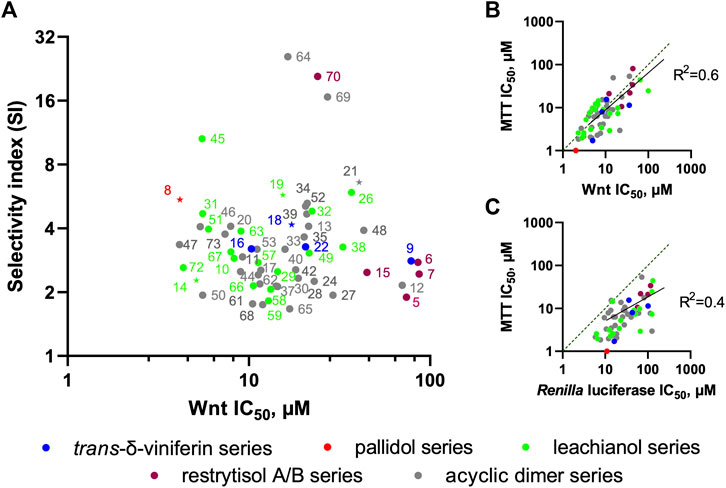
FIGURE 8. Relations between derivative scaffolds and anti-Wnt activity. (A) Distribution between the selectivity index (SI) and anti-Wnt potency of the investigated compounds. The compounds selected for further biochemical analysis are highlighted as stars (B) Correlation between anti-Wnt IC50 and IC50 of long-term (3 days) inhibition of the cell growth measured by MTT assay (C) Correlation between anti-Wnt IC50 and IC50 of acute cytotoxicity.
To identify key features that distinguish actives from inactive compounds in 3D space, we developed a pharmacophore model hypothesis. Pharmacophore models consider common electronic and shape features that would otherwise be difficult to assess in 2D space. The compounds were first split in three groups based on their SI and IC50 (Supplementary Figure S7A): we defined the “specific-active” group for compounds with a high selectivity index (>10) and a low IC50 (<35 μM, 4 compounds in total), the “low specificity-active” group (SI < 10, IC50 < 35 μM, 46 compounds in total) and the “low specificity-low activity” group (SI < 10, IC50 > 35 μM, 18 compounds in total). As the stereochemistry of these compounds is relative and not absolute, the enantiomers for each compound were considered to build the model. The pharmacophore analysis of the “specific-activity” group lead to a seven-features model that overlap with the asymmetric centers part of 64 and 69 from the acyclic dimer series (Figure 7B). This model describes also key features of 70 from the restrytisol series and 45 from the leachianol series (Figure 7B). To validate and assess the predictiveness of this model, we analyzed how the whole library fits to the model. Besides the true positives, false positive compounds from the acyclic dimer series appeared (Supplementary Figure S7B). The overlap of the model on false positives as well as the absence of excluded volumes in the model suggests additional requirements for a specific activity: the acyclic dimer must be in a threo and not in an erythro configuration, and the substituent in R3 must be smaller than an ethyl group and not branched.
The model derived from the “low specificity-active” compounds describes the pterostilbene moiety when overlapped with the acyclic dimer series such as compound 62 (Figure 7C). This model has to be considered as descriptive but non-predictive as shown by the poor correlation between the IC50 and the fitness (Supplementary Figure S7C). The two “specific-activity” compounds from this series (64 and 69) fit to this model, leading to the hypothesis that their IC50 might be the result of both unspecific and specific activity (Supplementary Figure S7C). Finally, the analysis of the “low specificity-low activity” group did not generate any model. This suggests that their low activity is not linked to a particular structure but rather a lack of the key features depicted in the model built on the “specific-active” compounds (Figure 7B) or the presence of substituents in hypothetical excluded volumes. It should be noted that some “low specificity-low activity” compounds still display features from the “low specificity-active” model.
Although these pharmacophore models suffer from limitations such as lack of excluded volumes, they rationally explain how different backbones can display the same activity and emphasize the crucial role of stereochemistry in natural products. The models built on defined groups identified discriminative key features for the specificity and descriptive common features for the activity. They lay the ground for analog synthesis or virtual screening.
We next analyzed whether these compounds are capable of inhibiting the proliferation of TNBC cell lines (BT-20, HCC1395, MDA-MB-231, and MDA-MB-468). Indeed, as reported in the corresponding columns of Table 1, the majority of the compounds efficient against the Wnt pathway were also excellent inhibitors of cancer cell proliferation. However, since the majority of the compounds demonstrated an unspecific inhibition of Renilla luciferase, which might be both manifestation of direct cytotoxicity or other interference with the cell well-being leading to the decrease in proliferation as well, we wanted to test independently whether this activity is indeed attributable to Wnt signaling.
To this end, we analyzed the relationships between the observed effect in MTT assay (taken as an average value of IC50s across all 4 TNBC cell lines, which are very close) and the potency of the compounds against the Wnt pathway (Figure 8B) and the Renilla luciferase reporter (Figure 8C). It is clearly evident that the long-term proliferation IC50s correlate much better (linear fit R2 = 0.6) with these against the Wnt signaling than with the Renilla reporter (R2 = 0.4. Data distribution indicates a relatively good linear correlation between anti-Wnt activity and long-term inhibition activity, while this correlation for Renilla is strongly right-shifted, indicating that, in general, the IC50s obtained from the MTT assay are lower. It should be also noted, that IC50 values for some compounds are either lower or higher than those obtained against Wnt signaling, which can be attributed to the different lengths of the assays, and thus effects of the tested compounds stability or subcellular accumulation might influence these values.
We also confirmed the Wnt inhibitory activity of the compounds in a different assay from TopFlash. A set of five representative compounds with different scaffolds was chosen on the grounds of both addressing the most prominent structural families and capacity to produce a sufficient quantity of the compound. We have analyzed this set for its ability to inhibit basal levels of the Wnt target genes Axin2 and c-Myc (Figure 9) (Koval et al., 2018; Xu et al., 2020). All the tested compounds demonstrated the capacity to decrease both target genes expression with slightly different efficacies. These levels of the Wnt target genes inhibition are in line with our and other’s findings, e.g., for clofazimine (Ahmed et al., 2019; Xu et al., 2020), a compound capable of suppressing tumor growth in vitro and in vivo.
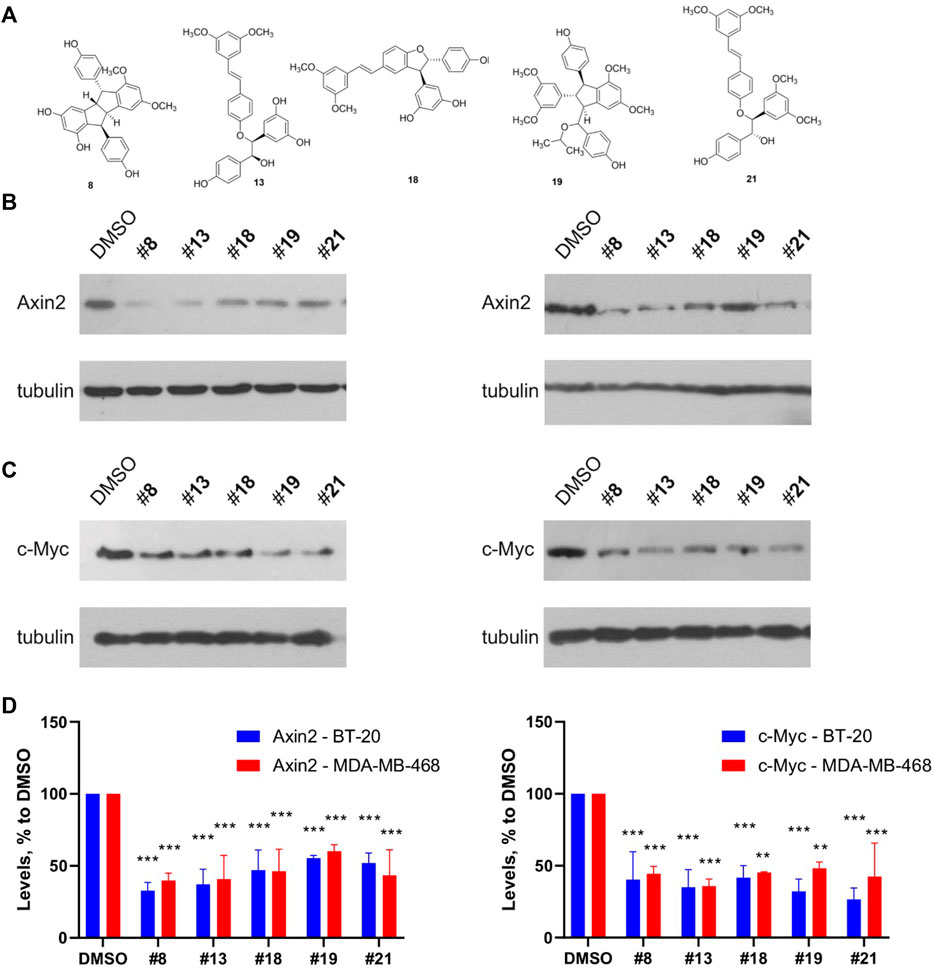
FIGURE 9. Representative compounds from the different structural groups of the derivatives (A) inhibit expression of the basal levels of Wnt target genes Axin2 (B) and c-Myc (C) in the TNBC cell lines BT-20 (left) and MDA-MB-468 (right). Quantification of the Western blots is presented on (D). Statistical significance was assessed by two-way ANOVA followed by multiple comparisons with DMSO-treated values, p values are shown as **p < 0.01, ***p < 0.001.
We next aimed at dissecting the potential mechanism of compounds’ activity within the Wnt pathway, analyzing whether the compounds are capable of inhibiting the signaling upstream or downstream of its symbolical “watershed”—the β-catenin destruction complex. For this purpose, we assessed the levels of β-catenin induced in L-cells (murine fibroblasts) by addition of Wnt3a (Koval et al., 2014; Koval et al., 2018). These cells have virtually no endogenous β-catenin, but it starts to accumulate rapidly upon the addition of the Wnt ligand Wnt3a. However, the compounds were not able to suppress this effect (Supplementary Figures S8A,B). At the same time, they were capable to suppress the Wnt pathway when it was stimulated by CHIR99021, a specific inhibitor of GSK3β kinase which can strongly stimulate the pathway downstream of Wnt3a ligand (Supplementary Figures S8C,D) (An et al., 2010). These results indicate that the target of the compound is located well downstream of the destruction complex.
We also assessed whether the compounds might affect two other important steps in Wnt signaling—translocation of β-catenin to the nucleus and its phosphorylation. To this end, we performed double staining of total and phosphorylated β-catenin in the BT-20 and MDA-MB-468 TNBC cell lines, stimulated or not by Wnt3a and treated by the compound 8 on the background of Wnt3a activation. Accumulation of the total β-catenin in the cytoplasm and nucleus of many cells is evident upon treatment with Wnt3a, but this effect was not reversed by addition of the compound 8 (Supplementary Figure S9). Similarly, in the MDA-MB-468 cells addition of Wnt3a result in a visible strong reduction of phosphorylated β-catenin, and this effect is again unsusceptible to the action of the compound 8.
With the strong inhibition of the target genes that we observe, we can therefore conclude that the Wnt inhibitory activity of the compounds studied is probably due to interference with the nuclear effectors of the pathway and thus with Wnt-induced transcription. Considering that selected compounds represent all the main scaffolds produced by biotransformation, this mechanism of action can be extrapolated to the entire set of the active compounds reported here.
Conclusion
Biotransformation reactions of the mixture of pterostilbene and resveratrol with the B. cinerea secretome generated a large diversity of 73 dimeric stilbene analogs organized into six scaffolds. All compounds were obtained by dimerization of the stilbene units by coupling of phenoxy radicals generated by oxidation with laccases (Langcake and Pryce, 1977; Keylor et al., 2015). The use of five different organic solvents in the biotransformation reactions enriched the chemodiversity of the library generated. These compounds (representing 78% of the library) were produced by a nucleophilic solvent attack (solvolysis) on a sp2 carbon of an intermediate structure (Figure 4) (Righi et al., 2020).
The structural sources of these derivatives (resveratrol and pterostilbene) were reported to both inhibit or activate Wnt pathway in various contexts, including cancers, such as gastric (Aquilano et al., 2009), lung (Langcake and Pryce, 1977), colon (Hope et al., 2008; Khan et al., 2020), endometrial (Wang et al., 2012; Mineda et al., 2019), and breast (Wang et al., 2012). Typically for PAINS (Pan-Assay INterfering compoundS), no clear target was identified, and likely the observed effects on Wnt pathway are the results of interaction with multiple targets and overlapping of various mechanisms of action. Such effects obviously depend on the context and the exact way of stimulating the pathway, thus we first verified to which extent our source compounds might inhibit Wnt signaling induced in BT-20 TNBC cells by addition of ligand in order to take into account inheritance of the same unspecific activity trend by the derivatives. Our results demonstrate no specific interference of the resveratrol and pterostilbene with Wnt3a-induced activity in TNBC cells.
The generated derivatives obtained by the enzymatic biotransformation of these two stilbenoids, however, result from a significant modification of the structures of the original compounds. None of the classes of the identified derivatives are described as PAINS, and overall, one might expect that the structural rearrangements in the derived molecules will result in the loss of the main part of the PAINS features of the original compounds—such as high reactivity, low structure complexity and ability to interfere with the redox balance, features that lead to the promiscuity due to oxidation and etherification. We also identified that none of the classes of investigated derivatives was reported as Wnt inhibitor before.
Nevertheless, despite the extent of structural variation across the large set of compounds studied, we identified that the anti-Wnt activity of the derivatives is not restricted to any particular scaffold, although some trends can be observed. It should be stressed that certain chemical scaffolds, such as the pallidol, the viniferin and the restrytisol series are represented by only one or several derivatives with results highly divergent among them and thus provide us only very limited information about their potential as Wnt inhibitors. The leachianol and acyclic dimer series have many more representatives and are therefore more adapted to in-depth analysis.
The acyclic dimer series shows little or no structure-activity relationship. Indeed, neither the nature of the monomers nor the erythto/threo configuration properly correlates with the observed IC50s. Two compounds from this series stand out concerning their selectivity indexes (64 and 69). Both are pterostilbene dimers and have a threo configuration. However, other derivatives with the same characteristics (40 and 53) have much lower SI values. This lack of structure-activity relationship could be explained by the high flexibility of this scaffold compared to the other ones. The absence of cyclization during the dimerization process leads to a high number of free-rotating bonds and therefore to a higher diversity of conformers.
A clearer correlation between structure and activity can be observed for the leachianol series. In general, the resveratrol dimers show the lowest activities, whereas the resveratrol-pterostilbene dimers show the highest ones. This could indicate that the presence of one more apolar side (with methoxy groups) is favorable to the interaction with the target. We however couldn’t correlate the observed activities within this scaffold with the F or G configuration, neither with the type of side chain introduced by the solvent. The pharmacophore models did not give further insight for this series in particular.
The in-depth investigations of the potential mechanism of action of these derivatives suggest that the compounds act on the transcriptional machinery associated with the Wnt pathway. The determination of the exact target protein(s) is clearly a task for future work and is expected to explain why so structurally divergent derivatives are all capable of specific inhibition of the pathway. Currently, it can be speculated in light of the different pharmacophore models that this is due to the presence of highly similar structural determinants in these compounds, present in optimal positions on different backbones. A significant input in activity can be also expected from differential stability, permeability and compartmentalization of the compounds, which is especially affected by the solvent-derived moieties.
Experimental Section
General Experiment Procedures
The UV spectra were recorded on an Agilent Cary 60 UV-Vis (Santa-Clara, CA, United States) spectrometer in MeCN, using a 1 cm cell. ECD spectra were recorded on a JASCO J-815 spectrometer (Loveland, CO, United States) in MeCN, using a 1 cm cell. NMR data were recorded on a Bruker Avance Neo 600 MHz NMR spectrometer equipped with a QCI 5 mm cryoprobe and a SampleJet automated sample changer (Bruker BioSpin, Rheinstetten, Germany). 1D and 2D NMR experiments (1H, COSY, ROESY, HSQC, and HMBC) were recorded in DMSO-d6. Chemical shifts are reported in parts per million (δ) and coupling constants (J) in Hz. The residual DMSO-d6 signal (δH 2.50; δC 39.5) were used as internal standards for 1H and 13C NMR, respectively. Resveratrol (99% purity) and pterostilbene (99% purity) were purchased from Biopurify Phytochemicals Ltd. (Chengdu, China).
Secretome Isolation From Botrytis cinerea Culture
The B. cinerea Pers., isolate K16, was obtained from naturally sporulated grape berries from the Changins Agroscope experimental vineyards in 2015. The strain was purified, determined phenotypically as well as by molecular tools (sequencing of the ITS regions), and maintained by regular transplanting. The fungus was grown on oatmeal agar medium (Difco), and conidia were sampled by vacuum aspiration and stored dry at −80°C until use. B. cinerea was cultivated in 1.5 L liquid medium (in 5 L bottles) at 22°C under alternating light and dark (12 h/12 h). The medium was filtrated through a double layer of folded filter paper (diameter 500 mm, Prat Dumas, France). The filtrate was brought to 80% saturation [(NH4)2SO4, 24 h at 4°C] and centrifuged (4,200 g, 4°C, 2 h). The supernatant was discarded, and the resulting pellet was solubilized in nanopure water (Evoqua Water Technologies, 4.21 μS cm−1) (the protein crude extract) and submitted to a dialysis (Spectra/Por 1 dialysis membrane, 6–8 kDa, diameter 14.6 mm) against nanopure water overnight at 4°C. The resulting protein extract was concentrated against polyethylene glycol beads (PEG 20,000) in the dialyzed tube. The protein content was determined by the method of Bradford, using a Bio-Rad protein assay kit with BSA as a standard. The volume of the extract was adjusted to obtain a final protein concentration of 2 μg/μl. The resulting extract (considered as secretome) was aliquoted to 1 ml and stored at −20°C until use.
Biotransformation of the Resveratrol/Pterostilbene Mixture at Analytical Scale
The biotransformation reactions of the resveratrol/pterostilbene mixtures at analytical scale were performed in 2 ml Eppendorf tubes. The conditions were kept constant for each reaction as follows: 1 ml of total volume, 1 mg/ml of total substrate concentration (0.5 mg/ml of each compound), 50% of alcohol (i-PrOH, n-BuOH, i-BuOH, MeOH or EtOH, 500 µl), 48% of water (480 µl), and 2% of secretome (20 µl). Once prepared, the mixtures were incubated at room temperature in the dark under constant gentle agitation. Reactions were stopped after 24 h by removing the solvent under vacuum in a centrifugal evaporator. 1 ml of MeOH was then added on the dry deposit. The sample was centrifugated and the supernatant was analyzed by UHPLC-PDA-ELSD-MS.
UHPLC-PDA-ELSD-MS Analysis
The crude reaction mixtures were analyzed on an Ultra-high-performance liquid chromatography system equipped with a photodiode array, an evaporative light-scattering detector, and a single quadrupole detector using heated electrospray ionization (UHPLC-PDA-ELSD-MS) (Waters, Milford, MA, United States). The ESI parameters were the following: capillary voltage 800 V, cone voltage 15 V, source temperature 120°C, and probe temperature 600°C. Acquisition was done in negative ionization mode (NI) with an m/z range of 150–1,000 Da. The chromatographic separation was performed on an Acquity UPLC BEH C18 column (50 × 2.1 mm i.d., 1.7 μm; Waters) at 0.6 ml/min, 40°C with H2O (A) and MeCN (B) both containing 0.1% formic acid as solvents. The gradient was carried out as follow: 5–100% B in 7 min, 1 min at 100% B, and a re-equilibration step at 5% B for 2 min. The ELSD temperature was fixed at 45°C, with a gain of 9. The PDA data were acquired from 190 to 500 nm, with a resolution of 1.2 nm. The sampling rate was set at 20 points/s. Every data was processed with the MassLynx software (Waters, Milford, MA, United States).
UHPLC-PDA-CAD-HRMS Analysis
The pure compounds were analyzed on a Waters Acquity UHPLC system equipped with a Q-Exactive Focus mass spectrometer (Thermo Scientific, Bremen, Germany), using heated electrospray ionization source (HESI-II). The chromatographic separation was carried out on an Acquity UPLC BEH C18 column (50 × 2.1 mm i.d., 1.7 μm; Waters) at 0.6 ml/min, 40°C with H2O (A) and MeCN (B) both containing 0.1% formic acid as solvents. The gradient was carried out as follow: 5%–100% B in 7 min, 1 min at 100% B, and a re-equilibration step at 5% B in 2 min. The ionization parameters were the same as used in Rutz et al. (2019).
Biotransformation of the Resveratrol/Pterostilbene Mixture at Large Scale
The biotransformation reactions at large scale were performed by keeping the same parameters as the analytical scale (substrate concentration, solvent and secretome percentages) but increasing the amount of starting materials. Typically, reactions were performed in Schott bottles of 200–500 ml using 200–400 mg of starting material (resveratrol + pterostilbene). Substrates were first dissolved in 100–200 ml of alcohol (i-PrOH, n-BuOH, i-BuOH, MeOH or EtOH), water was then added (50% of the total volume) and the secretome volume (2% of the total volume) was added last. This mixture was incubated for 24 h at room temperature in the dark under gentle magnetic stirring. After evaporation of the solvent with a rotatory evaporator, the dry deposit was resuspended in MeOH and filtered through 0.45 µm filters (13 mm Syringe filter, PVDF, BGB Analytik, Böckten, Switzerland). These crude reaction mixtures were analyzed by UHPLC-PDA-ELSD-MS for comparison with analytical scale reactions before moving to the gradient optimization and semi-preparative HPLC fractionation.
Purification of the Crude Reaction Mixtures by Semi-preparative HPLC-UV
The separation conditions of the 5 crude reaction mixtures were optimized on the UHPLC-PDA-ELSD-MS system with an Acquity UPLC BEH C18 column (100 × 2.1 mm i.d., 1.7 μm; Waters) at 0.4 ml/min, 40°C with H2O (A) and MeOH (B) both containing 0.1% formic acid as solvents. The optimized gradient conditions for each reaction were geometrically transferred by gradient transfer to the analytical and semi-preparative HPLC scale (Supplementary Table S1, for each chromatographic conditions). The geometrically transferred gradients were first tested on a HP 1260 Agilent High-Performance liquid chromatography equipped with a photodiode array detector (HPLC-PDA) (Agilent technologies, Santa Clara, CA, United States). The chromatographic separation was performed on an XBridge C18 column (250 × 4.6 mm i.d., 5 μm; Waters) equipped with a C18 pre-column at 1 ml/min, with H2O (A) and MeOH (B) both containing 0.1% formic acid as solvents. The UV absorbance was measured at 210 and 254 nm and the UV-PDA spectra were recorded between 190 and 600 nm (step 2 nm). The geometrically transferred gradients were used at the semi-preparative scale on a Shimadzu system equipped with an LC-20 A module pumps, an SPD-20 A UV/VIS, a 7725I Rheodyne® valve, and an FRC-40 fraction collector (Shimadzu, Kyoto, Japan). The separation was performed on a XBridge C18 column (250 mm × 19 mm i.d., 5 μm; Waters) equipped with a C18 pre-column cartridge holder (10 mm × 19 mm i.d., 5 μm; Waters) at 17 ml/min, with H2O (A) and MeOH (B) both containing 0.1% formic acid as solvents. The UV detection was set at 210, 254 and 280 nm. The mixtures were injected on the semi-preparative HPLC column using a dry-load methodology developed in our laboratory (Queiroz et al., 2019).
Biotransformation Reaction N°1 (With i-PrOH as Cosolvent)
Six semi-preparative HPLC dry-load injections of about 60 mg each yielded 60 fractions each. Fractions were analyzed by UHPLC-PDA-ELSD-MS and mixed together according to their composition. 24 compounds were obtained pure: 1 (7.3 mg, tR 5.1 min), 23 (0.2 mg, tR 6.9 min), 2 (7.7 mg, tR 8.0 min), 3 (1.1 mg, tR 9.2 min), 4 (6.6 mg, tR 12.3 min), 5 (10.0 mg, tR 14.7 min), 6 (1.1 mg, tR 17.5 min), 7 (0.3 mg, tR 19.0 min), 8 (1.4 mg, tR 19.8 min), 9 (60.7 mg, tR 21.0 min), 10 (0.7 mg, tR 25.5 min), 11 (8.5 mg, tR 27.0 min), 12 (4.6 mg, tR 29.6 min), 13 (6.1 mg, tR 33.5 min), 14 (21 mg, tR 35.2 min), 15 (0.4 mg, tR 37.7 min), 16 (38.4 mg, tR 39.9 min), 17 (2.7 mg, tR 44.9 min), 18 (49.9 mg, tR 46.0 min), 24 (0.4 mg, tR 47.1 min), 19 (1.2 mg, tR 49.5 min), 20 (1.8 mg, tR 50.3 min), 21 (5.4 mg, tR 51.2 min), and 22 (28.3 mg, tR 64.0 min).
Biotransformation Reaction N°2 (With n-BuOH as Cosolvent)
16 compounds were purified based on 2 injections of 55 mg each: 25 (5.1 mg, tR 13.2 min), 26 (0.5 mg, tR 14.1 min), 27 (0.3 mg, tR 20.5 min), 28 (0.4 mg, tR 28.0 min), 29 (0.2 mg, tR 29.0 min), 30 (1.8 mg, tR 31.3 min), 31 (3.8 mg, tR 36.9 min), 32 (0.1 mg, tR 38.8 min), 33 (1.0 mg, tR 46.7 min), 34 (0.7 mg, tR 48.0 min), 35 (0.3 mg, tR 48.8 min), 36 (0.2 mg, tR 51.7 min), 37 (1.0 mg, tR 52.8 min), 38 (0.4 mg, tR 53.4 min), 39 (0.3 mg, tR 65.8 min), and 40 (0.4 mg, tR 66.5 min).
Biotransformation Reaction N°3 (With i-BuOH as Cosolvent)
13 compounds purified based on 1 injection of 55 mg: 41 (2.1 mg, tR 13.8 min), 42 (0.4 mg, tR 29.0 min), 43 (0.7 mg, tR 29.8 min), 44 (0.6 mg, tR 32.8 min), 45 (1.9 mg, tR 38.1 min), 46 (0.4 mg, tR 48.1 min), 47 (0.1 mg, tR 49.2 min), 48 (0.1 mg, tR 49.7 min), 49 (0.1 mg, tR 53.0 min), 50 (0.3 mg, tR 53.9 min), 51 (0.2 mg, tR 54.8 min), 52 (0.2 mg, tR 66.5 min), and 53 (0.5 mg, tR 67.5 min).
Biotransformation Reaction N°4 (With MeOH as Cosolvent)
11 compounds were purified based on 2 injections of 50 mg: 54 (0.2 mg, tR 22.2 min), 55 (0.1 mg, tR 24.4 min), 56 (0.6 mg, tR 24.6 min), 57 (0.6 mg, tR 25.6 min), 58 (1.1 mg, tR 30.2 min), 59 (6 mg, tR 31.5 min), 60 (1.3 mg, tR 37.9 min), 61 (0.2 mg, tR 38.8 min), 62 (1 mg, tR 42.6 min), 63 (2.7 mg, tR 44.1 min), and 64 (0.4 mg, tR 54.1 min).
Biotransformation Reaction N°5 (With EtOH as Cosolvent)
6 compounds were purified based on 2 injections of 40 mg: 70 (0.5 mg, tR 22.5 min), 65 (1.6 mg, tR 35.8 min), 66 (0.5 mg, tR 41.6 min), 67 (5.4 mg, tR 42.5 min), 68 (0.4 mg, tR 54.8 min), 69 (0.7 mg, tR 70.6 min). The fractions collected from 17.5 to 19.0 min (3.6 mg) were further purified on a smaller XBridge C18 column (250 × 10 mm i.d., 5 μm; Waters) by using isocratic conditions at 30% MeOH to give compound 71 (0.7 mg, tR 50 min). The fractions collected from 33.5 to 35.0 min (2.3 mg) were further purified on a smaller XBridge C18 column (250 × 10 mm i.d., 5 μm; Waters) by using isocratic conditions at 42% MeOH to give compound 72 (1.1 mg,, tR 68 min) and 73 (0.2 mg, tR 92.5 min).
Description of the Generated Compounds
Pallidol (1) UV (MeCN) λmax (log ε) 228 (sh) (4.29), 283 (3.58) nm. 1H NMR (DMSO, 600 MHz) δ 3.60 (2H, s, H-8, H-8′), 4.30 (2H, s, H-7, H-7′), 6.05 (2H, d, J = 2.0 Hz, H-12, H-12′), 6.38 (2H, d, J = 2.0 Hz, H-10, H-10′), 6.60 (4H, d, J = 8.5 Hz, H-3, H-3′, H-5, H-5′), 6.84 (4H, d, J = 8.5 Hz, H-2, H-2′, H-6, H-6′), 8.88 (2H, s), 9.01 (2H, s), 9.05 (2H, s). HR-ESI/MS analysis: m/z 453.1346 [M-H]-, (calcd for C28H21O6, 453.1338, ∆ = 1.7 ppm). MS/MS spectrum: CCMSLIB00009918856.
Threo-resveratrol acyclic dimer (2) UV (MeCN) λmax (log ε) 228 (sh) (4.25), 285 (4.00), 306 (4.14), 323 (4.15), 340 (sh) (3.85) nm. 1H NMR (DMSO, 600 MHz) δ 4.66 (1H, dd, J = 6.4, 4.5 Hz, H-7′), 5.00 (1H, d, J = 6.4 Hz, H-8′), 5.41 (1H, d, J = 4.5 Hz, 7′OH), 5.97 (1H, t, J = 2.2 Hz, H-12′), 6.03 (2H, d, J = 2.2 Hz, H-10′, H-14′), 6.10 (1H, t, J = 2.1 Hz, H-12), 6.35 (2H, d, J = 2.1 Hz, H-10, H-14), 6.58 (2H, d, J = 8.6 Hz, H-3′, H-5′), 6.81 (1H, d, J = 16.2 Hz, H-8), 6.83 (2H, d, J = 8.6 Hz, H-3, H-5), 6.89 (1H, d, J = 16.2 Hz, H-7), 6.99 (2H, d, J = 8.5 Hz, H-2′, H-6′), 7.38 (2H, d, J = 8.5 Hz, H-2, H-6), 9.02 (2H, s, 11′OH, 13′OH), 9.18 (3H, m, 4′OH, 11OH, 13OH). HR-ESI/MS analysis: m/z 471.1451 [M-H]−, (calcd for C28H23O7, 471.1444, ∆ = 1.5 ppm). MS/MS spectrum: CCMSLIB00009918857.
Erythro-resveratrol acyclic dimer (3) UV (MeCN) λmax (log ε) 229 (sh) (4.33), 285 (4.00), 308 (4.10), 324 (4.11) nm. 1H NMR (DMSO, 600 MHz) δ 4.63 (1H, t, J = 6.3, 4.8 Hz, H-7′), 5.00 (1H, d, J = 6.3 Hz, H-8′), 5.30 (1H, d, J = 4.8 Hz, 7′OH), 6.05 (1H, t, J = 2.2 Hz, H-12′), 6.09 (1H, t, J = 2.2 Hz, H-12), 6.18 (2H, d, J = 2.2 Hz, H-10′, H-14′), 6.34 (2H, d, J = 2.2 Hz, H-10, H-14), 6.65 (2H, d, J = 8.5 Hz, H-3′, H-5′), 6.71 (2H, d, J = 8.7 Hz, H-3, H-5), 6.79 (1H, d, J = 16.2 Hz, H-8), 6.87 (1H, d, J = 16.2 Hz, H-7), 7.13 (2H, d, J = 8.5 Hz, H-2′, H- 6′), 7.34 (2H, d, J = 8.7 Hz, H-2, H-6), 9.07 (2H, s, 11′OH, 13′OH), 9.17 (2H, s, 11OH, 13OH), 9.20 (1H, s, 4′OH). 1H NMR (Acetone, 600 MHz) δ 4.28 (1H, d, J = 4.7 Hz, 7′OH), 4.86 (1H, d, J = 5.8 Hz, H-7′), 5.13 (1H, d, J = 5.8 Hz, H-8′), 6.23 (1H, t, J = 2.2 Hz, H-12′), 6.25 (1H, t, J = 2.2 Hz, H-12), 6.34 (2H, d, J = 2.2 Hz, H-10′, H-14′), 6.50 (2H, d, J = 2.2 Hz, H-10, H-14), 6.74 (2H, d, J = 8.6 Hz, H-3′, H-5′), 6.80 (1H, d, J = 8.8 Hz, H-3, H-5), 6.84 (1H, d, J = 16.3 Hz, H-8), 6.95 (1H, d, J = 16.3 Hz, H-7), 7.23 (2H, d, J = 8.6 Hz, H-2′, H-6′), 7.35 (2H, d, J = 8.8 Hz, H-2, H-6); 13C NMR (Acetone, 151 MHz) δ 77.3 (C-7′), 84.9 (C-8′), 102.7 (C-12), 102.8 (C-12′), 105.7 (C-10, C-14), 107.1 (C-10′, C-14′), 115.2 (C-3′, C-5′), 117.0 (C-3, C-5), 127.5 (C-8), 128.3 (C-2, C-6), 128.8 (C-7), 129.5 (C-2′, C-6′), 131.0 (C-1), 133.7 (C-1′), 140.7 (C-9), 142.1 (C-9′), 157.5 (C-4′), 158.9 (C-4), 159.0 (C-11′, C-13′), 159.6 (C-11, C-13). HR-ESI/MS analysis: m/z 471.1451 [M-H]-, (calcd for C28H23O7, 471.1444, ∆ = 1.5 ppm). MS/MS spectrum: CCMSLIB00009918858.
7-O-Isopropylleachianol G (4) UV (MeCN) λmax (log ε) 228 (sh) (4.36), 281 (3.60) nm. 1H NMR (DMSO, 600 MHz) δ 0.60 (3H, d, J = 6.0 Hz, CH3-7c), 0.83 (3H, d, J = 6.0 Hz, CH3-7b), 3.02 (1H, dd, J = 9.2, 2.1 Hz, H-8), 3.06 (1H, hept, J = 6.0 Hz, H-7a), 3.35 (1H, t, J = 2.1 Hz, H-8′), 3.89 (1H, d, J = 9.2 Hz, H-7), 4.06 (1H, d, J = 2.1 Hz, H-7′), 5.17 (1H, d, J = 2.1 Hz, H-14), 5.93 (2H, d, J = 2.2 Hz, H-10′, H-14′), 5.98 (1H, t, J = 2.2 Hz, H-12′), 6.07 (1H, d, J = 2.1 Hz, H-12), 6.64 (2H, d, J = 8.5 Hz, H-3′, H-5′), 6.67 (2H, d, J = 8.4 Hz, H-3, H-5), 6.78 (2H, d, J = 8.5 Hz, H-2′, H-6′), 6.88 (2H, d, J = 8.4 Hz, H-2, H-6), 8.74 (1H, s, 13OH), 8.77 (1H, s, 11OH), 8.99 (2H, s, 11′OH, 13′OH), 9.04 (1H, s, 4′OH), 9.29 (1H, s, 4OH); 13C NMR (DMSO, 151 MHz) δ 20.2 (CH3-7c), 23.4 (CH3-7b), 53.9 (C-7′), 57.6 (C-8′), 60.3 (C-8), 67.1 (C-7a), 81.1 (C-7), 100.0 (C-12′), 101.4 (C-12), 104.3 (C-14), 104.6 (C-10′, C-14′), 114.6 (C-3, C-3′, C-5, C-5′), 121.3 (C-10), 128.1 (C-2′, C-6′), 128.9 (C-2, C-6), 131.6 (C-1), 136.8 (C-1′), 145.6 (C-9), 150.5 (C-9′), 154.0 (C-11), 155.1 (C-4′), 156.6 (C-4), 157.1 (C-13), 158.2 (C-11′, C-13′). HR-ESI/MS analysis: m/z 513.1910 [M-H]−, (calcd for C31H29O7, 513.1913, ∆ = 0.6 ppm). MS/MS spectrum: CCMSLIB00009918859.
Di-O-methyl-restrytisol A (5) UV (MeCN) λmax (log ε) 228 (sh) (4.15), 281 (3.40) nm. 1H NMR (DMSO, 600 MHz) δ 3.53 (6H, s, CH3O-11′, CH3O-13′), 3.90 (1H, t, J = 6.2, 4.3 Hz, H-8′), 3.96 (1H, dd, J = 10.3, 6.2 Hz, H-8), 5.39 (1H, d, J = 10.3 Hz, H-7), 5.76 (1H, d, J = 4.3 Hz, H-7′), 5.87 (1H, t, J = 2.1 Hz, H-12), 5.89 (2H, d, J = 2.2 Hz, H-10, H-14), 6.05 (1H, t, J = 2.3 Hz, H-12′), 6.09 (2H, d, J = 2.3 Hz, H-10′, H-14′), 6.54 (2H, d, J = 8.6 Hz, H-3′, H-5′), 6.70 (2H, d, J = 8.5 Hz, H-3, H-5), 7.04 (2H, d, J = 8.6 Hz, H-2′, H-6′), 7.26 (2H, d, J = 8.6 Hz, H-2, H-6), 8.88 (2H, s, 11OH, 13OH), 9.08 (1H, s, 4′OH), 9.33 (1H, s, 4OH); 13C NMR (DMSO, 151 MHz) δ 54.7 (CH3O-11′, CH3O-13′), 56.6 (C-8′), 58.6 (C-8), 81.2 (C-7), 83.3 (C-7′), 97.6 (C-12′), 100.4 (C-12), 106.7 (C-12, C-14), 108.5 (C-10′, C-14′), 114.3 (C-3′, C-5′), 115.0 (C-3, C-5), 126.8 (C-2′, C-6′), 127.8 (C-2, C-6), 130.1 (C-1′), 133.0 (C-1), 155.2 (C-4′), 156.6 (C-4), 157.5 (C-11, C-13), 159.1 (C-11′, C-13′). HR-ESI/MS analysis: m/z 499.1760 [M-H]-, (calcd for C30H27O7, 499.1757, ∆ = 0.6 ppm). MS/MS spectrum: CCMSLIB00009918860.
11′,13′-Di-O-methyltricuspidatol A (6) UV (MeCN) λmax (log ε) 228 (sh) (4.35), 281 (3.74), 323 (3.44) nm. 1H NMR (DMSO, 600 MHz) δ 3.44 (2H, m, H-8, H-8′), 3.63 (6H, s, CH3O-11′, CH3O-13′), 5.06 (1H, m, H-7), 5.12 (1H, m, H-7′), 5.97 (1H, t, J = 2.2 Hz, H-12), 6.01 (2H, d, J = 2.1 Hz, H-10, H-14), 6.24 (2H, d, J = 2.3 Hz, H-10′, H-14′), 6.27 (1H, t, J = 2.2 Hz, H-12′), 6.69 (1H, d, J = 8.5 Hz, H-3′, H-5′), 6.70 (2H, d, J = 8.5 Hz, H-3, H-5), 7.13 (2H, d, J = 8.5 Hz, H-2′, H-6′), 7.16 (2H, d, J = 8.5 Hz, H-2, H-6), 9.01 (2H, s, 11OH, 13OH), 9.31 (1H, s, 4′OH), 9.32 (1H, s, 4OH); 13C NMR (DMSO, 151 MHz) δ 54.7 (CH3O-11′, CH3O-13′), 60.5 (C-8), 61.1 (C-8′), 86.0 (C-7′), 86.5 (C-7), 97.7 (C-12′), 100.9 (C-12), 105.8 (C-10, C-14), 106.0 (C-10′, C-14′), 114.6 (C-3, C-5, C-3′, C-5′), 127.8 (C-2, C-6, C-2′, C-6′), 131.5 (C-1, C-1′), 156.6 (C-4, C-4′), 157.8 (C-11, C-13), 160.0 (C-11′, C-13′). HR-ESI/MS analysis: m/z 499.1760 [M-H]-, (calcd for C30H27O7, 499.1757, ∆ = 0.6 ppm). MS/MS spectrum: CCMSLIB00009918861.
11,13-Di-O-methylrestrytisol B (7) UV (MeCN) λmax (log ε) 228 (sh) (4.21), 281 (3.47) nm. 1H NMR (DMSO, 600 MHz) δ 3.39 (1H, t, J = 10.2, 8.9 Hz, H-8), 3.64 (6H, s, CH3O-13, CH3O-11), 3.95 (1H, t, J = 10.2, 9.7 Hz, H-8′), 4.93 (1H, d, J = 9.7 Hz, H-7), 5.39 (1H, d, J = 8.9 Hz, H-7′), 5.78 (1H, t, J = 2.2 Hz, H-12′), 5.85 (2H, d, J = 2.2 Hz, H-10′, H-14′), 6.27 (1H, t, J = 2.2 Hz, H-12), 6.34 (2H, d, J = 2.2 Hz, H-10, H-14), 6.51 (2H, d, J = 8.5 Hz, H-3′, H-5′), 6.73 (2H, d, J = 8.7 Hz, H-3, H-5), 6.95 (2H, d, J = 8.5 Hz, H-2′, H-6′), 7.21 (2H, d, J = 8.7 Hz, H-2, H-6), 8.78 (2H, s, 11′OH, 13′OH), 9.08 (1H, s, 4′OH), 9.38 (1H, s, 4OH); 13C NMR (DMSO, 151 MHz) δ 54.7 (CH3O-11, CH3O-13), 57.5 (C-8), 57.6 (C-8′), 82.4 (C-7′), 85.4 (C-7), 97.6 (C-12), 100.2 (C-12′), 106.2 (C-10, C-14), 107.1 (C-10′, C-14′), 113.9 (C-3′, C-5′), 114.8 (C-3, C-5), 127.6 (C-2′, C-6′), 128.0 (C-2, C-6), 129.7 (C-1), 130.6 (C-1′), 141.6 (C-9), 141.8 (C-9′), 155.5 (C-4′), 156.7 (C-4), 157.1 (C-11′, C-13′), 160.0 (C-11, C-13). HR-ESI/MS analysis: m/z 499.1758 [M-H]−, (calcd for C30H27O7, 499.1757, ∆ = 0.2 ppm). MS/MS spectrum: CCMSLIB00009918862.
11, 13-Di-O-methylisopallidol (8) UV (MeCN) λmax (log ε) 229 (sh) (4.29), 282 (3.64) nm. 1H NMR (DMSO, 600 MHz) δ 3.57 (3H, s, CH3O-13), 3.64 (1H, d, J = 6.2 Hz, H-8′), 3.69 (1H, d, J = 6.2 Hz, H-8), 3.80 (3H, s, CH3O-11), 4.33 (1H, s, H-7′), 4.41 (1H, s, H-7), 6.05 (1H, d, J = 2.0 Hz, H-12′), 6.31 (1H, d, J = 2.1 Hz, H-12), 6.40 (1H, d, J = 2.0 Hz, H-10′), 6.60 (2H, d, J = 8.5 Hz, H-3, H-5), 6.61 (2H, d, J = 8.5 Hz, H-3′, H-5′), 6.75 (1H, d, J = 2.1 Hz, H-10), 6.82 (2H, d, J = 8.5 Hz, H-2′, H-6′), 6.90 (2H, d, J = 8.5 Hz, H-2, H-6), 8.92 (1H, s), 9.03 (1H, s), 9.06 (1H, s), 9.09 (1H, s). HR-ESI/MS analysis: m/z 481.1657 [M-H]−, (calcd for C30H25O6, 481.1651, ∆ = 1.2 ppm). MS/MS spectrum: CCMSLIB00009918863.
Trans-δ-viniferin (9) UV (MeCN) λmax (log ε) 227 (sh) (4.46), 285 (4.08), 312 (4.32), 333 (4.127), 350 (sh) (3.99) nm. 1H NMR (DMSO, 600 MHz) δ 4.44 (1H, d, J = 7.8 Hz, H-8′), 5.39 (1H, d, J = 7.8 Hz, H-7′), 6.03 (2H, d, J = 2.1 Hz, H-10′, H-14′), 6.10 (1H, t, J = 2.1 Hz, H-12′), 6.11 (1H, t, J = 2.2 Hz, H-12), 6.37 (2H, d, J = 2.2 Hz, H-10, H-14), 6.76 (2H, d, J = 8.6 Hz, H-3′, H-5′), 6.83 (1H, d, J = 16.3 Hz, H-8), 6.89 (1H, d, J = 8.3 Hz, H-5), 6.98 (1H, d, J = 16.3 Hz, H-7), 7.18 (2H, d, J = 8.6 Hz, H-2′, H-6′), 7.23 (1H, d, J = 1.9 Hz, H-2), 7.42 (1H, dd, J = 8.3, 1.9 Hz, H-6), 9.17 (2H, s), 9.21 (2H, s), 9.53 (1H, s, 4′OH). HR-ESI/MS analysis: m/z 453.1348 [M-H]-, (calcd for C28H21O6, 453.1338, ∆ = 2.2 ppm). MS/MS spectrum: CCMSLIB00009918864.
7-O-Isopropyl-11′,13′-di-O-methylleachianol F (10) UV (MeCN) λmax (log ε) 228 (sh) (4.30), 284 (3.76), 323 (3.70) nm. 1H NMR (DMSO, 600 MHz) δ 0.91 (6H, d, J = 6.1 Hz, CH3-7b, CH3-7c), 2.71 (1H, t, J = 3.4 Hz, H-8′), 3.05 (1H, dd, J = 9.1, 3.4 Hz, H-8), 3.21 (1H, hept, J = 6.1 Hz, H-7a), 3.57 (6H, s, CH3O-13′, CH3O-11′), 4.00 (1H, d, J = 9.1 Hz, H-7), 4.06 (1H, d, J = 3.1 Hz, H-7′), 5.73 (2H, d, J = 2.2 Hz, H-10′, H-14′), 6.16 (1H, d, J = 2.2 Hz, H-12), 6.20 (1H, t, J = 2.2 Hz, H-12′), 6.47 (1H, d, J = 2.2 Hz, H-14), 6.57 (2H, d, J = 8.5 Hz, H-3, H-5), 6.64 (1H, d, J = 8.5 Hz, H-3′, H-5′), 6.64 (2H, d, J = 8.5 Hz, H-2, H-6), 6.69 (2H, d, J = 8.5 Hz, H-2′, H-6′), 8.77 (1H, s, 11OH), 8.95 (1H, s, 13OH), 9.10 (1H, s, 4′OH), 9.25 (1H, s, 4OH); 13C NMR (DMSO, 151 MHz) δ 53.6 (C-7′), 54.7 (CH3O-11′, CH3O-13′), 57.5 (C-8′), 59.8 (C-8), 67.7 (C-7a), 81.2 (C-7), 96.9 (C-12′), 101.2 (C-12), 104.2 (C-10′, C-14′), 144.4 (C-3, C-5, C-3′, C-5′), 120.5 (C-10), 128.0 (C-2′, C-6′), 128.2 (C-2, C-6), 131.7 (C-1), 136.0 (C-1′), 147.0 (C-9), 149.2 (C-9′), 153.5 (C-11), 155.1 (C-4′), 156.2 (C-4), 157.3 (C-13), 159.8 (C-11′, C-13′). HR-ESI/MS analysis: m/z 541.2225 [M-H]−, (calcd for C33H33O7, 541.2226, ∆ = 0.2 ppm). MS/MS spectrum: CCMSLIB00009918865.
Erythro-7′-O-isopropylresveratrol acyclic dimer (11) UV (MeCN) λmax (log ε) 229 (sh) (4.32), 284 (3.73), 325 (3.67) nm. 1H NMR (DMSO, 600 MHz) δ 0.87 (3H, d, J = 6.0 Hz, H-7′c), 0.91 (3H, d, J = 6.2 Hz, H-7′b), 3.33 (1H, overlapped, H-7′a), 4.43 (1H, d, J = 6.6 Hz, H-7′), 5.02 (1H, d, J = 6.6 Hz, H-8′), 6.06 (1H, t, J = 2.2 Hz, H-12′), 6.09 (1H, t, J = 2.1 Hz, H-12), 6.21 (2H, d, J = 2.2 Hz, H-10′, H-14′), 6.34 (2H, d, J = 2.1 Hz, H-10, H-14), 6.67 (1H, d, J = 8.6 Hz, H-3′, H5′), 6.70 (2H, d, J = 8.8 Hz, H-3, H-5), 6.78 (1H, d, J = 16.6 Hz, H-8), 6.86 (1H, d, J = 16.6 Hz, H-7), 7.16 (2H, d, J = 8.6 Hz, H-2′, H-6′), 7.33 (2H, d, J = 8.8 Hz, H-2, H-6), 9.08 (2H, s, 11′OH, 13′OH), 9.17 (2H, s, 11OH, 13OH), 9.26 (1H, s, 4′OH); 13C NMR (DMSO, 151 MHz) δ 21.2 (C-7′c), 23.0 (C-7’b), 69.4 (C-7’a), 81.4 (C-7′), 82.1 (C-8′), 101.8 (C-12, C-12′), 104.2 (C-10, C-14), 105.7 (C-10′, C-14′), 114.3 (C-3′, C-5′), 115.9 (C-3, C-5), 126.6 (C-8), 127.4 (C-2, C-6, C-7), 128.9 (C-2′, C-6′), 129.4 (C-1), 129.8 (C-1′), 138.8 (C-9), 141.1 (C-9′), 156.6 (C-4′), 157.0 (C-4), 157.7 (C-11′, C-13′), 158.4 (C-11, C-13). HR-ESI/MS analysis: m/z 513.1913 [M-H]-, (calcd for C31H29O7, 513.1913, ∆ = 0 ppm). MS/MS spectrum: CCMSLIB00009918866.
Threo-7′-O-isopropylresveratrol acyclic dimer (12) UV (MeCN) λmax (log ε) 228 (sh) (4.29), 285 (4.01), 305 (4.14), 324 (4.15), 343 (sh) (3.84) nm. 1H NMR (DMSO, 600 MHz) δ 0.96 (3H, d, J = 6.2 Hz, H-7′c), 1.03 (3H, d, J = 6.2 Hz, H-7'b), 3.47 (1H, hept, J = 6.2 Hz, H-7′a), 4.51 (1H, d, J = 6.3 Hz, H-7′), 5.06 (1H, d, J = 6.3 Hz, H-8′), 5.97 (1H, t, J = 2.2 Hz, H-12′), 6.08 (2H, d, J = 2.2 Hz, H-10′, H-14′), 6.10 (1H, t, J = 2.1 Hz, H-12), 6.35 (2H, d, J = 2.1 Hz, H-10, H-14), 6.60 (2H, d, J = 8.5 Hz, H-3′, H-5′), 6.80 (2H, d, J = 8.9 Hz, H-3, H-5), 6.81 (1H, d, J = 16.3 Hz, H-8), 6.89 (1H, d, J = 16.3 Hz, H-7), 7.03 (2H, d, J = 8.5 Hz, H-2′, H-6′), 7.38 (2H, d, J = 8.9 Hz, H-2, H-6), 9.01 (2H, s, 11′OH, 13′OH), 9.17 (2H, s, 11OH, 13OH), 9.22 (1H, s, 4′OH). 13C NMR (DMSO, 151 MHz) δ 21.3 (C-7'c), 23.0 (C-7'b), 69.5 (C-7'a), 81.9 (C-7′), 82.9 (C-8′), 101.6 (C-12′), 101.7 (C-12), 104.2 (C-10, C-14), 105.5 (C-10′, C-14′), 114.3 (C-3′, C-5′), 115.7 (C-3, C-5), 126.5 (C-8), 127.3 (C-2, C-6, C-7), 128.6 (C-2′, C-6′), 129.3 (C-1), 129.8 (C-1′), 138.9 (C-9), 140.3 (C-9′), 156.4 (C-4′), 157.7 (C-11′, C-13′), 157.8 (C-4), 158.4 (C-11, C-13). HR-ESI/MS analysis: m/z 513.1918 [M-H]-, (calcd for C31H29O7, 513.1913, ∆ = 1.0 ppm). MS/MS spectrum: CCMSLIB00009918867.
Threo-resveptero acyclic dimer (13) UV (MeCN) λmax (log ε) 228 (sh) (4.37), 285 (4.01), 305 (4.12), 326 (4.10), 343 (sh) (3.80) nm. 1H NMR (DMSO, 600 MHz) δ 3.75 (6H, s, CH3O-11, CH3O-13), 4.67 (1H, dd, J = 6.4, 2.9 Hz, H-7′), 5.02 (1H, d, J = 6.4 Hz, H-8′), 5.41 (1H, d, J = 2.9 Hz, 7′OH), 5.98 (1H, t, J = 2.2 Hz, H-12′), 6.04 (2H, d, J = 2.2 Hz, H-10′, H-14′), 6.36 (1H, t, J = 2.3 Hz, H-12), 6.59 (2H, d, J = 8.4 Hz, H-3′, H-5′), 6.69 (2H, d, J = 2.3 Hz, H-10, H-14), 6.85 (2H, d, J = 9.0 Hz, H-3, H-5), 6.95 (1H, d, J = 16.3 Hz, H-8), 7.00 (2H, d, J = 8.4 Hz, H-2′, H-6′), 7.13 (1H, d, J = 16.3 Hz, H-7), 7.41 (2H, d, J = 9.0 Hz, H-2, H-6), 9.02 (2H, s, 11′OH, 13′OH), 9.18 (1H, s, 4′OH); 13C NMR (DMSO, 151 MHz) δ 55.2 (CH3O-11, CH3O-13), 76.1 (C-7′), 84.0 (C-8′), 99.5 (C-12), 101.7 (C-12′), 104.1 (C-10, C-14), 105.6 (C-10′, C-14′), 114.3 (C-3′, C-5′), 116.0 (C-3, C-5), 126.1 (C-8), 127.5 (C-2, C-6), 128.3 (C-2′, C-6′), 128.5 (C-7), 129.3 (C-1), 131.9 (C-1′), 139.4 (C-9), 140.6 (C-9′), 156.2 (C-4′), 157.7 (C-11′, C-13′), 157.9 (C-4), 160.6 (C-11, C-13). HR-ESI/MS analysis: m/z 499.1763 [M-H]−, (calcd for C30H27O7, 499.1757, ∆ = 1.2 ppm). MS/MS spectrum: CCMSLIB00009918868.
7-O-Isopropyl-11′,13′-di-O-methylleachianol G (14) UV (MeCN) λmax (log ε) 228 (sh) (4.42), 283 (3.75), 323 (3.44) nm. 1H NMR (DMSO, 600 MHz) δ 0.65 (3H, d, J = 6.1 Hz, CH3-7c), 0.85 (3H, d, J = 6.1 Hz, CH3-7b), 3.08 (1H, dd, J = 8.6, 2.6 Hz, H-8), 3.11 (1H, hept, J = 6.1 Hz, H-7a), 3.43 (1H, t, J = 2.6 Hz, H-8′), 3.66 (6H, s, CH3O-11′, CH3O-13′), 4.02 (1H, d, J = 8.6 Hz, H-7), 4.09 (6H, d, J = 2.6 Hz, H-7′), 5.31 (1H, d, J = 2.1 Hz, H-14), 6.08 (1H, d, J = 2.1 Hz, H-12), 6.12 (3H, d, J = 2.3 Hz, H-10′, H-14′), 6.30 (1H, t, J = 2.3 Hz, H-12′), 6.63 (2H, d, J = 8.5 Hz, H-3′, H-5′), 6.66 (2H, d, J = 8.4 Hz, H-3, H-5), 6.80 (2H, d, J = 8.5 Hz, H-2′, H-6′), 6.91 (2H, d, J = 8.4 Hz, H-2, H-6), 8.78 (1H, s, 11OH or 13OH), 8.79 (1H, s, 11OH or 13OH), 9.04 (1H, s, 4′OH), 9.28 (1H, s, 4OH); 13C NMR (DMSO, 151 MHz) δ 20.3 (CH3-7c), 23.4 (CH3-7b), 54.0 (C-7′), 54.9 (CH3O-11′, CH3O-13′), 57.5 (C-8′), 60.5 (C-8), 67.1 (C-7a), 80.6 (C-7), 96.9 (C-12′), 101.4 (C-12), 104.0 (C-14), 104.9 (C-10′, C-14′), 114.6 (C-3′, C-5′), 114.6 (C-3, C-5), 121.3 (C-10), 128.2 (C-2′, C-6′), 128.8 (C-2, C-6), 131.4 (C-1), 136.5 (C-1′), 145.4 (C-9), 150.4 (C-9′), 154.0 (C-11), 155.1 (C-4′), 156.6 (C-4), 157.3 (C-13), 160.3 (C-11′, C-13′). HR-ESI/MS analysis: m/z 541.2222 [M-H]−, (calcd for C33H33O7, 541.2226, ∆ = 0.7 ppm). MS/MS spectrum: CCMSLIB00009918869.
11,11′,13,13′-Tetra-O-methylrestrytisol B (15) UV (MeCN) λmax (log ε) 228 (sh) (4.43), 283 (3.88), 323 (3.84) nm. 1H NMR (DMSO, 600 MHz) δ 3.53 (6H, s, CH3O-11′, CH3O-13′), 3.57 (1H, t, J = 10.6, 9.8 Hz, H-8), 3.64 (6H, s, CH3O-11, CH3O-13), 4.12 (1H, t, J = 10.6, 9.1 Hz, H-8′), 4.99 (1H, d, J = 9.8 Hz, H-7), 5.43 (1H, d, J = 9.1 Hz, H-7′), 6.04 (1H, t, J = 2.3 Hz, H-12′), 6.10 (2H, d, J = 2.3 Hz, H-10′, H-14′), 6.25 (1H, t, J = 2.3 Hz, H-12), 6.42 (2H, d, J = 2.3 Hz, H-10, H-14), 6.52 (2H, d, J = 8.5 Hz, H-3′, H-5′), 6.74 (2H, d, J = 8.4 Hz, H-3, H-5), 6.96 (2H, d, J = 8.5 Hz, H-2′, H-6′), 7.25 (2H, d, J = 8.4 Hz, H-2, H-6), 9.10 (1H, s, 4′OH), 9.37 (1H, s, 4OH); 13C NMR (DMSO, 151 MHz) δ 54.6 (CH3O-11′, CH3O-13′), 54.7 (CH3O-11, CH3O-13), 56.3 (C-8), 57.7 (C-8′), 82.3 (C-7′), 85.5 (C-7), 97.7 (C-12, C-12′), 106.2 (C-10, C-14), 107.0 (C-10′, C-14′), 113.9 (C-3′, C-5′), 114.8 (C-3, C-5), 127.7 (C-2′, C-6′), 128.0 (C-2, C-6), 129.4 (C-1), 130.7 (C-1′), 141.0 (C-9′), 141.2 (C-9), 155.5 (C-4′), 156.7 (C-4), 159.1 (C-11′), 159.9 (C-11). HR-ESI/MS analysis: m/z 527.2075 [M-H]−, (calcd for C32H31O7, 527.2070, ∆ = 0.9 ppm). MS/MS spectrum: CCMSLIB00009918870.
11′,13′-Di-O-methyl-trans-δ-viniferin (16) UV (MeCN) λmax (log ε) 227 (sh) (4.45), 285 (4.06), 312 (4.30), 333 (4.25), 350 (sh) (3.97) nm. 1H NMR (DMSO, 600 MHz) δ 3.70 (6H, s, CH3O-11′, CH3O-13′), 4.58 (1H, d, J = 8.1 Hz, H-8′), 5.56 (1H, d, J = 8.1 Hz, H-7′), 6.11 (1H, t, J = 2.2 Hz, H-12), 6.36 (4H, 2d, J = 2.3 Hz, H-10, H-10′, H-14, H-14′), 6.43 (1H, t, J = 2.3 Hz, H-12′), 6.76 (2H, d, J = 8.7 Hz, H-3′, H-5′), 6.82 (1H, d, J = 16.3 Hz, H-8), 6.90 (1H, d, J = 8.3 Hz, H-5), 6.97 (1H, d, J = 16.3 Hz, H-7), 7.19 (2H, d, J = 8.7 Hz, H-2′, H-6′), 7.21 (1H, d, J = 1.9 Hz, H-2), 7.43 (1H, dd, J = 8.3, 1.9 Hz, H-6), 9.17 (2H, s, 11OH, 13OH), 9.53 (1H, s, 4′OH). HR-ESI/MS analysis: m/z 481.1652 [M-H]-, (calcd for C30H25O6, 481.1651, ∆ = 0.2 ppm). MS/MS spectrum: CCMSLIB00009918871.
Threo-7′-O-isopropylisoresveptero acyclic dimer (17) UV (MeCN) λmax (log ε) 228 (sh) (4.37), 285 (4.05), 305 (4.18), 323 (4.19), 343 (sh) (3.89) nm. 1H NMR (DMSO, 600 MHz) δ 0.97 (3H, d, J = 6.2 Hz, CH3-7′c), 1.03 (3H, d, J = 6.1 Hz, CH3-7′b), 3.49 (2H, hept, J = 6.1 Hz, H-7′a), 3.61 (6H, s, CH3O-11′, CH3O-13′), 4.60 (1H, d, J = 6.2 Hz, H-7′), 5.24 (1H, d, J = 6.2 Hz, H-8′), 6.10 (1H, t, J = 2.2 Hz, H-12), 6.25 (1H, t, J = 2.3 Hz, H-12′), 6.34 (2H, d, J = 2.3 Hz, H-10′, H-14′), 6.35 (2H, d, J = 2.2 Hz, H-10, H-14), 6.60 (2H, d, J = 8.4 Hz, H-3′, H-5′), 6.82 (1H, d, J = 16.3 Hz, H-8), 6.84 (2H, d, J = 8.7 Hz, H-3, H-5), 6.89 (1H, d, J = 16.3 Hz, H-7), 7.04 (2H, d, J = 8.4 Hz, H-2′, H-6′), 7.37 (2H, d, J = 8.7 Hz, H-2, H-6), 9.17 (2H, s, 11OH, 13OH), 9.23 (1H, s, 4′OH); 13C NMR (DMSO, 151 MHz) δ 21.4 (CH3-7′b), 23.1 (CH3-7'c), 54.9 (CH3O-11′, CH3O-13′), 69.5 (C-7′a), 81.8 (C-7′), 82.9 (C-8′), 99.0 (C-12′), 101.8 (C-12), 104.3 (C-10, C-14), 105.6 (C-10′, C-14′), 114.4 (C-3′, C5′), 115.9 (C-3, C-5), 126.6 (C-8), 127.3 (C-7), 127.5 (C-2, C-6), 128.8 (C-2′, C-6′), 129.5 (C-1), 129.6 (C-1′), 138.9 (C-9), 140.7 (C-9′), 156.5 (C-4′), 158.4 (C-11, C-13), 159.7 (C-11′, C-13′). HR-ESI/MS analysis: m/z 541.2231 [M-H]−, (calcd for C33H33O7, 541.2226, ∆ = 0.9 ppm). MS/MS spectrum: CCMSLIB00009918872.
11,13-Di-O-methyl-trans-δ-viniferin (18) UV (MeCN) λmax (log ε) 227 (sh) (4.48), 285 (4.10), 312 (4.34), 333 (4.30), 350 (sh) (4.00) nm. 1H NMR (DMSO, 600 MHz) δ 3.75 (6H, s, CH3O-11, CH3O-13), 4.46 (1H, d, J = 8.3 Hz, H-8′), 5.41 (1H, d, J = 8.3 Hz, H-7′), 6.05 (2H, d, J = 2.1 Hz, H-10′, H-14′), 6.11 (1H, t, J = 2.2 Hz, H-12′), 6.35 (1H, t, J = 2.2 Hz, H-12), 6.73 (2H, d, J = 2.2 Hz, H-10, H-14), 6.76 (2H, d, J = 8.7 Hz, H-3′, H-5′), 6.90 (1H, d, J = 8.3 Hz, H-5), 6.96 (1H, d, J = 16.4 Hz, H-8), 7.20 (2H, d, J = 8.7 Hz, H-2′, H-6′), 7.22 (1H, d, J = 16.4 Hz, H-7), 7.24 (1H, d, J = 1.8 Hz, H-2), 7.44 (1H, dd, J = 8.3, 1.8 Hz, H-6), 9.22 (2H, s, 11′OH, 13′OH), 9.53 (1H, s, 4′OH). HR-ESI/MS analysis: m/z 481.1654 [M-H]−, (calcd for C30H25O6, 481.1651, ∆ = 0.6 ppm). MS/MS spectrum: CCMSLIB00009918873.
7-O-Isopropyl-11,11′,13,13′-tetra-O-methylleachianol G (19) UV (MeCN) λmax (log ε) 228 (sh) (4.48), 283 (3.91), 324 (3.89) nm. 1H NMR (DMSO, 600 MHz) δ 0.67 (3H, d, J = 6.1 Hz, CH3-7c), 0.88 (3H, d, J = 6.1 Hz, CH3-7b), 3.13 (1H, dd, J = 8.8, 2.4 Hz, H-8), 3.16 (1H, overlapped, H-7a), 3.46 (1H, t, J = 2.4 Hz, H-8′), 3.48 (3H, s, CH3O-13), 3.56 (3H, s, CH3O-11), 3.66 (6H, s, CH3O-13′, CH3O-11′), 4.06 (1H, d, J = 8.8 Hz, H-7), 4.14 (1H, d, J = 2.4 Hz, H-7′), 5.41 (1H, d, J = 2.2 Hz, H-14), 6.10 (2H, d, J = 2.3 Hz, H-10′, H-14′), 6.31 (1H, t, J = 2.3 Hz, H-12′), 6.34 (1H, d, J = 2.2 Hz, H-12), 6.65 (2H, d, J = 8.5 Hz, H-3′, H-5′), 6.69 (2H, d, J = 8.4 Hz, H-3, H-5), 6.78 (2H, d, J = 8.5 Hz, H-2′, H-6′), 6.92 (2H, d, J = 8.4 Hz, H-2, H-6), 9.09 (1H, s, 4′OH), 9.32 (1H, s, 4OH); 13C NMR (DMSO, 151 MHz) δ 20.0 (CH3-7c), 23.2 (CH3-7b), 48.3 (C-7a), 54.1 (C-7′), 54.5 (CH3O-13), 54.7 (CH3O-11′, CH3O-13′), 54.8 (CH3O-11), 57.3 (C-8′), 60.6 (C-8), 80.5 (C-7), 96.7 (C-12′), 97.4 (C-12), 101.8 (C-14), 104.6 (C-10′, C-14′), 114.4 (C-3, C-5), 114.6 (C-3′, C-5′), 124.1 (C-10), 127.8 (C-2′, C-6′), 128.7 (C-2, C-6), 131.0 (C-1), 136.0 (C-1′), 145.0 (C-9), 149.9 (C-9′), 154.8 (C-4′), 156.0 (C-11), 156.6 (C-4), 159.6 (C-13), 160.2 (C-11′, C-13′). HR-ESI/MS analysis: m/z 569.2543 [M-H]−, (calcd for C35H37O7, 569.2539, ∆ = 0.7 ppm). MS/MS spectrum: CCMSLIB00009918874.
Threo-7′-O-isopropylresvepterol acyclic dimer (20) UV (MeCN) λmax (log ε) 228 (sh) (4.42), 285 (4.04), 306 (4.15), 326 (4.12), 343 (sh) (3.87) nm. 1H NMR (DMSO, 600 MHz) δ 0.96 (3H, d, J = 6.1 Hz, CH3-7'c), 1.03 (3H, d, J = 6.1 Hz, CH3-7'b), 3.47 (1H, hept, J = 6.1 Hz, H-7'a), 3.75 (6H, s, CH3O-11, CH3O-13), 4.51 (1H, d, J = 6.3 Hz, H-7′), 5.07 (1H, d, J = 6.3 Hz, H-8′), 5.98 (1H, t, J = 2.2 Hz, H-12′), 6.08 (2H, d, J = 2.2 Hz, H-10′, H-14′), 6.36 (1H, t, J = 2.3 Hz, H-12), 6.60 (2H, d, J = 8.5 Hz, H-3′, H-5′), 6.69 (3H, d, J = 2.3 Hz, H-10, H-14), 6.82 (2H, d, J = 8.8 Hz, H-3, H-5), 6.95 (1H, d, J = 16.4 Hz, H-8), 7.03 (2H, d, J = 8.5 Hz, H-2′, H-6′), 7.13 (1H, d, J = 16.4 Hz, H-7), 7.40 (2H, d, J = 8.8 Hz, H-2, H-6), 9.02 (2H, s, 11′OH, 13′OH), 9.23 (1H, s, 4′OH); 13C NMR (DMSO, 151 MHz) δ 21.2 (CH3-7′b), 22.8 (CH3-7′c), 54.8 (CH3O-11, CH3O-13), 69.4 (C-7′a), 81.8 (C-7′), 82.8 (C-8′), 99.3 (C-12), 101.4 (C-12′), 103.9 (C-10, C-14), 105.3 (C-10′, C-14′), 114.3 (C-3′, C-5′), 115.7 (C-3, C-5), 125.9 (C-8), 127.3 (C-2, C-6), 128.3 (C-7), 128.5 (C-2′, C-6′), 129.0 (C-1), 129.7 (C-1′), 139.1 (C-9), 140.2 (C-9′), 156.2 (C-4′), 157.5 (C-11′, C-13′), 157.8 (C-4), 160.5 (C-11,C-13). HR-ESI/MS analysis: m/z 541.2224 [M-H]−, (calcd for C33H33O7, 541.2226, ∆ = 0.4 ppm). MS/MS spectrum: CCMSLIB00009918875.
Erythro-pterostilbene acyclic dimer (21) UV (MeCN) λmax (log ε) 228 (sh) (4.34), 285 (4.09), 306 (4.25), 326 (4.22), 344 (sh) (3.81) nm. 1H NMR (DMSO, 600 MHz) δ 3.60 (6H, s, CH3O-11′, CH3O-13′), 3.75 (6H, s, CH3O-11, CH3O-13), 4.73 (1H, t, J = 6.2, 4.6 Hz, H-7′), 5.20 (1H, d, J = 6.2 Hz, H-8′), 5.48 (1H, d, J = 4.6 Hz, 7′OH), 6.26 (1H, t, J = 2.3 Hz, H-12′), 6.29 (2H, d, J = 2.3 Hz, H-10′, H-14′), 6.37 (2H, t, J = 2.3 Hz, H-12), 6.59 (2H, d, J = 8.5 Hz, H-3′, H-5′), 6.69 (2H, d, J = 2.3 Hz, H-10, H-14), 6.89 (2H, d, J = 8.8 Hz, H-3, H-5), 6.95 (1H, d, J = 16.4 Hz, H-8), 7.01 (2H, d, J = 8.5 Hz, H-2′, H-6′), 7.13 (2H, d, J = 16.4 Hz, H-7), 7.41 (2H, d, J = 8.8 Hz, H-2, H-6), 9.19 (1H, s, 4′OH); 13C NMR (DMSO, 126 MHz) δ 54.6 (CH3O-11′, CH3O-13′), 54.8 (CH3O-11, CH3O-13), 75.7 (C-7′), 83.5 (C-8′), 98.6 (C-12′), 99.2 (C-12), 103.8 (C-10, C-14), 105.4 (C-10′, C-14′), 113.9 (C-3′, C-5′), 115.7 (C-3, C-5), 125.8 (C-8), 127.2 (C-2, C-6), 128.0 (C-2′, C-6′), 128.1 (C-7), 129.3 (C-1), 131.5 (C-1′), 139.1 (C-9), 156.1 (C-4′), 157.5 (C-4), 159.5 (C-11′, C-13′), 160.3 (C-11, C-13). HR-ESI/MS analysis: m/z 527.2078 [M-H]−, (calcd for C32H31O7, 527.2070, ∆ = 1.5 ppm). MS/MS spectrum: CCMSLIB00009918876.
11,11′,13,13′-Tetra-O-methyl-trans-δ-viniferin (22) UV (MeCN) λmax (log ε) 227 (sh) (4.56), 285 (4.17), 311 (4.41), 333 (4.37), 350 (sh) (4.12) nm. 1H NMR (DMSO, 600 MHz) δ 3.63 (6H, s, CH3O-11′, CH3O-13′), 3.67 (6H, s, CH3O-11, CH3O-13), 4.54 (1H, d, J = 8.5 Hz, H-8′), 5.52 (1H, d, J = 8.5 Hz, H-7′), 6.28 (1H, t, J = 2.3 Hz, H-12), 6.31 (2H, d, J = 2.3 Hz, H-10′, H-14′), 6.36 (1H, t, J = 2.3 Hz, H-12′), 6.65 (2H, d, J = 2.3 Hz, H-10, H-14), 6.69 (2H, d, J = 8.6 Hz, H-3′, H-5′), 6.85 (1H, d, J = 8.3 Hz, H-5), 6.88 (1H, d, J = 16.4 Hz, H-8), 7.12 (1H, d, J = 1.8 Hz, H-2), 7.13 (2H, d, J = 8.6 Hz, H-2′, H-6′), 7.14 (1H, d, J = 16.4 Hz, H-7), 7.38 (1H, dd, J = 8.3, 1.8 Hz, H-6), 9.49 (1H, s, 4′OH). HR-ESI/MS analysis: m/z 509.1968 [M-H]−, (calcd for C32H29O6, 509.1964, ∆ = 0.8 ppm). MS/MS spectrum: CCMSLIB00009918877.
Parthenostilbenin A+B (23) Parthenostilbenin A (7S) 1H NMR (DMSO, 600 MHz) δ 2.59 (1H, t, J = 2.6 Hz, H-8′), 2.92 (3H, s, CH3O-7), 3.10 (1H, dd, J = 9.1, 2.6 Hz, H-8), 3.75 (1H, d, J = 9.1 Hz, H-7), 3.96 (1H, d, J = 2.6 Hz, H-7′), 5.60 (2H, d, J = 2.2 Hz, H-10′, H-14′), 5.91 (1H, t, J = 2.2 Hz, H-12′), 6.17 (1H, d, J = 2.1 Hz, H-12), 6.41 (1H, d, J = 2.1 Hz, H-14), 6.59–6.64 (8H, m, H-2, H-3, H-5, H-6, H-2′, H-3′, H-5′, H-6′), 8.80 (1H, s, 11OH), 8.92 (2H, s, 11′OH, 13′OH), 9.00 (1H, s, 13OH), 9.10 (1H, s, 4′OH), 9.30 (1H, s, 4OH); 13C NMR (DMSO, 151 MHz) δ 54.1 (C-7′), 55.5 (CH3O-7), 57.1 (C-8′), 58.9 (C-8), 85.9 (C-7), 99.7 (C-12′), 101.1 (C-12), 103.9 (C-10′, C-14′), 104.2 (C-14), 114.2 (C-3, C-5, C-3′, C-5′), 120.5 (C-10), 128.2 (C-2, C-6), 135.8 (C-1′), 152.6 (C-13), 153.7 (C-11), 156.2 (C-4), 157.8 (C-11′, C-13′). Parthenostilbenin B (7R) UV (MeCN) λmax (log ε) 227 (sh) (4.46), 283 (3.74) nm. 1H NMR (DMSO, 600 MHz) δ 2.83 (3H, s, CH3O-7), 3.20 (2H, m, H-8, H-8′), 3.72 (1H, d, J = 8.9 Hz, H-7), 4.01 (1H, d, J = 2.7 Hz, H-7′), 5.42 (1H, d, J = 2.1 Hz, H-14), 5.92 (2H, d, J = 2.2 Hz, H-10′, H-14′), 5.99 (1H, t, J = 2.2 Hz, H-12′), 6.06 (1H, d, J = 2.1 Hz, H-12), 6.60 (2H, d, J = 8.6 Hz, H-3′, H-5′), 6.68 (4H, 2xd, J = 8.5 Hz, H-2′, H-3, H-5, H-6′), 6.90 (2H, d, J = 8.5 Hz, H-2, H-6), 8.74 (1H, s, 11OH), 8.79 (1H, s, 13OH), 9.00 (2H, s, 11′OH, 13′OH), 9.04 (1H, s, 4′OH), 9.35 (1H, s, 4OH); 13C NMR (DMSO, 151 MHz) δ 54.1 (C-7′), 55.4 (CH3O-7), 57.3 (C-8′), 59.4 (C-8), 85.7 (C-7), 99.7 (C-12′), 101.1 (C-12), 103.5 (C-14), 104.4 (C-10′, C-14′), 114.2 (C-3, C-5, C-3′, C-5′), 121.2 (C-10), 127.6 (C-2′, C-6′), 128.8 (C-2, C-6), 129.5 (C-1), 136.3 (C-1′), 153.7 (C-11), 154.7 (C-4′), 156.4 (C-4), 156.9 (C-13), 157.8 (C-11′, C-13′). HR-ESI/MS analysis: m/z 485.1606 [M-H]−, (calcd for C29H25O7, 485.1600, ∆ = 1.2 ppm). MS/MS spectrum: CCMSLIB00009918878.
Erythro-7′-O-isopropylresvepterol acyclic dimer (24) UV (MeCN) λmax (log ε) 229 (sh) (4.08), 285 (3.75), 306 (3.92), 327 (3.91), 345 (sh) (3.62) nm. 1H NMR (DMSO, 600 MHz) δ 0.87 (3H, d, J = 6.0 Hz, CH3-7′c), 0.91 (3H, d, J = 6.2 Hz, CH3-7′b), 3.33 (1H, overlapped, H-7a), 3.75 (6H, s, CH3O-11, CH3O-13), 4.43 (1H, d, J = 6.5 Hz, H-7′), 5.04 (1H, d, J = 6.5 Hz, H-8′), 6.06 (1H, t, J = 2.2 Hz, H-12′), 6.22 (2H, d, J = 2.2 Hz, H-10′, H-14′), 6.36 (1H, t, J = 2.3 Hz, H-12), 6.67 (2H, d, J = 8.5 Hz, H-3′, H-5′), 6.68 (2H, d, J = 2.3 Hz, H-10, H-14), 6.73 (2H, d, J = 8.7 Hz, H-3, H-5), 6.92 (1H, d, J = 16.3 Hz, H-8), 7.10 (1H, d, J = 16.3 Hz, H-7), 7.16 (2H, d, J = 8.5 Hz, H-2′, H-6′), 7.36 (2H, d, J = 8.7 Hz, H-2, H-6), 9.09 (2H, s, 11′OH, 13′OH), 9.27 (1H, s, 4′OH); 13C NMR (DMSO, 151 MHz) δ 21.1 (CH3-7′c), 22.8 (CH3-7′b), 54.8 (CH3O-11, CH3O-13), 69.2 (C-7'a), 81.4, (C-7′), 81.7 (C-8′), 99.3 (C-12), 101.6 (C-12′), 103.9 (C-10, C-14), 105.5 (C-10′, C-14′), 114.2 (C-3′, C-5′), 115.7 (C-3, C-5), 125.9 (C-8), 127.3 (C-2, C-6), 128.3 (C-7), 128.7 (C-2′, C-6′), 129.2 (C-1), 130.1 (C-1′), 139.1 (C-9), 156.4 (C-4′), 157.1 (C-4), 157.5 (C-11′, C-13′), 160.5 (C-11, C-13). HR-ESI/MS analysis: m/z 541.2229 [M-H]−, (calcd for C33H33O7, 541.2226, ∆ = 0.6 ppm). MS/MS spectrum: CCMSLIB00009918879.
7-O-Butylleachianol G (25) UV (MeCN) λmax (log ε) 229 (sh) (4.39), 280 (3.64) nm. 1H NMR (DMSO, 600 MHz) δ 0.74 (3H, t, J = 7.2 Hz, CH3-7d), 1.18 (4H, m, H-7b, H-7c), 2.75 (1H, dt, J = 9.8, 5.9 Hz, H-7a″), 3.01 (1H, dt, J = 9.8, 6.0 Hz, H-7a′), 3.13 (1H, dd, J = 8.8, 2.8 Hz, H-8), 3.32 (1H, overlapped, H-8′), 3.80 (1H, d, J = 8.8 Hz, H-7), 4.03 (1H, d, J = 2.5 Hz, H-7′), 5.31 (1H, d, J = 2.1 Hz, H-14), 5.92 (2H, d, J = 2.2 Hz, H-10′, H-14′), 5.98 (1H, t, J = 2.2 Hz, H-12′), 6.07 (1H, d, J = 2.1 Hz, H-12), 6.62 (2H, d, J = 8.6 Hz, H-3′, H-5′), 6.67 (2H, d, J = 8.5 Hz, H-3, H-5), 6.73 (2H, d, J = 8.6 Hz, H-2′, H-6′), 6.89 (2H, d, J = 8.5 Hz, H-2, H-6), 8.76 (1H, s, 11OH), 8.78 (1H, s, 13OH), 9.00 (2H, s, 11′OH, 13′OH), 9.04 (1H, s, 4′OH), 9.32 (1H, s, 4OH); 13C NMR (DMSO, 151 MHz) δ 13.6 (CH3-7d), 18.9 (C-7c), 31.0 (C-7b), 54.3 (C-7′), 57.5 (C-8′), 60.1 (C-8), 66.8 (C-7a), 83.9 (C-7), 100.1 (C-12′), 101.4 (C-12), 104.1 (C-14), 104.6 (C-10′, C-14′), 114.6 (C-3′, C-5′), 114.7 (C-3, C-5), 121.5 (C-10), 128.0 (C-2′, C-6′), 129.0 (C-2, C-6), 130.8 (C-1), 136.7 (C-1′), 145.5 (C-9), 150.2 (C-9′), 154.0 (C-11), 155.1 (C-4′), 156.7 (C-4), 157.2 (C-13), 158.2 (C-11′, C-13′). HR-ESI/MS analysis: m/z 527.2071 [M-H]−, (calcd for C32H31O7, 527.2070, ∆ = 0.2 ppm). MS/MS spectrum: CCMSLIB00009918880.
7-O-Butylleachianol F (26) UV (MeCN) λmax (log ε) 229 (sh) (4.33), 282 (3.56) nm. 1H NMR (DMSO, 600 MHz) δ 0.78 (3H, t, J = 7.3 Hz, CH3-7d), 1.22 (2H, m, H-7c), 1.35 (2H, m, H-7b), 2.63 (1H, overlapped, H-8′), 2.95 (1H, dt, J = 9.5, 6.2 Hz, H-7a″), 3.05 (1H, m, H-7a′), 3.07 (1H, dd, J = 9.1, 2.7 Hz, H-8), 3.82 (1H, d, J = 9.1 Hz, H-7), 3.97 (1H, d, J = 2.4 Hz, H-7′), 5.61 (2H, d, J = 2.1 Hz, H-10′, H-14′), 5.92 (1H, t, J = 2.1 Hz, H-12′), 6.16 (1H, d, J = 2.1 Hz, H-12), 6.41 (1H, d, J = 2.1 Hz, H-14), 6.58 (4H, s, H-2, H-3, H-5, H-6), 6.63 (2H, d, J = 8.6 Hz, H-3′, H-5′), 6.68 (2H, d, J = 8.6 Hz, H-2′, H-6′), 8.78 (1H, s, 11OH), 8.92 (2H, s, 11′OH, 13′OH), 8.95 (1H, s, 13OH), 9.10 (1H, s, 4′OH), 9.27 (1H, s, 4OH); 13C NMR (DMSO, 151 MHz) δ 13.5 (CH3-7d), 18.6 (C-7c), 31.1 (C-7b), 54.1 (C-7′), 57.1 (C-8′), 59.2 (C-8), 67.1 (C-7a), 83.8 (C-7), 99.8 (C-12′), 101.1 (C-12), 104.1 (C-10′, C-14′), 104.6 (C-14), 114.4 (C-3′, C-5′), 114.5 (C-3, C-5), 120.5 (C-10), 127.8 (C-2′, C-6′), 128.0 (C-2, C-6), 131.1 (C-1), 136.0 (C-1′), 147.5 (C-9), 149.5 (C-9′), 153.8 (C-11), 154.9 (C-4′), 156.2 (C-4), 157.3 (C-13), 157.8 (C-11′, C-13′). HR-ESI/MS analysis: m/z 527.2074 [M-H]−, (calcd for C32H31O7, 527.2070, ∆ = 0.8 ppm). MS/MS spectrum: CCMSLIB00009918881.
Erythro-isoresveptero acyclic dimer (27) UV (MeCN) λmax (log ε) 228 (sh) (4.21), 285 (3.95), 307 (4.08), 322 (4.08), 340 (sh) (3.81) nm. 1H NMR (DMSO, 600 MHz) δ 3.59 (6H, s, CH3O-11′, CH3O-13′), 4.73 (1H, dd, J = 6.3, 4.5 Hz, H-7′), 5.19 (1H, d, J = 6.3 Hz, H-8′), 5.48 (1H, d, J = 4.5 Hz, 7′OH), 6.10 (1H, t, J = 2.2 Hz, H-12), 6.25 (1H, t, J = 2.3 Hz, H-12′), 6.28 (2H, d, J = 2.3 Hz, H-10′, H-14′), 6.35 (2H, d, J = 2.2 Hz, H-10, H-14), 6.59 (2H, d, J = 8.6 Hz, H-3′, H-5′), 6.82 (1H, d, J = 16.3 Hz, H-8), 6.86 (2H, d, J = 8.9 Hz, H-3, H-5), 6.89 (1H, d, J = 16.3 Hz, H-7), 7.01 (2H, d, J = 8.6 Hz, H-2′, H-6′), 7.38 (2H, d, J = 8.9 Hz, H-2, H-6), 9.18 (2H, s, 11OH, 13OH), 9.20 (1H, s, 4′OH); 13C NMR (DMSO, 126 MHz) δ 54.7 (CH3O-11′, CH3O-13′), 75.7 (C-7′), 83.7 (C-8′), 98.6 (C-12′), 101.6 (C-12), 104.1 (C-10, C-14), 105.5 (C-10′, C-14′), 114.1 (C-3′, C-5′), 115.8 (C-3, C-5), 126.5 (C-8), 127.1 (C-7), 127.3 (C-2, C-6), 128.1 (C-2′, C-6′), 129.5 (C-1), 131.7 (C-1′), 138.9 (C-9), 156.3 (C-4′), 157.4 (C-4), 158.3 (C-11, C-13), 159.6 (C-11′, C-13′). HR-ESI/MS analysis: m/z 499.1769 [M-H]−, (calcd for C30H27O7, 499.1757, ∆ = 2.4 ppm). MS/MS spectrum: CCMSLIB00009918882.
Erythro-7′-O-butylresveratrol acyclic dimer (28) UV (MeCN) λmax (log ε) 228 (sh) (4.29), 285 (4.02), 308 (4.18), 324 (4.18), 340 (sh) (3.90) nm. 1H NMR (DMSO, 600 MHz) δ 0.74 (3H, t, J = 7.4 Hz, CH3-7′d), 1.15 (2H, m, H-7′c), 1.32 (2H, m, H-7′b), 3.10 (1H, dt, J = 9.5, 6.3 Hz, H-7′a″), 3.21 (1H, dt, J = 9.5, 6.3 Hz, H-7′a′), 4.34 (1H, d, J = 6.4 Hz, H-7′), 5.10 (1H, d, J = 6.4 Hz, H-8′), 6.06 (1H, t, J = 2.2 Hz, H-12′), 6.09 (1H, t, J = 2.2 Hz, H-12), 6.20 (2H, d, J = 2.2 Hz, H-10′, H-14′), 6.34 (2H, d, J = 2.2 Hz, H-10, H-14), 6.68 (2H, d, J = 8.6 Hz, H-3′, H-5′), 6.71 (2H, d, J = 8.9 Hz, H-3, H-5), 6.79 (1H, d, J = 16.2 Hz, H-8), 6.87 (1H, d, J = 16.2 Hz, H-7), 7.13 (2H, d, J = 8.6 Hz, H-2′, H-6′), 7.33 (2H, d, J = 8.9 Hz, H-2, H-6), 9.08 (2H, s, 11′OH, 13′OH), 9.17 (2H, s, 11OH, 13OH), 9.29 (1H, s, 4′OH); 13C NMR (DMSO, 126 MHz) δ 13.2 (CH3-7'd), 18.3 (C-7'c), 30.8 (C-7'b), 67.5 (C-7'a), 81.4 (C-8′), 83.5 (C-7′), 101.4 (C-12, C-12′), 103.9 (C-10, C-14), 105.3 (C-10′, C-14′), 114.1 (C-3′, C-5′), 115.5 (C-3, C-5), 126.3 (C-8), 127.0 (C-7), 127.1 (C-2, C-6), 128.7 (C-2′, C-6′), 129.0 (C-1′), 129.2 (C-1), 138.5 (C-9), 156.2 (C-4′), 156.7 (C-4), 157.3 (C-11′, C-13′), 158.0 (C-11, C-13). HR-ESI/MS analysis: m/z 527.2076 [M-H]−, (calcd for C32H31O7, 527.2070, ∆ = 1.1 ppm). MS/MS spectrum: CCMSLIB00009918883.
7-O-Butyl-11′,13′-di-O-methylleachianol F (29) UV (MeCN) λmax (log ε) 229 (sh) (4.16), 283 (3.51), 322 (3.33) nm. 1H NMR (DMSO, 600 MHz) δ 0.79 (3H, t, J = 7.4 Hz, CH3-7d), 1.24 (2H, m, J = 6.7 Hz, H-7c), 1.38 (2H, m, H-7b), 2.70 (1H, t, J = 3.6 Hz, H-8′), 3.01 (1H, dt, J = 9.3, 6.2 Hz, H-7a″), 3.08 (1H, dt, J = 9.3, 6.7 Hz, H-7a′), 3.12 (1H, dd, J = 9.1, 3.6 Hz, H-8), 3.91 (1H, d, J = 9.1 Hz, H-7), 4.05 (1H, d, J = 3.6 Hz, H-7′), 5.73 (2H, d, J = 2.3 Hz, H-10′, H-14′), 6.16 (1H, d, J = 2.1 Hz, H-12), 6.20 (1H, t, J = 2.3 Hz, H-12′), 6.45 (1H, d, J = 2.1 Hz, H-14), 6.58 (2H, d, J = 8.6 Hz, H-3, H-5), 6.62 (2H, d, J = 8.5 Hz, H-3′, H-5′), 6.65 (2H, d, J = 8.6 Hz, H-2, H-6), 6.67 (2H, d, J = 8.5 Hz, H-2′, H-6′), 8.77 (1H, s, 11OH), 8.97 (1H, s, 13OH), 9.09 (1H, s, 4′OH), 9.28 (1H, s, 4OH); 13C NMR (DMSO, 151 MHz) δ 13.5 (CH3-7d), 18.6 (C-7c), 31.1 (C-7b), 53.8 (C-7′), 54.6 (CH3O-11′, CH3O-13′), 57.5 (C-8′), 59.6 (C-8), 67.1 (C-7a), 84.0 (C-7), 97.0 (C-12′), 107.1 (C-12), 104.2 (C-10′, C-14′), 104.4 (C-14), 114.4 (C-3, C-5, C-3′, C-5′), 120.6 (C-10), 128.0 (C-2′, C-6′), 128.3 (C-2, C-6), 130.7 (C-1), 135.9 (C-1′), 146.7 (C-9), 148.9 (C-9′), 154.8 (C-4′), 156.3 (C-4), 157.5 (C-13), 159.7 (C-11′, C-13′). HR-ESI/MS analysis: m/z 555.2392 [M-H]−, (calcd for C34H35O7, 555.2383, ∆ = 1.6 ppm). MS/MS spectrum: CCMSLIB00009918884.
Threo-7′-O-butylresveratrol acyclic dimer (30) UV (MeCN) λmax (log ε) 228 (sh) (4.34), 285 (4.12), 307 (4.30), 325 (4.28), 341 (sh) (4.03) nm. 1H NMR (DMSO, 600 MHz) δ 0.80 (3H, t, J = 7.4 Hz, CH3-7′d), 1.26 (2H, m, H-7′c), 1.41 (2H, m, H-7′b), 3.26 (2H, t, J = 6.4 Hz, H-7′a), 4.42 (1H, d, J = 6.6 Hz, H-7′), 5.13 (1H, d, J = 6.6 Hz, H-8′), 5.96 (1H, t, J = 2.2 Hz, H-12′), 6.05 (2H, d, J = 2.2 Hz, H-10′, H-14′), 6.10 (1H, t, J = 2.2 Hz, H-12), 6.36 (2H, d, J = 2.2 Hz, H-10, H-14), 6.60 (2H, d, J = 8.6 Hz, H-3′, H-5′), 6.81 (2H, d, J = 16.3 Hz, H-8), 6.82 (2H, d, J = 8.9 Hz, H-3, H-5), 6.89 (1H, d, J = 16.3 Hz, H-7), 7.00 (2H, d, J = 8.6 Hz, H-2′, H-6′), 7.38 (2H, d, J = 8.9 Hz, H-2, H-6), 9.02 (2H, s, 11′OH, 13′OH), 9.18 (2H, s, 11OH, 13OH), 9.26 (1H, s, 4′OH); 13C NMR (DMSO, 151 MHz) δ 13.7 (CH3-7′d), 18.8 (C-7′c), 31.4 (C-7′b), 68.1 (C-7′a), 82.8 (C-8′), 84.4 (C-7′), 101.7 (C-12′), 101.9 (C-12), 104.3 (C-10, C-14), 105.7 (C-10′, C-14′), 114.6 (C-3′, C-5′), 115.9 (C-3, C-5), 126.6 (C-8), 127.4 (C-7), 127.5 (C-2, C-6), 128.8 (C-1′), 128.9 (C-2′, C-6′), 129.5 (C-1), 139.0 (C-9), 140.2 (C-9′), 156.6 (C-4′), 157.7 (C-11′, C-13′), 157.8 (C-4), 158.5 (C-11, C-13). HR-ESI/MS analysis: m/z 527.2076 [M-H]−, (calcd for C32H31O7, 527.2070, ∆ = 1.3 ppm). MS/MS spectrum: CCMSLIB00009918885.
7-O-Butyl-11′,13′-di-O-methylleachianol G (31) UV (MeCN) λmax (log ε) 229 (sh) (4.44), 282 (3.69) nm. 1H NMR (DMSO, 600 MHz) δ 0.75 (3H, t, J = 7.2 Hz, CH3-7d), 1.18 (2H, m, H-7c), 1.24 (2H, m, H-7b), 2.81 (1H, dt, J = 9.7, 5.9 Hz, H-7a″), 3.05 (1H, dt, J = 9.7, 6.1 Hz, H-7a′), 3.19 (1H, dd, J = 8.2, 3.3 Hz, H-8), 3.38 (1H, t, J = 3.3 Hz, H-8′), 3.65 (6H, s, CH3O-13′, CH3O-11′), 3.92 (1H, d, J = 8.2 Hz, H-7), 4.07 (1H, d, J = 3.3 Hz, H-7′), 5.45 (1H, d, J = 2.1 Hz, H-14), 6.08 (1H, d, J = 2.1 Hz, H-12), 6.10 (2H, d, J = 2.3 Hz, H-10′, H-14′), 6.29 (1H, t, J = 2.3 Hz, H-12′), 6.61 (2H, d, J = 8.5 Hz, H-3′, H-5′), 6.66 (2H, d, J = 8.5 Hz, H-3, H-5), 6.75 (2H, d, J = 8.5 Hz, H-2′, H-6′), 6.91 (2H, d, J = 8.5 Hz, H-2, H-6), 8.77 (1H, s, 11OH), 8.82 (1H, s, 13OH), 9.05 (1H, s, 4′OH), 9.31 (1H, s, 4OH); 13C NMR (DMSO, 151 MHz) δ 13.6 (CH3-7d), 18.9 (C-7c), 31.1 (C-7b), 54.3 (C-7′), 54.9 (CH3O-11′, CH3O-13′), 57.6 (C-8′), 60.2 (C-8), 67.0 (C-7a), 83.6 (C-7), 97.0 (C-12′), 101.5 (C-12), 103.8 (C-14), 104.9 (C-10′, C-14′), 114.6 (C-3′, C-5′), 114.7 (C-3, C-5), 121.4 (C-10), 128.1 (C-2′, C-6′), 128.9 (C-2, C-6), 130.7 (C-1), 136.5 (C-1′), 145.4 (C-9), 150.1 (C-9′), 154.0 (C-11), 155.1 (C-4′), 156.6 (C-4), 157.3 (C-13), 160.3 (C-11′, C-13′). HR-ESI/MS analysis: m/z 555.2390 [M-H]−, (calcd for C34H35O7, 555.2383, ∆ = 1.3 ppm). MS/MS spectrum: CCMSLIB00009918886.
7-O-Butyl-11′,13′-di-O-methylisoleachianol G (32) UV (MeCN) λmax (log ε) 229 (sh) (4.41), 283 (3.80), 322 (3.66) nm. 1H NMR (DMSO, 600 MHz) δ 0.67 (3H, t, J = 7.3 Hz, CH3-7d), 1.03 (1H, m, H-7c″), 1.09 (1H, m, H-7c′), 1.19 (2H, m, H-7b), 2.39 (1H, overlapped, H-7a″), 2.73 (1H, dt, J = 8.3, 7.0 Hz, H-7a′), 3.48 (1H, d, J = 6.8 Hz, H-8′), 3.62 (1H, d, J = 10.4 Hz, H-7), 3.66 (6H, s, CH3O-13′, CH3O-11′), 3.82 (1H, dd, J = 10.4, 6.8 Hz, H-8), 4.11 (1H, overlapped, H-7′), 5.11 (1H, d, J = 2.0 Hz, H-14), 6.08 (1H, d, J = 2.0 Hz, H-12), 6.19 (2H, d, J = 2.3 Hz, H-10′, H-14′), 6.35 (1H, t, J = 2.3 Hz, H-12′), 6.65 (2H, d, J = 8.5 Hz, H-3′, H-5′), 6.73 (2H, d, J = 8.5 Hz, H-3, H-5), 6.79 (2H, d, J = 8.5 Hz, H-2′, H-6′), 7.02 (2H, d, J = 8.5 Hz, H-2, H-6), 8.84 (1H, s, 11OH), 8.85 (1H, s, 13OH), 9.13 (1H, s, 4′OH), 9.43 (1H, s, 4OH); 13C NMR (DMSO, 151 MHz) δ 13.3 (CH3-7d), 18.6 (C-7c), 31.1 (C-7b), 51.8 (C-7′), 52.2 (C-8), 54.6 (CH3O-11′, CH3O-13′), 58.7 (C-8′), 65.6 (C-7a), 80.1 (C-7), 96.9 (C-12′), 100.7 (C-12), 103.4 (C-14), 106.3 (C-10′, C-14′), 114.6 (C-3′, C-5′), 114.9 (C-3, C-5), 122.2 (C-10), 127.8 (C-2′, C-6′), 129.2 (C-2, C-6), 130.8 (C-1), 134.4 (C-1′), 144.9 (C-9), 146.2 (C-9′), 153.5 (C-11 or C-13), 155.3 (C-4′), 156.8 (C-4), 159.5 (C-11′, C-13′). HR-ESI/MS analysis: m/z 555.2395 [M-H]−, (calcd for C34H35O7, 555.2383, ∆ = 2.2 ppm). MS/MS spectrum: CCMSLIB00009918887.
Threo-7′-O-butylisoresveptero acyclic dimer (33) UV (MeCN) λmax (log ε) 228 (sh) (4.13), 287 (3.95), 309 (4.09), 325 (4.08), 340 (sh) (3.84) nm. 1H NMR (DMSO, 600 MHz) δ 0.80 (3H, t, J = 7.4 Hz, CH3-7′d), 1.26 (2H, m, H-7′c), 1.42 (2H, m, H-7′b), 3.28 (2H, t, J = 6.4 Hz, H-7'a), 3.60 (6H, s, CH3O-11′, CH3O-13′), 4.50 (1H, d, J = 6.5 Hz, H-7′), 5.30 (1H, d, J = 6.5 Hz, H-8′), 6.10 (1H, t, J = 2.2 Hz, H-12), 6.24 (1H, t, J = 2.3 Hz, H-12′), 6.32 (2H, d, J = 2.3 Hz, H-10′, H-14′), 6.36 (2H, d, J = 2.2 Hz, H-10, H-14), 6.61 (2H, d, J = 8.5 Hz, H-3′, H-5′), 6.82 (1H, d, J = 16.3 Hz, H-8), 6.86 (2H, d, J = 8.8 Hz, H-3, H-5), 6.89 (1H, d, J = 16.3 Hz, H-7), 7.01 (2H, d, J = 8.5 Hz, H-2′, H-6′), 7.38 (2H, d, J = 8.8 Hz, H-2, H-6), 9.18 (2H, s, 11OH, 13OH), 9.27 (1H, s, 4′OH); 13C NMR (DMSO, 151 MHz) δ 13.5 (CH3-7′d), 18.6 (C-7′c), 31.1 (C-7'b), 54.7 (CH3O-11′, CH3O-13′), 67.8 (C-7′a), 82.4 (C-8′), 84.0 (C-7′), 98.8 (C-12′), 101.6 (C-12), 104.1 (C-10, C-14), 105.5 (C-10′, C-14′), 114.3 (C-3′, C-5′), 115.7 (C-3, C-5), 126.6 (C-8), 127.1 (C-7), 127.3 (C-2, C-6), 128.6 (C-1′), 128.7 (C-2′, C-6′), 129.5 (C-1), 138.7 (C-9), 140.5 (C-9′), 156.4 (C-4′), 157.3 (C-4), 158.2 (C-11, C-13), 159.6 (C-11′, C-13′). HR-ESI/MS analysis: m/z 555.2401 [M-H]−, (calcd for C34H35O7, 555.2383, ∆ = 3.2 ppm). MS/MS spectrum: CCMSLIB00009918888.
Erythro-7′-O-butylisoresveptero acyclic dimer (34) UV (MeCN) λmax (log ε) 228 (sh) (3.99), 288 (3.75), 314 (3.90), 340 (sh) (3.63) nm. 1H NMR (DMSO, 600 MHz) δ 0.73 (3H, t, J = 7.4 Hz, CH3-7′d), 1.14 (2H, h, J = 7.3 Hz, H-7′c), 1.32 (2H, m, H-7′b), 3.12 (1H, dt, J = 9.5, 6.2 Hz, H-7′a″), 3.23 (1H, dt, J = 9.5, 6.2 Hz, H-7′a′), 3.67 (6H, s, CH3O-11′, CH3O-13′), 4.42 (1H, d, J = 6.5 Hz, H-7′), 5.28 (1H, d, J = 6.5 Hz, H-8′), 6.10 (1H, t, J = 2.2 Hz, H-12), 6.34 (2H, d, J = 2.2 Hz, H-10, H-14), 6.35 (1H, t, J = 2.2 Hz, H-12′), 6.48 (2H, d, J = 2.2 Hz, H-10′, H-14′), 6.69 (2H, d, J = 8.5 Hz, H-3′, H-5′), 6.77 (2H, d, J = 8.8 Hz, H-3, H-5), 6.80 (1H, d, J = 16.6 Hz, H-8), 6.87 (1H, d, J = 16.6 Hz, H-7), 7.15 (2H, d, J = 8.5 Hz, H-2′, H-6′), 7.34 (2H, d, J = 8.8 Hz, H-2, H-6), 9.17 (2H, s, 11OH, 13OH), 9.31 (1H, s, 4′OH). HR-ESI/MS analysis: m/z 555.2403 [M-H]−, (calcd for C34H35O7, 555.2383, ∆ = 3.6 ppm). MS/MS spectrum: CCMSLIB00009918889.
Erythro-7′-O-butylresveptero acyclic dimer (35) UV (MeCN) λmax (log ε) 228 (sh) (4.46), 288 (4.23), 308 (4.38), 325 (4.38), 340 (sh) (4.12) nm. 1H NMR (DMSO, 600 MHz) δ 0.74 (3H, t, J = 7.4 Hz, CH3-7′d), 1.15 (2H, m, H-7′c), 1.32 (2H, m, H-7′b), 3.10 (1H, dt, J = 9.4, 6.3 Hz, H-7′a″), 3.21 (1H, dt, J = 9.7, 6.4 Hz, H-7′a′), 3.75 (6H, s, CH3O-11, CH3O-13), 4.33 (1H, d, J = 6.5 Hz, H-7′), 5.12 (1H, d, J = 6.5 Hz, H-8′), 6.06 (1H, t, J = 2.2 Hz, H-12′), 6.21 (2H, d, J = 2.2 Hz, H-10′, H-14′), 6.36 (1H, t, J = 2.2 Hz, H-12), 6.68 (2H, d, J = 8.3 Hz, H-3′, H-5′), 6.68 (2H, d, J = 2.2 Hz, H-10, H-14), 6.74 (2H, d, J = 8.8 Hz, H-3, H-5), 6.92 (1H, d, J = 16.3 Hz, H-8), 7.10 (1H, d, J = 16.3 Hz, H-7), 7.13 (2H, d, J = 8.3 Hz, H-2′, H-6′), 7.36 (2H, d, J = 8.8 Hz, H-2, H-6), 9.09 (2H, s, 11′OH, 13′OH), 9.30 (1H, s, 4′OH); 13C NMR (DMSO, 126 MHz) δ 13.3 (CH3-7′d), 18.4 (C-7′c), 30.9 (C-7′b), 55.0 (CH3O-11, CH3O-13), 67.7 (C-7'a), 81.6 (C-8′), 83.7 (C-7′), 99.1 (C-12), 101.6 (C-12′), 103.9 (C-10, C-14), 105.3 (C-10′, C-14′), 114.3 (C-3′, C-5′), 115.7 (C-3, C-5), 125.9 (C-8), 127.3 (C-2, C-6), 128.3 (C-7), 128.9 (C-2′, C-6′), 129.2 (C-1, C-1′), 139.1 (C-9), 156.4 (C-4′), 157.1 (C-4), 157.5 (C-11′, C-13′), 160.4 (C-11, C-13). HR-ESI/MS analysis: m/z 555.2400 [M-H]−, (calcd for C34H35O7, 555.2383, ∆ = 3.1 ppm). MS/MS spectrum: CCMSLIB00009918891.
7-O-Butyl-11,11′,13,13′-tetra-O-methylleachianol G (36) UV (MeCN) λmax (log ε) 229 (sh) (3.87), 253 (3.56), 322 (3.49) nm. 1H NMR (DMSO, 600 MHz) δ 0.75 (3H, t, J = 7.2 Hz, CH3-7d), 1.19 (2H, m, H-7c), 1.25 (2H, m, H-7b), 2.86 (1H, dt, J = 9.8, 5.8 Hz, H-7a″), 3.10 (1H, dt, J = 9.6, 6.1 Hz, H-7a′), 3.24 (1H, dd, J = 8.4, 3.0 Hz, H-8), 3.40 (1H, t, J = 3.0 Hz, H-8′), 3.51 (3H, s, CH3O-13), 3.55 (3H, s, CH3O-11), 3.66 (6H, s, CH3O-11′, CH3O-13′), 3.96 (1H, d, J = 8.5 Hz, H-7), 4.12 (1H, d, J = 3.0 Hz, H-7′), 5.53 (1H, d, J = 2.2 Hz, H-14), 6.09 (2H, d, J = 2.3 Hz, H-10′, H-14′), 6.31 (1H, t, J = 2.3 Hz, H-12′), 6.34 (1H, d, J = 2.2 Hz, H-12), 6.63 (2H, d, J = 8.5 Hz, H-3′, H-5′), 6.70 (2H, d, J = 8.4 Hz, H-3, H-5), 6.73 (2H, d, J = 8.5 Hz, H-2′, H-6′), 6.93 (2H, d, J = 8.4 Hz, H-2, H-6), 9.10 (1H, s, 4′OH), 9.35 (1H, s, 4OH); 13C NMR (DMSO, 151 MHz) δ 13.3 (CH3-7d), 18.6 (C-7c), 30.7 (C-7b), 54.5 (C-7′), 54.5 (CH3O-13), 54.6 (CH3O-11′, CH3O-13′), 54.8 (CH3O-11), 57.5 (C-8′), 60.3 (C-8), 66.8 (C-7a), 83.5 (C-7), 96.8 (C-12′), 97.4 (C-12), 101.8 (C-14), 104.6 (C-10′, C-14′), 114.4 (C-3, C-5), 114.6 (C-3′, C-5′), 127.6 (C-2′, C-6′), 128.7 (C-2, C-6). HR-ESI/MS analysis: m/z 583.2706 [M-H]−, (calcd for C36H39O7, 583.2696, ∆ = 1.7 ppm). MS/MS spectrum: CCMSLIB00009918890.
Threo-7′-O-butylresveptero acyclic dimer (37) UV (MeCN) λmax (log ε) 228 (sh) (4.32), 286 (4.12), 308 (4.28), 326 (4.27), 343 (sh) (3.97) nm. 1H NMR (DMSO, 600 MHz) δ 0.80 (3H, t, J = 7.4 Hz, CH3-7′d), 1.26 (2H, m, H-7'c), 1.41 (2H, m, H-7′b), 3.26 (2H, t, J = 6.4 Hz, H-7'a), 3.75 (6H, s, CH3O-11, CH3O-13), 4.42 (1H, d, J = 6.6 Hz, H-7′), 5.14 (1H, d, J = 6.6 Hz, H-8′), 5.97 (1H, t, J = 2.2 Hz, H-12′), 6.06 (2H, d, J = 2.2 Hz, H-10′, H-14′), 6.36 (1H, t, J = 2.3 Hz, H-12), 6.61 (2H, d, J = 8.5 Hz, H-3′, H-5′), 6.70 (2H, d, J = 2.3 Hz, H-10, H-14), 6.84 (2H, d, J = 8.8 Hz, H-3, H-5), 6.95 (1H, d, J = 16.3 Hz, H-8), 7.00 (2H, d, J = 8.5 Hz, H-2′, H-6′), 7.13 (1H, d, J = 16.3 Hz, H-7), 7.41 (2H, d, J = 8.8 Hz, H-2, H-6), 9.03 (2H, s, 11′OH, 13′OH), 9.27 (1H, s, 4′OH); 13C NMR (DMSO, 151 MHz) δ 13.5 (CH3-7′d), 18.6 (C-7′c), 31.1 (C-7′b), 55.0 (CH3O-11, CH3O-13), 68.0 (C-7′a), 82.6 (C-8′), 84.2 (C-7′), 99.3 (C-12), 101.4 (C-12′), 103.9 (C-10, C-14), 105.5 (C-10′, C-14′), 114.3 (C-3′, C-5′), 115.7 (C-3, C-5), 125.9 (C-8), 127.3 (C-2, C-6), 128.3 (C-7), 128.5 (C-1′), 128.7 (C-2′, C-6′), 129.1 (C-1), 139.1 (C-9), 140.0 (C-9′), 156.4 (C-4′), 157.5 (C-11′, C-13′), 157.8 (C-4), 160.5 (C-11, C-13). HR-ESI/MS analysis: m/z 555.2394 [M-H]−, (calcd for C34H35O7, 555.2383, ∆ = 2.0 ppm). MS/MS spectrum: CCMSLIB00009918892.
7-O-Butyl-11,11′,13,13′-tetra-O-methylleachianol F (38) UV (MeCN) λmax (log ε) 229 (sh) (4.02), 285 (3.46), 320 (3.39) nm. 1H NMR (DMSO, 600 MHz) δ 0.79 (3H, t, J = 7.4 Hz, CH3-7d), 1.26 (2H, m, H-7c), 1.39 (2H, m, H-7b), 2.76 (1H, t, J = 3.7 Hz, H-8′), 3.02 (1H, dt, J = 9.4, 6.8 Hz, H-7a″), 3.12 (1H, dt, J = 9.5, 6.4 Hz, H-7a′), 3.24 (1H, dd, J = 9.1, 3.7 Hz, H-8), 3.57 (3H, s, CH3O-11), 3.57 (6H, s, CH3O-11′, CH3O-13′), 3.78 (3H, s, CH3O-13), 3.97 (1H, d, J = 9.1 Hz, H-7), 4.09 (1H, overlapped, H-7′), 5.71 (2H, d, J = 2.2 Hz, H-10′, H-14′), 6.21 (1H, t, J = 2.3 Hz, H-12′), 6.45 (1H, d, J = 2.1 Hz, H-12), 6.60 (2H, d, J = 8.5 Hz, H-3, H-5), 6.62 (2H, d, J = 8.7 Hz, H-3′, H-5′), 6.65 (2H, d, J = 8.7 Hz, H-2′, H-6′), 6.69 (2H, d, J = 8.5 Hz, H-2, H-6), 6.72 (1H, d, J = 2.1 Hz, H-14), 9.13 (1H, s, 4′OH), 9.30 (1H, s, 4OH); 13C NMR (DMSO, 151 MHz) δ 13.5 (CH3-7d), 18.6 (C-7c), 31.1 (C-7b), 54.1 (C-7′), 54.6 (CH3O-11, CH3O-11′, CH3O-13′), 54.8 (CH3O-13), 57.5 (C-8′), 59.6 (C-8), 67.1 (C-7a), 84.2 (C-7), 97.0 (C-12′), 97.5 (C-12), 102.5 (C-14), 104.2 (C-10′, C-14′), 114.4 (C-3, C-5), 114.7 (C-3′, C-5′), 127.6 (C-2′, C-6′), 128.3 (C-2, C-6). HR-ESI/MS analysis: m/z 583.2699 [M-H]−, (calcd for C36H39O7, 583.2696, ∆ = 0.5 ppm). MS/MS spectrum: CCMSLIB00009918893.
Erythro-7′-O-butylpterostilbene acyclic dimer (39) UV (MeCN) λmax (log ε) 228 (sh) (3.98), 285 (3.68), 314 (3.87), 344 (sh) (3.46) nm. 1H NMR (DMSO, 600 MHz) δ 0.73 (3H, t, J = 7.4 Hz, CH3-7′d), 1.14 (2H, h, J = 7.3 Hz, H-7′c), 1.32 (2H, dq, J = 8.2, 6.3 Hz, H-7′b), 3.12 (1H, dt, J = 9.5, 6.3 Hz, H-7′a″), 3.23 (1H, dt, J = 9.5, 6.3 Hz, H-7′a′), 3.67 (6H, s, CH3O-11′, CH3O-13′), 3.75 (6H, s, CH3O-11, CH3O-13), 4.42 (1H, d, J = 6.4 Hz, H-7′), 5.30 (1H, d, J = 6.4 Hz, H-8′), 6.36 (1H, t, J = 2.3 Hz, H-12′), 6.36 (1H, t, J = 2.4 Hz, H-12), 6.48 (2H, d, J = 2.3 Hz, H-10′, H-14′), 6.68 (2H, d, J = 2.4 Hz, H-10, H-14), 6.69 (2H, d, J = 8.6 Hz, H-3′, H-5′), 6.79 (2H, d, J = 8.8 Hz, H-3, H-5), 6.93 (1H, d, J = 16.3 Hz, H-8), 7.10 (1H, d, J = 16.3 Hz, H-7), 7.15 (2H, d, J = 8.6 Hz, H-2′, H-6′), 7.36 (2H, d, J = 8.8 Hz, H-2, H-6), 9.31 (1H, s, 4′OH); 13C NMR (DMSO, 126 MHz) δ 13.3 (CH3-7′d), 18.4 (C-7′c), 31.1 (C-7′b), 54.8 (CH3O-11, CH3O-13, CH3O-11′, CH3O-13′), 67.7 (C-7'a), 81.4 (C-8′), 83.3 (C-7′), 99.0 (C-12′), 99.2 (C-12), 103.9 (C-10, C-14), 105.6 (C-10′, C-14′), 114.3 (C-3′, C-5′), 115.8 (C-3, C-5), 126.0 (C-8), 127.3 (C-2, C-6), 128.2 (C-7), 128.9 (C-2′, C-6′). HR-ESI/MS analysis: m/z 583.2711 [M-H]−, (calcd for C36H39O7, 583.2696, ∆ = 2.6 ppm). MS/MS spectrum: CCMSLIB00009918894.
Threo-7′-O-butylpterostilbene acyclic dimer (40) UV (MeCN) λmax (log ε) 228 (sh) (4.56), 285 (4.35), 308 (4.53), 324 (4.53), 341 (sh) (4.27) nm. 1H NMR (DMSO, 600 MHz) δ 0.79 (3H, t, J = 7.4 Hz, CH3-7′d), 1.25 (2H, m, H-7′c), 1.42 (2H, m, H-7′b), 3.28 (2H, t, J = 6.4 Hz, H-7′a), 3.60 (6H, s, CH3O-11′, CH3O-13′), 3.75 (6H, s, CH3O-11, CH3O-13), 4.50 (1H, d, J = 6.5 Hz, H-7′), 5.31 (1H, d, J = 6.5 Hz, H-8′), 6.25 (1H, t, J = 2.3 Hz, H-12′), 6.33 (2H, d, J = 2.3 Hz, H-10′, H-14′), 6.37 (1H, t, J = 2.3 Hz, H-12), 6.61 (2H, d, J = 8.5 Hz, H-3′, H-5′), 6.70 (2H, d, J = 2.3 Hz, H-10, H-14), 6.89 (2H, d, J = 8.8 Hz, H-3, H-5), 6.96 (1H, d, J = 16.3 Hz, H-8), 7.01 (2H, d, J = 8.5 Hz, H-2′, H-6′), 7.13 (1H, d, J = 16.3 Hz, H-7), 7.41 (2H, d, J = 8.8 Hz, H-2, H-6), 9.27 (1H, s, 4′OH); 13C NMR (DMSO, 151 MHz) δ 13.5 (CH3-7′d), 18.6 (C-7′c), 31.1 (C-7′b), 54.6 (CH3O-11′, CH3O-13′), 55.0 (CH3O-11, CH3O-13), 67.8 (C-7′a), 82.4 (C-8′), 84.0 (C-7′), 98.8 (C-12′), 99.3 (C-12), 103.9 (C-10, C-14), 105.5 (C-10′, C-14′), 114.3 (C-3′, C-5′), 115.8 (C-3, C-5), 126.0 (C-8), 127.4 (C-2, C-6), 128.3 (C-7), 128.5 (C-1′), 128.7 (C-2′, C-6′), 129.2 (C-1), 139.1 (C-9), 140.4 (C-9′), 156.4 (C-4′), 157.5 (C-4), 159.5 (C-11′, C-13′), 160.4 (C-11, C-13). HR-ESI/MS analysis: m/z 583.2713 [M-H]−, (calcd for C36H39O7, 583.2696, ∆ = 2.9 ppm). MS/MS spectrum: CCMSLIB00009918895.
7-O-Isobutylleachianol G (41) UV (MeCN) λmax (log ε) 229 (sh) (4.37), 282 (3.62) nm. 1H NMR (DMSO, 600 MHz) δ 0.70 (3H, d, J = 6.6 Hz, CH3-7d), 0.72 (3H, d, J = 6.6 Hz, CH3-7c), 1.52 (1H, hept, J = 6.6 Hz, H-7b), 2.77 (1H, dd, J = 9.1, 6.5 Hz, H-7a′), 3.15 (1H, dd, J = 8.9, 2.7 Hz, H-8), 3.36 (1H, t, J = 2.7 Hz, H-8′), 3.77 (1H, d, J = 8.9 Hz, H-7), 4.03 (1H, d, J = 2.7 Hz, H-7′), 5.33 (1H, d, J = 2.1 Hz, H-14), 5.93 (2H, d, J = 2.2 Hz, H-10′, H-14′), 5.99 (1H, t, J = 2.2 Hz, H-12′), 6.07 (1H, d, J = 2.1 Hz, H-12), 6.61 (2H, d, J = 8.4 Hz, H-3′, H-5′), 6.67 (2H, d, J = 8.5 Hz, H-3, H-5), 6.72 (2H, d, J = 8.4 Hz, H-2′, H-6′), 6.88 (2H, d, J = 8.5 Hz, H-2, H-6), 8.76 (1H, s, 11OH), 8.78 (1H, s, 13OH), 9.00 (2H, s, 11′OH, 13′OH), 9.04 (1H, s, 4′OH), 9.32 (1H, s, 4OH); 13C NMR (DMSO, 151 MHz) δ 19.4 (C-7d), 19.4 (C-7c), 27.8 (C-7b), 54.2 (C-7′), 57.4 (C-8′), 60.2 (C-8), 74.6 (C-7a), 84.3 (C-7), 100.1 (C-12′), 101.4 (C-12), 104.1 (C-14), 104.6 (C-10′, C-14′), 114.6 (C-3′, C-5′), 114.7 (C-3, C-5), 121.5 (C-10), 128.0 (C-2′, C-6′), 129.0 (C-2, C-6), 130.7 (C-1), 136.7 (C-1′), 145.5 (C-9), 150.2 (C-9′), 154.0 (C-11), 155.1 (C-4′), 156.7 (C-4), 157.2 (C-13), 158.2 (C-11′, C-13′). HR-ESI/MS analysis: m/z 527.2072 [M-H]-, (calcd for C32H31O7, 527.2070, ∆ = 0.4 ppm). MS/MS spectrum: CCMSLIB00009918896.
Erythro-7′-O-isobutylresveratrol acyclic dimer (42) UV (MeCN) λmax (log ε) 227 (sh) (4.26), 286 (3.95), 308 (4.08), 325 (4.06), 341 (sh) (3.81) nm. 1H NMR (DMSO, 600 MHz) δ 0.69 (3H, d, J = 6.6 Hz, CH3-7′c), 0.69 (3H, d, J = 6.6 Hz, CH3-7′d), 1.63 (1H, hept, J = 6.6 Hz, H-7′b), 2.85 (1H, dd, J = 9.1, 6.6 Hz, H-7′a″), 2.99 (1H, dd, J = 9.1, 6.6 Hz, H-7′a′), 4.33 (1H, d, J = 6.6 Hz, H-7′), 5.09 (1H, d, J = 6.6 Hz, H-8′), 6.06 (1H, t, J = 2.2 Hz, H-12′), 6.09 (1H, t, J = 2.1 Hz, H-12), 6.22 (2H, d, J = 2.2 Hz, H-10′, H-14′), 6.34 (2H, d, J = 2.1 Hz, H-10, H-14), 6.69 (2H, d, J = 8.5 Hz, H-3′, H-5′), 6.72 (2H, d, J = 8.8 Hz, H-3, H-5), 6.79 (1H, d, J = 16.3 Hz, H-8), 6.86 (1H, d, J = 16.3 Hz, H-7), 7.14 (2H, d, J = 8.5 Hz, H-2′, H-6′), 7.33 (2H, d, J = 8.8 Hz, H-2, H-6), 9.07 (2H, s, 11′OH, 13′OH), 9.17 (2H, s, 11OH, 13OH), 9.30 (1H, s, 4′OH); 13C NMR (DMSO, 151 MHz) δ 19.1 (CH3-7′d), 19.1 (CH3-7′c), 27.9 (C-7′b), 75.1 (C-7'a), 81.9 (C-8′), 84.0 (C-7′), 101.8 (C-12′), 101.9 (C-12), 104.3 (C-10, C-14), 105.7 (C-10′, C-14′), 114.6 (C-3′, C-5′), 115.9 (C-3, C-5), 126.7 (C-8), 127.3 (C-7), 127.4 (C-2, C-6), 129.0 (C-2′, C-6′), 129.4 (C-1′), 129.6 (C-1), 139.0 (C-9), 141.0 (C-9′), 156.8 (C-4′), 157.2 (C-4), 157.8 (C-11′, C-13′), 158.5 (C-11, C-13). HR-ESI/MS analysis: m/z 527.2076 [M-H]−, (calcd for C32H31O7, 527.2070, ∆ = 1.1 ppm). MS/MS spectrum: CCMSLIB00009918897.
7-O-Isobutyl-11′,13′-di-O-methylleachianol F (43) UV (MeCN) λmax (log ε) 229 (sh) (4.44), 285 (3.81), 320 (3.62) nm. 1H NMR (DMSO, 600 MHz) δ 0.77 (3H, d, J = 6.7 Hz, CH3-7d), 0.78 (3H, d, J = 6.7 Hz, CH3-7c), 1.70 (1H, dp, J = 13.3, 6.6 Hz, H-7b), 2.70 (1H, t, J = 3.4 Hz, H-8′), 2.82 (2H, m, H-7a), 3.12 (1H, dd, J = 9.1, 3.4 Hz, H-8), 3.57 (5H, s, CH3O-13′, CH3O-11′), 3.90 (1H, d, J = 9.1 Hz, H-7), 4.06 (1H, d, J = 3.4 Hz, H-7′), 5.73 (2H, d, J = 2.3 Hz, H-10′, H-14′), 6.17 (1H, d, J = 2.1 Hz, H-12), 6.20 (1H, t, J = 2.3 Hz, H-12′), 6.47 (1H, d, J = 2.1 Hz, H-14), 6.59 (2H, d, J = 8.5 Hz, H-3, H-5), 6.62 (2H, d, J = 8.5 Hz, H-3′, H-5′), 6.65 (1H, d, J = 8.5 Hz, H-2, H-6), 6.67 (2H, d, J = 8.5 Hz, H-2′, H-6′), 8.79 (1H, s, 11OH), 8.97 (1H, s, 13OH), 9.10 (1H, s, 4′OH), 9.29 (1H, s, 4OH); 13C NMR (DMSO, 151 MHz) δ 19.1 (CH3-7d), 19.1 (CH3-7c), 27.7 (C-7b), 53.8 (C-7′), 54.6 (CH3O-11′, CH3O-13′), 57.3 (C-8′), 59.7 (C-8), 74.5 (C-7a), 84.2 (C-7), 97.0 (C-12′), 101.2 (C-12), 104.2 (C-10′, C-14′), 104.6 (C-14), 114.4 (C-3, C-5, C-3′, C-5′), 120.7 (C-10), 128.1 (C-2, C-6, C-2′, C-6′), 130.8 (C-1), 135.7 (C-1′), 146.7 (C-9), 149.2 (C-9′), 153.7 (C-11), 154.8 (C-4′), 156.4 (C-4), 157.5 (C-13), 159.8 (C-11′, C-13′). HR-ESI/MS analysis: m/z 555.2393 [M-H]−, (calcd for C34H35O7, 555.2383, ∆ = 1.8 ppm). MS/MS spectrum: CCMSLIB00009918898.
Threo-7′-O-isobutylresveratrol acyclic dimer (44) UV (MeCN) λmax (log ε) 229 (sh) (4.36), 285 (4.15), 307 (4.31), 326 (4.30), 341 (sh) (4.05) nm. 1H NMR (DMSO, 600 MHz) δ 0.79 (3H, d, J = 6.6 Hz, CH3-7′d), 0.79 (3H, d, J = 6.6 Hz, CH3-7'c), 1.73 (1H, hept, J = 6.6 Hz, H-7′b), 3.03 (2H, d, J = 6.6 Hz, H-7'a), 4.41 (1H, d, J = 6.6 Hz, H-7′), 5.13 (1H, d, J = 6.6 Hz, H-8′), 5.97 (1H, t, J = 2.2 Hz, H-12′), 6.06 (2H, d, J = 2.2 Hz, H-10′, H-14′), 6.10 (1H, t, J = 2.1 Hz, H-12), 6.36 (2H, d, J = 2.1 Hz, H-10, H-14), 6.61 (2H, d, J = 8.6 Hz, H-3′, H-5′), 6.81 (2H, d, J = 16.3 Hz, H-8), 6.82 (2H, d, J = 8.8 Hz, H-3, H-5), 6.89 (1H, d, J = 16.3 Hz, H-7), 6.99 (2H, d, J = 8.6 Hz, H-2′, H-6′), 7.39 (2H, d, J = 8.8 Hz, H-2, H-6), 9.02 (2H, s, 11′OH, 13′OH), 9.18 (2H, s, 11OH, 13OH), 9.26 (1H, s, 4′OH); 13C NMR (DMSO, 151 MHz) δ 19.2 (CH3-7′d), 19.3 (CH3-7′c), 28.0 (C-7'b), 75.4 (C-7′a), 82.9 (C-8′), 84.6 (C-7′), 101.7 (C-12′), 101.9 (C-12), 104.3 (C-10, C-14), 105.6 (C-10′, C-14′), 114.6 (C-3′, C-5′), 115.8 (C-3, C-5), 126.6 (C-8), 127.4 (C-7), 127.5 (C-2, C-6), 128.7 (C-1′), 128.9 (C-2′, C-6′), 129.4 (C-1), 139.0 (C-9), 140.2 (C-9′), 156.6 (C-4′), 157.7 (C-11′, C-13′), 157.8 (C-4), 158.5 (C-11, C-13). HR-ESI/MS analysis: m/z 527.2077 [M-H]−, (calcd for C32H31O7, 527.2070, ∆ = 1.3 ppm). MS/MS spectrum: CCMSLIB00009918899.
7-O-Isobutyl-11′,13′-di-O-methylleachianol G (45) UV (MeCN) λmax (log ε) 228 (sh) (4.43), 285 (3.65) nm. 1H NMR (DMSO, 600 MHz) δ 0.71 (3H, d, J = 6.7 Hz, CH3-7d), 0.73 (3H, d, J = 6.7 Hz, CH3-7c), 1.56 (1H, hept, J = 6.7 Hz, H-7b), 2.57 (1H, dd, J = 9.0, 6.7 Hz, H-7a″), 2.81 (1H, dd, J = 9.0, 6.7 Hz, H-7a′), 3.21 (1H, dd, J = 8.3, 3.3 Hz, H-8), 3.42 (1H, t, J = 3.3 Hz, H-8′), 3.65 (6H, s, CH3O-11′, CH3O-13′), 3.89 (1H, d, J = 8.3 Hz, H-7), 4.08 (1H, d, J = 3.3 Hz, H-7′), 5.46 (1H, d, J = 2.1 Hz, H-14), 6.08 (1H, d, J = 2.1 Hz, H-12), 6.11 (2H, d, J = 2.3 Hz, H-10′, H-14′), 6.30 (1H, t, J = 2.3 Hz, H-12′), 6.61 (2H, d, J = 8.5 Hz, H-3′, H-5′), 6.66 (2H, d, J = 8.5 Hz, H-3, H-5), 6.74 (2H, d, J = 8.5 Hz, H-2′, H-6′), 6.91 (2H, d, J = 8.5 Hz, H-2, H-6), 8.77 (1H, s, 11OH), 8.82 (1H, s, 13OH), 9.04 (1H, s, 4′OH), 9.30 (1H, s, 4OH); 13C NMR (DMSO, 151 MHz) δ 19.3 (CH3-7d), 19.4 (CH3-7c), 27.9 (C-7b), 54.2 (C-7′), 54.9 (CH3O-11′, CH3O-13′), 57.5 (C-8′), 60.3 (C-8), 74.7 (C-7a), 84.0 (C-7), 97.0 (C-12′), 101.5 (C-12), 103.9 (C-14), 104.9 (C-10′, C-14′), 114.6 (C-3′, C-5′), 114.7 (C-3, C-5), 121.5 (C-10), 128.1 (C-2′, C-6′), 128.9 (C-2, C-6), 130.6 (C-1), 136.4 (C-1′), 145.3 (C-9), 150.1 (C-9′), 154.0 (C-11), 155.1 (C-4′), 156.7 (C-4), 157.3 (C-13), 160.3 (C-11′, C-13′). HR-ESI/MS analysis: m/z 555.2390 [M-H]−, (calcd for C34H35O7, 555.2383, ∆ = 1.3 ppm). MS/MS spectrum: CCMSLIB00009918900.
Threo-7′-O-isobutylisoresveptero acyclic dimer (46) UV (MeCN) λmax (log ε) 229 (sh) (4.36), 285 (4.15), 307 (4.33), 326 (4.30), 341 (sh) (4.08) nm. 1H NMR (DMSO, 600 MHz) δ 0.79 (3H, d, J = 6.6 Hz, CH3-7′d), 0.80 (3H, d, J = 6.6 Hz, CH3-7′c), 1.74 (1H, hept, J = 6.6 Hz, H-7′b), 3.06 (2H, d, J = 6.6 Hz, H-7′a), 3.60 (6H, s, CH3O-11′, CH3O-13′), 4.50 (1H, d, J = 6.5 Hz, H-7′), 5.30 (1H, d, J = 6.5 Hz, H-8′), 6.10 (1H, t, J = 2.1 Hz, H-12), 6.25 (1H, t, J = 2.3 Hz, H-12′), 6.32 (2H, d, J = 2.3 Hz, H-10′, H-14′), 6.36 (2H, d, J = 2.1 Hz, H-10, H-14), 6.61 (2H, d, J = 8.7 Hz, H-3′, H-5′), 6.82 (1H, d, J = 16.3 Hz, H-8), 6.87 (2H, d, J = 8.8 Hz, H-3, H-5), 6.90 (1H, d, J = 16.3 Hz, H-7), 7.00 (2H, d, J = 8.4 Hz, H-2′, H- 6′), 7.39 (2H, d, J = 8.7 Hz, H-2, H-6), 9.18 (2H, s, 11OH, 13OH), 9.28 (1H, s, 4′OH); 13C NMR (DMSO, 151 MHz) δ 19.2 (CH3-7′d), 19.2 (CH3-7′c), 28.1 (C-7′b), 55.0 (CH3O-11′, CH3O-13′), 75.3 (C-7′a), 82.7 (C-8′), 84.4 (C-7′), 99.1 (C-12′), 101.9 (C-12), 104.4 (C-10, C-14), 105.6 (C-10′, C-14′), 114.5 (C-3′, C-5′), 115.9 (C-3, C-5), 126.7 (C-8), 127.4 (C-7), 127.5 (C-2, C-6), 128.6 (C-1′), 129.0 (C-2′, C-6′), 129.6 (C-1), 139.0 (C-9), 140.6 (C-9′), 156.7 (C-4′), 157.7 (C-4), 158.5 (C-11, C-13), 159.8 (C-11′, C-13′). HR-ESI/MS analysis: m/z 555.2402 [M-H]−, (calcd for C34H35O7, 555.2383, ∆ = 3.4 ppm). MS/MS spectrum: CCMSLIB00009918901.
Erythro-7′-O-isobutylisoresveptero acyclic dimer (47) UV (MeCN) λmax (log ε) 229 (sh) (4.41), 285 (4.16), 307 (4.32), 327 (4.27), 342 (sh) (4.01) nm. 1H NMR (DMSO, 600 MHz) δ 0.69 (3H, d, J = 6.6 Hz, CH3-7′d), 0.70 (3H, d, J = 6.6 Hz, CH3-7′c), 1.64 (1H, hept, J = 6.6 Hz, CH3-7′b), 2.88 (1H, dd, J = 9.0, 6.3 Hz, CH3-7′a″), 3.01 (1H, dd, J = 9.1, 6.4 Hz, CH3-7′a′), 3.67 (6H, s, CH3O-11′, CH3O-13′), 4.42 (1H, d, J = 6.5 Hz, H-7′), 5.27 (1H, d, J = 6.5 Hz, H-8′), 6.10 (1H, t, J = 2.2 Hz, H-12), 6.34 (2H, d, J = 2.2 Hz, H-10, H-14), 6.35 (1H, t, J = 2.3 Hz, H-12′), 6.50 (2H, d, J = 2.3 Hz, H-10′, H-14′), 6.70 (2H, d, J = 8.5 Hz, H-3′, H-5′), 6.77 (2H, d, J = 8.9 Hz, H-3, H-5), 6.80 (1H, d, J = 16.2 Hz, H-8), 6.87 (1H, d, J = 16.2 Hz, H-7), 7.15 (2H, d, J = 8.6 Hz, H-2′, H-6′), 7.34 (2H, d, J = 8.9 Hz, H-2, H-6), 9.17 (2H, s, 11OH, 13OH), 9.32 (1H, s, 4′OH); 13C NMR (DMSO, 151 MHz) δ 18.9 (CH3-7′d, CH3-7′c), 27.7 (C-7′b), 74.9 (C-7′a), 81.6 (C-8′), 83.5 (C-7′), 99.0 (C-12′), 101.6 (C-12), 104.1 (C-10, C-14), 105.6 (C-10′, C-14′), 114.3 (C-3′, C-5′), 115.8 (C-3, C-5), 126.7 (C-8), 127.1 (C-7), 127.3 (C-2, C-6), 128.9 (C-2′, C-6′), 129.0 (C-1′), 129.5 (C-1), 138.7 (C-9), 156.7 (C-4′), 156.9 (C-4), 158.3 (C-11, C-13), 159.6 (C-11′, C-13′). HR-ESI/MS analysis: m/z 555.2402 [M-H]−, (calcd for C34H35O7, 555.2383, ∆ = 3.4 ppm). MS/MS spectrum: CCMSLIB00009918902.
Erythro-7′-O-isobutylresveptero acyclic dimer (48) UV (MeCN) λmax (log ε) 229 (sh) (4.63), 286 (4.36), 307 (4.51), 326 (4.51), 343 (sh) (4.19) nm. 1H NMR (DMSO, 600 MHz) δ 0.69 (4H, d, J = 6.6 Hz, CH3-7′d), 0.70 (4H, d, J = 6.6 Hz, CH3-7′c), 1.63 (1H, m, H-7′b), 2.86 (1H, dd, J = 9.1, 6.4 Hz, H-7′a″), 2.99 (1H, dd, J = 9.1, 6.5 Hz, H-7′a′), 3.75 (6H, s, CH3O-11, CH3O-13), 4.32 (1H, d, J = 6.7 Hz, H-7′), 5.10 (1H, d, J = 6.7 Hz, H-8′), 6.06 (1H, t, J = 2.2 Hz, H-12′), 6.23 (2H, d, J = 2.2 Hz, H-10′, H-14′), 6.36 (1H, t, J = 2.3 Hz, H-12), 6.68 (2H, d, J = 2. Hz, H-10, H-14), 6.69 (2H, d, J = 8.6 Hz, H-3′, H-5′), 6.74 (2H, d, J = 8.8 Hz, H-3, H-5), 6.92 (1H, d, J = 16.4 Hz, H-8), 7.10 (1H, d, J = 16.4 Hz, H-7), 7.14 (2H, d, J = 8.6 Hz, H-2′, H-6′), 7.36 (2H, d, J = 8.8 Hz, H-2, H-6), 9.08 (2H, s, 11′OH, 13′OH), 9.30 (1H, s, 4′OH); 13C NMR (DMSO, 151 MHz) δ 19.0 (CH3-7′d, CH3-7′c), 27.7 (C-7′b), 54.8 (CH3O-11, CH3O-13), 74.9 (C-7′a), 81.6 (C-8′), 83.9 (C-7′), 99.3 (C-12), 101.6 (C-12′), 103.9 (C-10, C-14), 105.5 (C-10′, C-14′), 114.3 (C-3′, C-5′), 115.7 (C-3, C-5), 125.9 (C-8), 127.3 (C-2, C-6), 128.1 (C-7), 128.9 (C-2′, C-6′), 129.3 (C-1, C-1′), 139.1 (C-9), 140.9 (C-9′), 156.4 (C-4′), 157.1 (C-4), 157.6 (C-11′, C-13′), 160.5 (C-11, C-13). HR-ESI/MS analysis: m/z 555.2398 [M-H]−, (calcd for C34H35O7, 555.2383, ∆ = 2.7 ppm). MS/MS spectrum: CCMSLIB00009918903.
7-O-Isobutyl-11,11′,13,13′-tetra-O-methylleachianol G (49) UV (MeCN) λmax (log ε) 229 (sh) (4.30), 285 (3.70), 321 (3.56) nm. 1H NMR (DMSO, 600 MHz) δ 0.72 (3H, d, J = 6.3 Hz, CH3-7d), 0.73 (3H, d, J = 6.3 Hz, CH3-7c), 1.58 (1H, m, J = 6.6 Hz, H-7b), 2.63 (1H, dd, J = 9.1, 6.6 Hz, H-7a″), 2.87 (1H, dd, J = 9.1, 6.6 Hz, H-7a′), 3.26 (1H, dd, J = 8.4, 3.0 Hz, H-8), 3.43 (1H, t, J = 3.0 Hz, H-8′), 3.51 (3H, s, CH3O-13), 3.55 (3H, s, CH3O-11), 3.66 (6H, s, CH3O-13′, CH3O-11′), 3.94 (1H, d, J = 8.4 Hz, H-7), 4.13 (1H, d, J = 3.0 Hz, H-7′), 5.57 (1H, d, J = 2.1 Hz, H-14), 6.10 (2H, d, J = 2.3 Hz, H-10′, H-14′), 6.31 (1H, t, J = 2.3 Hz, H-12′), 6.35 (1H, d, J = 2.1 Hz, H-12), 6.62 (2H, d, J = 8.5 Hz, H-3′, H-5′), 6.69 (3H, d, J = 8.4 Hz, H-3, H-5), 6.71 (3H, d, J = 8.5 Hz, H-2′, H-6′), 6.92 (2H, d, J = 8.4 Hz, H-2, H-6), 9.09 (1H, s, 4′OH), 9.35 (1H, s, 4OH); 13C NMR (DMSO, 151 MHz) δ 19.2 (CH3-7d), 19.3 (CH3-7c), 27.9 (C-7b), 54.5 (C-7′), 54.7 (CH3O-13), 54.9 (CH3O-11′, CH3O-13′), 55.0 (CH3O-11), 57.7 (C-8′), 60.6 (C-8), 74.7 (C-7a), 84.1 (C-7), 97.1 (C-12′), 97.6 (C-12), 102.0 (C-14), 104.8 (C-10′, C-14′), 114.7 (C-3, C-5), 114.8 (C-3′, C-5′), 124.4 (C-10), 127.9 (C-2′, C-6′), 129.0 (C-2, C-6), 130.4 (C-1), 136.0 (C-1′), 145.1 (C-9), 149.8 (C-9′), 155.2 (C-4′), 156.3 (C-11), 156.9 (C-4), 159.8 (C-13), 160.4 (C-11′, C-13′). HR-ESI/MS analysis: m/z 583.2704 [M-H]−, (calcd for C36H39O7, 583.2696, ∆ = 1.4 ppm). MS/MS spectrum: CCMSLIB00009918904.
Threo-7′-O-isobutylresveptero acyclic dimer (50) UV (MeCN) λmax (log ε) 228 (sh) (4.35), 286 (4.13), 307 (4.28), 326 (4.27), 343 (sh) (3.96) nm. 1H NMR (DMSO, 600 MHz) δ 0.78 (3H, d, J = 6.7 Hz, CH3-7′c), 0.79 (3H, d, J = 6.7 Hz, CH3-7′d), 1.73 (1H, hept, J = 6.6 Hz, H-7′b), 3.03 (2H, d, J = 6.6 Hz, H-7′a), 3.76 (6H, s, CH3O-11, CH3O-13), 4.41 (1H, d, J = 6.6 Hz, H-7′), 5.14 (1H, d, J = 6.6 Hz, H-8′), 5.97 (1H, t, J = 2.2 Hz, H-12′), 6.07 (2H, d, J = 2.2 Hz, H-10′, H-14′), 6.36 (1H, t, J = 2.2 Hz, H-12), 6.61 (2H, d, J = 8.5 Hz, H-3′, H-5′), 6.70 (2H, d, J = 2.2 Hz, H-10, H-14), 6.85 (2H, d, J = 8.8 Hz, H-3, H-5), 6.95 (1H, d, J = 16.4 Hz, H-8), 7.00 (2H, d, J = 8.5 Hz, H-2′, H-6′), 7.13 (1H, d, J = 16.4 Hz, H-7), 7.41 (2H, d, J = 8.8 Hz, H-2, H-6), 9.02 (2H, s, 11′OH, 13′OH), 9.26 (1H, s, 4′OH); 13C NMR (DMSO, 151 MHz) δ 19.2 (CH3-7′d), 19.3 (CH3-7′c), 28.0 (C-7′b), 55.2 (CH3O-11, CH3O-13), 75.4 (C-7'a), 82.8 (C-8′), 84.6 (C-7′), 99.5 (C-12), 101.7 (C-12′), 104.1 (C-10, C-14), 105.6 (C-10′, C-14′), 114.6 (C-3′, C-5′), 115.8 (C-3, C-5), 126.1 (C-8), 127.6 (C-2, C-6), 128.5 (C-7), 128.7 (C-1′), 128.9 (C-2′, C-6′), 129.3 (C-1), 139.4 (C-9), 140.2 (C-9′), 156.6 (C-4′), 157.7 (C-11′, C-13′), 158.0 (C-4), 160.6 (C-11, C-13). HR-ESI/MS analysis: m/z 555.2395 [M-H]−, (calcd for C34H35O7, 555.2383, ∆ = 2.2 ppm). MS/MS spectrum: CCMSLIB00009918905.
7-O-Isobutyl-11,11′,13,13′-tetra-O-methylleachianol F (51) UV (MeCN) λmax (log ε) 228 (sh) (4.43), 285 (3.75), 321 (3.56) nm. 1H NMR (DMSO, 600 MHz) δ 0.78 (3H, d, J = 6.7 Hz, CH3-7c), 0.80 (3H, d, J = 6.7 Hz, CH3-7d), 1.70 (1H, m, H-7b), 2.76 (1H, t, J = 3.7 Hz, H-8′), 2.82 (1H, dd, J = 9.0, 6.2 Hz, H-7a″), 2.87 (1H, dd, J = 9.0, 6.5 Hz, H-7a′), 3.23 (1H, dd, J = 9.2, 3.7 Hz, H-8), 3.57 (6H, s, CH3O-13′, CH3O-11′), 3.58 (3H, s, CH3O-11), 3.78 (3H, s, CH3O-13), 3.96 (1H, d, J = 9.2 Hz, H-7), 4.10 (1H, d, J = 3.7 Hz, H-7′), 5.70 (2H, d, J = 2.3 Hz, H-10′, H-14′), 6.20 (1H, t, J = 2.3 Hz, H-12′), 6.45 (1H, d, J = 2.1 Hz, H-12), 6.60 (2H, d, J = 8.4 Hz, H-3, H-5), 6.62 (2H, d, J = 8.7 Hz, H-3′, H-5′), 6.65 (2H, d, J = 8.7 Hz, H-2′, H-6′), 6.68 (2H, d, J = 8.4 Hz, H-2, H-6), 6.75 (1H, d, J = 2.1 Hz, H-14), 9.14 (1H, s, 4′OH), 9.31 (1H, s, 4OH); 13C NMR (DMSO, 151 MHz) δ 19.1 (CH3-7c, CH3-7d), 27.9 (C-7b), 53.9 (C-7′), 54.6 (CH3O-11, CH3O-11′, CH3O-13′), 54.8 (CH3O-13), 57.3 (C-8′), 59.7 (C-8), 74.3 (C-7a), 84.4 (C-7), 97.0 (C-12′), 97.6 (C-12), 102.7 (C-14), 104.2 (C-10′, C-14′), 114.4 (C-3, C-5), 114.6 (C-3′, C-5′), 123.6 (C-10), 127.6 (C-2′, C-6′), 128.2 (C-2, C-6), 130.3 (C-1), 135.5 (C-1′), 146.7 (C-9), 148.8 (C-9′), 155.1 (C-4′), 156.0 (C-11), 156.4 (C-4), 159.8 (C-11′, C-13′), 160.0 (C-13). HR-ESI/MS analysis: m/z 583.2704 [M-H]−, (calcd for C36H39O7, 583.2696, ∆ = 1.4 ppm). MS/MS spectrum: CCMSLIB00009918906.
Erythro-7′-O-isobutylpterostilbene acyclic dimer (52) UV (MeCN) λmax (log ε) 227 (sh) (4.32), 285 (4.04), 307 (4.22), 324 (4.23), 344 (sh) (3.82) nm. 1H NMR (DMSO, 600 MHz) δ 0.69 (3H, d, J = 6.7 Hz, CH3-7′d), 0.70 (3H, d, J = 6.6 Hz, CH3-7′c), 1.64 (1H, m, H-7'b), 2.88 (1H, dd, J = 9.1, 6.4 Hz, H-7′a″), 3.01 (1H, dd, J = 9.1, 6.4 Hz, H-7′a′), 3.67 (6H, s, CH3O-11′, CH3O-13′), 3.75 (6H, s, CH3O-11, CH3O-13), 4.42 (1H, d, J = 6.5 Hz, H-7′), 5.29 (1H, d, J = 6.5 Hz, H-8′), 6.36 (1H, t, J = 2.3 Hz, H-12′), 6.36 (1H, t, J = 2.3 Hz, H-12), 6.50 (2H, d, J = 2.3 Hz, H-10′, H-14′), 6.68 (2H, d, J = 2.3 Hz, H-10, H-14), 6.70 (2H, d, J = 8.6 Hz, H-3′, H-5′), 6.80 (2H, d, J = 8.8 Hz, H-3, H-5), 6.93 (1H, d, J = 16.3 Hz, H-8), 7.10 (1H, d, J = 16.3 Hz, H-7), 7.15 (2H, d, J = 8.6 Hz, H-2′, H-6′), 7.36 (2H, d, J = 8.8 Hz, H-2, H-6), 9.32 (1H, s, 4′OH); 13C NMR (DMSO, 151 MHz) δ 19.1 (CH3-7′c, CH3-7′d), 55.0 (CH3O-11′, CH3O-13′), 55.2 (CH3O-11, CH3O-13), 75.1 (C-7′a), 81.7 (C-8′), 83.7 (C-7′), 99.1 (C-12′), 99.5 (C-12), 104.1 (C-10, C-14), 105.9 (C-10′, C-14′), 114.6 (C-3′, C-5′), 116.0 (C-3, C-5), 126.3 (C-8), 127.6 (C-2, C-6), 128.4 (C-7), 129.0 (C-2′, C-6′), 129.2 (C-1′), 129.7 (C-1), 139.3 (C-9), 141.3 (C-9′), 156.8 (C-4′), 157.2 (C-4), 159.8 (C-11′, C-13′), 160.6 (C-11, C-13). HR-ESI/MS analysis: m/z 583.2715 [M-H]−, (calcd for C36H39O7, 583.2696, ∆ = 3.3 ppm). MS/MS spectrum: CCMSLIB00009918907.
Threo-7′-O-isobutylpterostilbene acyclic dimer (53) UV (MeCN) λmax (log ε) 227 (sh) (4.34), 286 (4.13), 307 (4.29), 327 (4.28), 342 (sh) (3.99) nm. 1H NMR (DMSO, 600 MHz) δ 0.79 (3H, d, J = 6.7 Hz, CH3-7′d), 0.80 (3H, d, J = 6.7 Hz, CH3-7′c), 1.74 (1H, m, J = 6.6 Hz, H-7′b), 3.05 (2H, d, J = 6.5 Hz, H-7′a), 3.60 (6H, s, CH3O-11′, CH3O-13′), 3.75 (6H, s, CH3O-11, CH3O-13), 4.50 (1H, d, J = 6.5 Hz, H-7′), 5.32 (1H, d, J = 6.5 Hz, H-8′), 6.25 (1H, t, J = 2.3 Hz, H-12′), 6.33 (2H, d, J = 2.3 Hz, H-10′, H-14′), 6.37 (1H, t, J = 2.2 Hz, H-12), 6.61 (2H, d, J = 8.6 Hz, H-3′, H-5′), 6.70 (2H, d, J = 2.2 Hz, H-10, H-14), 6.89 (2H, d, J = 8.9 Hz, H-3, H-5), 6.96 (1H, d, J = 16.3 Hz, H-8), 7.01 (2H, d, J = 8.6 Hz, H-2′, H-6′), 7.13 (1H, d, J = 16.3 Hz, H-7), 7.41 (2H, d, J = 8.9 Hz, H-2, H-6), 9.27 (1H, s, 4′OH); 13C NMR (DMSO, 151 MHz) δ 19.2 (CH3-7′c, CH3-7′d), 28.1 (C-7′b), 55.0 (CH3O-11′, CH3O-13′), 55.2 (CH3O-11, CH3O-13), 75.3 (C-7′a), 82.6 (C-8′), 84.4 (C-7′), 99.1 (C-12′), 99.5 (C-12), 104.1 (C-10, C-14), 105.6 (C-10′, C-14′), 114.5 (C-3′, C-5′), 116.0 (C-3, C-5), 126.2 (C-8), 127.6 (C-2, C-6), 128.5 (C-7), 128.6 (C-1′), 129.0 (C-2′, C-6′), 129.5 (C-1), 139.4 (C-9), 140.6 (C-9′), 156.7 (C-4′), 157.9 (C-4), 159.8 (C-11′, C-13′), 160.6 (C-11, C-13). HR-ESI/MS analysis: m/z 583.2715 [M-H]−, (calcd for C36H39O7, 583.2696, ∆ = 3.3 ppm). MS/MS spectrum: CCMSLIB00009918908.
Resvepterodimer A (54) UV (MeCN) λmax (log ε) 228 (sh) (4.14), 282 (3.52) nm. 1H NMR (DMSO, 600 MHz) δ 2.84 (3H, s, CH3O-7′a), 3.00 (1H, dd, J = 9.5, 2.5 Hz, H-8′), 3.36 (3H, s, CH3O-7′b), 3.57 (6H, s, CH3O-11′, CH3O-13′), 3.74 (1H, d, J = 12.7 Hz, H-7), 4.06 (2H, dd, J = 12.7, 2.5 Hz, H-8), 4.43 (1H, d, J = 9.5 Hz, H-7′), 5.38 (2H,br s, H-10, H-14), 5.60 (2H, brs, H-10′, H-14′), 5.88 (1H, t, J = 2.2 Hz, H-12), 6.28 (1H, t, J = 2.3 Hz, H-12′), 6.37 (2H, d, J = 8.5 Hz, H-3, H-5), 6.74 (2H, d, J = 8.4 Hz, H-3′, H-5′), 6.88 (2H, d, J = 8.5 Hz, H-2, H-6), 7.20 (2H, d, J = 8.4 Hz, H-2′, H-6′). HR-ESI/MS analysis: m/z 545.2160 [M-H]−, (calcd for C32H33O8, 545.2175, ∆ = 2.8 ppm). MS/MS spectrum: CCMSLIB00009918909.
Erythro-7′-O-methylresveratrol acyclic dimer (55) UV (MeCN) λmax (log ε) 228 (sh) (4.46), 286 (4.13), 324 (4.19), 341 (sh) (3.96) nm. 1H NMR (DMSO, 600 MHz) δ 3.03 (3H, s, CH3O-7′), 4.29 (1H, d, J = 6.2 Hz, H-7′), 5.15 (1H, d, J = 6.2 Hz, H-8′), 6.06 (1H, t, J = 2.2 Hz, H-12′), 6.09 (1H, t, J = 2.2 Hz, H-12), 6.18 (2H, d, J = 2.2 Hz, H-10′, H-14′), 6.34 (2H, d, J = 2.2 Hz, H-10, H-14), 6.69 (2H, d, J = 8.6 Hz, H-3′, H-5′), 6.72 (2H, d, J = 8.8 Hz, H-3, H-5), 6.79 (1H, d, J = 16.3 Hz, H-8), 6.87 (1H, d, J = 16.3 Hz, H-7), 7.12 (2H, d, J = 8.6 Hz, H-2′, H-6′), 7.34 (2H, d, J = 8.8 Hz, H-2, H-6), 9.11 (2H, s, 11′OH, 13′OH), 9.18 (2H, s, 11OH, 13OH), 9.33 (1H, s, 4′OH); 13C NMR (DMSO, 151 MHz) δ 56.1 (CH3O-7′), 81.6 (C-8′), 85.6 (C-7′), 101.9 (C-12, C-12′), 104.2 (C-10, C-14), 105.7 (C-10′, C-14′), 114.6 (C-3′, C-5′), 115.9 (C-3, C-5), 126.8 (C-8), 127.3 (C-7), 127.5 (C-2, C-6), 128.4 (C-1′), 129.2 (C-2′, C-6′), 129.6 (C-1), 138.8 (C-9), 156.8 (C-4′), 157.0 (C-4), 157.7 (C-11′, C-13′), 158.3 (C-11, C-13). HR-ESI/MS analysis: m/z 485.1593 [M-H]−, (calcd for C29H25O7, 485.1600, ∆ = 1.4 ppm). MS/MS spectrum: CCMSLIB00009918910.
Threo-7′-O-methylresveratrol acyclic dimer (56) UV (MeCN) λmax (log ε) 228 (sh) (4.32), 286 (4.05), 308 (4.14), 325 (4.17), 342 (sh) (3.92) nm. 1H NMR (DMSO, 600 MHz) δ 3.12 (3H, s, CH3O-7′), 4.35 (1H, d, J = 6.6 Hz, H-7′), 5.16 (1H, d, J = 6.6 Hz, H-8′), 5.96 (1H, t, J = 2.2 Hz, H-12′), 6.04 (2H, d, J = 2.2 Hz, H-10′, H-14′), 6.10 (1H, t, J = 2.3 Hz, H-12), 6.36 (2H, d, J = 2.3 Hz, H-10, H-14), 6.62 (2H, d, J = 8.5 Hz, H-3′, H-5′), 6.81 (1H, d, J = 16.3 Hz, H-8), 6.83 (2H, d, J = 8.7 Hz, H-3, H-5), 6.89 (1H, d, J = 16.3 Hz, H-7), 6.99 (2H, d, J = 8.5 Hz, H-2′, H-6′), 7.38 (2H, d, J = 8.7 Hz, H-2, H-6), 9.03 (2H, s, 11′OH, 13′OH), 9.18 (2H, s, 11OH, 13OH), 9.30 (1H, s, 4′OH); 13C NMR (DMSO, 151 MHz) δ 56.1 (CH3O-7′), 82.3 (C-8′), 85.8 (C-7′), 101.6 (C-12, C-12′), 104.0 (C-10, C-14), 105.5 (C-10′, C-14′), 114.5 (C-3′, C-5′), 115.7 (C-3, C-5), 126.5 (C-8), 127.2 (C-7), 127.3 (C-2, C-6), 127.8 (C-1′), 128.9 (C-2′, C-6′), 129.2 (C-1), 138.8 (C-9), 139.8 (C-9′), 156.4 (C-4′), 157.3 (C-4), 157.5 (C-11′, C-13′), 158.2 (C-11, C-13). HR-ESI/MS analysis: m/z 485.1593 [M-H]−, (calcd for C29H25O7, 485.1600, ∆ = 1.4 ppm). MS/MS spectrum: CCMSLIB00009918911.
7,11′,13′-Tri-O-methylleachianol F (57) UV (MeCN) λmax (log ε) 228 (sh) (4.46), 285 (3.71) nm. 1H NMR (DMSO, 600 MHz) δ 2.67 (1H, t, J = 4.2, 3.4 Hz, H-8′), 2.96 (3H, s, CH3O-7), 3.15 (1H, dd, J = 9.5, 4.2 Hz, H-8), 3.57 (6H, s, CH3O-11′, CH3O-13′), 3.81 (1H, d, J = 9.5 Hz, H-7), 4.04 (1H, d, J = 3.4 Hz, H-7′), 5.71 (2H, d, J = 2.3 Hz, H-10′, H-14′), 6.17 (1H, d, J = 2.1 Hz, H-12), 6.20 (1H, t, J = 2.3 Hz, H-12′), 6.45 (1H, d, J = 2.1 Hz, H-14), 6.59 (3H, d, J = 8.6 Hz, H-3, H-5), 6.62 (2H, d, J = 8.6 Hz, H-3′, H-5′), 6.66 (2H, d, J = 8.6 Hz, H-2, H-6), 6.66 (2H, d, J = 8.6 Hz, H-2′, H-6′), 8.80 (1H, s, OH), 9.01 (1H, s, OH), 9.09 (1H, s, OH), 9.31 (1H, s, OH); 13C NMR (DMSO, 151 MHz) δ 54.2 (C-7′), 54.8 (CH3O-11′, CH3O-13′), 55.9 (CH3O-7), 57.8 (C-8′), 59.6 (C-8), 86.6 (C-7), 97.2 (C-12′), 101.5 (C-12), 104.4 (C-14), 104.5 (C-10′, C-14′), 114.7 (C-3′, C-5′), 114.7 (C-3, C-5), 120.9 (C-10), 128.1 (C-2′, C-6′), 128.7 (C-2, C-6), 130.3 (C-1), 136.0 (C-1′), 147.1 (C-9), 149.3 (C-9′), 154.0 (C-11), 155.2 (C-4′), 156.8 (C-4), 157.8 (C-13), 160.1 (C-11′, C-13′). HR-ESI/MS analysis: m/z 513.1899 [M-H]−, (calcd for C31H29O7, 513.1913, ∆ = 2.7 ppm). MS/MS spectrum: CCMSLIB00009918912.
7,11,13-Tri-O-methylleachianol F (58) UV (MeCN) λmax (log ε) 228 (sh) (4.43), 284 (3.78), 322 (3.39) nm. 1H NMR (DMSO, 600 MHz) δ 2.67 (1H, t, J = 2.8 Hz, H-8′), 2.96 (3H, s, CH3O-7), 3.21 (1H, dd, J = 8.9, 2.8 Hz, H-8), 3.58 (3H, s, CH3O-11), 3.79 (3H, s, CH3O-13), 3.83 (1H, d, J = 8.9 Hz, H-7), 4.00 (1H, d, J = 2.8 Hz, H-7′), 5.59 (2H, d, J = 2.1 Hz, H-10′, H-14′), 5.93 (1H, t, J = 2.2 Hz, H-12′), 6.45 (1H, d, J = 2.1 Hz, H-12), 6.62 (9H, m, H-2, H-2′, H-3, H-3′, H-5, H-5′, H-6, H-6′, H-14), 8.95 (2H, s, 11′OH, 13′OH), 9.13 (1H, s, OH), 9.33 (1H, s, OH); 13C NMR (DMSO, 151 MHz) δ 54.8 (C-7′), 55.1 (CH3O-11), 55.2 (CH3O-13), 56.1 (CH3O-7), 57.3 (C-8′), 59.5 (C-8), 86.2 (C-7), 97.3 (C-12), 100.2 (C-12′), 103.0 (C-14), 104.2 (C-10′, C-14′), 114.8 (C-3, C-5, C-3′, C-5′), 123.9 (C-10), 127.8 (C-2′, C-6′), 128.4 (C-2, C-6), 130.1 (C-1), 135.8 (C-1′), 147.3 (C-9), 149.4 (C-9′), 155.2 (C-4′), 156.4 (C-11), 156.7 (C-4), 158.1 (C-11′, C-13′), 160.3 (C-13). HR-ESI/MS analysis: m/z 513.1901 [M-H]−, (calcd for C31H29O7, 513.1913, ∆ = 2.3 ppm). MS/MS spectrum: CCMSLIB00009918913.
7,11′,13′-Tri-O-methylleachianol G (59) UV (MeCN) λmax (log ε) 227 (sh) (4.45), 285 (3.68) nm. 1H NMR (DMSO, 600 MHz) δ 2.86 (3H, s, CH3O-7), 3.30 (2H, m, H-8, H-8′), 3.66 (6H, s, CH3O-11′, CH3O-13′), 3.83 (1H, d, J = 7.2 Hz, H-7), 4.03 (1H, d, J = 3.3 Hz, H-7′), 5.57 (1H, d, J = 2.1 Hz, H-14), 6.07 (1H, d, J = 2.1 Hz, H-12), 6.12 (2H, d, J = 2.3 Hz, H-10′, H-14′), 6.29 (1H, t, J = 2.3 Hz, H-12′), 6.60 (2H, d, J = 8.5 Hz, H-3′, H-5′), 6.66 (2H, d, J = 8.5 Hz, H-3, H-5), 6.70 (2H, d, J = 8.5 Hz, H-2′, H-6′), 6.92 (2H, d, J = 8.5 Hz, H-2, H-6), 8.75 (1H, s, OH), 8.85 (1H, s, OH), 9.06 (1H, s, OH), 9.34 (1H, s, OH); 13C NMR (DMSO, 151 MHz) δ 54.7 (C-7′), 55.0 (CH3O-11′, CH3O-13′), 55.8 (CH3O-7), 57.7 (C-8′), 59.4 (C-8), 85.6 (C-7), 97.2 (C-12′), 101.5 (C-12), 103.8 (C-14), 105.0 (C-10′, C-14′), 114.6 (C-3′, C-5′), 114.7 (C-3, C-5), 121.4 (C-10), 128.1 (C-2′, C-6′), 129.1 (C-2, C-6), 129.8 (C-1), 136.3 (C-1′), 145.3 (C-9), 149.6 (C-9′), 154.1 (C-11), 155.1 (C-4′), 156.8 (C-4), 157.4 (C-13), 160.3 (C-11′, C-13′). HR-ESI/MS analysis: m/z 513.1897 [M-H]−, (calcd for C31H29O7, 513.1913, ∆ = 3.1 ppm). MS/MS spectrum: CCMSLIB00009918914.
Threo-7′-O-methylisoresveptero acyclic dimer (60) UV (MeCN) λmax (log ε) 228 (sh) (4.37), 285 (4.09), 308 (4.21), 325 (4.19), 342 (sh) (3.91) nm. 1H NMR (DMSO, 600 MHz) δ 3.15 (3H, s, CH3O-7′), 3.59 (6H, s, CH3O-11′, CH3O-13′), 4.43 (1H, d, J = 6.9 Hz, H-7′), 5.33 (1H, d, J = 6.9 Hz, H-8′), 6.10 (1H, t, J = 2.1 Hz, H-12), 6.23 (1H, t, J = 2.3 Hz, H-12′), 6.29 (2H, d, J = 2.3 Hz, H-10′, H-14′), 6.36 (2H, d, J = 2.1 Hz, H-10, H-14), 6.62 (2H, d, J = 8.5 Hz, H-3′, H-5′), 6.82 (1H, d, J = 16.3 Hz, H-8), 6.88 (2H, d, J = 8.8 Hz, H-3, H-5), 6.89 (1H, d, J = 16.3 Hz, H-7), 7.00 (2H, d, J = 8.5 Hz, H-2′, H-6′), 7.38 (2H, d, J = 8.8 Hz, H-2, H-6), 9.18 (2H, s, 11OH, 13OH), 9.31 (1H, s, 4′OH); 13C NMR (DMSO, 151 MHz) δ 55.0 (CH3O-11′, CH3O-13′), 56.3 (CH3O-7′), 82.3 (C-8′), 85.9 (C-7′), 99.0 (C-12′), 101.9 (C-12), 104.4 (C-10, C-14), 105.7 (C-10′, C-14′), 114.6 (C-3′, C-5′), 116.0 (C-3, C-5), 126.7 (C-8), 127.4 (C-7), 127.5 (C-2, C-6), 127.9 (C-1′), 129.1 (C-2′, C-6′), 129.7 (C-1), 139.0 (C-9), 140.4 (C-9′), 156.8 (C-4′), 157.4 (C-4), 158.5 (C-11, C-13), 159.8 (C-11′, C-13′). HR-ESI/MS analysis: m/z 513.1905 [M-H]−, (calcd for C31H29O7, 513.1913, ∆ = 1.6 ppm). MS/MS spectrum: CCMSLIB00009918915.
Erythro-7′-O-methylisoresveptero acyclic dimer (61) UV (MeCN) λmax (log ε) 228 (sh) (4.31), 285 (3.91), 308 (3.99), 324 (3.96), 342 (sh) (3.67) nm. 1H NMR (DMSO, 600 MHz) δ 3.05 (3H, s, CH3O-7′), 3.66 (6H, s, CH3O-11′, CH3O-13′), 4.37 (1H, d, J = 6.1 Hz, H-7′), 5.34 (1H, d, J = 6.1 Hz, H-8′), 6.10 (1H, t, J = 2.2 Hz, H-12), 6.34 (2H, d, J = 2.2 Hz, H-10, H-14), 6.35 (1H, t, J = 2.3 Hz, H-12′), 6.44 (2H, d, J = 2.3 Hz, H-10′, H-14′), 6.70 (2H, d, J = 8.6 Hz, H-3′, H-5′), 6.77 (2H, d, J = 8.8 Hz, H-3, H-5), 6.80 (1H, d, J = 16.2 Hz, H-8), 6.87 (1H, d, J = 16.2 Hz, H-7), 7.13 (2H, d, J = 8.6 Hz, H-2′, H-6′), 7.34 (2H, d, J = 8.8 Hz, H-2, H-6), 9.18 (2H, s, 11OH, 13OH), 9.35 (1H, s, 4′OH); 13C NMR (DMSO, 151 MHz) δ 54.9 (CH3O-11′, CH3O-13′), 55.9 (CH3O-7′), 81.2 (C-8′), 85.1 (C-7′), 98.8 (C-12′), 101.6 (C-12), 104.1 (C-10, C-14), 105.7 (C-10′, C-14′), 114.5 (C-3′, C-5′), 115.7 (C-3, C-5), 126.7 (C-8), 127.1 (C-7), 127.3 (C-2, C-6), 128.3 (C-1′), 129.1 (C-2′, C-6′), 129.6 (C-1), 138.8 (C-9), 156.8 (C-4′), 157.0 (C-4), 158.4 (C-11, C-13), 159.8 (C-11′, C-13′). HR-ESI/MS analysis: m/z 513.1905 [M-H]−, (calcd for C31H29O7, 513.1913, ∆ = 1.6 ppm). MS/MS spectrum: CCMSLIB00009918916.
Threo-7′-O-methylresveptero acyclic dimer (62) UV (MeOH) λmax (log ε) 226 (sh) (4.24), 285 (3.74), 305 (3.80), 322 (3.79) nm. 1H NMR (DMSO, 600 MHz) δ 3.13 (3H, s, CH3O-7′), 3.75 (6H, s, CH3O-11, CH3O-13), 4.35 (1H, d, J = 6.6 Hz, H-7′), 5.17 (1H, d, J = 6.6 Hz, H-8′), 5.96 (1H, t, J = 2.2 Hz, H-12′), 6.05 (2H, d, J = 2.2 Hz, H-10′, H-14′), 6.36 (1H, t, J = 2.3 Hz, H-12), 6.62 (2H, d, J = 8.5 Hz, H-3′, H-5′), 6.69 (2H, d, J = 2.3 Hz, H-10, H-14), 6.85 (2H, d, J = 8.8 Hz, H-3, H-5), 6.95 (1H, d, J = 16.4 Hz, H-8), 7.00 (2H, d, J = 8.5 Hz, H-2′, H-6′), 7.13 (1H, d, J = 16.4 Hz, H-7), 7.41 (2H, d, J = 8.8 Hz, H-2, H-6); 13C NMR (DMSO, 151 MHz) δ 55.2 (CH3O-11, CH3O-13), 56.4 (CH3O-7′), 82.4 (C-8′), 86.0 (C-7′), 99.5 (C-12), 101.8 (C-12′), 104.1 (C-10, C-14), 105.7 (C-10′, C-14′), 114.6 (C-3′, C-5′), 115.9 (C-3, C-5), 126.1 (C-8), 127.6 (C-2, C-6), 128.0 (C-1′), 128.5 (C-7), 129.1 (C-2′, C-6′), 129.4 (C-1), 139.4 (C-9), 140.1 (C-9′), 156.7 (C-4′), 157.7 (C-4, C-11′, C-13′), 160.6 (C-11, C-13). HR-ESI/MS analysis: m/z 513.1897 [M-H]−, (calcd for C31H29O7, 513.1913, ∆ = 3.1 ppm). MS/MS spectrum: CCMSLIB00009918917.
7,11,11′,13,13′-Penta-O-methylleachianol F (63) UV (MeCN) λmax (log ε) 228 (sh) (4.55), 284 (3.79) nm. 1H NMR (DMSO, 600 MHz) δ 2.74 (1H, t, J = 3.9 Hz, H-8′), 3.00 (3H, s, CH3O-7), 3.26 (1H, dd, J = 9.0, 4.1 Hz, H-8), 3.56 (3H, s, CH3O-11), 3.58 (6H, s, CH3O-11′, CH3O-13′), 3.78 (3H, s, CH3O-13), 3.90 (1H, d, J = 8.9 Hz, H-7), 4.09 (1H, d, J = 3.7 Hz, H-7′), 5.72 (2H, d, J = 2.3 Hz, H-10′, H-14′), 6.21 (1H, t, J = 2.3 Hz, H-12′), 6.46 (1H, d, J = 2.1 Hz, H-12), 6.62 (6H, m, H-2′, H-3, H-3′, H-5, H-5′, H-6′), 6.69 (1H, d, J = 2.2 Hz, H-14), 6.70 (2H, m, H-2, H-6); 13C NMR (DMSO, 151 MHz) δ 54.6 (C-7′), 54.8 (CH3O-11′, CH3O-13′), 55.1 (CH3O-11), 55.2 (CH3O-13), 56.0 (CH3O-7), 57.8 (C-8′), 59.6 (C-8), 86.3 (C-7), 97.3 (C-12′), 97.5 (C-12), 102.9 (C-14), 104.5 (C-10′, C-14′), 114.8 (C-3′, C-5′), 114.8 (C-3, C-5), 123.9 (C-10), 127.9 (C-2′, C-6′), 128.7 (C-2, C-6), 130.0 (C-1), 135.7 (C-1′), 146.9 (C-9), 148.9 (C-9′), 155.3 (C-4′), 156.3 (C-13), 156.9 (C-4), 160.1 (C-11′, C-13′), 160.4 (C-11). HR-ESI/MS analysis: m/z 541.2210 [M-H]−, (calcd for C33H33O7, 541.2226, ∆ = 3.0 ppm). MS/MS spectrum: CCMSLIB00009918918.
Threo-7′-O-methylpterostilbene acyclic dimer (64) UV (MeCN) λmax (log ε) 227 (sh) (4.51), 285 (4.28), 308 (4.44), 324 (4.43), 341 (sh) (4.13) nm. 1H NMR (DMSO, 600 MHz) δ 3.15 (3H, s, CH3O-7′), 3.59 (6H, s, CH3O-11′, CH3O-13′), 3.75 (6H, s, CH3O-11, CH3O-13), 4.44 (1H, d, J = 6.8 Hz, H-7′), 5.34 (1H, d, J = 6.8 Hz, H-8′), 6.24 (1H, t, J = 2.3 Hz, H-12′), 6.31 (2H, d, J = 2.3 Hz, H-10′, H-14′), 6.37 (1H, t, J = 2.3 Hz, H-12), 6.62 (2H, d, J = 8.5 Hz, H-3′, H-5′), 6.69 (2H, d, J = 2.3 Hz, H-10, H-14), 6.91 (2H, d, J = 8.8 Hz, H-3, H-5), 6.96 (1H, d, J = 16.3 Hz, H-8), 7.01 (2H, d, J = 8.5 Hz, H-2′, H-6′), 7.13 (1H, d, J = 16.3 Hz, H-7), 7.41 (2H, d, J = 8.8 Hz, H-2, H-6); 13C NMR (DMSO, 151 MHz) δ 54.6 (CH3O-11′, CH3O-13′), 55.0 (CH3O-11, CH3O-13), 56.0 (CH3O-7′), 82.1 (C-8′), 85.6 (C-7′), 98.6 (C-12′), 99.1 (C-12), 103.9 (C-10, C-14), 105.5 (C-10′, C-14′), 114.4 (C-3′, C-5′), 115.7 (C-3, C-5), 126.0 (C-8), 127.4 (C-2, C-6), 127.6 (C-1′), 128.3 (C-7), 128.8 (C-2′, C-6′), 129.2 (C-1), 139.1 (C-9), 156.6 (C-4′), 157.2 (C-4), 159.5 (C-11′, C-13′), 160.4 (C-11, C-13). HR-ESI/MS analysis: m/z 541.2218 [M-H]−, (calcd for C33H33O7, 541.2226, ∆ = 1.8 ppm). MS/MS spectrum: CCMSLIB00009918919.
Threo-7′-O-ethylresveratrol acyclic dimer (65) UV (MeCN) λmax (log ε) 230 (sh) (4.40), 285 (4.16), 306 (4.27), 322 (4.27), 340 (sh) (4.00) nm. 1H NMR (DMSO, 600 MHz) δ 1.04 (3H, t, J = 7.0 Hz, CH3-7′b), 3.32 (2H, overlapped, H-7′a), 4.44 (1H, d, J = 6.6 Hz, H-7′), 5.14 (1H, d, J = 6.6 Hz, H-8′), 5.96 (1H, t, J = 2.2 Hz, H-12′), 6.05 (2H, d, J = 2.2 Hz, H-10′, H-14′), 6.10 (1H, t, J = 2.2 Hz, H-12), 6.36 (2H, d, J = 2.2 Hz, H-10, H-14), 6.61 (2H, d, J = 8.4 Hz, H-3′, H-5′), 6.81 (1H, d, J = 16.3 Hz, H-8), 6.83 (2H, d, J = 8.8 Hz, H-3, H-5), 6.89 (1H, d, J = 16.3 Hz, H-7), 7.00 (2H, d, J = 8.4 Hz, H-2′, H-6′), 7.38 (2H, d, J = 8.8 Hz, H-2, H-6), 9.04 (2H, s, 11′OH, 13′OH), 9.20 (2H, s, 11OH, 13OH), 9.28 (1H, s, 4′OH); 13C NMR (DMSO, 151 MHz) δ 15.3 (CH3-7′b), 63.9 (C-7'a), 82.7 (C-8′), 84.2 (C-7′), 101.8 (C-12′), 101.9 (C-12), 104.4 (C-10, C-14), 105.7 (C-10′, C-14′), 114.6 (C-3′, C-5′), 115.9 (C-3, C-5), 126.6 (C-8), 127.4 (C-7), 127.5 (C-2, C-6), 128.7 (C-1′), 128.9 (C-2′, C-6′), 129.5 (C-1), 139.1 (C-9), 140.2 (C-9′), 156.6 (C-4′), 157.7 (C-4, C-11′, C-13′), 158.5 (C-11, C-13). HR-ESI/MS analysis: m/z 499.1788 [M-H]−, (calcd for C30H27O7, 499.1757, ∆ = 6.2 ppm). MS/MS spectrum: CCMSLIB00009918920.
7-O-Ethyl-11,13-di-O-methylleachianol F (66) UV (MeCN) λmax (log ε) 228 (sh) (4.42), 283 (3.79), 323 (3.48) nm. 1H NMR (DMSO, 600 MHz) δ 1.01 (3H, t, J = 6.9 Hz, CH3-7b), 2.67 (1H, t, J = 2.7 Hz, H-8′), 3.02 (1H, dq, J = 9.5, 7.0 Hz, H-7a″), 3.17 (2H, m, H-7a′, H-8), 3.58 (3H, s, CH3O-11), 3.78 (3H, s, CH3O-13), 3.90 (1H, d, J = 9.0 Hz, H-7), 4.01 (1H, d, J = 2.7 Hz, H-7′), 5.59 (2H, d, J = 2.2 Hz, H-10′, H-14′), 5.93 (1H, t, J = 2.2 Hz, H-12′), 6.45 (1H, d, J = 2.1 Hz, H-12), 6.60 (2H, d, J = 8.7 Hz, H-3, H-5), 6.63 (4H, m, H-2, H-3′, H-5′, H-6), 6.65 (1H, overlapped, H-14), 6.66 (2H, d, J = 8.8 Hz, H-2′, H-6′), 8.94 (2H, s, 11′OH, 13′OH), 9.14 (1H, s, 4′OH), 9.31 (1H, s, 4OH); 13C NMR (DMSO, 151 MHz) δ 15.1 (CH3-7b), 54.7 (C-7′), 55.1 (CH3O-11, CH3O-13), 57.3 (C-8′), 59.5 (C-8), 63.4 (C-7a), 84.2 (C-7), 97.5 (C-12), 100.2 (C-12′), 102.8 (C-14), 104.1 (C-10′, C-14′), 114.8 (C-3, C-5, C-3′, C-5′), 123.9 (C-10), 127.8 (C-2′, C-6′), 128.3 (C-2, C-6), 130.9 (C-1), 135.9 (C-1′), 147.4 (C-9), 149.4 (C-9′), 155.2 (C-4′), 156.3 (C-11), 156.6 (C-4), 158.1 (C-11′, C-13′), 160.2 (C-13). HR-ESI/MS analysis: m/z 527.2099 [M-H]−, (calcd for C32H31O7, 527.2070, ∆ = 5.5 ppm). MS/MS spectrum: CCMSLIB00009918921.
7-O-Ethyl-11′,13′-di-O-methylleachianol G (67) UV (MeCN) λmax (log ε) 228 (sh) (4.43), 282 (3.71) nm. 1H NMR (DMSO, 600 MHz) δ 0.89 (3H, t, J = 7.0 Hz, CH3-7b), 2.90 (1H, dq, J = 9.6, 7.0 Hz, H-7a″), 3.12 (1H, dq, J = 9.7, 7.0 Hz, H-7a′), 3.23 (1H, dd, J = 8.0, 3.7 Hz, H-8), 3.33 (1H, overlapped, H-8′), 3.66 (6H, s, CH3O-13′, CH3O-11′), 3.96 (1H, d, J = 8.0 Hz, H-7), 4.06 (1H, d, J = 3.4 Hz, H-7′), 5.52 (1H, d, J = 2.1 Hz, H-14), 6.08 (1H, d, J = 2.1 Hz, H-12), 6.11 (2H, d, J = 2.3 Hz, H-10′, H-14′), 6.29 (1H, t, J = 2.3 Hz, H-12′), 6.60 (2H, d, J = 8.5 Hz, H-3′, H-5′), 6.67 (2H, d, J = 8.5 Hz, H-3, H-5), 6.73 (2H, d, J = 8.5 Hz, H-2′, H-6′), 6.93 (2H, d, J = 8.5 Hz, H-2, H-6), 8.75 (1H, s, 11OH), 8.83 (1H, s, 13OH), 9.05 (1H, s, 4′OH), 9.31 (1H, s, 4OH); 13C NMR (DMSO, 151 MHz) δ 14.9 (CH3-7b), 54.5 (C-7′), 54.9 (CH3O-13′, CH3O-11′), 57.7 (C-8′), 59.9 (C-8), 62.8 (C-7a), 83.3 (C-7), 97.1 (C-12′), 101.5 (C-12), 103.8 (C-14), 105.0 (C-10′, C-14′), 114.6 (C-3′, C-5′), 114.7 (C-3, C-5), 121.5 (C-10), 128.2 (C-2′, C-6′), 128.9 (C-2, C-6), 130.6 (C-1), 136.5 (C-1′), 145.3 (C-9), 150.0 (C-9′), 154.0 (C-11), 155.1 (C-4′), 156.7 (C-4), 157.4 (C-13), 160.3 (C-11′, C-13′). HR-ESI/MS analysis: m/z 527.2098 [M-H]−, (calcd for C32H31O7, 527.2070, ∆ = 5.3 ppm). MS/MS spectrum: CCMSLIB00009918922.
Erythro-7′-O-ethylresveptero acyclic dimer (68) UV (MeCN) λmax (log ε) 228 (sh) (4.47), 285 (4.16), 306 (4.26), 325 (4.26), 340 (sh) (4.00) nm. 1H NMR (DMSO, 600 MHz) δ 0.96 (3H, t, J = 7.0 Hz, CH3-7′b), 3.18 (1H, m, H-7′a″), 3.27 (1H, m, H-7′a′), 3.75 (6H, s, CH3O-11, CH3O-13), 4.37 (1H, d, J = 6.1 Hz, H-7′), 5.15 (1H, d, J = 6.1 Hz, H-8′), 6.06 (1H, t, J = 2.2 Hz, H-12′), 6.18 (2H, d, J = 2.2 Hz, H-10′, H-14′), 6.36 (1H, t, J = 2.3 Hz, H-12), 6.68 (2H, d, J = 8.5 Hz, H-3′, H-5′), 6.68 (3H, d, J = 2.3 Hz, H-10, H-14), 6.74 (2H, d, J = 8.9 Hz, H-3, H-5), 6.93 (1H, d, J = 16.4 Hz, H-8), 7.11 (1H, d, J = 16.4 Hz, H-7), 7.12 (2H, d, J = 8.5 Hz, H-2′, H-6′), 7.37 (2H, d, J = 8.9 Hz, H-2, H-6), 9.11 (2H, s, 11′OH, 13′OH), 9.30 (1H, s, 4′OH); 13C NMR (DMSO, 151 MHz) δ 15.1 (CH3-7'b), 55.2 (CH3O-11, CH3O-13), 63.7 (C-7′a), 81.6 (C-8′), 83.7 (C7′), 99.5 (C-12), 101.8 (C-12′), 104.1 (C-10, C-14), 105.6 (C-10′, C-14′), 114.5 (C-3′, C-5′), 115.9 (C-3, C-5), 126.2 (C-8), 127.5 (C-2, C-6), 128.4 (C-7), 129.1 (C-2′, C-6′), 129.2 (C-1′), 129.5 (C-1), 139.4 (C-9), 140.9 (C-9′), 156.7 (C-4′), 157.3 (C-4), 157.8 (C-11′, C-13′), 160.6 (C-11, C-13). HR-ESI/MS analysis: m/z 527.2103 [M-H]−, (calcd for C32H31O7, 527.2070, ∆ = 6.3 ppm). MS/MS spectrum: CCMSLIB00009918923.
Threo-7′-O-ethylpterostilbene acyclic dimer (69) UV (MeCN) λmax (log ε) 228 (sh) (4.51), 285 (4.29), 306 (4.43), 325 (4.42), 340 (sh) (4.14) nm. 1H NMR (DMSO, 600 MHz) δ 1.05 (3H, t, J = 7.0 Hz, CH3-7′b), 3.35 (2H, t, J = 7.0 Hz, H-7′a), 3.60 (6H, s, CH3O-11′, CH3O-13′), 3.75 (6H, s, CH3O-11, CH3O-13), 4.52 (1H, d, J = 6.7 Hz, H-7′), 5.32 (1H, d, J = 6.7 Hz, H-8′), 6.24 (1H, t, J = 2.3 Hz, H-12′), 6.33 (2H, d, J = 2.3 Hz, H-10′, H-14′), 6.37 (1H, t, J = 2.3 Hz, H-12), 6.61 (2H, d, J = 8.6 Hz, H-3′, H-5′), 6.70 (2H, d, J = 2.3 Hz, H-10, H-14), 6.90 (2H, d, J = 8.8 Hz, H-3, H-5), 6.96 (1H, d, J = 16.3 Hz, H-8), 7.01 (2H, d, J = 8.6 Hz, H-2′, H-6′), 7.13 (1H, d, J = 16.3 Hz, H-7), 7.41 (2H, d, J = 8.8 Hz, H-2, H-6), 9.29 (1H, s, 4′OH); 13C NMR (DMSO, 151 MHz) δ 15.3 (CH3-7′b), 55.0 (CH3O-11′, CH3O-13′), 55.2 (CH3O-11, CH3O-13), 63.9 (C-7′a), 82.5 (C-8′), 84.0 (C-7′), 99.0 (C-12′), 99.5 (C-12), 104.1 (C-10, C-14), 105.7 (C-10′, C-14′), 114.5 (C-3′, C-5′), 116.1 (C-3, C-5), 126.2 (C-8), 127.6 (C-2, C-6), 128.5 (C-7), 128.7 (C-1′), 128.9 (C-2′, C-6′), 129.6 (C-1), 139.4 (C-9), 140.6 (C-9′), 156.7 (C-4′), 157.7 (C-4), 159.8 (C-11′, C-13′), 160.6 (C-11, C-13). HR-ESI/MS analysis: m/z 555.2426 [M-H]−, (calcd for C34H35O7, 555.2383, ∆ = 7.7 ppm). MS/MS spectrum: CCMSLIB00009918924.
11′,13′-Di-O-methylrestrytisol B (70) UV (MeCN) λmax (log ε) 228 (sh) (4.30), 282 (3.65) nm. 1H NMR (DMSO, 600 MHz) δ 3.38 (1H, overlapped, H-8), 3.54 (6H, s, CH3O-11′, CH3O-13′), 3.98 (1H, t, J = 9.0 Hz, H-8′), 4.90 (1H, d, J = 9.7 Hz, H-7), 5.41 (1H, d, J = 9.0 Hz, H-7′), 5.98 (1H, t, J = 2.2 Hz, H-12), 6.05 (1H, t, J = 2.3 Hz, H-12′), 6.07 (1H, d, J = 2.3 Hz, H-10′, H-14′), 6.08 (2H, d, J = 2.2 Hz, H-10, H-14), 6.51 (2H, d, J = 8.5 Hz, H-3′, H-5′), 6.73 (1H, d, J = 8.5 Hz, H-3, H-5), 6.95 (2H, d, J = 8.5 Hz, H-2′, H-6′), 7.22 (1H, d, J = 8.5 Hz, H-2, H-6), 9.07 (2H, s, 11OH, 13OH), 9.12 (1H, s, 4′OH), 9.40 (1H, s, 4OH); 13C NMR (DMSO, 151 MHz) δ 54.7 (CH3O-11′, CH3O-13′), 57.1 (C-8), 58.3 (C-8′), 82.5 (C-7′), 85.7 (C-7), 97.7 (C-12′), 100.9 (C-12), 106.1 (C-10, C-14), 107.1 (C-10′, C-14′), 114.1 (C-3′, C-5′), 114.9 (C-3, C-5), 127.8 (C-2′, C-6′), 128.0 (C-2, C-6), 129.9 (C-1), 130.9 (C-1′), 141.0 (C-9′), 141.7 (C-9), 155.7 (C-4′), 156.9 (C-4), 158.1 (C-11, C-13), 159.3 (C-11′, C-13′). HR-ESI/MS analysis: m/z 499.1789 [M-H]−, (calcd for C30H27O7, 499.1757, ∆ = 6.4 ppm). MS/MS spectrum: CCMSLIB00009918925.
7-O-Ethylleachianol G (71) UV (MeCN) λmax (log ε) 228 (sh) (4.45), 282 (3.74) nm. 1H NMR (DMSO, 600 MHz) δ 0.86 (3H, t, J = 7.0 Hz, CH3-7b), 2.84 (1H, dq, J = 9.8, 6.9 Hz, H-7a″), 3.09 (1H, m, H-7a′), 3.15 (1H, dd, J = 8.5, 3.2 Hz, H-8), 3.25 (1H, t, J = 3.2 Hz, H-8′), 3.84 (1H, d, J = 8.5 Hz, H-7), 4.03 (1H, d, J = 3.2 Hz, H-7′), 5.38 (1H, d, J = 2.1 Hz, H-14), 5.92 (2H, d, J = 2.2 Hz, H-10′, H-14′), 5.98 (1H, t, J = 2.2 Hz, H-12′), 6.06 (1H, d, J = 2.1 Hz, H-12), 6.61 (2H, d, J = 8.3 Hz, H-3′, H-5′), 6.67 (2H, d, J = 8.2 Hz, H-3, H-5), 6.71 (2H, d, J = 8.1 Hz, H-2′, H-6′), 6.90 (2H, d, J = 8.1 Hz, H-2, H-6); 13C NMR (DMSO, 151 MHz) δ 14.6 (CH3-7b), 54.1 (C-7′), 57.5 (C-8′), 59.7 (C-8), 62.4 (C-7a), 83.4 (C-7), 99.8 (C-12′), 101.2 (C-12), 103.9 (C-14), 104.4 (C-10′, C-14′), 114.4 (C-3, C-5, C-3′, C-5′), 121.3 (C-10), 127.7 (C-2′, C-6′), 128.8 (C-2, C-6), 130.3 (C-1), 136.6 (C-1′), 145.1 (C-9), 149.8 (C-9′), 153.7 (C-11), 154.8 (C-4′), 156.4 (C-4), 156.8 (C-13), 157.9 (C-11′, C-13′). HR-ESI/MS analysis: m/z 499.1788 [M-H]−, (calcd for C30H27O7, 499.1757, ∆ = 6.3 ppm). MS/MS spectrum: CCMSLIB00009918926.
7-O-Ethyl-11′,13′-di-O-methylleachianol F (72) UV (MeCN) λmax (log ε) 228 (sh) (4.40), 282 (3.70) nm. 1H NMR (DMSO, 600 MHz) δ 1.02 (3H, t, J = 7.0 Hz, CH3-7b), 2.68 (1H, t, J = 4.0, 3.4 Hz, H-8′), 3.07 (1H, dq, J = 9.5, 7.0 Hz, H-7a″), 3.13 (1H, dd, J = 9.1, 4.0 Hz, H-8), 3.16 (1H, m, H-7a′), 3.57 (6H, s, CH3O-11′, CH3O-13′), 3.92 (1H, d, J = 9.1 Hz, H-7), 4.05 (1H, d, J = 3.4 Hz, H-7′), 5.72 (2H, d, J = 2.2 Hz, H-10′, H-14′), 6.16 (1H, d, J = 2.2 Hz, H-12), 6.20 (1H, t, J = 2.3 Hz, H-12′), 6.48 (1H, d, J = 2.1 Hz, H-14), 6.57 (2H, d, J = 8.5 Hz, H-3, H-5), 6.62 (2H, d, J = 8.7 Hz, H-3′, H-5′), 6.66 (2H, d, J = 8.5 Hz, H-2, H-6), 6.67 (2H, d, J = 8.7 Hz, H-2′ H-6′); 13C NMR (DMSO, 151 MHz) δ 15.1 (CH3-7b), 54.2 (C-7′), 54.8 (CH3O-11′, CH3O-13′), 57.8 (C-8′), 59.5 (C-8), 63.2 (C-7a), 84.5 (C-7), 97.2 (C-12′), 101.5 (C-12), 104.5 (C-14), 104.5 (C-10′, C-14′), 114.6 (C-3, C-3′, C-5, C-5′), 120.9 (C-10), 128.1 (C-2′, C-6′), 128.5 (C-2, C-6), 131.0 (C-1), 136.1 (C-1′), 147.2 (C-9), 149.3 (C-9′), 153.9 (C-11), 155.2 (C-4′), 156.7 (C-4), 157.8 (C-13), 160.1 (C-11′, C-13′). HR-ESI/MS analysis: m/z 527.2100 [M-H]−, (calcd for C32H31O7, 527.2070, ∆ = 5.7 ppm). MS/MS spectrum: CCMSLIB00009918927.
Erythro-7′-O-ethylresveratrol acyclic dimer (73) UV (MeCN) λmax (log ε) 226 (sh) (4.15), 285 (3.88), 306 (3.94), 325 (3.92) nm. 1H NMR (DMSO, 600 MHz) δ 0.96 (3H, t, J = 7.0 Hz, CH3-7'b), 3.18 (1H, m, H-7′a″), 3.27 (1H, m, H-7′a′), 4.37 (1H, d, J = 6.2 Hz, H-7′), 5.14 (1H, d, J = 6.2 Hz, H-8′), 6.06 (1H, t, J = 2.2 Hz, H-12′), 6.10 (1H, t, J = 2.1 Hz, H-12), 6.18 (2H, d, J = 2.2 Hz, H-10′, H-14′), 6.34 (2H, d, J = 2.1 Hz, H-10, H-14), 6.68 (2H, d, J = 8.6 Hz, H-3′, H-5′), 6.72 (2H, d, J = 8.8 Hz, H-3, H-5), 6.79 (1H, d, J = 16.3 Hz, H-8), 6.87 (1H, d, J = 16.3 Hz, H-7), 7.12 (2H, d, J = 8.6 Hz, H-2′, H-6′), 7.34 (2H, d, J = 8.8 Hz, H-2, H-6); 13C NMR (DMSO, 151 MHz) δ 14.9 (CH3-7′b), 63.4 (C-7′a), 81.4 (C-8′), 83.4 (C7′), 101.6 (C-12, C-12′), 104.1 (C-10, C-14), 105.3 (C-10′, C-14′), 114.3 (C-3′, C-5′), 115.7 (C-3, C-5), 126.4 (C-8), 127.1 (C-7), 127.3 (C-2, C-6), 128.8 (C-2′, C-6′), 129.2 (C-1), 156.4 (C-4′), 156.9 (C-4), 157.5 (C-11′, C-13′), 158.2 (C-11, C-13). HR-ESI/MS analysis: m/z 499.1791[M-H]−, (calcd for C30H27O7, 499.1757, ∆ = 6.8 ppm). MS/MS spectrum: CCMSLIB00009918928.
Wnt Activity Measurements
The Wnt-induced luciferase activity was analyzed as described. (Koval et al., 2014; Shaw et al., 2019) Briefly, 30 μl of BT-20 cells stably transfected with the TopFlash luciferase reporter (Koval et al., 2014) at 150,000 cells/ml were distributed in a white opaque 384-well plate. The cells were maintained in DMEM containing 10% FBS and incubated at 37°C, 5% CO2 overnight for attachment. Afterwards, the BT-20 cells were additionally transfected with the plasmid constitutively (under the CMV promoter) expressing Renilla luciferase (Addgene, Cambridge, MA, United States). Transfection was carried out as described in manufacturer’s protocol using 12 µg/ml of DNA and 40 µl/ml XtremeGENE 9 reagent (Roche). Next day, the medium of each well was replaced by 30 μL fresh medium containing Wnt3a (500 ng/ml) [purified as described (Koval et al., 2011] or CHIR99021 (1 µM) and the compounds at 6-8 different concentrations (following 1h of pre-incubation with the compound). After overnight incubation, the supernatant in each well was removed and the cells were harvested and analyzed for activities of both firefly and Renilla luciferase as described (Koval et al., 2014).
MTT Assay
50 μl of indicated TNBC cell lines were added into each well of a transparent 384-well plate at density 1,500 cells/well. The cells were maintained in DMEM containing 10% FBS and incubated at 37°C, 5% CO2 overnight. The next day, the medium of each well was replaced by 50 μl fresh medium containing indicated concentrations of compounds. After incubation for 3–4 days, the medium in each well was replaced by 50 μl of 0.5 mg/ml Thiazolyl blue (Carl Roth) solution in 1xPBS. The plates were incubated for 3 h at 37°C. Then the solution was removed, and 25 μl DMSO was added into each well. Absorbance at 570 nm was measured in the Tecan Infinite 200 Pro reader.
Western Blotting
TNBC cell lines and L-cells were seeded at 100,000 cells/well in 24 well plates. The next day, the medium was replaced with fresh medium pre-warmed at 37°C containing the indicated compounds. After further 24h incubation, the medium was removed, followed by washing with 500 μl of 1x PBS twice per well. The cells were lysed in the well by addition of 30 μl of ice-cold RIPA buffer (1x TBS, 4 mM EDTA, 1% Triton, 0.1% SDS, 1x Protease inhibitor cocktail (Roche)) and incubated on ice for 10 min. The samples were resuspended and then centrifuged at 18,000 g at 4°C to remove debris. 15 μl of the supernatants each were further analyzed by Western blot with antibodies against active β-catenin (Millipore), Axin2 (Abcam), c-Myc (Abcam) and α-Tubulin (Sigma) at 1:1,000 dilutions.
Immunofluorescent Analysis
The indicated cell lines were seeded at 200,000 cells/well in the 6-well plates with poly-L-lysine-coated coverslips on the bottom. After 5 h of treatment with or without Wnt3a in presence or absence of 8, the cells were washed 2x by PBS, fixed by 4%PFA at room temperature and stained and visualized as described above for tumor samples. The antibodies used for staining were against β-catenin (BD Biosciences) and pS33/37/T41-β-catenin (CST) at 1:200 and 1:100 dilution respectively.
Similarity Analysis
The similarity matrix was calculated with all 73 compounds with the “Similarity and clustering” utility of Canvas (Schrödinger, LLC, New York, NY, release 2021-1) (Duan et al., 2010). A dendritic fingerprint of 64-bit was calculated with the daylight invariant atom types and bonds distinguished by bond order. The Tanimoto metric was chosen with the average linkage method to compute the matrix.
Pharmacophore Model Hypothesis Development
The model was built with Phase and Maestro (Schrödinger, LLC, New York, NY, release 2021-1). The compounds were prepared with the LigPrep utility at pH 7.0 and the OPLS4 force field (Lu et al., 2021), then split into 3 groups: the “specific-activity” group (SI > 10, IC50 <35 μM), the “low specificity-active” group (SI < 10, IC50 < 35 μM) and the “low specificity-low activity” group (SI < 10, IC50 > 35 μM). The enantiomer for each compound was also generated. The pharmacophore method was to find the best alignment and common features, with a number of features in the hypothesis between 5 and 7, and a match of 100% of the input compounds. For the non-specific actives, matching 100% of the input compounds failed to produce any model, the matching percentage was then reduced to 75%. The output conformers were minimized. All other settings were the default ones. The model with the highest Phase Hypo Score was used for subsequent hypothesis validation with “Phase Ligand Screening” tab. The fitness score was used to assess the quality of the modes. Model images were rendered with PyMOL Molecular Graphics System (Schrödinger, LLC, New York, NY).
Data Availability Statement
The raw data files for the UHPLC-PDA-ELSD-MS analysis of the biotransformation reactions and NMR and HRMS data of the isolated compounds are available at the following link: https://doi.org/10.26037/yareta:zla4rtnowzfcdo6fqhqfec47jq. The MS/MS spectrum of each isolated compound has its own accession number from CCMSLIB00009918856 to CCMSLIB00009918928 on the Global Natural Product Social Molecular Networking (GNPS) (accessed via: https://gnps.ucsd.edu/ProteoSAFe/static/gnps-splash.jsp).
Author Contributions
Conceived and designed the experiments: EFQ, KG, MH, RH, VLK, LM and J-LW. Performed the experiments: RH, AK, EM, LM and MH. Analyzed the data: RH, MH, LS, AK, EFQ, KG and LM. Contributed reagents/materials/analysis tools: VLK, KG, J-LW and EFQ. Contributed to the writing of the manuscript: RH, AK, VLK, SS, J-LW, KG, LM, LS and EFQ.
Funding
Swiss National Science Foundation for providing financial support for this project, which aims to improve natural products chemical biodiversity for drug discovery by fungal secretome assisted biotransformation (grant 205321_182438/1 EFQ and KG) MH is supported by the SNF grant no: 186405.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The School of Pharmaceutical Sciences of the University of Geneva (J-LW) is thankful to the Swiss National Science Foundation for the support in the acquisition of the NMR 600 MHz (SNF R’Equip grant 316030_164095).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2022.881298/full#supplementary-material. NMR spectra of compounds 1 to 25 can be found in data sheet 2, those of compounds 26 to 47 in data sheet 3 and those of compounds 48 to 73 in data sheet 4.
References
Abdullah, L. N., and Chow, E. K. H. (2013). Mechanisms of Chemoresistance in Cancer Stem Cells. Clin. Translational Med. 2. doi:10.1186/2001-1326-2-3
Adesanya, S. A., Nia, R., Martin, M.-T., Boukamcha, N., Montagnac, A., and Païs, M. (1999). Stilbene Derivatives from Cissus quadrangularis. J. Nat. Prod. 62, 1694–1695. doi:10.1021/np9902744
Ahmed, K., Koval, A., Xu, J., Bodmer, A., and Katanaev, V. L. (2019). Towards the First Targeted Therapy for Triple-Negative Breast Cancer: Repositioning of Clofazimine as a Chemotherapy-Compatible Selective Wnt Pathway Inhibitor. Cancer Lett. 449, 45–55. doi:10.1016/j.canlet.2019.02.018
An, W. F., Germain, A. R., Bishop, J. A., Nag, P. P., Metkar, S., Ketterman, J., et al. (2010). “Discovery of Potent and Highly Selective Inhibitors of GSK3b,” in Probe Reports from the NIH Molecular Libraries Program (Bethesda (MD: National Center for Biotechnology Information).
Aquilano, K., Baldelli, S., Rotilio, G., and Ciriolo, M. R. (2009). trans-Resveratrol Inhibits H2O2-Induced Adenocarcinoma Gastric Cells Proliferation via Inactivation of MEK1/2-Erk1/2-C-Jun Signalling axis. Biochem. Pharmacol. 77, 337–347. doi:10.1016/j.bcp.2008.10.034
Ashrafizadeh, M., Ahmadi, Z., Farkhondeh, T., and Samarghandian, S. (2020). Resveratrol Targeting the Wnt Signaling Pathway: A Focus on Therapeutic Activities. J. Cel. Physiol. 235, 4135–4145. doi:10.1002/jcp.29327
Baell, J., and Walters, M. A. (2014). Chemistry: Chemical con artists foil drug discovery. Nature 513, 481–483. doi:10.1038/513481a
Barker, N., and Clevers, H. (2006). Mining the Wnt Pathway for Cancer Therapeutics. Nat. Rev. Drug Discov. 5, 997–1014. doi:10.1038/nrd2154
Bianchini, G., De Angelis, C., Licata, L., and Gianni, L. (2021). Treatment Landscape of Triple-Negative Breast Cancer - Expanded Options, Evolving Needs. Nat. Rev. Clin. Oncol. 19, 91–113. doi:10.1038/s41571-021-00565-2
Blagodatski, A., Klimenko, A., Jia, L., and Katanaev, V. L. (2020). Small Molecule Wnt Pathway Modulators from Natural Sources: History, State of the Art and Perspectives. Cells 9, 589. doi:10.3390/cells9030589
Blagodatski, A., Poteryaev, D., and Katanaev, V. L. (2014). Targeting the Wnt Pathways for Therapies. Mol. Cell Therapies 2, 28. doi:10.1186/2052-8426-2-28
Dey, N., Barwick, B. G., Moreno, C. S., Ordanic-Kodani, M., Chen, Z., Oprea-Ilies, G., et al. (2013). Wnt Signaling in Triple Negative Breast Cancer Is Associated with Metastasis. BMC Cancer 13, 537. doi:10.1186/1471-2407-13-537
Duan, J., Dixon, S. L., Lowrie, J. F., and Sherman, W. (2010). Analysis and Comparison of 2D Fingerprints: Insights into Database Screening Performance Using Eight Fingerprint Methods. J. Mol. Graphics Model. 29, 157–170. doi:10.1016/j.jmgm.2010.05.008
Gindro, K., Schnee, S., Righi, D., Marcourt, L., Nejad Ebrahimi, S., Codina, J. M., et al. (2017). Generation of Antifungal Stilbenes Using the Enzymatic Secretome of Botrytis cinerea. J. Nat. Prod. 80, 887–898. doi:10.1021/acs.jnatprod.6b00760
Giordano, S. H., Buzdar, A. U., Smith, T. L., Kau, S.-W., Yang, Y., and Hortobagyi, G. N. (2004). Is Breast Cancer Survival Improving? Cancer 100, 44–52. doi:10.1002/cncr.11859
Guillarme, D., Nguyen, D. T.-T., Rudaz, S., and Veuthey, J.-L. (2007). Method Transfer for Fast Liquid Chromatography in Pharmaceutical Analysis: Application to Short Columns Packed with Small Particle. Part I: Isocratic Separation. Eur. J. Pharmaceutics Biopharmaceutics 66, 475–482. doi:10.1016/j.ejpb.2006.11.027
Guillarme, D., Nguyen, D. T. T., Rudaz, S., and Veuthey, J.-L. (2008). Method Transfer for Fast Liquid Chromatography in Pharmaceutical Analysis: Application to Short Columns Packed with Small Particle. Part II: Gradient Experiments. Eur. J. Pharmaceutics Biopharmaceutics 68, 430–440. doi:10.1016/j.ejpb.2007.06.018
Gurney, A., Axelrod, F., Bond, C. J., Cain, J., Chartier, C., Donigan, L., et al. (2012). Wnt Pathway Inhibition via the Targeting of Frizzled Receptors Results in Decreased Growth and Tumorigenicity of Human Tumors. Proc. Natl. Acad. Sci. U.S.A. 109, 11717–11722. doi:10.1073/pnas.1120068109
Herrera Braga, A. C., Zacchino, S., Badano, H., Sierra, M. G., and Rúveda, E. A. (1984). 13C NMR Spectral and Conformational Analysis of 8-O-4′ Neolignans. Phytochemistry 23, 2025–2028. doi:10.1016/s0031-9422(00)84963-8
Hope, C., Planutis, K., Planutiene, M., Moyer, M. P., Johal, K. S., Woo, J., et al. (2008). Low Concentrations of Resveratrol Inhibit Wnt Signal Throughput in colon-derived Cells: Implications for colon Cancer Prevention. Mol. Nutr. Food Res. 52 (Suppl. 1), S52–S61. doi:10.1002/mnfr.200700448
Huber, R., Marcourt, L., Koval, A., Schnee, S., Righi, D., Michellod, E., et al. (2021). Chemoenzymatic Synthesis of Complex Phenylpropanoid Derivatives by the Botrytis cinerea Secretome and Evaluation of Their Wnt Inhibition Activity. Front. Plant Sci. 12, 805610. doi:10.3389/fpls.2021.805610
Jang, G.-B., Kim, J.-Y., Cho, S.-D., Park, K.-S., Jung, J.-Y., Lee, H.-Y., et al. (2015). Blockade of Wnt/β-Catenin Signaling Suppresses Breast Cancer Metastasis by Inhibiting CSC-like Phenotype. Sci. Rep. 5, 12465. doi:10.1038/Srep12465
Kassam, F., Enright, K., Dent, R., Dranitsaris, G., Myers, J., Flynn, C., et al. (2009). Survival Outcomes for Patients with Metastatic Triple-Negative Breast Cancer: Implications for Clinical Practice and Trial Design. Clin. Breast Cancer 9, 29–33. doi:10.3816/CBC.2009.n.005
Katanaev, V. L., Blagodatski, A., Xu, J., Khotimchenko, Y., and Koval, A. (2021). Mining Natural Compounds to Target WNT Signaling: Land and Sea Tales. Handb Exp. Pharmacol. 269, 215–248. doi:10.1007/164_2021_530
Keylor, M. H., Matsuura, B. S., and Stephenson, C. R. J. (2015). Chemistry and Biology of Resveratrol-Derived Natural Products. Chem. Rev. 115, 8976–9027. doi:10.1021/cr500689b
Khan, S. H., Alhumaydhi, F. A., Khan, M. A., and Younus, H. (2021). Therapeutic Potential of Polyphenols and Their Nanoformulations in the Treatment of Colorectal Cancer. Acamc 21, 2117–2129. doi:10.2174/1871520621666201231144007
Khramtsov, A. I., Khramtsova, G. F., Tretiakova, M., Huo, D., Olopade, O. I., and Goss, K. H. (2010). Wnt/β-Catenin Pathway Activation Is Enriched in Basal-like Breast Cancers and Predicts Poor Outcome. Am. J. Pathol. 176, 2911–2920. doi:10.2353/ajpath.2010.091125
Kim, H. J., Saleem, M., Seo, S. H., Jin, C., and Lee, Y. S. (2005). Two New Antioxidant Stilbene Dimers, Parthenostilbenins A and B from Parthenocissus Tricuspidata. Planta Med. 71, 973–976. doi:10.1055/s-2005-871229
Klibanov, A. M. (2001). Improving Enzymes by Using Them in Organic Solvents. Nature 409, 241–246. doi:10.1038/35051719
Koval, A., Bassanini, I., Xu, J., Tonelli, M., Boido, V., Sparatore, F., et al. (2021). Optimization of the Clofazimine Structure Leads to a Highly Water-Soluble C3-Aminopyridinyl Riminophenazine Endowed with Improved Anti-Wnt and Anti-cancer Activity In Vitro and In Vivo. Eur. J. Med. Chem. 222, 113562. doi:10.1016/j.ejmech.2021.113562
Koval, A., and Katanaev, V. L. (2018). Dramatic Dysbalancing of the Wnt Pathway in Breast Cancers. Sci. Rep. 8, 7329. doi:10.1038/s41598-018-25672-6
Koval, A., Pieme, C. A., Queiroz, E. F., Ragusa, S., Ahmed, K., Blagodatski, A., et al. (2018). Tannins from Syzygium guineense Suppress Wnt Signaling and Proliferation of Wnt-dependent Tumors through a Direct Effect on Secreted Wnts. Cancer Lett. 435, 110–120. doi:10.1016/j.canlet.2018.08.003
Koval, A. V., Vlasov, P., Shichkova, P., Khunderyakova, S., Markov, Y., Panchenko, J., et al. (2014). Anti-leprosy Drug Clofazimine Inhibits Growth of Triple-Negative Breast Cancer Cells via Inhibition of Canonical Wnt Signaling. Biochem. Pharmacol. 87, 571–578. doi:10.1016/j.bcp.2013.12.007
Koval, A., Purvanov, V., Egger-Adam, D., and Katanaev, V. L. (2011). Yellow Submarine of the Wnt/Frizzled Signaling: Submerging from the G Protein Harbor to the Targets. Biochem. Pharmacol. 82 (10), 1311–1319. doi:10.1016/j.bcp.2011.06.005
Langcake, P., and Pryce, R. J. (1977). A New Class of Phytoalexins from Grapevines. Experientia 33, 151–152. doi:10.1007/BF02124034
Larasati, Y., Boudou, C., Koval, A., and Katanaev, V. L. (2022). Unlocking the Wnt Pathway: Therapeutic Potential of Selective Targeting FZD7 in Cancer. Drug Discov. Today 27, 777–792. doi:10.1016/j.drudis.2021.12.008
Le, P. N., Mcdermott, J. D., and Jimeno, A. (2015). Targeting the Wnt Pathway in Human Cancers: Therapeutic Targeting with a Focus on OMP-54F28. Pharmacol. Ther. 146, 1–11. doi:10.1016/j.pharmthera.2014.08.005
Lopes, G. R., Pinto, D. C. G. A., and Silva, A. M. S. (2014). Horseradish Peroxidase (HRP) as a Tool in green Chemistry. RSC Adv. 4, 37244–37265. doi:10.1039/c4ra06094f
Lu, C., Wu, C., Ghoreishi, D., Chen, W., Wang, L., Damm, W., et al. (2021). OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theor. Comput. 17, 4291–4300. doi:10.1021/acs.jctc.1c00302
Mineda, A., Nishimura, M., Kagawa, T., Takiguchi, E., Kawakita, T., Abe, A., et al. (2019). Resveratrol Suppresses Proliferation and Induces Apoptosis of Uterine Sarcoma Cells by Inhibiting the Wnt Signaling Pathway. Exp. Ther. Med. 17, 2242–2246. doi:10.3892/etm.2019.7209
Mukohara, T., Hosono, A., Mimaki, S., Nakayama, A., Kusuhara, S., Funasaka, C., et al. (2021). Effects of Ado-Trastuzumab Emtansine and Fam-Trastuzumab Deruxtecan on Metastatic Breast Cancer Harboring HER2 Amplification and the L755S Mutation. The Oncologist 26, 635–639. doi:10.1002/onco.13715
Newman, D. J., and Cragg, G. M. (2020). Natural Products as Sources of New Drugs Over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 83, 770–803. doi:10.1021/acs.jnatprod.9b01285
O'toole, S. A., Beith, J. M., Millar, E. K. A., West, R., Mclean, A., Cazet, A., et al. (2013). Therapeutic Targets in Triple Negative Breast Cancer. J. Clin. Pathol. 66, 530–542. doi:10.1136/jclinpath-2012-201361
Obrador, E., Salvador-Palmer, R., Jihad-Jebbar, A., López-Blanch, R., Dellinger, T. H., Dellinger, R. W., et al. (2021). Pterostilbene in Cancer Therapy. Antioxidants 10, 492. doi:10.3390/antiox10030492
Ohyama, M., Tanaka, T., and Iinuma, M. (1995). Five Resveratrol Oligomers from Roots of Sophora leachiana. Phytochemistry 38, 733–740. doi:10.1016/0031-9422(94)00722-6
O’Reilly, E. A., Gubbins, L., Sharma, S., Tully, R., Guang, M. H. Z., Weiner-Gorzel, K., et al. (2015). The Fate of Chemoresistance in Triple Negative Breast Cancer (TNBC). BBA Clin. 3, 257–275. doi:10.1016/j.bbacli.2015.03.003
Ponzoni, C., Beneventi, E., Cramarossa, M. R., Raimondi, S., Trevisi, G., Pagnoni, U. M., et al. (2007). Laccase-catalyzed Dimerization of Hydroxystilbenes. Adv. Synth. Catal. 349, 1497–1506. doi:10.1002/adsc.200700043
Quarta, A., Gaballo, A., Pradhan, B., Patra, S., Jena, M., and Ragusa, A. (2021). Beneficial Oxidative Stress-Related Trans-resveratrol Effects in the Treatment and Prevention of Breast Cancer. Appl. Sci. 11, 11041. doi:10.3390/app112211041
Queiroz, E. F., Alfattani, A., Afzan, A., Marcourt, L., Guillarme, D., and Wolfender, J.-L. (2019). Utility of Dry Load Injection for an Efficient Natural Products Isolation at the Semi-preparative Chromatographic Scale. J. Chromatogr. A 1598, 85–91. doi:10.1016/j.chroma.2019.03.042
Ren, B., Kwah, M. X.-Y., Liu, C., Ma, Z., Shanmugam, M. K., Ding, L., et al. (2021). Resveratrol for Cancer Therapy: Challenges and Future Perspectives. Cancer Lett. 515, 63–72. doi:10.1016/j.canlet.2021.05.001
Righi, D., Huber, R., Koval, A., Marcourt, L., Schnee, S., Le Floch, A., et al. (2020). Generation of Stilbene Antimicrobials against Multiresistant Strains of Staphylococcus aureus through Biotransformation by the Enzymatic Secretome of Botrytis cinerea. J. Nat. Prod. 83, 2347–2356. doi:10.1021/acs.jnatprod.0c00071
Rutz, A., Dounoue-Kubo, M., Ollivier, S., Bisson, J., Bagheri, M., Saesong, T., et al. (2019). Taxonomically Informed Scoring Enhances Confidence in Natural Products Annotation. Front. Plant Sci. 10, 1329. doi:10.3389/fpls.2019.01329
Schlam, I., and Swain, S. M. (2021). HER2-positive Breast Cancer and Tyrosine Kinase Inhibitors: the Time Is Now. npj Breast Cancer 7. doi:10.1038/s41523-021-00265-1
Seligson, J. M., Patron, A. M., Berger, M. J., Harvey, R. D., and Seligson, N. D. (2020). Sacituzumab Govitecan-Hziy: An Antibody-Drug Conjugate for the Treatment of Refractory, Metastatic, Triple-Negative Breast Cancer. Ann. Pharmacother. 55, 921–931. doi:10.1177/1060028020966548
Shaw, H. V., Koval, A., and Katanaev, V. L. (2019). A High-Throughput Assay Pipeline for Specific Targeting of Frizzled GPCRs in Cancer. Methods Cel Biol 149, 57–75. doi:10.1016/bs.mcb.2018.08.006
Shen, T., Wang, X.-N., and Lou, H.-X. (2009). Natural Stilbenes: an Overview. Nat. Prod. Rep. 26, 916–935. doi:10.1039/b905960a
Shingai, Y., Fujimoto, A., Nakamura, M., and Masuda, T. (2011). Structure and Function of the Oxidation Products of Polyphenols and Identification of Potent Lipoxygenase Inhibitors from Fe-Catalyzed Oxidation of Resveratrol. J. Agric. Food Chem. 59, 8180–8186. doi:10.1021/jf202561p
Simmons, G. E., Pandey, S., Nedeljkovic-Kurepa, A., Saxena, M., Wang, A., and Pruitt, K. (2014). Frizzled 7 Expression Is Positively Regulated by SIRT1 and β-Catenin in Breast Cancer Cells. Plos One 9, e98861. doi:10.1371/journal.pone.0098861
Turashvili, G., Bouchal, J., Burkadze, G., and Kolar, Z. (2006). Wnt Signaling Pathway in Mammary Gland Development and Carcinogenesis. Pathobiology 73, 213–223. doi:10.1159/000098207
Veeman, M. T., Slusarski, D. C., Kaykas, A., Louie, S. H., and Moon, R. T. (2003). Zebrafish Prickle, a Modulator of Noncanonical Wnt/Fz Signaling, Regulates Gastrulation Movements. Curr. Biol. 13, 680–685. doi:10.1016/s0960-9822(03)00240-9
Wang, X., Jung, Y.-S., Jun, S., Lee, S., Wang, W., Schneider, A., et al. (2016). PAF-wnt Signaling-Induced Cell Plasticity Is Required for Maintenance of Breast Cancer Cell Stemness. Nat. Commun. 7, 10633. doi:10.1038/Ncomms10633
Wang, Y., Ding, L., Wang, X., Zhang, J., Han, W., Feng, L., et al. (2012). Pterostilbene Simultaneously Induces Apoptosis, Cell Cycle Arrest and Cyto-Protective Autophagy in Breast Cancer Cells. Am. J. Transl. Res. 4, 44–51.
Wend, P., Runke, S., Wend, K., Anchondo, B., Yesayan, M., Jardon, M., et al. (2013). WNT10B/β‐catenin Signalling Induces HMGA2 and Proliferation in Metastatic Triple‐negative Breast Cancer. EMBO Mol. Med. 5, 264–279. doi:10.1002/emmm.201201320
Xu, J., Koval, A., and Katanaev, V. L. (2020). Beyond TNBC: Repositioning of Clofazimine against a Broad Range of Wnt-dependent Cancers. Front. Oncol. 10, 602817. doi:10.3389/fonc.2020.602817
Yang, L., Perez, A. A., Fujie, S., Warden, C., Li, J., Wang, Y., et al. (2014). Wnt Modulates MCL1 to Control Cell Survival in Triple Negative Breast Cancer. BMC Cancer 14, 124. doi:10.1186/1471-2407-14-124
Yang, L., Wu, X., Wang, Y., Zhang, K., Wu, J., Yuan, Y.-C., et al. (2011). FZD7 Has a Critical Role in Cell Proliferation in Triple Negative Breast Cancer. Oncogene 30, 4437–4446. doi:10.1038/onc.2011.145
Keywords: chemoenzymatic synthesis, Botrytis cinerea, enzymatic secretome, stilbene dimers, high-resolution semi-preparative HPLC, dry load introduction, Wnt inhibition, triple negative breast cancer cells
Citation: Huber R, Koval A, Marcourt L, Héritier M, Schnee S, Michellod E, Scapozza L, Katanaev VL, Wolfender J-L, Gindro K and Ferreira Queiroz E (2022) Chemoenzymatic Synthesis of Original Stilbene Dimers Possessing Wnt Inhibition Activity in Triple-Negative Breast Cancer Cells Using the Enzymatic Secretome of Botrytis cinerea Pers.. Front. Chem. 10:881298. doi: 10.3389/fchem.2022.881298
Received: 22 February 2022; Accepted: 18 March 2022;
Published: 19 April 2022.
Edited by:
Simone Brogi, University of Pisa, ItalyReviewed by:
Barbara De Filippis, University of Studies G. d’Annunzio Chieti and Pescara, ItalyGabriella Massolini, University of Pavia, Italy
Letizia Giampietro, University G. d’Annunzio, Italy
Paulo Castro, University of Santiago, Chile
Copyright © 2022 Huber, Koval, Marcourt, Héritier, Schnee, Michellod, Scapozza, Katanaev, Wolfender, Gindro and Ferreira Queiroz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vladimir L. Katanaev, VmxhZGltaXIuS2F0YW5hZXZAdW5pZ2UuY2g= Katia Gindro, a2F0aWEuZ2luZHJvQGFncm9zY29wZS5hZG1pbi5jaA== Emerson Ferreira Queiroz, ZW1lcnNvbi5mZXJyZWlyYUB1bmlnZS5jaA==
†These authors have contributed equally to this work and share last authorship